
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical deposition of gold nanoparticles on carbon ultramicroelectrode arrays†
Courtney J.
Weber
,
Natalie E.
Strom
and
Olja
Simoska
 *
*
Department of Chemistry and Biochemistry, University of South Carolina, 631 Sumter Street, Columbia, SC 29208, USA. E-mail: osimoska@mailbox.sc.edu
First published on 9th August 2024
Abstract
Electrode surfaces functionalized with gold nanoparticles (AuNP) are widely used in electroanalysis, electrocatalysis, and electrochemical biosensing due to their increased surface area and conductivity. Electrochemical deposition of AuNPs offers advantages over chemical synthesis, including better control over AuNP size, dispersion, and morphology. This study examines the electrodeposition of AuNPs on carbon ultramicroelectrode arrays (CUAs) focusing on electrodeposition parameters, such as deposition potential, deposition time, and gold ion concentration. Detailed analysis based on scanning electron microscopy revealed that higher reductive potentials and shorter deposition times result in smaller AuNP particle sizes and greater particle counts. Unlike previous studies using planar, macro-sized electrodes and millimolar concentrations of gold ion, as well as longer deposition times (e.g., 100–300 s), this research employed micromolar concentration ranges (25–50 μM) of gold ion solution and shorter deposition times (5–60 s) for successful electrodeposition of AuNPs on the array-based CUAs. This is attributed to the physical properties of the ultramicroelectrodes in the array geometry and the distinct material composition of the CUAs. The gold amounts deposited on the CUA electrodes were determined (88.73 ± 0.06 nmol cm−2), which were in correlation with the electrocatalytic responses for the hydrogen evolution reaction (HER) measured on AuNP-modified CUAs. Overall, the array-based geometry, nanometer-scale electrode sizes, and unique material composition of the CUAs significantly influence AuNP electrodeposition. This study underscores the importance of systematically characterizing the electrodeposition parameters on novel electrode surfaces.
 Olja Simoska | Olja Simoska is an assistant professor in the Department of Chemistry and Biochemistry at the University of South Carolina. Her research focuses on the development of electrochemical sensing devices, modified electrodes for sensing and catalysis, and engineering electrode interfaces. Prof. Simoska received her Bachelor's degree in Chemistry from Bard College in 2015 and her Ph.D. in Analytical Chemistry from the University of Texas at Austin in 2019. Before joining the University of South Carolina, she was the 13th ACS Irving S. Sigal Postdoctoral Fellow in the Department of Chemistry at the University of Utah (2020–2022). |
1. Introduction
Nanoparticles (NPs) are materials characterized by their dimensions in the nanoscale (1–100 nm) and can be comprised of various materials, including metals, inorganic semiconductor molecules, natural or artificial polymers, and carbon.1–3 Given that they are made up of only a few atoms, NPs exhibit unique chemical and physical properties, different than their parent bulk material, which can be attributed to their increased surface area-to-volume ratio (SA/V).2,4–7 Specifically, metal NPs are associated with desirable properties for applications in catalysis,8–11 drug delivery and gene systems,12,13 environmental remediation,14 electroanalysis,15,16 as well as the development of optical and electrochemical sensors and biosensors for health monitoring or medical analysis,17–21 fuel cells,22,23 and batteries.24 In the area of electrochemistry and electroanalysis, electrode surfaces are often decorated with NPs to enhance catalytic activity toward redox-active analytes due to the greater SA/V and higher conductivity of these nano-sized materials.15,25–29 Namely, gold nanoparticles (AuNPs) are frequently utilized in the fabrication of electrochemical sensing devices due to the chemical inertness and conductivity of the metal.6,30–34AuNPs are traditionally synthesized by chemical means, which involves particle formation in a colloidal solution upon the addition of reducing agents and capping agents, which are steps performed under high-temperature conditions.33,35,36 Although these chemical synthesis methods have proven to be effective in achieving particle formation, these approaches require extensive sample preparation and involve usage of toxic solvents, contamination from precursor chemicals, and production of hazardous derivatives.1,35,37 Furthermore, chemical AuNP syntheses have limitations in the control of NP size, morphology, and dispersion on the electrode surface. On the other hand, electrochemical means of AuNP formation via electrodeposition represent an attractive alternative to the chemical-based approaches, addressing the challenges of synthetic steps. In electrodeposition methods, the reduction of gold ions in an electrolyte solution is initiated by applying a reductive potential to the working electrode, resulting in the formation of solid AuNPs on the electrode surface. In general, the electrodeposition of AuNPs utilizes a solution of (1) tetrachloroaurate ion (AuCl4−), usually in the form of chloroauric acid (HAuCl4), as the gold ion (Au3+) source and (2) a supporting electrolyte, such as sulfuric acid (H2SO4), perchloric acid (HClO3), nitric acid (HNO3), or citric acid (C6H8O7), to allow for ion mobility in the electrochemical cell and establish electrical conductivity in solution.38–41 Electrodeposition enables the control of NP physical properties through the variance of experimental parameters, including electrode material, deposition time, deposition potential, tetrachloroaurate ion concentration, and supporting electrolyte composition and concentration.2,26,28
Several fundamental studies on the effects of electrochemical deposition parameters on AuNP formation have been previously reported. These research works have reported a wide variety of electrode materials and modifications, including single-walled carbon nanotube (SWNT)-modified electrodes,26 platinum electrodes,41 glassy carbon electrodes,25,38 graphite electrodes,42 boron-doped diamond electrodes,36 fluorine-doped tin oxide electrodes,43 and indium tin oxide (ITO) electrodes.39 Namely, Zamborini and co-workers reported several results on the electrodeposition of NPs, including a specific study analyzing the effects of particle size on oxidation potential in brominated supporting electrolyte solution.39 It was observed that applying more reductive potentials resulted in the formation of a larger collection of smaller AuNPs on planar ITO electrodes. Furthermore, the authors reported that increasing particle diameter yielded a positive shift in oxidation potential, which was in agreement with theoretical predictions.39 Dudin et al. discussed the influence of both electrochemical driving force (e.g., deposition potential), and deposition time on AuNP formation on SWNTs via microcapillary electrodeposition. The authors found that a higher driving force (i.e., more reductive deposition potential), yielded increased particle density. Additionally, this study revealed that longer time regimes produced particles of larger size with a broader size distribution. This phenomenon was attributed to Ostwald ripening, in which the growth of smaller particles is hindered and overtaken by larger-sized particles.26,44 More recently, Zakaria and co-workers presented new insights on the effects of supporting electrolyte composition on AuNP morphology and formation. Their findings indicated that sodium nitrate (NaNO3) produced the most uniform, homogenous AuNPs of approximately 50 nm in diameter.38 Although these studies provide valuable fundamental insight into how supporting electrolyte composition, deposition time, and deposition potential affect AuNP formation, each of the aforementioned studies utilizes planar, macro-sized electrodes. To date, there are few reports of AuNP electrodeposition on electrodes and ultramicroelectrodes with an array-based geometry, which have applications in environmental trace analysis, electrophoresis detectors, and chemical and biological sensors.45–47 Hariri et al. reported the electrodeposition mechanism of gold nanowires and nanotubes by analyzing the system in a recessed cylindrical ultramicroelectrode diffusional model.48 While Hariri's work presented relevant information on the transition from radial to planar diffusion upon deposition of the cylindrical gold components in an array-based geometry, it is limited with regard to the discussion of gold nanomaterial deposition on ultramicroelectrodes fashioned in an array design.
Herein, we demonstrate a fundamental electrochemical study on the electrodeposition of AuNPs on a novel, array-based electrode platform, based on carbon ultramicroelectrode arrays (CUAs). CUAs consist of a unique combination of carbon and metal oxide materials, resulting in an array-based geometry of carbon nanometer-sized electrodes (∼90 nm radius) encircled by a metal oxide layer of aluminum oxide (Al2O3).49–52 The carbon material employed in these electrodes is a pyrolyzed photoresist film (PPF), which exhibits similar electrochemical behavior to glassy carbon.49,50 This PPF material is highly conductive, hard, and inert with a wide window of electrochemical stability.50 The individual electrodes in the array are wired in a parallel circuit, yielding the advantages of improved signal-to-noise ratio for the detection of electrochemical events (amplified currents).49 Most of the exposed electrode surface is the Al2O3 layer, which also limits adsorption on the overall carbon surface.49,50,53,54 Furthermore, the combination of carbon and alumina material contributes to significantly lowering the background current responses at the electrode surface.50 Most of the total capacitance (proportional to noise) on the CUA electrodes is a result of the Al2O3 metal oxide layer, which acts as a dielectric that stores and reorganizes charge. On the other hand, a Macro electrode consisting of only PPF film and composed entirely of carbon material has capacitance associated with double-layer charging on the electrode surface. The Al2O3 layer on the CUA array-based electrodes greatly diminishes the double-layer capacitance, thereby resulting in significantly lower noise and improved S/N ratios compared to the Macro electrodes. As a result, CUAs offer highly sensitive analyte detection with fast response times and have been successfully employed for electrochemical detection of various analytes in complex biological matrices with adequate analytical figures of merit.51–56 These electrodes are fabricated using facile and highly reproducible methods reported previously.49,50,52 Although photoresist-derived carbon is used for CUA fabrication in this study, it should be noted that this versatile process could be adapted to design ultramicroelectrode arrays on other conductive materials, such as gold and platinum, allowing for the creation of customizable electrode platforms suited for various electrochemical methods. Moreover, the CUA electrodes can be made transparent, making them particularly valuable for spectro-electrochemical studies. Finally, CUAs can also be easily modified with nanomaterials, such as AuNPs, for desired electrochemical applications,52 ranging from electroanalytical (bio)sensors to electrocatalysis to energy storage and conversion devices.
