
Universal pH electrocatalytic hydrogen evolution with Au-based high entropy alloys†
Sangmin
Jeong
 ,
Anthony J.
Branco
,
Silas W.
Bollen
,
Connor S.
Sullivan
and
Michael B.
Ross
*
,
Anthony J.
Branco
,
Silas W.
Bollen
,
Connor S.
Sullivan
and
Michael B.
Ross
*
Department of Chemistry, University of Massachusetts Lowell, Lowell, MA 01854, USA. E-mail: michael_ross@uml.edu
First published on 4th June 2024
Abstract
The creation of electrocatalysts with reduced concentrations of platinum-group metals remains a critical challenge for electrochemical hydrogen production. High-entropy alloys (HEAs) offer a distinct type of catalyst with tunable compositions and engineered surface activity, significantly enhancing the hydrogen evolution reaction (HER). We present the synthesis of AuPdFeNiCo HEA nanoparticles (NPs) using a wet impregnation method. The composition and structure of the AuPdFeNiCo HEA NPs are characterized by X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), and high-resolution transmission electron microscopy (HR-TEM). These nanoparticles exhibit robust HER performance quantified over a broad pH range, with higher activity than any of the unary metal counterparts in all pHs. In comparison to a commercial 10%Pt/C electrocatalyst, AuPdFeNiCo HEA NPs exhibit enhanced electrochemical activity in both acidic and alkaline electrolytes at a current density of 10 mA cm−2. Additionally, these nanoparticles achieve a current density of 100 mA cm−2 at a voltage of 540 mV in neutral electrolytes, outperforming Pt/C which requires 570 mV. These findings help enable broad use of reduced precious metal electrocatalysts for water electrolysis in a variety of water and pH conditions.
Introduction
Nanoscale electrocatalysts are critical materials for efficient electrochemical water splitting that can provide carbon-free generation of fuels and energy.1–3 Presently, platinum-group metals (PGMs) exhibit the highest efficiency in water splitting.4–8 Nonetheless, their commercialization and growth are impeded by cost and durability concerns.3,9 Therefore, to establish cost-effective electrolysis systems, developing highly active electrocatalysts with reduced noble metal loadings is imperative, such as the alloy structure strategy recently reported in studies.10,11 In the realm of water splitting, optimizing the interplay between electrocatalysts and electrolytes remains pivotal to enhancing overall catalytic activity.12,13 While significant breakthroughs have propelled innovations in acidic and alkaline electrolytes, understanding of the water-splitting in neutral to near-neutral electrolytes remains comparatively nascent.1 It also remains challenging to identify electrocatalysts that will operate well over a range of pHs.The use of Au in hydrogen electrocatalysis research is limited in part due to its low intrinsic hydrogen evolution activity.14 However, Au generally has high quality electron transport and is oxidatively and structurally stable, making it an attractive component for pH-versatile electrocatalysts.15–18 Several studies have reported the use of Au as an electrocatalyst in universal-pH electrolytes, encompassing not only well-known acidic and alkaline conditions but also neutral electrolytes.19,20 These distinct attributes have driven scientific exploration in this field, resulting in reported research on various compositions involving Au–Pd, Au–Ni, Au–Cu and others.15,19,21–23 However, the extent of modification in the electronic properties is confined by the range of compositional tunability, limiting the performance.24
High-entropy alloys (HEAs) are compositionally complex solid solutions that contain five or more elements; they provide an attractive approach to controlling composition in electrocatalysis.25–29 Specifically, the compositional complexity of HEAs, resulting from the random distribution of atoms with different radii and its irregular surface which closely mimics an almost continuous distribution, differentiates it from binary/ternary alloys. As such, HEAs provide a wide diversity of electronic and physical structures for electrochemical reactions.30–32 Furthermore, adding more components to HEAs, compared to binary alloy catalysts, significantly reduces the use of noble metals by dilution. In principle, these unique attributes of HEAs have an opportunity to enhance the efficacy of electrochemical hydrogen evolution reaction (HER) over a broad pH range due to their durability and versatile morphologies.
