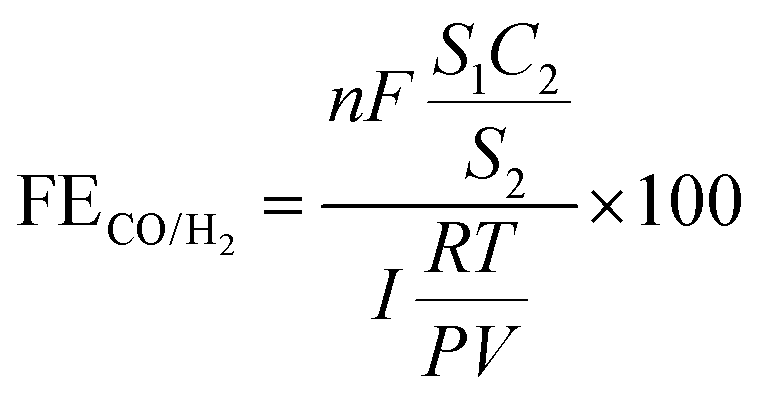DOI:
10.1039/D4NR00340C
(Paper)
Nanoscale, 2024,
16, 10628-10636
Easily constructed porous silver films for efficient catalytic CO2 reduction and Zn–CO2 batteries†
Received
23rd January 2024
, Accepted 22nd April 2024
First published on 23rd April 2024
Abstract
For the electroreduction of carbon dioxide into high value-added chemicals, highly active and selective catalysts are crucial, and metallic silver is one of the most intriguing candidate materials available at a reasonable cost. Herein, through a novel two-step operation of Ag paste/SBA-15 coating and HF etching, porous silver films on a commercial carbon paper with a waterproofer (p-Ag/CP) could be easily fabricated on a large scale as highly efficient carbon dioxide reduction reaction (CO2RR) electrocatalysts with a CO Faraday efficiency (FECO) as high as 96.7% at −1.0 V vs. the reversible hydrogen electrode (RHE), and it still reaches up to 90% FECO over applied potentials ranging from −0.8 to −1.1 V vs. the RHE. Meanwhile, the membrane electrode assembly (MEA) utilizing the p-Ag/CP catalyst has achieved a current density, FECO, and stability of ∼60 mA cm−2, >91%, and 11 h, respectively. Furthermore, the assembled aqueous Zn–CO2 battery using p-Ag/CP cathode yielded a peak power density of 0.34 mW cm−2, 75 charge–discharge cycles for 25 h, and 64% FECO at 2.5 mA cm−2. Compared with flat Ag/CP, the significant enhancement in the CO2RR activity of p-Ag/CP was mainly attributed to the distinctive porous structure and an improved three-phase boundary, which is capable of inducing the stabilization of *COOH intermediates, increased active specific surface areas, fast electron transfer kinetic and mass transportation. Further, theoretical calculations revealed that p-Ag/CP possessed an optimized energy barrier for *COOH intermediates.
1. Introduction
The electrochemical carbon dioxide reduction reaction (CO2RR) represents an attractive avenue for converting greenhouse gases into value-added fuels and chemicals, contributing to an affordable carbon-neutral energy cycle.1–3 Owing to the multi-electronic reaction mechanism of the CO2RR, the products are diverse ranging from C1 to C2 (e.g., CO, CH4, CH3OH, HCOOH; C2H4, C2H6, C2H5OH, and CH3COOH).4 Among those electroreduction derivatives, liquid products normally experience lower Faraday efficiencies and complicated separation processes, whereas gaseous CO is one of the more favorable derivative since it is more efficiently separated and purified and further serves as a starting material for high-value-added fuels or chemicals.5 Meanwhile, in order to overcome higher energy barriers and the undesired hydrogen evolution reaction (HER) as a side reaction during CO2-to-CO conversion, the development of a type of electrocatalyst that is easy to prepare, economical, stable, and highly active is crucial for achieving the practical application of this technology.6,7
In 1994, Hori et al. found a series of bulk metal electrodes with a higher CO selectivity, i.e., Au > Ag > Cu > Zn.8 Through comparison of the volcano plot of their *COOH binding energy, which usually describes the possibility of CO production, it can be further observed that Au and Ag appear at the top, implying that they have the most selectivity for CO generation.9 Therefore, mechanistic investigations and experimental data co-corroborate this essential result that Au/Ag are preeminent candidates. For polycrystalline Au and its derivatives, several kinds of catalytic materials have been identified for CO2 conversion. For instance, Au nanoparticles measuring 8 nm exhibited a faradaic efficiency (FE) up to 90% (−0.67 V vs. the reversible hydrogen electrode (RHE)),10 the coralloid Au constructed via electrodeposition achieved 94.2% CO faradaic efficiency (FECO),11 and 5-mercapto-1-methyltetrazole modified 4H/fcc Au nanorods delivered an excellent CO selectivity of 95.6%.12 Compared to the scarcity and high expense of metallic gold, metallic silver has garnered significant interest owing to its cost-effectiveness and inherent reactivity towards the CO2RR.13 Crucially, the morphology optimization or nanostructural features of Ag catalysts have a remarkable contribution to the electrocatalytic CO2RR.14 Ma et al. discovered that nanostructured Ag tends to create a high local pH on its surface, which is believed to aid in promoting the catalytic activity (∼80% FECO) as well as hindering HER.15 To achieve greater precision in regulating nanostructures, Luan et al. and colleagues designed Ag nanowire arrays with 200 nm in diameter, which displayed over 90% FECO, much higher than that of the original Ag foil.16 Apart from the above-mentioned, various other nanostructures, including dealloying nanoporous Ag,17 plasma engineering microrod array,18 nanoparticles,19 and alloyed Pd–Ag nanowires,20 have also demonstrated outstanding electrocatalytic performances for CO or other hydrocarbons.
In addition, enhancing CO2 electroreduction activity entails considering the three-phase boundary at the interface of the electrolyte and catalyst as another critical operating parameter, except the intrinsic activity of the catalyst, including the mass diffusion of the reactants (i.e., CO2, H+, and H2O) near the catalyst interface, the construction of gaseous diffusion layer (GDL), and application of membrane electrode assembly (MEA) and flow cell electrolyzers, etc.21 The artificial alveolar bilayer structure with high CO2 gas permeability engineered by Li et al. exhibited the capacity to extensively suppress the generation of H2 and a remarkable FECO of 92% at −0.6 V.22 In the CO2RR work reported by Dinh et al., the high selectivity and activity (70% FE of C2H4, 150 h) were achieved at the abrupt reaction interface (a polymer-based GDL for optimizing the diffusion CO2 into the catalytic sites).23 Catalyst-coated GDLs are arranged on both sides of a membrane in MEA electrolyzers, with one side acting as an anode and the other as a cathode. Liquid electrolytes or CO2 gas reactants flow over the corresponding electrodes to take part in the reaction. The porous GDL structure enhances the effective contact between CO2 molecules, the catalyst layer, and the proton source, leading to an increase in current density.24
Considering the significance of nanostructured Ag-based materials and the three-phase interfacial structure of electrodes (CO2 (gas), H2O (liquid), and catalyst (solid)) in enhancing CO2RR performance, we used commercial carbon paper with a hydrophobic layer as a conductive substrate to prepare porous Ag-coated electrodes (p-Ag/CP). The constructed p-Ag/CP demonstrates high efficiency for CO2 reduction to CO with remarkable selectivity (FECO > 90%) across a broad potential region (−0.8 to −1.1 V vs. RHE) as well as chronopotentiometry stability of 27 h. Meanwhile, the FECO of the MEA can be enhanced up to approximately >91% at ∼60 mA cm−2. The rechargeable Zn–CO2 cell demonstrates a power density of 0.34 mW cm−2 and achieves stability throughout over 75 charge–discharge cycles. Contrary to the conventional method of fabricating nano-sized Ag-based materials, self-supporting electrodes produced through a straightforward coating-etching procedure not only mitigate superfluous waste of the precious metal but also avoid the covering of active sites by the Nafion binder. In comparison to that of the flat Ag/CP, the higher catalytic activity of p-Ag/CP might be related to a highly curved surface (stabilized CO2− intermediates),17 the construction of porous structure (increased active specific surface areas) and three-phase boundary (improved mass transportation).