To the best of our knowledge, this work provides, for the first time, a detailed fundamental understanding and characterization of AuNP electrodeposition on CUAs. This study establishes experimental electrodeposition parameters to achieve optimal AuNP formation with small, narrow size distribution and consistent dispersion across each individual electrode in the CUA electrode platform. Cyclic voltammetry was employed to evaluate the redox activity of gold across a wide range of potentials at the CUA surface. AuNPs were electrodeposited on CUAs via amperometric methods under specific experimental conditions of varying deposition potential, deposition time, and AuCl4− concentration. The AuNP-modified CUA samples were characterized with scanning electron microscopy (SEM), providing information on AuNP particle size and dispersion, as well as the average number of particles per individual electrode within the CUA for quantitative comparison of the experimental parameters. As a proof-of-concept study, we demonstrate the electrocatalytic activity of the AuNP-modified CUAs for the hydrogen evolution reaction (HER). Control studies were performed with carbon Macro electrodes (planar electrodes comprised of PPF electrode surface without the array), displaying the CUA advantages of array-based geometry and the insulating alumina layer. Our experimental results indicate that each experimental electrodeposition parameter, including the electrochemical driving force (i.e., applied potential), electrodeposition time regime, and gold ion source concentration, significantly impacts the formation of AuNPs on the CUAs. Furthermore, the physical characteristics resulting from the unique carbon-based CUA array shape and electrode material composition influence the concentration of the gold ion solution and deposition times necessary to yield consistent particle formation across the electrode surface. Namely, compared to previous studies performed on planar, macro-sized electrodes employing minute-long deposition times and millimolar gold source concentration ranges for AuNP electrodeposition,5,39,44,57,58 our results show that we can successfully electrodeposit AuNPs on CUA surfaces with seconds-long deposition times and lower, micromolar concentrations of gold ion solution.
2. Experimental methods
2.1 Chemicals and materials
All chemicals and materials were used as received. Polystyrene spheres (Polybead®) with a diameter of 1.54 μm were purchased from Polysciences, Inc. Photoresist AZ1518 from Integrated Micro Materials (iMicromaterials). Quartz microscopic slides (with an area of 6.45 cm2 and thickness of 1 mm) for electrode fabrication were acquired from Technical Glass Products. Gold(III) chloride solution (HAuCl4), and sulfuric acid (H2SO4) were purchased from Sigma-Aldrich. The water for all experiments was dispensed from a Millipore Sigma Milli-Q® EQ 7000 ultrapure water purification system (resistivity of 18.2 MΩ cm at 25 °C).2.2 Electrochemical deposition and characterization of gold nanoparticles (AuNPs)
All electrochemical experimentation utilized a three-electrode cell setup with either carbon ultramicroelectrode arrays (CUAs) or Macro electrodes as the working electrode (WE), platinum mesh (Pt) as the counter electrode (CE), and a saturated calomel electrode (SCE) as the reference electrode (RE). CUAs were fabricated according to previously established procedures from the Stevenson Research Group.49–56 Briefly, quartz substrates were cleaned with piranha solution (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2SO4:30% H2O2) to remove organic contamination and residual organics. Undiluted AZ1518 photoresist was spun on the piranha-cleaner quartz slides at 6000 rpm for 1 min (WS-650Mz-23NPPB spin coater, Laurell Technologies), which was then pyrolyzed via tube furnace procedures detailed elsewhere.59–63 Namely, after purging for 15 s with forming gas consisting of 5% H2:95% N2 (∼100 mL min−1), the photoresist quartz slides were pyrolyzed by heating to 1000 °C at 5 °C min−1 and holding at that temp for 1 h before allowing them to cool slowly back to room temperature at 5 °C min−1. Following this tube furnace procedure, pyrolyzed photoresist film (PPF) formed, yielding the carbon Macro electrode (carbon electrode without the array). The PPFs were then stored for 3 days prior to use to allow for the oxide layer to stabilize.64 As reported previously, the PPFs prepared by this approach have a thickness of 250 ± 20 nm, roughness of 0.39 ± 0.07 nm, and sheet resistance of 97 ± 3 Ω □−1.65 Following the pyrolysis step, polystyrene spheres (PSSs) with 1.54 μm diameter were then drop cast onto the PPF film, and a 10 nm-layer of alumina (Al2O3) was deposited onto the PSS via atomic layer deposition (ALD) (AT410 bench-top ALD system, Anric Technologies). Following the ALD step, the PSSs were removed via a series of sonication steps in methanol, acetone, ethanol, and water, leaving an ordered array of individual carbon ultramicroelectrodes where the spheres contacted the PPF, forming the CUAs. In the final step, the electrodes were dried using a stream of N2 gas prior to experimentation.
:1 H2SO4:30% H2O2) to remove organic contamination and residual organics. Undiluted AZ1518 photoresist was spun on the piranha-cleaner quartz slides at 6000 rpm for 1 min (WS-650Mz-23NPPB spin coater, Laurell Technologies), which was then pyrolyzed via tube furnace procedures detailed elsewhere.59–63 Namely, after purging for 15 s with forming gas consisting of 5% H2:95% N2 (∼100 mL min−1), the photoresist quartz slides were pyrolyzed by heating to 1000 °C at 5 °C min−1 and holding at that temp for 1 h before allowing them to cool slowly back to room temperature at 5 °C min−1. Following this tube furnace procedure, pyrolyzed photoresist film (PPF) formed, yielding the carbon Macro electrode (carbon electrode without the array). The PPFs were then stored for 3 days prior to use to allow for the oxide layer to stabilize.64 As reported previously, the PPFs prepared by this approach have a thickness of 250 ± 20 nm, roughness of 0.39 ± 0.07 nm, and sheet resistance of 97 ± 3 Ω □−1.65 Following the pyrolysis step, polystyrene spheres (PSSs) with 1.54 μm diameter were then drop cast onto the PPF film, and a 10 nm-layer of alumina (Al2O3) was deposited onto the PSS via atomic layer deposition (ALD) (AT410 bench-top ALD system, Anric Technologies). Following the ALD step, the PSSs were removed via a series of sonication steps in methanol, acetone, ethanol, and water, leaving an ordered array of individual carbon ultramicroelectrodes where the spheres contacted the PPF, forming the CUAs. In the final step, the electrodes were dried using a stream of N2 gas prior to experimentation.
All electrochemical experiments were performed on CH650E, CH660E, and CH440C potentiostats (CH Instruments). Gold nanoparticle (AuNP) electrochemical deposition experiments employed single-potential-step chronoamperometry to reduce gold cations in solution to solid gold at the surface of the CUA WE. The potential was held at an initial value of 0.75 V vs. SCE for 1 s, then stepped to the associated cathodic deposition potential (0.25, 0, −0.25, or −0.5 V vs. SCE) and held at this electrodeposition potential for a specified deposition time. The sample interval was 0.001 s with a sensitivity of 0.001 A V−1. Cyclic voltammetric characterization studies were conducted at a scan rate of 0.1 V s−1 with a sample interval of 0.001 V, quiet time of 2 s, and sensitivity of 0.001 A V−1. Experimental data was plotted and analyzed in Excel and OriginLab Graphing and Analysis Software. All electrochemical results were plotted using the classic (or polarographic, Texas) convention, with oxidation potentials plotted in the negative “x-axis” direction and reduction potentials plotted in the positive “x-axis” direction, with cathodic currents in the positive “y-axis” direction and anodic currents in the negative “y-axis” direction.