Herein, we report the synthesis of AuPdFeNiCo HEA nanoparticles (NPs) using a wet chemical impregnation method. Five elements were chosen for alloying based on their known properties in the unary phase: Au and Pd provide desirable mechanical properties, thermal stability, and compatibility in alloy formation while the 3d metals Fe, Ni, and Co were chosen due to their abundance and their reported desirable HER activity.33,34 The as-prepared AuPdFeNiCo HEA NPs have enhanced electrochemical HER activity in acidic, neutral, and basic electrolytes, 0.5 M H2SO4, 1 M PBS, and 1 M KOH, respectively. Particularly, HEA NPs have favorable overpotential values of 45 (acidic) and 43 (alkaline) mV vs. reversible hydrogen electrode (RHE) at 10 mA cm−2; these compare favorably to those measured for commercial Pt/C 10%, 53 and 43 mV respectively. Additionally, in neutral electrolytes, the AuPdFeNiCo HEA NPs (132 mV dec−1) demonstrated faster electron transfer capability than Pt (150 mV dec−1), suggesting desirable electrochemical properties for hydrogen evolution in the as-synthesized AuPdFeNiCo HEA NPs. This work provides a comprehensive overview of the design of Au-based HEA NPs and quantification of their potential at pH-versatile HER electrocatalysts.
Experimental
Nanoparticle synthesis
AuPdFeNiCo HEA NPs were synthesized from Au, Pd, Fe, Ni, and Co precursor salts via a wet-chemical process. The precursors were prepared by dissolving 0.5 mmol of AuCl3 (99.99%, Alfa Aesar), 0.5 mmol of PdCl2 (99.99%, Acros Organics), 5 mmol of FeCl2H8O4 (99%, Acros Organics), 5 mmol of NiCl2 (99.995%, Alfa Aesar), and 5 mmol of CoCl2H12O6 (99.998%, Alfa Aesar) in 40 mL ultrapure water. Then, 0.1 g of poly(N-vinyl-2-pyrrolidone) (PVP, M.W. 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Alfa Aesar) powder was added to the metal solution to prevent aggregation. The temperature of the mixture solution was maintained at 230 °C under reflux for 1hr. After that, the temperature was allowed to cool to room temperature. The black powder product was purified by centrifugation and resuspension in a mixture of ethanol and water (equal ratio). Finally, the AuPdFeNiCo powder was collected after centrifugation and dried under vacuum.
000, Alfa Aesar) powder was added to the metal solution to prevent aggregation. The temperature of the mixture solution was maintained at 230 °C under reflux for 1hr. After that, the temperature was allowed to cool to room temperature. The black powder product was purified by centrifugation and resuspension in a mixture of ethanol and water (equal ratio). Finally, the AuPdFeNiCo powder was collected after centrifugation and dried under vacuum.
Characterization of materials
The morphology and structural analysis were recorded on a high-resolution/scanning transmission electron microscope (HR-STEM, JEOL, JEM-2100Plus, 200 kV) and energy dispersive X-ray spectroscopy (EDS) with elemental mapping feature. The crystal phase and structure were characterized by X-ray diffraction (XRD, Rigaku Miniflex X-ray diffractometer, CuKα, (λ = 1.5406 Å)) and synchrotron wide-angle X-ray scattering (WAXS, NSLS-II, λ = 0.1665 Å) measurements. X-ray photoelectron spectroscopy (XPS) was performed on a PHI Versaprobe II using an Al Kα X-ray radiation, and XPS peaks were calibrated to the C 1s peak at 284.8 eV. Inductively coupled plasma with an optical emission spectrometer (ICP-OES) was carried out on an Agilent 5110 using Agilent ICP Expert software.Electrochemical hydrogen evolution reaction measurements
All electrochemical measurements were performed at ambient temperature (∼25 °C) using an electrochemical potentiostat (Pine research, Wavedriver 100) connected to a commercial three-electrode system in various electrolytes: 0.5 M H2SO4, 1 M PBS, and 1 M KOH. A Pt mesh and silver/silver chloride electrode (Ag/AgCl) were used as counter and reference electrodes, respectively. For preparing the working electrode, an electrocatalyst ink was prepared by sonicating a mixture solution containing 1 mg of AuPdFeNiCo nanoparticles, 3 mg of vulcanized carbon (Carbon Black, XC-72, Fuel Cell Earth), 50 μL Nafion (5 wt% of dispersion, Thermo Scientific), and 300 μL isopropyl alcohol (IPA, Ricca). Then, 10 μL of ink solution was drop cast onto a glassy carbon electrode (active area 0.07 cm2). To compare the HER activity of the prepared working electrode, the commercial Pt/C (10%) and various bulk single metal foils such as Au (25 μm, 99.95%, Sigma-Aldrich), Pd (25 μm, 99.9%, Thermo Scientific), Fe (100 μm, 99.995%, Thermo Scientific), Ni (100 μm, 99.994%, Thermo Scientific), and Co (100 μm, 99.995%, Alfa Aesar) were respectively prepared for individual evaluation. All the potentials vs. Ag/AgCl reference values were converted to the reversible hydrogen electrode (RHE) using the Nernst equation as below:| ERHE = EAg/AgCl + (0.0591 × pH) + E0Ag/AgCl |
where ja and jc are the anodic and cathodic current densities, respectively, and v is the scan rate in mV s−1 from CV. EIS measurements were performed using a Nyquist plot in the range of 100 kHz to 0.1 Hz at 10 mA cm−2. The experimental data were fitted with the Pine-view software and Origin software using appropriate equivalent circuits.