2. Experimental section
Synthesis of p-Ag/CP and Ag/CP
First, the carbon paper (CP, HCP120, Hensen) with 1.0 × 2.0 cm2 geometric areas was ultrasonically treated with acetone and ethanol for 3 h in turn and then dried at 100 °C in the oven for 24 h to completely remove adsorbed impurities. Next, conductive silver paste (DJ002, mechanic) was evenly coated on the hydrophilic surface (black, 1.0 × 1.0 cm2) of CP, while the hydrophobic side (gray) of the CP was retained to ensure the diffusion of CO2 molecules. Third, SBA-15 (XFF01, XFNANO) template powder was continued to be covered in the silver film, and the 50 mL autoclave was placed on the CP for 10 min to ensure that the two were fully mixed. Fourth, Ag-SBA-15/CP was transferred to an oven and cured at 160 °C for 12 h, and after natural cooling to room temperature, the SiO2 template was removed by etching with 5 vol% HF (≥40.0%, AR, Sinopharm Chemical Reagent Co., Ltd) for 2 h to obtain p-Ag/CP catalysts. For comparison, using a similar preparation procedure, Ag/CP without the SBA-15 template was also constructed.
Physical characterizations
The microstructure and morphology features of as-prepared catalysts were investigated via scanning electron microscopy (SEM, Quanta FEG 250) equipped with energy dispersive X-ray (EDX) spectroscopy modules and transmission electron microscopy (TEM, Talos F200X).
The BET-specific surface area was obtained by N2 adsorption at 77 K using a Micromeritics ASAP 2020 specific surface area analyzer. Based on the Barrett–Joyner–Halenda method, the corresponding pore size distribution plots can be achieved. X-ray diffraction (XRD, Bruker D8 instrument) characterization was carried out to obtain the crystalline structure information. The surface elemental composition of the samples was further characterized through the application of X-ray photoelectron spectroscopy (XPS, ESCALAB 250Xi, Thermo Scientific).
Electrochemical tests
Through the CHI760e electrochemical workstation, all CO2RR electrochemical experiments were conducted on a classic three-electrode system in an H-type reactor with a Nafion 117 membrane. The prepared p-Ag/CP was utilized as the working electrode directly, with the Ag/AgCl (saturated KCl) and a Pt foil (1.0 × 1.0 cm2) as the reference electrode and the counter electrode, respectively. The two separated compartments were filled with 30 mL of 0.1 M KHCO3 electrolyte, and CO2 or Ar (99.999%) with a rate of 30 ml min−1 was introduced into the catholyte at least 30 min prior to CO2RR tests. The linear sweep voltammetry (LSV) curves were recorded with a sweep rate of 5 mV s−1 without IR compensation. Electrochemical impedance spectroscopy (EIS) analysis at an open circuit potential was performed in the frequency range of 10−1 Hz–105 Hz. The electrochemical double-layer capacitance (Cdl) is calculated by recording cyclic voltammetry (CV) curves in the non-faradaic region (0.25–0.35 V vs. RHE) with different scan rates from 20–120 mV s−1. The calculation formula of the electrochemical surface areas (ECSA) is ECSA = (Cdl/Cs) × A, where Cs represents the specific capacitance and A represents the area of the working electrode. Therefore, the Cdl values are positively correlated to the ECSA.25,26 All the potentials in this work have been calculated for RHE based on the following equation, and the pH of CO2-saturated 0.1 M KHCO3 was 6.8:
| ERHE = EAg/AgCl + 0.059 × pH + 0.198 |
The faradaic efficiency for gaseous products of CO/H2 was obtained using the equation:5
where
n is the number of transferred electrons (
n = 2),
V is the gas flow rate,
S1 is the product peak area,
C2 is the standard gas product concentration, and
S2 is the standard gas product peak area.
I is the total current measured during gas collection.
P = 101
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
325 Pa,
F = 96
485 C mol
−1,
R = 8.314 J mol
−1 K
−1, and
T = 298.15 K. The CO/H
2 gas in the cathodic compartment was vented into gas chromatography (GC9790 Plus, FULI instruments) with a flame ionization detector (FID) for CO and a thermal conductivity detector (TCD) for H
2. Moreover, to evaluate the oxygen evolution reaction (OER) performance of samples in CO
2-saturated 0.8 M KHCO
3, the three-electrode setup similar to CO
2RR was applied, and the corresponding data was corrected using 90% IR compensation.
Zn–CO2 battery tests
An aqueous rechargeable Zn–CO2 electrochemical cell was assembled by placing p-Ag/CP as the cathode into the catholyte (CO2-saturated 0.8 M KHCO3) and Zn foil (2.0 × 5.0 cm2) as the anode into anolyte (6 M KOH + 0.2 M Zn(CH3COO)2) in an H-cell. The various electrolytes mentioned above were separated by a bipolar membrane (FBM-PK, Fumasep). Before the test, the 0.5 M KHCO3 catholyte was saturated with CO2, and the CO2 (30 ml min−1) was continuously injected into the cathode chamber during the measurement.
The overall reaction of the discharge and charge process could be described as:27
| Zn + CO2 + 2H+ + 4OH− → Zn(OH)42− + CO + H2O, |
| Zn(OH)42− + H2O → Zn + 1/2O2 + 2H+ + 4OH− |
MEA electrolyzer tests
For the MEA test, this device (working area: 1.0 cm2) was purchased from Shanghai ChuXi. An anion exchange membrane (FAS-PET-130) was sandwiched between the cathode (p-Ag/CP with GDS3250) and anode (stainless steel mesh). During electrolysis, the CO2 ingredient (70 mL min−1) and 1.0 M KOH aqueous solution were successively imported to the cathode and anode regions, respectively.28
Density functional theory (DFT) calculations
The Ag crystal structure information from the ICSD database and the Device Studio program (Hongzhiwei Technology (Shanghai) Co. Ltd) provides a number of functions for performing visualization, modeling, and simulation.29,30 Calculations are then carried out via the DS-PAW software,30 which employs the generalized gradient approximation (GGA) in the Perdew–Burke–Ernzerhof (PBE) generalization to deal with exchange–correlation interactions between electrons.30 During the configuration optimization, a 6 × 6 × 1 Gamma-centered k-point grid was used, and the energy cut-off was set to be 500 eV for energy and 0.02 eV Å−1 for force. The Ag (111) and (200) model was constructed by a 2 × 2 × 1 supercell with a vacuum region of 15 Å. Free energy for each reaction step was calculated as G = EDFT + EZPE − TΔS, where EDFT is the DFT calculated energy.