2.3 Scanning electron microscopy (SEM) imaging and sample preparation
Following amperometric electrochemical deposition, the AuNP-modified CUA and Macro electrode samples were prepared for SEM by rinsing with MilliQ ultrapure water and drying with nitrogen. SEM experiments were performed in the Electron Microscopy Center, a shared facility at the University of South Carolina. A Zeiss Gemini500 FESEM model was used for all gold nanoparticle (AuNP) imaging experiments. ImageJ software was then utilized to analyze the obtained SEM images and perform AuNP size measurements. For SEM analysis, each SEM image was imported and analyzed in ImageJ software by first setting and defining the scale (known scale bar in nanometers). The area option was selected as the analysis object to measure the square of the particles. For the ImageJ analysis of certain irregularly shaped nanoparticles, Feret's diameter was used to obtain a solid value for nanoparticle diameter. Each SEM image was prepared by enhancing the contrast and thresholding to improve the nanoparticle visibility and ensure that nanoparticles are highlighted, respectively. Following these ImageJ steps, nanoparticle analyses were performed.3. Results and discussion
3.1 Fundamental understanding of gold nanoparticle (AuNP) electrodeposition on carbon ultramicroelectrode arrays (CUAs)
The electrochemical behavior of gold nanoparticle (AuNP) formation has been well-characterized in previous research studies.25,26,39,41 The complete, three-electron redox mechanism of gold reduction is displayed in eqn (1). In this reaction, tetrachloroaurate ion (AuCl4−) is reduced to solid gold at the electrode surface. Previous reports indicate that the full reaction mechanism for gold reduction is a three-step CEE process, involving a rate-determining chemical step (C) followed by two successive electron transfer steps (E, E).41 Goolsby and co-workers previously proposed that the first, chemical step converts AuCl4− to the cationic, intermediate species AuCl2+. This initial step is followed by a two-electron transfer reaction, resulting in an anionic intermediate species of AuCl2−. The final step is suggested to be a single-electron transfer reaction, forming the product of solid gold (Au0). The majority of studies pertaining to mechanistic analysis of Au electrodeposition have been performed in organic media because Au1+ is stable in these organic solvents via solvent interactions. However, in aqueous environments, gold tends to form Au0 or Au3+ and intermediate species, such as AuCl2−, which are extremely short-lived and unstable.41 In aqueous environments, it is likely that these reaction steps occur simultaneously, explaining the single cathodic peak observed in the cyclic voltammogram (CV) in Fig. 1, demonstrating typical behaviors for an irreversible electrochemical system.48| AuCl4(aq)− + 3e− → Au(s)0 + 4Cl(aq)− E° = 0.994 V vs. NHE | (1) |
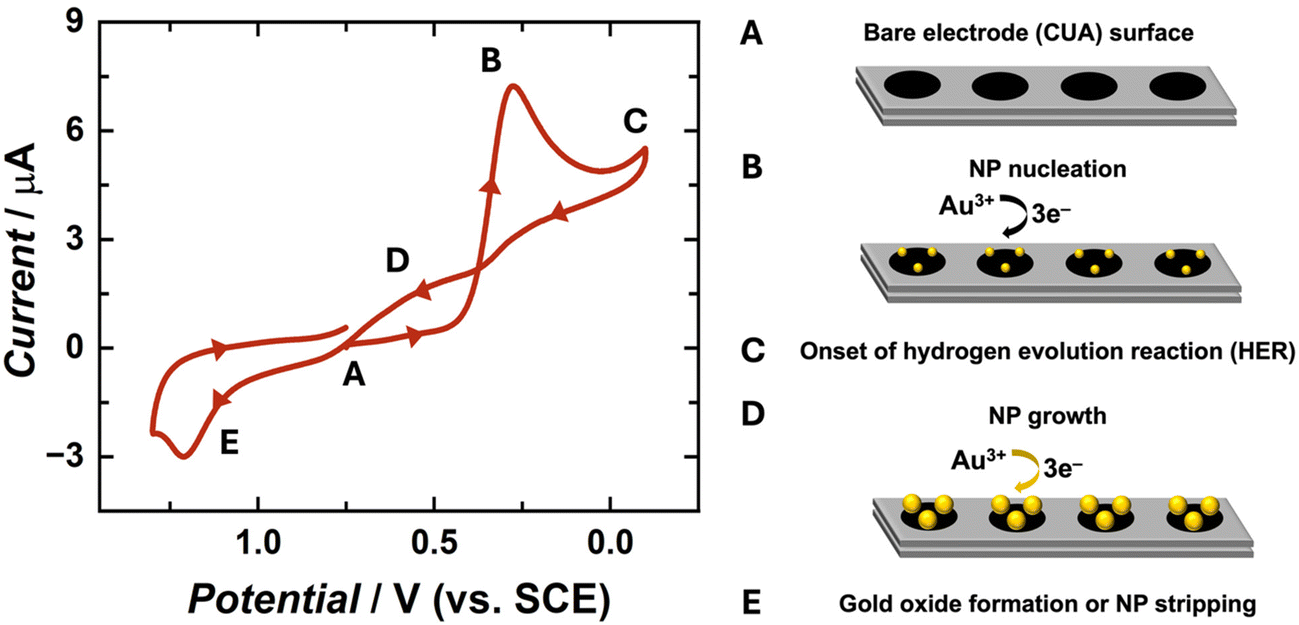 | ||
| Fig. 1 Cyclic voltammogram (CV) of 50 μM HAuCl4 in 0.5 M H2SO4 under nitrogen (oxygen-free) conditions at a scan rate of 100 mV s−1 on the carbon ultramicroelectrode arrays (CUAs). The potential was swept from 0.75 V to −0.1 V, then to 1.3 V, and back to 0.75 V vs. SCE, as indicated by the arrows on the CV plot. Points A–E on the CV trace represent various potential-dependent gold redox processes occurring throughout the sweep, as briefly outlined on the right side. | ||
Voltammetric studies were performed to obtain fundamental information on the electrochemical behavior of AuCl4− on the carbon ultramicroelectrode arrays (CUAs), specifically, over a wide range of potentials (1.3 V to −0.1 V vs. SCE). The unique CV shape (Fig. 1) is governed by the diffusion of the redox-active analyte towards the working electrode, or CUA, surface resulting from applied potential. The CV trace began at an initial potential of 0.75 V vs. SCE (Fig. 1A), associated with minimal faradaic current response. The potential was then swept in the negative direction, initiating the three-electron reduction of gold according to eqn (1), with the cathodic current peaking at approximately 0.25 V vs. SCE (Fig. 1B). At −0.1 V vs. SCE, the potential polarity changes, initiating the positive potential sweep. The cycle does not extend beyond this potential to avoid inducing the hydrogen evolution reaction (HER) at the electrode surface (Fig. 1C and Fig. S1†).39,48 Further discussion of electrode material influence on HER is included in subsequent sections. As the potential is swept in the positive direction, a looped region is formed in the cyclic voltammogram in a potential window ranging from 0.3 to 0.75 V vs. SCE (Fig. 1D). This is a well-understood AuNP electrochemical deposition phenomenon, previously described as the “nucleation loop” or “current loop”.25,43 The nucleation loop is the result of current response differences at the same potential values between the negative and positive sweep. In the negative sweep, more thermodynamic driving force, with regard to the electrochemical potential, is needed for the formation of AuNP nucleation sites on the carbon electrode surface. Once these sites are formed, the deposition of Au-on-Au is more favorable than Au-on-carbon. Because of this difference in substrate surface energy, less overpotential is required to deposit Au on other AuNPs, rather than on the carbon electrode surface, yielding higher electrical current responses on the reverse sweep. This growth mechanism is also known as Volmer–Weber growth, in which atom-to-atom interaction is more favorable than atom-to-substrate interaction, inducing the formation of three-dimensional particles, rather than a thin gold film at the electrode surface.25,42 The CV trace continues in the positive direction, reaching the peak anodic current at approximately 1.2 V vs. SCE (Fig. 1E). Any associated oxidative current for this reaction can be attributed to two processes, specifically, either (1) the dissolution of the solid gold particles back into solution as gold ions (stripping) or (2) the formation of a gold oxide film on the NP surfaces. The oxidative peak in Fig. 1 is most likely associated with the latter of the two processes, given that the gold oxide formation occurs at more oxidative potentials than AuNP stripping.25,48 Furthermore, most anodic stripping studies of AuNPs have been performed in bromide-containing solutions.39 This voltammetric data reveals that gold exhibits similar electrochemical behavior on CUAs in comparison to planar, macro-sized electrodes of carbon and other materials.5,39,44
3.2 Impact of electrodeposition parameters on gold nanoparticle (AuNP) formation on carbon ultramicroelectrode arrays (CUAs)