Results and discussion
AuPdFeNiCo HEA nanoparticles (NPs) were synthesized via a wet-chemical reaction, where five metal chloride precursors and poly(N-vinyl-2-pyrrolidone) (PVP, Mw ≈ 40000) were mixed in ultrapure water at 230 °C (Scheme 1, see details in Experimental section). PVP facilitates the formation of metal alloys by augmenting stability and preventing aggregation.28,35
 | ||
| Scheme 1 The synthesis method for AuPdFeNiCo HEA nanoparticles. | ||
Morphological, structural, and compositional analysis of AuPdFeNiCo HEA NPs was conducted using high-resolution transmission electron microscopy (HR-TEM), scanning transmission electron microscopy (STEM) with energy dispersive spectroscopy (EDS) mapping, and powder X-ray diffraction (XRD). Examination of STEM and HR-TEM images revealed an average particle size of ∼13.45 nm (Fig. S1†). Higher magnification imaging reveals a lattice spacing of 2.40 Å (Fig. 1 and Fig. S1†). This spacing corresponds to the (111) plane, as corroborated by fast Fourier transformation (FFT) (Fig. 1b) and XRD patterns (Fig. 2) associated with HEA NPs. Additionally, the crystalline structure of AuPdFeNiCo HEA NPs exhibits distinct diffraction peaks corresponding to the (111), (200), and (220) facets. Fig. 1c–h and Fig. S3† show the EDS mapping and line scan profiles of the AuPdFeNiCo HEA NPs, demonstrating a uniform dispersion of the five elements. One noteworthy aspect is the formation of a seemingly Fe-rich shell (Fig. S2 and S3†). This was verified through the edge FFT and the distribution pattern observed in the low-magnification EDS Mapping. Fig. S2† shows the HR-TEM image of AuPdFeNiCo HEA NPs and Fe NPs with an interplanar spacing of 2.4 Å and 2.57 Å, including different FFT images