
Improved electrochemical reduction of CO2 to syngas with a highly exfoliated Ti3C2Tx MXene–gold composite†
Murugan
Krishnan
a,
Aathilingam
Vijayaprabhakaran
ab and
Murugavel
Kathiresan
 *a
*a
aElectro Organic & Materials Electrochemistry Division, CSIR-Central Electrochemical Research Institute, Karaikudi – 630003, Tamil Nadu, India. E-mail: kathiresan@cecri.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad – 201002, India
First published on 31st July 2024
Abstract
Transforming carbon dioxide (CO2) into valuable chemicals via electroreduction presents a sustainable and viable approach to mitigating excess CO2 in the atmosphere. This report provides fresh insights into the design of a new titanium-based MXene composite as a catalyst for the efficient conversion of CO2 in a safe aqueous medium. Despite its excellent electrocatalytic activity towards CO2 reduction and high selectivity for CO production, the high cost of Au and the decline in catalytic activity on a larger scale hinder its large-scale CO2 conversion applications. In this research, we have successfully prepared an Au/Ti3C2Tx composite and tested its catalytic activity in the electrochemical CO2 reduction reaction (ECRR). The as-prepared composite features strong interactions between gold atoms and the MXene support, achieved through the formation of metal–oxygen/carbon bonds. The Au/Ti3C2Tx electrode demonstrated a significant current density of 17.3 mA cm−2 at a potential of −0.42 V vs. RHE, in a CO2 saturated atmosphere (faradaic efficiency: CO = 48.3% and H2 = 25.6%). Nyquist plots further indicated a reduction in the charge-transfer resistance of the Au/Ti3C2Tx layer, signifying rapid charge transfer between the Au and Ti3C2Tx. Furthermore, it is known that liquid crossover through the Gas Diffusion Electrode (GDE) significantly improves CO2 diffusion to catalyst active sites, thereby enhancing CO2 conversion efficiency. The goal of this work is to design an interface between metal and MXene so that CO2 can be electroreduced to fuels and other useful chemical compounds with great selectivity.
Introduction
The amount of carbon dioxide (CO2) in the Earth's atmosphere has been rising over the past few decades; in 2016, it surpassed and remained above 400 parts per million (ppm) for the first time in human history.1,2 There is a correlation between the rise in global mean temperature anomalies, the consequent impacts of climate change and the increase in atmospheric CO2 levels.3 It is therefore still a major task for the twenty-first century to develop cost-effective technologies that can reduce, trap, or use excessive anthropogenic CO2 emissions.4–6 One of the carbon capture and utilization technologies that has attracted interest recently is the electrochemical CO2 reduction reaction (ECRR), which uses both water and electricity to convert CO2 into useful compounds.7,8 CO2 may be reduced in the cathodic compartment to a number of products, including syngas, formate (HCOO−), methane (CH4), and other hydrocarbons, depending on the type of modified catalyst.9,10 However, the electrochemical CO2 transformation system has a relatively low energy efficiency for conversion resulting from its high overpotential to form transition states and poor selectivity among multiple competing processes. Therefore, it is crucial to develop catalytic electrodes11 that can facilitate the transfer of electrons to the thermodynamically inert CO2 molecule in order to create reduced products selectively. Research indicates that gold (Au) is a particularly efficient electrocatalyst12 that has a high Faraday efficiency (FE) and can convert CO2 to carbon monoxide (CO) at low overpotentials.13–15 Nevertheless, the high cost and enormous potential for poisoning of Au restrict its widespread use.16,17 Research using novel nanostructures18,19 has shown that even small changes in the size, shape, or surface characteristics of metal nanoparticles (NPs)20–22 have a significant impact on the ECRR activity. Size dependence on selectivity has been particularly proposed to be caused by the simultaneous competition of active centres for the ECRR. The intrinsic characteristics and the interaction that exists between the loaded catalyst and the support can have a major impact on the binary system's final catalytic activity. Porous or nanostructured architectures23 provide a large surface area for the ECRR and improve support/substrate conductivity, which prevents resistance losses. These are examples of well-known strategies for improving electrocatalytic performances.24,25 In contrast to other supports, Ti3C2Tx-MXene can improve ECRR performance because of its active and abundant adsorption sites, high electron density, high carrier mobility, tunable Fermi level, and 2D multi-layered structure.26–29 Moreover, Ti3C2Tx can speed up the kinetics of the process and electron transfer by acting as a promoter.30 Because Ti3C2Tx can be easily synthesized in large quantities from a variety of inexpensive, readily available precursors, and its degradation produces simply TiO2 and CO2, it is more viable for use in large-scale applications.31–33 Ti3C2Tx enhanced with terminal functionalities (Tx = O2, OH, and F) can provide readily available catalytic sites for reactant adsorption and reduce the binding energies of ECRR intermediates and products. Despite the fact that Ti3C2Tx has been used for ECRR applications for a long time, we anticipate that coupling Au NPs with it can dramatically enhance the ECRR performance. In order to overcome the main challenges of liquid reduction processes and enable upscaling, researchers have turned to gas-diffusion electrode34 (when a porous, conductive support exists to hold the catalyst) based systems. A higher CO2 concentration and a shorter diffusion path can result in commercially-relevant current densities. Unexpectedly, the Au/Ti3C2Tx catalyst functions as a gas-diffusion electrode (GDE) and presents a promising avenue for enhancing the overall performance of the ECRR under mild operating conditions.35,36 However, the doping of Au NPs into Ti3C2Tx for the ECRR has not been investigated yet.In this study, we report the selective and stable electroreduction of CO2 to syngas using Au decorated MXene. To achieve high conversion efficiency, a gas-diffusion layer was prepared with Au/Ti3C2Tx and used as a cathode. The Ti3C2Tx catalyst was prepared by a simple etching method using HF/HCl and the etched MXene was composited with Au (Fig. 1). We report a binary system that uses Ti3C2Tx as the substrate and a secondary catalyst, with well-dispersed Au NPs acting as an ECRR catalyst. This thin-layered, exfoliated Ti3C2Tx is perfect for adding well-dispersed Au NPs since Au/Au NPs exhibit remarkable efficacy in a variety of catalytic processes. The binary catalytic system, i.e., Au NPs incorporated into Ti3C2Tx, was demonstrated to be an excellent catalyst for the co-conversion of CO2 and H2O to CO and H2 with varying ratios over a wide potential range.