A significant advantage of utilizing electrochemical methods for nanoparticle deposition is the control over multiple experimental parameters, including deposition time, deposition potential, metal ion concentration, electrode material, as well as supporting electrolyte composition and concentration. This study employed single-step chronoamperometry for AuNP electrodeposition this electrochemical technique enables control of the electrodeposition potential by holding it at a single value over a specified time period while measuring the associated current response.66,67 Herein, several values of three main parameters were tested in the electrodeposition of AuNPs on CUAs, namely (1) deposition times of 5, 15, 30, and 60 s, (2) electrodeposition potentials of 0.25, 0, −0.25, and −0.5 V vs. SCE, and (3) AuCl4− concentrations of 25, 50, and 100 μM. Combinations of the values for each electrodeposition parameter resulted in extensive scanning electron microscopy (SEM) imaging and analysis of the AuNPs electrodeposited on the CUA electrodes. Energy dispersive X-ray spectroscopy (EDX) was utilized to confirm the presence of gold metal (Au) on the surface of the CUAs (Fig. S2†). The AuNP particle size data is displayed in Fig. 2 and Fig. S3, S4,† organized by deposition time and potential for gold ion solution concentrations of 50, 25, and 100 μM, respectively. These figures contain representative SEM images with the associated particle size histogram measurements for direct comparison between parameters. For particle count, Fig. 3 displays the average number of AuNPs on each individual electrode, organized by parameter. Due to the array-based CUA electrode geometry, AuNPs from n = 9 individual ultramicroelectrodes were measured and plotted to maintain consistency for both particle size and count. It should be noted that certain nanoparticles demonstrate irregular shapes. The mechanism of these irregularities in the nanoparticle shapes should be investigated in a future study by using several microscopy approaches, such as transmission electron microscopy (TEM) and calcite-assisted localization and kinetics (CLocK) microscopy.68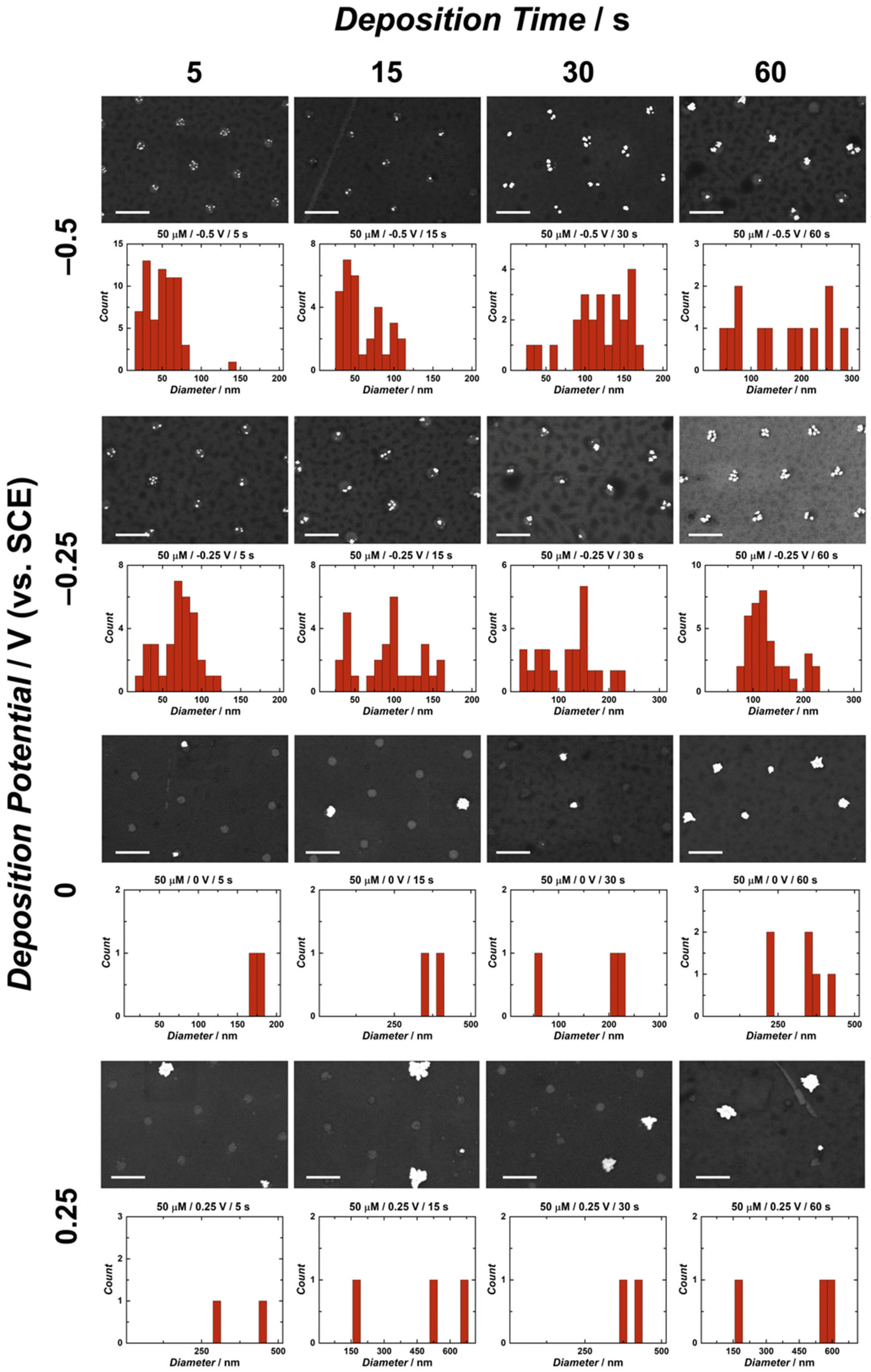 | ||
| Fig. 2 Scanning electron microscopy (SEM) images and particle size distribution histograms for all time and potential parameters for the electrodeposition of gold nanoparticles (AuNPs) on carbon ultramicroelectrode arrays (CUAs) in 50 μM HAuCl4 and 0.5 M H2SO4 solution. The SEM micrographs and histogram plots are arranged in order of increasing deposition time (from left to right) and decreasing reductive potential (from top to bottom). The scale bars represent 1 μm for all SEM images. Particle diameter measurements were taken from nine individual electrodes in the CUA of the same area as shown in the respective SEM images for each. | ||
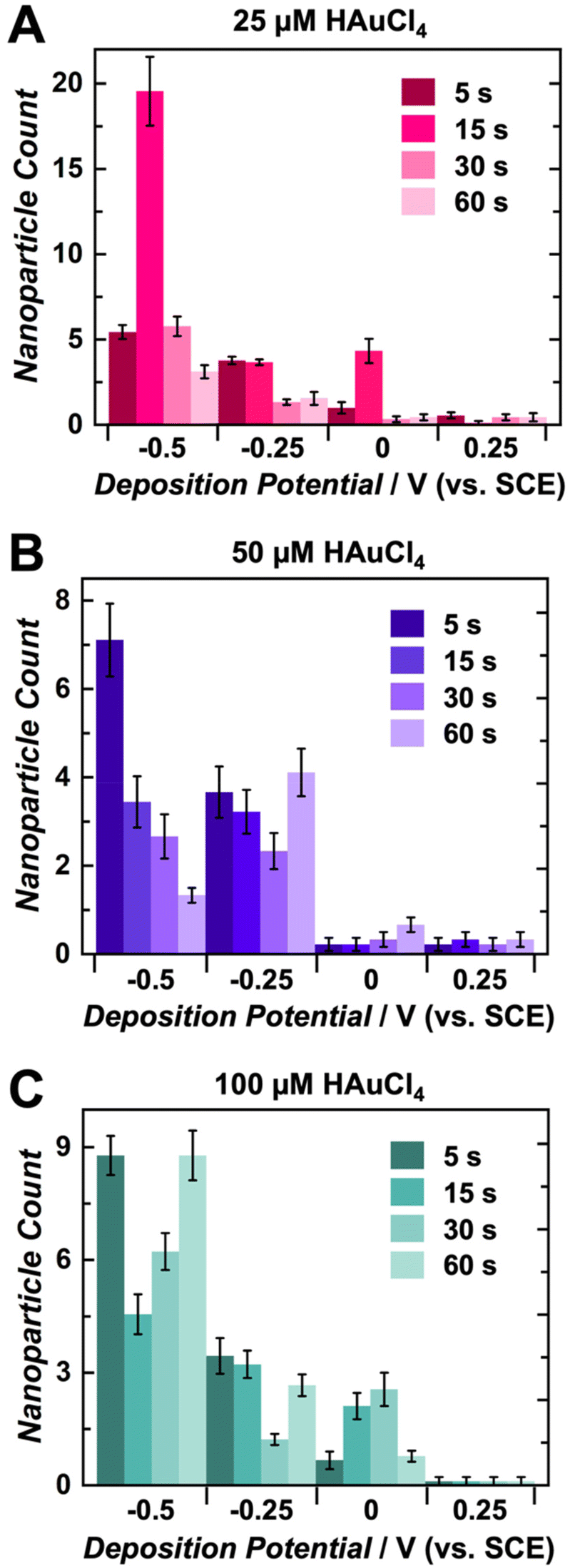 | ||
| Fig. 3 Gold nanoparticle (AuNP) count per individual electrode within the carbon ultramicroelectrode array (CUA) at various deposition times and potentials in solutions of (A) 25 μM, (B) 50 μM, and (C) 100 μM HAuCl4 in 0.5 M H2SO4, displayed in pink, purple, and green, respectively. Particles were counted from nine individual electrodes (n = 9) within the CUA. | ||
In terms of AuNP sizes, the SEM and histogram data exhibit clear trends for all electrodeposition parameters. For the single-potential step chronoamperometry electrodeposition methods, the cyclic voltammetry data in Fig. 1 was utilized to establish the deposition potentials. As previously explained, the gold reduction on CUAs is initiated at approximately 0.25 V vs. SCE, thus establishing the least reductive deposition potential used in this study. The determination of the most reductive potential value of −0.5 V vs. SCE required further cyclic voltammetry tests, given the prevalence of HER at the electrode surface in acid-containing analyte solutions (here, 0.5 M H2SO4). This known reaction occurs at reductive potentials in acidic conditions, where protons in solution are reduced to form hydrogen gas bubbles (H2) at the electrode surface (eqn (2)).69–71
| 2H(aq)+ + 2e− ↔ H2(g) E° = 0.000 V vs. NHE | (2) |
The HER reaction should be avoided in the electrodeposition of AuNPs due to the hindering of mass transport to the CUA working electrode, resulting from H2 bubble generation on the electrode surface.39,72 The data in Fig. S1† shows cyclic voltammograms of the supporting electrolyte solution, namely 0.5 M H2SO4, on the CUA electrode surface before AuNP deposition (i.e., bare CUA) and after AuNP deposition (i.e., AuNP-CUA). Upon the comparison of these cyclic voltammograms in the presence and absence of AuCl4−, significant differences in HER current response and associated potential are observed (Fig. S1†). This change in kinetics (relating to the electrical current) can be attributed to the formation of AuNPs on the carbon CUA electrode surface in a solution of gold ions at reductive potentials. Fig. S1A† displays the CV data in a wide potential window, revealing a large increase in the resulting current signal on the AuNP-modified CUA compared to the current signal on the bare CUA. Gold, in the form of nanoparticles, on the CUA electrode platform acts as an electrocatalyst for HER.69 This catalytic phenomenon will be further explored in AuNP characterization studies in a subsequent section. In addition to the greater current response, the electrocatalytic properties of AuNPs also result in a positive shift in the onset HER potential, due to the lower driving force required to initiate this reaction (Fig. S1B†). The onset of HER occurs at about −0.1 V vs. SCE on the AuNP-modified CUAs, which is over 400 mV more positive than the potential of HER onset on the bare CUAs (beyond −0.5 V vs. SCE). Overall, the cyclic voltammograms in Fig. S1† indicate that the HER exhibits sluggish kinetics on the bare carbon CUA electrode surface, thus requiring greater applied potential to drive the reaction. In contrast, the AuNP-modified CUA shows greater current, indicative of enhanced HER kinetics, and requires a smaller applied potential, as evidenced by the potential shift to more positive values. Considering the results of this voltammetric experiment on bare and AuNP-modified CUAs, the most reductive deposition potential chosen for this study was −0.5 V vs. SCE.
Based on the SEM and histogram results shown in Fig. 2 and Fig. S3, S4,† there is a clear trend in the AuNP sizes with regard to the deposition potentials. Namely, the electrodeposition of AuNPs on CUAs performed at more reductive values (−0.5 V vs. SCE) yields smaller particle sizes for all deposition times and gold ion solution concentrations. In contrast, larger particle sizes were observed at less reductive potential values (0.25 V vs. SCE) for all deposition periods and AuCl4− concentrations. These electrodeposition trends well-agree with findings from previous studies,25,26,39,43,48 indicating that more reductive potentials result in the formation of higher amounts of smaller AuNPs.26,39 At more reductive potentials, a greater electrochemical driving force is applied to the working electrode, rendering the deposition of gold onto the carbon electrode surface more favorable, producing more nucleation sites and smaller particle sizes. In contrast, at less reductive potentials, less thermodynamic driving force is applied to the working electrode, resulting in limited nucleation and more particle growth, yielding a smaller number of larger-sized particles with a lower surface area. Furthermore, this trend is also displayed in the lower-magnitude SEM images of AuNP-modified CUAs showing particle surface coverage. Fig. S5† shows the effects of deposition potentials on the AuNP coverage on the electrode surfaces, with more reductive potentials resulting in particle nucleation on every individual electrode and less reductive potentials yielding sparse, stochastic particle growth on the CUA surfaces. In summary, a greater driving force, achieved by applying a more negative potential, enhances the nucleation kinetics, resulting in more nucleation sites and smaller nanoparticle sizes.
