
Advances in MXene surface functionalization modification strategies for CO2 reduction
Hailong
Li†
a,
Linhao
Liu†
ab,
Tianbin
Yuan
ab,
Jianwen
Zhang
a,
Tiantian
Wang
b,
Juan
Hou
*a and
Jiangzhao
Chen
 *ac
*ac
aCollege of Sciences/Xinjiang Production & Construction Corps Key Laboratory of Advanced Energy Storage Materials and Technologies, Shihezi University, Shihezi, 832003, China. E-mail: hjuan05@sina.com; jzchen@kust.edu.cn
bKey Laboratory for Green Process of Chemical Engineering of Xinjiang Bingtuan, School of Chemistry and Chemical Engineering, Shihezi University, Shihezi, 832003, China
cFaculty of Materials Science and Engineering, Kunming University of Science and Technology, Kunming 650093, China
First published on 7th June 2024
Abstract
MXenes, 2D transition metal carbides and nitrides, show great potential in electrocatalytic CO2 reduction reaction (ECO2RR) applications owing to their tunable structure, abundant surface functional groups, large specific surface area and remarkable conductivity. However, the ECO2RR has a complex pathway involving various reaction intermediates. The reaction process yields various products alongside a competitive electrolytic water-splitting reaction. These factors limit the application of MXenes in ECO2RRs. Therefore, this review begins by examining the functionalized modification of MXenes to enhance their catalytic activity and stability via the regulation of interactions between carriers and the catalytic centre. The review firstly covers the synthesis methods and characterisation techniques for functionalized MXenes reported in recent years. Secondly, it presents the methods applied for the functionalized modification of carriers through surface loading of single atoms, clusters, and nanoparticles and construction of composites. These methods regulate the stability, active sites, and metal-carrier electronic interactions. Finally, the article discusses the challenges, opportunities, pressing issues, and future prospects related to MXene-based electrocatalysts.
1. Introduction
Layered transition metal carbides/nitrides, namely MXenes, as emerging two-dimensional nanomaterials, have a wide range of applications in electrical energy storage,1,2 optoelectronic functional devices,3 biomedicine,4 photo-catalysis,5 electro-catalysis,6–12etc., compared with graphene, MOFs, and TMDs owing to the unique nanostructures and excellent electrical properties of MXenes.13–15 The structural formula of MXenes is Mn+1AXn, where M is a transition metal (e.g., Cr, Ti, V, Mn, Nb, and Mo), A is usually a III or IV main group element (e.g., Al, Ga, Si, and Ge), and X is C, N, etc. Based on the different n values, MXenes are divided into three types, namely M2X1Tx, M3X2Tx, and M4X3Tx, with 211, 312, and 413 phases; Mn+1Xn layers are sequentially interleaved with an A-atom layer to form a layer-stacked MAX phase structure.16 To date, more than 100 MXenes have been experimentally and theoretically predicted, of which up to forty have been prepared (Ti3C2, Ti2C, Mo2C, V2C, etc.).17,18 Among them, Ti3C2Tx, the most common MXene material, is more conducive to proton and electron transport in the electrocatalytic CO2 reduction process because of its excellent electrical conductivity,19 low energy barrier for electron transport,20 abundant ionic diffusion pathways,21 and abundant active sites,22 which provide necessary prerequisites for CO2 activation and adsorption of key intermediates. Meanwhile, due to its multilayered lamellar structure and abundant surface end groups (–O, –OH, and –F), it not only increases the possibility of adsorption of CO2 but also interacts with CO2 to enhance C–H, C–O, and C–C coupling, which further facilitates the catalytic reaction.23The application of MXenes in the electrocatalytic reduction of CO2, although promising, presents several problems and challenges.24,25 This is mainly because catalytic CO2 reduction is itself a complex process involving multiple steps and reaction intermediates. Firstly, the high dissociation energy of CO2 molecules (750 kJ mol−1) makes it difficult to capture and activate them by MXenes, resulting in a high potential barrier for electrocatalytic reduction.26 Secondly, since CO2 reduction is a multi-electron transfer process,27 the diversity of reduction products makes the selectivity of the catalyst to target products a key issue. The selectivity of MXenes in the catalytic process may be affected by various factors such as their surface functional groups, active sites, and interactions with other reactants, and the relationship between the qualities of MXenes themselves (end groups, intrinsic defects, etc.) and the true active sites of the dominant products needs to be elucidated.28 In addition, since the standard potential for CO2 reduction is similar to that of the hydrogenolysis reaction, the involvement of a competing hydrogenolysis reaction (HER) in the catalytic process of MXenes decreases the efficiency of catalytic reduction of CO2.29 Therefore, in order to improve the adsorption and activation of CO2 molecules, the selectivity of target products and the reduction efficiency during the electrocatalytic reduction process of MXenes, the functionalization and modification of the surface of MXenes to improve their CO2 reduction performance have become a hot spot in current research.
Currently, researchers have altered the type of CO2 adsorption by modulating the type of central metal,30 surface substitution functional groups,31 and the type of vacancies to achieve physical (–O and –F) and chemical (–OH) adsorption, controlling the residence time of CO2 on the surface of MXenes,32 and modulating the reaction steps to improve the selectivity of the products and the catalytic efficiency,33 as shown in Fig. 1. In addition to the surface functional groups, the introduced non-metallic elements can provide an unsaturated coordination environment for the active sites of metals to produce electron synergistic catalysis, but the content of the non-metallic elements plays a crucial role.28 By regulating the content of non-metallic elements and their configurations on MXenes under the premise of defining the active center and providing an unsaturated coordination environment for the metal elements, the adsorption and electron transfer process of MXenes to the key intermediates can be effectively enhanced. The selectivity and efficiency of generating a single carbon product can be improved.34 All of the above-mentioned modification methods are modulated based on MXenes, and although they can play a role in promoting CO2 reduction, they always start from MXene materials, which makes it difficult to make a breakthrough. Therefore, the researchers proposed loading other materials onto MXenes such as single atoms,35 clusters,36 nanoparticles,37 and other bulk-phase materials.38 As MXenes have complex surface chemical groups, these groups make their surface rich in functionalization sites, which is conducive to compounding with other materials, thus increasing its potential for application in the catalysis of CO2.39 When the material is loaded onto the surface of MXene materials, the good metal conductivity of MXenes can make up for the disadvantage of the slow carrier migration rate of the material itself, which is conducive to the electron transport and electron transfer process, and improves the catalytic efficiency and activity of the loaded material. Meanwhile, atomic and metal cluster loading can also change the electronic state density and energy level distribution on the surface of MXenes, thus modulating the selectivity of the reduction products of CO2. The dispersion of single atoms or clusters can effectively inhibit the cluster aggregation and coalescence phenomena on the MXene surface, preventing the masking and deactivation of the active sites.40 This helps maintain the high activity and stability of the catalyst and keep the good catalytic performance during a long reaction time.
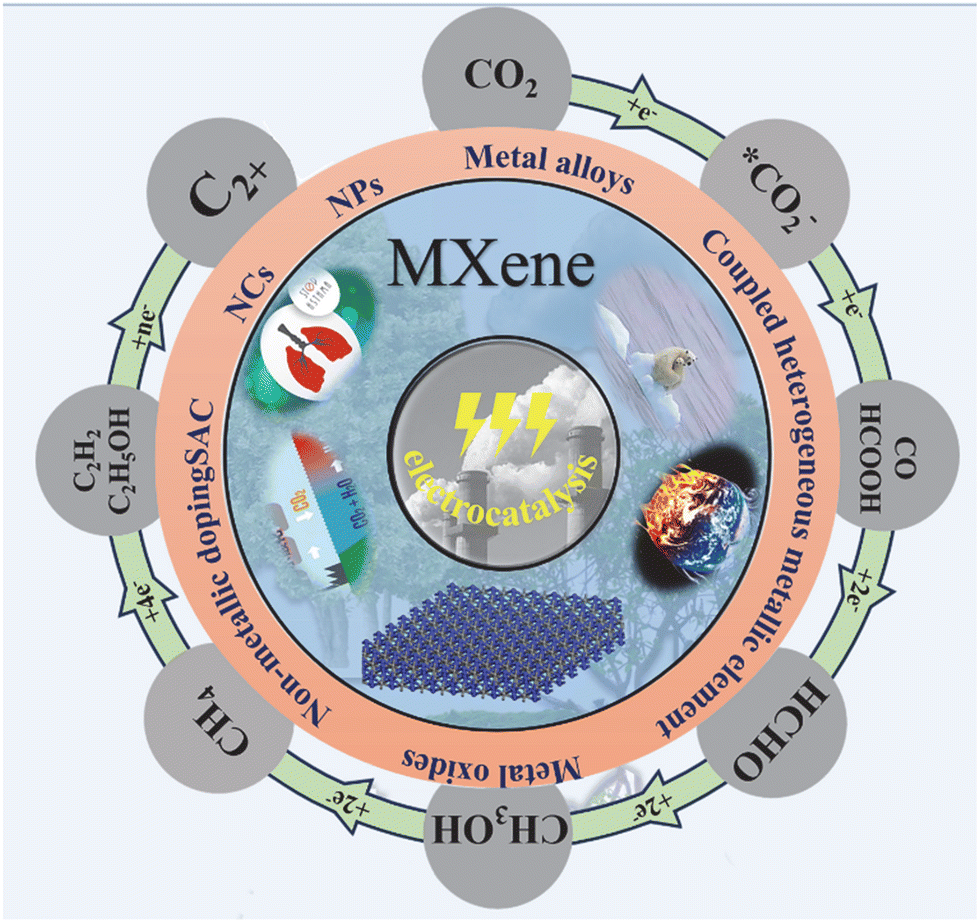 | ||
| Fig. 1 Progress in the study of MXenes for electrocatalytic carbon dioxide reduction. | ||
Therefore, to improve the potential of MXenes in ECO2 reduction, the understanding of the mechanism of ECO2 reduction by functionalized modifications is strengthened. In this review, first, the catalysts synthesized based on single atom, cluster, or nanoparticle-loaded MXenes and the catalysts synthesized by compositing MXenes with other materials are systematically compared, the modulation of loaded MXene-based catalysts is discussed, and an overview of the electrocatalytic reduction of CO2 into C1 as well as C2 abducts with loaded MXene-based catalysts is presented in terms of both theoretical calculations and experiments. Meanwhile, the shortcomings of the current CO2ERR of MXene-based materials are addressed one by one to address the existing key scientific issues, to increase the catalytic activity to break through the CO2 reduction barrier by increasing the number of active sites through fictionalized modification methods and to inhibit the HER and improve the selectivity of the products by changing the type of active sites. Finally, the prospects and challenges of MXene-based catalysts for electrocatalytic CO2RRs are discussed.