in the inserted images, respectively. These images appear to depict iron or iron oxide nanoparticles formed either as side products or as nuclei during the reaction. Additionally, to mitigate potential nanoparticle damage from prolonged exposure to the electron beam during mapping characterization, the mapping duration was restricted to less than 10 min.36
 | ||
| Fig. 1 HR-TEM and STEM-EDS characterization of AuPdFeNiCo HEA NPs. (a) Representative HR-TEM image of AuPdFeNiCo HEA NPs and (b) zoomed in view of the HR-TEM image with the insert shows the fast Fourier transformation (FFT). (c) STEM image of the corresponding energy dispersive spectroscopy (EDS) elemental mapping of (d) Au, (e) Pd, (f) Fe, (g) Ni, and (h) Co. | ||
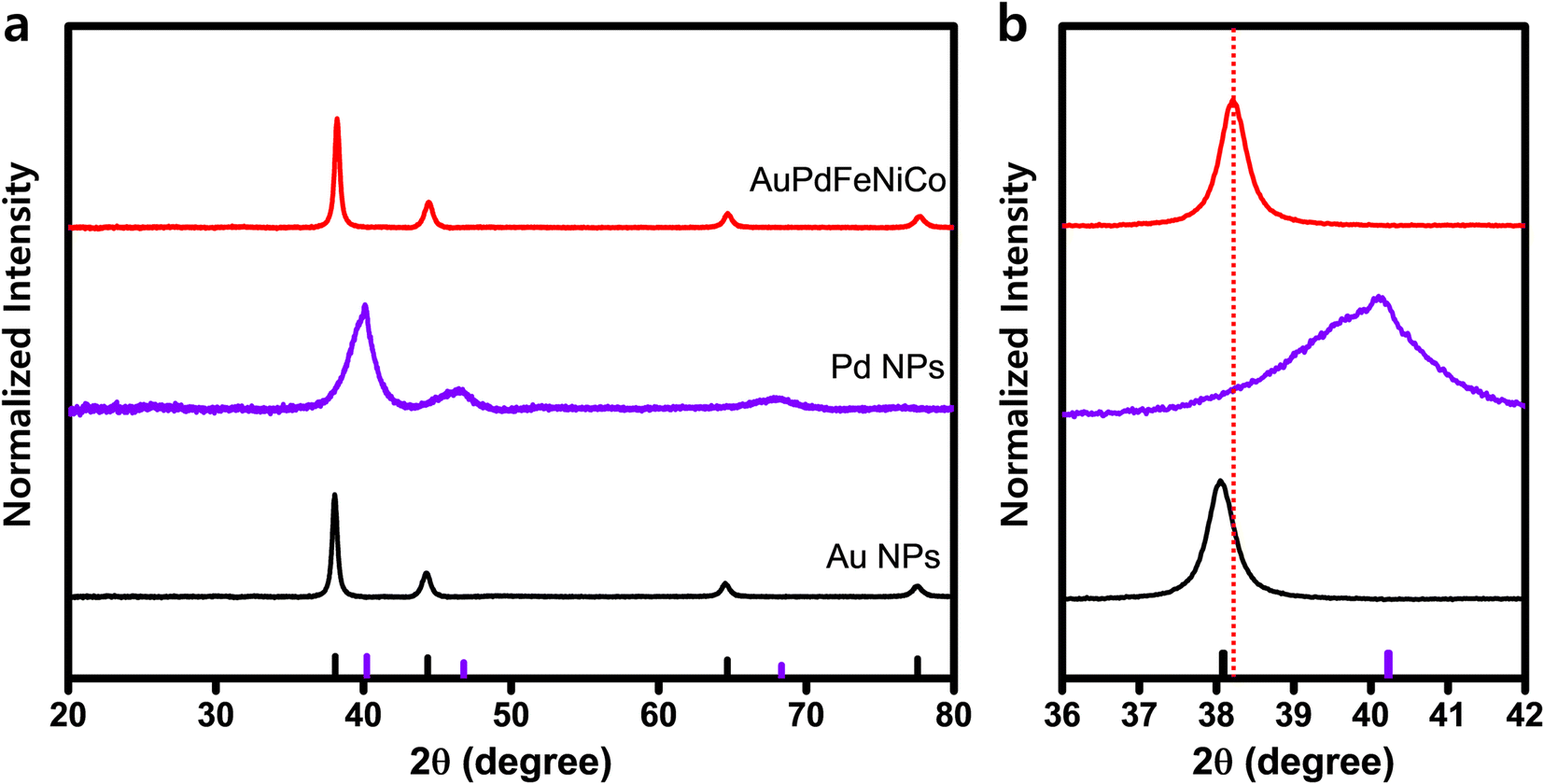 | ||
| Fig. 2 X-ray diffraction (XRD) characterization of AuPdFeNiCo HEA NPs. (a) XRD patterns of Au NPs, Pd NPs and AuPdFeNiCo HEA NPs with JCPDS data (Au: 04–0748, Pd: 46–1043), (b) XRD patterns of (111) fcc peak. | ||
XRD patterns show that the AuPdFeNiCo HEA NPs exhibit a fcc structure, with a lattice constant of 4.15 Å (Fig. 2). For comparison, Fig. 2 shows the XRD patterns of Au and Pd NPs, exhibiting clearly defined crystalline phases of the (111), (200), and (220) planes. In contrast, the diffraction pattern of AuPdFeNiCo HEA NPs is positioned between that of Au NPs and Pd NPs, indicating crystalline phases at 38.3°, 44.5°, and 64.9°, which corresponds to the (111), (200), and (220) planes, respectively. Moreover, we have validated the long-range order of the AuPdFeNiCo HEA NPs using high-resolution synchrotron wide-angle X-ray scattering (WAXS), which showcases analogous results to XRD but with additional high quality diffraction orders (Fig. S4†).
