
Multi-atomic loaded C2N1 catalysts for CO2 reduction to CO or formic acid†
Yimeng
Sun
a,
Lin
Tao
 *a,
Mingjie
Wu
*b,
Davoud
Dastan
c,
Javed
Rehman
d,
Lixiang
Li
a and
Baigang
An
*a
*a,
Mingjie
Wu
*b,
Davoud
Dastan
c,
Javed
Rehman
d,
Lixiang
Li
a and
Baigang
An
*a
aSchool of Chemical Engineering, University of Science and Technology Liaoning, Anshan 114051, China. E-mail: taolin@ustl.edu.cn; bgan@ustl.edu.cn
bState Key Laboratory of New Textile Materials and Advanced Processing Technologies, Wuhan Textile University, Wuhan 430200, China. E-mail: mjwu@wtu.edu.cn
cDepartment of Materials Science and Engineering, Cornell University, Ithaca, NY 14850, USA
dState Key Laboratory of Metastable Materials Science and Technology, Yanshan University, Qinhuangdao 066004, China
First published on 19th April 2024
Abstract
In recent years, the development of highly active and selective electrocatalysts for the electrochemical reduction of CO2 to produce CO and formic acid has aroused great interest, and can reduce environmental pollution and greenhouse gas emissions. Due to the high utilization of atoms, atom-dispersed catalysts are widely used in CO2 reduction reactions (CO2RRs). Compared with single-atom catalysts (SACs), multi-atom catalysts have more flexible active sites, unique electronic structures and synergistic interatomic interactions, which have great potential in improving the catalytic performance. In this study, we established a single-layer nitrogen–graphene-supported transition metal catalyst (TM-C2N1) based on density functional theory, facilitating the reduction of CO2 to CO or HCOOH with single-atom and multi-atomic catalysts. For the first time, the TM-C2N1 monolayer was systematically screened for its catalytic activity with ab initio molecular dynamics, density of states, and charge density, confirming the stability of the TM-C2N1 catalyst structure. Furthermore, the Gibbs free energy and electronic structure analysis of 3TM-C2N1 revealed excellent catalytic performance for CO and HCOOH in the CO2RR with a lower limiting potential. Importantly, this work highlights the moderate adsorption energy of the intermediate on 3TM-C2N1. It is particularly noteworthy that 3Mo-C2N1 exhibited the best catalytic performance for CO, with a limiting potential (UL) of −0.62 V, while 3Ti-C2N1 showed the best performance for HCOOH, with a corresponding UL of −0.18 V. Additionally, 3TM-C2N1 significantly inhibited competitive hydrogen evolution reactions. We emphasize the crucial role of the d-band center in determining products, as well as the activity and selectivity of triple-atom catalysts in the CO2RR. This theoretical research not only advances our understanding of multi-atomic catalysts, but also offers new avenues for promoting sustainable CO2 conversion.
1. Introduction
The increase in fossil fuel consumption leads to the accumulation of CO2 in the atmosphere, causing serious environmental problems such as the greenhouse effect and ocean acidification, forcing people to find effective ways to reduce the concentration of CO2 in the atmosphere and promote green and low-carbon development.1–5 In recent years, a variety of efficient CO2 conversion technologies have been developed, including biocatalysis, photocatalysis, and electrocatalysis.6–8 Among them, the electrocatalytic CO2 reduction reaction (CO2RR) has attracted extensive attention as a promising approach.3,9 However, the excellent thermodynamic stability and high activation barrier of CO2 make the reaction more difficult.10 In addition, the hydrogen evolution reaction (HER) is a competitive side reaction of the CO2RR because it has a low overpotential.11 Therefore, it is of great significance to design a novel CO2RR electrocatalyst with a low overpotential and HER inhibition.In recent years, due to the development of nanotechnology, it is possible to fine-tune the structure of an electrocatalyst at the nanoscale to control the structure of the electrocatalyst, making it have better stability, catalytic activity, and product selectivity. Single-atom catalysts (SACs) with isolated transition metal (TM) atoms stabilized by carbon-based materials have attracted more and more attention due to their special electronic structure and platform utilization12 and have become promising candidate materials in the nitrogen reduction reaction,13 HER,14–16 oxygen reduction reaction,15,17 oxygen evolution reaction,18,19 CO2RR,20,21 and other reactions.22 Previous studies have indicated that catalysts such as Mn, Mo, Ru, and Ti offer distinct advantages in the CO2RR compared to the HER, requiring lower overpotentials for the generation of intermediate products (*COOH or *OCHO).23–29 Unfortunately, the stability of SACs is not satisfactory,30 because when the bond of individual atoms to the carrier is weak, they tend to aggregate into nanoparticles, which inevitably affects their catalytic activity and further hinders their practical application.
In contrast, single-cluster catalysts (SCCs) supported in single-layer nitrogen–graphene have great potential to advance the design and manufacture of high-performance catalysts due to their tunable electronic structure and adaptable active sites and have shown superiority in terms of stability, and are widely regarded as promising alternatives to SACs.31 The C3N1 monolayer is a two-dimensional material with periodic uniform cavities, and is a derivative of nitrogen-doped graphene. Its cavity is surrounded by 6 nitrogen atoms and is considered an ideal carrier for transition metal loading. Its high thermal stability facilitates the synthesis of stable transition metal catalysts.32–34
Among SCCs, dual-atomic catalysts (DACs) have been extensively studied and are considered a new frontier.35–38 Chen et al.37 reported that DACs have the smallest metal alloy size and the largest atom utilization and exhibit excellent performance in various electrocatalytic reactions. In DACs, the binding strength of several intermediates can be optimized simultaneously to achieve multi-intermediate reactions. Zhang et al.9 found that the combination of diatomic metal and non-metal sites disrupted the electron density symmetry of the local coordination environment, and the formed symmetrical break sites improved the adsorption of key intermediates (such as *COOH and *OCHO). Wang et al.39 demonstrated that a unique dual Fe2–N6 catalyst showed the electrocatalytic reduction of CO2 to CO configuration, outstanding intrinsic activity, selectivity, and durability compared to SACs. Additionally, Mn and Mo, serving as DACs, are situated at the center and exhibit a polarized bond interaction with CO2, underscoring the role of metal-centered catalysts in CO2 and substrate interactions.40,41 DACs break the structural relationship constructed in traditional metal catalysts and improve the catalytic activity of the catalyst to form higher-order carbon products.42–44
Furthermore, triple-atom catalysts (TACs) have attracted much attention as a deeper extension of SACs. First, they exhibit a higher loading capacity for metal atoms and are able to get closer to the active site.45 The formation of metal–metal bonds or metal–bridge atomic bonds provides a strategic means to control the electron synergistic adsorption of various reactants and intermediates.46,47 TACs can provide a wider variety of asymmetric adsorption sites. These sites are particularly suitable for more complex catalytic processes involving multiple reaction steps and intermediates, and the synergy between the three atoms can also modulate the d-band electronic structure, thereby enhancing the catalytic performance.45,48,49 Jia et al.50 reported that TACs, as powerful electrocatalysts for the CO2RR, provide a large number of active sites, and also have a large electrochemical surface area and rapid charge transfer, which improves the reaction rate. Liu et al.51 found that TACs have abundant active sites for the CO2RR. their catalytic performance can be enhanced by the interaction of adjacent active sites. In particular, metals such as Mn, Mo, Ru, etc., as catalysts, demonstrate promising experimental feasibility and stable catalytic performance. They effectively suppress the HER while exhibiting excellent selectivity in the CO2RR.45,52,53
As excellent CO2RR catalysts, both single atoms and single clusters demonstrate outstanding catalytic performance. However, the effect of the number of metal atoms on the structure–activity relationship of the catalyst is still unclear. In addition, previous studies have inconsistently described the catalytic properties of catalysts loaded with Mn, Mo, Ru, and Ti. Therefore, it is necessary to systematically analyze the catalytic performance of CO2 using the above metal atoms and their numbers and provide theoretical guidance for the rational design of low-cost high-performance catalysts.