2. Functional modification methods for MXenes
MXenes have been widely used in electrocatalysis due to their typical features such as excellent electrical conductivity and abundant surface end groups. However, the further development of MXenes still faces some challenges and difficulties such as the presence of solid-phase impurities, stability, and scalability. To solve the above-mentioned problems, many researchers have utilized MXene surface functionalization modification methods such as loading single atoms and nanoclusters and constructing composite heterojunctions on the MXene surface, to improve the application of MXenes in electrocatalytic CO2 reduction. Several synthesis strategies and methods of typical MXene surface functionalization modifications are briefly described below.2.1 Synthesis strategy for MXene-based single-atom catalysts
Recently, single-atom-loaded MXene catalysts have been considered as potential catalysts for CO2 reduction conversion due to their catalytic activities such as high activity, high selectivity, and high stability. However, the dispersion of metal atoms on the atomic scale and the formation of strong coordination bonds between isolated metal atoms and carrier ligand atoms have become the key to single-atom-loaded MXene catalysts. Therefore, to overcome the competition between metal-carrier atomic coordination and metal–metal bonding coordination, the current synthesis strategies for MXene-loaded metal single-atom catalysts mainly include wet chemical impregnation, defective vacancy anchoring, and metal-carrier interactions.Wet-chemical impregnation, as a synthetic method with a relatively simple preparation process, allows the metal atoms to be uniformly dispersed in MXenes, ensuring good homogeneity and consistency of the catalyst. This helps improve the properties and stability of the material, resulting in excellent catalytic activity. For example, Zou et al.40 successfully prepared Ru-SA@Ti3C2Tx catalysts by anchoring Ru-SA on Ti3C2Tx by the wet chemical impregnation method. It was demonstrated by advanced optical imaging techniques (SEM, AFM, TEM SAED, etc.) that RuSA@Ti3C2Tx not only retains the hexagonal crystal structure of Ti3C2Tx but also has a two-dimensional shaped structure with a thickness of 4 nm, as shown in Fig. 2a. Meanwhile, due to the presence of Ti vacancies in the defective state and the reducing nature of MXenes themselves, the introduction of metal monoatoms is stably dispersed on the Ti vacancies during wet chemical impregnation. For example, Zhou et al.41 successfully synthesized a series of single-atom catalysts, Ni-SA@Ti3C2Tx, enriched with Ti defects (Ni loading of 2.93 wt%) by combining wet chemical impregnation with defect trapping and “self-reduction” process and confirmed the substitution of Ti vacancies by Ni atoms with three neighboring C atoms by HAADF-STEM and EXAFS, which effectively improved its electrocatalytic performance (Fig. 2b).
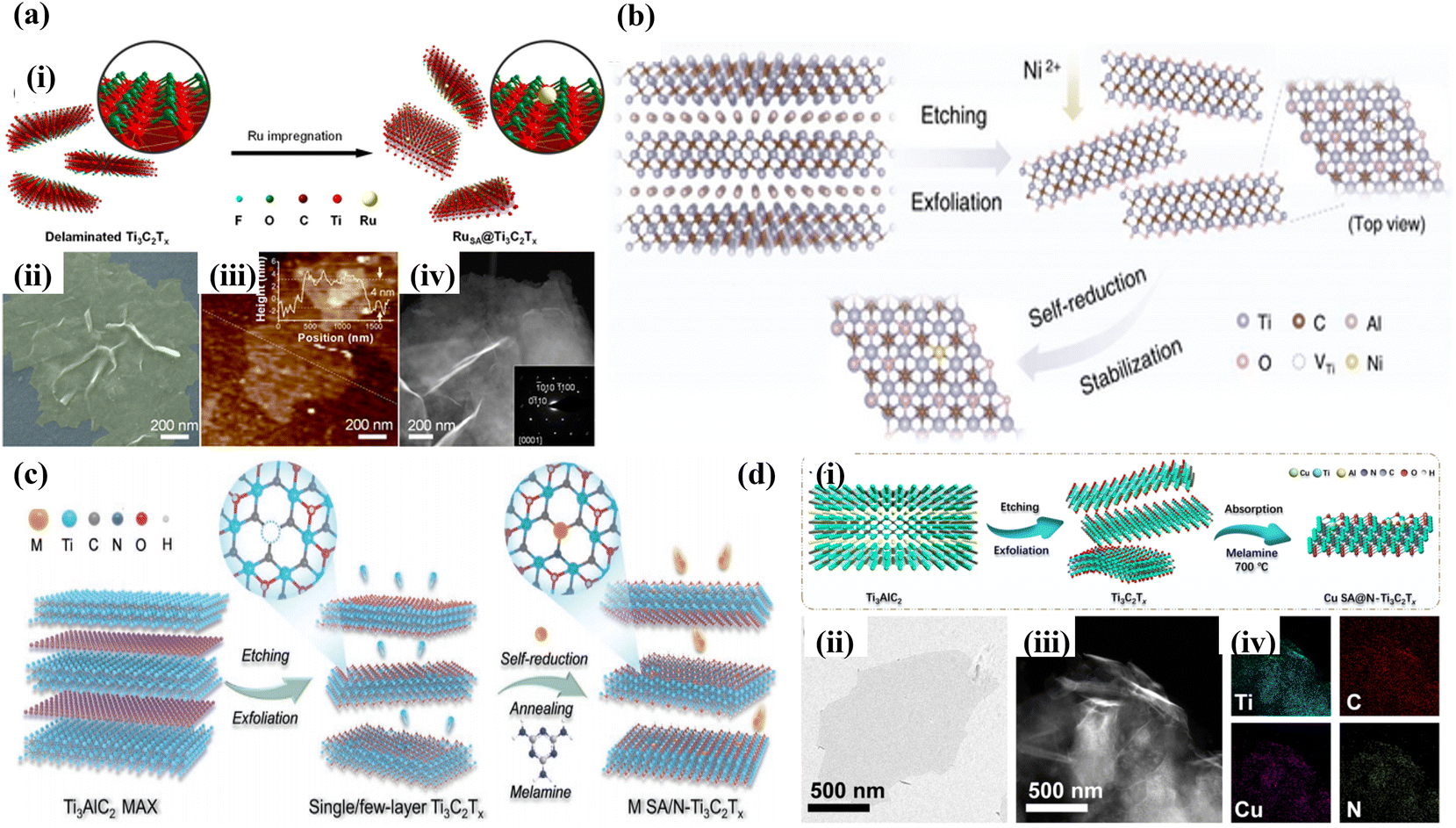 | ||
| Fig. 2 (a) (i) Schematic illustrating the fabrication procedure of RuSA@Ti3C2Tx. (ii) Colored scanning electron microscopy (SEM) image, (iii) atomic force microscopy (AFM) image, and (iv) dark-field transmission electron microscopy (TEM) image. Reproduced from ref. 40 with permission from Wiley & Sons Inc., copyright 2023. (b) Schematic of the synthesis process of Ni SACs/Ti3C2Tx. Reproduced from ref. 41 with permission from Wiley & Sons Inc., copyright 2022. (c) Schematic illustration of the fabrication procedure of Cu SA/N-Ti3C2Tx. Reproduced from ref. 42 with permission from Wiley & Sons Inc., copyright 2023. (d) (i) Schematic illustration of Cu SA@N-Ti3C2Tx synthesis, (ii) TEM image of Ti3C2Tx, (iii) HAADF-STEM image of Cu SA@N-Ti3C2Tx, and (iv) corresponding EDS analysis. Reproduced from ref. 43 with permission from Springer-Verlag, copyright 2023. | ||
Unlike wet chemical impregnation, defect vacancy anchoring exploits the lattice defects of MXenes to anchor the hetero-metal atoms by atomic coordination. For example, Gu et al.42 employed a vacancy-heteroatom fixation strategy to anchor asymmetrically coordinated metal single atoms (M SA/N-Ti3C2Tx, M = Cu, Co, Ni, Mn, Zn, In, Sn, Pb, and Bi) on N-doped Ti3C2Tx nanosheets by using an N2 annealing (500 °C) method and a self-reducing process of the metal atoms, as shown in Fig. 2c. Meanwhile, the distribution of Cu atoms and asymmetric coordination configurations on Ti3C2Tx nanosheets were confirmed by the DFT and XANES. Liu et al.43 also successfully synthesized a single-atom Cu catalyst (Cu-SA@N-Ti3C2Tx) with an asymmetric synergistic structure by using defect vacancy anchoring, whose Cu-N1C1 synergistic structure was anchored on N-doped Ti3C2Tx to form an asymmetric C–Cu–N bridge coordination structure, which effectively ensured the stability of the material, as illustrated in Fig. 2d.
Meanwhile, strong metal–support interaction (SMSI) serves as the main driving force for single-atom loading, where atoms are immobilized by strong interactions with carbon atoms around defects on the MXene surface. For example, Ramalingam et al.44 successfully synthesized two-dimensional materials of Ru SA-N/S-Ti3C2Tx with N- or S-coordinated Ru atoms using Ti3C2Tx as a carrier by taking advantage of the SMSI action. HAADF-STEM characterization reveals that Ru is uniformly distributed with sub-nanometre-sized heavy metal bright spots, suggesting that the Ru atoms are uniformly monodispersed on the Ti3C2Tx surface. The formation of Ru–N and Ru–S bonds and the absence of Ru–Ru bimetallic bonds were demonstrated by XPS and FT-EXAFS, indicating that monoatomic Ru–N/S-MXenes mediated by N and S heteroatoms were successfully synthesized. Meanwhile, Lin et al.45 synthesized a porous material of Ir monoatoms confined to heteroatom (N, S) co-doped Ti3C2Tx. The formation of new bridging structures between Ir atoms and N and S atoms enabled the Ir atoms to be well dispersed in porous Ti3C2Tx, and the N and S-doped Ti3C2Tx carriers captured electrons from the Ir monoatom via highly coordinated Ir–N and Ir–S interactions, leading to charge redistribution in the interfacial region, providing a new strategy to regulate the atom-dispersed active sites for various electrocatalytic processes. N,S-co-doped Ti3C2Tx carriers capture electrons from the Ir monoatoms via highly coordinated Ir–N and Ir–S interactions, leading to charge redistribution in the interfacial region of the Ti3C2Tx carriers, which provides a new strategy to regulate the atomically dispersed active sites for various electrocatalytic processes.