To understand the electronic structure and chemical state of the AuPdFeNiCo HEA NPs, X-ray photoelectron spectroscopy (XPS) was performed. Fig. 3a shows XPS survey spectra for AuPdFeNiCo HEA NPs in the range of 0–1100 eV, revealing the presence of each constituent element state. A high resolution scan of the Au 4f region (Fig. 3b) shows well-resolved peaks with binding energies (BEs) of 83.8 eV and 87.4 eV, which can be assigned to metallic gold (Au0 4f7/2 and Au0 4f5/2). Similarly, the corresponding Pd 3d peaks were located at 335.2 and 340.5 eV, which can be assigned to metallic palladium (Pd0 3d5/2 and Pd0 3d3/2) (Fig. 3c).21,37,38Fig. 3d shows the resolved peaks in the Fe 2p region with BEs of 707.3 eV, 710.6 eV, and 713.2 eV, which correspond with metallic iron, Fe2+, and Fe3+.34,39,40 In the Co 2p region (Fig. 3e), the spectrum has a broad feature that can be deconvoluted into peaks at 780.7 eV (Co°) and 784.0 eV (Co2+).34,41 For the Ni 2p region (Fig. 3f), the presence of metallic nickel and nickel oxides as Ni0 and Ni2+ are attributed to BEs at 854.5 eV and 856.7 eV.27
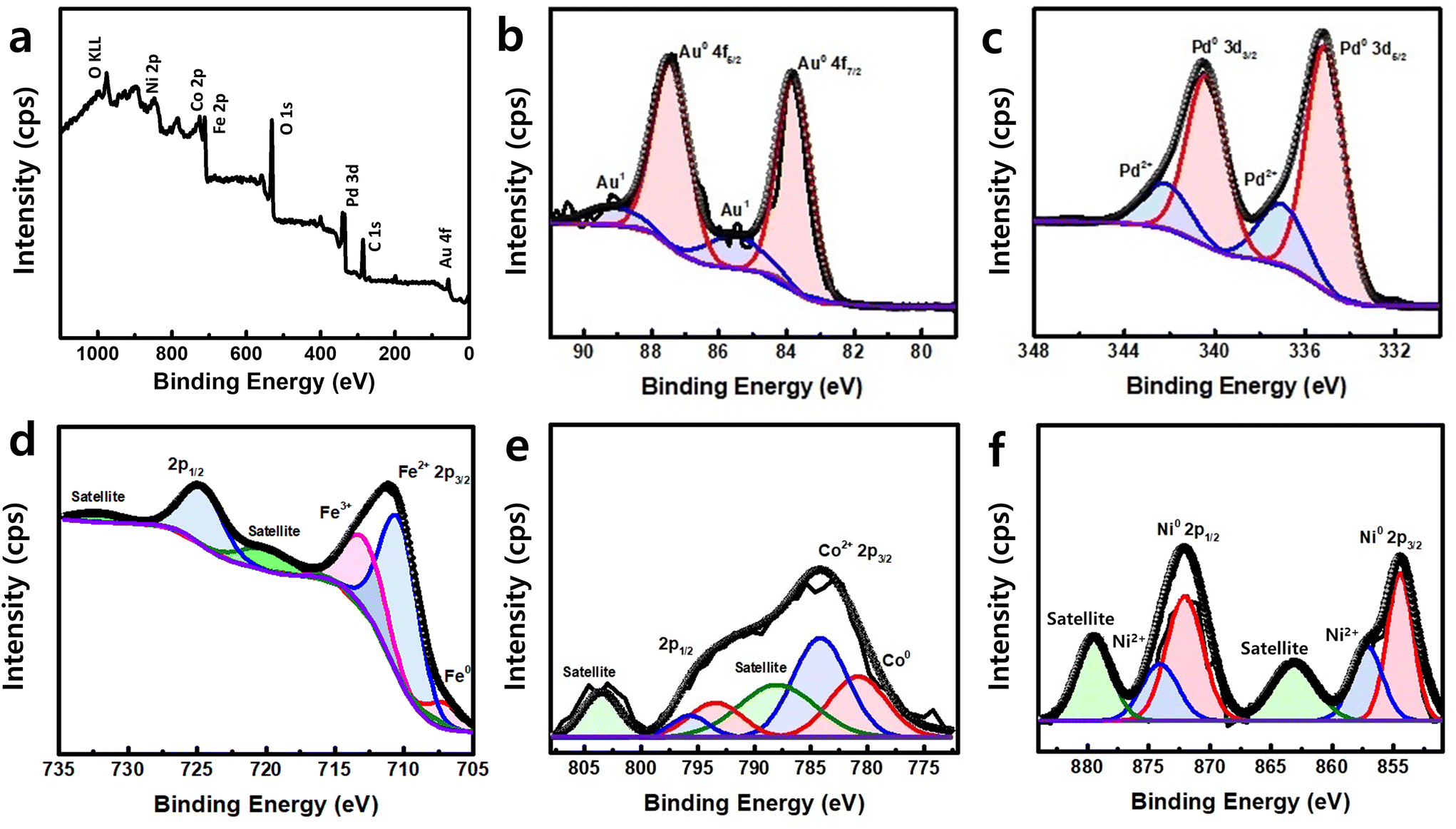 | ||
| Fig. 3 X-ray photoelectron spectroscopy (XPS) characterization. (a) XPS survey spectrum of the synthesized AuPdFeNiCo HEA NPs. High-resolution XPS spectra of (b) Au 4f, (c) Pd 3d, (d) Fe 2p, (e) Co 2p, and (f) Ni 2p. | ||
Interestingly, the BEs of each metal exhibited noticeable shifts in the AuPdFeNiCo HEA NPs compared to the bulk (Table 1). Specifically, there are lower shifts observed in the BEs of metallic gold and palladium. Conversely, the core levels of metallic iron, cobalt, and nickel showed a higher shift compared to their respective bulk metal values. These core level shifts of each element indicate the electron charge transfer between elements