
Perovskite CoSn(OH)6 nanocubes with tuned d-band states towards enhanced oxygen evolution reactions†
Mingwei
Sun
ab,
Baopeng
Yang
c,
Jiaxing
Yan
b,
Yulong
Zhou
b,
Zhencong
Huang
b,
Ning
Zhang
 *b,
Rong
Mo
*a and
Renzhi
Ma
*d
*b,
Rong
Mo
*a and
Renzhi
Ma
*d
aHunan Key Laboratory for Micro–Nano Energy Materials and Devices, School of Physics and Optoelectronics, Xiangtan University, Hunan 411105, P. R. China. E-mail: morong@xtu.edu.cn
bSchool of Materials Science and Engineering, Central South University, Changsha 410083, China. E-mail: nzhang@csu.edu.cn
cSchool of Physics and Electronics, Central South University, Changsha 410083, China
dResearch Center for Materials Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan. E-mail: ma.renzhi@nims.go.jp
First published on 2nd May 2024
Abstract
The CoSn(OH)6 perovskite hydroxide is a structure stable and inexpensive electrocatalyst for the oxygen evolution reaction (OER). However, the OER activity of CoSn(OH)6 is still unfavorable due to its limited active sites. In this work, an Fe3+ doping strategy is used to optimize the d-band state of the CoSn(OH)6 perovskite hydroxide. The CoSn(OH)6 catalyst with slightly Fe3+ doped nanocubes is synthesized by a facile hydrothermal method. Structure characterization shows that Fe3+ ions are incorporated into the crystal structure of CoSn(OH)6. Owing to the regulation of the electronic structure, CoSn(OH)6-Fe1.8% exhibits an OER overpotential of 289 mV at a current density of 10 mA cm−2 in OER electrochemical tests. In situ Raman spectroscopy shows that no obvious re-construction occurred during the OER for both CoSn(OH)6 and CoSn(OH)6-Fe1.8%. DFT calculations show that the introduction of Fe3+ into CoSn(OH)6 can shift the d-band center to a relatively high position, thus promoting the OER intermediates’ adsorption ability. Further DFT calculations suggest that incorporation of an appropriate amount of Fe3+ into CoSn(OH)6 significantly reduces the rate-determining Gibbs free energy during the OER. This work offers valuable insights into tuning the d-band center of perovskite hydroxide materials for efficient OER applications.
1. Introduction
The intensifying energy crisis, driven by excessive fossil fuel consumption, is becoming more serious.1,2 In response to this challenge, electrochemical water decomposition has gained widespread attention as a sustainable energy conversion technology.3,4 The electrocatalytic process of water decomposition to produce hydrogen is seen as a promising technique achieving green and sustainable hydrogen energy.5,6 The main challenge in this process lies in the high dynamic barrier of the four-electron-transfer oxygen evolution reaction (OER) at the anode.7 Currently, precious metal-based catalysts like RuO2 and IrO2 are considered benchmarks for industrial OER catalysts.5,8 However, their limited reserves and high costs constrain their commercial application.9–11 Therefore, the development of durable, stable, and highly active non-noble metal electrocatalysts holds great significance in addressing these challenges.12,13Non-precious transition-metal-based catalysts such as phosphates, sulfides, nitrides, and hydroxides have received much attention due to their inexpensiveness, abundance, and unique electrochemical properties.14–18 Among them, transition metal hydroxides, such as layered transition metal hydroxides (e.g. Co(OH)2, Ni(OH)2, NixFe1−x(OH)2etc.), have been widely studied as electrocatalysts for the OER.19–21 However, the layered transition metal hydroxides are easily converted into oxyhydroxides during the OER process, which possibly causes their instability and reduced activity.22,23 The perovskite type hydroxides, such as CoSn(OH)6, MgSn(OH)6, CdSn(OH)6, etc., also have attracted attention.24 The perovskite type hydroxides are chemically stable, and usually show relatively inert phase transformation to oxyhydroxides during the OER process.25 Furthermore, the perovskite structure and the contained special metals such as Sn, Ti, Fe, and Co can bring special electronic structures, which will enable unique electrochemical behaviors.26 As a typical perovskite hydroxide, CoSn(OH)6 has electrochemical activity and a stable chemical structure, and is widely studied as an electrocatalyst for the OER. For example, Chen et al. reported that the CoSn(OH)6 perovskite hydroxide with Sn vacancies shows improved adsorption and electron transfer during the electrocatalytic OER process.27 Wang et al. reported single-crystal CoSn(OH)6 nanoboxes with a porous shell for improved OER properties.28 Liu and co-workers demonstrated that CoSn(OH)6 can be transformed into amorphous CoSnOx materials, which have promoted the OER process.24 Although these processes have been achieved with perovskite structure materials, the OER activity of perovskite CoSn(OH)6 materials is still too low to be applied in practical water splitting.29
Modulating the electronic d-band center of catalysts has been realized as one of the most effective strategies to optimize the energy of the binding intermediates.30–34 Precisely controlling the location, width, and electron count of the d-band center enables adjustment of intermediates’ adsorption strength and reactivity, thereby optimizing the performance of OER catalysts.35,36 For instance, NiO exogenously doped with Li+,37 Ni3Ge2O5(OH)4 with doped Fe3+,38 and Ni–Fe layered double hydroxides (LDH) with doped Ce3+ have been developed to optimize the d-band states for efficient OER applications.39 Unfortunately, tuning the d-band center of perovskite based electrocatalytic materials has been seldom reported yet.
In this work, CoSn(OH)6 nanocubes with a tuned d-band center were synthesized for promoting OER behaviors. The CoSn(OH)6 nanocubes with different amounts of doped Fe3+ were synthesized by a simple hydrothermal process. X-Ray diffraction (XRD) spectroscopy, scanning electron microscopy (SEM), transmission electron microscopy (TEM), energy dispersive X-ray spectroscopy (EDX), and X-Ray photoelectron spectroscopy (XPS) prove that the crystal structure, morphology and chemical composition of the CoSn(OH)6 nanocubes with proper Fe3+ doped. During the electrocatalytic measurements, CoSn(OH)6 doped with 1.8 wt% Fe3+ exhibited optimized OER activity to deliver 10 mA cm−2 at an overpotential of 289 mV. Such a performance is much superior to that of the corresponding pristine CoSn(OH)6. To further reveal the intrinsic mechanism of the OER behaviors of these materials, the electronic states of the d-band are calculated using density functional theory (DFT). With the increase of the doping content of Fe3+, the d-band center of CoSn(OH)6 increased. Such a change brought easier adsorption of OER intermediates. The work reported here has given a facile and efficient strategy to promote the OER activity of perovskite transition metal-based hydroxides.