2.2 Cluster and nanoparticle synthesis strategies
Nanoclusters (NCs) and nanoparticles (NPs) can provide optimal coordination environments depending on the ratio of the number of metal atoms to the number of ligands (size effect) interacting with the carriers, thereby significantly increasing the catalytic activity of the catalysts. For instance, Liu et al.46 utilized the strong interaction between MXenes and (NH4)9[Ag9(MBA)9] nanoclusters to produce Ag9/MXene catalysts. This was achieved by loading silver nanoclusters onto the surface of MXenes through wet chemical impregnation. The method avoids the aggregation and growth of Ag NCs in the catalytic process (particle size of 1.16 nm), and the clusters are more uniformly distributed on the MXene surface, as shown in Fig. 3a and b. At the same time, since the MXene carrier can not only serve as an excellent light-absorbing carrier to make up for the lack of cluster light absorption but also effectively stabilize and disperse the metal clusters, so that a stable interfacial structure will be formed between the MXenes and the clusters. For example, the MXene-loaded ruthenium cluster catalysts were prepared by an adsorption–reduction method by Wu et al.,47 and the TEM image of Ru/Mo2TiC2 shows that the ruthenium clusters are well dispersed on the surface of molybdenum carbon titanium and the structure of the MXene material is not damaged. A comparison of the morphology and crystal structure of molybdenum carbide before and after loading shows that the original densely packed hexagonal structure is still maintained after loading the ruthenium clusters, which proves the stability of the MXene carriers. Combining XPS, XANES, and EXAFS characterizations, it was found that both ruthenium and molybdenum in the MXene-loaded ruthenium cluster catalysts were biased towards the oxidation state, and the strong electronic interactions between the ruthenium particles and the molybdenum carbon–titanium carriers caused the ruthenium to lose some of its electrons to form the stable Ru/RuOx/MoOx/Mo2TiC2 interfacial structure, which anchors the cluster catalysts efficiently and enhances the stability of the catalysts.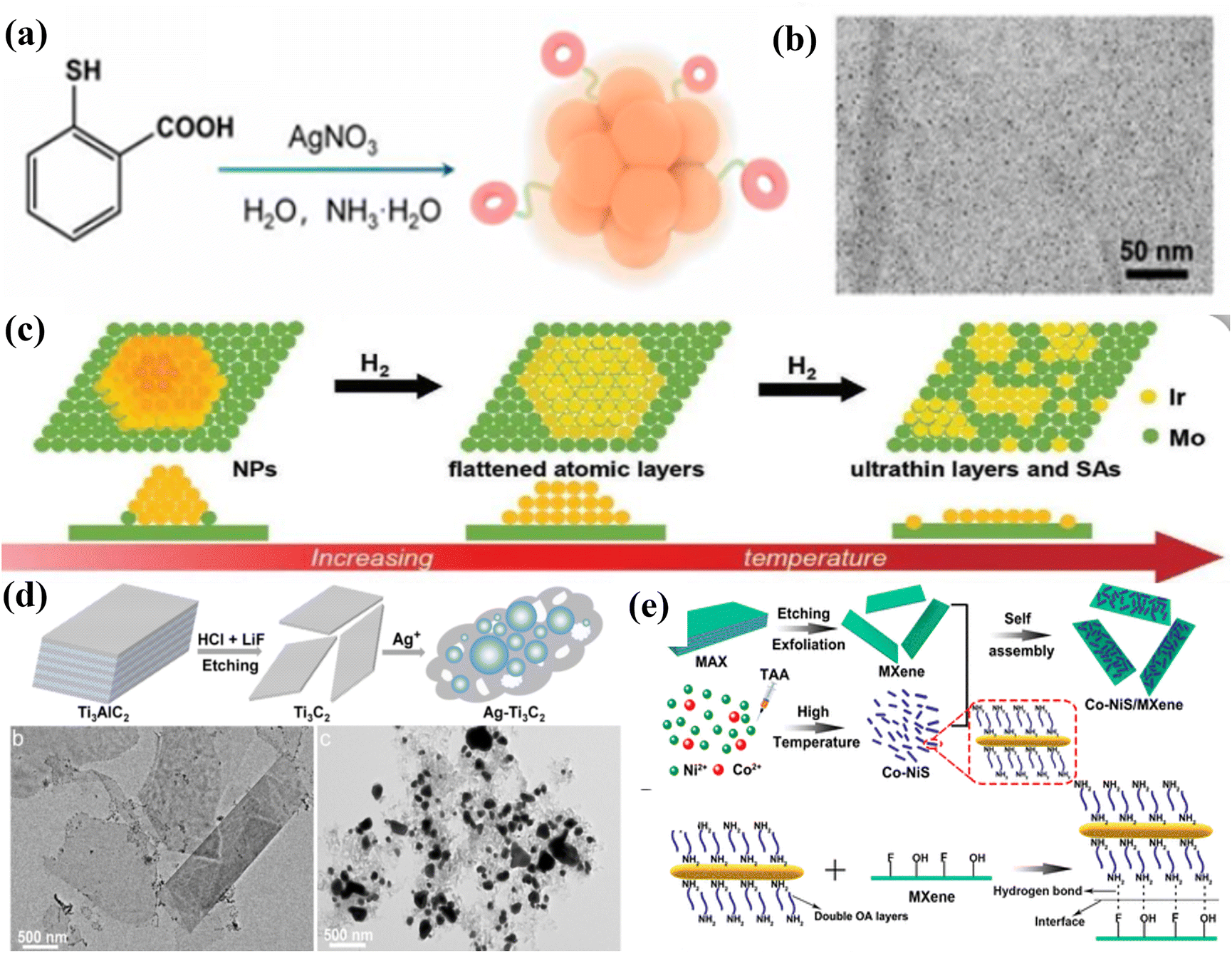 | ||
| Fig. 3 (a) Schematic illustration of the preparation of Ag9 NCs. (b) Typical TEM image. Reproduced from ref. 46 with permission from Wiley & Sons Inc., copyright 2024. (c) Scheme of the NP-flattened layer-ultrathin layer transformation of Ir on Mo2TiC2Tx. Reproduced from ref. 49 with permission from Elsevier, copyright 2023. (d) Schematic illustration of the preparation of Ag-Ti3C2 nanohybrids, and TEM images of Ti3C2 MXenes before (b) and after (c) reaction with Ag+. Reproduced from ref. 48 with permission from Wiley & Sons Inc., copyright 2022. (e) Synthesis and structural characterization of the Co-NiS/MXene composite. Reproduced from ref. 50 with permission from the American Chemical Society, copyright 2021. | ||
At the same time, the presence of OH terminal groups on the surface of Ti3C2 gives it a high adsorption capacity and reducibility, which, in turn, enables the spontaneous reduction of noble metal cations to metal NPs without the addition of a reducing agent or post-treatment. Remarkably, the atomic-level modulation of the structure of metal components and their substrate interactions are crucial for enhancing the catalytic performance of metal catalysts. For example, Dai et al.48 reported a composite catalyst loaded with Ir on Mo2TiC2TxMXenes with controllable morphology from the atomic level to nanoparticles, culminating in the formation of ultrathin monoatomic layers dispersed on MXene nanosheets, as the temperature is increased (shown in Fig. 3c). Jin et al.49 synthesized Ag-NPs/Ti3C2 nano complexes by the in situ reduction of Ag+ by Ti3C2 with the OH-terminal groups on the surface (with a maximum Ag NP loading of 75.0 wt%), and a large number of Ag NPs with diameters ranging from 20 to 400 nm were uniformly immobilized on the surface of Ti3C2 (shown in Fig. 3d). Meanwhile, in combination with the IR spectral data of Ag+-OH functional groups, the possible Ti–O–Ag bonding interactions between Ti3C2 and Ag NPs resulted in the homogeneous dispersion of Ag NPs on Ti3C2 nanosheets.
2.3 Synthesis strategies for another MXene-based composite catalyst
The composites are important for the adsorption and transfer of electrons in the key intermediate of the CO2 reduction process due to the strong interfacial coupling, which is important for the improvement of the selectivity of single carbon products. Currently, for the preparation and synthesis of MXene-based composites, the main self-assembly method, electrodeposition method, and molten salt method were used. For example, Zou et al.50 prepared Co-NiS/MXene composites by self-assembly at the interface of water and cyclohexane using the interfacial self-assembly method via non-covalent interactions (e.g., hydrogen bonding, van der Waals forces, and electrostatic interactions) between the molecules of Co-NiS and MXenes. The Co-NiS nanorods were uniformly dispersed on the surface of MXenes, which effectively inhibited the self-stacking and agglomeration of Co-NiS and MXene self-stacking and agglomeration, ensuring good electrocatalytic performance (shown in Fig. 3e). The method can be completed quickly (10 min) at room temperature, which can effectively solve the oxidation problem of MXenes under hydrothermal and high-temperature calcination conditions, and opens up a new method for the preparation of MXene-based composites. Meanwhile, to drive the spontaneous interfacial reduction process of MXene-based composites, Yun et al.51 took advantage of the difference in intrinsic reduction potentials between the negatively charged functional groups on the surface of MXenes and the substrate to induce the spontaneous transfer of electrons from Zn to Ti3C2Tx nanosheets, which weakened the electrostatic repulsion between the nanosheets and effectively improved the stability and electrocatalytic CO2 performance of Ti3C2Tx. In order to avoid the influence of intra-particle and inter-particle charge transport in MXenes on the electrocatalytic CO2 performance of MXenes and the modulation of the interface, Li et al.52 used electrodeposition to uniformly deposit nickel–aluminium layered double hydroxide (Ni–Al-LDH) on MXene particles, and the uniformly grown MWCNT not only acted as a spacer preventing the re-obstruction of the MXene membrane but also acted as an intra-particle and inter-particle charge collector, effectively enhancing the mechanical and chemical stability and the adsorption of key intermediates (electron transfer) of the composite electrode. The uniformly grown MWCNT not only acted as a spacer to prevent the MXene film from re-obstructing but also as an intra-particle and inter-particle charge collector, which effectively improved the mechanical and chemical stability of the composite electrode and the adsorption of key intermediates (electron transfer). Xia et al.53 synthesized nitrogen-doped graphene and ReSe2 complexes by chemical vapor deposition using 3D MXenes as the structural support, and the ReSe2 nanoparticles exhibited good electrocatalytic performance for CO2 reduction via strong interfacial interactions between nitrogen-doped graphene and 3D porous structure.3. MXenes in ECO2 RRs
The electrochemical reduction of CO2 is an important method for converting it into high-value-added hydrocarbon products such as CH4 and CH3OH. However, due to its high thermodynamic stability (750 kJ mol−1 energy required to break the CO2 bond), the process faces a large reaction potential barrier (−1.9 V vs. NHE) for catalysts to capture CO2 and react with a single electron.54 However, the complex reaction path of CO2 results in poor catalyst selectivity for the reduction products, which affects the isolation of single carbon products and high-value-added applications. Furthermore, the similarity between the theoretical potential for reducing CO2 and the potential for the hydrogen evolution reaction (HER) can lead to significant side reactions of HERs in aqueous reaction systems, resulting in poor Faraday efficiency of the target products. MXenes, which are typical electrocatalytic materials, not only possess excellent electrical conductivity properties but also has abundant surface groups and defects that provide reactive active sites for CO2 adsorption and reduction. During the etching process of MXenes, the Al layer is expected, resulting in the loss of Ti atoms and the formation of a large number of Ti vacancies in the interlayer. This leads to the creation of a high concentration of low-valent Ti ions on the surface, which exhibit good self-reducing properties. Furthermore, the Ti 3d orbitals can be redistributed, resulting in the formation of a ‘dangling bond’. This can induce the transformation of Ti–Al bonds into localized Ti–Ti-like metal bonds, which appear near the Fermi energy level of Ti3C2, thus presenting good electrical conductivity. However, the lower electrocatalytic efficiency and selectivity for CO2 reduction, as well as the higher occurrence of HERs, limit its application.55–57 Therefore, researchers have used various functionalization modification methods to enhance the efficiency of electrocatalytic CO2 reduction in MXene-based materials, as illustrated in Table 1.| Catalyst | E/[V vs. RHE] | Production | FE/% | Electrolyte | JC2+/(mA cm−2) | Ref. |
|---|---|---|---|---|---|---|
| Cu-meso-crystal | −0.99 | CH4 | 18 | 0.1 M KHCO3 | — | 58 |
| Prism-Cu | −1.1 | C2+ | 35 | 0.1 M KHCO3 | 10 | 59 |
| Cu-nanowire-array | −1.1 | C2H4 | 17.4 | 0.1 M KHCO3 | 60 | |
| Cu-cubic-NPs | −1.1 | C2H4 | 41 | 0.1 M KHCO3 | 1.4 | 61 |
| Cu-thin-film (100) | −0.97 | C2+ | 60 | 0.1 M KHCO3 | 2 | 62 |
| Cu2O-film | −0.99 | C2H4/C2H5OH | 34.26/16.37 | 0.1 M KHCO3 | 17.8 | 63 |
| CuOx-Vo | −1.4 | C2H4 | 63 | 0.1 M KHCO3 | 64 | |
| Plasma-oxidized-Cu | −0.9 | C2H4 | 60 | 0.1 M KHCO3 | 12 | 65 |
| Cu Ag-core–shell | −1.06 | C2H4 | 25 | 0.1 M KHCO3 | 66 | |
| Cu Ag-wire | −0.68 | C2H4 | 60 | 1.0 M KOH | 26.5 | 67 |
| Cu28Ag72 | Pulse | C2H4O | 24.1 | 0.1 M KHCO3 | 68 | |
| Oxide-derived-Cu4Zn | −1.05 | C2+ | 51 | 0.1 M KHCO3 | 15 | 69 |
| Cu SA-Ti3C2Clx | −1.4 | CH3OH | 59.1 | 0.1 M KHCO3 | 21.3 | 70 |
| Cu/Ti3C2Tx-nanosheets | −0.7 | C2H4 | 71 | 1 M KOH | 24.8 | 71 |
| Cu–Pd/Ti3C2Tx | −0.5 | HCOOH | 79 | 0.1 M KHCO3 | 25 | 72 |
| MXenes@Por-COF-Co | −0.6 | CO | 97.28 | 0.5 M KHCO3 | 9.33 | 73 |
| FePc/MXenes | −0.6 | CO | 98.23 | 0.5 M KHCO3 | 5.24 | 74 |
| N-Ti3C2-VTi | −0.8 | CO | 92 | 0.5 M KHCO3 | 16.2 | 75 |
| SnO2/MXenes | −0.8 | Formate | 94 | 0.1 M KHCO3 | 57.8 | 76 |
| Ag-ZnO/Ti3C2Tx | −0.87 | CO | 98 | 0.5 M KHCO3 | 22.5 | 77 |
3.1 Increasing selectivity by reducing reaction energy barriers
 | ||
| Fig. 4 (a) Influence of vacancies on the binding energy of intermediates and the limit potential of CO2RRs. (b) Cross-sectional view of the ULCO2 contour map at a *H2CO binding energy of −0.634 eV. (c) Free energy diagram of pristine Hf2NO2 (black line), VHf-Hf2NO2 (red line) and VN-Hf2NO2 (blue line). Faded lines indicate non-preferred alternative pathways. Reproduced from ref. 57 with permission from Wiley & Sons Inc., copyright 2020. | ||
Functional groups on the surface of active MXenes have been shown to effectively influence the hydrogen desorption process in the field of electrolytic hydrogen production. This plays an important role in inhibiting HER and improving CO2RR selectivity. However, the rich defects and multiple functional groups of MXenes often render the ideal model based on calculations ineffective in guiding experiments. Handoko et al.78 investigated the ECO2RR process of Ti2CTx and Mo2CTx by combining experiments with theories. Fig. 5 demonstrates that MXenes with different contents of –F end-groups were obtained by HF and KF-HCl etching methods. In particular, Ti2CTx (HF) (F/Ti = 0.36) and Ti2CTx (KF-HCl) (F/Ti = 0.21) were obtained. At a potential of 1.8 V (V vs. SHE), FEHCOOH of Ti2CTx (KF-HCl) and Ti2CTx (HF) were 56.1% and 20.7%, respectively. To compare the effects of different end groups, the CO2 → HCOOH process was studied on the surface of the fully –O end-group Ti2CTx. The process involves CO2 adsorption, two consecutive proton-coupled electron transfers (PCET), and HCOOH desorption. While the adsorption of *HCOOH is not significantly affected, the *COOH conformation becomes increasingly unstable with a higher –F content. Therefore, Ti2CTx enriched with more –O end-groups demonstrate superior CO2RR activity and selectivity compared to Ti2CTx enriched with more –F end-groups. The same phenomenon also appeared in the reduction of CO2 by Mo2CTx with different end-group contents, and higher CO2RR activity was exhibited on the Mo2CTx catalyst with the smallest –F end-group content. Meanwhile, to clarify the effect of –OH end-groups. Meng et al.79 conducted a systematic study on the impact of different surface end-groups and end-group fragment distributions of Ti3C2 on CO2RR performance using DFT. They analyzed the possibility of catalytic synthesis of a range of CO2RR products, including CO, HCOOH, and CH3OH. Among all the surface-terminated models, the one fully terminated with –OH exhibited the strongest CO2 adsorption and CH4 desorption. When the –F end group is present, the –OH and –F end groups play similar roles. However, the electron density at the –F end is more localized, leading to stronger repulsion. This affects the MXene activity at different stages of the reaction. The UL decreases as the content of –O end group increases. This is because –O and –OH groups play a dual role as H acceptors and donors, respectively. This lowers the energy barrier of the hydrogenation process, increasing the likelihood of simultaneous hydrogenation of the reacting material by H+ reduction and H transfer from the surface –OH group. As a result, the energy barrier of the reduction reaction is lowered.