In this work, density functional theory (DFT) calculations were employed to reveal the structures of single atoms and single clusters composed of four metal elements (Mn, Mo, Ru, and Ti), which were loaded onto C2N1 monolayer materials as catalysts for CO2 electrochemical reduction. First, the stability and conductivity of TM-C2N1 were investigated through the analysis of its electronic structure, density of states (DOS), and ab initio molecular dynamics (AIMD). Second, the adsorption and activation effect of TM-C2N1 on CO2 were explored, focusing on the CO2 configuration, adsorption energy comparison with H2O, and the competitive adsorption relationship between the CO2RR and HER. Finally, to better understand the preferred pathways for CO2 reduction to CO or HCOOH on TM-C2N1, calculations were conducted for Gibbs free energy, limiting potential, overpotential, and Crystal Orbital Hamiltonian Population (COHP). The results show that the 3TM-C2N1 catalyst has a stable structure, can effectively adsorb and activate CO2, and has optimal inhibition performance for the HER. Among them, 3Mo-C2N1 and 3Ti-C2N1 had the best catalytic activity for CO and HCOOH, in the CO2RR with limiting potentials of −0.62 V and −0.42 V, respectively. These findings not only provide a theoretical basis for the experimental regulation of C2N1-based catalysts, but also have guiding significance for the development of other high-efficiency CO2RR electrocatalysts.
2. Results and discussion
2.1 Structural stability
In this section, first, in order to clearly understand the electrocatalytic process of CO2 for materials with different types of atoms and different numbers of atoms, a schematic diagram is shown, as shown in Fig. 1a. It shows the reduction pathways of CO2 at different catalytic sites. Second, all transition metal single-atom catalysts and single-cluster catalyst structures supported on N-doped graphene were constructed, and the optimized structures are shown in Fig. S1–S4.† Then, in order to evaluate the stability of the TM-C2N1 structure, the cohesion energy, charge density, and AIMD were calculated to study the stable structure. Finally, the heights of DOS curves for the TM-C2N1 and C3N1 monolayers were calculated as the key parameters for the conductivity of the catalyst. The TM-C2N1 model uses a 5 × 5 supercell of the C3N1 monolayer, and there are a total of 54 C atoms and 18 N atoms in the model, as shown in Fig. S1–S4,† and we set a vacuum layer of 20 Å in the z direction to avoid the influence between adjacent lattices. The transition metal was placed in the N6 cavity to form the TM-C2N1 configuration.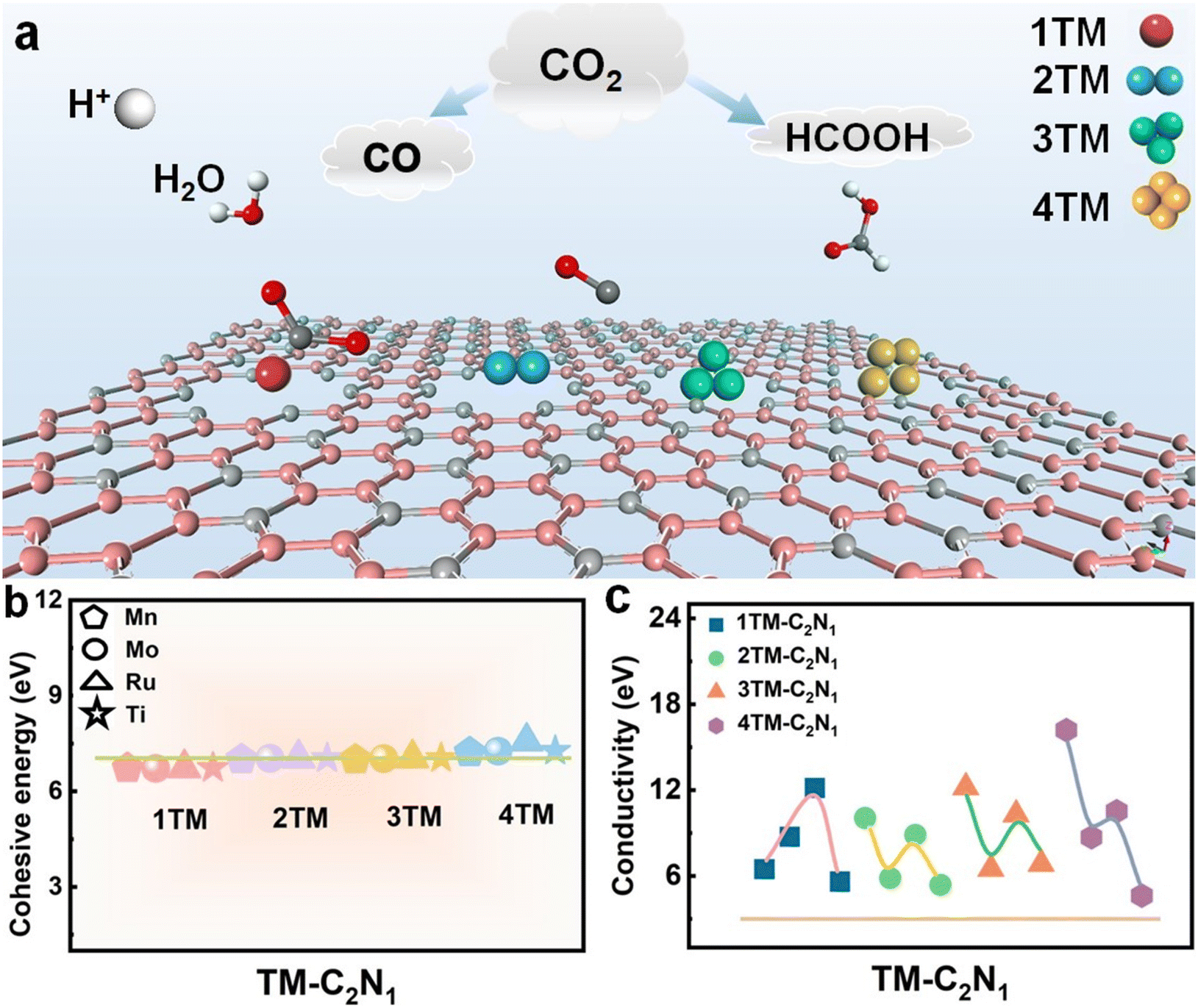 | ||
| Fig. 1 (a) Schematics of C2N1 with TM loads facilitates CO2 conversion. (b) Cohesion energy of the TM-C2N1 monolayer. (c) The height of the DOS curve for TM-C2N1 at the Fermi level. The order of data points in (b) and (c) is Mn, Mo, Ru, and Ti. | ||
High stability is a prerequisite for catalyst applications, so we evaluated the stability of TM-C2N1 catalysts with cohesive energy. The higher the cohesion energy, the more stable the corresponding structure.54–56 Cohesion energy (Ecoh) can be calculated according to the following formula:2
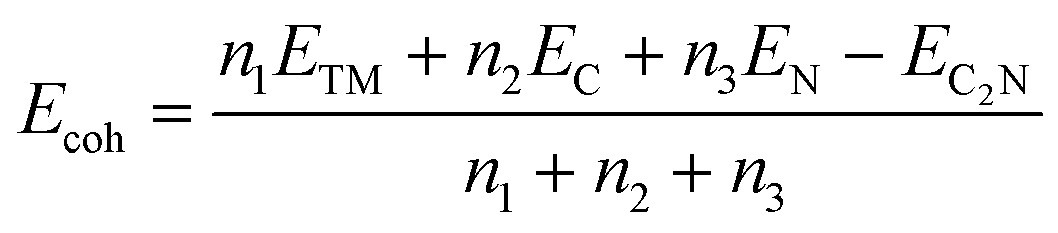 | (1) |
To better understand the interaction between the TM and the monolayer, we calculated the charge density of TM-C2N1, and the charge density diagram in Fig. S5–S8† show that the positive charge accumulates around the metal atom and decreases around the N atom, indicating that the electrons are transferred from the metal atom to the N atom, allowing the N atom to bind the TM atom. In addition, in order to judge the stability of the TM-C2N1 structure from the thermodynamic properties, AIMD simulation was carried out. It was calculated that the AIMD runs at 500 K for 10 ps and outputs a structure file every 10 fs. The detailed calculations are shown in Fig. S9–S12.† The results show that these materials are able to maintain good stability.