2. Experimental
2.1 Synthesis of CoSn(OH)6
Nanocrystals of the perovskite hydroxide CoSn(OH)6 were prepared by a hydrothermal method. In a typical procedure, Na2SnO3 (1.0 mmol) and CoCl2·6H2O (1.5 mmol) were dissolved in 20 mL of deionized water separately. Then, the above solutions were mixed and stirred for 10 min. Afterward, the mixed solution was transferred into a sealed Teflon-lined stainless-steel autoclave and subsequently heated in an electronic oven at 200 °C for 24 h. After cooling to room temperature, the products were washed several times using distilled water and ethanol. Finally, the products were dried at 60 °C for 12 h. The obtained powders were collected and ground into a fine powder.2.2 Synthesis of Fe3+ doped CoSn(OH)6
The CoSn(OH)6 doped with Fe3+ was prepared by the same process as that for CoSn(OH)6. The difference was that the CoCl2·6H2O solution was replaced by different amounts of mixed solution of FeCl3·6H2O and CoCl2·6H2O in 15 mL of deionized water with the Fe to Co ratios of 0 at%, 2 at%, 5 at%, and 8 at%. The exact composition of Fe, Sn, and Co was confirmed by inductively coupled plasma-optical emission spectrometry (ICP-OES) results as shown in Table S1†. According to the ICP-OES measurements, the as-prepared products were denoted as CoSn(OH)6, CoSn(OH)6-Fe0.9%, CoSn(OH)6-Fe1.8%, and CoSn(OH)6-Fe2.7%, respectively.More experimental details are available in the ESI.†
3. Results and discussion
3.1 Structure characterization
A series of perovskite hydroxides doped with different amounts of Fe3+ were synthesized by hydrothermal methods. The crystal composition of the prepared products was further analyzed by the XRD technique. As shown in Fig. 1a, the XRD peaks of the products are finely indexed to the perovskite CoSn(OH)6 (JCPDS no. 13-0356), suggesting that CoSn(OH)6 materials have been successfully synthesized. The enlarged figure displays the high-resolution image of (200) peaks. It is shown that the (200) peak is shifted to a smaller angle as the Fe3+ doping concentration increased. Such a phenomenon is caused because Fe3+ has a larger anion radius than Co3+ and the cell lattice volume is expanded after doping.40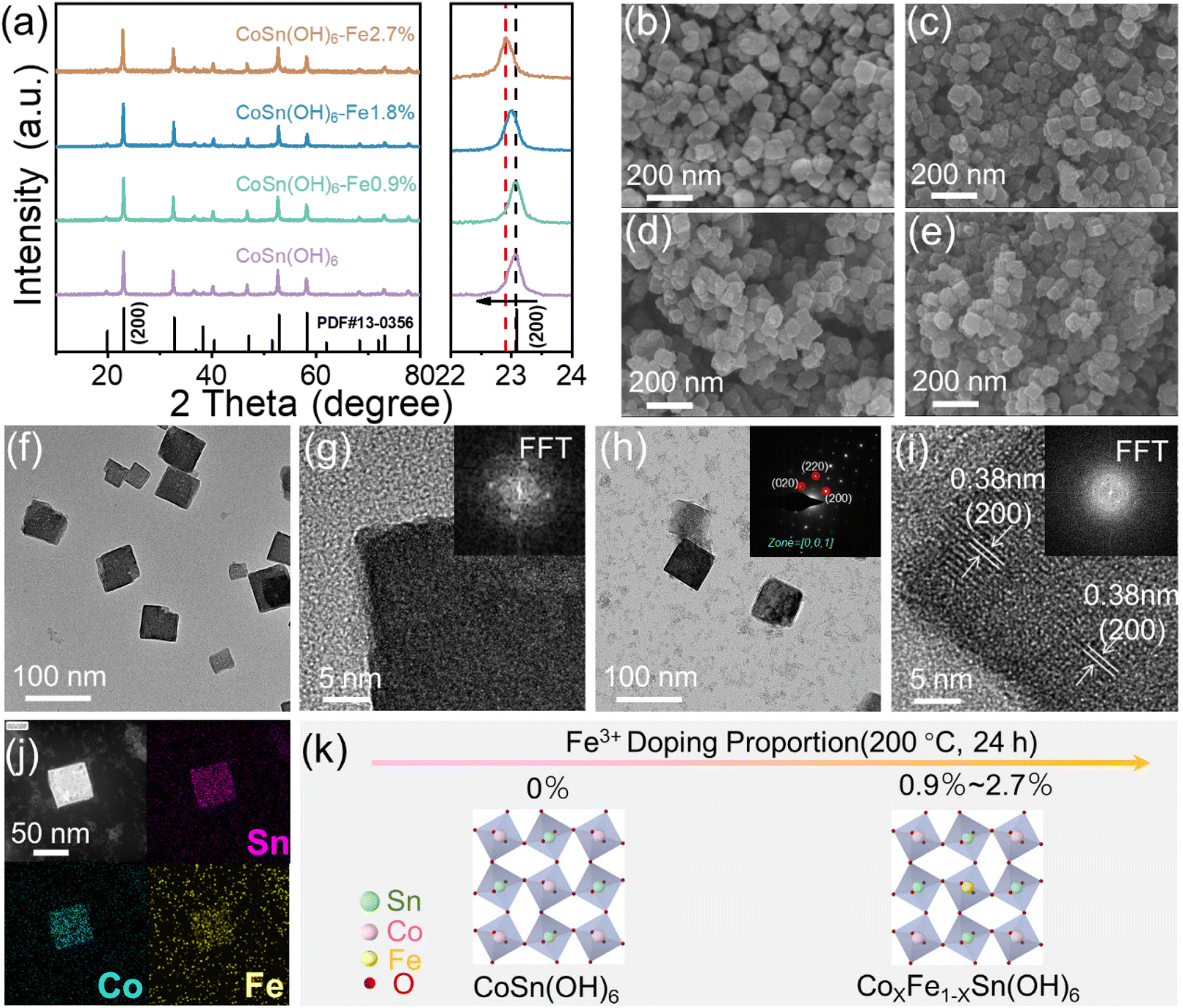 | ||
| Fig. 1 (a) XRD patterns and SEM (b–e) images of CoSn(OH)6, CoSn(OH)6-Fe0.9%, CoSn(OH)6-Fe1.8%, and CoSn(OH)6-Fe2.7%; (f) TEM image of CoSn(OH)6; (g) HR-TEM image of CoSn(OH)6. The inset is the corresponding FFT diffraction. (h) TEM image and SAED, (i) HR-TEM image (inset is the FFT patterns), and (j) HAADF-STEM images of CoSn(OH)6-Fe1.8% and the EDX elemental maps of Sn, Co, and Fe; (k) the scheme of the structure evolution process of CoSn(OH)6 nanocubes doped with Fe at different proportions. | ||
Fig. 1b shows the scanning electron microscopy (SEM) image of pure CoSn(OH)6. It is observed that the pristine CoSn(OH)6 material was cube shaped with a size of about 60 nm. After Fe3+ doping, the SEM images display that CoSn(OH)6-Fe0.9%(Fig. 1c), CoSn(OH)6-Fe1.8%(Fig. 1d), and CoSn(OH)6-Fe2.7% (Fig. 1e) have overall maintained the cube-like morphology.24 With the amount of doped Fe3+ increasing, the cube-like morphology becomes more irregular (Fig. S1†). Moreover, the corresponding energy dispersive spectrometry (EDX) was performed to further analyze the elemental percentage data of the as-prepared materials. As the EDX spectra in Fig. S2 and 3† exhibit, the elemental proportions of Co, Sn and Fe are extremely similar to the results of ICP-OES.