 | ||
| Fig. 5 (a) *HCOOH binding energy plot against *COOH binding energy showing deviation from linear scaling relations. (b) Limiting CO2RR potentials for elementary steps. The lines represent the calculated potential, where the most negative reaction steps are neutral as a function of *COOH binding energy. (c) ULCO2–ULH2 plots of ULCO2 for theoretical models of Ti2CTx and Mo2CTx with different distributions of number-F functional groups. Reproduced from ref. 78 with permission from Cell Press, copyright 2020. (d) Graphical abstract. Reproduced from ref. 79 with permission from the Royal Society of Chemistry, copyright 2024. | ||
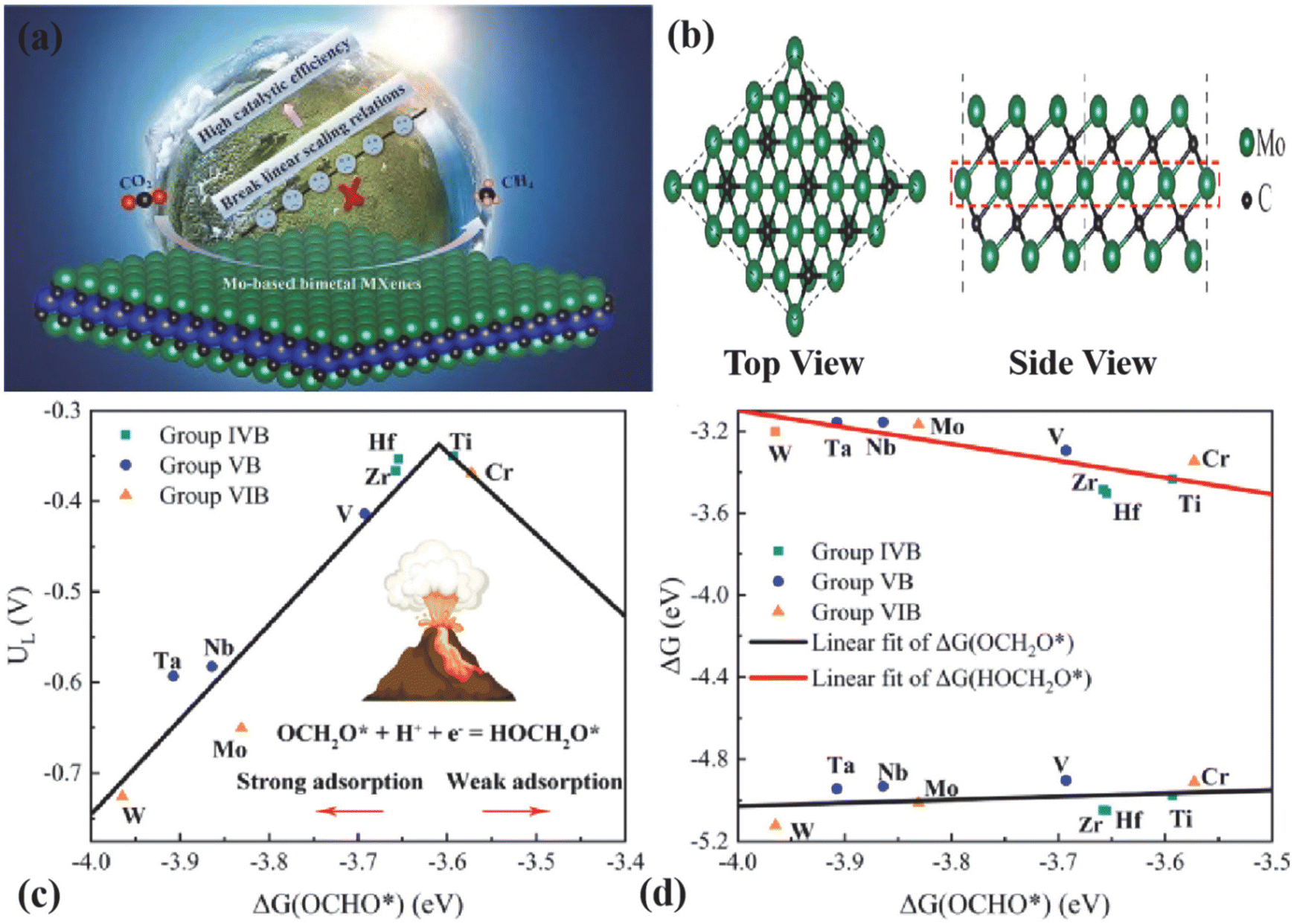 | ||
| Fig. 6 (a) Graphical abstract. (b) Top and side views of the stable structure of Mo3C2. (c) CO2RR volcano plot of Mo3C2 and TM-substituted bimetal MXenes, in which adsorption free energy of OCHO*, i.e., ΔG(OCHO*), acts as the descriptor of UL. (d) Linear relation of adsorption free energy ΔG(OCH2O*) and ΔG(HOCH2O*) versus the d-band center Ed. Reproduced from ref. 80 with permission from Elsevier, copyright 2022. | ||
The catalyst with a strong single-atom metal–support interaction (SMSI) effect was constructed by utilizing SMSI.81 This prevents the atomic diffusion of single metal atoms from aggregating into particles, achieving catalyst stability and high selectivity. Zhao et al.82 used surface metal Ti vacancy defects generated during the etching of MXenes (Ti3−xC2Ty) to anchor Pt atoms, as shown in Fig. 7a. They took advantage of the self-reducing property of Pt/Ti3−xC2Ty and the unsaturated coordination environment of the Pt atoms to reduce the adsorption and activation energies of silane, CO2, and aniline. This reduced the energy barrier of the catalytic reaction of CO2. To verify the effect of transition metal loading on the selectivity of single carbon products, Li et al.83 designed a series of transition-metal-loaded Ti2C-based MXenes (TM@Ti2CTx, TM = V, Cr, Mn, Fe, Co, and Ni). These electrocatalysts not only activated CO2 molecules but also reduced the first step of the hydrogenation reaction due to stronger charge transfer lowering. As shown in Fig. 7b, Niu et al.84 found that the energy barriers of CO in V@Ti2CO2, Mn@Ti2CO2, and Fe@Ti2CO2 surfaces were reduced, resulting in smaller adsorption Gibbs free energy. This helped avoid *COOH cleavage and improve the HCOOH selectivity of Co@Ti2CS2 and Ni@Ti2CS2. Bi-based catalysts were developed and anchored onto eight MXenes, namely, Mo2C, Nb2C, Nb4C3, Ti2C, Ti3C2, V2C, V4C3, and Zr3C2. The results showed that Bi monoatomic-loaded V2C exhibited high activity for HCOO formation, improved selectivity towards formic acid and methanol, and introduced a new reaction pathway for HCOOH formation, as illustrated in Fig. 7c.