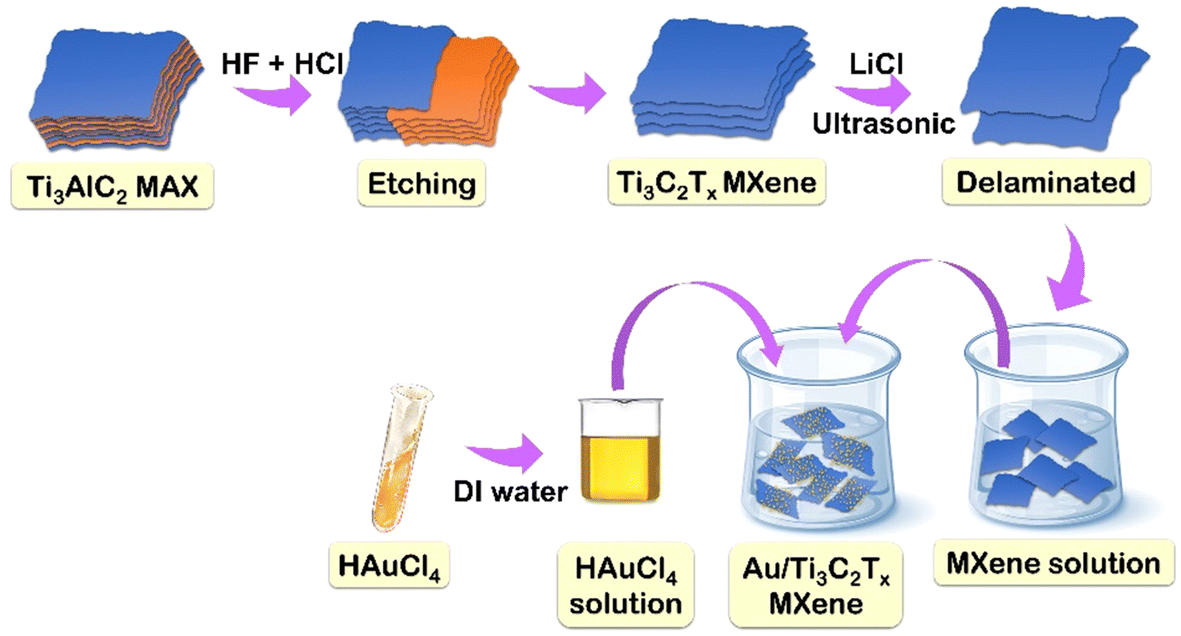 | ||
| Fig. 1 Schematic illustration of the preparation of Au/Ti3C2Tx. | ||
Results and discussion
Powder X-ray diffraction (PXRD) was conducted to analyse the crystal structure of the Ti3AlC2, Ti3C2Tx and Au/Ti3C2Tx nanocomposites, as illustrated in Fig. 2. It depicts the PXRD patterns of Ti3AlC2 and Ti3C2Tx (before and after HF etching), respectively. It is evident that the Al present in the Ti3AlC2 max phase was successfully etched to yield Ti3C2Tx MXene (JCPDS no: 04-8738), as apparent from the disappearance of the Al peak at 2θ ∼39°. Furthermore, it is evident from Fig. 2 that the diffraction peak, which corresponds to the (002) plane at ∼9.5°, shifted slightly towards lower angles, suggestive of the fading of Al intercalation.37 The XRD spectrum of Ti3C2Tx obtained after the HF-etching of the Ti3AlC2 sample correlated well with the literature.38,39 PXRD reveals the crystalline nature of Au/Ti3C2Tx nanocomposites, composed primarily of Au and functional Ti3C2Tx phases. The (111), (002), (022), and (113) crystal planes are linked to the broad and highly intense diffraction peaks at 38.3°, 44.5°, 64.7° and 77.8° that Au displays in the diffraction pattern. Ti3C2Tx showed a shift in the positions of the peaks as a result of functionalization and composite effects. Previous studies have shown that Ti3C2Tx possesses a substantial surface energy and very rich surface functional groups, such as –O, –OH and –F, which are advantageous to some substances to be adsorbed on its surface.40,41 Finding all the key peaks of both Au and Ti3C2Tx in the composite materials confirms the successful formation of the catalytically functionalised material.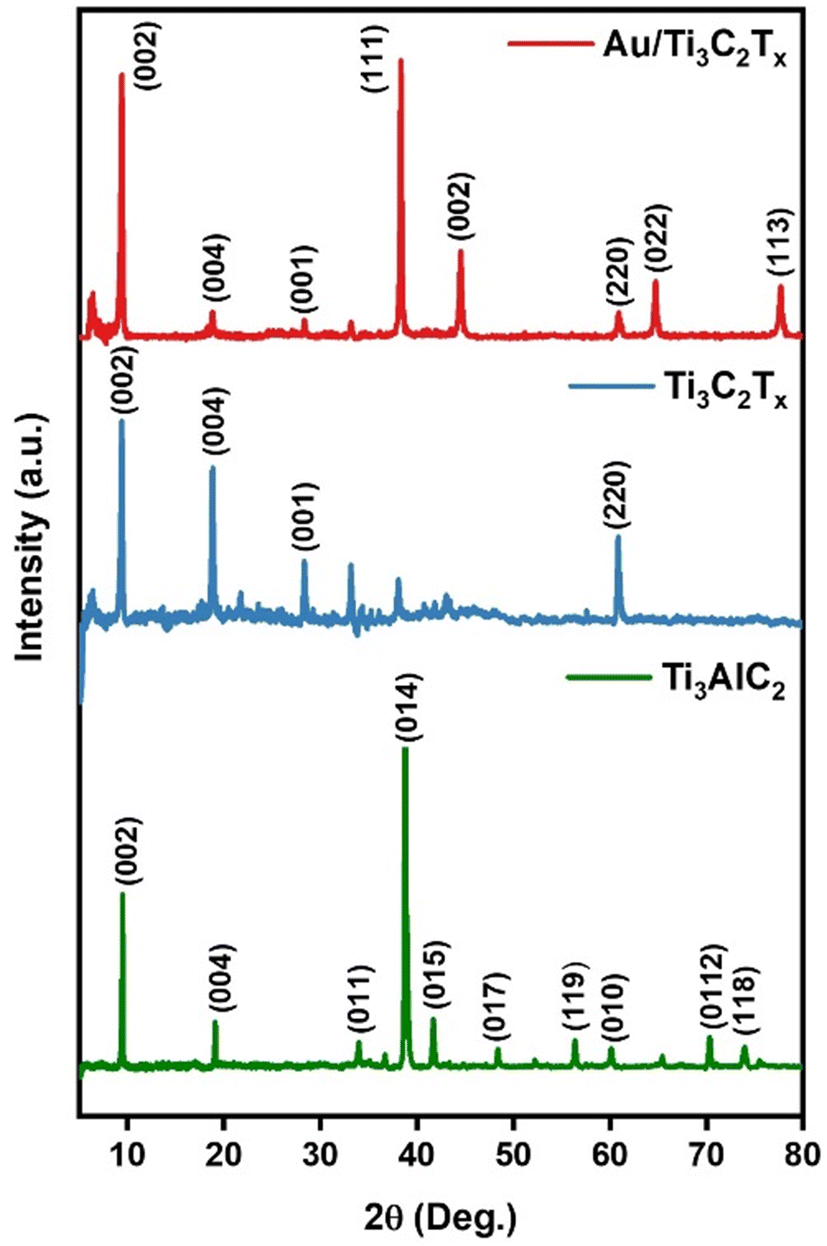 | ||
| Fig. 2 The PXRD of Ti3AlC2, Ti3C2Tx and Au/Ti3C2Tx composites. | ||
To understand the valence bond states of Au/Ti3C2Tx and Ti3C2Tx, XPS analysis was performed (Fig. 3 and Fig. S3†). The peaks between 454 and 457 eV indicate the potential presence of Ti low valence state species, including Ti(III) (∼456.2 eV) (Fig. S3†).42Fig. 3a shows a complete survey spectrum of Au/Ti3C2Tx and illustrates the simultaneous existence of Au, C, Ti, O, and F elements. The high-resolution XPS spectra of Ti 2p, C 1s, Au 4f, O 1s, and F 1s are given in Fig. 3a–f respectively.43 The two typical peaks of Ti 2p were observed at 459.4 eV and 465.1 eV, which are ascribed to Ti(IV) 2p3/2 and Ti(IV) 2p1/2, respectively (Fig. 3b).44 This suggests that during the synthesis of Au/Ti3C2Tx, a transition from low valence levels of Ti(II and III) to the high valence level of Ti occurs, i.e., Ti(II and III) were converted to Ti(IV) (TiO2) (Fig. S3†). Fig. 3c displays the deconvoluted C 1s spectrum of Au/Ti3C2Tx. The peaks at 289.4 eV, 286.5 eV, 285.4 eV, and 281.8 eV are attributed to O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O, C–C, and C–Ti.45 The deconvoluted Au 4f spectrum displays