Finally, in order to explore the conductivity of the catalyst, the height of the DOS curve at the Fermi level for the TM-C2N1 monolayer represents the conductivity of the catalyst.2,57–62 DOSs were calculated as the key parameters of catalyst conductivity. As shown in Fig. 1c, the DOS height for TM-C2N1 is higher than that of C3N1, demonstrating that the conductivity is improved after the introduction of loaded metals. The detailed calculations are shown in Fig. S5–S8,† and the overall DOS shows that after the introduction of the TM, the curve around the Fermi level increases slightly, indicating that the conductivity of the material has improved. In addition, partial density of states (PDOS) calculations were performed to study the behavior of the doped TM in depth. There were obvious overlapping peaks between the d orbital of the TM and the p orbital of the N of the substrate, indicating that the interaction between the TM and the substrate was stronger. The TM can be firmly attached to the C3N1 monolayer. This indicates that there is an interaction between the loaded TM atoms and the substrate, ensuring its structural stability.
2.2 Activation and selectivity
In this section, to understand the catalytic performance of catalysts and the advantages of CO2RR development, the d-band center and activation degree of CO2 were studied first, and second, the adsorption energies of CO2 and H2O were calculated as important references. In addition, we further compared ΔG(*COOH) and ΔG(*OCHO) with ΔG(*H). Further screening of the good TM-C2N1 catalyst ensures the normal CO2RR.CO2 adsorption is the first and crucial step in the CO2RR. Stable CO2 adsorption is a prerequisite for the continuation of the CO2RR. The degree of CO2 activation reflects the catalytic performance of the catalyst to a certain extent. As shown in Fig. S13† (CO2 planar three-dimensional structure display), CO2 can be stably adsorbed on the surface of the catalyst, and the molecular configuration of CO2 has changed significantly, indicating that CO2 has been initially activated. In addition, in order to more intuitively observe the degree of activation and conductivity, as well as charge accumulation and consumption during the interaction between CO2 and TM-C2N1, the results of charge density analysis are shown in Fig. S13† and the DOSs are shown in Fig. S14–S17.† The results show that TM-C2N1 transfers a large amount of charge during gas adsorption. A distinct red color is observed around the CO2, which clearly indicates that the CO2 acquires electrons from TM-C2N1, indicating that there is an interaction between CO2 and TM-C2N1. Moreover, the p-orbital anchoring of C in the activated CO2 and the d-orbital of the transition metal in TM-C2N1 have a hybridization effect, indicating that there is an interaction between CO2 and TM-C2N1. For quantitative analysis, information such as the CO2 adsorption energy and CO2 bond length angle are listed in Table S1.† The smaller the bond angle, the stronger the degree of activation. The change of C–O bond length from 1.18–1.32 Å and the bending of O–C–O bond angle from 127.19°–163.22° (Fig. 2a) indicate that CO2 has been effectively activated.
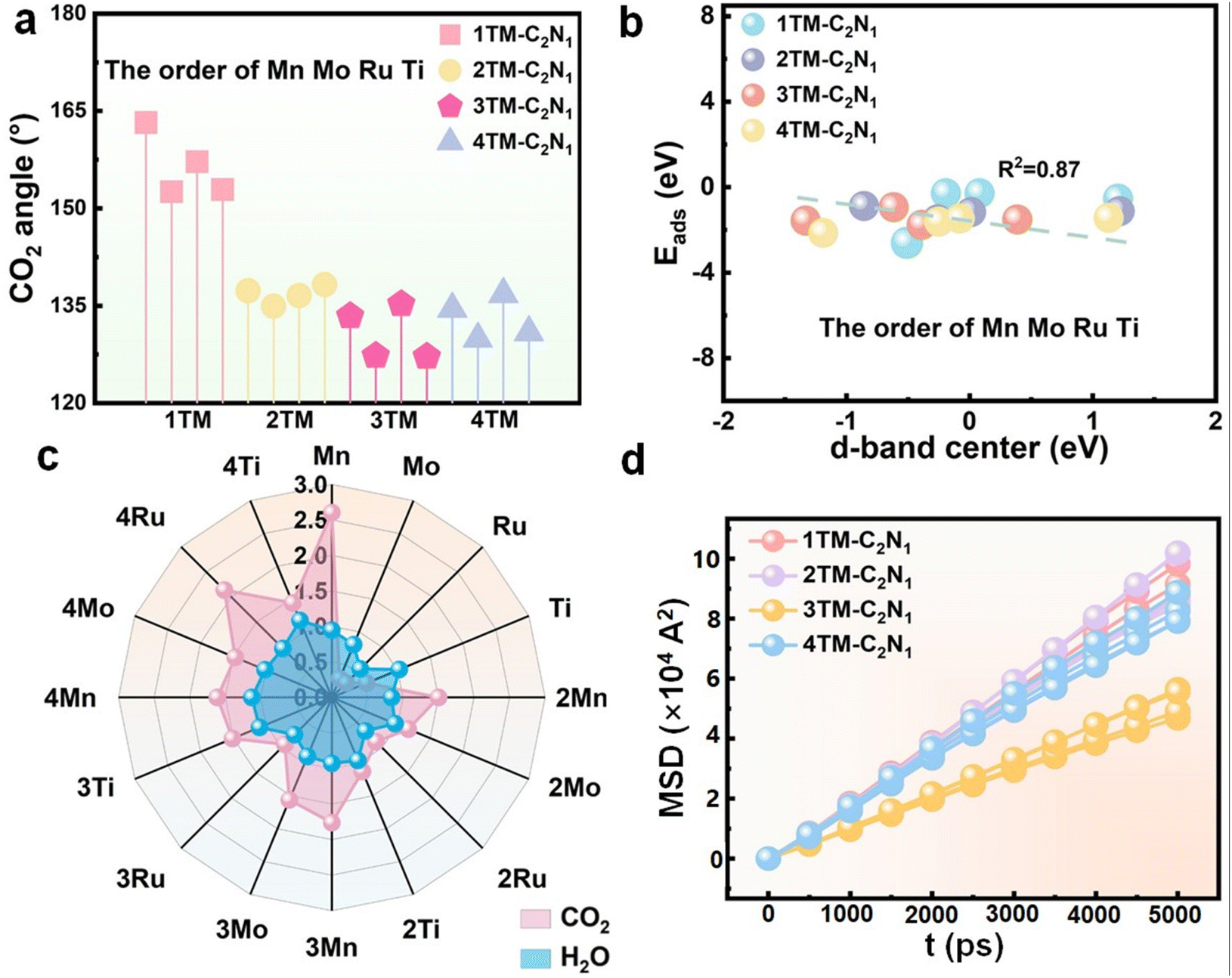 | ||
| Fig. 2 (a) CO2 activation angle on TM-C2N1. (b) Linear relationship between the d-band center of TM-C2N1 and the adsorption energy of CO2. (c) Comparison of H2O and CO2 adsorption energies on the TM-C2N1 monolayer. (d) Diffusion of CO2 on the TM-C2N1 monolayer. | ||
In order to explore the potential mechanism of the adsorption reaction of CO2 on the surface of the catalyst, we calculated the d-band center of TM-C2N1 because it can reflect the interaction between the gas molecules and the TM atoms on the surface. Fig. 2b shows that the d-band center of TM-C2N1 has a linear relationship with the adsorption energy, and the correlation coefficient is in good agreement with the theoretical model of the d-band center. Thus, the elevation of antibonding orbitals contributes to an increase in the adsorption energy.
It is well known that the kinetically prone HER hinders the progress of the CO2RR as a side reaction. So a good catalyst to inhibit the HER is necessary. First, we calculated the reaction mechanism of the HER by adsorbing H2O, because the CO2RR tends to be performed in neutral or alkaline electrolytes. Fig. 2c shows the stark contrast between the adsorption energies of H2O and CO2. Due to |Eads(H2O)| < |Eads(CO2)|, *CO2 is more easily adsorbed on the surface of TM-C2N1 catalysts than *H, thereby inhibiting the HER. Therefore, CO2 rather than H2O first occupies the active sites on the surface of TM-C2N1, promoting the CO2RR as the main reaction on these 13 catalysts. Please refer to Tables S1 and S2† for details on the adsorption energy of H2O and CO2.Moreover, during the first protonation process, the hydrogenation of CO2 produces two intermediates *COOH and *OCHO. As shown in Fig. S18,† by comparing ΔG(*COOH) and ΔG(*OCHO) with ΔG(*H), the 2/3TM-C2N1 catalysts all fall in the lower right half region (CO2RR selective), which tends to produce *COOH or *OCHO. Combining the above two points, it can be concluded that the 2/3TM-C2N1 catalyst is selective to the CO2RR and can well inhibit the occurrence of the HER. Therefore, the CO2RR has a clear advantage over the HER under electrochemical conditions.