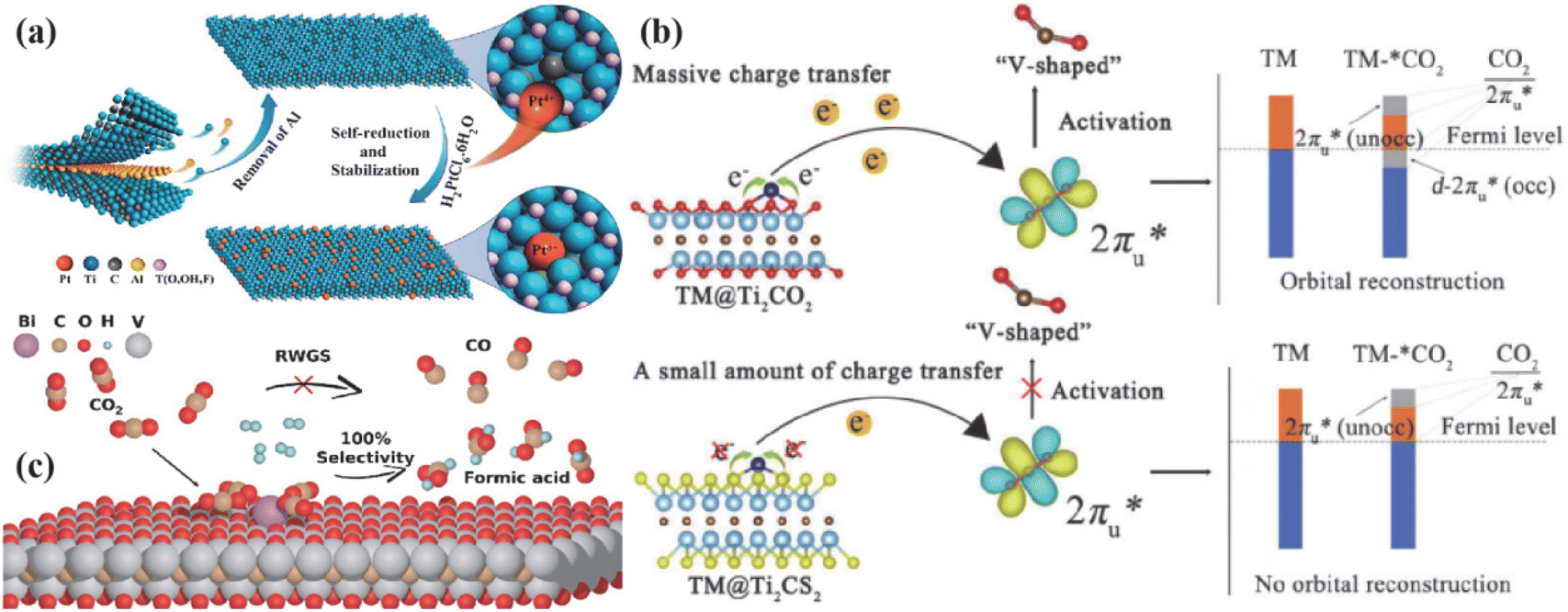 | ||
| Fig. 7 (a) Abstract structural morphology of Pt/Ti3−-xC2Ty. Reproduced from ref. 82 with permission from the American Chemical Society, copyright 2019. (b) Schematic diagram of CO2 activation mechanism on TM/Ti2CTx. Reproduced from ref. 83 with permission from Elsevier, copyright 2022. (c) Reaction process mechanism of Bi@V2CO2 to achieve 100% formic acid pathway. Reproduced from ref. 84 with permission from the American Chemical Society, copyright 2024. | ||
In addition, the transition metal increases effective binding to key intermediates of the ECO2RR reduction due to its empty d-orbitals and off-domain d-orbital electrons. According to Sabatier's principle, the *CO adsorbed on the surface of TM-MXenes would neither instantaneously desorb due to too weak adsorption force nor poison the catalyst due to too strong adsorption force, thus achieving the goal of lowering the reaction energy barriers and improving the catalytic selectivity. Zhao et al.85 achieved a maximum FE (methanol) of 59.1% at −1.4 V (V vs. RHE) by selectively etching the mixed MAX phase (Ti3(Al1−xCux)C2) of Al with preservation of the Cu single atom. Meanwhile, DFT calculations showed that the Cu monoatoms on the MXenes preferred to convert CO2* into absorbed OCHO* intermediates (not COOH* intermediates), reducing the free energy (0.53 eV), as the rate-determining step (HCOOH* → CHO*) of the SA-Cu MXenes and significantly lowering the potential barrier of the electrocatalytic CO2 reduction to the CH3OH reaction. Meanwhile, Eid et al.86 found that the Cu-loaded Ti3C2Tx surface has a high electron density of polarisation sites, as shown in Fig. 8a, which allows the formation of formic acid (HCOOH) from CO2RRs via the *HCOOH intermediate, leading to a three times higher catalytic efficiency of HCOOH on Cu/Ti3C2Tx (58.1%) than on Ti3C2Tx (18.7%). In addition to Cu, the transition metal Co also showed unique selectivity for the ECO2RR process,87 and the loaded Co catalysts were highly active for C–O dissociation and C–C coupling. For example, Ma processed the interaction of Ti3C2Tx with Co by NH3 and synchronized N-doping and surface modification (the presence of TiO2 in amorphous form appeared), which not only preserved the structure of the original MXenes but also enhanced the strong interactions between the support and Co nanoparticle, increased the CO desorption energy barrier and promoted the further reduction of chemisorbed CO to CH4, as shown in Fig. 8.
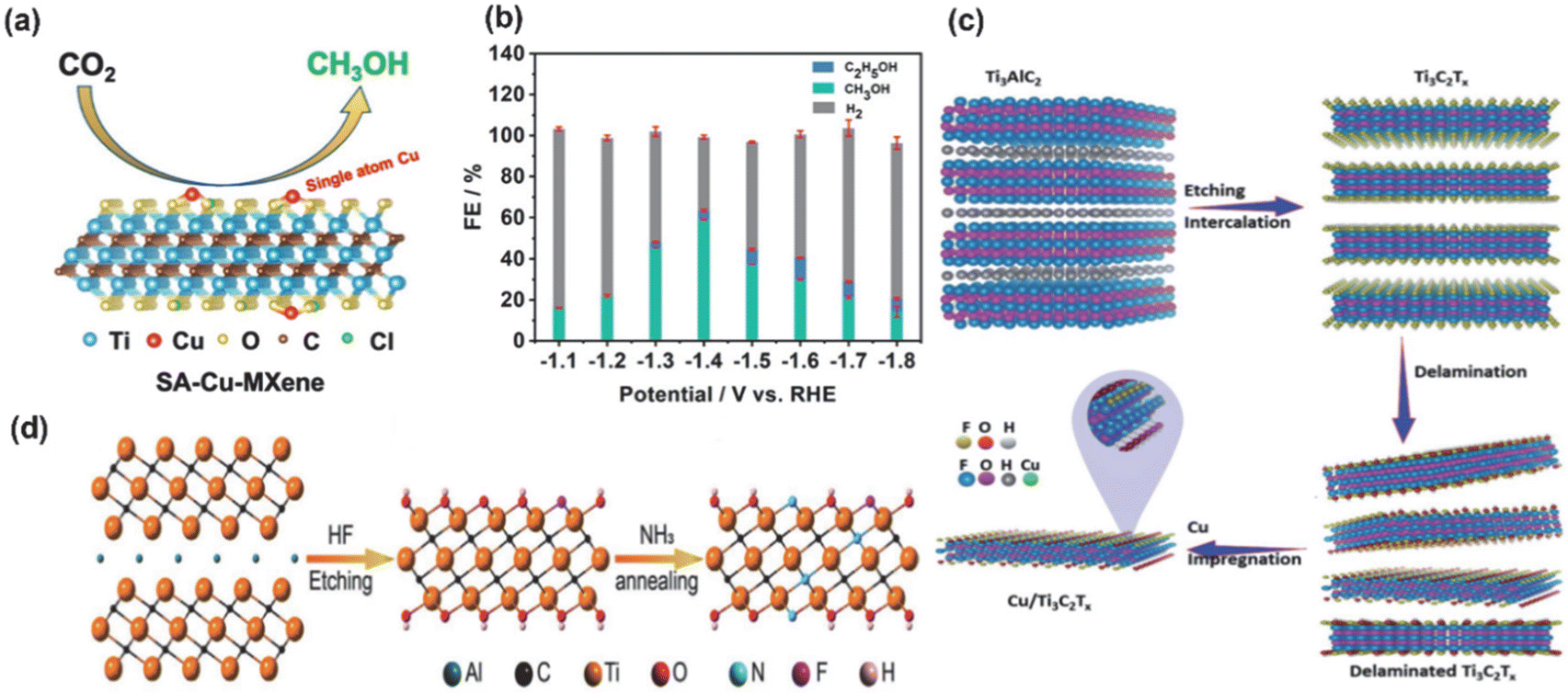 | ||
| Fig. 8 (a) Catalytic process and schematic structure of SA-Cu-MXenes (Tx = Cl). (b) Histogram of the product selectivity of SA-Cu-MXenes at different potentials. Reproduced from ref. 85 with permission from the American Chemical Society, copyright 2021. (c) Proposed scheme of the fabrication process of Cu/Ti3C2Tx. Reproduced from ref. 86 with permission from the Royal Society of Chemistry, copyright 2022. (d) Schematic illustration of the MXene-NH3 samples. Reproduced from ref. 87 with permission from John Wiley & Sons, Inc., copyright 2021. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) compared to MXenes, which extremely exposed the reaction center accelerating the charge transfer and thus promoting the CO2RR.
:1) compared to MXenes, which extremely exposed the reaction center accelerating the charge transfer and thus promoting the CO2RR.
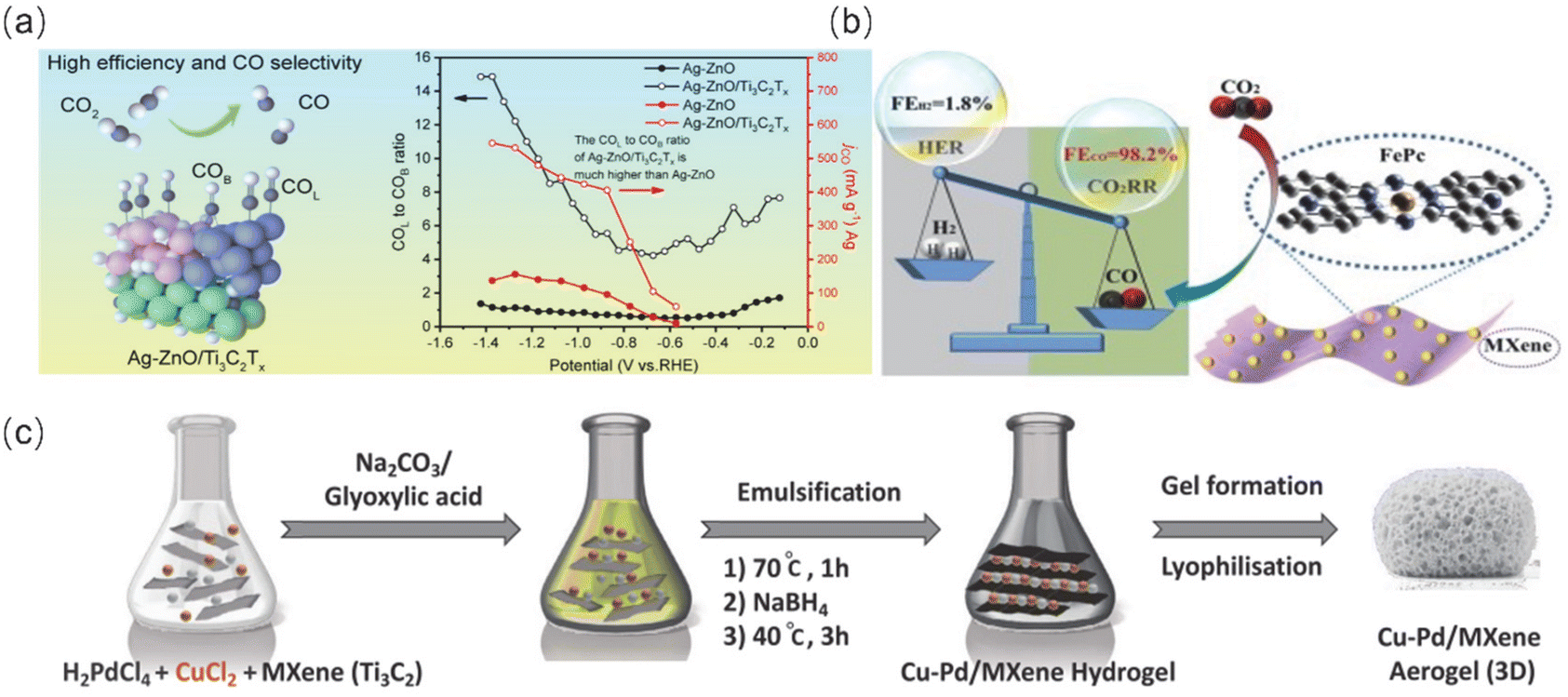 | ||
| Fig. 9 (a) Highly efficient electrocatalytic reduction of CO2 to CO achieved by Ag nanodomains loaded on ZnO porous nanobelts coupled with high-electronic-conductivity MXenes (Ag-ZnO/Ti3C2Tx). Reproduced from ref. 90 with permission from John Wiley & Sons, Inc., copyright 2023. (b) Schematic structure and Faraday efficiency of Fe-Pc/MXenes. Reproduced from ref. 91 with permission from Elsevier, copyright 2021. (c) Schematic of Cu–Pd/MXene preparation. Reproduced from ref. 89 with permission from John Wiley & Sons Inc., copyright 2023. | ||
Furthermore, covalent organic frameworks (COFs) are a type of organic functional material that can effectively adsorb CO2 and facilitate rapid proton transfer. This is due to their controllable active sites and large specific surface area. Additionally, the heterojunctions formed after complexation with MXenes provide more active sites and rapid ion transport channels, which can improve the selectivity of single-carbon products. For instance, MXenes@Por-COF-Co-7 was synthesized by utilizing exposed amine groups on triethoxysilane surface-modified Ti3C2 nanosheets as reactive sites to promote the in situ growth of imine COF (Por-COF) layer.92 This method achieved a CO FE of up to 97.28% at a voltage of −0.6 V (V vs. RHE). The successful functionalization of the amino group on the MXene nanosheets with the active center metal coordination promotes the transfer of more electrons along the porphyrin-benzene ring to the MXene surface. This improves the selective adsorption of key intermediates and the proton transfer process.