within the HEA. This phenomenon confirms that electron redistribution reduces atom size disparity, suggesting local lattice distortion and indicating diverse chemical environments for each element. Taken together, HR-TEM/STEM, XRD, and XPS results support the formation of HEA nanoparticles based on the measured morphology, crystalline, structure, and chemical BEs of Au, Pd, Fe, Co, and Ni.
| Au 4f (eV) | Pd 3d (eV) | Fe 2p (eV) | Ni 2p (eV) | Co 2p (eV) | |
|---|---|---|---|---|---|
| Bulk metal | 84.2 | 336.0 | 706.6 | 852.6 | 778.1 |
| AuPdFeNiCo HEA NPs | 83.8 | 335.2 | 707.7 | 854.5 | 780.7 |
| Core level shift from bulk | −0.4 | −0.8 | 1.1 | 1.9 | 2.6 |
For investigation as HER electrocatalysts, AuPdFeNiCo HEA NPs were stabilized on glassy carbon (GC). Working electrodes were prepared for quantification in alkaline, neutral, and acidic pH electrolytes in a three-electrode cell. Their activity was compared with the intrinsic activity of each pure metal foil. Compared with the HER activity of bulk single metals, the linear sweep voltammetry (LSV) polarization curve of AuPdFeNiCo HEA NPs showed superior activity toward HER with an overpotential (η) of ∼45 mV, 279 mV, and 43 mV at a geometric current density of 10 mA cm−2 in 0.5 M H2SO4, 1 M PBS, and 1 M KOH, respectively (Fig. 4). The individual metals exhibit lower HER activity compared to AuPdFeNiCo HEA NPs in all pH electrolytes, with Au notably displaying the lowest HER activity.17,19 The activity sequentially increases from Au < Ni < Fe < Co < Pd < HEA in 0.5M H2SO4. The superior HER activity of AuPdFeNiCo HEA NPs over bulk single metal foils is attributed to inducing favorable energy binding of hydrogen formed on the uneven surface during the HER process, such as the ensemble effect, thereby requiring lower overpotentials.