The diffusion performance of CO2 on the catalyst material surface dictates the reaction rate.63,64 Therefore, to explore the performance of catalysts in practical applications, molecular dynamics simulations were employed to calculate the diffusion coefficients of CO2 on different material surfaces, thereby obtaining diffusion performance. Fig. S19–S22† depict snapshots of all catalysts capturing CO2 at equilibrium. First, the mean-squared displacement (MSD) of TM-C2N1 was calculated, as shown in Fig. 2d. The results display that 3TM-C2N1 has the smallest diffusion performance based on the slopes. Furthermore, diffusion coefficients can be obtained through the Einstein diffusion equation. The detailed data are shown in Table S3.† The diffusion coefficients of 3Mn-C2N1, 3Mo-C2N1, 3Ru-C2N1 and 3Ti-C2N1 were 2.91, 2.19, 3.86 and 2.78, respectively. Among which, 3Mo-C2N1 and 3Ti-C2N1 had the best diffusion performance. The diffusion coefficient has an effect on the diffusion of CO2 on the surface, and the smaller the diffusion coefficient, the more stable the CO2 adsorption and the better the catalytic effect.
2.3 CO2 reduction to CO or HCOOH
In this section, we first present the intermediate structure of the CO2 reduction process to CO and HCOOH, followed by the calculation of the free energy for each corresponding structure. Finally, we conducted free energy calculations for the reaction pathway. The path of CO2 to CO on TM-C2N1 is: CO2 → *CO2 → *COOH → *CO → CO. For HCOOH generation, the CO2 to HCOOH generation path on TM-C2N1 is CO2 → *CO2 → *OCHO → *HCOOH → HCOOH. Fig. 3 shows the adsorption structure and free energy distribution of CO2 intermediates to generate CO and HCOOH via a 2-electron path on the 3TM-C2N1 catalyst. | ||
| Fig. 3 Gibbs free energy distribution diagram of the CO2 reduction pathway toward CO or HCOOH on (a) 3Mn-C2N1, (b) 3Mo-C2N1, (c) 3Ru-C2N1, and (d) 3Ti-C2N1. | ||
Regarding the CO product, 3Mn-C2N1 has a CO2 → *COOH speed-limiting step and the Gibbs free energy variation is 0.63 eV. For the 3Mo-C2N1 catalyst, the speed-limiting step is CO2 → *COOH and the maximum Gibbs free energy variation is 0.62 eV. Besides, for the 3Ru-C2N1 monolayer, the speed-limiting step is CO2 → *COOH and the maximum Gibbs free energy variation is 0.92 eV. Finally, 3Ti-C2N1 has a CO2 → *COOH speed-limiting step and the maximum Gibbs free energy variation is 1.15 eV. For the production of HCOOH, first, 3Mn-C2N1 has a rate-limiting step of CO2 → *OCHO and a Gibbs free energy variation maximum of 0.91 eV. Then, 3Mo-C2N1 has a CO2 → *OCHO rate-limiting step and a maximum Gibbs free energy variation of 0.93 eV. 3Ru-C2N1 has a CO2 → *OCHO rate-limiting step and a maximum Gibbs free energy variation of 1.39 eV. Finally, for 3Ti-C2N1, the rate-limiting step is *OCHO → *HCOOH and the maximum variation of Gibbs free energy is 0.42 eV. Furthermore, these results have also been validated by the dynamic barrier of elementary steps. Thus, the rate-limiting step was elucidated based on both thermodynamic and kinetic processes.
It is worth noting that 1TM-C2N1, 2TM-C2N1, and 4TM-C2N1 generate CO or HCOOH paths and rate-limiting steps are presented in Fig. S23–S25.† The Gibbs free energy of 1TM-C2N1 varies greatly overall, and for the CO product, Mn-C2N1 has a CO2 → *COOH rate-limiting step with a maximum free energy difference of 4.83 eV. For HCOOH products, Ru-C2N1 has a CO2 → *OCHO rate-limiting step with a maximum free energy difference of 3.78 eV. For the CO product, 4Ti-C2N1 has a CO2 → *COOH speed-limiting ladder, and the Gibbs free energy becomes 2.18 eV. With regard to the HCOOH product, 4Mn-C2N1 has a CO2→*OCHO rate-limiting step and a Gibbs free energy change of 1.28 eV. Among them, for CO, 3Mo-C2N1 has the smallest free energy difference, making it the best CO catalyst. For HCOOH, 3Ti-C2N1 has the smallest free energy difference and is the most effective catalyst for HCOOH generation, so 3Mo-C2N1 and 3Ti-C2N1 exhibit excellent performance.
2.4 Catalytic performance
For a quantitative analysis and clear comparison of catalytic performance, Fig. S26 and S27† show the limiting potential (UL) and overpotential (η) of CO2 reduction to CO or HCOOH on TM-C2N1. The results demonstrate that TACs exhibit the best catalytic performance, based on the smallest overpotential, compared to other single-atom and multi-atom catalysts. Among them, the 3Mo/3Ti-C2N1 catalyst shows the optimal catalytic performance, as indicated by the minimal η for the formation of CO and HCOOH products. This parameter determines the energy cost in experiments. The impact of product separation on catalytic rates cannot be overlooked, as higher adsorption energy makes desorption more challenging. Therefore, establishing the relationship between the catalytic performance (overpotential) and the product adsorption energy is of significant importance for a deeper understanding of the structure–performance relationship of catalysts.As shown in Fig. 4a, for CO, 3Mo-C2N1 exhibits the best performance with a limiting potential of CO generation at 0.62 V. This indicates good catalytic activity and the lowest energy requirement. The vertices of the volcano plot represent the moderate adsorption energy of the product and therefore have the best catalytic performance. Conversely, as illustrated in Fig. 4b, 3Ti-C2N1 demonstrates the best overall performance for HCOOH generation. The limiting potential for the formation of HCOOH on 3Ti-C2N1 is 0.42 V; the results show that moderate adsorption can ensure a high yield of HCOOH. When |UL| > 0.9 V, it suggests that the reaction requires a significant amount of energy absorption, making it less likely to occur. The analysis suggests that the CO2RR catalyzed by TM-C2N1 is feasible, with 3TM-C2N1 being the optimal catalyst for achieving efficient CO2RR performance.
 | ||
| Fig. 4 Relationship between the adsorption energy and the overpotential of products on TM-C2N1. (a) CO adsorbed on TM-C2N1. (b) HCOOH adsorbed on TM-C2N1. | ||
The catalytic performance of 3Mn-C2N1 is also very good, and the UL of CO generation is 0.63 V, which is only 0.01 V higher than the that of 3Mo-C2N1. The catalyst with the best performance to generate HCOOH is 3Ti-C2N1. The UL for generating HCOOH on 3Ti-C2N1 is 0.42 V, which exhibits good catalytic activity and is the lowest energy required among the four catalysts. The analysis shows that the 3TM-C2N1-catalyzed CO2RR is achievable. Our work explores the excellent performance of TM-C2N1 for deep hydrogenation of CO2 under electrocatalysis at lower electrode potentials. In the formation of CO and HCOOH, 3Mo-C2N1 and 3Ti-C2N1 have the best performance among the 13 catalysts. This result demonstrates the optimal potential of TAC catalysts supported by the C2N1 monolayer for the CO2RR. Therefore, when CO2 is reduced to CO and HCOOH, 3Mo-C2N1 and 3Ti-C2N1 have the best catalytic performance.