peaks at 83.9 and 87.6 eV, which are indicative of the reduced form of Au(0) with a distinct spin–orbit component (Δ = 3.7 eV), Fig. 3d.46,47 The result therefore suggests further that the Au NPs were successfully synthesized on the surfaces of Ti3C2Tx. As seen in Fig. 3e, the deconvoluted O 1s spectrum shows peaks at 530.6 eV and 532.1 eV, which can be matched with metal oxygen bonds (Ti–O and Ti–OH).48 Moreover, oxygenated functional groups play a role in enhancing surface functionality and improving wettability and they provide a negatively charged surface. Similarly, Fig. 3f shows the deconvoluted F 1s spectrum, in which the peak at 685.0 eV was assigned to Ti–F. These results further indicate that AuNPs exist on the Ti3C2Tx substrate, which has oxide, hydroxide and fluoride functionalities on the surface.
O, C–O, C–C, and C–Ti.45 The deconvoluted Au 4f spectrum displays peaks at 83.9 and 87.6 eV, which are indicative of the reduced form of Au(0) with a distinct spin–orbit component (Δ = 3.7 eV), Fig. 3d.46,47 The result therefore suggests further that the Au NPs were successfully synthesized on the surfaces of Ti3C2Tx. As seen in Fig. 3e, the deconvoluted O 1s spectrum shows peaks at 530.6 eV and 532.1 eV, which can be matched with metal oxygen bonds (Ti–O and Ti–OH).48 Moreover, oxygenated functional groups play a role in enhancing surface functionality and improving wettability and they provide a negatively charged surface. Similarly, Fig. 3f shows the deconvoluted F 1s spectrum, in which the peak at 685.0 eV was assigned to Ti–F. These results further indicate that AuNPs exist on the Ti3C2Tx substrate, which has oxide, hydroxide and fluoride functionalities on the surface.
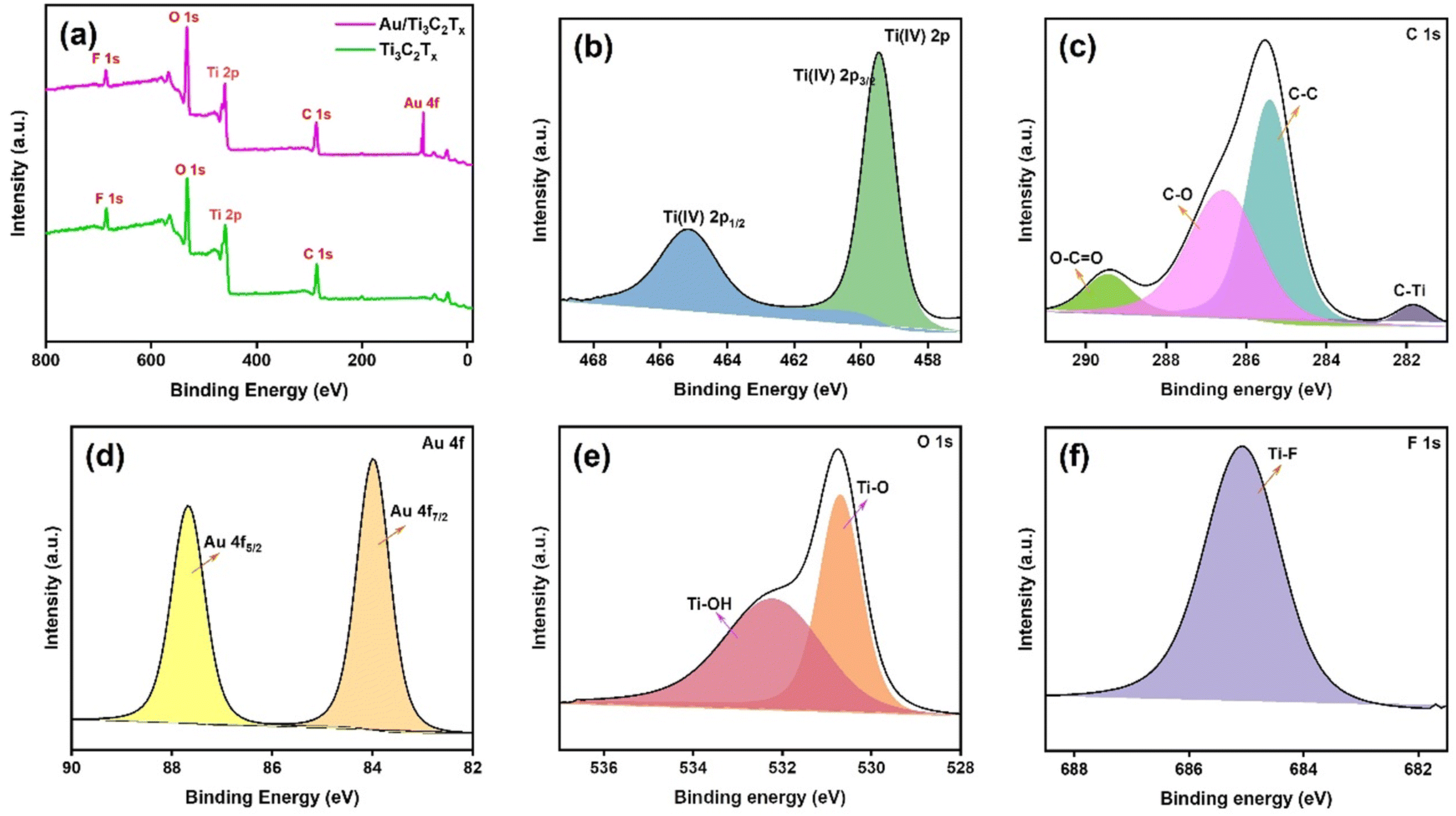 | ||
| Fig. 3 (a) The full survey XPS spectrum, and (b) Ti 2p, (c) C 1s, (d) Au 4f, (e) O 1s and (f) F 1s spectra of Au/Ti3C2Tx. | ||
The morphological features of Ti3C2Tx MXene nanosheets are shown in Fig. 4. The SEM image of etched Ti3C2Tx-MXene (Fig. 4a) indicated that Ti3C2Tx was well delaminated and exhibited an “accordion-like” morphology. Furthermore, the SEM image showed that the as-prepared Ti3C2Tx was stacked with a few layers of nanosheets (Fig. 4b). The AuNPs were successfully integrated into the exfoliated MXene nanosheets, as shown by the SEM image in Fig. 4c. Furthermore, efficient etching and integration are clearly seen in the EDX spectrum (Fig. S1†). Fig. 4d shows the remarkable transparency contrast of the 2D Ti3C2Tx thin sheets with the super carbon membrane, demonstrating their ultrathin nature. Fig. 4e shows the HR-TEM picture, which shows a lattice fringe spacing of 0.41 nm with the corresponding SAED pattern in the inset. The surface of 2D MXene thin sheets has uniformly dispersed AuNPs, as seen from the TEM image in Fig. 4f and the lattice fringe spacing is 0.61 nm, as seen from the HR-TEM picture in Fig. 4g. Additionally, the SAED pattern in the inset and the TEM image show that AuNPs were adhered to the MXene surface. EDS mapping analysis was performed to further validate the formation of the AuNPs and MXene assemblies (Fig. S2†). Fig. 4h shows the uniform distribution of AuNPs on the Ti3C2Tx thin sheets and the existence of Au, Ti, and C. According to the statistical TEM analysis of the Au/Ti3C2Tx composite (Fig. 4i), the average particle size was found to be 70 nm.