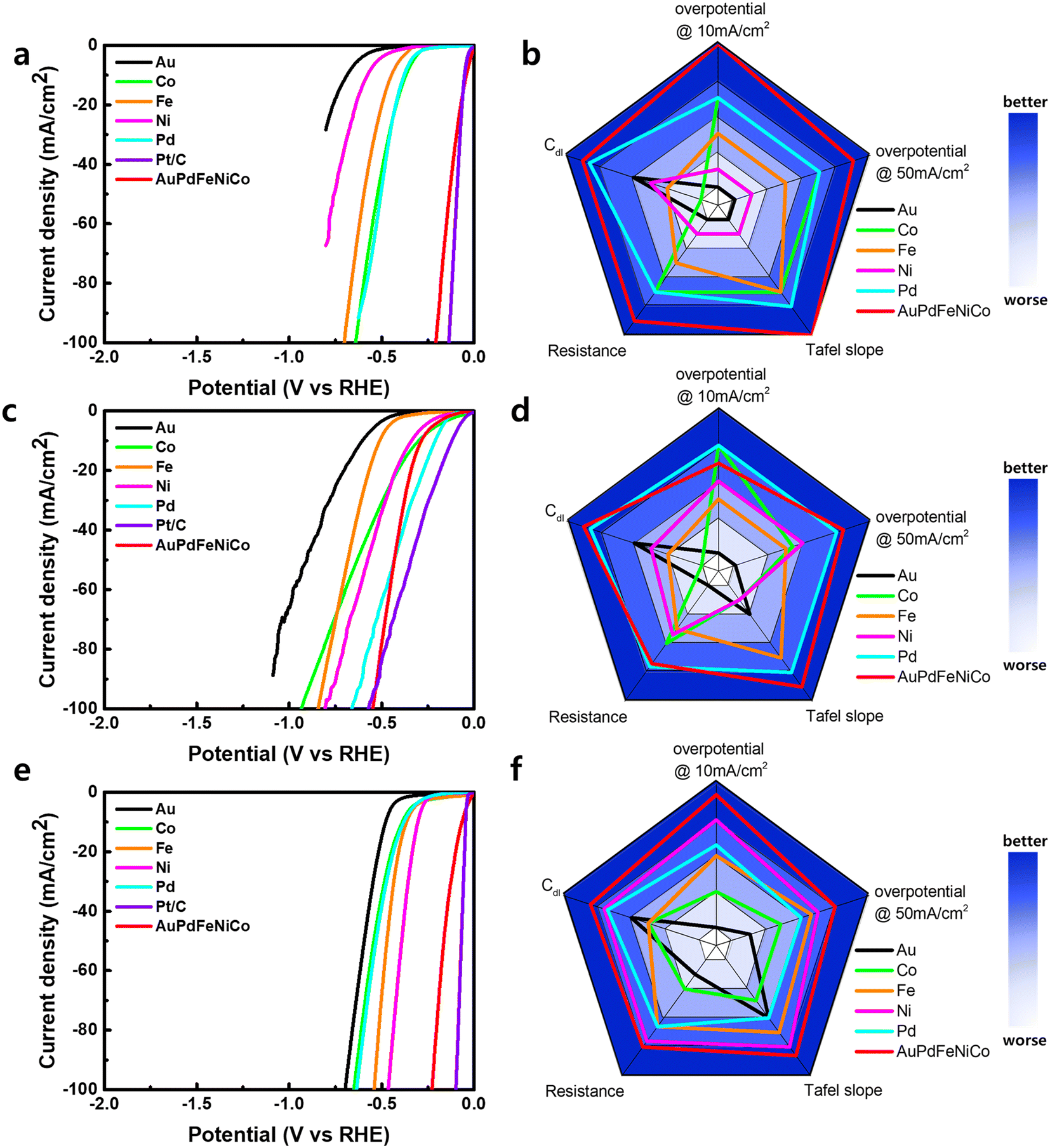 | ||
| Fig. 4 HER electrocatalysis in acidic, neutral, and alkaline electrolyte. HER polarization curves of Au, Pd, Fe, Ni, Co, AuPdFeNiCo, and Pt/C in (a) 0.5M H2SO4, (c) 1M PBS, (e) 1M KOH, corresponding radar chart comparison of the potential characteristics of the electrocatalysts in(b) 0.5M H2SO4, (d) 1M PBS, and (f) 1M KOH. | ||
For a direct comparison of activity with a known electrocatalyst, HER activity was compared with a commercial 10% Pt/C. As shown in Fig. 4, Pt/C requires higher overpotentials compared to AuPdFeNiCo HEA NPs to produce a current density of 10 mA cm−2 in acidic and alkaline electrolytes (Fig. S5†), indicating greater HER activity for AuPdFeNiCo HEA NPs. The electrocatalytic activity and mechanism aspects of HER are further supported by Tafel slope quantification (Fig. S6†). The Tafel slope of AuPdFeNiCo HEA NPs exhibited the lowest values of 32, 132, and 55 mV dec-1 under acidic, neutral, and alkaline conditions, respectively. Among these, the low Tafel slope of AuPdFeNiCo HEA NPs (32 and 55 mV dec−1) suggests the Volmer–Heyrovsky mechanism under acidic and alkaline electrolytes.3,42 However, under neutral conditions, HER necessitates a higher overpotential compared to acidic and alkaline electrolytes. This heightened requirement can be attributed to the inherent complexity of the reaction mechanism in neutral or near-neutral conditions, which contrasts with the relatively straightforward processes observed in acidic or alkaline environments.43 Unlike the conventional single-step reduction (Volmer reaction) predominant in acidic or alkaline electrolytes, HER in neutral or near-neutral electrolytes entails a multifaceted two-step reduction process, characterized by the involvement of both water molecules and dissociated H3O+ ions.1,44
The electrical double-layer capacitance (Cdl) was quantified by cyclic voltammetry (CV) to estimate the surface area of the AuPdFeNiCo HEA NPs. Furthermore, we determined the electrocatalytic specific activity, electrochemical surface area, and resistance by analyzing the Nyquist plots obtained using electrochemical impedance spectroscopy (EIS). Fig. S7† shows the CV and Cdl values for AuPdFeNiCo HEA NPs were 25.6 mF cm−2, 25.5 mF cm−2, and 55.81 mF cm−2 based on different scan rates as 10–200 mV s−1 and different pH electrolytes. The Nyquist plot was utilized to assess the charge transfer resistance of