3.2 Inhibition of hydrogen production
The faradaic efficiency of the ECO2RR is lower due to a competing HER, as the product potential of ECO2RR has a more negative potential than the theoretical value. Additionally, the negative ΔGH indicates that the adsorption of H on the active site is very stable, which can hinder the occurrence of CO2RRs. To promote ECO2RRs, it is ideal to increase ΔGH at the active site. Therefore, increasing ΔGH at the active site is an ideal way to promote ECO2RR. Studies have confirmed that modulating suitable defects can limit the kinetic desorption process of hydrogen and exert an inhibitory effect on the HER process. Lv et al.93 employed a bismuth edge defect modulation strategy by coordinating the edge defects of bismuth (Bi) and sulfur to enhance the selectivity of ECO2RRs and inhibit the competing HER. This approach exhibited a high formic acid faradaic efficiency (∼95%). Meanwhile, the surface functional groups of MXenes can promote the ECO2RR kinetic process while inhibiting the HER. The presence of the –F end group can inhibit the HER kinetic process of Ti3C2Tx, resulting in decreased HER performance of the catalyst. Handoko et al.78 found that even with –OH and –O end groups, the competitive reaction with HER could not be avoided in the selective generation of CH4. However, a small amount of –F substitution could balance the individual limiting potentials of the PCET-1 and PCET-2 steps, resulting in a small over-potential in the generation of formic acid. Functionalized modification through the introduction of transition metal group elements is considered as an effective means of inhibiting HER. Additionally, Cu exhibited higher electrocatalytic activity for the CO2 → CH4 reaction process compared to Pt, Rh, Pd, Ni, Au, and Ag, with higher protonation of CO* and lower activity for HERs. For instance, Li et al. discovered that substituting TM in the Mo layer of Mo3C2 could effectively inhibit HER performance. Deng et al.94 constructed electrocatalysts with superhydrophobic surface microenvironments by one-step co-electrodeposition of Cu and polytetrafluoroethylene (PTFE) on carbon paper (CP), and the superhydrophobic Cu-based electrode exhibited high selectivity for ethylene (C2H4), with a faradaic efficiency of 67.3% at −1.25 V (V vs. RHE), effectively enhancing the ECO2RR activity and inhibiting the hydrogen precipitation reaction.4. Challenges and perspectives for ECO2RR
For MXene-based catalysts, the abundance of surface defects and end-groups can be both an advantage in facilitating the kinetic conversion process and a disadvantage in not being able to focus to enhance the highest selectivity. Unlike other two-dimensional materials, the simultaneous presence of –O end groups, –F end groups, and –OH end groups on the surface all have an impact on the faradaic efficiency of MXenes for single-carbon products, in which Ti3C2Tx showed high selectivity for CO, methanol, and ethanol, suggesting that the biggest challenge for MXene materials is still the selectivity for single carbon products, and it is not high enough to decide the control step of the desorption rate of key intermediates of single-carbon products, as shown in Fig. 10.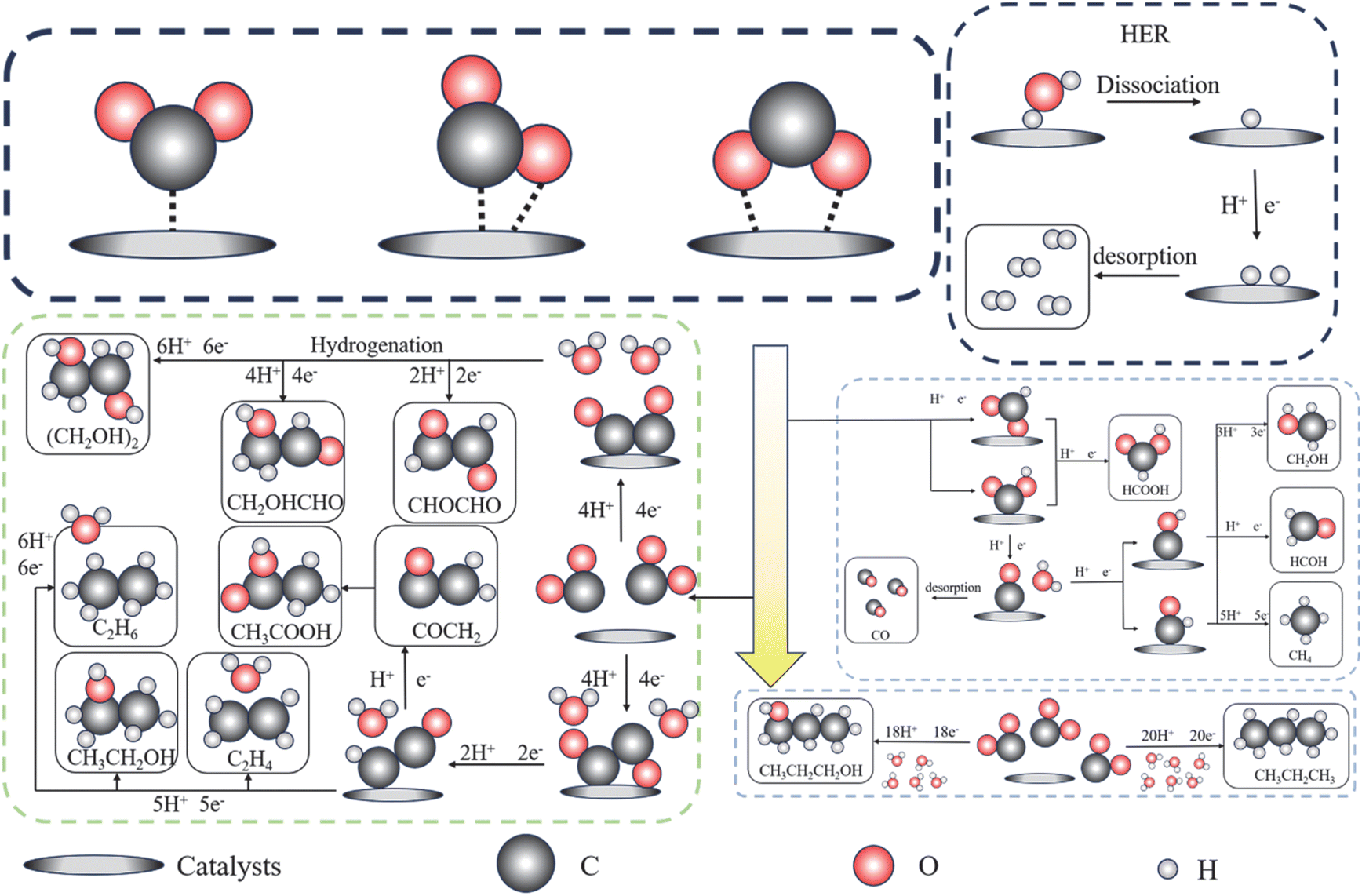 | ||
| Fig. 10 Reaction mechanism process diagram of electrocatalytic carbon dioxide reduction. | ||
4.1 C1 product pathway
Due to the uncertainty of the surface functionalization modification of MXenes due to their characteristics, it is necessary to select suitable objects in the large MXene family to verify their theoretical feasibility based on the construction of active sites affecting the rate-control step by MXenes themselves. For MXenes to achieve high selectivity for C1 products, tuning the adsorption strength for key intermediates while inhibiting the HER and constructing active sites with high selectivity properties for a particular intermediate are the keys to the development of ideal electrocatalysts for the reduction of CO2 to CO. CO, as the basis of high-value-added applications of CO2, involves in the electrocatalytic reduction process not only the selectivity for key intermediates but also important C–O, C–H and C–C coupling processes. In addition to the noble metal catalysts (Au, Ag, and Pd), Cu, Ni, and Zn are excellent for CO production via synergistic proton–electron-transfer (CO2 → *COOH → *CO) reactions with strong binding of the intermediate *COOH and weak binding of *CO.95,96 Unlike CO, the *HCOO pathway when forming COOH is the key intermediate, while *COH is the key intermediate shared by CH4 and CH3OH, the number of electrons gained in the rate-control step determines the selectivity of the products (CHO, CH4, and CH3OH). Ti3C2Tx exhibits selectivity for two key intermediates, COOH* and HCOOH*. Therefore, determining the reaction path by adjusting the adsorption strength of COOH* or HCOOH* is the most critical factor in achieving high product selectivity, and the surface functionalization modifies MXenes to achieve the high Faraday efficiency of the C1 product, the main way to achieve high Faraday efficiency, which summarizes a few ideas: (1) the use of intrinsic defects and heteroatom doping. For example, Chen,57 Handoko78et al. systematically verified that the intrinsic defects and end groups of MXenes can serve as the key factors in modulating C1 products. Meanwhile, heteroatom doping can effectively change the electronic structure of MXenes, increase their conductivity, and form an off-domain conjugated system, which, in turn, improves their performance in the electrocatalytic reduction of CO2. For example, Wang et al.97 used the controllability of nitrogen coordination to modulate the electronic structure of the active Ti-site, which led to a decrease in the free energy for the formation and desorption of *CO and *COOH intermediates and significantly improved the ECO2RR performance. In the study of other materials, N, S, and other hetero-elements have demonstrated the special role of the ECO2RR process, from the overall research, the heteroatom engineering on the basic application of MXenes in the study of ECO2RR is still in its infancy, and there exists a great development of the research space to form powerful descriptive symbols to describe the hetero-atoms, and intrinsic defects for the MXene ECO2RR process of kinetic influence process. (2) Metal loading and construction of heterojunctions. Metal substitution controls the ECO2RR performance by modulating the coordination environment of the metal atoms in the center of the catalyst. However, the loading of noble metals and transition metal group elements on MXenes has a large uncertainty on the ECO2RR process due to the quantum effect caused by the size feature and the unsaturated electronic state of the single atoms prone to causing higher hydrogen precipitation reactions. It has been confirmed that the heterostructures formed by MXenes with other metals or metal oxides can better solve the problem of HER competition reaction, in particular the heterojunctions formed by electron coupling with metal oxides not only exhibit excellent formic acid selectivity but also effectively inhibit the HER. Therefore, the materials constructing heterojunctions with MXenes not only satisfy the adsorption capacity of the key intermediates of the target product and lower hydrogen precipitation capacity but also further enhance the faradaic efficiency of the target product while lowering the required reaction potential.4.2 C2+ product pathway
Since the electrocatalytic reduction of CO2 to C2+ products undergoes a series of proton-assisted electron transfer processes involving a variety of reaction intermediates and transition states, it becomes a key issue in achieving efficient selectivity of C2+ products. Therefore, regulating the interaction of reaction intermediates with the catalytic site and the proton transfer process is an effective way to improve the selectivity of C2+ products. Kim et al.98 found that not only did CO* act as a key process in C–C coupling, but its coverage was also an important factor influencing the selectivity of the C2+ products by monitoring the key intermediates during the reduction process through ATR-SEIRAS. Bao et al.71 demonstrated that CO* is not only the key process in C–C coupling, but also that its coverage is an important factor affecting the selectivity of C2+ products. They used facile MXenes catalysts prepared by the wet chemical impregnation method, and achieved 71% ethylene Faraday efficiency (−0.7 V vs. RHE) under the atmosphere of CO. This demonstrates the excellent potential of MXenes to achieve C2+ products.99 In order to enhance the selectivity of MXene-based catalysts for producing C2+ products, it's essential to first improve the selectivity for CO* and also increase the coverage of CO* key intermediates at the catalytic active site. This can be accomplished by adjusting the desorption free energy of the CO* intermediates through appropriate surface interface modifications, and by ensuring a smooth C–C coupling step in the subsequent reaction pathway.5. Conclusions
The application of surface functionalization modification of MXenes in CO2 reduction has progressed very rapidly, and significant results have been achieved, opening up new pathways for CO2 capture and conversion. First, surface functionalization modification can significantly enhance the adsorption capacity of MXenes for CO2. By introducing specific functional groups, metal atoms, and other active sites on the surface of MXenes, the effective capture of CO2 molecules can be achieved, which can help improve the rate and efficiency of the CO2 reduction reaction. Second, the surface functionalization modification can regulate the electronic structure, surface active sites, and the stability of the reaction intermediates of MXenes to optimize their catalytic performance in the CO2 reduction reaction, which enables MXenes to realize the efficient conversion of CO2 under different conditions. In addition, by precisely controlling the type and concentration of modifiers via functionalization modification, the selectivity to single-carbon products is improved, while the stability and durability of the catalyst are enhanced to achieve efficient and sustainable conversion of CO2. Therefore, to improve the adsorption capacity, catalytic performance, and selectivity of MXenes to CO2 during the CO2 reduction reaction, more new and efficient modifiers can be explored for the future functionalized modification of MXenes to achieve a finer regulation of the surface properties of MXenes, and to improve the catalytic performance and selectivity of MXenes in the CO2 reduction reaction. Meanwhile, combining theoretical calculations and simulation methods, an in-depth understanding of the effects of surface functionalization modification on the electronic structure and catalytic performance of MXenes can provide strong guidance for the design of more efficient MXene-based catalysts and improve the application of MXene-based catalysts in the field of CO2 reduction and energy conversion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (62274018), the Xinjiang Production and Construction Corps Key Areas of Science and Technology Research Project (2023AB029), Xinjiang Production and Construction Corps Science and Technology Plan Project (2023CB008-06), and the new faculty startup funds from Shihezi University (2022ZK006).References
- Z. Cao, Y. Zhu, K. Chen, Q. Wang, Y. Li, X. Xing, J. Ru, L. Meng, J. Shu, N. Shpigel and L. Chen, Adv. Mater., 2024, e2401271 CrossRef PubMed.