3. Results and discussion
Synthesis and structural characterization
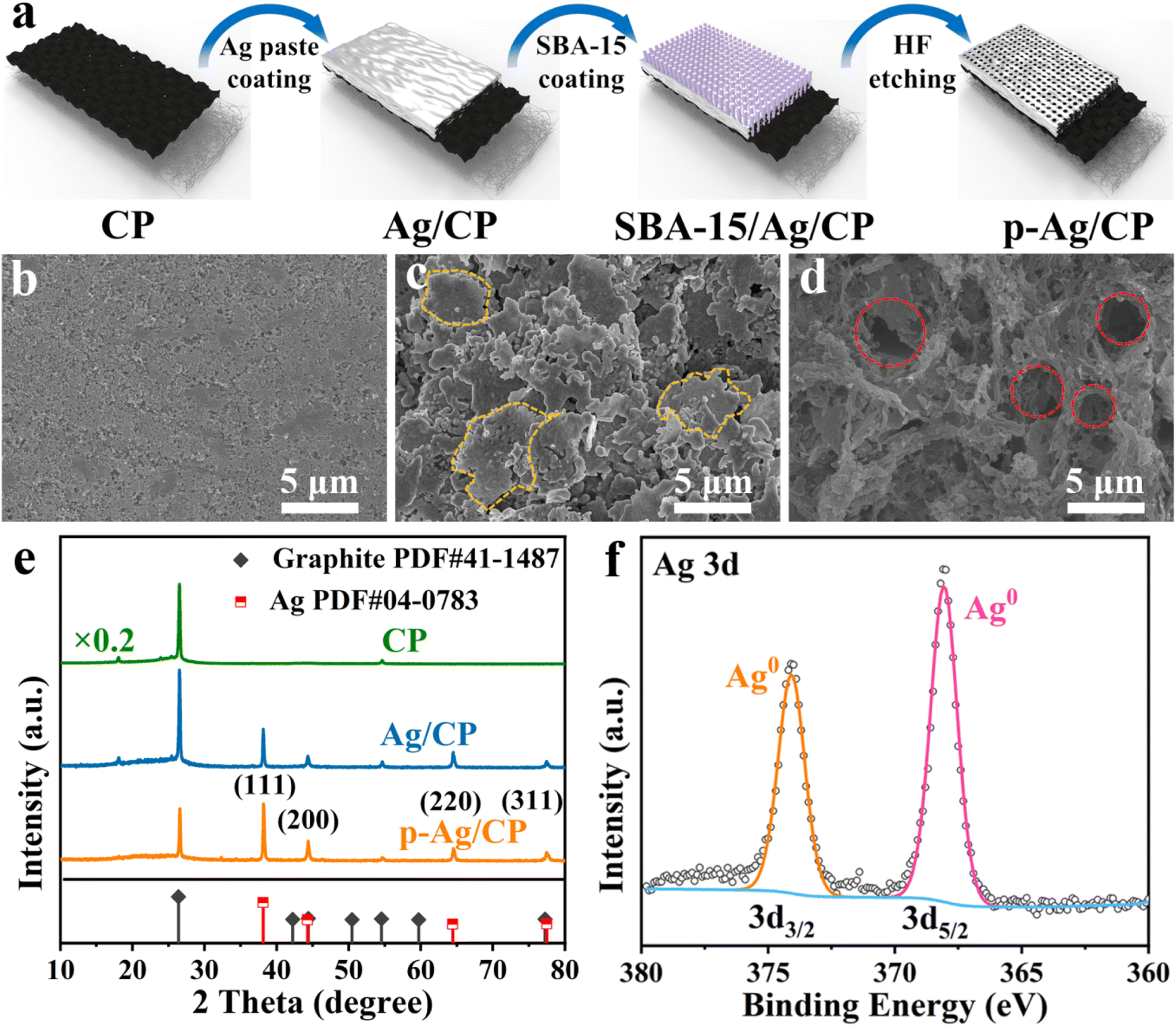
Fig. 1a depicts a straightforward preparation route. Initially, the conductive silver paste and SBA-15 template are coated in succession to the hydrophilic side of CP and then cured at high temperatures. Subsequently, the templates are eliminated using HF to produce p-Ag/CP with optimized porous architecture and a three-phase interface. The corresponding optical photos of them can be acquired in Fig. S1.† Through typical SEM images (Fig. 1b–d and S2†), the micro-morphological evolution of these as-synthesized samples is further investigated. Images of the hydrophilic side of the commercial CP (Fig. 1b and S2a, d†) reveal that the uneven surface comprises aggregated nanoparticles with diameters ranging from ∼55 to ∼78 nm, while the existing channels or crevices (diameter: 60–120 nm) between the nanoparticles serve to facilitate quick gas transfer to the hydrophobic side (i.e., the waterproofing layer) and to enable effective contact between the catalyst and conductive substrate.31 As expected, the silver paste of Ag/CP is able to adhere well to the CP without shedding, and where the Ag films consist mainly of a number of micron-sized unevenly sized flakes stacked on top of each other (Fig. 1c and S2b, e†). On the contrary, in p-Ag/CP, under the influence of the SBA-15 template, the Ag films are transformed into another loose porous structure (pore size: 2–4 μm) composed of nanofibres with diameters of several hundred nanometres (Fig. 1d and S2c, f†). The porosity features of the p-Ag/CP samples were analyzed by N2 adsorption measurements (Fig. S3†), which showed a type-IV isotherm with hysteresis loops, demonstrating a BET-specific surface area of 4.53 m2 g−1 and a rich mesoporous distribution. The above results indicate that the porous Ag film can form a balanced three-phase interface (catalyst (solid), H2O (liquid), and CO2 (gas)) with the original gas transfer channel of the CP, thus improving the CO2RR performance.32 The nanofibres of p-Ag/CP were further characterized by TEM (Fig. S4†) and found to contain nano-sized particles, while the elemental distribution confirms that these particles are mainly composed of Ag and that the uniformly distributed F, C, N and O may originate from the residues of HF and the organic curing agent of silver paste. The corresponding Ag loading of p-Ag/CP was obtained as 94.758 wt% via EDX spectroscopy (Table S1†). XRD and XPS are employed to acquire the crystalline phase as well as electronic structure information, respectively. Fig. 1e provides the co-existence of single-phase metallic Ag (PDF#04-0783) and Graphite (PDF#41-1487) toward p-Ag/CP and Ag/CP, where the diffraction peaks at 38.1°, 44.3°, 64.4° and 77.5° corresponds to the (111), (200), (220), and (311) planes of metallic Ag, while the three remaining peaks located at 18.1°, 26.4°, and 54.5° originate from the CP substrate. Before determining the elemental chemical state and composition of the surface, the C 1s peak at 284.8 eV is ascribed to calibrate all the original XPS data (Fig. S5a†).33 In the Ag 3d spectra of Fig. 1f and a pair of peaks with bond energies of 