The detailed structural information was further elucidated by transmission electron microscopy (TEM). The TEM image shows that the produced CoSn(OH)6 perovskite hydroxide has a cube-like shape with a size of approximately 60 nm (Fig. 1f). The CoSn(OH)6 perovskite hydroxide exhibits a closed perpendicular morphology with a uniform contrast, as seen in the high-resolution TEM (HRTEM) image (Fig. 1g). The corresponding fast Fourier transform (FFT) pattern in the inset of Fig. 1g reflects that the obtained CoSn(OH)6 exhibits crystallinity.27 The TEM image of CoSn(OH)6-Fe1.8% shows that the obtained CoSn(OH)6-Fe1.8% perovskite hydroxide has the same morphology as CoSn(OH)6 (Fig. 1h). The corresponding selected area electron diffraction (SAED) pattern shows a set of clear diffraction spots, which can be indexed to the (020), (220), and (200) crystal facets of the perovskite CoSn(OH)6 (inset of Fig. 1h), suggesting the single crystal property of the as-prepared CoSn(OH)6 materials.41 Furthermore, the HR-TEM image of CoSn(OH)6-Fe1.8% shows that the lattice spacing value is calculated to be 0.38 nm, matching the (200) lattice plane of CoSn(OH)6 (200) (Fig. 1i).28 According to the FFT pattern, the resulting CoSn(OH)6-Fe1.8% has an octahedral structure (inset of Fig. 1i).40 In addition, the elemental mapping image of CoSn(OH)6-Fe1.8% from EDX and the high-angle annular dark-field scanning TEM (HAADF-STEM) image in Fig. 1j show that the Co, Sn and Fe elements are homogeneously distributed in CoSn(OH)6-Fe1.8% nanocubes, suggesting that the CoSn(OH)6 nanocubes are evenly doped with Fe3+. The above investigations confirm that we have successfully synthesized the Fe3+ doped CoSn(OH)6 nanocubes. The proposed crystal scheme is shown in Fig. 1k, and the Sn, Co, Fe, and O atoms were arranged in the form of a cubic like perovskite structure. The doping of Fe replaces a slight amount of Co.27
3.2 Elemental states
The elemental states of the as-prepared CoSn(OH)6 materials were further investigated by X-ray photoelectron spectroscopy (XPS). The C 1s from adventitious carbon (284.8 eV) was used to calibrate the XPS spectra. Fig. 2a displays the Co 2p spectra of CoSn(OH)6 and CoSn(OH)6-Fe1.8%. The centers of Co2+ and Co3+ states’ signal peaks of Co 2p in CoSn(OH)6 are 783.1 and 781 eV, respectively.42 The centers of Co2+ and Co3+ states’ signal peaks of Co 2p in CoSn(OH)6-Fe1.8% are 782.8 and 780.8 eV, respectively.43 It demonstrates that CoSn(OH)6-Fe1.8%'s Co 2p binding energy peaks move to a lower energy than that of CoSn(OH)6, which indicates that the electron cloud around the Co atom becomes more concentrated after the incorporation of Fe.44Fig. 2b shows the Sn 3d XPS spectra of CoSn(OH)6 and CoSn(OH)6-Fe1.8%. The signal peaks of Sn 3d5/2 and Sn 3d3/2 of CoSn(OH)6 are mainly concentrated at 486.6 and 495 eV. In addition, the Sn 3d5/2 and Sn 3d3/2 signal peaks of CoSn(OH)6-Fe1.8% are mainly concentrated at 486.5 and 494.9 eV. Such a result indicates that the main characteristics of Sn4+ states exist in CoSn(OH)6 and CoSn(OH)6-Fe1.8% samples.45 Meanwhile, in comparison with pristine CoSn(OH)6, the 3d orbital energy of Sn4+ in the CoSn(OH)6-Fe1.8% sample was negatively shifted, indicating that the electron cloud around the Sn atom becomes thicker after the incorporation of Fe. As shown in Fig. 2c, the peak of fitted curves of CoSn(OH)6-Fe1.8% in the O 1s XPS spectrum is positively shifted in comparison with those of CoSn(OH)6, indicating that the electron cloud around O atom becomes thinner after the incorporation of Fe3+.46 The existence of the element Fe is thus confirmed by the high-resolution Fe 2p XPS spectrum of the CoSn(OH)6-Fe1.8% sample (Fig. 2d). Th Fe 2p3/2 and Fe 2p1/2 of Fe3+ are represented by the peaks at 712.3 and 720.7 eV, suggesting the existence of the Fe3+ state.47 Based on the above analysis, it could be deduced that the Fe3+ doping efficiently modulated the electronic structure of CoSn(OH)6.48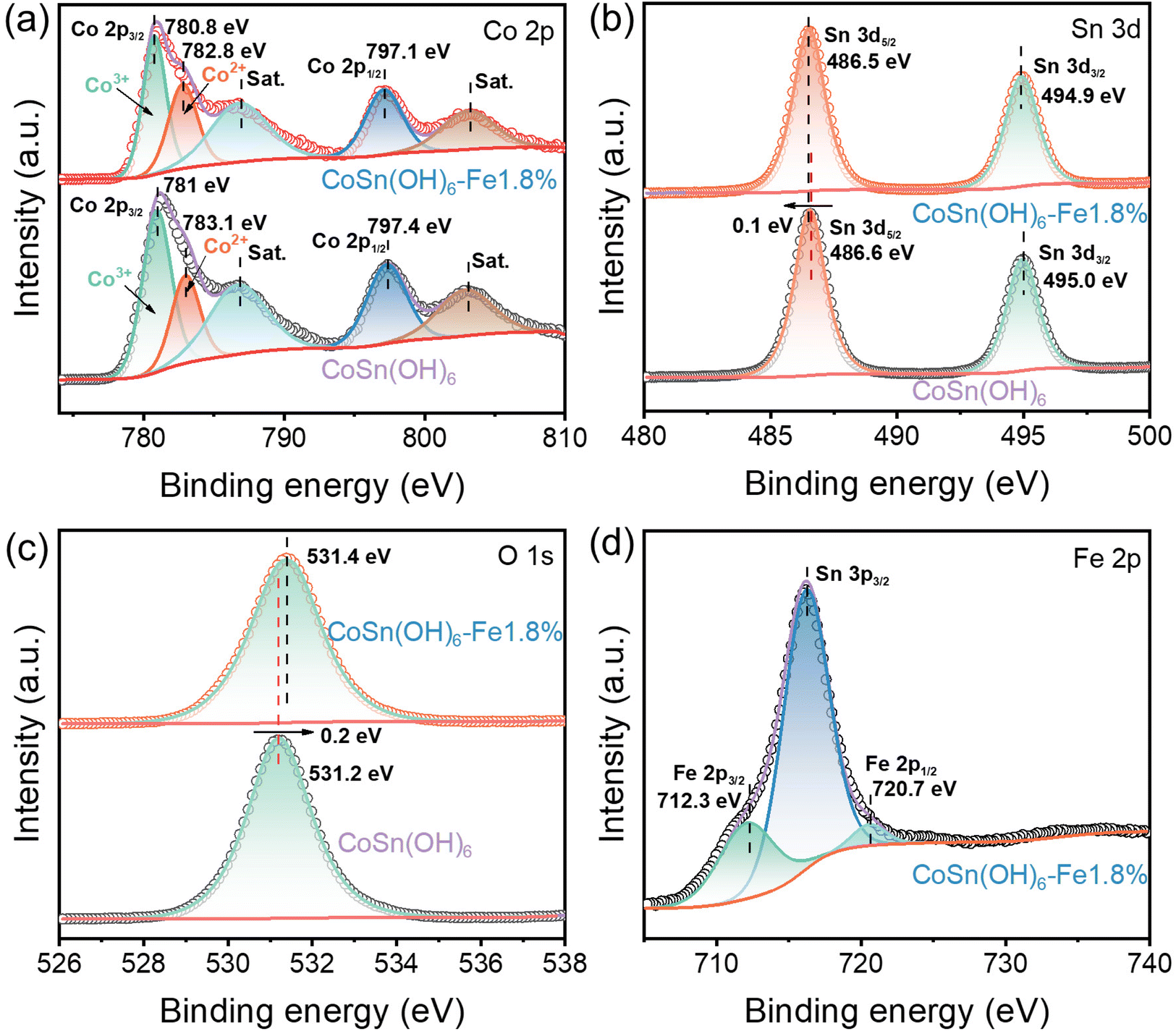 | ||