- H. Xue, P. Huang, L. Lai, Y. Su, A. Strömberg, G. Cao, Y. Fan, S. Khartsev, M. Göthelid and Y. Sun, Carbon Energy, 2023, e442 Search PubMed.
- J. Chen, X. Liu, Z. Li, F. Cao, X. Lu and X. Fang, Adv. Funct. Mater., 2022, 32, 2201066 CrossRef CAS.
- W. Yu, Y. Yang, Y. Wang, L. Hu, J. Hao, L. Xu and W. Liu, Nano-Micro Lett., 2024, 16, 94 CrossRef CAS PubMed.
- J. Shen, Z. Wu, C. Li, C. Zhang, A. Genest and G. Rupprechter, FlatChem, 2021, 28, 100252 CrossRef CAS.
- J. Cai, J. Huang, A. Cao, Y. Wei, H. Wang, X. Li, Z. Jiang, G. I. N. Waterhouse, S. Lu and S. Q. Zang, Appl. Catal., B, 2023, 328, 122473 CrossRef CAS.
- R. Ramírez-Grau, M. Cabrero-Antonino, H. García and A. Primo, Appl. Catal., B, 2024, 341, 123316 CrossRef.
- Z. W. Seh, K. D. Fredrickson, B. Anasori, J. Kibsgaard, A. L. Strickler, M. R. Lukatskaya and Y. Gogotsi, ACS Energy Lett., 2016, 1, 589–594 CrossRef CAS.
- T. Amrillah, A. R. Supandi and V. Puspasari, Trans. Tianjin Univ., 2022, 28, 307–322 CrossRef CAS.
- Z. Li, N. H. Attanayake and J. L. Blackburn, Energy Environ. Sci., 2022, 15(4), 1696 RSC.
- Q. Tang, T. Li, W. Tu, H. Wang, Y. Zhou and Z. Zou, Adv. Funct. Mater., 2024, 2311609 CrossRef CAS.
- S. Cao, H. Chen and Y. Hu, Chem. Eng. J., 2023, 461, 141936 CrossRef CAS.
- E. S. Muckley, M. Naguib, H. Wang, L. Vlcek, N. C. Osti, R. L. Sacci, X. Sang, R. R. Unocic and Y. Xie, ACS Nano, 2017, 11, 11118–11126 CrossRef CAS PubMed.
- K. R. G. Lim, M. Shekhirev and B. C. Wyatt, Nat. Synth., 2022, 1(8), 601–614 CrossRef.
- G. Murali, J. K. Modigunta and Y. H. Park, ACS Nano, 2022, 16(9), 13370–13429 CrossRef CAS.
- X. Wang, X. Fan, M. Li, W. Zhu, J. Xue, F. Ye and L. Cheng, Ceram. Int., 2021, 47, 13628–13634 CrossRef CAS.
- T. L. Tan, H. M. Jin, M. B. Sullivan, B. Anasori and Y. Gogotsi, ACS Nano, 2017, 11, 4407–4418 CrossRef CAS PubMed.
- I. Hussain, U. Amara, F. Bibi, A. Hanan, M. N. Lakhan, I. A. Soomro, A. Khan, I. Shaheen and U. Sajjad, J. Colloid Interface Sci., 2024, 324, 103077 CrossRef CAS PubMed.
- L. Jia, S. Zhou, A. Ahmed, Z. Yang, S. Liu, H. Wang, F. Li, M. Zhang, Y. Zhang and L. Sun, Chem. Eng. J., 2023, 475, 146361 CrossRef CAS.
- E. Balcı, Ü. Ö. Akkuş and S. Berber, ACS Appl. Mater. Interfaces, 2019, 11, 3609–3616 CrossRef.
- Y. Zhang, D. Li, J. Li, Y. Li, L. Wang, H. Xu and W. Han, J. Colloid Interface Sci., 2024, 657, 550–558 CrossRef CAS.
- V. Sharma, S. Sardana, R. Dhiman and A. Mahajan, Appl. Phys. Lett., 2023, 122, 191601 CrossRef CAS.
- H. Chen, A. D. Handoko, J. Xiao, X. Feng, Y. Fan, T. Wang, D. Legut, Z. W. Seh and Q. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 36571–36579 CrossRef CAS PubMed.
- D. Gao, Y. Xu, Z. Liu, Y. Yu, C. Yu, Y. Fang, Y. Huang, C. Tang and Z. Guo, Appl. Surf. Sci., 2024, 654, 159501 CrossRef CAS.
- K. Huang, P. Qu, Y. Wang, C. Lian, J. Li, H. Su and H. Liu, Ind. Eng. Chem. Res., 2023, 62, 20716–20726 CrossRef CAS.
- Y. Hao, F. Hu, S. Zhu, Y. Sun, H. Wang, L. Wang, Y. Wang, J. Xue, Y. Liao, M. Shao and S. Peng, Angew. Chem., Int. Ed., 2023, 62, e202304179 CrossRef CAS PubMed.
- J. Peng, Z. Zhang, H. Wang, P. Zhang, X. Zhao, Y. Jia, Y. Yue and N. Li, Small, 2023, 2308528 Search PubMed.
- D. Qu, X. Peng, Y. Mi, H. Bao, S. Zhao and X. Liu, Nanoscale, 2020, 12(33), 17191–17195 RSC.
- B. M. Abraham, O. Piqué, M. A. Khan, F. Viñes, F. Illas and J. K. Singh, ACS Appl. Mater. Interfaces, 2023, 15, 30117–30126 CrossRef CAS PubMed.
- T. A. Le, Q. V. Bui, N. Q. Tran, Y. Cho, Y. Hong, Y. Kawazoe and H. Lee, ACS Sustainable Chem. Eng., 2019, 7, 16879–16888 CrossRef CAS.
- X. Zhang, Z. Zhang, J. Li, X. Zhao, D. Wu and Z. Zhou, J. Mater. Chem. A, 2017, 5, 12899–12903 RSC.
- D. Zhao, Z. Chen, W. Yang, S. Liu, X. Zhang, Y. Yu, W.C. Cheong, L. Zheng, F. Ren, G. Ying, X. Cao, D. Wang, Q. Peng, G. X. Wang and C. Chen, J. Am. Chem. Soc., 2019, 141, 4086–4093 CrossRef CAS PubMed.
- F. Q. Liu, X. Liu, L. Sun, R. Li, C. X. Yin and B. Wu, J. Mater. Chem. A, 2021, 9, 12763–12771 RSC.
- H. Zhou, Z. Chen, A. V. López, E. D. López, E. Lam, A. Tsoukalou, E. Willinger, D. A. Kuznetsov, D. Mance, A. Kierzkowska, F. Donat, P. M. Abdala, A. Comas-Vives, C. Copéret, A. Fedorov and C. R. Müller, Nat. Catal., 2021, 4, 860–871 CrossRef CAS.
- K. Niu, L. Chen, J. Rosen and J. Björk, ACS Catal., 2024, 14, 1824–1833 CrossRef CAS.
- G. R. Weal, K. I. Guðmundsson, F. D. Mackenzie, J. R. Whiting, N. B. Smith, E. Skúlason and A. L. Garden, Nanoscale, 2024, 16, 5242–5256 RSC.
- Y. Lu, D. Fan, Z. Chen, W. Xiao, C. Cao and X. Yang, Sci. Bull., 2020, 65, 460–466 CrossRef CAS.
- H. Chen, A. D. Handoko, T. Wang, J. Qu, J. Xiao, X. Liu, D. Legut, Z. W. Seh and Q. Zhang, ChemSusChem, 2020, 13, 5690–5698 CrossRef CAS PubMed.
- S. Cao, Y. Liu, Y. Hu, J. Li, C. Yang, Z. Chen, Z. Wang, S. Wei, S. Liu and X. Lu, J. Colloid Interface Sci., 2023, 642, 273–282 CrossRef CAS PubMed.
- Y. Zou, S. A. Kazemi, G. Shi, J. Liu, Y. Yang, N. M. Bedford, K. Fan, Y. Xu, H. Fu and M. Dong, EcoMat, 2023, 5(1), e12274 CrossRef CAS.
- S. Zhou, Y. Zhao, R. Shi, Y. Wang, A. Ashok, F. Héraly, T. Zhang and J. Yuan, Adv. Mater., 2022, 34(36), 2204388 CrossRef CAS PubMed.
- H. Gu, W. Yue, J. Hu, X. Niu, H. Tang, F. Qin, Y. Li, Q. Yan, X. Liu, W. Xu, Z. Sun, Q. Liu and W. Yan, Adv. Energy Mater., 2023, 13(20), 2204014 CrossRef CAS.
- Z. Liu, Y. Liu, J. Zhang, T. Cao, Z. Sun, J. Liu and H. Shang, Nano Res., 2024, 17, 3911–3918 CrossRef CAS.
- V. Ramalingam, P. Varadhan, H. C. Fu, H. Kim, D. Zhang, S. Chen, L. Song, D. Ma and Y. Wang, Adv. Mater., 2019, 31(48), 1903841 CrossRef CAS PubMed.