For the deposition times, longer deposition times (30–60 seconds) result in the formation of larger-sized nanoparticles due to prolonged AuNP growth periods.26 In contrast, shorter times (5–15 seconds) employed for AuNP electrodeposition yield smaller nanoparticle sizes. These deposition time trends, observed in Fig. 2 and Fig. S3, S4,† suggest that longer deposition times result in increased particle growth, which is consistent with observations from previous studies.26,43,48 In addition to individual AuNP size, deposition time also influences nanoparticle size distribution. For instance, the histogram data of several CUA samples demonstrate bimodal particle size distributions at the longest deposition time of 60 seconds. This broad nanoparticle size distribution could be attributed to Ostwald ripening, a well-known phenomenon in which larger particles grow at the expense of smaller NPs.44 Furthermore, longer deposition times could lead to broader nanoparticle size distribution from the Ostwald ripening phenomenon, causing particles to grow at different rates.44 Future work needs to focus on studying and characterizing the ripening effects on CUAs to provide mechanistic insights into this phenomenon.
The trends shown in Fig. 2 and Fig. S3, S4† indicate that the average AuNP count per individual electrode agrees with particle size data. Overall, more negative potentials and shorter deposition times yield higher particle counts on each CUA electrode. The particle number is also influenced by AuNP nucleation, with more nucleation sites resulting in higher counts of smaller-sized particles, due to limited growth. The opposite is true for less reductive potentials and longer deposition times. Under these conditions, fewer AuNP nucleation sites are formed, yielding lower particle counts. Although the particle numbers in Fig. 2, 3 and Fig. S3, S4† apply to nine individual ultramicroelectrodes in the array, the CUA surface (with a total exposed carbon area of 1.4%, resulting in a CUA electrode area of 0.0071 cm2) consists of approximately 3 × 109 ultramicroelectrodes per cm2, which was calculated from SEM images of CUA electrodes. Based on exhaustive SEM analysis, one electrode in the CUA array was determined an average radius of 91 ± 7 nm, which agrees with previous works using CUAs.49 Therefore, assuming nearly perfect and uniform AuNP distribution on all individual CUA wells in the electrode array, the total number of AuNPs deposited for each set of electrodeposition parameters can be estimated by multiplying the average particle count by the total number of individual ultramicroelectrodes in the CUA array.
Differences in AuCl4− concentration also impact the electrodeposition of AuNPs on CUAs. Considering the particle size, the median solution concentration of 50 μM AuCl4− yields small-sized particles at more reductive potentials and short deposition times (Fig. 2 and 3). The slightly lower concentration of 25 μM displayed similar results (Fig. S3†), indicating there is a range of acceptable working concentrations for the electrodeposition on CUAs (25–50 μM). At higher concentrations (100 μM and greater), the AuNP growth is more pronounced, producing larger-sized particles and aggregates, due to the greater availability of gold ions in solution. Previous studies employing planar, macro-sized electrodes for AuNP electrodeposition have used gold ion concentrations in the millimolar range from 1 to 5 mM.26,39,48 Compared to these previous studies, the electrodeposition of gold nanoparticles on CUA surfaces requires significantly lower gold ion concentrations (25–100 μM) for adequate AuNP formation, which is both advantageous and cost-effective. These micromolar concentration ranges result from the array-based geometry and the unique combination of carbon and Al2O3 materials in the design of the CUA surface.
To illustrate the differences in gold ion concentration for the electrodeposition of AuNPs on CUAs, control studies were performed with the planar Macro electrodes. The CUA electrode surface is only 1.4% PPF carbon film as the majority of the electrode area encircling the individual electrodes in the array consists of the 10 nm Al2O3 layer,49 which acts as an insulating material to store charge.50 On the other hand, the planar Macro electrodes consist of 100% PPF carbon film (Fig. 4). Furthermore, a control AuNP electrodeposition experiment was performed with samples with the 10 nm Al2O3 layer only. To compare the effects of electrode geometry and material on particle size, count, and surface coverage, AuNPs were deposited with the same parameters on each electrode surface in a solution of 25 μM AuCl4−. Fig. 4 displays the resulting SEM micrographs and representative schematics of each electrode surface, revealing significant differences in particle coverage between the CUA electrode, the planar Macro electrode, and the surface consisting of the 10 nm Al2O3 layer. The representative SEM image for the array-based CUA electrode displays desirable AuNP coverage across nine individual electrodes in the array (shown in the SEM window), with high counts of small-size particles. Contrastingly, SEM data for the planar Macro electrode under the same electrodeposition conditions shows poor particle coverage, with a low number of small particles randomly dispersed across the electrode surface. This data also suggests that for a more desirable AuNP electrodeposition on the planar Macro electrode, higher AuCl4− concentrations are necessary. The SEM image for the Al2O3 layer shows no gold nanoparticles electrodeposited on the surface.
 | ||
| Fig. 4 Representative scanning electron microscopy (SEM) images of gold nanoparticle (AuNP) electrodeposition on the (A) carbon ultramicroelectrode array (AuNP-CUA), (B) the carbon Macro electrode (AuNP-Macro), and (C) the aluminum oxide (AuNP-Al2O3) surfaces. The Al2O3 layer surface is a control surface, as 10 nm Al2O3 is used for the fabrication of the CUA electrodes. Deposition times, potentials, and HAuCl4 concentrations were the same for all three samples at 15 s, −0.5 V vs. SCE, and 25 μM, respectively. Scale bars represent 1 μm for all SEM images. | ||
The differences in AuNP formation at AuCl4− concentration of 25 μM between the planar electrodes and the CUAs can be attributed to several physical properties and electrode surface processes. Specifically, there are differences in the electrode geometry and size between the planar macro-sized electrode with respect to the array-based configuration and the nanometer size of the CUA electrode. This array-based geometry of the CUA electrodes results in radial diffusion profiles (Fig. S6A†),48,49,73 characteristic of ultramicroelectrodes.49 It is well-known that increasing the hemispherical radial diffusion via decreasing the electrode size results in enhanced rates of mass transport, and therefore increased flux of species toward the electrode surface. On the other hand, the planar Macro electrode has characteristic linear diffusion profiles (Fig. S6B†). These differences in diffusion-based mass transport profiles on the CUA electrodes compared to the Macro electrodes result in greater AuNP surface coverage on the CUAs with enhanced flux of species. In addition, the unique combination of electrode materials comprising the CUA electrode surface influences AuNP electrodeposition with regard to the gold ion concentration needed for successful AuNP electrodeposition. Namely, the insulating metal oxide layer consisting of 10 nm Al2O3 and the conductive PPF electrode material exhibit different electrochemical properties, including the potential of zero charge (EPZC) value. The EPZC corresponds to the potential at which there is no surface free charge on the electrode.66,74,75 The electrodeposition potential to the working electrode (Edep) influences the charge established on the electrode surface, which is dictated by the EPZC of the electrode material. In the case of CUAs, the EPZC for Al2O3 is significantly more negative (e.g., more reductive) compared to the EPZC of the carbon PPF film.74,76–78 When the electrodeposition potential is applied to the CUA, with respect to the SCE reference electrode, the EPZC of Al2O3 is more negative than the applied electrodeposition potential (−0.5 V vs. SCE), resulting in a positively charged Al2O3 layer on the CUAs. On the other hand, the EPZC of carbon is more positive with respect to the applied electrodeposition potential (−0.5 V vs. SCE), yielding a negatively charged electrode surface for the planar Macro electrode. The tetrachloroaurate anion is attracted to the positively charged Al2O3 surface, due to coulombic attraction, on the CUAs, creating pre-concentration AuCl4− zones on the CUA surface. These phenomena combined with the radial diffusion profiles allow for facile mass transport, which ultimately results in successful electrodeposition of AuNPs on the CUAs at lower gold ion concentrations (Fig. 5). In contrast, for the planar Macro electrode, the negative charge established on the carbon surface repels the AuCl4− species, hindering diffusion to the electrode surface. Fig. 5 graphically illustrates these processes. Therefore, both the diffusional mass transport and electrode material can theoretically be attributed to the differences in AuNP formation on CUAs in comparison to planar Macro electrodes. In the context of CUA electrode materials, the metal oxide Al2O3 layer could play a role in lowering the barrier for Au nucleation and increasing nucleation kinetics, resulting in greater Au deposition. Other aspects, such as nucleation kinetics changes in more confined regions, charge effects, and/or AuCl4− concentrations, need to also be considered in future studies. However, further modeling and theoretical studies are necessary to confirm these phenomena.75 With regard to the diffusion mass transport processes, it is crucial to note that the CUA electrode platform is rather complex. Namely, the diffusion over the CUAs can be categorized into several types: (1) planar diffusion over individual ultramicroelectrodes, (2) a mixed diffusion layer over individual ultramicroelectrodes with a transition between planar and hemispherical diffusion layers, (3) hemispherical diffusion layers over individual ultramicroelectrodes, and (4) a mixed diffusion layer resulting from overlapping individual diffusion layers.73 Thus, studying mass transport in the recessed ultramicroelectrodes in an array requires the numerical simulation of diffusional processes and the development of accurate simulation models, similar to previous models on microelectrode arrays.73,79–81 These intriguing aspects will be modeled and examined in future studies.
 | ||
| Fig. 5 Representative scanning electron microscopy (SEM) images of gold nanoparticle (AuNP) electrodeposition on the carbon ultramicroelectrode array (AuNP-CUA, left) and the Macro carbon electrode (AuNP-Macro, right). Deposition times, potentials, and HAuCl4 concentrations were the same for all three samples at 15 s, −0.5 V vs. SCE, and 25 μM, respectively. Below the SEM images is a schematic showing the effects of potential of zero charge (EPZC) on the electrodeposition of AuNPs on the CUA in comparison to the Macro electrode. Scale bars represent 1 μm for both SEM images. | ||
Overall, these results demonstrate the importance of methodically establishing electrodeposition potentials on novel electrode surfaces to obtain ideal deposition of AuNP size, surface coverage, and dispersion. This favorable AuNP formation involves a larger number of well-dispersed, smaller-sized particles, covering each individual electrode in the array to maximize Au surface area. Based on the extensive analysis of SEM micrographs on AuNPs on various electrodeposition parameters, the described AuNP deposition on the CUA electrodes can be achieved using a reductive deposition potential of −0.5 vs. SCE, a deposition time of 15 s, and AuCl4− concentration of 25 μM (Fig. 4A and Fig. S3†).