2.5 Catalytic mechanism
To gain insight into why the catalyst performance of the 3Mo-C2N1 and 3Ti-C2N1 monolayers is the best, we analyzed the key intermediates, which interact with the catalytically active site, from a molecular orbital perspective. It is the bonding orbital that plays a key role in the adsorption according to previous studies.36,65–69 First of all, the interaction should not be too strong, which will lead to a sudden increase in the energy barrier of the next step, forming a restrictive link. Second, the interaction should not be too weak, which will lead to a decrease in the catalytic effect of the catalyst and make the catalyst fail. Hence, maintaining a moderate interaction is essential to obtain high-performance catalysts. To achieve this, we conducted COHP calculations for reducing the intermediates of the TM catalysts exhibiting the best performance.Fig. S29 and S30† depict the most stable adsorption configuration, charge density distribution, and corresponding DOS of *COOH and *OCHO adsorption on 3Mo-C2N1 and 3Ti-C2N1, respectively. In Fig. S29,† the interaction between the intermediate products (*COOH and *OCHO) and 3Mo-C2N1 demonstrates pronounced charge accumulation or depletion, particularly evident in Fig. S29b,† indicating strong adsorption between the surfaces of OCHO and 3Mo-C2N1. Additionally, the DOS reveals numerous overlapping peaks between the p orbital of O and the d orbital of Mo, signifying a strong hybrid effect. Consequently, desorption and reduction to CO are facilitated at 3Mo-C2N1. Similarly, in Fig. S30,† the interaction between *COOH and *OCHO and 3Ti-C2N1 exhibits notable charge accumulation or depletion, particularly evident in Fig. S30b† with strong adsorption observed between *COOH and the surfaces of 3Ti-C2N1. Additionally, the DOS highlights numerous overlapping peaks between the p orbital of C and the d orbital of Ti, indicating a strong hybrid effect. Hence, desorption and reduction to HCOOH are facilitated at 3Ti-C2N1. Consequently, 3Mo-C2N1 and 3Ti-C2N1 demonstrate the best catalytic performance for CO2 reduction to CO and HCOOH, respectively.
To further elucidate the interactions between atoms, we conducted COHP calculations. As depicted in Fig. 5, the bonding and anti-bonding regions between the 3TM and gas atoms are illustrated at the Fermi level, and the ICOHP values (absolute values) were plotted accordingly. For the CO product, it delineates a bonding region spanning from −2.3 eV to 1.2 eV and an anti-bonding region ranging from −12.5 eV to −16 eV between the metal Mo atom and the gas C atom with an ICOHP value of 0.02 eV. Furthermore, the bonding region between the metal Ti atom and the gas C atom spans from −6.6 eV to 2.4 eV with an anti-bonding region from −14.8 eV to −7.1 eV and an ICOHP value of 0.78 eV. As depicted in Fig. S28,† there is an ICOHP value of 0.01 eV between the metal Mn atom and the gas C atom and an ICOHP value of 0.53 eV between the metal Ru atom and the gas C atom. This indicates that the interaction between *C in the intermediate product *COOH and Mo atoms is moderate, thus ensuring the optimal performance of the 3Mo-C2N1 catalyst for CO generation.
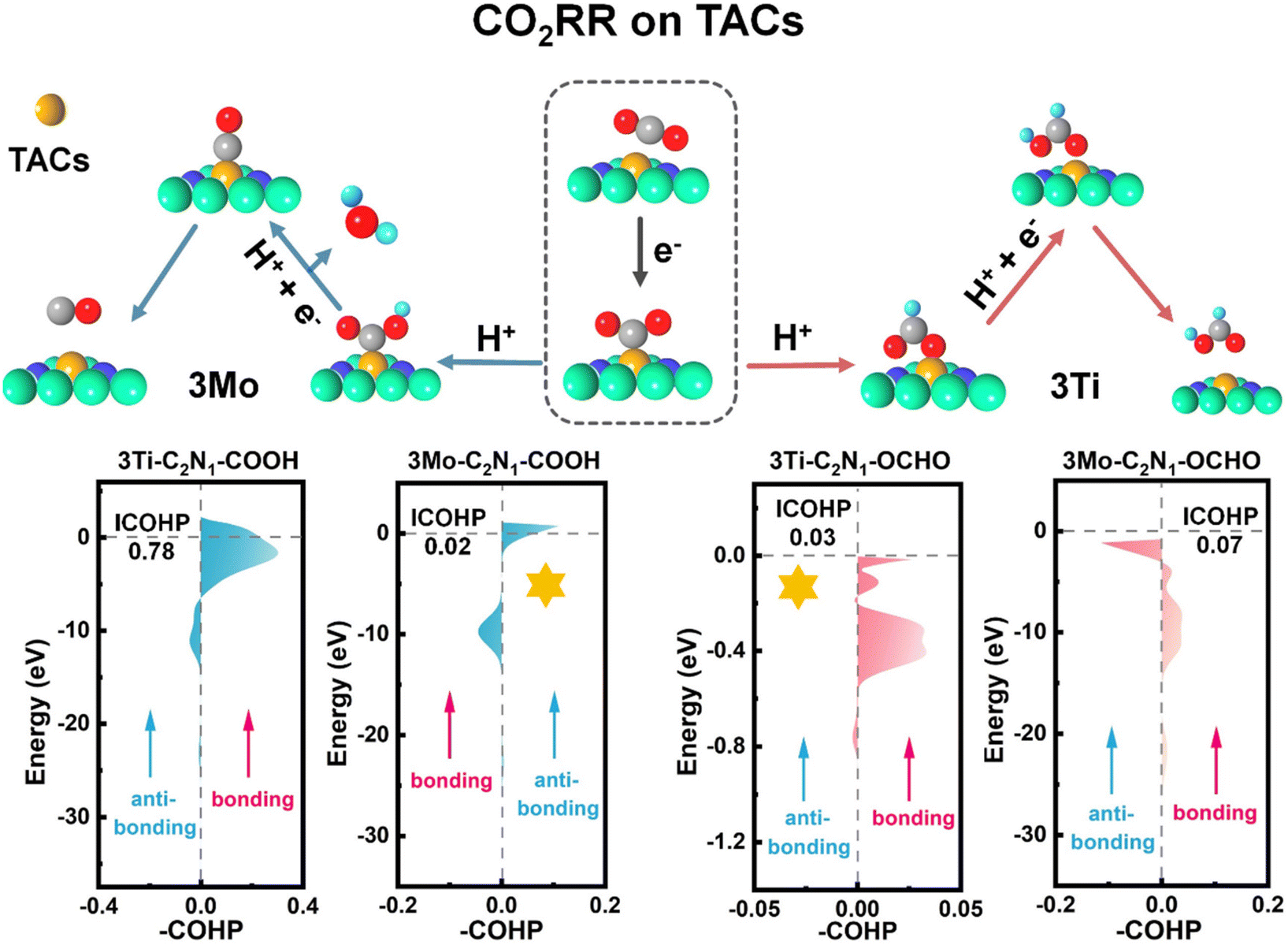 | ||
| Fig. 5 ICOHP values of *COOH and *OCHO on 3TM-C2N1. | ||
For the HCOOH product, the bonding region between the metal Mo atom and the gas O atom extends from −13.8 eV to −2.7 eV, while the anti-bonding region ranges from −2.8 eV to −0.12 eV with an ICOHP value of 0.07 eV. Meanwhile, the bonding region between the metal Ti atom and the gas O atom ranges from −0.53 eV to 0 eV with an anti-bonding region from −0.88 eV to −0.67 eV and an ICOHP value of 0.03 eV. As depicted in Fig. S28,† there is an ICOHP value of 0.007 eV between the metal Mn atom and the gas O atom, and an ICOHP value of 0.25 eV between the metal Ru atom and the gas O atom. This indicates that the interaction between *O in the intermediate product *OCHO and the Ti atoms is moderate, thus ensuring the optimal performance of the 3Ti-C2N1 catalyst for HCOOH generation.
In addition, based on the relationship between the d-band center and the adsorption energy (as shown in Fig. 2b), the height of the d-band center of the catalyst material can be best regulated after the doping of three atoms. Therefore, TACs have a moderate adsorption energy for the intermediate products (*COOH and *OCHO), thus ensuring their optimal catalytic performance. In summary, these results demonstrate that 3Mo-C2N1 and 3Ti-C2N1 are the best catalysts for the formation of CO and HCOOH, respectively.