| Fig. 2 XPS spectra of (a) Co 2p, (b) Sn 3d, (c) O 1s, and (d) Fe 2p. | ||
3.3 Electrocatalytic OER performance
At a scan rate of 10 mV s−1, the catalysts’ OER activity was assessed in O2-saturated 1.0 M KOH (see details in the ESI†). The linear sweep voltammetry (LSV) curves scanned at 10 mV s−1 are displayed in Fig. 3a. It can be seen that the Fe3+ doped CoSn(OH)6 shows reduced OER overpotentials compared to pristine CoSn(OH)6. The OER activity of Fe3+ doped CoSn(OH)6 thus improved gradually along with the Fe3+ doping ratio and CoSn(OH)6-Fe1.8% achieved the relatively optimized activity. At a current density of 10 mA cm−2, the overpotential of CoSn(OH)6-Fe1.8% for the OER was determined to be 289 mV. The results show that the electrochemical OER performance of CoSn(OH)6 is significantly influenced by the doping amount of Fe3+. For comparison purpose, the CoFe-LDH (Co0.982Fe0.018(OH)x) was also investigated (Fig. S4†), and CoSn(OH)6-Fe1.8% also shows relatively reduced overpotentials at 10 and 50 mA cm−2, indicating that the perovskite exhibits superior OER activity compared to the corresponding layered hydroxides. The overpotentials at 10 and 50 mA cm−2 were compared (Fig. 3b). The highest overpotential of 383 mV was observed in pristine CoSn(OH)6 at a current density of 10 mA cm−2. Otherwise, the OER overpotentials of 325, 289, and 293 mV at 10 mA cm−2 were much lower for CoSn(OH)6-Fe0.9%, CoSn(OH)6-Fe1.8%, and CoSn(OH)6-Fe2.7%, respectively. At a current density of 50 mA cm−2, the trend remains the same, where CoSn(OH)6, CoSn(OH)6-Fe0.9%, CoSn(OH)6-Fe1.8%, and CoSn(OH)6-Fe2.7% showed overpotentials of 479, 415, 366, and 436 mV, respectively. According to this analysis, CoSn(OH)6-Fe1.8% possesses optimized electrochemical activity when compared to pristine CoSn(OH)6.24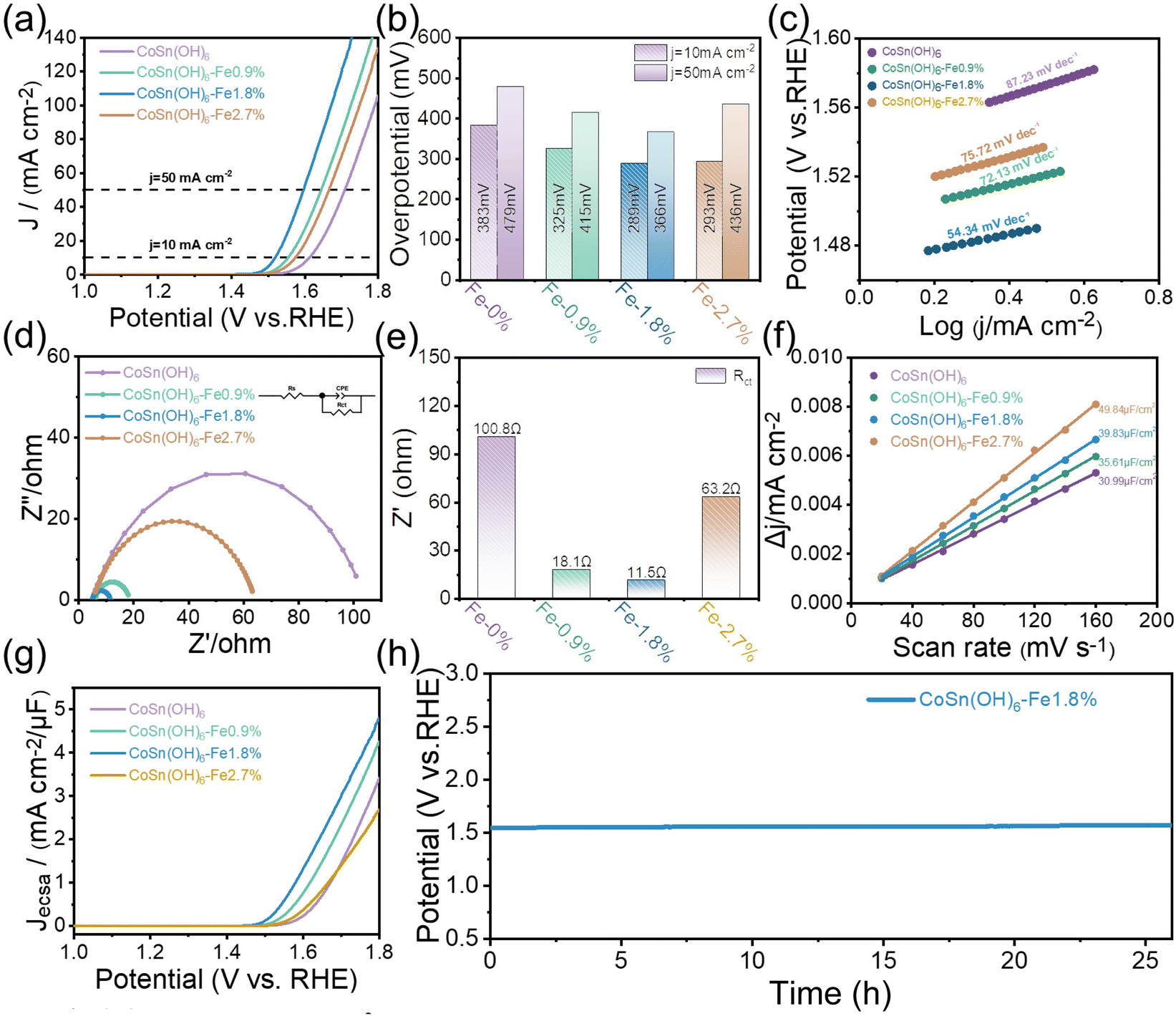 | ||
| Fig. 3 (a) LSV curves; (b) calculated overpotentials at 10 mA cm−2 and 50 mA cm−2; (c) the LSV-derived Tafel slopes; (d) EIS analysis at 1.6 V vs. RHE (the inset is the model of the equivalent circuit); (e) impedance values of different samples; (f) current density difference plotted against scan rates; (g) LSV curves normalized with ESCA. (h) Chronopotentiometry curves of CoSn(OH)6-Fe1.8% at 10 mA cm−2. | ||
The Tafel slope (Fig. 3c) was used to determine the OER kinetics of these samples. CoSn(OH)6-Fe1.8% showed the lowest Tafel slope (54.34 mV dec−1) in comparison with CoSn(OH)6-Fe0.9% (72.13 mV dec−1), CoSn(OH)6-Fe2.7% (75.72 mV dec−1), and pristine CoSn(OH)6 (87.23 mV dec−1), confirming that the optimal synthesis conditions are those of the CoSn(OH)6-Fe1.8% product. The electrochemical impedance spectroscopy (EIS) results were obtained at 1.60 V vs. RHE (Fig. 3d). Fitting the EIS spectra with an equivalent circuit yields the solution resistance (Rs) and charge transfer resistance (Rct) (inset of Fig. 3d).27 Every sample has an Rs value of around 6.2 Ω (Table S2†), indicating comparable electrochemical test conditions.38 The Rct value was obtained by fitting the semicircle at low frequency.39 As shown in Fig. 3e, the Rct value which represents the charge transfer surface intermediate resistance is ranked in the following order in alkaline electrolyte: CoSn(OH)6-Fe1.8% (11.5 Ω) < CoSn(OH)6-Fe0.9% (18.1 Ω) < CoSn(OH)6-Fe2.7% (63.2 Ω) < CoSn(OH)6 (100.8 Ω). The CoSn(OH)6-Fe1.8% anode thus shows the smallest bulk charge transport resistance. The optimal Rct of CoSn(OH)6-Fe1.8% is beneficial for the charge and mass transfer and during the OER process. In addition, the OER performance of CoSn(OH)6-Fe1.8% and that of already reported Fe, Co, and Sn based materials are compared. The OER activity of the as-prepared CoSn(OH)6-Fe1.8% is comparatively favorable (Table S3†).
The electrochemically active surface area (ECSA) calculated using double-layer capacitance (Cdl) is studied to reveal the intrinsic electrocatalytic activity of the Fe3+ doped CoSn(OH)6. The CV test conducted between 1.35 and 1.45 V (vs. RHE) was used to determine the Cdl value (Fig. S5†). As shown in Fig. 3f, the capacitance of CoSn(OH)6, CoSn(OH)6-Fe0.9%, CoSn(OH)6-Fe1.8%, and CoSn(OH)6-Fe2.7% was determined to be 30.99, 35.61, 39.83, and 49.84 μF cm−2, respectively. Then we normalized the LSV curves of Fe3+ doped CoSn(OH)6 samples using ECSA, and the Fe3+ doped sample still has higher intrinsic activity with the lowest overpotential observed for CoSn(OH)6-Fe1.8% as shown in Fig. 3g. Therefore, Fe3+ doping significantly increases the intrinsic activity of CoSn(OH)6 rather than only enhancing the ECSA. Further evaluation of the CoSn(OH)6-Fe1.8% catalyst's durability was conducted using chronopotentiometric (CP) measurement at room temperature and a fixed current density of 10 mA cm−2 (Fig. 3h). The potential of CoSn(OH)6-Fe1.8% increases by just 20 mV over 26 hours to maintain a current density of 10 mA cm−2. Meanwhile, the durability of the CoSn(OH)6-Fe1.8% catalyst was further tested by a chronopotentiometric test at a fixed current density of 100 mA cm−2 at room temperature (Fig. S6†), which also shows favorable stability. A comparison of the morphology before and after the electrochemical reactions is shown in Fig. S7.† After the long-term electrochemical measurement, the morphology is almost maintained.
3.4 In situ electrochemical characterization
CoSn(OH)6-Fe1.8% and pristine CoSn(OH)6 were characterized by in situ Raman spectroscopy in order to gain insights into the structural changes in the electrochemical OER test (Fig. 4a). The carbon fiber paper substrate shows three identical Raman bands at 416, 575, and 749 cm−1 (Fig. 4b–c and Fig. S8†). The Co–O vibration in CoSn(OH)6 could be assigned to the other Raman band at 479 cm−1, as demonstrated by the in situ Raman spectra of pristine CoSn(OH)6.49,50 Furthermore, there was no obvious shift in the peak's position nor the appearance of new bands as exhibited in the whole applied potential range. The structure of the CoSn(OH)6 material is stable under the OER process. The CoSn(OH)6-Fe1.8% sample shows the same result, suggesting that the structure is stable during the OER test. There is an extra Raman band (601 cm−1) for the CoSn(OH)6-Fe1.8% sample (Fig. 4c), which is attributed to the Raman band corresponding to the Fe–O vibration from the produced Fe–O(OH).50,51 The XRD spectra before and after the OER were also examined, which are also consistent with the in situ Raman results (Fig. S9†). It is reported that most layered hydroxides materials such as Ni(OH)2 and Co(OH)2 converted into oxyhydroxides (e.g. NiOOH and CoOOH) during the OER process.19,52 For the CoSn(OH)6 material, such a re-construction is not obviously observed, suggesting the superior stability of the perovskite structure during the electrochemical OER process.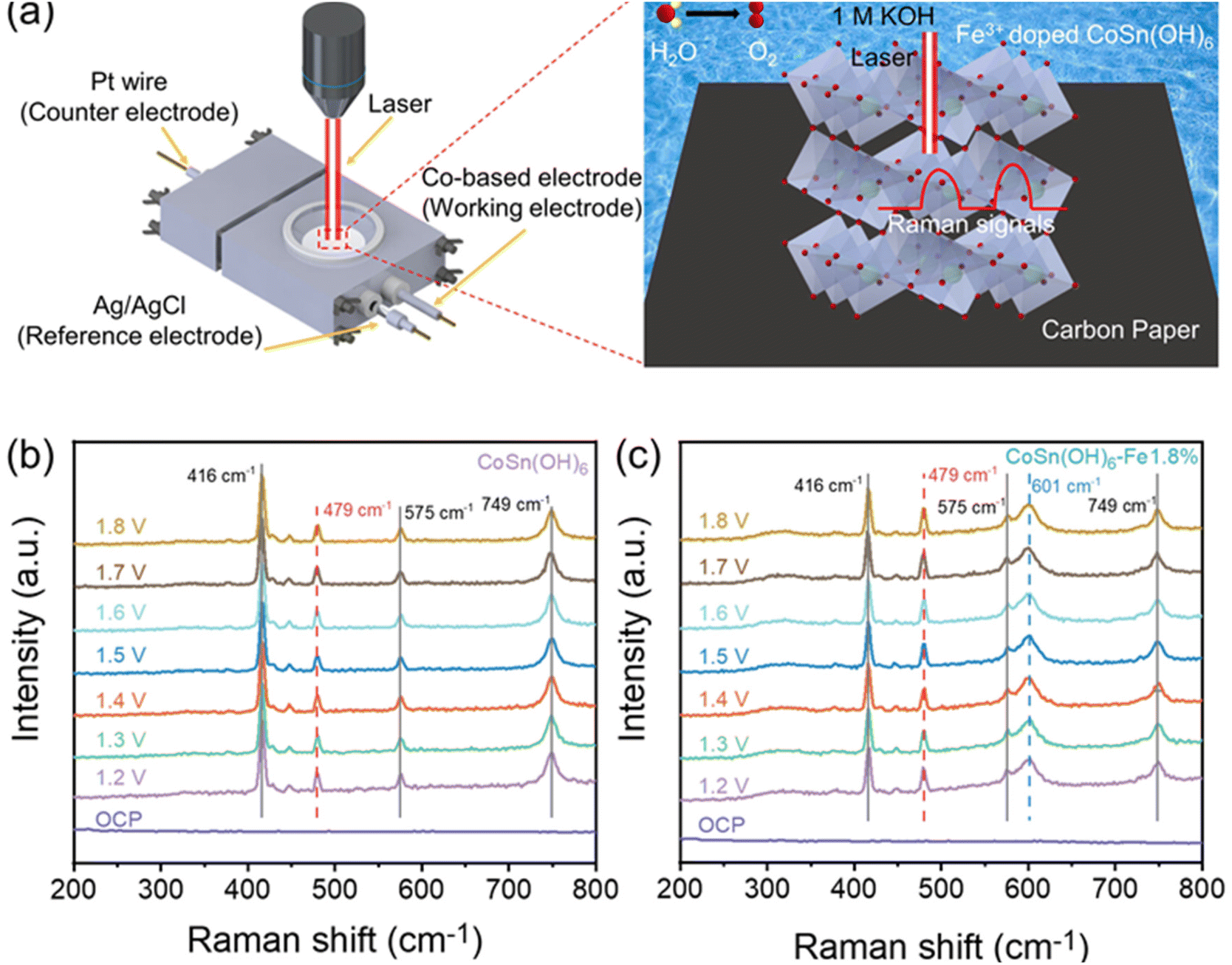 | ||