- W. Lin, Y. R. Lu, W. Peng, M. Luo, T. S. Chan and Y. Tan, J. Mater. Chem. A, 2022, 10(18), 9878–9885 RSC.
- L. Liu, S. Zheng, H. Chen, J. Cai and S. Zang, Angew. Chem., Int. Ed., 2024, 63(8), e202316910 CrossRef CAS PubMed.
- Z. Wu, J. Shen, C. Li, C. Zhang, K. Feng, Z. Wang, X. Wang, D. M. Meira, M. Cai, D. Zhang and S. Wang, ACS Nano, 2022, 17(2), 1550–1559 CrossRef.
- L. Dai, Y. Shen, J. Chen, L. Zhou, X. Wu, Z. Li, J. Wang, W. Huang, J. T. Miller, Q. Wang and A. Cao, Small, 2022, 18(14), 2105226 CrossRef CAS PubMed.
- P. Jin, P. Han, X. Li and K. Li, Appl. Surf. Sci., 2023, 612, 155850 CrossRef CAS.
- Z. Zou, Q. Wang, J. Yan, K. Zhu, K. Ye, G. Wang and D. Cao, ACS Nano, 2021, 15(7), 12140–12150 CrossRef CAS PubMed.
- T. Yun, G. S. Lee, J. Choi, H. Kim, G. Yang, H. J. Lee, J. G. Kim, H. M. Lee, C. M. Koo, J. Lim and S. O. Kim, ACS Nano, 2021, 15(6), 10058–10066 CrossRef CAS PubMed.
- H. Li, R. Chen, M. Ali, H. Lee and M. J. Ko, Adv. Funct. Mater., 2020, 30(47), 2002739 CrossRef CAS.
- Z. Xia, X. Chen, H. Ci, Z. Fan, Y. Yi, W. Yin and N. Wei, J. Energy Chem., 2021, 53, 155–162 CrossRef CAS.
- P. Saha, S. Amanullah and A. Dey, Acc. Chem. Res., 2022, 55(2), 134–144 CrossRef CAS PubMed.
- S. Bai, M. Yang, J. Jiang, X. He, J. Zou and Z. Xiong, npj 2D Mater. Appl., 2021, 5(1), 78 CrossRef CAS.
- Z. Kang, M. A. Khan, Y. Gong, R. Javed, Y. Xu, D. Ye, H. Zhao and J. Zhang, J. Mater. Chem. A, 2021, 9(10), 6089–6108 RSC.
- H. Chen, A. D. Handoko, T. Wang, J. Qu, J. Xiao, X. Liu, D. Legut, Z. Wei Seh and Q. Zhang, ChemSusChem, 2020, 13(21), 5690–5698 CrossRef CAS PubMed.
- C. S. Chen, A. D. Handoko, J. H. Wan, L. Ma, D. Ren and B. S. Yeo, Catal. Sci. Technol., 2015, 5(1), 161–168 RSC.
- H. S. Jeon, S. Kunze, F. R. Scholten and B. Cuenya, ACS Catal., 2017, 8(1), 531–535 CrossRef.
- M. Ma, K. Djanashvili and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55(23), 6680–6684 CrossRef CAS.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55(19), 5789–5792 CrossRef CAS PubMed.
- T. Hatsukade, Y. G. Kim, A. Vailionis, J. H. Baricuatro, D. C. Higgins, S. A. Nitopi, M. P. Soriaga and T. F. Jaramillo, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(23), 5918–5923 CrossRef PubMed.
- Y. Ren, A. D. Deng, C. S. Handoko, S. M. Chen and B. S. Yeo, ACS Catal., 2015, 5, 2814–2821 CrossRef.
- Z. Gu, N. Yang, P. Han, M. Kuang, B. Mei, Z. Jiang, J. Zhong, L. Li and G. Zheng, Small Methods, 2018, 3(2), 1800449 CrossRef.
- H. Mistry, A. S. Varela, C. S. Bonifacio, I. Zegkinoglou, I. Sinev, Y. W. Choi, K. Kisslinger, E. A. Stach, J. C. Yang, P. Strasser and B. R. Cuenya, Nat. Commun., 2016, 7, 12123 CrossRef PubMed.
- Z. Chang, S. Huo, W. Zhang, J. Fang and H. Wang, J. Phys. Chem. C, 2017, 121(21), 11368–11379 CrossRef CAS.
- T. T. H. Hoang, S. Verma, S. Ma, T. T. Fister, J. Timoshenko, A. I. Frenkel, P. J. A. Kenis and A. A. Gewirth, J. Am. Chem. Soc., 2018, 140(17), 5791–5797 CrossRef CAS PubMed.
- S. Ishimaru, R. Shiratsuchi and G. Nogami, J. Electrochem. Soc., 2000, 147(5), 1864–1867 CrossRef CAS.
- D. Ren, B. S. H. Ang and B. S. Yeo, ACS Catal., 2016, 6(12), 8239–8247 CrossRef CAS.
- Q. Zhao, C. Zhang, R. Hu, Z. Du, J. Gu, Y. Cui, X. Chen, W. Xu, Z. Cheng, S. Li, B. Li, Y. Liu, W. Chen, C. Liu, J. Shang, L. Song and S. Yang, ACS Nano, 2021, 15(3), 4927–4936 CrossRef CAS PubMed.
- H. Bao, Y. Qiu, X. Peng, J. A. Wang, Y. Mi, S. Zhao, X. Liu, Y. Liu, R. Cao, L. Zhuo, J. Ren, J. Sun, J. Luo and X. Sun, Nat. Commun., 2021, 12(1), 238 CrossRef CAS PubMed.
- M. Abdinejad, S. Subramanian, M. K. Motlagh, M. Noroozifar, S. Duangdangchote, I. Neporozhnii, D. Ripepi, D. Pinto, M. Li, K. Tang, J. Middelkoop, A. Urakawa, O. Voznyy, H. B. Kraatz and T. Burdyny, Adv. Energy Mater., 2023, 13(19), 2300402 CrossRef CAS.
- L. Zhou, Q. Tian, X. Shang, Y. Zhao, W. Yao, H. Liu and Q. Xu, Green Chem., 2024, 26(3), 1454–1461 RSC.
- M. Gao, Y. Sun, K. Zhao, M. Zhang, X. Wang and W. Wang, J. Environ. Chem. Eng., 2024, 12(1), 111802 CrossRef CAS.
- D. Qu, X. Peng, Y. Mi, H. Bao, S. Zhao, X. Liu and J. Luo, Nanoscale, 2020, 12(33), 17191–17195 RSC.
- L. Han, X. Peng, H. T. Wang, P. Ou, Y. Mi, C. W. Pao, J. Zhou, J. Wang, X. Liu, W. F. Pong and J. Song, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(42), e2207326119 CrossRef CAS PubMed.
- Y. Hao, F. Hu, S. Zhu, Y. Sun, H. Wang, L. Wang, Y. Wang, J. Xue, Y. F. Liao, M. Shao and S. Peng, Angew. Chem., Int. Ed., 2023, 62(35), e202304179 CrossRef CAS PubMed.
- A. D. Handoko, H. Chen, Y. Lum, Q. Zhang, B. Anasori and Z. W. Seh, iScience, 2020, 23(6), 101181 CrossRef CAS.
- L. Meng, L.K. Yan, F. Viñes and F. Illas, J. Mater. Chem. A, 2024, 12, 7856–7874 RSC.
- Y. Li, Y. Chen, Z. Guo, C. Tang, B. Sa, N. Miao, J. Zhou and Z. Sun, Chem. Eng. J., 2022, 429, 132171 CrossRef CAS.
- Z. Luo, G. Zhao, H. Pan and W. Sun, Adv. Energy Mater., 2022, 12(37), 2201395 CrossRef CAS.
- D. Zhao, Z. Chen, W. Yang, S. Liu, X. Zhang, Y. Yu, W. C. Cheong, L. Zheng, F. Ren, G. Ying and X. Cao, J. Am. Chem. Soc., 2019, 141(9), 4086–4093 CrossRef CAS.
- N. Li, J. Peng, Z. Shi, P. Zhang and X. Li, Chin. J. Catal., 2022, 43(7), 1906–1917 CrossRef CAS.
- K. Niu, L. Chen, J. Rosen and J. Björk, ACS Catal., 2024, 14, 1824–1833 CrossRef CAS.
- Q. Zhao, C. Zhang, R. Hu, Z. Du, J. Gu, Y. Cui, X. Chen, W. Xu, Z. Cheng, S. Li, B. Li, Y. Liu and W. Chen, ACS Nano, 2021, 15(3), 4927–4936 CrossRef CAS PubMed.
- K. Eid, Q. Lu, S. Abdel-Azeim, A. Soliman, A. M. Abdullah, A. M. Abdelgwad and R. P. Forbes, J. Mater. Chem. A, 2022, 10(4), 1965–1975 RSC.
- J. Ma, Q. Jiang, Y. Zhou, W. Chu, S. Perathoner, C. Jiang, K. H. Wu, G. Centi and Y. Liu, Small, 2021, 17(26), 2007509 CrossRef CAS.
- B. Ren, G. Wen, R. Gao, D. Luo, Z. Zhang, W. Qiu, Q. Ma, X. Wang, Y. Cui and L. Ricardez-Sandoval, Nat. Commun., 2022, 13(1), 2486 CrossRef CAS PubMed.
- M. Abdinejad, S. Subramanian, M. K. Motlagh, M. Noroozifar, S. Duangdangchote and I. Neporozhnii, Adv. Energy Mater., 2023, 13(19), 2300402 CrossRef CAS.
- Y. Hao, F. Hu, S. Zhu, Y. Sun, H. Wang, L. Wang, Y. Wang, J. Xue, Y. Liao, M. Shao and S. Peng, Angew. Chem., 2023, 135(35), e202304179 CrossRef.
- L. Zhou, Q. Tian, X. Shang, Y. Zhao, W. Yao, H. Liu and Q. Xu, Green Chem., 2024, 26(3), 1454–1461 RSC.
- M. Gao, Y. Sun, K. Zhao, M. Zhang, X. Wang and W. Wang, J. Environ. Chem. Eng., 2024, 12(1), 111802 CrossRef CAS.
- L. Lv, R. Lu, J. Zhu, R. Yu, W. Zhang, E. Cui, X. Chen, Y. Dai, L. Cui, J. Li, L. Zhou, W. Chen and Z. Wang, Angew. Chem., 2023, 135(25), e202303117 CrossRef.
- T. Deng, S. Jia, C. Chen, J. Jiao, X. Chen, C. Xue, W. Xia, X. Xing, Q. Zhu, H. Wu, M. He and B. Han, Angew. Chem., 2024, 136(2), e202313796 CrossRef.
- F. Franco, C. Rettenmaier, H. S. Jeon and B. R. Cuenya, Chem. Soc. Rev., 2020, 49(19), 6884–6946 RSC.
- F. Lv, N. Han, Y. Qiu, X. Liu, J. Luo and Y. Li, Coord. Chem. Rev., 2020, 422, 213435 CrossRef CAS.
- Z. Wang, C. Wang, Y. Hu, S. Yang, J. Yang, W. Chen, H. Zhou, F. Zhou, L. Wang, J. Du, Y. Li and Y. Wu, Nano Res., 2021, 14, 2790–2796 CrossRef CAS.
- Y. Kim, S. Park, S. J. Shin, W. Choi, B. K. Min, H. Kim, W. Kim and Y. J. Hwang, Energy Environ. Sci., 2020, 13(11), 4301–4311 RSC.
- W. Quan, Y. Lin, Y. Luo and Y. Huang, Adv. Sci., 2021, 8(23), 2101597 CrossRef CAS PubMed.
Footnote |
| † These authors are co-first authors and contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |