DOI:
10.1039/D4NR00665H
(Paper)
Nanoscale, 2024,
16, 9827-9835
Rapid targeting and imaging of mitochondria via carbon dots using an amino acid-based amphiphile as a carrier†
Received
16th February 2024
, Accepted 6th April 2024
First published on 11th April 2024
Abstract
Green-fluorescent biocompatible carbon dots with a quantum yield of 40% were successfully synthesized through a solvothermal process and then they are comprehensively characterized. The carbon dots showed a negatively charged surface owing to the presence of carboxylic groups. This negative surface charge hinders the effective targeting and imaging of mitochondria. To address this limitation, a new approach is developed in this study. An amphiphile containing phenylalanine, with a positively charged polar head consisting of triphenylphosphine and a hydrophobic aliphatic tail, was designed, synthesized, purified, and characterized. This amphiphile formed spherical micelle-type nanostructures in an aqueous medium in the aggregated state. Although these nanoprobes lack inherent fluorescence, they exhibited the capability to image mitochondria when their spherical micelle-type nanostructures were decorated with negatively charged fluorescent nanocarbon dots in both cancerous (KB cells) and non-cancerous (CHO cells) cell lines. Notably, carbon dots without the amphiphile failed to penetrate the cell membrane as they exhibited significantly low emission inside the cell. This study extensively explored the cell entry mechanism of the hybrid nanoprobes. The photophysical changes and the interaction between the negatively charged carbon dots and the positively charged nanospheres of the amphiphile were also analyzed in this study.
Introduction
Mitochondria have substantial influence in the realms of human evolution and the varieties of diseases in humans.1 Over an evolutionary time-scale, mitochondria have exerted influence in establishing pivotal organelles within eukaryotic cells.2,3 As a cellular powerhouse, mitochondria adeptly transmute oxygen and nutrients into adenosine triphosphate (ATP), subsequently fuelling an array of biochemical and physiological processes, encompassing growth, mobility, and homeostasis.4 Mitochondria also play a crucial role in numerous physiologically significant processes, encompassing the generation of reactive oxygen species (ROS) and adenosine triphosphate (ATP), storage and signalling of calcium ions, synthesis of steroids, initiation of apoptosis, and regulation of cellular proliferation.5,6 Moreover, recent research has confirmed the strong correlation between mitochondrial dysfunction and a spectrum of metabolic diseases and neurodegenerative disorders.7 This association extends to conditions such as Parkinson's disease (PD), Alzheimer's disease (AD), and Huntington's disease (HD).8,9 Although advances in cellular biology have allowed greater knowledge of the morphology of mitochondria, there are still many unknown facts that hinder attempts to solve the many problems associated with mitochondria.10 Therefore, a proper mapping and understanding of the morphology of mitochondria still need close attention even today. Several microscopic techniques, such as electron microscopy (EM), transmission electron microscopy (TEM), cryo electron microscopy (Cryo-EM), and scanning electron microscopy (SEM), have been applied as potent tools in studies to reveal the intricate structure of mitochondria since the 1950s.10 However, nowadays the in situ observation of mitochondrial dynamics has become popular using fluorophore molecules through mapping them with an appropriate MitoTracker dye, revealing a clear morphology of the mitochondria in living cells.11,12
Materials designed for mitochondrial targeting include small organic fluorophore molecules, peptides, polymer–micelles, dendrimers, and several organic and inorganic nanoparticles, and these provide many opportunities to know more about this organelle through their selective imaging, monitoring, drug delivery, and therapeutic studies.13–15 Organic fluorescent molecules, particularly commercial probes, are widely favoured for their easily modifiable characteristics to target diverse subcellular organelles and identify specific analytes.16 Notable examples include the cationic probe rhodamine 123 (Rho 123) and chloromethyl-based probes, like MitoTracker Green (MTG) and MitoTracker Deep Red (MTDR), which are utilized for mitochondria targeting and labelling.17,18 However, despite their popularity, these molecules come with some inherent limitations, such as high background noise, intricate synthetic processes, and poor photostability, which hinder their application in wash-free imaging, real-time monitoring, and long-term tracking.10 Thus, many research efforts have been recently dedicated to the development of many analogous fluorophores that can be used as an efficient MitoTracker for eliminating those limitations.19,20 As mitochondria carry a highly negative charge21 (−150 to −200 mV), there is always a restriction in designing the fluorophore in such a way that the fluorophore must contain positively charged heads, such as triphenylphosphine, arginine, or other positively charged species.22–24 In this work, we attempted to address this gap by developing a strategy involving synthesizing a positively charged non-fluorescent triphenyl phosphine containing amphiphile and using this as a carrier of a negatively charged carbon dot-based fluorophore to the mitochondria and labelling them for their rapid bioimaging. To the best of our knowledge, no reports are available where a fluorophore with a negatively charged surface has been used as a MitoTracker. Carbon dots (C-dots), being fluorescent nanomaterials with promising characteristics, such as strong biocompatibility, outstanding cell permeability, vivid fluorescence, high photostability, and straightforward synthetic pathways, have emerged as compelling contenders for bioimaging applications.25–35 To date, few examples of C-dots modified by positively charged surface groups have been reported as mitochondria targeting agents, but C-dots with a negatively charge have been barely reported.36,37 Therefore, C-dots with negatively charged surfaces, which are easily available in the literature, have not been utilized as a MitoTracker till now.
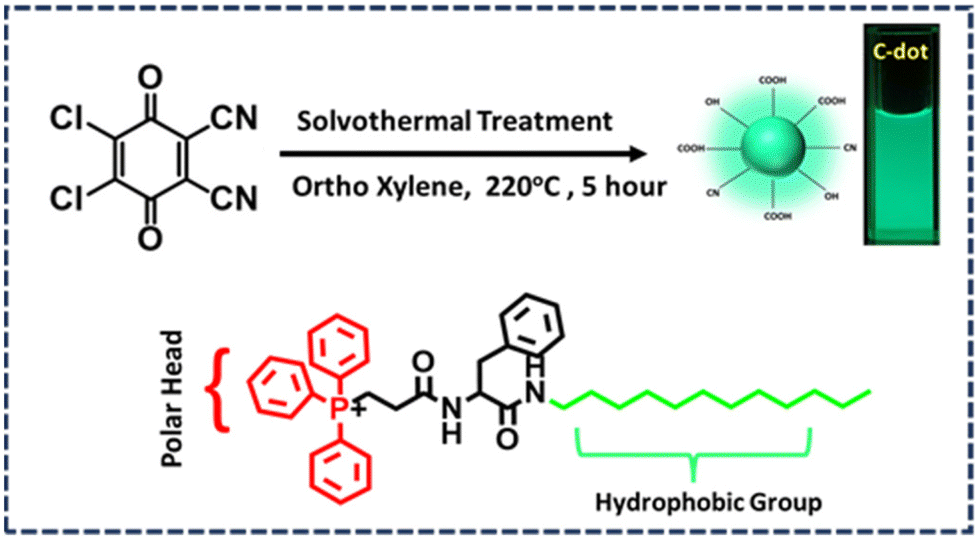
Herein, we synthesized bright green emitting photostable C-dots by a solvothermal process from 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) according to our previously reported work with a slight modification. The only difference regarding the synthesis was the higher temperature used in this study.25 We found that the C-dots had a negatively charged surface, which was probably due to the presence of carboxylic groups on their surface. These fluorescent particles could not penetrate the cell line, as no emission was seen in the microscopic image after incubation with the cell, indicating that they were able to penetrate the cell line properly. To solve this problem, we synthesized an amphiphile containing phenylalanine amino acid, with a positively charged polar head consisting of triphenylphosphine and a hydrophobic aliphatic tail (P). These amphiphiles form spherical micelle-type nanostructures when they are aggregated in an aqueous medium. These spherical micelle-type nanostructures do not contain any inherent fluorescence, and thus they are not capable of being used in imaging any organelle inside the cell through fluorescence microscopy. Surprisingly, when those spherical micelle-type nanostructures of the aggregated amphiphile were decorated with negatively charged nanoC-dots, those C-dots tagged nanospheres (CDTP) exhibited the capability to image mitochondria in both of cancerous (KB cell) and non-cancerous (CHO cell) cell lines. The endocytosis process through the caveolae pathway was found to be the mechanistic pathway of their cell entry. Furthermore, in this work, the interaction between the C-dots and P was analyzed in detail through several optical spectroscopic and microscopic techniques.
Experimental
NMR experiments
All NMR studies were carried out on a Bruker DPX400 MHz or Bruker DPX500 MHz spectrometer at 300 K.
Mass spectrometry
Mass spectra were recorded on a Q-Tof micro-TM (Waters Corp.) mass spectrometer by a positive-mode electrospray ionization process.
Fourier transform infrared study
All FT-IR spectra were recorded using the KBr pellet technique in a Nicolet 380 FT-IR spectrophotometer (Thermo Scientific).
UV/Vis spectroscopy
UV/vis absorption spectra were recorded on a Hewlett–Packard (model 8453) UV/vis spectrophotometer (Varian Carry 50.bio) at 0.05 mM concentration.
PL spectroscopy
The photoluminescence (PL) spectra of the C-dot solution was completed on a Fluoromax-3 instrument (HORIBA Jovin Yvon).
Transmission electron microscopy
The morphology was investigated using a transmission electron microscope (JEOL, 2010EX). Cryo-TEM images were recorded on a JEOL JEM-210 PLUS Cryogenic transmission electron microscope at an accelerating voltage of 200 kV. In vitro cell images were captured by an Olympus IX 81 microscope and confocal microscope (ZEISS LSM 880).
Time-correlated single-photon counting methodology (TCSPC) study
TCSPC measurements were done on a HORIBA join Yvon IBH instrument having an MCPPMT Hamamatsu R3809 detector.
Material and methods
Materials
2,3-Dichloro-5,6-dicyano-1,4-benzoquinone, phenyl alanine, triphenyl phosphine, and 1-bromo propionic acid were purchased from Aldrich. Ditert-butyl decarbonates (BOC anhydride) and 1-hydroxy benzotriazole (HOBt) were purchased from Merck, Germany. N,N′-Dicyclohexyl carbodiimide (DCC), NaOH pallets, methanol, silica gel (100–200 mesh), petroleum ether (60–80 °C), ethyl acetate, and N,N dimethylformamide were purchased from Sisco Research Laboratory, India. The water used in all experiments was Millipore Milli-Q grade. Furthermore, Dulbecco's modified Eagle's culture medium (DMEM), 2-deoxy-D-glucose (deoxy-glucose), sodium azide (NaN3), genistein, amiloride hydrochloride, sucrose, and methyl-βcyclodextrin were purchased from Sigma Aldrich and used as received. Fetal bovine serum (FBS), penicillin–streptomycin antibody, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Hi-media, while MitoTracker Red was purchased from Invitrogen.
Synthesis of the C-dots
In our previous work, we synthesized green emitting carbon dot (C-dots) from the DDQ (2,3-dichloro-5,6-dicyano-1,4 benzoquinone).25 Here, we used a somewhat similar procedure but with a change in the temperature (Fig. 1). Previously the temperature was kept at 180 °C, but in this study a 40 °C higher temperature (220 °C) was used. First, 20 mg of DDQ was taken in a 100 ml beaker and 50 ml of ortho-xylene was added followed by 5 min of sonication to obtain a homogeneous dark red solution. Then the solution was transferred into a Teflon-lined autoclave and heated at 220 °C for 5 h in an oven. After that, the autoclave was kept outside the oven for a few hours to cool down and then the reaction mixture was poured into a flat surface container, like a Petri plate. Then the solvent was evaporated from the reaction mixture in a vacuum oven at 90 °C for 3 h to obtain dry C-dots. These C-dots were dispersed into Milli-Q water and centrifuged for 10 min. Then the supernatant C-dots solution was further purified via dialysis (Av. size of the membrane was 23.8 mm). The fraction inside the dialysis membrane was taken carefully and was further freeze dried to obtain pure C-dots powder (Fig. 1) The detailed synthetic procedure, purification, and characterization methods for triphenylphosphine containing the amino acid-based amphiphile used in this study are provided in the ESI.†
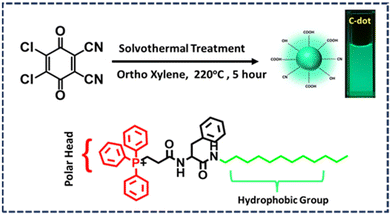 |
| | Fig. 1 Synthetic route for the C-dots and the molecular structure of the P. | |
Preparation of the C-dots and amphiphile conjugates (CDTP)
First, 100 μg of the C-dots was dissolved in Milli-Q water and then the needed amount of the aqueous solution of P was added into the C-dot solution fixing the weight percentage ratio from 5 to 20 for the different experiments. For the UV–visible analysis, fluorescence study, and the DLS zeta potential analysis the aqueous solution of P was added to the aqueous solution of the carbon dots to fix the concentration of the P starting from 0.1 mM to 1 mM. For the mitochondrial targeting, CDTP was prepared by dissolving 50 μg C-dots in 1 ml water and the concentration of the P was fixed at 0.2 mM in this solution. The cell labelling and the cytotoxicity of the CDTP were also measured in that combination of the concentration.
Cell labelling study
Chinese hamster ovary (CHO) cells and KB (oral cancer cells) cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% FBS and 1% penicillin–streptomycin at 37 °C and 5% CO2 atmosphere. For the cell labelling study, the cells were sub-cultured overnight in a 12-well plate with the medium. Next, 20 μL nanoprobe samples (CDTP) were added in DMEM medium followed by 45 min incubation. Then, the cells were washed with 7.4 PBS buffer to remove the unbound particles and were fixed by 4% paraformaldehyde solution for confocal microscope imaging.
For the co-localization study, the cells were incubated with CDTP for 45 min in DMEM medium. Then, the cells were washed with 7.4 PBS buffer and incubated with MitoTracker Red for a further 20 min. Next, the washed cells were fixed with 4% paraformaldehyde solution and the fixed cells were imaged under 488 nm excitation for CDTP and 561 nm excitation for MitoTracker Red imaging under a confocal microscope.
Cell uptake mechanism study
For the cell uptake mechanism study, KB cells were sub-cultured in DMEM medium at 37 °C with 5% CO2 atmosphere and then incubated with different inhibitors (or conditions) for 45 min, including 10 mM sodium azide (NaN3), 50 mM deoxy-glucose, 4 °C temperature (inhibits energy-dependent pathway), 450 mM sucrose (inhibits clathrin-mediated endocytosis), 200 μM genistein (inhibits caveolae-mediated endocytosis), 50 μM amiloride (inhibits macropinocytosis), and 10 mM methyl-βcyclodextrin (inhibits lipid-raft-mediated endocytosis). Then, the cells were incubated with CDTP for 45 min and the washed cells were detached from the wells using a cell scraper. The isolated cells were centrifuged and finally dispersed in PBS buffer at pH 7.4 for the flow-cytometric study.
Cytotoxicity assay
KB cells were sub-cultured overnight in a 24-well plate containing DMEM medium at 37 °C with a 5% CO2 atmosphere. After that, the cells were incubated with different doses of CDTP solution in DMEM medium for 24 h, washed with PBS buffer at pH 7.4, and then fresh DMEM medium was added to it. Next, 50 μL of freshly prepared MTT solution (5 mg MTT dissolved in 1 mL of deionized water) was added to each well and incubated for the next 4 h. Then, the supernatant solution was removed and the produced violet formazan was dissolved in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DMF–H2O solution of sodium-dodecyl sulfate. Finally, the absorbance was measured at 570 nm and cell viability was estimated using 100% viability of the control cells without any sample treatment.
:1 DMF–H2O solution of sodium-dodecyl sulfate. Finally, the absorbance was measured at 570 nm and cell viability was estimated using 100% viability of the control cells without any sample treatment.
Results and discussion
Characterization of the C-dots
First, the formation of the C-dots was confirmed by UV–visible spectroscopy as peaks at 266 nm and 380 nm were found for the π–π* and n–π* transitions of the chromophore moiety of the C-dots in the water medium.38 The optical analysis of the C-dots is described in the next part of the manuscript. To examine the surface functional groups of the C-dots, we carried out FT-IR spectroscopy in the dry state (Fig. S2†). Peaks were found at 3346 and 2969 cm−1, which were assigned to O–H and ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H stretching vibrations of the C-dots. Other peaks were found at 1645 cm−1 for the CO stretching frequency of the carboxylic groups (COO−). Another peak at 2213 cm−1 was due to the –CN present of the surface of the C-dots.39 It could be found from the high-resolution C 1s spectrum in the XPS analysis that there were two peaks located at 282.29 and 285.48 eV, which were due to the presence of CO, and C–O bonds respectively in the C-dots (Fig. S3a†). In the N 1s spectrum, the peaks at 399.66 and 402.19 eV were assigned to CN and OCNH bonds (Fig. S3c†). In the O 1s spectrum, peaks were found at 531.77 and 533.22 eV, which were assigned to C–O and CO bonds, respectively (Fig. S3c†).40 From the FEG-TEM images of the C-dots (Fig. 3f), we could find that the average particle size was 5 nm in the histogram plot.
C–H stretching vibrations of the C-dots. Other peaks were found at 1645 cm−1 for the CO stretching frequency of the carboxylic groups (COO−). Another peak at 2213 cm−1 was due to the –CN present of the surface of the C-dots.39 It could be found from the high-resolution C 1s spectrum in the XPS analysis that there were two peaks located at 282.29 and 285.48 eV, which were due to the presence of CO, and C–O bonds respectively in the C-dots (Fig. S3a†). In the N 1s spectrum, the peaks at 399.66 and 402.19 eV were assigned to CN and OCNH bonds (Fig. S3c†). In the O 1s spectrum, peaks were found at 531.77 and 533.22 eV, which were assigned to C–O and CO bonds, respectively (Fig. S3c†).40 From the FEG-TEM images of the C-dots (Fig. 3f), we could find that the average particle size was 5 nm in the histogram plot.
Optical properties of CDTP
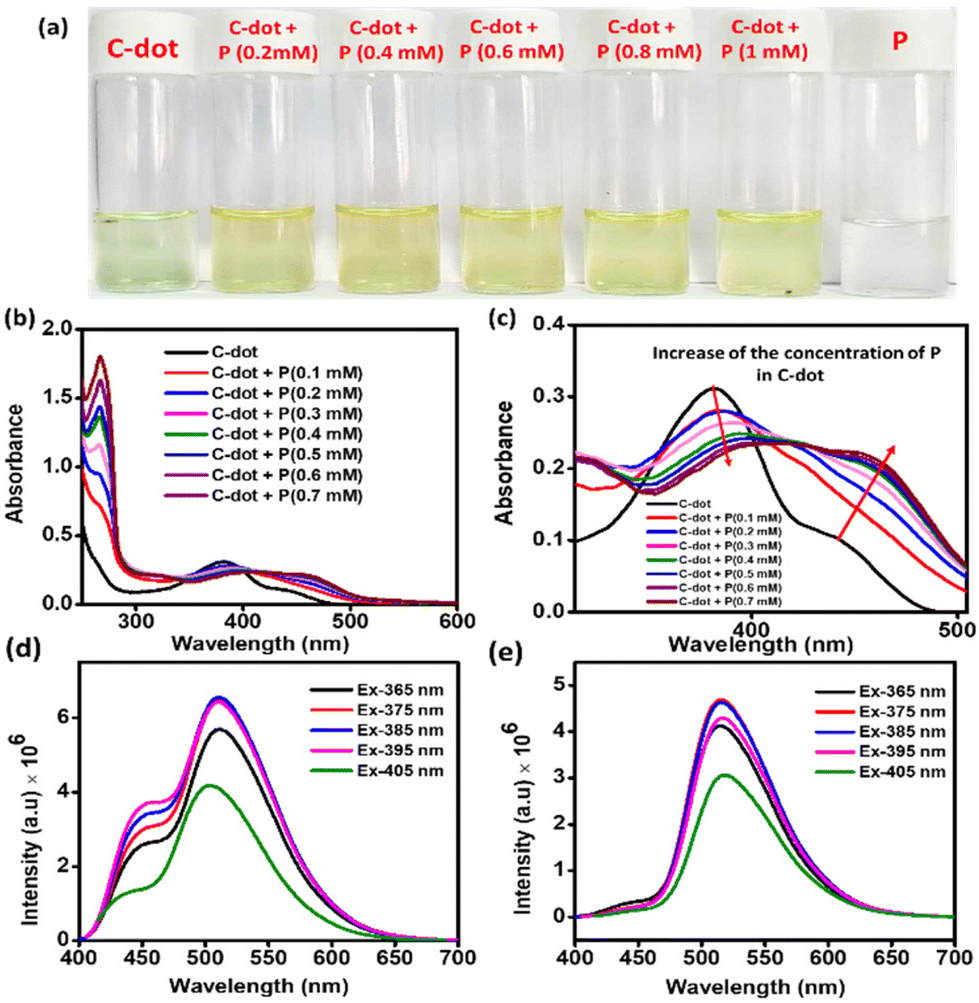
First, UV–visible analysis of the C-dots was performed in the water medium. Peaks at 266 and 380 nm were found, corresponding to π–π* and the n–π* transactions due to the presence of different functional groups, like CC, CO, and CN (Fig. 2a).41 A peak with low intensity was also observed at 440 nm, which was due to the surface state of the C-dots. This type of peak above 400 nm is mainly generated due to the presence of oxygen-containing functional groups at the surface of the C-dots.42 Furthermore, to know the interaction between the molecule P and C-dots, UV–visible studies were conducted with increasing the amount of P in the C-dot aqueous solution while maintaining the same concentration of C-dots (Fig. 2b and c). Here a very interesting observation was found whereby the intensity of the n–π* transition of the C-dots decreased and the surface peak at 440 nm was gradually red-shifted to 462 nm with the increasing amount of P (Fig. 2c). It was also evident that the intensity of this new surface peak located around 462 nm also gradually increased with increasing the amount of P. This 22 nm red-shift of the electron-rich surface band of the C-dots was probably due to photoexcited electron transfer from the surface of the C-dots to the electronically vacant positively charged triphenyl phosphine head of the nanospherical nanodots of the amphiphilic P in the water medium.43 This observation directly suggests noncovalent complex formation between the C-dots and the P associated with the electron-transfer process.43 Furthermore, this noncovalent complex formation between the C-dots and the P was also verified by the TCSPC (time-correlated single-photon counting) lifetime measurements. A gradual decay profile of the lifetime of the C-dots (Fig. S4†) at the emission wavelength of 520 nm with the increase in P (wt%) was evidence of the electron-transfer process from the C-dots to the micelle of the P in the water medium.44 This observation confirmed the probable surface-to-surface interaction between the micelle of the P and the C-dot in the aqueous solution. The light greenish colour (under daylight) solution of the C-dots (100 μg ml−1) in water (Fig. 2a) turned to yellow with the addition of 0.2 mM of P into the solution. This colour change also suggested a surface interaction between the C-dots and the P nanospheres. The fluorescence analysis also showed very interesting results. The aqueous solution of the C-dots emitted at 510 nm with a corresponding shoulder peak at 451 nm at the excitation wavelength of 365 nm. Fig. 2d shows that at the excitation wavelength starting from 365 nm to 405, the C-dots showed dual emission centres. The shoulder peak at 450 nm completely vanished when we added the P in to the C-dots solution (Fig. 2e). Furthermore, to examine the photostable character of the C-dots, we performed a photoluminescence study. At the excitation wavelength of 425 nm, the emission wavelength and the intensity were not different after exposing the aqueous C-dot solution under broad daylight for 3 h (Fig. S5†). This experiment indicates its stability under the photoradiative conditions.
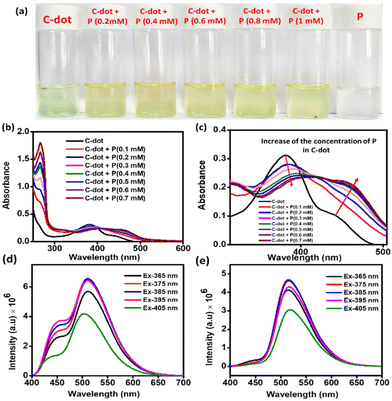 |
| | Fig. 2 (a) Photographs of an aqueous solution of carbon dots, and the colour change of the solution after the gradual addition of P amphiphile. (b) Change in the absorbance spectra of the C-dots with increasing P, (c) zoomed-in version of the change. (d) Excitation-dependent emission of the C-dots (e) in CDTP. | |
Morphological and size-distribution analysis of CDTP
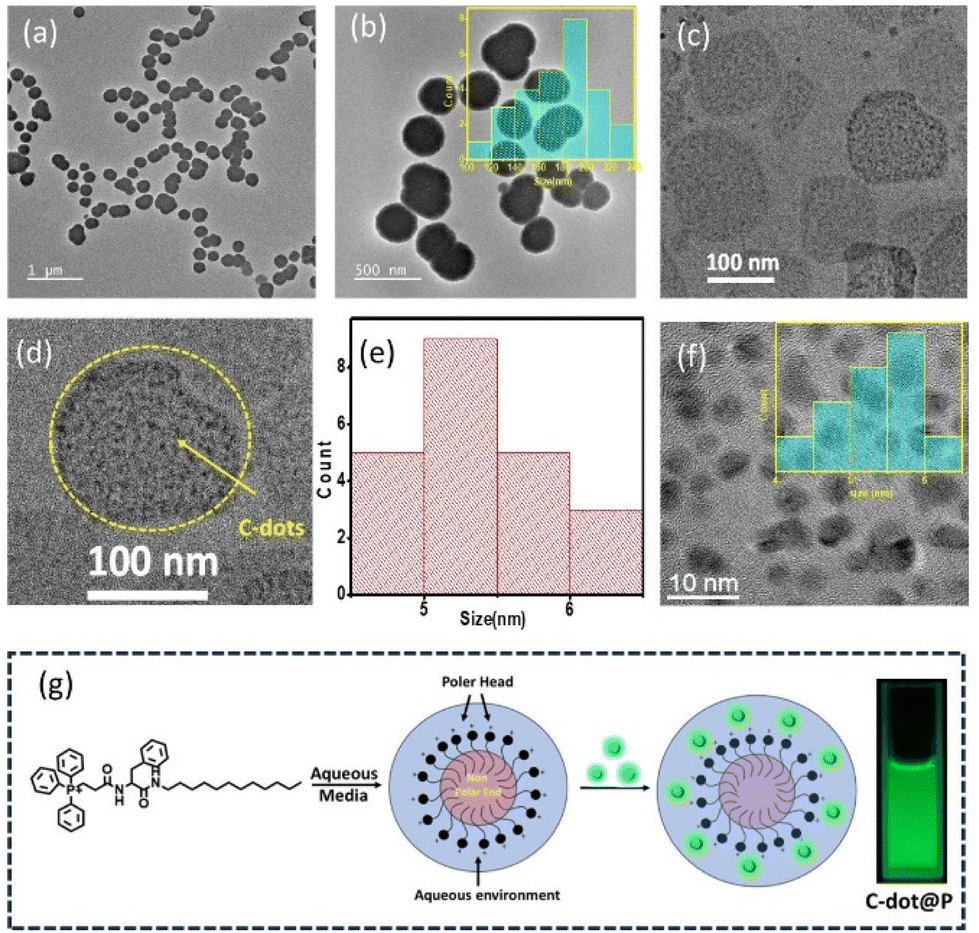
The amphiphilic P leads to spherical micelle-type nanostructure formation in the aggregated state in water, as confirmed in the FEG-TEM and CRYO-TEM images (Fig. 3a and b). The size distribution of the micelles was found to be from 120 to 220 nm with an average size of 180 nm, as shown in the histogram size-distribution plot (Fig. 3c). On the other hand, the FEG-TEM images also confirmed the average size of the carbon dot was 5 nm, as we previously noted (Fig. 3f). Next, we also captured FEG-TEM images after the addition of the C-dots with the aggregated P in the water medium. Fig. 3d and the zoomed-in photograph in Fig. 3e confirmed the perfect decoration of C-dots on the surface of the nanospherical dots of the P. We already observed the noncovalent complex formation between the C-dots and the P in the UV–visible spectrum, which was probably due to the electrostatic interaction between the two oppositely charged surfaces of the P micelle and the C-dots. P was designed in such a way that the positively charged polar head group should be present on the outer side of the nanospherical dots in the water medium. This expectation was further confirmed by measuring the emission intensity of the 0.01 mM concertation of pyrene (Py) in water with gradually increasing the concentration of P. The hydrophobic pocket inside the nanosphere helped to increase the solubility of the hydrophobic Py in the water by encapsulating the Py inside a hydrophobic pocket, as noted by the gradual increase in the emission intensity of Py with the gradual addition of the P (Fig. S6†). So, this result confirmed the outer sphere presence of the positively charged polar group of the amphiphile when they formed such nanospheres.45 Furthermore, the average size of C-dots decorated on the nanospheres of P was also found to be 5 nm, which was exactly the same as the purified C-dots before the addition of the aqueous solution of P (Fig. 3f).
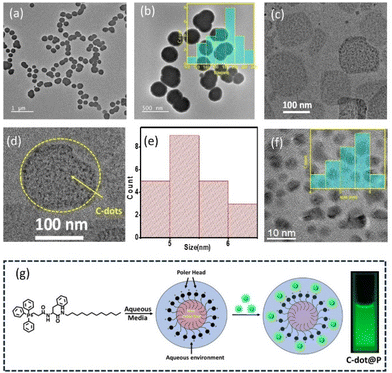 |
| | Fig. 3 (a) Cryo-TEM and (b) FEG-TEM images of the P (inset, size-distribution plot). (c) FEG-TEM images of the CDTP and the zoomed-in version (d). (e) Size-distribution plot of the C-dots in CDTP (f) FEG-TEM images of the C-dots and size-distribution plot (inset). (g) Graphical presentation of the P in water and in the CDTP. | |
Surface charge
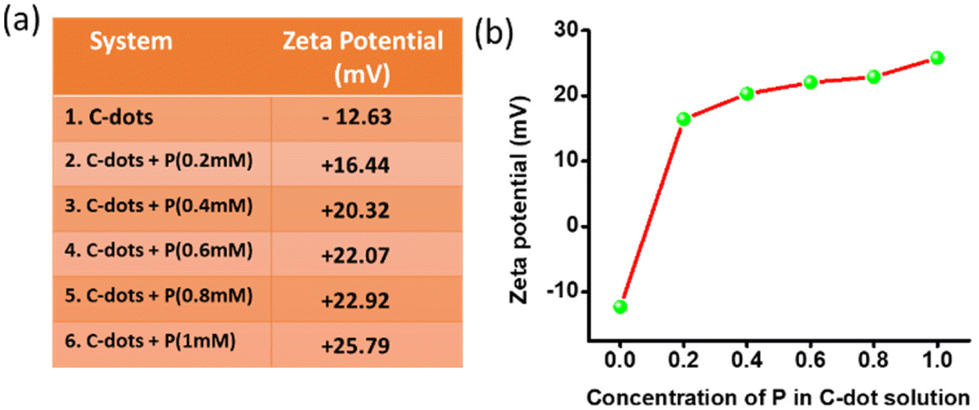
The zeta potential of the aqueous solution of the C-dots was found to be −12.63 mV (100 μg ml−1). The gradual addition of an aqueous solution of P in the C-dots increased the overall surface charge. In Fig. 4a, it could be observed that the zeta potential of the C-dots solution gradually increased with the addition of P. The solution of carbon dots with a negative surface charge surprisingly changed to +16.44 mV as soon as we added the aqueous solution of P, with the concentration of P fixed at 0.2 mM. At a 1 mM concentration of P, the CDTP displayed a zeta potential of +25.79 mV. We checked the zeta potential of the aqueous P (2.5 mM) too, which was +50.86 mV. Here the presence of the –COOH and –OH groups on the surface of the C-dots was the main reason for the negative surface charge of the C-dots.46,47 There was an increase in surface charge of almost 29 mV in the presence of 0.2 mM P in the C-dots solution, due to the availability of the positively charged surface of the spherical nanodots of the P. The positively charged nanospheres of P electrostatically attract the negatively charged C-dots and the C-dots are then decorated on the surface of the nanospheres, as it was evident from the respective TEM images (Fig. 3d and e).
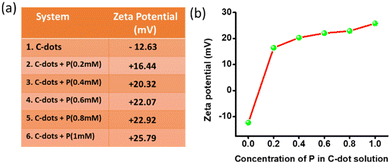 |
| | Fig. 4 (a) and (b) Change in the surface charge of the C-dot aqueous solution with adding P. | |
Rapid mitochondria labelling by the CDTP
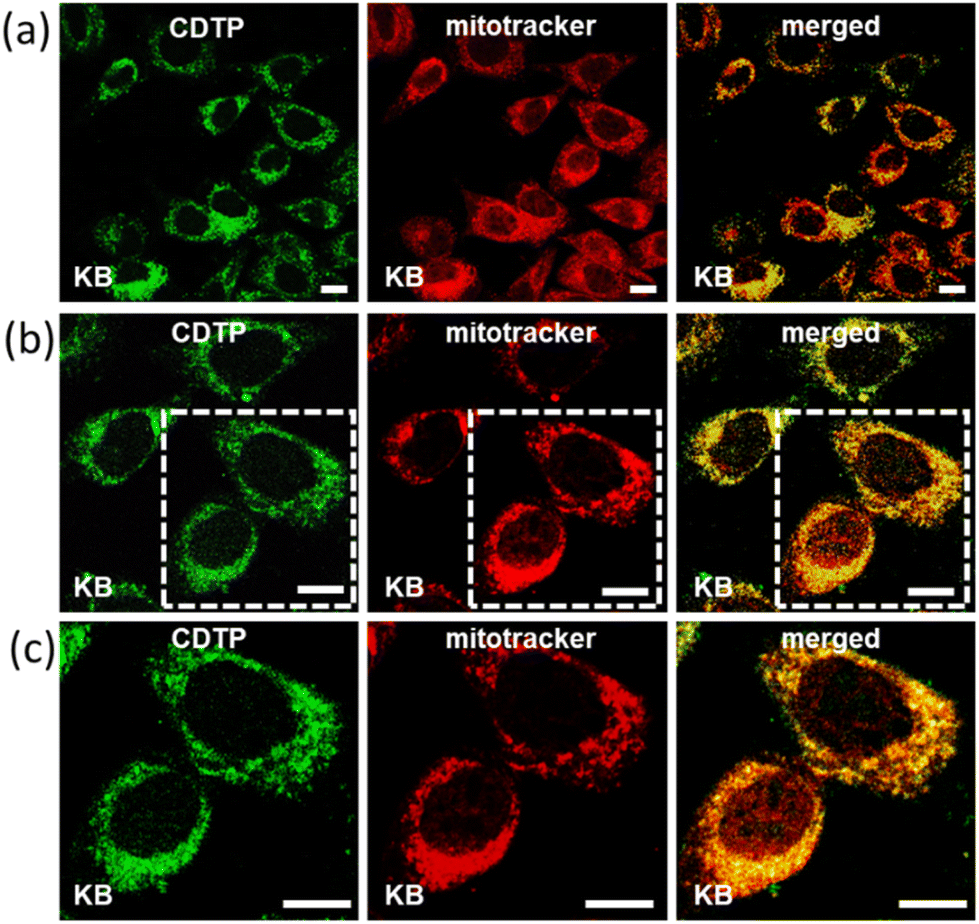
Mitochondria targeting and imaging of both the cancerous and non-cancerous cell lines were performed by our C-dots-tagged nanosphere of P (CDTP). The KB cell line (oral cancer cells) and CHO cell line (Chinese hamster ovary cells) were used respectively as the model cancerous and non-cancerous cell lines in this study. First, we checked the performance for mitochondria targeting in the KB cell line by incubating them with C-dots (50 μg ml−1). After an incubation time of 45 min, we did not find any emission of the C-dots from those incubated KB cells at excitation wavelengths from 400 nm to 480 nm (Fig. S7†), confirming that the control C-dots were not able to enter in to cells. However, when the KB cells were incubated with CDTP (0.2 mM P in 50 μg ml−1 C-dots) for 45 min and the incubated cells were observed by microscope at the excitation wavelength of 488 nm, we found a strong green emission, indicating that the CDTP was successfully able to label those cells by entering inside the cells. To know whether the CDTP were labelling the mitochondria or not, those cells were further incubated with MitoTracker Red for the next 20 min. Interestingly, we observed that our green flourishing CDTP was efficiently targeting the mitochondria within 1 h, which makes this system more desirable over many mitochondrial-targeting fluorophores reported in the literature.48–50 The co-localization of the C-dots in the incubated KB cells, with both the CDTP and MitoTracker Red, was confirmed by confocal microscopy. First, the excitation wavelength at 488 nm was carefully monitored to image the carbon dots as they emitted green emission at that excitation wavelength. Furthermore, the MitoTracker Red was excited at the excitation wavelength of 561 nm, which is the reported excitation wavelength for MitoTracker Red. Finally, the two separated images were merged with each other and a yellow merged image with a good Pearson coefficient was obtained, confirming the co-localization of the CDTP within the mitochondria (Fig. 5). The same procedure was applied for the CHO cell line to investigate the targeting of mitochondria by our reported nanoprobes to see if CDTP would be able to be used to image the mitochondria in a non-cancerous cell line. Here the CHO cell line was incubated with CDTP nanoprobes for 45 min and then they were washed with a subsequent incubation with MitoTracker Red. In this case, the same monitoring of the excitation wavelength of both the C-dots and MitoTracker Red was performed as for the KB cell and a perfect merging of images with a good Pearson coefficient was also obtained in this case too. Like the KB cell, 1 h incubation of the CDTP nanoprobes with the CHO cell was enough for co-localization of the carbon dots with the MitoTracker dye (Fig. 6). The nontoxic nature of the CDTP was checked with the KB cell, verifying this with different concentrations of CDTP. Fig. S8a† shows the good cell viability of the KB cells with the increasing concentration of CDTP, confirming the nontoxic behaviour of CDTP towards the KB cell. The IC50 value was found to be 30 μM P present in the CDTP solution from Fig. S8b.† Herein, the observed results for mitochondria labelling with this hybrid nanomaterial (CDTP) is significant due to allowing a rapid and fast targeting, compared to the literature, where the targeting agents typically need 4 to 24 h for mitochondria labelling.51–53 Herein, this system could label the mitochondria within 45 min. Additionally, in this study, negatively charged particles were used as mitochondrial-targeting agents with a positively charged carrier.
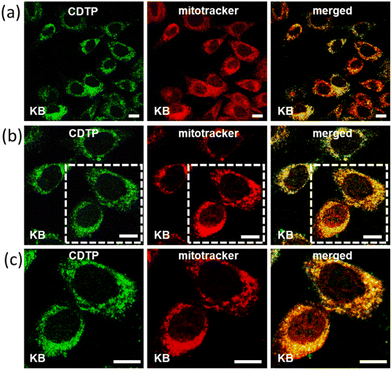 |
| | Fig. 5 Evidence of the mitochondria targeting of CDTP within 45 minutes. Typically, KB cells were incubated with CDTP for 30 min, washed, and then incubated with MitoTracker Red for 15 min. Next, the washed cells were fixed with 4% paraformaldehyde and imaged under a confocal microscope. An excitation wavelength of 488 nm was used for imaging the green-emissive carbon dots and 561 nm excitation wavelength for imaging MitoTracker red (red emission). The yellow colour and high Pearson correlation coefficient (PCC: 85%–92%) values in the merged images indicate the predominate co-localization of the probe with MitoTracker Red. Scale bar represents 10 μm. Low-magnification images are shown in (a), and high-resolution images are shown in (b) and (c). | |
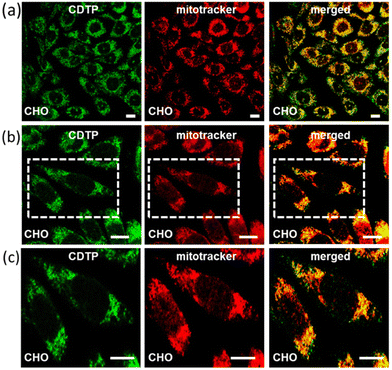 |
| | Fig. 6 Co-localization study of CDTP with MitoTracker Red within 45 min in CHO cells. Typically, CHO cells were incubated with CDTP for 45 min followed by washing and a subsequent incubation with MitoTracker Red for the next 15 min. Next, the washed cells were fixed with 4% paraformaldehyde followed by imaging under a confocal microscope. The green-emissive C-dots were imaged under blue excitation (488 nm) and MitoTracker Red under green excitation (561 nm) using a confocal microscope. The yellow colour and high Pearson correlation coefficient (PCC: 72%85%) values in the merged images suggest the predominate co-localization of the probe with MitoTracker Red. Scale bar represents 10 μm. Low-magnification images are shown in (a) and high-resolution images are shown in (c). | |
Path of the cellular uptake of the nanoprobes
The surface charge and the size of the nanoprobes are the major factors that determine the process of their cellular uptake pathway, whereby either they will follow the direct membrane penetration pathway or the endocytosis technique pathway.54 The positively charged surface and the larger size of the nanoprobe favours cell entry through the endocytosis process.54 The different pathway of the endocytosis process demands different sizes; for instance, endocytosis through the phagocytosis pathway needs the size of the nano probes to be larger than 500 nm, while endocytosis through the Clathrin-mediated pathway needs the size of the nanoprobes to be between 50–150 nm, and endocytosis through the micropinocytosis pathway also demands a larger size of particles (>500 nm). On the other hand, nanoparticles with a positive charged surface are more favourable for internalization through the endocytosis process in cells.55,56 He et al. published an important study that emphasized that larger particles with higher positive surface charges of the nanoprobes are much more efficient for cellular uptake through endocytosis.57 It has long been a serious problem with carbon dots that they are usually associated with a negatively charged surface, which is not perfectly suitable for their cellular uptake through the endocytosis mechanism. On the other hand, positively charged particles ranging in size between 100–200 nm fulfil the all-fundamental necessities for cellular cell entry through the endocytosis mechanism.58
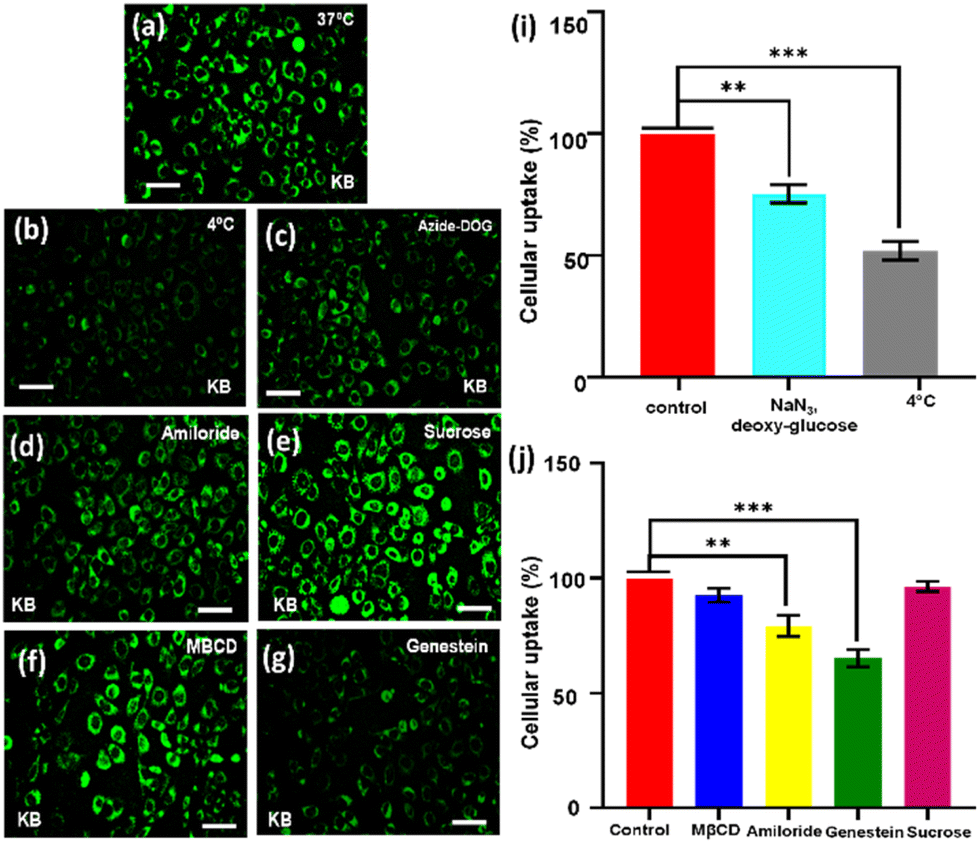
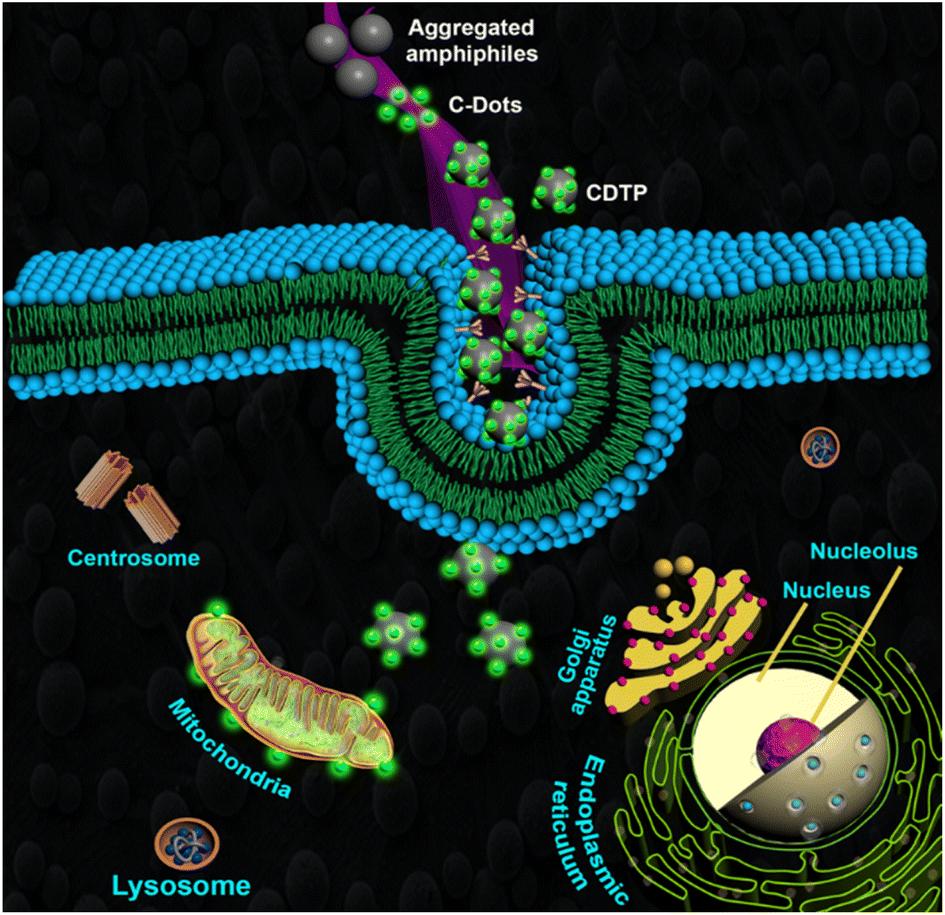
Next, we were curious to understand how the CDTP penetrates inside the cell. We had already observed that carbon dots without the P amphiphile were not able to penetrate the cell. However, when they were tagged with the nanospherical dots of the P, it was evident that the energy-dependent endocytic procedure for cell uptake was probable and was highly influenced by the ATP and temperature.59,60 This was confirmed by treating KB cells with CDTP nanoprobes and these were incubated at 4 °C for 45 min. A drastic fall in emission of the nanoprobes was observed (Fig. 7c), which was due to the cellule uptake inhibition of the probes at lower temperature. Next, NaN3/deoxy-glucose were used in another test, as they are well known for disturbance of the production of the ATP and for blocking the endocytic cell entry.59 This time, it was also seen that there was a considerable decrease in the emission for the nanoprobes (Fig. 7b). These two results indicate that the cell entry of the nanoprobe was highly influenced by the temperature and ATP. Further, to know the exact endocytosis pathway, we used other endocytosis inhibitors, such as the MβCD (inhibitor of lipid raft-mediated endocytosis), amiloride (inhibitor of caveolae-mediated endocytosis), genestein (inhibitor of caveolae-mediated endocytosis), and sucrose. In this case, genestein was able to inhibit the cell entry of the nanoprobes unlike the other three inhibitors (Fig. 7h). So, it could be concluded that the endocytosis of the probes very much followed the caveolae pathway. It has been well reported that the mitochondrial membrane has a very high negative potential (−150 mV to −200 mV) with a dense hydrophobic layer.61,62 So, either cationic charge (for electrostatic binding with the negatively charged mitochondrial membrane) or lipophilicity (for binding with hydrophobic part of the membrane) is required to bind with the mitochondrial membrane. As we have already mentioned, the C-dots alone could not penetrate the cell, but once they had adhered to the surface of the spherical aggregated amphiphile, they were able to enter inside the cell. This was because of the fact that the overall positive charge of the conjugate system (CDTP) helped to carry the fluorescent C-dots inside the cell through the endocytosis process.22–24 So, after entering into the cell, the negatively charged C-dots were probably capable of targeting the mitochondria due to a hydrophobic effect, although they have a small negative charge (−12.6 mV) at the surface. C-dots have a strong hydrophobic conjugated aromatic core.63 Here, in this carbon dots system, the conjugated aromatic core was confirmed by 1H-NMR spectroscopy. The peak around 7.2 ppm was due to the presence of the aromatic core of the carbon dots (Fig. S8†). Another possible mechanism for targeting and labelling the mitochondria for these C-dots is, after entering inside the cell, the micellar structure of the amphiphile P may be broken and the molecular P becomes electrostatically attached with the negatively charged C-dots. Finally, the electrostatically conjugated systems of both the C-dots (with a negative charge) and the amphiphile (with a positive charge) successfully attach to the surface of the mitochondria for the labelling. The C-dots without amphiphiles were not able to enter properly into the cell, but once they are tagged with the micelle, huge number of carbon dot fluorophore particles become capable of entering into the cell. This large number of negatively charged C-dots with the presence of positively charged amphiphile inside the cell is probably the cause of them adhering on the surface of the mitochondrial membrane (which is negatively charged) to enable imaging of the mitochondria (Fig. 8).