3.3 Amount of gold deposited on the gold nanoparticle-modified carbon ultramicroelectrode arrays (AuNP-CUAs)
The amount of electrodeposited gold (in moles) was determined from the electrodeposition chronoamperograms obtained with the optimal electrodeposition parameters. Upon comparison of the chronoamperometric trace obtained at a greater reductive deposition potential (−0.5 V vs. SCE) compared to the one obtained at a less reductive potential (0.25 V vs. SCE), distinct differences in the area under the responding current–time (i–t) curves were observed (Fig. S7†). The total charge passed through the system was calculated via integration of the area under each chronoamperometric trace ranging from 0.5 s to 5 s. The total amount of gold deposited on the CUAs was then estimated from the charge using Faraday's law (eqn (S1)†).66,67Table 1 summarizes the determined values for the gold amounts electrodeposited on both the AuNP-modified CUAs, as well as the carbon Macro electrode. Furthermore, the amount of deposited gold was normalized to the exposed carbon electrode area for the CUA and planar Macro electrodes (0.0071 cm2 for the CUA vs. 0.495 cm2 for the carbon Macro electrode), providing comparative information regarding the AuNP density at each electrode surface. Namely, using an electrodeposition potential of −0.5 V vs. SCE, the gold amount deposited on the CUA surface per electrode area was determined to be 88.73 ± 0.06 nmol cm−2. In contrast, the electrodeposition of AuNPs using a more positive, oxidative potential of 0.25 V vs. SCE yielded 10.6 ± 0.1 nmol of gold per electrode area (cm−2). These results indicate that the amount of gold electrodeposited on CUAs using more reductive electrodeposition potential is approximately nine times higher compared to the amount of gold deposited on the CUA surfaces at more oxidative electrodeposition potential. Thus, our data suggests that the amount of gold electrodeposited on CUAs is governed and greatly affected by deposition potential, reiterating the importance of establishing optimal electrodeposition methodology prior to experimentation. Furthermore, a control experiment was performed using the planar carbon Macro electrode, lacking the array-based electrode geometry, where AuNPs were electrodeposited at the more reductive potential of −0.5 V vs. SCE (Fig. S8†). The amount of gold per electrode area for the planar Macro electrode was estimated to be 3.64 ± 0.02 nmol cm−2, which is almost 24 times less compared to the gold electrodeposited on the CUAs under the same electrodeposition parameters. Further work is needed to mathematically determine the theoretical values of gold amount electrodeposited on the recessed cylindrical ultramicroelectrodes in the CUA array and the planar Macro electrodes, allowing for comparison with the quantitative experimental values reported here.| Electrode | Deposition potential (V vs. SCE) | Charge (μC) | Amount of gold (nmol) | Amount of gold per electrode area (nmol cm−2) |
|---|---|---|---|---|
| AuNP-CUA | 0.25 | 22 ± 2 | 0.075 ± 0.008 | 10.6 ± 0.1 |
| AuNP-CUA | −0.5 | 180 ± 11 | 0.63 ± 0.04 | 88.73 ± 0.06 |
| AuNP-Macro | −0.5 | 520 ± 11 | 1.80 ± 0.04 | 3.64 ± 0.02 |
As discussed in the previous section, these differences in gold amounts electrodeposited on the CUAs vs. the planar Macro electrodes are due to the differences in physical properties, electrode material, and electrode geometry, all of which govern the diffusion profiles, and charges established on the electrode surfaces relating to the potential of zero charge. Overall, our results demonstrate that the electrodeposition potential, as well as the electrode material, size, and geometry, greatly influence the amount of gold deposited on the electrode surfaces.
3.4 Gold nanoparticle-modified carbon ultramicroelectrode arrays for the catalytic enhancement of the hydrogen evolution reaction (HER)
AuNP-modified electrodes have been shown to display increased catalytic activity for various electrochemical applications.1,7 Specifically, AuNP-modified electrodes have been implemented in enhancing the HER reaction towards applications in green energy production and storage.69,70 AuNP size, shape, count, and dispersion affect electrode catalytic performance, with higher particle surface area yielding increased current responses.5 To further characterize AuNP electrodeposition on CUAs, cyclic voltammograms of 0.5 M H2SO4 were performed to induce the HER on the electrode surface. Two sets of AuNP-modified CUA samples were compared, namely one with desirable AuNP formation obtained at more reductive electrodeposition potential (−0.5 V vs. SCE), and the other with sparse AuNP deposition due to less reductive applied potential (0.25 V vs. SCE). In addition, a control study was performed with the planar Macro electrodes at the same electrodeposition parameters, namely deposition time of 5 s and HAuCl4 concentration of 50 μM, as the desirable AuNP-modified CUA (−0.5 V vs. SCE).Fig. 6 and Fig. S9† display the cyclic voltammetry results of the HER proof-of-concept studies, comparing the resulting current responses of the AuNP-modified CUAs and AuNP-modified Macro electrodes to bare electrodes of the same type. The associated voltammetric peak cathodic currents for the HER at a potential of −1 V vs. SCE are summarized in Table 2. The resulting peak cathodic currents from Fig. 6 and Fig. S9† divided by the exposed carbon areas of each electrode (0.0071 cm2 for the CUA vs. 0.495 cm2 for the carbon Macro electrode)49 to obtain the catalytic current densities, thus allowing for comparison of catalytic HER activity on each AuNP-functionalized electrode and also a comparison with the bare electrodes (without any AuNPs). Based on the cyclic voltammograms in Fig. 6, the HER current density at the AuNP-CUA obtained with more reductive electrodeposition potential (−0.5 V vs. SCE) yielded approximately a 90-fold increase in the current density response compared to the HER current density at a potential of −1 V vs. SCE for the bare CUA not modified with AuNPs (4.8 ± 0.1 mA cm−2). In contrast, the AuNP-decorated CUAs obtained with the less reductive electrodeposition potential (0.25 V vs. SCE) produced only a 20-fold increase in the catalytic current in comparison with the bare CUAs. The planar Macro electrode modified with AuNPs resulted in HER current density of 18.38 ± 0.02 mA cm−2 (Fig. S9†), which is only a 25-fold increase compared to the catalytic cathodic current density obtained at the non-modified, bare, Macro electrode (0.7 ± 0.1 mA cm−2).
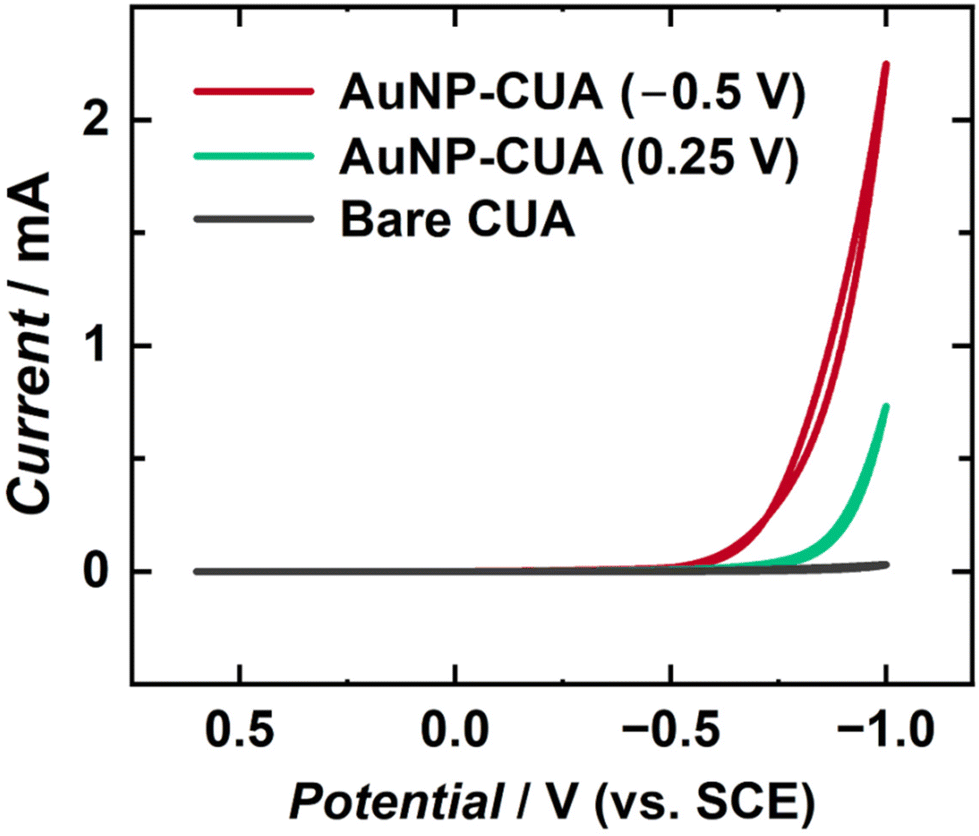 | ||
| Fig. 6 Cyclic voltammetric current–potential traces of the hydrogen evolution reaction (HER) in 0.5 M H2SO4. The bare CUA (gray trace) does not contain AuNPs. For the AuNP-modified CUAs, both deposition times and HAuCl4 concentrations were 5 s and 50 μM, respectively. Differences in the deposition potential contribute to HER catalytic current increase, with the red trace representing AuNPs deposition at −0.5 V vs. SCE and the green trace electrodeposition at 0.25 V vs. SCE. The voltammograms are one representative plot from n = 3 replicates. | ||
| Electrode | Deposition potential (V vs. SCE) | HER cathodic current (mA) | HER cathodic current density (mA cm−2) |
|---|---|---|---|
| CUA | N/A | 0.034 ± 0.004 | 4.8 ± 0.1 |
| Macro | N/A | 0.36 ± 0.05 | 0.7 ± 0.1 |
| AuNP-CUA | 0.25 | 0.71 ± 0.04 | 100.0 ± 0.1 |
| AuNP-CUA | −0.5 | 3 ± 2 | 422.5 ± 0.7 |
| AuNP-Macro | −0.5 | 9.1 ± 0.3 | 18.38 ± 0.02 |
Using the cyclic voltammograms in Fig. 6, the HER catalytic current density for the CUAs with AuNPs deposited at the less reductive deposition potential (0.25 V vs. SCE) was estimated to be 100.0 ± 0.1 mA cm−2. On the other hand, the AuNP-functionalized CUAs using the more reductive deposition potential (−0.5 V vs. SCE) resulted in a catalytic current density of 422.5 ± 0.7 mA cm−2 for the HER (Fig. 6), which is a 4-fold increase compared to the CUA decorated with AuNPs at deposition potential of 0.25 V vs. SCE. Additionally, the AuNP-functionalized CUA and planar Macro electrodes using an electrodeposition potential of −0.5 V vs. SCE were compared. The HER catalytic current density at the AuNP-modified Macro electrode was measured to be 18.38 ± 0.02 mA cm−2, which is 23 times lower than the catalytic current density obtained for the HER on the AuNP-CUA electrodes (Table 2). These results are also in agreement with the amounts of gold electrodeposited on the CUA and Macro electrode surfaces (Table 1). Comparison of the resulting current densities between these AuNP-decorated electrodes demonstrates that the AuNP-modified CUA with the more reductive electrodeposition potential of −0.5 V vs. SCE results in a significant enhancement of the rate of electron transfer for the HER reaction as observed by the resulting catalytic current responses in the cyclic voltammetry data. This improved catalytic performance is associated with the larger number of smaller particles formed at the CUA electrode surface, resulting in greater surface area with higher conductivity. It is important to note that the values of Au deposited at −0.5 V vs. SCE may be overestimated due to potential interference from the HER. Our cyclic voltammetry data in Fig. S1† shows a minimal current response of approximately 3 μA at −0.5 V vs. SCE with bare CUA, which is significantly lower than the expected current for the HER. Although the current is minimal, the possibility of HER interference during Au deposition at this potential cannot be excluded. This interesting aspect will be examined in future work. Generally, our results show that the electrodeposition potential critically impacts the AuNPs size, dispersion, and distribution on the CUA surfaces, which consequently affects both the amount of gold deposited, and thus, the catalytic activity for the HER reaction.