 | ||
| Fig. 4 (a) SEM image of pristine Ti3C2Tx, (b) FESEM image of exfoliated Ti3C2Tx, (c) the typical FESEM image of Au/Ti3C2Tx, (d) HRTEM image of exfoliated Ti3C2Tx, (e) TEM image of Ti3C2Tx, with the inset showing the SAED pattern, (f) HRTEM image of the Ti3C2Tx/AuNPs composite, (g) TEM image of the Ti3C2Tx/AuNP composite, with the inset showing the SAED pattern, (h) HAADF-TEM image of Au/Ti3C2Tx and the corresponding EDS mapping, and (i) particle size distribution. | ||
To assess the electrochemical performance of the MXene based catalysts in the co-conversion of CO2 and H2O to CO and H2, electrochemical experiments such as cyclic and linear sweep voltammetry were conducted in 0.1 M KHCO3 at ambient temperature and pressure. As shown in Fig. 5, an electrochemical cell was homemade and designed to collect the evolved gases from the working electrode compartment and inject them directly into the short-path of the gas chromatography valve for quantification. The catalyst's electrochemical behavior in the working solution was studied using Cyclic Voltammetry (CV) at 50 mV s−1 with N2. These voltammograms can be considered as blanks (Fig. S4a and S4b†). The corresponding electrocatalysts were tested over a range of potentials (0.6 to −1.4 VRHE). Electrocatalytic activity under a N2 flow can be attributed to either the hydrogen evolution reaction (HER) or catalyst reduction. Based on the voltammograms obtained with CO2 saturation and under a positive pressure of CO2, a reduction peak appeared at about −0.42 V vs. RHE (Fig. S4c†). This may be explained by the electron transfer mechanism caused by the adsorption intermediates formed during the CO2 reduction. In terms of current densities measured at the same potentials, for the same catalyst, slightly higher activity is noted in the presence of CO2 than in N2. This enhanced outcome could be attributed to the electrochemical CO2 reduction (Fig. 6a). The catalysts’ electrocatalytic activity was greatly influenced by their composition in the following order: Ti3AlC2 < Ti3C2Tx < Au/Ti3C2Tx. In this regard, the Ti3AlC2 and Ti3C2Tx electrocatalysts exhibited cathodic current densities of 17.0 mA cm−2 and 26.8 mA cm−2, respectively, at −1.2 V vs. RHE, whereas the Au/Ti3C2Tx composite displayed a current density of 43.9 mA cm−2 under identical conditions. When compared to the MAX phase Ti3AlC2 and pristine MXene Ti3C2Tx electrocatalysts, the Au/Ti3C2Tx composite showed significantly greater electrochemical activity.