368.1 eV for 3d5/2 and 374.1 eV for 3d3/2 are observed, implying its surface was metallic Ag without any Ag oxide, which is consistent with the XRD characterization of p-Ag/CP.34 Furthermore, the O 1s region in Fig. S5b† could be split into two peaks at 531.8 eV and 532.7 eV, and the lower binding energies are well matched to the oxygen chemisorbed on a silver surface or Ag–O–Ag bonds but not to pure Ag2O,35 while the higher binding energies can mainly be ascribed to contamination from the air (adsorbed O species, hydroxyl, H2O, etc.).36,37
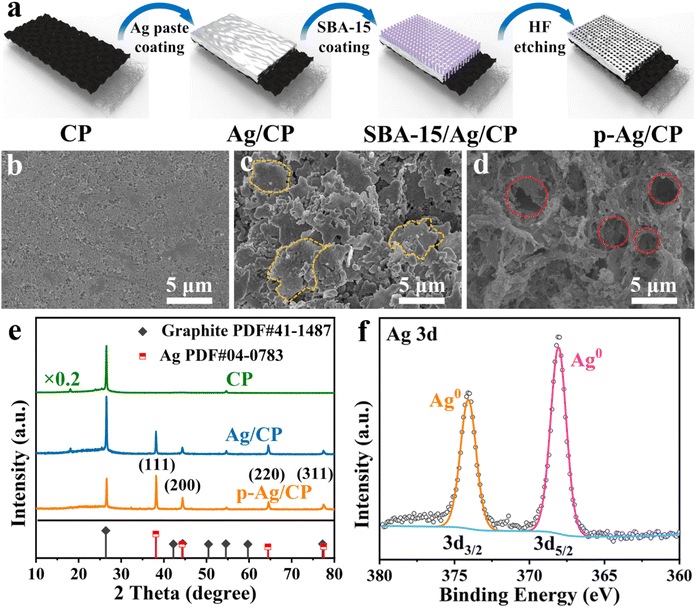 |
| | Fig. 1 (a) Schematic diagram of the synthetic process of p-Ag/CP. SEM images of (b) CP, (c) Ag/CP, and (d) p-Ag/CP. (e) XRD patterns of CP, Ag/CP, and p-Ag/CP. (f) High-resolution Ag 3d XPS spectrum of p-Ag/CP. | |
Electrochemical CO2RR performance
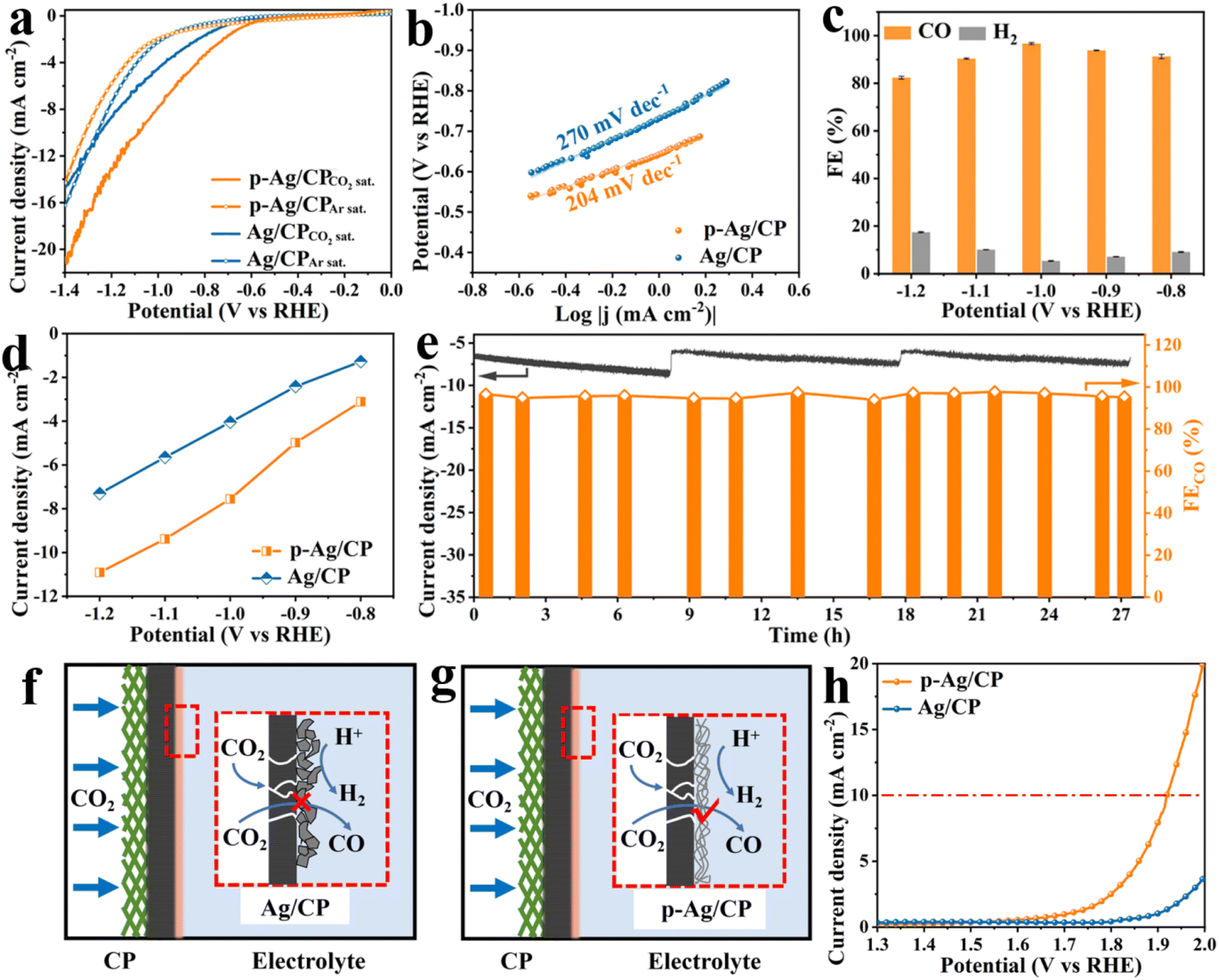
The CO2RR catalytic activities of p-Ag/CP were evaluated in a two-compartment H-cell with 0.1 M CO2-saturated KHCO3 aqueous solution, employing a three-electrode system. In LSV measurements (Fig. 2a), both p-Ag/CP and Ag/CP in CO2-saturated KHCO3 solution illustrate improved current densities as well as positively motivated onset potential, compared with that in Ar-saturated ones, whereas the current densities measured on the CP substrate are almost identical in CO2 as well as Ar (Fig. S6†), meaning that there is no CO2RR activity. Furthermore, the p-Ag/CP demands a lower potential (−1.08 V) than that of Ag/CP (−1.25 V) for reaching the same current density of 10 mA cm−2, suggesting that p-Ag/CP features a superior electrocatalytic activity in CO2RR. Meantime, p-Ag/CP also shows the lowest Tafel slope of 204 mV dec−1 (Fig. 2b) and the fastest electron-transfer capacity of 3.94 Ω (Fig. S7†), superior to that of Ag/CP (270 mV dec−1, 5.18 Ω), indicating that the former presents a favorable kinetics pathway toward CO formation.38,39 To estimate the reduction activity of the samples for CO, chronopotentiometry electrolysis is performed (Fig. S8†), and the corresponding generated gaseous reduction products at different applied potentials, e.g., CO and H2, are quantitatively determined via GC. As predicted, the FECO of p-Ag/CP achieves >90% across a broad application potential range of −0.8 to −1.1 V vs. RHE, peaking (96.7%) at −1.0 V vs. RHE (Fig. 2c), which is much superior to that of Ag/CP (89.1% at −1.0 V vs. RHE, Fig. S9a†), CP (1.78% at −1.2 V vs. RHE, Fig. S9b†), and comparable to or even better than previously reported catalysts (Table S3†). Meanwhile, GC and 1H nuclear magnetic resonance (1H NMR) spectroscopy were employed to confirm the reduction products of p-Ag/CP at −1.0 V. The GC (Fig. S10a†) showed peaks of CO, N2 + O2 and CO2, and no obvious H2 peaks were detected, whereas the 1H NMR spectrum (Fig. S10b†) only showed peaks of the internal standard dimethyl sulfoxide (DMSO) and H2O, both of them indicated that the main product of CO2RR is CO and the by-product is H2 and does not contain other liquid or gas phase products. As shown in Fig. 2d, the CO partial current density of 7.55 mA cm−2 at −1.0 V towards p-Ag/CP, which is ca. 1.86 times higher than that of Ag/CP (4.05 mA cm−2) at the same