4. Conclusions
To summarize, we reported the electrodeposition and characterization of gold nanoparticles (AuNPs) on carbon ultramicroelectrode arrays (CUAs). Specific electrodeposition parameters, including deposition time, potential, and gold ion concentration were varied to show the effects on AuNP particle formation. Based on extensive analysis of scanning electron micrographs, electrodeposition parameters were established to yield desirable AuNP sizes, count, and electrode surface coverage. Our results demonstrate that more reductive electrodeposition potentials and shorter deposition times yielded higher AuNP particle counts with smaller sizes. Furthermore, compared to previous AuNP electrodeposition studies utilizing millimolar concentrations of gold ion solution, this study employed lower gold ion concentrations in the micromolar ranges to successfully deposit and form AuNPs the CUAs, which was attributed to the physical properties governing the nanometer-sized electrodes organized in the array-based geometry and the distinct material composition of the CUA surfaces. Additionally, the amount of gold deposited was determined at AuNP-functionalized CUAs at electrodeposition potentials of −0.5 V and 0.25 V vs. SCE, giving 88.73 ± 0.06 nmol cm−2 and 10.6 ± 0.1 nmol cm−2, respectively, resulting in higher particle density for deposition performed at more reductive potentials. The control experiments performed with a planar Macro electrode decorated with AuNPs at an electrodeposition potential of −0.5 V vs. SCE resulted in 3.64 ± 0.02 nmol cm−2 gold deposited on the electrode surface. These results show that the amount of gold deposited on the electrode surfaces is a function of not only the electrodeposition potential but also the electrode size, geometry, and material. Furthermore, we demonstrated a proof-of-concept to characterize the electrocatalytic responses for the HER reaction on the AuNP-modified CUAs. The AuNP-functionalized CUA electrodes show an HER catalytic current density of 422.5 ± 0.7 mA cm−2, which is 23 times higher than the electrocatalytic current density obtained on the AuNP-modified planar Macro electrodes. These results are consistent with the determined amounts of gold deposited on each electrode surface. Overall, the array-based geometry, nanometer electrode sizes, and unique electrode material combination of the CUAs greatly impact the physical processes on the electrode surface for AuNP electrodeposition. As such, this fundamental study highlights the need to methodically characterize the impacts of AuNP electrodeposition parameters on novel electrode surfaces. Future studies need to focus on examining Ostwalt ripening at the CUA surface during longer deposition time regimes. Additional electrodeposition parameters, including capping agent composition and concentration, and supporting electrolyte composition and concentration, need to be examined in the AuNP formation on CUAs.Author contributions
O. S. conceived the study, designed and planned experiments, supervised and funded the research. C. J. W. performed electrode fabrication and all electrochemical measurements/characterization, electrochemical deposition experiments, and data processing. N. E. S. assisted C. J. W. in electrode fabrication, and electrodeposition experiments, as well as contributed to nanoparticle data work-up. C. J. W. and O. S. wrote the manuscript, and critically analyzed all data. All authors contributed to preparation and editing and have given approval to the final version of the manuscript.Data availability
Supporting data and figures.† See https://doi.org/10.1039/D4NR02326A.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by startup funding from the University of South Carolina (to O. S.). C. J. W. gratefully acknowledges the financial support from the 2024 Joseph W. Richards Summer Fellowship from the Electrochemical Society (ECS). N. E. S. is thankful for support from the Magellan Scholars Research Grant from the Office of Undergraduate Research at the University of South Carolina. The authors would like to acknowledge helpful research discussions with Prof. Seyyedamirhossein (Amir) Hosseini from the Department of Chemistry and Biochemistry at the University of South Carolina. The authors also acknowledge experimental assistance from Brienne Hingst and Emma Vagnoni, both undergraduate students in the Simoska Research Group. Finally, the authors are grateful for SEM training received from CJ Sturgill (from the Stefik Research Group at the University of South Carolina).References
- K. A. Altammar, Front. Microbiol., 2023, 14, 1–20 CrossRef PubMed.
- C. M. Welch and R. G. Compton, Anal. Bioanal. Chem., 2006, 384, 601–619 CrossRef CAS PubMed.
- A. Wiesenthal, L. Hunter, S. G. Wang, J. Wickliffe and M. Wilkerson, Int. J. Dermatol., 2011, 50, 247–254 CrossRef CAS PubMed.
- F. P. Zamborini, L. L. Bao and R. Dasari, Anal. Chem., 2012, 84, 541–576 CrossRef CAS.
- J. N. Sharma, D. K. Pattadar, B. P. Mainali and F. P. Zamborini, Anal. Chem., 2018, 90, 9308–9314 CrossRef CAS.
- R. Sardar, A. M. Funston, P. Mulvaney and R. W. Murray, Langmuir, 2009, 25, 13840–13851 CrossRef CAS PubMed.
- D. Guo, G. X. Xie and J. B. Luo, J. Phys. D: Appl. Phys., 2014, 47, 013001 CrossRef CAS.
- N. Narayan, A. Meiyazhagan and R. Vajtai, Materials, 2019, 12, 3602–3614 CrossRef CAS PubMed.
- C. Gao, F. Lyu and Y. Yin, Chem. Rev., 2021, 121, 834–881 CrossRef CAS PubMed.
- A. Bano, A. Dawood, F. S. Rida, A. Malik, M. Alkholief, H. Ahmad, M. A. Khan, Z. Ahmad and O. Bazighifan, Sci. Rep., 2023, 13, 12359–12372 CrossRef CAS PubMed.
- C. Li, O. J. H. Chai, Q. Yao, Z. Liu, L. Wang, H. Wang and J. Xie, Mater. Horiz., 2021, 8, 1657–1682 RSC.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- S. K. Murthy, Int. J. Nanomed., 2007, 2, 129–141 CAS.
- A. R. Dhanapal, M. Thiruvengadam, J. Vairavanathan, B. Venkidasamy, M. Easwaran and M. Ghorbanpour, ACS Omega, 2024, 9, 13522–13533 CrossRef CAS PubMed.
- F. W. Campbell and R. G. Compton, Anal. Bioanal. Chem., 2010, 396, 241–259 CrossRef CAS PubMed.
- F. S. Ligler and H. S. White, Anal. Chem., 2013, 85, 11161–11162 CrossRef CAS PubMed.
- C. J. Weber, O. M. Clay, R. E. Lycan, G. K. Anderson and O. Simoska, Anal. Bioanal. Chem., 2024, 416, 87–106 CrossRef CAS PubMed.
- O. Simoska and K. J. Stevenson, Sens. Actuators Rep., 2022, 4, 100072–100085 CrossRef.
- C. Zhu, G. Yang, H. Li, D. Du and Y. Lin, Anal. Chem., 2015, 87, 230–249 CrossRef CAS PubMed.
- A. Mejri, G. Mandriota, H. Elfil, M. L. Curri, C. Ingrosso and A. Mars, Molecules, 2022, 27, 8490–8508 CrossRef CAS.
- L. Fritea, F. Banica, T. O. Costea, L. Moldovan, L. Dobjanschi, M. Muresan and S. Cavalu, Materials, 2021, 14, 6319–6356 CrossRef CAS.