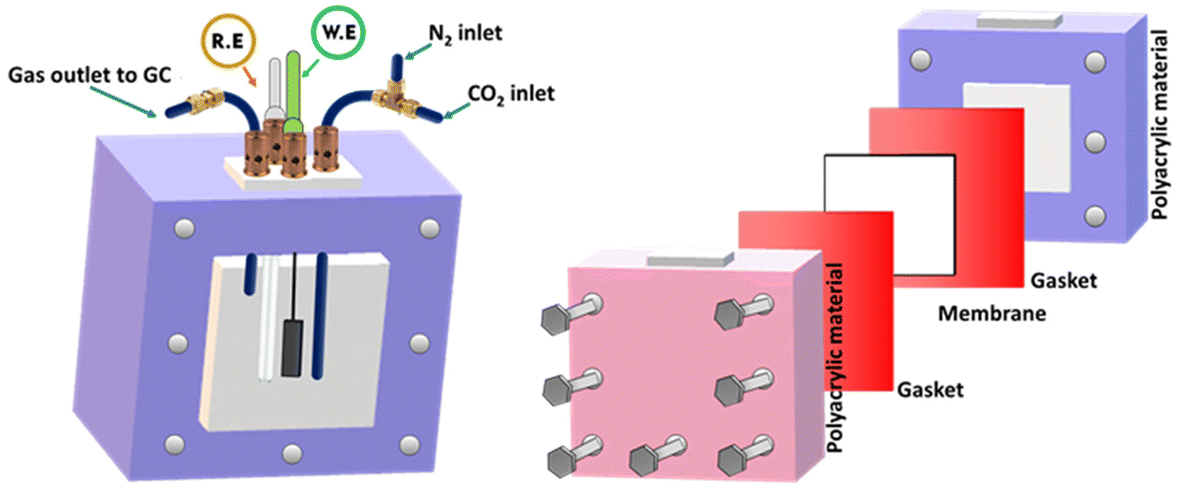 | ||
| Fig. 5 Electrochemical cell used in the ECRR experiment. | ||
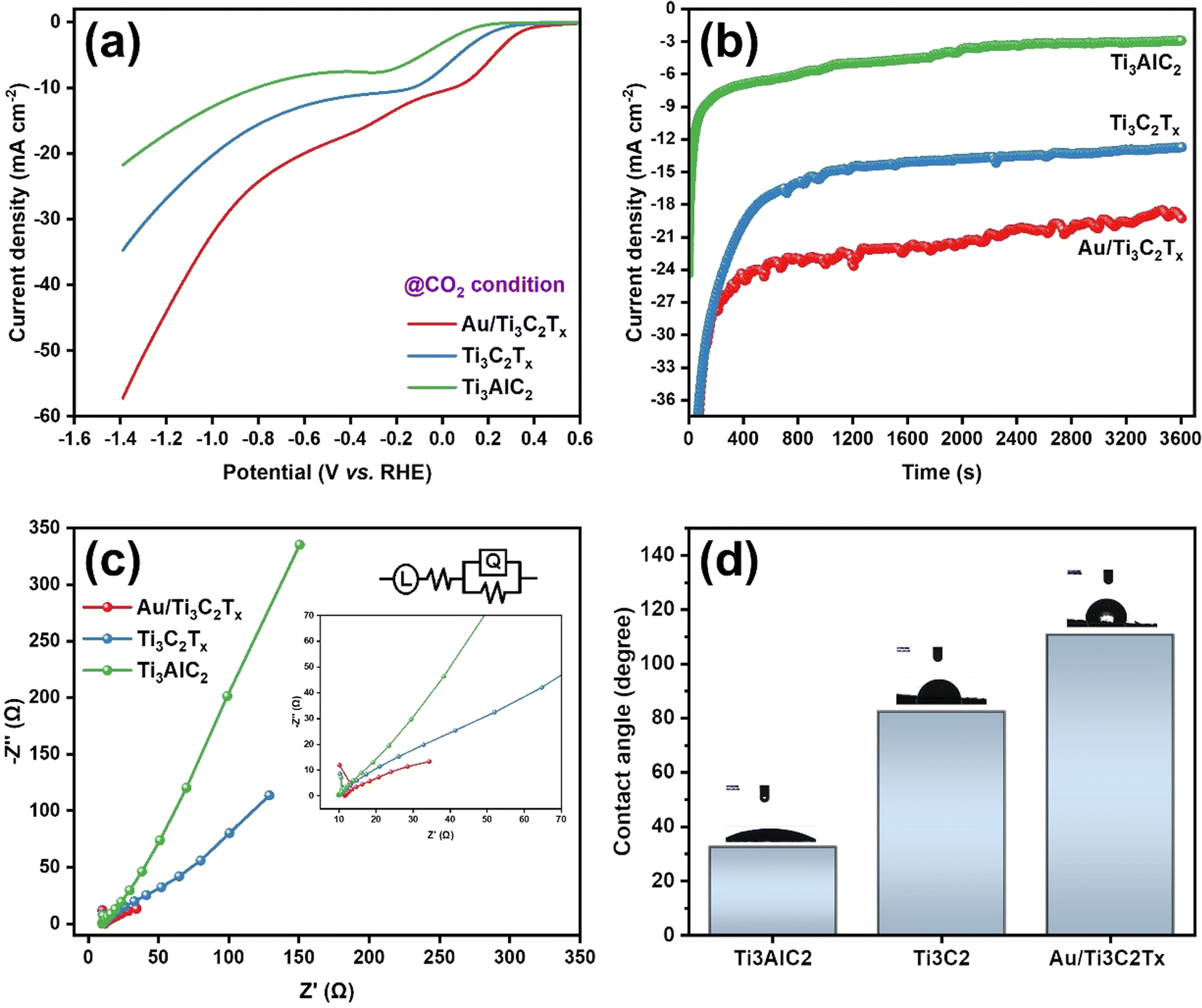 | ||
| Fig. 6 Assessment of the electrocatalytic efficiency of catalysts composed of Ti3AlC2, Ti3C2Tx and Au/Ti3C2Tx in the ECRR: (a) LSV plots; (b) chronoamperometric responses at −0.62 V vs. RHE; (c) EIS conducted on Au/Ti3C2Tx, Ti3C2Tx, and Ti3AlC2. Inset: equivalent electrical circuit; and (d) contact angle. | ||
Chronoamperometry (CA) was used to further investigate the electrochemical activity of these catalysts under ECRR conditions. The current–time transients are differentiated in Fig. 6b, where the CA was recorded for 60 minutes at E = −0.62 V vs. RHE. The CA curves followed the same trend as the LSV curves, which confirms that the formed Au/Ti3C2Tx composite displays the highest activity among the prepared catalysts. Ensuring the durability of the electrocatalyst over extended periods is a crucial aspect and a primary requirement for commercialization. In this aspect, a thorough examination was conducted to assess the long-term stability of the Au/Ti3C2Tx composite over a span of 36 hours, at a constant potential of −0.62 V vs. RHE (Fig. S4d†). Following the 36 hours electroreduction period, no noticeable alteration in the current density was detected, indicating the exceptional stability of the Au/Ti3C2Tx composite. The minor fluctuations observed in the chronoamperometry curve can be attributed to the formation and bursting of bubbles during continuous electrolysis, a phenomenon previously documented in similar investigations.
Furthermore, the ECSA of the Au/Ti3C2Tx composite was determined by measuring its double layer capacitance (Cdl) using cyclic voltammetry at various scan rates (10 to 50 mV s−1). The ECSA plot of the Au/Ti3C2Tx composite shows a maximum Cdl value of 153 μF cm−2, as shown in Fig. S5,† suggesting that there are more electrocatalytically active sites than usual. This further supports the ECRR's exceptional performance. To further understand the improved electrochemical behaviors, electrochemical impedance spectroscopy (EIS) was carried out in a CO2-saturated 0.1 M KHCO3 solution on Au/Ti3C2Tx, Ti3C2Tx, and Ti3AlC2 over a frequency spectrum ranging from 100 mHz to 1 MHz, using an AC voltage amplitude of 10 mV at the open circuit potential as shown in Fig. 6c. Au/Ti3C2Tx clearly exhibits a lower charge transfer resistance than Ti3AlC2 and Ti3C2Tx which is advantageous. This implies that Au/Ti3C2Tx has rapid charge transfer kinetics and the proposed electrical equivalent circuit could effectively fit all the EIS data (inset of Fig. 6c).