potential, emphasizing the key role of the porous structure in CO2RR activation. Moreover, the excellent CO2RR durability of the p-Ag/CP electrode is also assessed through a long-term chronopotentiometry test at an optimum potential of −1.0 V vs. RHE, and almost no significant attenuation of both current density and FECO are observed during the 27 h operation (Fig. 2e). After 27 h of continuous operation, its silver loading, specific surface area, microstructure, and crystal structure are nearly unchanged via EDX, N2 adsorption, SEM/TEM, and XRD measurement, respectively (Table S2, Fig. S11, S12, S13, and S14†). The p-Ag/CP catalyst's initial catalytic activity is further investigated using ECSA, which is positively correlated with Cdl from CV data (Fig. S15 and S16†).40,41 Compared with Ag/CP (1.08 mF cm−2), p-Ag/CP presents an enhanced Cdl value (2.49 mF cm−2, 2.31 folds that of Ag/CP), demonstrating the presence of abundant active sites that are exposed and accessible.42 Interestingly, the amount of active sites available might be closely interrelated to their microstructure.43,44 For this purpose, flat Ag/CP and porous p-Ag/CP are first compared to evaluate the influence of the electrode structure (three-phase interface) on electrochemical properties (Fig. 2f, g, and S17†). The hydrophobic layer (i.e., GDL) of CP ensures that the dissolved CO2 gas in solution allows timely diffusion to the catalyst surface, guaranteeing an adequate supply of reaction feedstock. In Ag/CP, the inter-stacked silver sheet structure hinders the mass transport of CO2, leaving most of the catalytic sites exposed only to the liquid electrolyte, which results in fewer desirable CO products and more HER-competitive reactions. In contrast, for p-Ag/CP, the loose and porous structure formed by silver nanofibres allows the gas diffusion path to be lengthened and the exposed active sites to function more efficiently, thus increasing the CO2 utilization, facilitating the escape of the gaseous products and limiting the occurrence of the HER side reactions.21,45,46
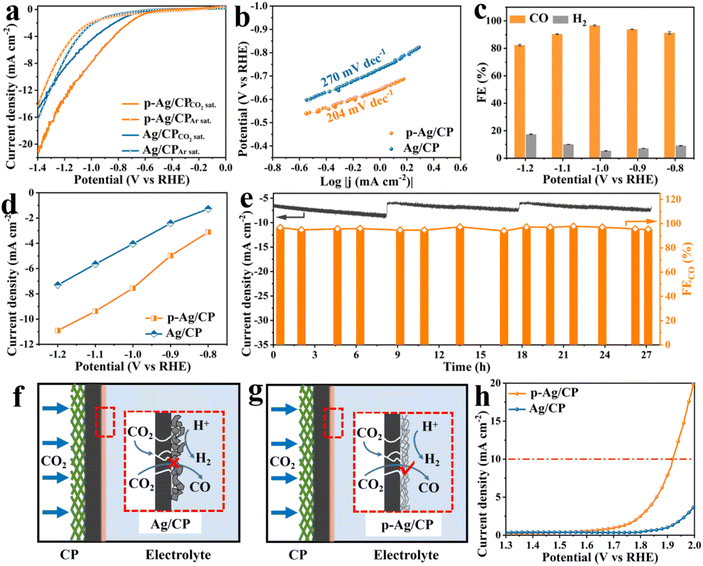 |
| | Fig. 2 (a) LSV curves (5 mV s−1) in Ar- and CO2-saturated 0.1 M KHCO3 solution. (b) Tafel plots, (c) FECO value, and (d) CO partial current densities of different electrodes at various applied potentials. (e) Stability test of p-Ag/CP at −1.0 V vs. the RHE over 27 h (electrolyte replacement at 8 h and 18 h). (f and g) Schematic of the three-phase interface in the CO2RR for Ag/CP and p-Ag/CP. (h) LSV curves (10 mV s−1) in CO2-saturated 0.8 M KHCO3 solution for the OER. | |
Zn–CO2 battery performance
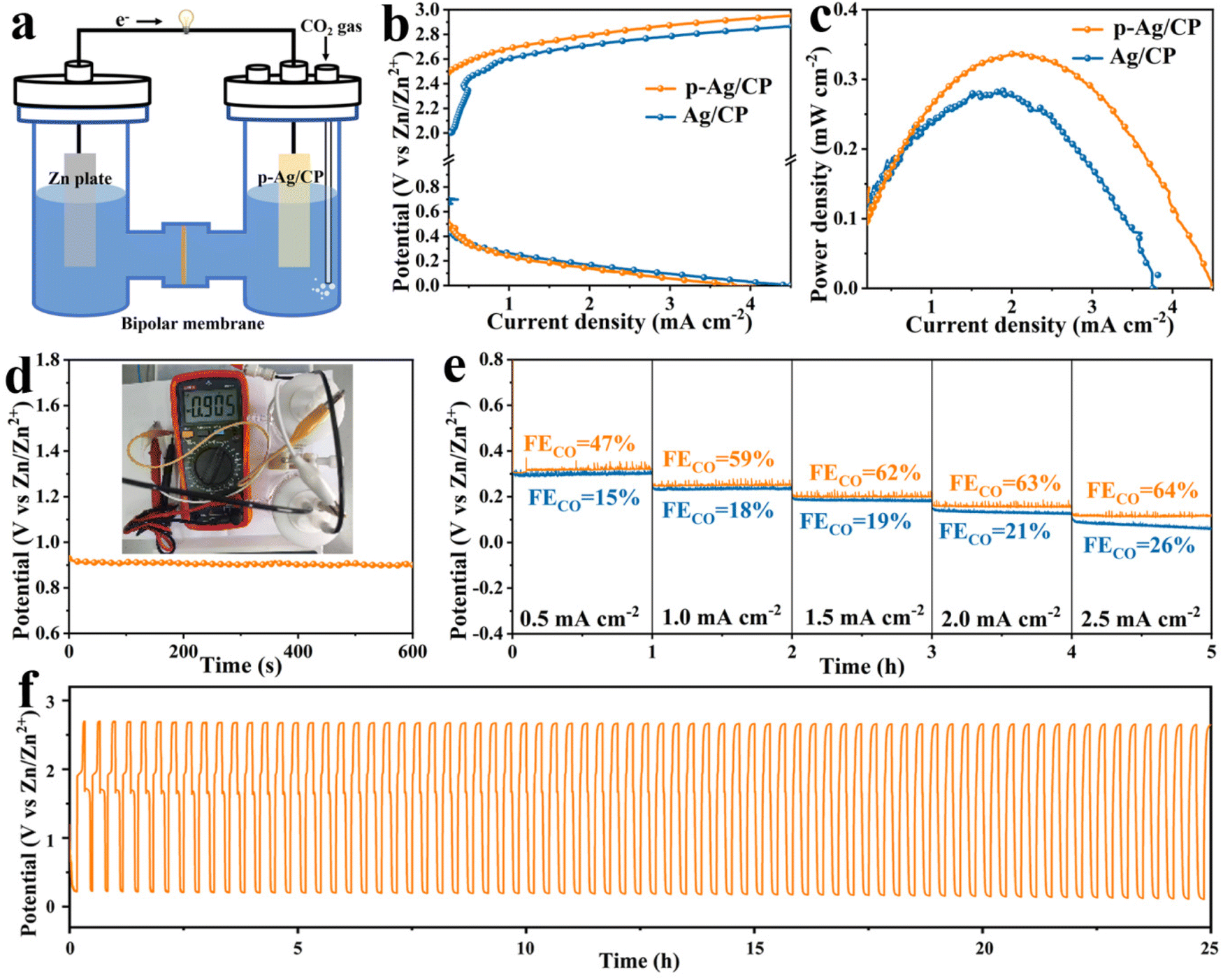
Considering the outstanding CO2RR capability of p-Ag/CP, aqueous rechargeable Zn–CO2 batteries with Zn foil as an anode and p-Ag/CP as a cathode are assembled. Through LSV curves, its OER activity is first investigated. As displayed in Fig. 2h, p-Ag/CP is able to achieve a higher current density of 19.9 mA cm−2 at 2.0 V vs. RHE, whereas Ag/CP only achieves a lower current of 3.6 mA cm−2 at the same potential. Therefore, the bifunctional p-Ag/CP with CO2RR and OER make it a promising cathode material for batteries. In assembled rechargeable Zn–CO2 batteries, the