3. Conclusions
In summary, the mechanism of the CO2RR on the SACs and SCCs of Mn, Mo, Ru and Ti anchored on the C3N1 monolayer was studied by DFT calculations. The results show that the TM-C2N1 catalysts are stable based on cohesion energy. Besides, AIMD was used to further verify the thermodynamic stability of the catalyst material. As an excellent electrocatalytic material, the conductivity of the TM-C2N1 monolayer was significantly improved based on the height of DOS at the Fermi level. Furthermore, the PDOS shows the obvious overlapping peaks between the d orbital of the TM and the p orbital of the N of the C2N1 monolayer, indicating that the TM can be stably anchored to the substrate. Importantly, CO2 is significantly activated on the TM-C2N1 monolayer and the change in the bond angle is from 127.19° to 163.22°. At the same time, the catalyst can effectively prevent the side reaction of the HER. The good diffusion of the gas reactants is the initial condition for the CO2RR, so the diffusion performance of CO2 on the TM-C2N1 catalyst was calculated by molecular dynamics simulations. The results show that the 3Mo-C2N1 and 3Ti-C2N1 catalysts have the smallest diffusion coefficients of 2.19 and 2.78, respectively, and could stably adsorb CO2 at active sites. Interestingly, 3Mo-C2N1 and 3Ti-C2N1 also exhibited the best catalytic activity for CO2 reduction to CO and HCOOH, respectively. The pathway for the reduction of CO2 to CO involves steps of CO2 → *COOH → *CO → CO with a UL of −0.62 V. The pathway for CO2 reduction to HCOOH was CO2 → *OCHO → *HCOOH → HCOOH and UL is −0.42 V. In order to elucidate why the 3TM-C2N1 catalyst has the best catalytic performance, we established a volcano diagram relationship curve between η and Eads. The results emphasize the importance of moderate adsorption energy. In particular, the COHPs of *COOH and *OCHO for 3Mo-C2N1 and 3Ti-C2N1 catalysts were investigated in detail to compare the bonding and antibonding contributions. ICOHP shows that for CO, the ICOHP value of 3Mo-C2N1-COOH is 0.02 eV, and for HCOOH, the ICOHP value of 3Ti-C2N1-OCHO is 0.03 eV. It is proved that the optimum catalytic effect can be guaranteed only when the intermediate products have moderate adsorption energy. Therefore, this work provides theoretical guidance for the rational design of high-performance CO2RR catalysts and offers new insights into understanding the catalytic mechanism of multi-atomic catalysts.Author contributions
Yimeng Sun: investigation, data curation, formal analysis, methodology, and writing – original draft. Lin Tao: investigation, data curation, methodology, supervision, funding acquisition, conceptualization, validation, and writing – review and editing. Mingjie Wu: methodology, resources, and writing – review and editing. Davoud Dastan: formal analysis and software. Javed Rehman: methodology and software. Lixiang Li: methodology and formal analysis. Baigang An: supervision, formal analysis, and writing – review and editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The funding from the National Natural Science Foundation of China (Grant No. 52304330, 52371224, and 22208331), the University of Science and Technology Liaoning Talent Project Grants (6003000317), the Outstanding Youth Fund of University of Science and Technology Liaoning (2023YQ11), and the Youth Fund of the Education Department of Liaoning Province (LJKQZ20222324) is gratefully acknowledged.References
- T. Liu, Y. Wang and Y. Li, Can Metal–Nitrogen–Carbon Single-Atom Catalysts Boost the Electroreduction of Carbon Monoxide?, JACS Au, 2023, 3, 943–952 CrossRef CAS PubMed.
- L. Tao, D. Dastan, W. Wang, P. Poldorn, X. Meng, M. Wu, H. Zhao, H. Zhang, L. Li and B. An, Metal-Decorated InN Monolayer Senses N2 against CO2, ACS Appl. Mater. Interfaces, 2023, 15, 12534–12544 CrossRef CAS PubMed.
- S. Liu, M. Ge, L. Meng, X. Liu and T. Fang, Mechanism analysis of CO2 separation from pvdf-supported deep eutectic solvent: A molecular dynamics simulation study, J. Mol. Liq., 2024, 403, 124806 CrossRef.
- Y. Ouyang, L. Shi, X. Bai, C. Ling, Q. Li and J. Wang, Selectivity of Electrochemical CO2 Reduction toward Ethanol and Ethylene: The Key Role of Surface-Active Hydrogen, ACS Catal., 2023, 15448–15456 CrossRef CAS.
- L. Tao, J. Huang, D. Dastan, T. Wang, J. Li, X. Yin and Q. Wang, New insight into absorption characteristics of CO2 on the surface of calcite, dolomite, and magnesite, Appl. Surf. Sci., 2021, 540, 148320 CrossRef CAS.
- X. Li, X. Yu and Q. Yu, Research progress on electrochemical CO2 reduction for Cu-based single-atom catalysts, Sci. China Mater., 2023, 66, 3765–3781 CrossRef CAS.
- L. Yu, F. Li, J. Huang, B. G. Sumpter, W. E. Mustain and Z. Chen, Double-Atom Catalysts Featuring Inverse Sandwich Structure for CO2 Reduction Reaction: A Synergetic First-Principles and Machine Learning Investigation, ACS Catal., 2023, 9616–9628 CrossRef CAS.
- L. Liu and H. Xiao, Inverted Region in Electrochemical Reduction of CO2 Induced by Potential-Dependent Pauli Repulsion, J. Am. Chem. Soc., 2023, 145, 14267–14275 CrossRef CAS PubMed.
- Y. Zhang, T. Liu, X. Wang, Q. Dang, M. Zhang, S. Zhang, X. Li, S. Tang and J. Jiang, Dual-Atom Metal and Nonmetal Site Catalyst on a Single Nickel Atom Supported on a Hybridized BCN Nanosheet for Electrochemical CO2 Reduction to Methane: Combining High Activity and Selectivity, ACS Appl. Mater. Interfaces, 2022, 14, 9073–9083 CrossRef CAS PubMed.
- Y. Yan, Z. Zhao, J. Zhao, W. Tang, W. Huang and J.-M. Lee, Atomic-thin hexagonal CuCo nanocrystals with d-band tuning for CO2 reduction, J. Mater. Chem. A, 2021, 9, 7496–7502 RSC.
- P. D. Pedersen, M. M. Melander, T. Bligaard, T. Vegge, K. Honkala and H. A. Hansen, Grand Canonical DFT Investigation of the CO2RR and HER Reaction Mechanisms on MoTe2 Edges, J. Phys. Chem. C, 2023, 127, 18855–18864 CrossRef CAS.
- Z. Ma, T. Wan, D. Zhang, J. A. Yuwono, C. Tsounis, J. Jiang, Y.-H. Chou, X. Lu, P. V. Kumar, Y. H. Ng, D. Chu, C. Y. Toe, Z. Han and R. Amal, Atomically Dispersed Cu Catalysts on Sulfide-Derived Defective Ag Nanowires for Electrochemical CO2 Reduction, ACS Nano, 2023, 17, 2387–2398 CrossRef CAS PubMed.
- G. Wang, R. Ma, N. Zhang, Y. Guo and K. Chu, Single-atom Cu anchored on Mo2C boosts nitrite electroreduction to ammonia, Chem. Commun., 2023, 59, 13887–13890 RSC.
- S. Ren, X. Cao, Z. Jiang, Z. Yu, T. Zhang, S. Wei, Q. Fan, J. Yang, J. Mao and D. Wang, Single-atom catalysts for electrochemical applications, Chem. Commun., 2023, 59, 2560–2570 RSC.
- H. Zhu, Y. Feng, D. Zheng, X. Zhao, Y. Zhou, X. Fu, L. Zhao and X. Chen, Theoretical prediction of emerging high-performance trifunctional ORR/OER/HER single-atom catalysts: transition metals anchored into π–π conjugated graphitic carbon nitride (g-C10N3), Phys. Chem. Chem. Phys., 2023, 25, 31983–31994 RSC.
- T. Zhang, L. Li, T. Huang, H. Wan, W.-Y. Chen, Z.-X. Yang, G.-F. Huang, W. Hu and W.-Q. Huang, Correlation between spin state and activity for hydrogen evolution of PtN2 monolayer, Appl. Phys. Lett., 2024, 124(6), 063903 CrossRef CAS.
- W. Zou, R. Lu, X. Liu, G. Xiao, X. Liao, Z. Wang and Y. Zhao, Theoretical insights into dual-atom catalysts for the oxygen reduction reaction: the crucial role of orbital polarization, J. Mater. Chem. A, 2022, 10, 9150–9160 RSC.