| Fig. 4 (a) Schematic illustration of the in situ Raman spectroscopy setting; in situ Raman spectra of (b) CoSn(OH)6 and (c) CoSn(OH)6-Fe1.8% at different potentials. | ||
3.5 DFT calculations
To further gain deep insight into the influence of Fe3+ on the OER performance of CoSn(OH)6, the d-band states were studied using density functional theory calculations (DFT). The corresponding atomic models of CoSn(OH)6 and Co0.83Fe0.17Sn(OH)6 are displayed in Fig. 5a and b. The (001) surfaces of CoSn(OH)6 and Co0.83Fe0.17Sn(OH)6 were constructed to simulate the surface of catalysts, which contained two octahedral layers (Fig. S10†). Fig. 5c and d show the partial density of states (PDOS) of 3d orbitals calculated on the corresponding atomic model.24,53 The d-band states of these catalysts are near the Fermi level.30 The electronic structure of the CoSn(OH)6 perovskite hydroxide was changed after Fe3+ doping. The calculated d-band centers of CoSn(OH)6, and Co0.83Fe0.17Sn(OH)6 are approximately −2.21 and −1.95 eV, respectively. The value of the d-band center increases after the increase of Fe3+ content. It is known that the filling degree of the antibonding state can be reflected from the location of the d-band center with respect to the Fermi level.31 With a higher center of the d band, the antibonding state is filled with fewer electrons, which will result in a stronger bond between the adsorbent and the catalyst.54 Compared to CoSn(OH)6, the d-band center of the Co0.83Fe0.17Sn(OH)6 perovskite hydroxide is higher, indicating a relatively strong adsorption ability between active sites and intermediates.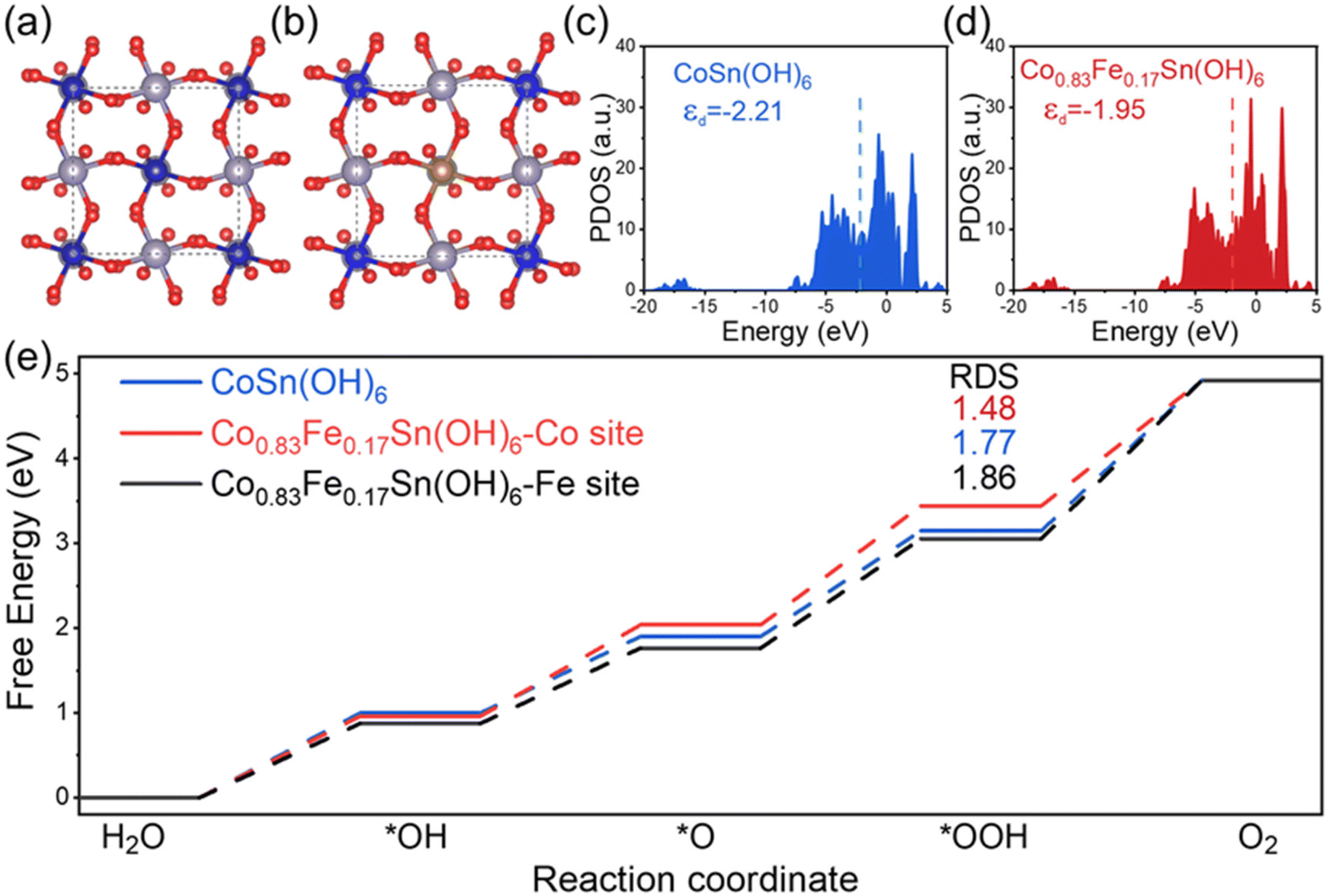 | ||
| Fig. 5 Schematic changes of the bond structure for (a) CoSn(OH)6 and (b) Co0.83Fe0.17Sn(OH)6; the PDOS of 3d orbitals for (c) CoSn(OH)6, and (d) Co0.83Fe0.17Sn(OH)6; (e) the reaction Gibbs free energy changes during the OER process. | ||
In addition, to study the influences of the d-band states on the intermediates’ adsorption on these materials, the reaction Gibbs free energies of *OH, *O, and *OOH intermediates adsorbed on the Co0.83Fe0.17Sn(OH)6 surface were calculated (Fig. S11 and 12†).55 The Gibbs free energies of *OH, *O, and *OOH at different active sites are displayed in Fig. 5e. The *O to *OOH is the rate-determining step for the various active sites. The Co and Fe sites in the Co0.83Fe0.17Sn(OH)6 perovskite hydroxide require an overpotential of 1.48 eV to adsorb OOH* intermediates following Fe3+ substitution, which is lower than that of the Co site in CoSn(OH)6 (1.77 eV). The Fe3+ dopants thus can reduce the rate-limiting step's reaction energy barrier and provide a comparatively simple reaction path for the OER.24,38 Therefore, DFT theoretical calculations show that appropriate Fe3+ doping amount can slightly improve the d-band center of the CoSn(OH)6 perovskite hydroxide, which will promote the adsorption ability and reduce the OER reaction energy barrier.