alkaline 6.0 M KOH + 0.2 M Zn(CH3COO)2 is employed as the anolyte, the catholyte employed is a near-neutral solution with 0.8 M of KHCO3, and a bipolar membrane is employed to separate the two compartments and maintain the pH difference between them (Fig. 3a and S18†).47–49 The discharge process entails the oxidation of zinc metal that promotes the reduction of CO2; meanwhile, during the charging process, zinc is deposited at the anode and OER occurs at the cathode.50Fig. 3b shows the charge–discharge curves of the battery based on p-Ag/CP and Ag/CP cathode, where the p-Ag/CP exhibits higher discharge current density and greater power density (0.37 mW cm−2 at 2.1 mA cm−2) than Ag/CP (0.28 mW cm−2 at 1.9 mA cm−2), demonstrating a favorable activation for generating power (Fig. 3c). The open-circuit voltage of ∼0.9 V is shown in Fig. 3d. During the discharge process with a wide current density range of 0.5 mA cm−2 to 2.5 mA cm−2, the corresponding gaseous products in the cathode chamber are quantitatively measured, and the maximum FECO of 64% suggests a promising CO2 to CO conversion capability (Fig. 3e). Furthermore, the rechargeable cycling durability of the Zn–CO2 battery is evaluated, which achieves 75 continuous charge–discharge cycles at 1.0 mA cm−2 over 25 h, demonstrating its stable behavior in Fig. 3f. Unfortunately, the p-Ag/CP electrode in the Zn–CO2 battery underwent a long period of charge/discharge test, with the silver-white surface turning grey and FECO decreasing from the original 96.7% to 20.0% at the optimal potential. This may be attributed to the electrochemical oxidation and irreversible destruction of the microstructure occurring on the catalyst surface as a result of the charging process.
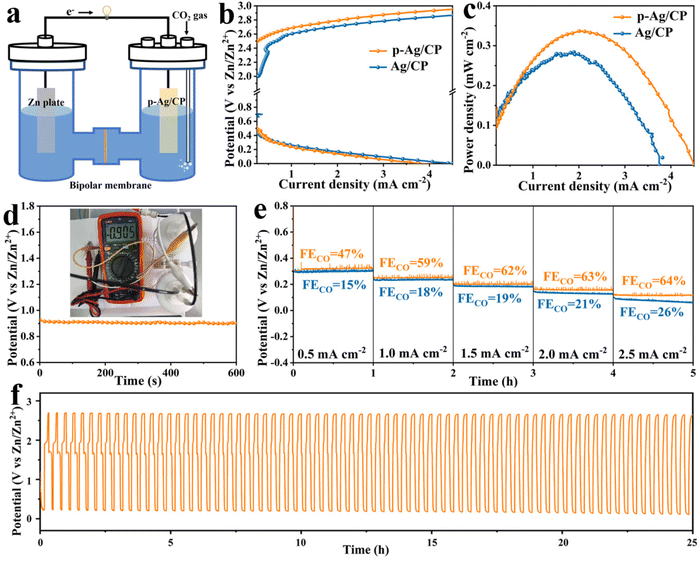 |
| | Fig. 3 (a) Schematic configuration of the Zn–CO2 battery. (b) Discharge and charge polarization curves (10 mV s−1) with the p-Ag/CP and Ag/CP cathode. (c) Power density plots with the p-Ag/CP and Ag/CP cathode. (d) The open-circuit voltage (inset: the corresponding photograph) with the p-Ag/CP cathode. (e) Galvanostatic discharge curves at different current densities and corresponding FECO with the p-Ag/CP and Ag/CP cathode. (f) Galvanostatic charge–discharge cycling curves (1.0 mA cm−2) with the p-Ag/CP cathode. | |
MEA performance and DFT calculations
In traditional H-type electrolytic cells, their overall effectiveness is restricted. To reach CO2RR with industrial CO current density and limited HER side reaction, the MEA system derived from fuel cells is employed to facilitate the electrochemical conversion of CO2 to CO (Fig. 4a and S19†).51 As depicted in Fig. 4b, the polarization curve of p-Ag/CP demonstrated a much higher current response in contrast to N2 ones under CO2 atmospheres and below 2.8 V cell voltage. On the other hand, p-Ag/CP possesses a FECO of almost 100.0% in the range of −2.4 to −2.6 V (Fig. 4c and S20†). The results of the long-term durability tests at −2.6 V for 11 h in Fig. 4d show that the FECO and current density were basically maintained at >91% and ∼60 mAcm−2 without significant degradation, indicating good CO2RR stability, and in consequence, meeting the practical requirements.