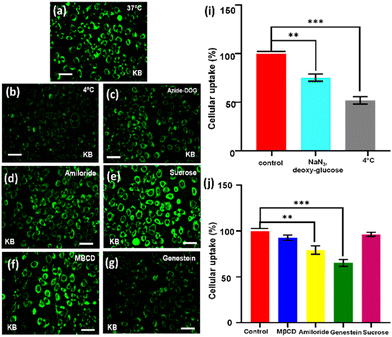 |
| | Fig. 7 Evidence of the energy-dependent endocytic cell uptake of CDTP as observed from fluorescence imaging and flow cytometry-based quantitative estimation analysis. Typically, KB cells were incubated with (a) endocytosis inhibitors, (b) at 4 °C, (c) Azide-DOG, (d) amiloride, (e) sucrose, (f) MBCD, (g) genestein along with incubation with CDTP for 30 min and then imaged by fluorescence microscopy. Significant uptake inhibition occurred at 4 °C, indicating the endocytic uptake of CDTP. Proof of the energy-dependent endocytic cell uptake of CDTP was obtained from flow cytometry-based quantitative analysis (i and j). Typically, KB cells were incubated with NaN3, deoxy-glucose or 4 °C for 45 min along with the incubation of CDTP for another 30 min. Flow cytometry-based quantitative estimation showed there was a significant uptake inhibition of CDTP in the presence of different endocytosis inhibitors (50% of cellular uptake at 4 °C). Additionally, uptake inhibition in the presence of genestein indicated a caveolae-mediated endocytic uptake for CDTP, where the other pharmacological inhibitors hardly affected the uptake mechanism. Error bar represents mean ± S.D.; asterisks denote significant differences between the values (**P < 0.01, ***P < 0.001); Dunnett's multiple comparison test. | |
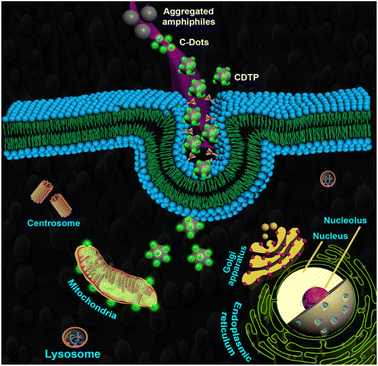 |
| | Fig. 8 Mechanistic endocytic pathway (caveolae pathway) of CDTP for imaging of the mitochondria. | |
Conclusion
This study demonstrates a solvothermal method for fabricating green-fluorescent C-dots carrying negative charges on their surfaces, but the C-dots were found to be incapable of penetrating the cell properly. To address this challenge, a non-fluorescent triphenylphosphine containing amphiphile with a positively charged head group was designed and synthesized and it formed a spherical micelle-type nanostructure in water in the aggregated state. These nanospheres-based cationic species acted as a carrier for taking the green-fluorescent negative C-dots inside the cells (both cancerous and non-cancerous) and they were then used to image the mitochondria in living cells. The nanohybrid system with the fluorescent C-dots and triphenylphosphine containing amphiphile with an amino acid residue were found to enter into the cells through the endocytosis process with the caveolae pathway.
Conflicts of interest
There is no conflict of interest.
Acknowledgements
N. H. acknowledges UGC for financial assistance. We acknowledge Prof. Nikhil R. Jana (IACS) for his helpful discussion.
References
- M. Tobler, N. Barts and R. Greenway, Integr. Comp. Biol., 2019, 59, 900–911 CrossRef CAS PubMed.
- M. Ashwell and T. Work, Annu. Rev. Biochem., 1970, 39, 251–290 CrossRef CAS PubMed.
- L. Ernster and G. Schatz, J. Cell Biol., 1981, 91, 227–255 CrossRef CAS PubMed.
- J. R. Friedman and J. Nunnari, Nature, 2014, 505, 335–343 CrossRef CAS PubMed.
- A. W. El-Hattab, W. J. Craigen and F. Scaglia, Biochim. Biophys. Acta, Mol. Basis Dis., 2017, 1863, 1539–1555 CrossRef CAS PubMed.
- M. Saki and A. Prakash, Free Radical Biol. Med., 2017, 107, 216–227 CrossRef CAS PubMed.
- C. Vásquez-Trincado, I. García-Carvajal, C. Pennanen, V. Parra, J. A. Hill, B. A. Rothermel and S. Lavandero, J. Physiol., 2016, 594, 509–525 CrossRef PubMed.
- Y. Yang, S. Karakhanova, W. Hartwig, J. G. D'Haese, P. P. Philippov, J. Werner and A. V. Bazhin, J. Cell. Physiol., 2016, 231, 2570–2581 CrossRef CAS PubMed.
- W. X. Zong, J. D. Rabinowitz and E. White, Mol. Cell, 2016, 61, 667–676 CrossRef CAS PubMed.
- R. Zhai, B. Fang, Y. Lai, B. Peng, H. Bai, X. Liu, L. Li and W. Huang, Chem. Soc. Rev., 2023, 52, 942–972 RSC.
- H. Blom and J. Widengren, Chem. Rev., 2017, 117, 7377–7427 CrossRef CAS PubMed.
- X. Q. Yi, Y. F. He, Y. S. Cao, W. X. Shen and Y. Y. Lv, ACS Sens., 2019, 4, 856–864 CrossRef CAS PubMed.
- Q. Li and Y. Huang, J. Pharm. Invest., 2020, 50, 271–293 CrossRef.
- K. L. Horton, K. M. Stewart, S. B. Fonseca, Q. Guo and S. O. Kelley, Cell Chem. Biol., 2008, 15, 375–382 CAS.
- J. Zielonka, J. Joseph, A. Sikora, M. Hardy, O. Ouari, J. Vasquez-Vivar, G. Cheng, M. Lopez and B. Kalyanaraman, Chem. Rev., 2017, 117, 10043–10120 CrossRef CAS PubMed.
- H. Zhu, J. Fan, J. Du and X. Peng, Acc. Chem. Res., 2016, 49, 2115–2126 CrossRef CAS PubMed.
- H. Wang, B. Fang, B. Peng, L. Wang, Y. Xue, H. Bai, S. Lu, N. H. Voelcker, L. Li, L. Fu and W. Huang, Front. Chem., 2021, 9, 683220 CrossRef CAS PubMed.
- J. Zielonka, J. Joseph, A. Sikora, M. Hardy, O. Ouari, J. VasquezVivar, G. Cheng, M. Lopez and B. Kalyanaraman, Chem. Rev., 2017, 117, 10043–10120 CrossRef CAS PubMed.
- J. Ahn, B. Lee, Y. Choi, H. Jin, N. Y. Lim, J. Park, J. H. Kim, J. Bae and J. H. Jung, J. Mater. Chem. B, 2018, 6, 5698–5707 RSC.
- M. T. Jeena, S. Kim, S. Jin and J. H. Ryu, Cancers, 2020, 12, 4 CrossRef CAS PubMed.
- W. Zhou, H. Yu, L. J. Zhang, B. Wu, C. X. Wang, Q. Wang, K. Deng, R. X. Zhuo and S. W. Huang, Nanoscale, 2017, 9, 17044–17053 RSC.