- P. Kumar, K. Kannimuthu, A. S. Zeraati, S. Roy, X. Wang, X. Wang, S. Samanta, K. A. Miller, M. Molina, D. Trivedi, J. Abed, M. A. Campos Mata, H. Al-Mahayni, J. Baltrusaitis, G. Shimizu, Y. A. Wu, A. Seifitokaldani, E. H. Sargent, P. M. Ajayan, J. Hu and M. G. Kibria, High-Density Cobalt Single-Atom Catalysts for Enhanced Oxygen Evolution Reaction, J. Am. Chem. Soc., 2023, 145, 8052–8063 CrossRef CAS PubMed.
- T. Huang, Z.-X. Yang, L. Li, H. Wan, C. Leng, G.-F. Huang, W. Hu and W.-Q. Huang, Dipole Effect on Oxygen Evolution Reaction of 2D Janus Single-Atom Catalysts: A Case of Rh Anchored on the P6m2-NP Configurations, J. Phys. Chem. Lett., 2024, 15, 2428–2435 CrossRef CAS PubMed.
- Q. Pan, Y. Chen, S. Jiang, X. Cui, G. Ma and T. Ma, Insight into the active sites of M–N–C single-atom catalysts for electrochemical CO2 reduction, Energy Chem., 2023, 100114 CrossRef CAS.
- Q. Chang, Y. Liu, J.-H. Lee, D. Ologunagba, S. Hwang, Z. Xie, S. Kattel, J. H. Lee and J. G. Chen, Metal-Coordinated Phthalocyanines as Platform Molecules for Understanding Isolated Metal Sites in the Electrochemical Reduction of CO2, J. Am. Chem. Soc., 2022, 144, 16131–16138 CrossRef CAS PubMed.
- S. Nie, J. Li, L. Tao, Y. He, D. Dastan, X. Meng, P. Poldorn and X. Yin, Insights into Selective Mechanism of NiO-TiO2 Heterojunction to H2 and CO, ACS Sens., 2023, 8, 4121–4131 CrossRef CAS PubMed.
- Y. Liu, L. Li, Q. Li, J. Lin, Z. Guo, X. Zhang, Z. Lu, Y. Ma, Y. Huang and C. Tang, Fluorine doped porous boron nitride for efficient CO2 capture and separation: A DFT study, Appl. Surf. Sci., 2021, 556, 149775 CrossRef CAS.
- X. Lu, Y. Hu, S. Cao, J. Li, C. Yang, Z. Chen, S. Wei, S. Liu and Z. Wang, Two-dimensional MBene: a comparable catalyst to MXene for effective CO2RR towards C1 products, Phys. Chem. Chem. Phys., 2023, 25, 18952–18959 RSC.
- Z. Wang, B. N. DiMarco, M. Z. Ertem, R. N. Sampaio and G. F. Manbeck, Electronic and Electrochemical Control of Isostructural Ruthenium Hydricities and the Implications for Catalytic Overpotentials, ACS Catal., 2024, 1419–1431 CrossRef CAS.
- M. Sadehvand, A. Amiri, A. Pazoki and F. F. Tirani, Electrochemically CO2 conversion using homogeneous and nanostructured heterogeneous Ru(II) electrocatalysts, Fuel, 2023, 335, 126712 CrossRef CAS.
- K. Peng, J. Ye, H. Wang, H. Song, B. Deng, S. Song, Y. Wang, L. Zuo and J. Ye, Natural halloysite nanotubes supported Ru as highly active catalyst for photothermal catalytic CO2 reduction, Appl. Catal., B, 2023, 324, 122262 CrossRef CAS.
- M. Pu, W. Guo and Y. Guo, Non-Noble Metal Incorporated Transition Metal Dichalcogenide Monolayers for Electrochemical CO2 Reduction: A First-Principles Study, ACS Appl. Mater. Interfaces, 2023, 15, 58388–58396 CrossRef CAS PubMed.
- C. Zhao, X. Su, S. Wang, Y. Tian, L. Yan and Z. Su, Single-atom catalysts on supported silicomolybdic acid for CO2 electroreduction: a DFT prediction, J. Mater. Chem. A, 2022, 10, 6178–6186 RSC.
- X. Wang, L. Xu, C. Li, C. Zhang, H. Yao, R. Xu, P. Cui, X. Zheng, M. Gu, J. Lee, H. Jiang and M. Huang, Developing a class of dual atom materials for multifunctional catalytic reactions, Nat. Commun., 2023, 14, 7210 CrossRef CAS PubMed.
- J. Pan, X. E. Li, Y. Zhu, J. Zhou, Z. Zhu, C. Li, X. Liu, X. Liang, Z. Yang, Q. Chen, P. Ren, X. D. Wen, X. Zhou and K. Wu, Clustering-Evolved Frontier Orbital for Low-Temperature CO2 Dissociation, J. Am. Chem. Soc., 2023, 145, 18748–18752 CrossRef CAS PubMed.
- Y. Zhao, D. Ma, J. Zhang, Z. Lu and Y. Wang, Transition metal embedded C3N monolayers as promising catalysts for the hydrogen evolution reaction, Phys. Chem. Chem. Phys., 2019, 21, 20432–20441 RSC.
- Q. Bing and J.-Y. Liu, Transition metal single atom anchored C3N for highly efficient formic acid dehydrogenation: A DFT study, Appl. Surf. Sci., 2021, 562, 150186 CrossRef CAS.
- M. Gu, L. Tao, D. Dastan, J. Dang, T. Fang and B. An, Metal-Modified C3N1 Monolayer Sensors for Battery Instability Monitoring, J. Mater. Chem. A, 2024 10.1039/d4ta00645c.
- Q. Zhang, D. Liu, Y. Zhang, Z. Guo, M. Chen, Y. Chen, B. Jin, Y. Song and H. Pan, Insight into coupled Ni-Co dual-metal atom catalysts for efficient synergistic electrochemical CO2 reduction, J. Energy Chem., 2023, 87, 509–517 CrossRef CAS.
- J. Wu, D. Wu, H. Li, Y. Song, W. Lv, X. Yu and D. Ma, Tailoring the coordination environment of double-atom catalysts to boost electrocatalytic nitrogen reduction: a first-principles study, Nanoscale, 2023, 15, 16056–16067 RSC.
- C. Chen, M. Sun, K. Wang and Y. Li, Dual–metal single–atomic catalyst: The challenge in synthesis, characterization, and mechanistic investigation for electrocatalysis, SmartMat, 2022, 3, 533–564 CrossRef CAS.
- D. Ma, Y. Wang, L. Liu and Y. Jia, Electrocatalytic nitrogen reduction on the transition-metal dimer anchored N-doped graphene: performance prediction and synergetic effect, Phys. Chem. Chem. Phys., 2021, 23, 4018–4029 RSC.
- Y. Wang, B. J. Park, V. K. Paidi, R. Huang, Y. Lee, K.-J. Noh, K.-S. Lee and J. W. Han, Precisely Constructing Orbital Coupling-Modulated Dual-Atom Fe Pair Sites for Synergistic CO2 Electroreduction, ACS Energy Lett., 2022, 7, 640–649 CrossRef CAS.
- Y. Li, Y. Chen, Z. Guo, C. Tang, B. Sa, N. Miao, J. Zhou and Z. Sun, Breaking the linear scaling relations in MXene catalysts for efficient CO2 reduction, Chem. Eng. J., 2022, 429, 132171 CrossRef CAS.
- H. T. D. Bui, V. Q. Bui, S. G. Kim, Y. Kawazoe and H. Lee, Revealing well-defined cluster-supported bi-atom catalysts for enhanced CO2 electroreduction: a theoretical investigation, Phys. Chem. Chem. Phys., 2021, 23, 25143–25151 RSC.
- S. Wang, M. Luo and S. Guo, Two atoms are better than one, Nat. Synth., 2022, 1, 676–677 CrossRef.
- M. Jiao, Z. Chen, N. Wang and L. Liu, DFT calculation screened CoCu and CoFe dual-atom catalysts with remarkable hydrogen evolution reaction activity, Appl. Catal., B, 2023, 324, 122244 CrossRef CAS.