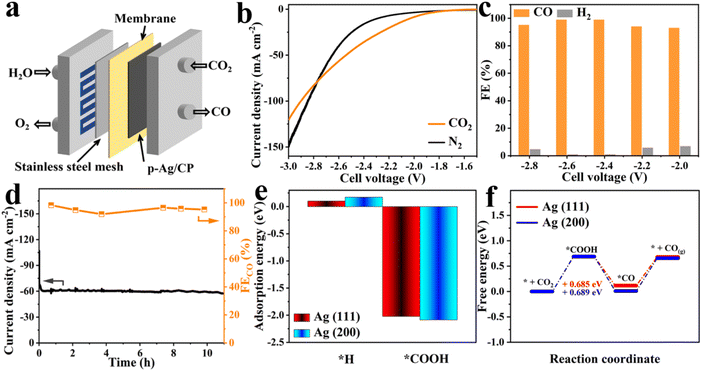 |
| | Fig. 4 (a) Schematic configuration of the MEA instrument. (b) LSV curves (10 mV s−1) with CO2/N2 supplies. (c) FECO at different applied potentials. (d) Stability test at −2.6 V over 11 h. (e) Calculated adsorption energies of *H and *COOH on Ag (111) and (200) surfaces. (f) Calculated free energy profile for the CO2RR of p-Ag/CP. | |
Moreover, the DFT calculation was performed to gain insight into the better CO2RR activity of p-Ag/CP using the DS-PAW package and the Device Studio program.29,30 Based on the XRD results (Fig. 1e), the (111) and (200) facets of Ag were selected as computational models. First, the adsorption energies of *H (ΔE*H) and *COOH (ΔE*COOH) onto the Ag (111) and (200) facets were calculated (Fig. 4e). As a result, the ΔE*COOH (−2.02 and −2.09 eV) has a significantly lower value than ΔE*H (0.10 and 0.17 eV), indicating that the COOH species adsorbed on the surface of the catalysts more efficiently, and thus inhibiting HER on Ag.52,53 The involved fundamental CO2-to-CO conversion mechanism is as follows:51
| CO2 + * + H+ + e− → *COOH, |
| *COOH + H+ + e− → *CO + H2O, |
The Gibbs free energies (ΔG) for CO2-to-CO conversion are illustrated in Fig. 4f, where the generation of the *COOH intermediate (ΔG*COOH) serves as the rate-determining step, which yields the corresponding favorable energy barrier values, i.e., 0.685 eV for Ag (111) and 0.689 eV for Ag (200). Although ΔG*COOH is higher than the ΔG*H values (0.338 eV for Ag (111) and 0.195 eV for Ag (200)) in Fig. S21,† based on the charge-density difference (Fig. S22†), enough electron exchange between the O atoms in *COOH and the Ag atoms took place to enhance the adsorption between them, effectively preventing the occurrence of the HER side reaction.54–56
4. Conclusion
In summary, we develop a strategy for the construction of highly active silver-based catalysts with porous structure (p-Ag/CP) in a facile and cost-effective manner. The p-Ag/CP exhibits superior CO2 conversion to CO over a wide potential range (FECO > 90% from −0.8 V to −1.1 V vs. RHE). By introducing the MEA electrolyzer, the corresponding FECO (>91% at ∼60 mA cm−2) is further enhanced. Moreover, an aqueous rechargeable Zn–CO2 battery with p-Ag/CP demonstrates a power density of 0.34 mW cm−2 and a continuous charge discharge of 75 cycles. Compared to flat Ag/CP, the improved CO2RR activity of p-Ag/CP originated from the porous structure of the silver catalyst and the three-phase interface constructed on the basis of CP. Meanwhile, the DFT calculations further revealed that p-Ag/CP has a lower thermodynamic energy barrier in the conversion of CO2 to CO. This research provides insights into microstructure optimization as an important avenue in devising high-efficient CO2RR electrocatalysts.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22075211), Shenzhen Science and Technology Program (JCYJ20210324115412035, JCYJ20210324123202008, JCYJ20210324122803009 and ZDSYS20210813095534001), Guangdong Foundation for Basic and Applied Basic Research Program (2021A1515110880). We also thank Shenbo Yang (Hongzhiwei Technology (Shanghai) Co. Ltd) for the help and discussions regarding this study.
References
- J. Wu, S. Ma, J. Sun, J. I. Gold, C. Tiwary, B. Kim, L. Zhu, N. Chopra, I. N. Odeh, R. Vajtai, A. Z. Yu, R. Luo, J. Lou, G. Ding, P. J. A. Kenis and P. M. Ajayan, Nat. Commun., 2016, 7, 13869 CrossRef CAS PubMed.
- T. R. Wei, S. S. Zhang, Q. Liu, Y. Qiu, J. Luo and X. J. Liu, Acta Phys.-Chim. Sin., 2023, 39, 2207026 Search PubMed.
- Z. Liu, D. Sun, C. Wang, B. You, B. Li, J. Han, S. Jiang, C. Zhang and S. He, Coord. Chem. Rev., 2024, 502, 215612 CrossRef CAS.
- Z. Gu, H. Shen, L. Shang, X. Lv, L. Qian and G. Zheng, Small Methods, 2018, 2, 1800121 CrossRef.
- W.-B. Wan, Y.-T. Zhou, S.-P. Zeng, H. Shi, R.-Q. Yao, Z. Wen, X.-Y. Lang and Q. Jiang, Small, 2021, 17, 2100683 CrossRef CAS PubMed.
- H. Zhang, G. Meng, Q. Liu, Y. Luo, M. Niederberger, L. Feng, J. Luo and X. Liu, Small, 2023, 19, 2303165 CrossRef CAS PubMed.
- C. Chen, J. F. Khosrowabadi Kotyk and S. W. Sheehan, Chem, 2018, 4, 2571–2586 CAS.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- D. L. Nguyen, Y. Kim, Y. J. Hwang and D. Won, Carbon Energy, 2020, 2, 72–98 CrossRef CAS.
- W. Zhu, R. Michalsky, Ö. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- S. Gao, M. Jin, J. Sun, X. Liu, S. Zhang, H. Li, J. Luo and X. Sun, J. Mater. Chem. A, 2021, 9, 21024–21031 RSC.
- J. Wang, J. Yu, M. Sun, L. Liao, Q. Zhang, L. Zhai, X. Zhou, L. Li, G. Wang, F. Meng, D. Shen, Z. Li, H. Bao, Y. Wang, J. Zhou, Y. Chen, W. Niu, B. Huang, L. Gu, C.-S. Lee and Z. Fan, Small, 2022, 18, 2106766 CrossRef CAS PubMed.
- Z. Xie, Y. Qiu, S. Gao, J. Sun, H. Cao, S. Zhang, J. Luo and X. Liu, ChemElectroChem, 2021, 8, 3579–3583 CrossRef CAS.
- C. Du, P. Li, Z. Zhuang, Z. Fang, S. He, L. Feng and W. Chen, Coord. Chem. Rev., 2022, 466, 214604 CrossRef CAS.
- M. Ma, B. J. Trześniewski, J. Xie and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 9748–9752 CrossRef CAS PubMed.
- C. Luan, Y. Shao, Q. Lu, S. Gao, K. Huang, H. Wu and K. Yao, ACS Appl. Mater. Interfaces, 2018, 10, 17950–17956 CrossRef CAS PubMed.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 3242 CrossRef PubMed.
- Q. Yu, C. Guo, J. Ge, Y. Zhao, Q. Liu, P. Gao, J. Xiao and H. Li, J. Power Sources, 2020, 453, 227846 CrossRef CAS.
- L. Zeng, J. Shi, J. Luo and H. Chen, J. Power Sources, 2018, 398, 83–90 CrossRef CAS.