3.6 Solar-driven water splitting
Direct electrolysis of water into hydrogen through the injection of renewable solar energy is the ideal method for producing sustainable energy.56 In the following study, we built a solar-driven water splitting reaction system by utilizing CoSn(OH)6-Fe1.8% nanocubes as the electrocatalyst for the OER (Fig. 6a). A commercial solar cell was coupled to a two-electrode system and the gas yield during the photovoltaic-electrocatalytic water splitting process was recorded using an online gas chromatograph (Fig. S13† displays the energy conversion system photograph). The light from a Xe lamp was used to simulate sunlight. Fig. S14† displays the Xe lamp's irradiative spectrum. The input solar energy was calculated from the value of light intensity measured using a spectroradiometer (0.309 W cm−2). The actual gas yield is comparable with the theoretical gas yield (Fig. S15†). As Fig. 6b illustrates, the real gas yields of O2 are approximately half of that of H2. Furthermore, the faradaic efficiency of evolved H2 can reach 98.21% (Fig. 6c), and it is still maintained at 87.17% after 160 min (Fig. S16†). Furthermore, about 8.87% was the calculated solar-to-hydrogen energy conversion efficiency (Fig. 6c). The above results show that CoSn(OH)6-Fe1.8% nanocubes are promising for photovoltaic-electrocatalytic water decomposition.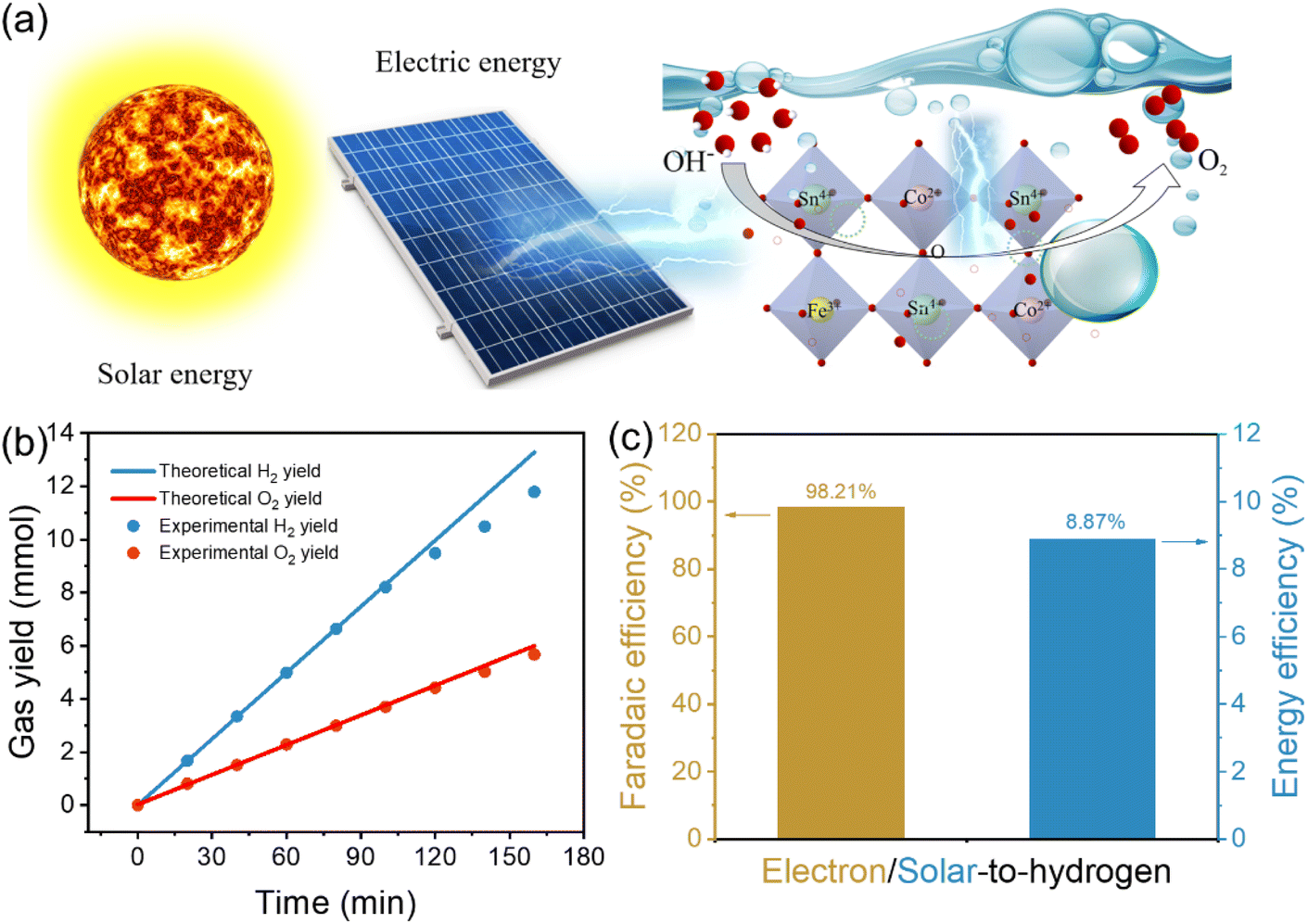 | ||
| Fig. 6 (a) Schematic of a solar-driven water splitting system; (b) H2 and O2 yields for the cathode and anode in 1 M KOH solution; (c) faradaic efficiency and the solar-to-hydrogen energy conversion efficiency at 80 min. | ||
4. Conclusions
In summary, CoSn(OH)6 perovskite hydroxides with tuned d band states by doping varying atomic ratios of Fe3+ were achieved. XRD, EDX, and XPS confirmed the successful doping of Fe3+ into the CoSn(OH)6 perovskite hydroxide. The optimal Fe3+ content was found to be 1.8 mol% (CoSn(OH)6-Fe1.8%), which led to the highest OER activity with an overpotential of 289 mV at 10 mA cm−2 in alkaline electrolyte. In situ Raman spectroscopy showed that no obvious re-construction occurred during the OER for both CoSn(OH)6 and CoSn(OH)6-Fe1.8%. The DFT-based density of states (DOS) analysis indicated an increase in the d-band center after Fe3+ doping. The DFT calculations suggested that incorporation of an appropriate amount of Fe3+ into CoSn(OH)6 significantly lowers the Gibbs free energy in the rate-determining step compared to that in pure-phase CoSn(OH)6. In order to evaluate the potential practical application of the as-prepared CoSn(OH)6-Fe1.8%, a photovoltaic-electrolysis water splitting system was established and a solar–hydrogen energy conversion efficiency of 8.87% can be achieved. This study provides an effective strategy to modulate the d-band center of Co-based perovskite hydroxides for a promoted electrocatalytic OER.Author contributions
Mingwei Sun: conceptualization, data curation, formal analysis, investigation, methodology, writing – original draft, writing – review and editing. Baopeng Yang: formal analysis. Jiaxing Yan: data curation – support, formal analysis – support. Yulong Zhou: validation – support. Zhencong Huang: validation – support. Ning Zhang: funding acquisition, project administration, resources, supervision, validation, writing – review and editing. Rong Mo: review and editing. Renzhi Ma: review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work received financial support from the National Natural Science Foundation of China (22072183) and the Natural Science Foundation of Hunan Province, China (2022JJ30690 and 2024JJ5378). This work was supported in part by the High Performance Computing Center of Central South University.References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed
.
- S. Wang, G. Liu and L. Wang, Chem. Rev., 2019, 119, 5192–5247 CrossRef CAS PubMed
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed
- K. Jiao, J. Xuan, Q. Du, Z. Bao, B. Xie, B. Wang, Y. Zhao, L. Fan, H. Wang, Z. Hou, S. Huo, N. P. Brandon, Y. Yin and M. D. Guiver, Nature, 2021, 595, 361–369 CrossRef CAS PubMed
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC
- Y. He, L. Jia, X. Lu, C. Wang, X. Liu, G. Chen, D. Wu, Z. Wen, N. Zhang, Y. Yamauchi, T. Sasaki and R. Ma, ACS Nano, 2022, 16, 4028–4040 CrossRef CAS PubMed
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef CAS PubMed
- Y. Chen, H. Li, J. Wang, Y. Du, S. Xi, Y. Sun, M. Sherburne, J. W. Ager, A. C. Fisher and Z. J. Xu, Nat. Commun., 2019, 10, 572 CrossRef CAS PubMed
- D. Wang and D. Astruc, Chem. Soc. Rev., 2017, 46, 816–854 RSC
- L. Wu, L. Yu, X. Xiao, F. Zhang, S. Song, S. Chen and Z. Ren, Research, 2020, 2020 DOI:10.34133/2020/3976278
- N. Sakai, K. Fukuda, R. Ma and T. Sasaki, Chem. Mater., 2018, 30, 1517–1523 CrossRef CAS
- Y. Hu, F. Li, Y. Long, H. Yang, L. Gao, X. Long, H. Hu, N. Xu, J. Jin and J. Ma, J. Mater. Chem. A, 2018, 6, 10433–10440 RSC
- X. Xu, F. Song and X. Hu, Nat. Commun., 2016, 7, 12324 CrossRef CAS PubMed
- W. Xin, B. Liu, Y. Zhao, G. Chen, P. Chen, Y. Zhou, W. Li, Y. Xu, Y. Zhong and Y. A. Nikolaevich, Electrochim. Acta, 2022, 404, 139748 CrossRef CAS
- H. Wan, R. Ma, X. Liu, J. Pan, H. Wang, S. Liang, G. Qiu and T. Sasaki, ACS Energy Lett., 2018, 3, 1254–1260 CrossRef CAS