The performance and long-term stability of GDEs are also controlled by their wetting behaviour, which determines the presence and durability of gas–liquid–solid triple-phase barriers, in addition to their catalytic activity and accessible surface area. Two possible configurations for Au/Ti3C2Tx composite-coated GDEs in a zero-gap gas diffusion electrolyzer seem feasible: because the surface of the Au/Ti3C2Tx composites and their interior nanopores are fully wetted, there is a possibility that the electrolyte will enter the GDE; alternatively, the Au/Ti3C2Tx composites will preferentially become wet due to their higher capillary forces. The composited Au/Ti3C2Tx surface may also exhibit hydrophobic properties when the surface structures of Au are at the micrometer scale. The hydrophobicity of Au/Ti3C2Tx composite-coated GDEs with nanoscale porosity achieved through compositing is significantly increased, as measured using contact angle measurements. Consequently, the hydrophobicity increased from ∼32.6° for the Ti3AlC2 coating to ∼110.7° for the Au/Ti3C2Tx composite coatings (Fig. 6d). Overall, the contact angle measurements exhibited high reproducibility across various regions of the Au/Ti3C2Tx composite-coated GDEs, indicating a high level of sample uniformity. It is noteworthy that the hydrophobic properties of Au/Ti3C2Tx composite coatings align with the finding that free-standing Au/Ti3C2Tx layers float on water. The hydrophobic GDE substrate may have a bigger influence on the Au/Ti3C2Tx composite coating due to its higher contact angle than Ti3C2Tx.
The catalytic performance of Au/Ti3C2Tx composites and that of the resulting products were analyzed in detail during the ECRR on the Au/Ti3C2Tx electrode. The catalytic efficiency of the Au/Ti3C2Tx catalyst in the ECRR was assessed by contrasting the current densities obtained from LSVs in a 0.1 M KHCO3 electrolyte under N2-saturated and CO2-saturated conditions. The cathodic current observed in the N2-saturated solution was assigned to the Hydrogen Evolution Reaction (HER), whereas the cathodic current recorded in the CO2-saturated solution encompassed contributions from both the HER and the ECRR (Fig. 7a). Consequently, the electrocatalytic activity for the ECRR decreased as the cathodic potential shifted from −0.22 V to −0.82 V vs. RHE.
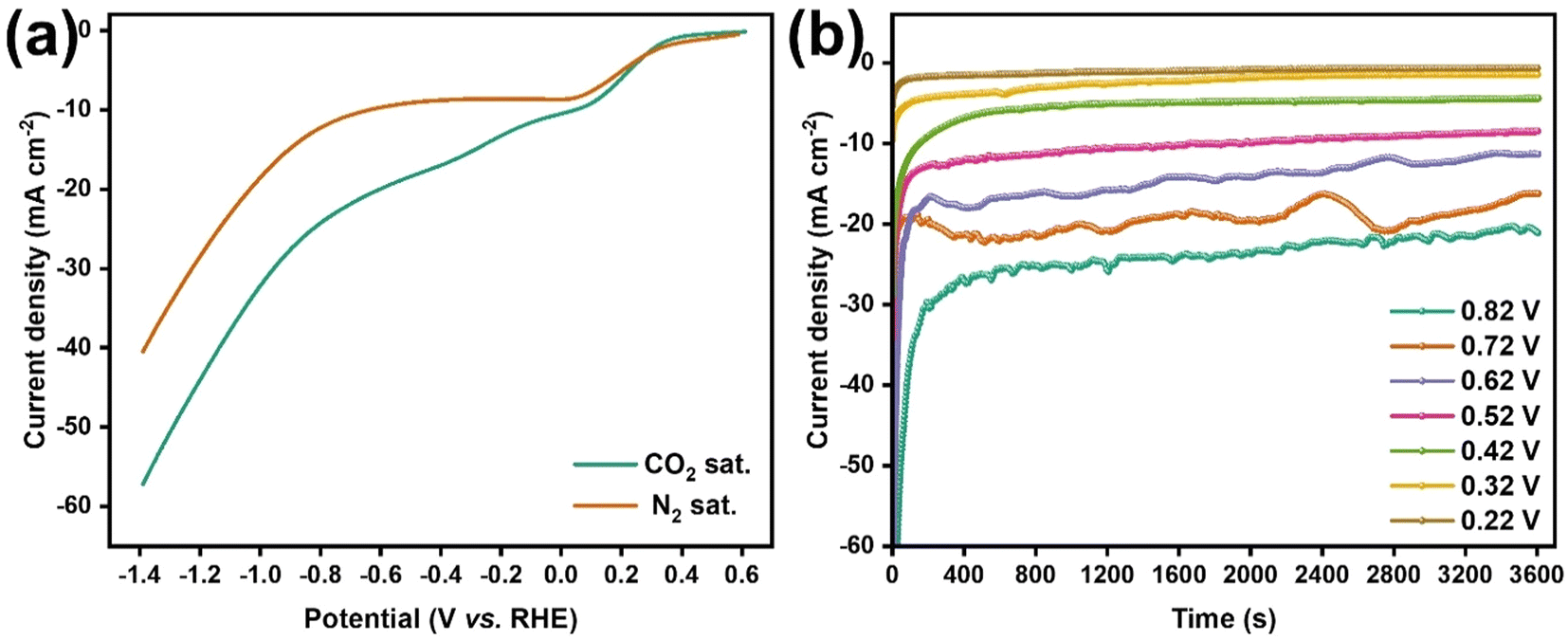 | ||
| Fig. 7 Electrocatalytic activity of the Au/Ti3C2Tx composites for the ECRR. (a) LSVs under CO2 saturated and N2 saturated conditions and (b) chronoamperometric curves at a potential of −0.22 V (brown) and −0.82 V (green) vs. RHE (colour on line). | ||
The influence of the applied electrode potential on the electrochemical performance of the Au/Ti3C2Tx catalyst was further explored using chronoamperometry. Fig. 7b displays the CA curves recorded at −0.22, −0.32, −0.42, −0.52, −0.62, −0.72, and −0.82 V vs. RHE for 60 min. The disturbance observed in the recorded CA curve was due to the gas formation on the catalyst surface and the subsequent detachment of the formed gas bubbles from the electrode surface. Once the gas bubbles reach a sufficient size, they separate from the catalyst surface, resulting in a subsequent increase in current density. The applied cathodic potential increased from −0.22 to −0.82 V vs. RHE, increasing the steady-state current density, which was consistent with the LSV curves shown in Fig. 7a. Furthermore, the CA tests demonstrated that the Au/Ti3C2Tx composite exhibited great stability during the electrochemical reduction of CO2.
Gaseous products generated in the course of the ECRR at the Au/Ti3C2Tx composites were analyzed using gas chromatography (GC), as shown in Fig. S6.† It was observed that CO and H2 were the major products, and that only <1% of CH4 was detected, as shown in Fig. S7.†Fig. 8 shows that the faradaic efficiency of the production of H2 and CO is strongly dependent on the applied electrode potential. It varied from −0.22 V to −0.82 V vs. RHE. At a cathodic potential of −0.42 V vs. RHE, analysis revealed that syngas was the predominant product obtained with a higher faradaic efficiency of 48.3% for CO and 25.6% for H2. Different ratios of syngas were generated and this variation in the ratio might be useful for the production of numerous industrial compounds. NMR was utilized for the analysis of the aqueous products, uncovering that formate was obtained as a major product in the aqueous phase, as shown in Fig. S8.† The findings from both GC and NMR analyses support the conclusion that the ECRR using the Au/Ti3C2Tx electrode primarily promotes the formation of syngas, a mixture of CO and H2, with a small amount of liquid products. The results further indicate that the ratio of syngas/liquid products may be adjusted by altering the applied cathodic potential.
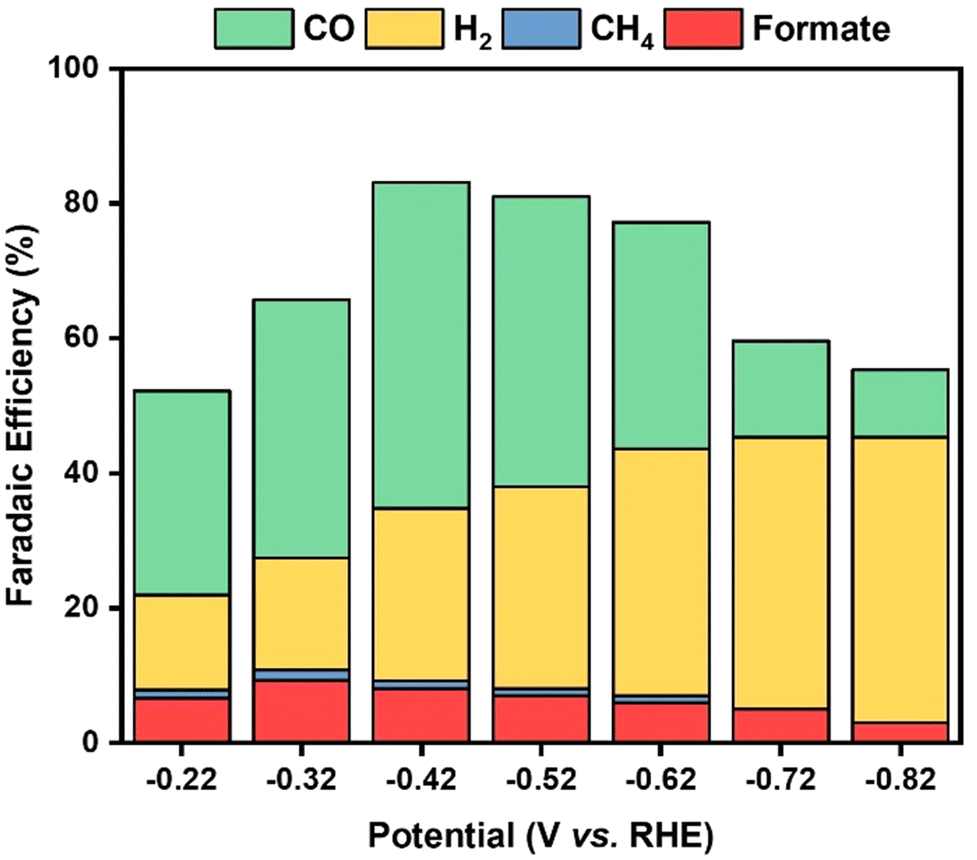 | ||
| Fig. 8 Faradaic efficiency of ECRR products. | ||
Conclusions
A new Au/Ti3C2Tx electrocatalyst was prepared for application in the ECRR through an etching/fabrication process. The elemental compositions revealed by XRD and EDS analyses of the Au/Ti3C2Tx composites closely matched their surface compositions as determined by XPS, indicating uniformity of composition across the catalyst materials. The morphologies demonstrate that Au/Ti3C2Tx composites were significantly formed. The Au/Ti3C2Tx composite exhibited the highest electrocatalytic activity for the ECRR compared to Ti3AlC2 and Ti3C2Tx examined in this study. Also, an Au/Ti3C2Tx based gas-diffusion electrode was prepared and used as a cathode for the conversion of CO2 to CO owing to its improved electrocatalytic activity. GC analysis revealed that syngas was the predominant product obtained with a higher faradaic efficiency of 48.3% for CO and 25.6% for H2 at a cathodic potential of −0.42 V vs. RHE. NMR analysis also revealed that formate was generated with a significant faradaic efficiency. Moreover, the EIS investigation demonstrated that Au/Ti3C2Tx composites exhibited significantly lower charge transfer resistance compared to the other materials. In summary, the Au/Ti3C2Tx composite presents itself as a compelling material with promising potential for applications in the production of synthesis gas from CO2. Further studies using this Au/Ti3C2Tx catalyst towards the large-scale production of syngas and further optimization are underway.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to thank the Director of CSIR-CECRI, Karaikudi, and staff members for their constant support and encouragement. Central Instrumentation Facility Division, CSIR-CECRI is acknowledged for characterization facilities. Dr M. K. acknowledges CSIR-FIRST (MLP-1004) for financial support. CECRI manuscript number: CECRI/PESVC/Pubs/2024-043.References
- D. R. Feldman, W. D. Collins, P. J. Gero, M. S. Torn, E. J. Mlawer and T. R. Shippert, Nature, 2015, 519, 339–343 CrossRef CAS PubMed.
- S. Pacala and R. Socolow, Science, 2004, 305, 968–972 CrossRef CAS PubMed.
- S. Verma and A. Yadav, Energy Fuels, 2023, 37, 5712–5742 CrossRef CAS.
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Chem. Soc. Rev., 2021, 50, 4993–5061 RSC.
- Z. Chen, X. Wang and L. Liu, Chem. Rec., 2019, 19, 1272–1282 CrossRef CAS PubMed.
- A. Prajapati, R. Sartape, M. T. Galante, J. Xie, S. L. Leung, I. Bessa, M. H. S. Andrade, R. T. Somich, M. V. Rebouças, G. T. Hutras, N. Diniz and M. R. Singh, Energy Environ. Sci., 2022, 15, 5105–5117 RSC.
- Y. Zhang, L.-Z. Dong, S. Li, X. Huang, J.-N. Chang, J.-H. Wang, J. Zhou, S.-L. Li and Y.-Q. Lan, Nat. Commun., 2021, 12, 6390 CrossRef CAS PubMed.
- Q. Lu, K. Eid and W. Li, Nanomaterials, 2022, 12, 2379 CrossRef CAS.
- T. N. Nguyen and C.-T. Dinh, Chem. Soc. Rev., 2020, 49, 7488–7504 RSC.
- F. P. García de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef.
- B. Ambrose, R. Naresh, S. Deshmukh, M. Kathiresan and P. Ragupathy, Energy Fuels, 2023, 37, 18226–18242 CrossRef CAS.
- H. Mistry, R. Reske, Z. Zeng, Z.-J. Zhao, J. Greeley, P. Strasser and B. R. Cuenya, J. Am. Chem. Soc., 2014, 136, 16473–16476 CrossRef CAS PubMed.
- W. Zhu, R. Michalsky, Ö. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- D. Gao, Y. Zhang, Z. Zhou, F. Cai, X. Zhao, W. Huang, Y. Li, J. Zhu, P. Liu, F. Yang, G. Wang and X. Bao, J. Am. Chem. Soc., 2017, 139, 5652–5655 CrossRef CAS PubMed.
- W. Zhu, R. Michalsky, Ö. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- S. Back, M. S. Yeom and Y. Jung, ACS Catal., 2015, 5, 5089–5096 CrossRef CAS.
- W. Zhu, Y.-J. Zhang, H. Zhang, H. Lv, Q. Li, R. Michalsky, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2014, 136, 16132–16135 CrossRef CAS PubMed.
- G. O. Larrazábal, A. J. Martín and J. Pérez-Ramírez, J. Phys. Chem. Lett., 2017, 8, 3933–3944 CrossRef.
- K. Sun, T. Cheng, L. Wu, Y. Hu, J. Zhou, A. Maclennan, Z. Jiang, Y. Gao, W. A. Goddard, III and Z. Wang, J. Am. Chem. Soc., 2017, 139, 15608–15611 CrossRef CAS PubMed.
- Y. Chen, C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS PubMed.
- X. Yan, C. Duan, S. Yu, B. Dai, C. Sun and H. Chu, Renewable Sustainable Energy Rev., 2024, 190, 114086 CrossRef CAS.
- B. Salah, K. Eid, A. M. Abdelgwad, Y. Ibrahim, A. M. Abdullah, M. K. Hassan and K. I. Ozoemena, Electroanalysis, 2022, 34, 677–683 CrossRef CAS.
- Y. He, W.-J. Jiang, Y. Zhang, L.-B. Huang and J.-S. Hu, J. Mater. Chem. A, 2019, 7, 18428–18433 RSC.
- L. Zhang, Z. Wang, N. Mehio, X. Jin and S. Dai, ChemSusChem, 2016, 9, 428–432 CrossRef CAS PubMed.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 3242 CrossRef PubMed.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- K. Rasool, M. Helal, A. Ali, C. E. Ren, Y. Gogotsi and K. A. Mahmoud, ACS Nano, 2016, 10, 3674–3684 CrossRef CAS PubMed.
- A. Vijayaprabhakaran and M. Kathiresan, Mater. Adv., 2023, 4, 3593–3602 RSC.
- Y. Ibrahim, M. Meslam, K. Eid, B. Salah, A. M. Abdullah, K. I. Ozoemena, A. Elzatahry, M. A. Sharaf and M. Sillanpää, Sep. Purif. Technol., 2022, 282, 120083 CrossRef CAS.
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS.
- X.-Q. Tan, W. Mo, X. Lin, J. Y. Loh, A. R. Mohamed and W.-J. Ong, Nanoscale, 2023, 15, 6536–6562 RSC.
- D. Qu, X. Peng, Y. Mi, H. Bao, S. Zhao, X. Liu and J. Luo, Nanoscale, 2020, 12, 17191–17195 RSC.
- Z. Chen, X. Wang, J. P. Mills, C. Du, J. Kim, J. Wen and Y. A. Wu, Nanoscale, 2021, 13, 19712–19739 RSC.
- S. Verma, X. Lu, S. Ma, R. I. Masel and P. J. A. Kenis, Phys. Chem. Chem. Phys., 2016, 18, 7075–7084 RSC.
- L.-C. Weng, A. T. Bell and A. Z. Weber, Phys. Chem. Chem. Phys., 2018, 20, 16973–16984 RSC.
- H. Rabiee, L. Ge, X. Zhang, S. Hu, M. Li and Z. Yuan, Energy Environ. Sci., 2021, 14, 1959–2008 RSC.
- W. Feng, H. Luo, Y. Wang, S. Zeng, L. Deng, X. Zhou, H. Zhang and S. Peng, RSC Adv., 2018, 8, 2398–2403 RSC.
- Z. Li, L. Wang, D. Sun, Y. Zhang, B. Liu, Q. Hu and A. Zhou, Mater. Sci. Eng., B, 2015, 191, 33–40 CrossRef CAS.
- H. Fang, Y. Pan, M. Yin and C. Pan, J. Mater. Sci.: Mater. Electron., 2019, 30, 14954–14966 CrossRef CAS.
- F. Zhang, W. Liu, S. Wang, C. Liu, H. Shi, L. Liang and K. Pi, Composites, Part B, 2021, 217, 108900 CrossRef CAS.
- K. Tan, S. Liu, Y. Liu, L. Hou and C. Yuan, Nanoscale Adv., 2022, 4, 5253–5256 RSC.
- S. S. Sangu, N. M. Illias, C. C. Ong, S. C. B. Gopinath and M. S. M. Saheed, BioNanoScience, 2021, 11, 314–323 CrossRef.
- Z. Wu, D.-W. Sun, H. Pu, Q. Wei and X. Lin, Food Chem., 2022, 372, 131293 CrossRef CAS.
- H. Medetalibeyoglu, G. Kotan, N. Atar and M. L. Yola, Talanta, 2020, 220, 121403 CrossRef CAS PubMed.
- B. Wang, Q. Shu, H. Chen, X. Xing, Q. Wu and L. Zhang, Metals, 2022, 12, 2022 CrossRef CAS.
- Z. Zheng, W. Wu, T. Yang, E. Wang, Z. Du, X. Hou, T. Liang and H. Wang, J. Adv. Ceram., 2021, 10, 1061–1071 CrossRef CAS.
- H. Zhang, Z. Wang, F. Wang, Y. Zhang, H. Wang and Y. Liu, Anal. Chem., 2020, 92, 5546–5553 CrossRef CAS PubMed.
- K. Eid, Q. Lu, S. Abdel-Azeim, A. Soliman, A. M. Abdullah, A. M. Abdelgwad, R. P. Forbes, K. I. Ozoemena, R. S. Varma and M. F. Shibl, J. Mater. Chem. A, 2022, 10, 1965–1975 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01122h |
| This journal is © The Royal Society of Chemistry 2024 |