- S. Das, A. Tripathi and M. M. Ghangrekar, Chemosphere, 2024, 352, 141392–141402 CrossRef CAS PubMed.
- B. Cao, Z. Zhao, L. Peng, H. Y. Shiu, M. Ding, F. Song, X. Guan, C. K. Lee, J. Huang, D. Zhu, X. Fu, G. C. L. Wong, C. Liu, K. Nealson, P. S. Weiss, X. Duan and Y. Huang, Science, 2021, 373, 1336–1340 CrossRef CAS PubMed.
- J. Lu, Z. Chen, Z. Ma, F. Pan, L. A. Curtiss and K. Amine, Nat. Nanotechnol., 2016, 11, 1031–1038 CrossRef CAS PubMed.
- L. Komsiyska and G. Staikov, Electrochim. Acta, 2008, 54, 168–172 CrossRef CAS.
- P. V. Dudin, P. R. Unwin and J. V. Macpherson, J. Phys. Chem. C, 2010, 114, 13241–13248 CrossRef CAS.
- S. R. Belding, F. W. Campbell, E. J. F. Dickinson and R. G. Compton, Phys. Chem. Chem. Phys., 2010, 12, 11208–11221 RSC.
- S. E. F. Kleijn, S. C. S. Lai, M. T. M. Koper and P. R. Unwin, Angew. Chem., Int. Ed., 2014, 53, 3558–3586 CrossRef CAS PubMed.
- J. E. Kellon, S. L. Young and J. E. Hutchison, Chem. Mater., 2019, 31, 2685–2701 CrossRef CAS.
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
- R. Suárez-López, V. F. Puntes, N. G. Bastús, C. Hervés and C. Jaime, Sci. Rep., 2022, 12, 13926 CrossRef.
- P. Yáñez-Sedeño and J. M. Pingarrón, Anal. Bioanal. Chem., 2005, 382, 884–886 CrossRef.
- G. Siciliano, A. Alsadig, M. S. Chiriacò, A. Turco, A. Foscarini, F. Ferrara, G. Gigli and E. Primiceri, Talanta, 2024, 268, 125280 CrossRef CAS PubMed.
- J. M. Pingarrón, P. Yáñez-Sedeño and A. González-Cortés, Electrochim. Acta, 2008, 53, 5848–5866 CrossRef.
- R. Herizchi, E. Abbasi, M. Milani and A. Akbarzadeh, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 596–602 CrossRef CAS PubMed.
- O. Matvieiev, R. Selesovská, M. Marton, M. Hatala, R. Metelka, M. Weis and M. Vojs, Sci. Rep., 2023, 13, 21525 CrossRef CAS PubMed.
- J. Ustarroz, X. X. Ke, A. Hubin, S. Bals and H. Terryn, J. Phys. Chem. C, 2012, 116, 2322–2329 CrossRef CAS.
- N. D. Zakaria, M. H. Omar, N. N. A. Kamal, K. A. Razak, T. Sönmez, V. Balakrishnan and H. H. Hamzah, ACS Omega, 2021, 6, 24419–24431 CrossRef CAS PubMed.
- O. S. Ivanova and F. P. Zamborini, Anal. Chem., 2010, 82, 5844–5850 CrossRef CAS PubMed.
- S. S. Zarei, S. Soleimanian-Zad and A. A. Ensafi, Microchim. Acta, 2018, 185, 1–9 CrossRef CAS PubMed.
- A. D. Goolsby and D. T. Sawyer, Anal. Chem., 1968, 40, 1978–1983 CrossRef CAS.
- R. M. Penner, J. Phys. Chem. B, 2002, 106, 3339–3353 CrossRef CAS.
- E. Sheridan, J. Hjelm and R. J. Forster, J. Electroanal. Chem., 2007, 608, 1–7 CrossRef CAS.
- D. K. Pattadar and F. P. Zamborini, Langmuir, 2019, 35, 16416–16426 CrossRef CAS PubMed.
- A. Berduque, Y. H. Lanyon, V. Beni, G. Herzog, Y. E. Watson, K. Rodgers, F. Stam, J. Alderman and D. W. Arrigan, Talanta, 2007, 71, 1022–1030 CrossRef CAS PubMed.
- R. Feeney and S. P. Kounaves, Electroanalysis, 2000, 12, 677–684 CrossRef CAS.
- R. D. Gardner, A. Zhou and N. A. Zufelt, Sens. Actuators, B, 2009, 136, 177–185 CrossRef CAS PubMed.
- M. B. Hariri, A. Dolati and R. S. Moakhar, J. Electrochem. Soc., 2013, 160, D279–D288 CrossRef CAS.
- J. Duay, J. M. Goran and K. J. Stevenson, Anal. Chem., 2014, 86, 11528–11532 CrossRef CAS PubMed.
- J. Duay, J. Elliott, J. B. Shear and K. J. Stevenson, Anal. Chem., 2015, 87, 10109–10116 CrossRef CAS PubMed.
- J. Elliott, O. Simoska, S. Karasik, J. B. Shear and K. J. Stevenson, Anal. Chem., 2017, 89, 6285–6289 CrossRef CAS PubMed.
- J. Elliott, J. Duay, O. Simoska, J. B. Shear and K. J. Stevenson, Anal. Chem., 2017, 89, 1267–1274 CrossRef CAS PubMed.
- O. Simoska, M. Sans, M. D. Fitzpatrick, C. M. Crittenden, L. S. Eberlin, J. B. Shear and K. J. Stevenson, ACS Sens., 2019, 4, 170–179 CrossRef CAS PubMed.
- O. Simoska, J. Duay and K. J. Stevenson, ACS Sens., 2020, 5, 3547–3557 CrossRef CAS PubMed.
- O. Simoska, M. Sans, L. S. Eberlin, J. B. Shear and K. J. Stevenson, Biosens. Bioelectron., 2019, 142, 111538 CrossRef CAS PubMed.
- O. Simoska and K. J. Stevenson, Analyst, 2019, 144, 6461–6478 RSC.
- N. D. Zakaria, M. H. Omar, N. N. A. Kamal, K. A. Razak, T. Sonmez, V. Balakrishnan and H. H. Hamzah, ACS Omega, 2021, 6, 24419–24431 CrossRef CAS PubMed.
- E. Verma, M.-H. Choi, N. Kar, L. A. Baker and S. E. Skrabalak, Nanoscale, 2024, 16, 8002–8012 RSC.
- E. K. Walker, D. A. Vanden Bout and K. J. Stevenson, J. Phys. Chem. C, 2011, 115, 2470–2475 CrossRef CAS.
- S. Donner, H. W. Li, E. S. Yeung and M. D. Porter, Anal. Chem., 2006, 78, 2816–2822 CrossRef CAS PubMed.
- H. Tian, A. J. Bergren and R. L. McCreery, Appl. Spectrosc., 2007, 61, 1246–1253 CrossRef CAS PubMed.
- S. Ranganathan, R. L. Mccreery, S. M. Majji and M. Madou, J. Electrochem. Soc., 2000, 147, 277–282 CrossRef CAS.
- S. Ranganathan and R. L. McCreery, Anal. Chem., 2001, 73, 893–900 CrossRef CAS PubMed.
- K. L. Johnson, K. Kendall and A. D. Roberts, Proc. R. Soc. A, 1971, 324, 301–313 CAS.
- C. Amatore, J. M. Savéant and D. J. Tessier, Electroanal. Chem. Interfacial Electrochem., 1983, 147, 39–51 CrossRef CAS.
- A. J. Bard, L. R. Faulkner and H. S. White, Electrochemical methods: fundamentals and applications, John Wiley & Sons, Inc., 3rd edn, 2020 Search PubMed.
- O. Simoska and S. D. Minteer, Techniques in electroanalytical chemistry, American Chemical Society, ACS InFocus, 2022 Search PubMed.
- J. W. Monaghan, Z. J. O'Dell, S. Sridhar, B. Paranzino, V. Sundaresan and K. A. Willets, J. Phys. Chem. Lett., 2022, 13, 10527 CrossRef CAS PubMed.
- Y. Yu, Y. Gao, K. K. Hu, P. Y. Blanchard, J. M. Noël, T. Nareshkumar, K. L. Phani, G. Friedman, Y. Gogotsi and M. V. Mirkin, ChemElectroChem, 2015, 2, 58–63 CrossRef CAS.
- G. K. Gebremariam, A. Z. Jovanovic and I. A. Pasti, Hydrogen, 2023, 4, 776–806 CrossRef CAS.
- S. D. Lubetkin, Chem. Soc. Rev., 1995, 24, 243–250 RSC.
- Q. J. Chen, L. Luo, H. Faraji, S. W. Feldberg and H. S. White, J. Phys. Chem. Lett., 2014, 5, 3539–3544 CrossRef CAS PubMed.
- J. Guo and E. Lindner, Anal. Chem., 2009, 81, 130–138 CrossRef CAS PubMed.
- H. R. Zebardast, S. Rogak and E. Asselin, J. Electroanal. Chem., 2014, 724, 36–42 CrossRef CAS.
- P. Li, J. Huang, Y. C. Hu and S. L. Chen, J. Phys. Chem. C, 2021, 125, 3972–3979 CrossRef CAS.
- K. Leung, J. Electrochem. Soc., 2022, 169, 081502 CrossRef CAS.
- A. Soffter and M. Folman, J. Electroanal. Chem., 1972, 38, 25–43 CrossRef.
- E. Bayram and E. Ayranci, Electrochim. Acta, 2011, 56, 2184–2189 CrossRef CAS.
- J. Guo and E. Lindner, J. Electroanal. Chem., 2009, 629, 180–184 CrossRef CAS PubMed.
- A. C. West and J. Newman, J. Electrochem. Soc., 1991, 138, 1620 CrossRef CAS.
- C. Amatore, A. I. Olenick and I. Svir, Anal. Chem., 2009, 81, 4397–4405 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02326a |
| This journal is © The Royal Society of Chemistry 2024 |