- N. Han, M. Sun, Y. Zhou, J. Xu, C. Cheng, R. Zhou, L. Zhang, J. Luo, B. Huang and Y. Li, Adv. Mater., 2021, 33, 2005821 CrossRef CAS PubMed.
- H. Jiang, R. Luo, Y. Li and W. Chen, EcoMat, 2022, 4, e12199 CrossRef CAS.
- J. Li, G. Chen, Y. Zhu, Z. Liang, A. Pei, C.-L. Wu, H. Wang, H. R. Lee, K. Liu, S. Chu and Y. Cui, Nat. Catal., 2018, 1, 592–600 CrossRef CAS.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- Y. J. Sa, C. W. Lee, S. Y. Lee, J. Na, U. Lee and Y. J. Hwang, Chem. Soc. Rev., 2020, 49, 6632–6665 RSC.
- Z. Liu, R. Wang, S. Li, Y. Gu, J. Lan, Q. Zhou and W. Xu, Electrochim. Acta, 2022, 412, 140134 CrossRef CAS.
- T. Xiong, Z. Zhu, Y. He, M. S. Balogun and Y. Huang, Small Methods, 2023, 7, 2201472 CrossRef CAS PubMed.
- S. Gao, Y. Liu, Z. Xie, Y. Qiu, L. Zhuo, Y. Qin, J. Ren, S. Zhang, G. Hu, J. Luo and X. Liu, Small Methods, 2021, 5, 2001039 CrossRef CAS PubMed.
- S. Liu, M. Jin, J. Sun, Y. Qin, S. Gao, Y. Chen, S. Zhang, J. Luo and X. Liu, Chem. Eng. J., 2022, 437, 135294 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- Hongzhiwei Technology, Device Studio, Version 2022B, China, (2022) available online: https://iresearch.net.cn/cloudSoftware (accessed on 24 Feb. 2022).
- R. Kas, K. K. Hummadi, R. Kortlever, P. de Wit, A. Milbrat, M. W. J. Luiten-Olieman, N. E. Benes, M. T. M. Koper and G. Mul, Nat. Commun., 2016, 7, 10748 CrossRef CAS PubMed.
- Y. Chen, D. Su, Y. Chen, Z. Zhu and W. Li, Cell Rep. Phys. Sci., 2021, 2, 100602 CrossRef CAS.
- D. Fang, F. He, J. Xie and L. Xue, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2020, 35, 711–718 CrossRef CAS.
- N. J. Firet, M. A. Blommaert, T. Burdyny, A. Venugopal, D. Bohra, A. Longo and W. A. Smith, J. Mater. Chem. A, 2019, 7, 2597–2607 RSC.
- L. Q. Zhou, C. Ling, M. Jones and H. Jia, Chem. Commun., 2015, 51, 17704–17707 RSC.
- Z.-Y. Luo, K.-X. Chen, J.-H. Wang, D.-C. Mo and S.-S. Lyu, J. Mater. Chem. A, 2016, 4, 10566–10574 RSC.
- C. Wang, Q. Zhang, Z. Liu, B. Li, W. Zhao, C. Zhang, S. Jiang, J. Wang, K. Liu and S. He, ChemSusChem, 2024, e202301703, DOI:10.1002/cssc.202301703.
- F. Lü, H. Bao, F. He, G. Qi, J. Sun, S. Zhang, L. Zhuo, H. Yang, G. Hu, J. Luo and X. Liu, Mater. Chem. Front., 2021, 5, 4225–4230 RSC.
- H. Zhang, G. Meng, T. Wei, J. Ding, Q. Liu, J. Luo and X. Liu, Chem. Commun., 2023, 59, 12144–12147 RSC.
- H. Shen, T. Wei, Q. Liu, S. Zhang, J. Luo and X. Liu, J. Colloid Interface Sci., 2023, 634, 730–736 CrossRef CAS PubMed.
- X. Xu, H. Ullah, M. Humayun, L. Li, X. Zhang, M. Bououdina, D. P. Debecker, K. Huo, D. Wang and C. Wang, Adv. Funct. Mater., 2023, 33, 2303986 CrossRef CAS.
- T. Hou, J. Ding, H. Zhang, S. Chen, Q. Liu, J. Luo and X. Liu, Mater. Chem. Front., 2023, 7, 4952–4960 RSC.
- W. Wang, L. Gan, J. P. Lemmon, F. Chen, J. T. S. Irvine and K. Xie, Nat. Commun., 2019, 10, 1550 CrossRef PubMed.
- Z. F. Wang, Y. J. Yan, Y. G. Zhang, Y. X. Chen, X. Y. Peng, X. Wang, W. M. Zhao, C. L. Qin, Q. Liu, X. J. Liu and Z. W. Chen, Carbon Energy, 2023, 5, e306 CrossRef CAS.
- T. Burdyny, P. J. Graham, Y. Pang, C.-T. Dinh, M. Liu, E. H. Sargent and D. Sinton, ACS Sustainable Chem. Eng., 2017, 5, 4031–4040 CrossRef CAS.
- Z. Xing, L. Hu, D. S. Ripatti, X. Hu and X. Feng, Nat. Commun., 2021, 12, 136 CrossRef CAS PubMed.
- X. Wang, J. Xie, M. A. Ghausi, J. Lv, Y. Huang, M. Wu, Y. Wang and J. Yao, Adv. Mater., 2019, 31, 1807807 CrossRef PubMed.
- W. Wu, Z. Han, F. Zhang, P. Liu and J. Li, China Powder Sci. Technol., 2024, 30, 56–65 Search PubMed.
- H. Wu, G. Fei, X. Gao, X. Guo, X. Gong, X. Ma, Q. Wang and S. Xu, China
Powder Sci. Technol., 2023, 29, 70–80 Search PubMed.
- T. Wang, X. Sang, W. Zheng, B. Yang, S. Yao, C. Lei, Z. Li, Q. He, J. Lu, L. Lei, L. Dai and Y. Hou, Adv. Mater., 2020, 32, 2002430 CrossRef CAS PubMed.
- H.-Y. Jeong, M. Balamurugan, V. S. K. Choutipalli, E.-s. Jeong, V. Subramanian, U. Sim and K. T. Nam, J. Mater. Chem. A, 2019, 7, 10651–10661 RSC.
- J. Ge, T. Wei, J. Ding, Z. Wang, Q. Liu, G. Qi, G. Hu, J. Luo and X. Liu, ChemCatChem, 2023, 15, e202300795 CrossRef CAS.
- L. Zhang, F. Mao, L. R. Zheng, H. F. Wang, X. H. Yang and H. G. Yang, ACS Catal., 2018, 8, 11035–11041 CrossRef CAS.
- B. Seger, M. Robert and F. Jiao, Nat. Sustain., 2023, 6, 236–238 CrossRef.
- Y. Ji, Z. Yu, L. Yan and W. Song, CHN Powder Sci. & Tec., 2023, 29, 100–107 Search PubMed.
- S. Chen, G. Qi, R. Yin, Q. Liu, L. Feng, X. Feng, G. Hu, J. Luo, X. Liu and W. Liu, Nanoscale, 2023, 15, 19577–19585 RSC.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Tong
Hou
b,
Weijia
Liu
b,
Tong
Hou
b,
Weijia
Liu