- J. Qin, N. Gong, Z. Liao, S. Zhang, P. Timashev, S. Huo and X. J. Liang, Nanoscale, 2021, 13, 7108 RSC.
- R. Ray, S. Ghosh and N. R. Jana, Phys. Chem. B, 2021, 125, 9186–9196 CrossRef CAS PubMed.
- Y. Zheng, X. Ji, B. Yu, K. Ji, D. Gallo, E. Csizmadia, M. Zhu, M. R. Choudhury, L. K. C. De La Cruz, V. Chittavong, Z. Pan, Z. Yuan, L. E. Otterbein and B. Wang, Nat. Chem., 2018, 10, 787–794 CrossRef CAS PubMed.
- N. Hazra, S. Hazra, S. Paul and A. Banerjee, Chem. Commun., 2023, 59, 4931 RSC.
- N. Hazra, S. Paul, S. Hazra and A. Banerjee, ChemistrySelect, 2023, 8, 1–7 Search PubMed.
- S. Paul, S. Panja, N. Hazra and A. Banerjee, J. Org. Chem., 2024, 89(1), 91–100 CrossRef CAS PubMed.
- N. C. Verma, C. Rao and C. K. Nandi, J. Phys. Chem. C, 2018, 122, 4704–4709 CrossRef CAS.
- A. Pal, M. P. Sk and A. Chattopadhyay, ACS Appl. Mater. Interfaces, 2016, 8, 5758–5762 CrossRef CAS PubMed.
- R. Atchudan, T. N. J. I. Edison, S. Perumal, R. Vinodh and Y. R. Lee, J. Mol. Liq., 2019, 296, 111817 CrossRef CAS.
- R. Atchudan, T. N. J. I. Edison, K. R. Aseer, S. Perumal, N. Karthik and Y. R. Lee, Biosens. Bioelectron., 2018, 99, 303–311 CrossRef CAS PubMed.
- P. Krishnaiah, R. Atchudan, S. Perumal, E. S. Salama, Y. R. Lee and B. H. Jeon, Chemosphere, 2022, 286, 131764 CrossRef CAS PubMed.
- R. Atchudana, T. N. J. I. Edisona, S. Perumalb, N. Muthuchamyc and Y. R. Lee, Fuel, 2020, 275, 1178 Search PubMed.
- R. Atchudan, S. C. Kishore, P. Gangadaran, T. N. J. I. Edison, S. Perumal, R. L. Rajendran, M. Alagan, S. A. Rashed, B. C. Ahn and Y. R. Lee, Environ. Res., 2022, 204, 112365 CrossRef CAS PubMed.
- S. T. Yang, L. Cao, P. G. Luo, F. Lu, X. Wang, H. Wang, M. J. Meziani, Y. Liu, G. Qi and Y. P. Sun, J. Am. Chem. Soc., 2009, 131(32), 11308–11309 CrossRef CAS PubMed.
- X. W. Hua, Y. W. Bao, Z. Chen and F. G. Wu, Nanoscale, 2017, 9, 10948–11096 RSC.
- J. H. Liu, R. S. Li, B. Yuan, J. Wang, Y. F. Li and C. Z. Huang, Nanoscale, 2018, 10, 17402–17408 RSC.
- H. Qi, M. Teng, M. Liu, S. Liu, J. Li, H. Yu, C. Teng, Z. Huang, H. Liu, Q. Shao, A. Umar, T. Ding, Q. Gao and Z. Guo, J. Colloid Interface Sci., 2019, 539, 332–341 CrossRef CAS PubMed.
- S. Paul, N. Hazra, S. Hazra and A. Banerjee, J. Mater. Chem. C, 2020, 8, 15735–15741 RSC.
- S. Paul, A. Bhattacharya, N. Hazra, K. Gayen, P. Sen and A. Banerjee, Langmuir, 2022, 38(29), 8829–8836 CrossRef CAS PubMed.
- Y. Zhou, K. J. Mintz, S. K. Sharma and R. M. Leblanc, Langmuir, 2019, 35(28), 9115–9132 CrossRef CAS PubMed.
- H. Ding, S. B. Yu, J. S. Wei and H. M. Xiong, ACS Nano, 2016, 10(1), 484–491 CrossRef CAS PubMed.
- S. Hazra, S. Paul, K. Basu, A. K. Nandi and A. Banerjee, J. Phys. Chem. C, 2022, 126(13), 5906–5915 CrossRef CAS.
- M. K. Barman, B. Jana, S. Bhattacharyya and A. Patra, J. Phys. Chem. C, 2014, 118, 20034–20041 CrossRef CAS.
- F. Behroozi, M. J. Abdkhodaie, H. S. Abandansari, L. Satarian, M. Molazem, K. T. A. Jamal and H. Baharvand, Acta Biomater., 2018, 76, 239–256 CrossRef CAS PubMed.
- J. C. Kung, I. Tseng, C. S. Chien, S. H. Lin, C. C. Wang and C. J. Shih, RSC Adv., 2020, 10, 41202–41208 RSC.
- C. Wu, Y. Zheng, W. Wang, Y. Liu, J. Yu and Y. Liu, Langmuir, 2022, 38(45), 13771–13781 CrossRef CAS PubMed.
- A. Chakraborty and N. R. Jana, Phys. Chem. C, 2015, 119, 2888–2895 CrossRef CAS.
- A. Mallick, A. Nandi and S. Basu, ACS Appl. Bio Mater., 2019, 2, 14–19 CrossRef CAS PubMed.
- P. Ning, L. Huang, Y. Bao, Y. Fu, C. Xu, Y. Shen, X. Zhou, X. Wen, Y. Cheng and Y. Qin, Bioconjugate Chem., 2020, 31, 2719–2725 CrossRef CAS PubMed.
- C. Li, W. Zhang, S. Liu, X. Hu and Z. Xie, ACS Appl. Mater. Interfaces, 2020, 12, 30077–30084 CrossRef CAS PubMed.
- J. L. Wang, L. Zhang, M. J. Zhao, T. Zhang, Y. Liu and F. L. Jiang, ACS Appl. Bio Mater., 2021, 4, 1760–1770 CrossRef CAS PubMed.
- P. Panja and N. R. J. Jana, Phys. Chem. Lett., 2020, 11, 2363–2368 CrossRef CAS PubMed.
- M. S. Almeida, E. Susnik, B. Drasler, P. T. Blanco, A. P. Fink and B. R. Rutishauser, Chem. Soc. Rev., 2021, 50, 5397 RSC.
- A. Akinc and G. Battaglia, Cold Spring Harbor Perspect. Biol., 2013, 5, a016980 CrossRef PubMed.
- S. Jambhrunkar, Z. Qu, A. Popat, J. Yang, O. Noonan, L. Acauan, Y. Ahmad Nor, C. Yu and S. Karmakar, Mol. Pharmaceutics, 2014, 11, 3642–3655 CrossRef CAS PubMed.
- C. He, Y. Hu, L. Yin, C. Tang and C. Yin, Biomaterials, 2010, 31, 3657–3666 CrossRef CAS PubMed.
- J. Rejman, V. Oberle, I. S. Zuhorn and D. Hoekstra, Biochem. J., 2004, 377, 159–169 CrossRef CAS PubMed.
- R. Ray, S. Ghosh, P. Panja and N. R. Jana, ACS Appl. Bio Mater., 2023, 6, 2338–2344 CrossRef CAS PubMed.
- N. Xin, D. Gao, B. Su, T. Zhou, Y. Zhu, C. Wu, D. Wei, J. Sun and H. Fan, ACS Sens., 2023, 8, 1161–1172 CrossRef CAS PubMed.
- S. R. Jean, M. Ahmed, E. K. Lei, S. P. Wisnovsky and S. O. Kelley, Acc. Chem. Res., 2016, 49, 1893–1902 CrossRef CAS PubMed.
- S. Kim, H. Y. Nam, J. Lee and J. Seo, Biochemistry, 2020, 59, 270–284 CrossRef CAS PubMed.
- H. Yang, Y. Liu, Z. Guo, B. Lei, J. Zhuang, X. Zhang, Z. Liu and C. Hu, Nat. Commun., 2019, 10, 1789 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis procedure,1H-NMR and 13C-NMR spectra, FT-IR spectrum, TCSPC data, MTT assay, FEG-TEM images. See DOI: https://doi.org/10.1039/d4nr00665h |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a
*a