- S. Liu, R.-T. Gao, M. Sun, Y. Wang, T. Nakajima, X. Liu, W. Zhang and L. Wang, Appl. Catal., B, 2021, 292, 120063 CrossRef CAS
- S.-Y. Lee, I.-S. Kim, H.-S. Cho, C.-H. Kim and Y.-K. Lee, Appl. Catal., B, 2021, 284, 119729 CrossRef CAS
- D. Zhong, T. Li, D. Wang, L. Li, J. Wang, G. Hao, G. Liu, Q. Zhao and J. Li, Nano Res., 2022, 15, 162–169 CrossRef CAS
- S. Anantharaj, K. Karthick and S. Kundu, Mater. Today Energy, 2017, 6, 1–26 CrossRef
- Z. He, J. Zhang, Z. Gong, H. Lei, D. Zhou, N. Zhang, W. Mai, S. Zhao and Y. Chen, Nat. Commun., 2022, 13, 2191 CrossRef CAS PubMed
- S. Liu, S. Geng, L. Li, Y. Zhang, G. Ren, B. Huang, Z. Hu, J.-F. Lee, Y.-H. Lai, Y.-H. Chu, Y. Xu, Q. Shao and X. Huang, Nat. Commun., 2022, 13, 1187 CrossRef CAS PubMed
- H. Ding, H. Liu, W. Chu, C. Wu and Y. Xie, Chem. Rev., 2021, 121, 13174–13212 CrossRef CAS PubMed
- M. A. Peña and J. L. G. Fierro, Chem. Rev., 2001, 101, 1981–2018 CrossRef PubMed
- D. Chen, M. Qiao, Y.-R. Lu, L. Hao, D. Liu, C.-L. Dong, Y. Li and S. Wang, Angew. Chem., Int. Ed., 2018, 57, 8691–8696 CrossRef CAS PubMed
- Z. Wang, Z. Wang, H. Wu and X. W. Lou, Sci. Rep., 2013, 3, 1391 CrossRef PubMed
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed
- Z. Chen, Y. Song, J. Cai, X. Zheng, D. Han, Y. Wu, Y. Zang, S. Niu, Y. Liu, J. Zhu, X. Liu and G. Wang, Angew. Chem., Int. Ed., 2018, 57, 5076–5080 CrossRef CAS PubMed
- S. Bhattacharjee, U. V. Waghmare and S.-C. Lee, Sci. Rep., 2016, 6, 35916 CrossRef CAS PubMed
- H. Zhang, F. Wan, X. Li, X. Chen, S. Xiong and B. Xi, Adv. Funct. Mater., 2023, 33, 2306340 CrossRef CAS
- X. Guo, J. Shi, M. Li, J. Zhang, X. Zheng, Y. Liu, B. Xi, X. An, Z. Duan, Q. Fan, F. Gao and S. Xiong, Angew. Chem., Int. Ed., 2023, 62, e202314124 CrossRef CAS PubMed
- X. Guo, J. Zhang, L. Yuan, B. Xi, F. Gao, X. Zheng, R. Pan, L. Guo, X. An, T. Fan and S. Xiong, Adv. Energy Mater., 2023, 13, 2204376 CrossRef CAS
- P. Li, S. Zhao, Y. Huang, Q. Huang, B. Xi, X. An and S. Xiong, Adv. Energy Mater., 2024, 14, 2303360 CrossRef CAS
- X. Guo, M. Duan, J. Zhang, B. Xi, M. Li, R. Yin, X. Zheng, Y. Liu, F. Cao, X. An and S. Xiong, Adv. Funct. Mater., 2022, 32, 2209397 CrossRef CAS
- Z. Xiao, W. Zhou, N. Zhang, C. Liao, S. Huang, G. Chen, G. Chen, M. Liu, X. Liu and R. Ma, Chem. Commun., 2021, 57, 6070–6073 RSC
- D. Luo, B. Yang, Z. Mei, Q. Kang, G. Chen, X. Liu and N. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 52857–52867 CrossRef CAS PubMed
- S. Chen, Z. Zheng, Q. Li, H. Wan, G. Chen, N. Zhang, X. Liu and R. Ma, J. Mater. Chem. A, 2023, 11, 1944–1953 RSC
- L. Kang, C. Ye, X. Zhao, X. Zhou, J. Hu, Q. Li, D. Liu, C. M. Das, J. Yang, D. Hu, J. Chen, X. Cao, Y. Zhang, M. Xu, J. Di, D. Tian, P. Song, G. Kutty, Q. Zeng, Q. Fu, Y. Deng, J. Zhou, A. Ariando, F. Miao, G. Hong, Y. Huang, S. J. Pennycook, K.-T. Yong, W. Ji, X. Renshaw Wang and Z. Liu, Nat. Commun., 2020, 11, 3729 CrossRef CAS PubMed
- F. Song, K. Schenk and X. Hu, Energy Environ. Sci., 2016, 9, 473–477 RSC
- N. S. McIntyre and M. G. Cook, Anal. Chem., 1975, 47, 2208–2213 CrossRef CAS
- H. Lyu, D. Yin, B. Zhu, G. Lu, Q.-Y. Liu, X. Zhang and X. Zhang, ACS Sustainable Chem. Eng., 2020, 8, 9404–9414 CrossRef CAS
- S. Riyajuddin, K. Azmi, M. Pahuja, S. Kumar, T. Maruyama, C. Bera and K. Ghosh, ACS Nano, 2021, 15, 5586–5599 CrossRef CAS PubMed
- R. Sahoo, A. K. Sasmal, C. Ray, S. Dutta, A. Pal and T. Pal, ACS Appl. Mater. Interfaces, 2016, 8, 17987–17998 CrossRef CAS PubMed
- L. Zhuang, L. Ge, Y. Yang, M. Li, Y. Jia, X. Yao and Z. Zhu, Adv. Mater., 2017, 29, 1606793 CrossRef PubMed
- X. Jiang, W. Xu, R. Tan, W. Song and J. Chen, Mater. Lett., 2013, 102–103, 39–42 CrossRef CAS
- M. Liu, K.-A. Min, B. Han and L. Y. S. Lee, Adv. Energy Mater., 2021, 11, 2101281 CrossRef CAS
- J.-Z. Zhang, Z. Zhang, H.-B. Zhang, Y. Mei, F. Zhang, P.-X. Hou, C. Liu, H.-M. Cheng and J.-C. Li, Nano Lett., 2023, 23, 8331–8338 CrossRef CAS PubMed
- W. H. Lee, M. H. Han, Y.-J. Ko, B. K. Min, K. H. Chae and H.-S. Oh, Nat. Commun., 2022, 13, 605 CrossRef CAS PubMed
- X. Xing, C. Wu, G. Yang, T. Tong, Y. Wang, D. Wang, F. C. Robles Hernandez, Z. Ren, Z. Wang and J. Bao, Mater. Today Chem., 2022, 26, 101110 CrossRef CAS
- S. She, Y. Zhu, H. A. Tahini, X. Wu, D. Guan, Y. Chen, J. Dai, Y. Chen, W. Tang, S. C. Smith, H. Wang, W. Zhou and Z. Shao, Small, 2020, 16, 2006800 CrossRef CAS PubMed
- W. Wu, S. Shi, Z. Zhang, X. Guo, L. Sun, R. Wei, J. Zhang, L. Gao, X. Pan and G. Xiao, J. Colloid Interface Sci., 2022, 619, 407–418 CrossRef CAS PubMed
- J. Liu, H. Liu, H. Chen, X. Du, B. Zhang, Z. Hong, S. Sun and W. Wang, Adv. Sci., 2020, 7, 1901614 CrossRef CAS PubMed
- N. Zhang, B. Yang, Y. He, Y. He, X. Liu, M. Liu, G. Song, G. Chen, A. Pan, S. Liang, R. Ma, S. Venkatesh and V. A. L. Roy, Small, 2018, 14, 1803015 CrossRef PubMed
- T. Faunce, S. Styring, M. R. Wasielewski, G. W. Brudvig, A. W. Rutherford, J. Messinger, A. F. Lee, C. L. Hill, H. deGroot, M. Fontecave, D. R. MacFarlane, B. Hankamer, D. G. Nocera, D. M. Tiede, H. Dau, W. Hillier, L. Wang and R. Amal, Energy Environ. Sci., 2013, 6, 1074–1076 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr00975d |
| This journal is © The Royal Society of Chemistry 2024 |