- C. Yang, Y. Wu, Y. Wang, H.-N. Zhang, L.-H. Zhu and X.-C. Wang, Electronic properties of double-atom catalysts for electrocatalytic oxygen evolution reaction in alkaline solution: a DFT study, Nanoscale, 2022, 14, 187–195 RSC.
- S. Li, L. Tong, Z. Peng, B. Zhang and X. Fu, Efficient CO2 reduction for CO production using triatomic catalyst screening: A DFT study, Appl. Surf. Sci., 2024, 644, 158804 CrossRef CAS.
- P. V. Bakharev, M. Huang, M. Saxena, S. W. Lee, S. H. Joo, S. O. Park, J. Dong, D. C. Camacho-Mojica, S. Jin, Y. Kwon, M. Biswal, F. Ding, S. K. Kwak, Z. Lee and R. S. Ruoff, Chemically induced transformation of chemical vapour deposition grown bilayer graphene into fluorinated single-layer diamond, Nat. Nanotechnol., 2019, 15, 59–66 CrossRef PubMed.
- M. Wu, F. Dong, Y. Yang, X. Cui, X. Liu, Y. Zhu, D. Li, S. Omanovic, S. Sun and G. Zhang, Emerging Atomically Precise Metal Nanoclusters and Ultrasmall Nanoparticles for Efficient Electrochemical Energy Catalysis: Synthesis Strategies and Surface/Interface Engineering, Electrochem. Energy Rev., 2024, 7, 10 CrossRef CAS.
- Y. Wang, J. Zhang, J. Zhao, Y. Wei, S. Chen, H. Zhao, Y. Su, S. Ding and C. Xiao, Strong Hydrogen-Bonded Interfacial Water Inhibiting Hydrogen Evolution Kinetics to Promote Electrochemical CO2 Reduction to C2+, ACS Catal., 2024, 14, 3457–3465 CrossRef CAS.
- S. Li, M. Shi, C. Wu, K. Nie, Z. Wei, X. Jiang, X. Liu, H. Chen, X. Tian, D. Wu and Y. Li, Surface addition of Ag on PbO2 to enable efficient oxygen evolution reaction in pH-neutral media, Chem. Eng. J., 2024, 485, 150043 CrossRef CAS.
- S. Jia, Q. Zhu, H. Wu, S. Han, M. Chu, J. Zhai, X. Xing, W. Xia, M. He and B. Han, Preparation of trimetallic electrocatalysts by one-step co-electrodeposition and efficient CO2 reduction to ethylene, Chem. Sci., 2022, 13, 7509–7515 RSC.
- W. Liu, J. Liu, Y. Yang, B. Xiong and H. Bai, Tunable trimetallic TM-NiFe catalysts for enhancing the products selectivity of CO2 electroreduction, Fuel, 2023, 335, 127026 CrossRef CAS.
- J.-C. Liu, H. Xiao, X.-K. Zhao, N.-N. Zhang, Y. Liu, D.-H. Xing, X. Yu, H.-S. Hu and J. Li, Computational Prediction of Graphdiyne-Supported Three-Atom Single-Cluster Catalysts, CCS Chem., 2023, 5, 152–163 CrossRef CAS.
- L. Yang, S. Feng and W. Zhu, Achieving reaction pathway separation for electrochemical nitrate fixation on triatomic catalysts: A new mechanism, J. Hazard. Mater., 2023, 441, 129972 CrossRef CAS.
- S. Nie, L. Tao, J. Li, W. Wang, P. Poldorn, Y. He, X. Yin and M. Wu, A single response to reducing gases by NiO-TiO2 heterojunction nanocrystals, Appl. Surf. Sci., 2024, 644, 158821 CrossRef CAS.
- Y. He, L. Tao, J. Li, M. Wu, P. Poldorn, D. Dastan, S. Abbasi, S. Nie, X. Yin and Q. Wang, Atomic-level insights into selective adsorption of H2 and CO on SnO2/CoO heterojunctions, Mater. Today Nano, 2023, 22, 100334 CrossRef CAS.
- L. Tao, J. Huang, D. Dastan, T. Wang, J. Li, X. Yin and Q. Wang, CO2 capture and separation on charge-modulated calcite, Appl. Surf. Sci., 2020, 530, 147265 CrossRef CAS.
- I. S. Amiinu, X. Liu, Z. Pu, W. Li, Q. Li, J. Zhang, H. Tang, H. Zhang and S. Mu, From 3D ZIF Nanocrystals to Co-Nx/C Nanorod Array Electrocatalysts for ORR, OER, and Zn-Air Batteries, Adv. Funct. Mater., 2018, 28, 1704638 CrossRef.
- M. Wang, W. Ma, C. Tan, Z. Qiu, L. Hu, X. Lv, Q. Li and J. Dang, Designing Efficient Non-Precious Metal Electrocatalysts for High-Performance Hydrogen Production: A Comprehensive Evaluation Strategy, Small, 2023, e2306631 Search PubMed.
- Y. He, J. Li, L. Tao, S. Nie, T. Fang, X. Yin and Q. Wang, First-principles calculations on the resistance and electronic properties of H2 adsorption on a CoO-SnO2 heterojunction surface, Phys. Chem. Chem. Phys., 2021, 24, 392–402 RSC.
- Z. Lv, W. Ma, M. Wang, J. Dang, K. Jian, D. Liu and D. Huang, Co–Constructing Interfaces of Multiheterostructure on MXene (Ti3C2Tx)–Modified 3D Self–Supporting Electrode for Ultraefficient Electrocatalytic HER in Alkaline Media, Adv. Funct. Mater., 2021, 31, 2102576 CrossRef CAS.
- K. Boonpalit, J. Kinchagawat, C. Prommin, S. Nutanong and S. Namuangruk, Efficient exploration of transition-metal decorated MXene for carbon monoxide sensing using integrated active learning and density functional theory, Phys. Chem. Chem. Phys., 2023, 25, 28657–28668 RSC.
- R. Dagan, Y. Vaknin and Y. Rosenwaks, Gap state distribution and Fermi level pinning in monolayer to multilayer MoS2 field effect transistors, Nanoscale, 2020, 12, 8883–8889 RSC.
- L. Tao, J. Huang, X. Yin, Q. Wang, Z. Li, G. Wang and B. Cui, Adsorption Kinetics of CO2 on a Reconstructed Calcite Surface: An Experiment-Simulation Collaborative Method, Energy Fuels, 2019, 33, 8946–8953 CrossRef CAS.
- L. Tao, J. Huang, D. Dastan, J. Li, X. Yin and Q. Wang, Flue gas separation at organic-inorganic interface under geological conditions, Surf. Interfaces, 2021, 27, 101462 CrossRef CAS.
- Y.-Q. Wang, X.-H. Dan, Z.-Y. Yi, X. Wang, Y.-C. Feng, Y. Feng, D. Wang and L.-J. Wan, Single-Molecule Study on the Catalytic Role of Co–O2 Binding in ORR by In Situ ECSTM, J. Phys. Chem. C, 2023, 127, 2929–2935 CrossRef CAS.
- P. Sun, S. Liu, X. Zheng, G. Hu, Q. Zhang, X. Liu, G. Zheng and Y. Chen, Challenges and opportunities of atomic-scales reactive sites in thriving electrochemical CO2 reduction reaction, Nano Today, 2024, 55, 102152 CrossRef CAS.
- G. Yasin, A. Kumar, S. Ajmal, M. Asim Mushtaq, M. Tabish, A. Saad, M. A. Assiri, M. Tariq Nazir and Q. Zhuo, Advances and perspectives on heteronuclear dual-atomic catalysts for prevailing the linear scaling relationship in electrocatalytic CO2 reduction, Coord. Chem. Rev., 2024, 501, 215589 CrossRef CAS.
- R. Zhu, Y. Qin, T. Wu, S. Ding and Y. Su, Insights into local coordination environment of main group metal-nitrogen-carbon catalysts for enhanced oxygen reduction reaction, Appl. Surf. Sci., 2023, 631, 157581 CrossRef CAS.
- L. Diao, B. Zhang, Q. Sun, N. Wang, N. Zhao, C. Shi, E. Liu and C. He, An in-plane Co9S8@MoS2 heterostructure for the hydrogen evolution reaction in alkaline media, Nanoscale, 2019, 11, 21479–21486 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01082e |
| This journal is © The Royal Society of Chemistry 2024 |