AuPdFeNiCo HEA NPs, indicating values of 1.65 Ω in 0.5M H2SO4, 2.4 Ω in 1 M PBS, and 2.4 Ω in 1 M KOH at an overpotential of 10 mA cm−2, respectively (Fig. S8†). These results support the trend in electrochemical reactions, indicating that AuPdFeNiCo HEA NPs demonstrate higher HER activity compared to other single metals. In addition, the HER stability of AuPdFeNiCo HEA NPs was evaluated using XRD analysis after chronopotentiometry studies in all pH-electrolytes at the fixed current densities of 50 mA cm−2, and all results showed no significant change during 12 h in all electrolyte conditions (Fig. S9 and S10†).
All experimental results regarding the electrochemical HER activity were summarized to compare the key quantitative performance parameters in radar charts as shown in Fig. 4b, d, and f, where AuPdFeNiCo HEA NPs exhibit desirable HER properties in all pHs compared to their constituent metals, as observed by the breadth across the radar. Therefore, the superior electrochemical activity compared to single metal catalysts confirms that AuPdFeNiCo HEA NPs serve as an efficient pH-universal HER electrocatalyst.
Above all, reducing the quantity of precious metal catalysts is crucial to producing cost-effective and efficient HER catalysts for practical use. In this context, the gold and palladium content determined by ICP-OES is 1.5 wt% and 2.4 wt%, respectively, significantly lower than that of commercial Pt/C (10 wt%) as shown in Table S1.† Even more intriguingly, despite the generally recognized low HER activity of Au, to our knowledge, HEAs designs based on Au have exhibited exceptional electrocatalytic performance compared with existing reports of their HER activity, suggesting the potential for achieving commercially viable electrocatalyst performance levels.17,19
Conclusion
We have synthesized AuPdFeNiCo HEA nanoparticles (NPs) by a facile wet impregnation method. Based on transmission electron microscopy, X-ray photoelectron spectroscopy, and electrochemical analysis, a combined approach was employed to characterize and reveal the properties of AuPdFeNiCo HEA NPs, suggesting its potential application for pH-universal hydrogen evolution. The AuPdFeNiCo HEA NPs showed notably high HER activity compared with bulk single metals and were comparable with commercial Pt/C catalysts across a wide range of pH 0 to pH 14 electrolytes, which can be an approach to enable straightforward modification of alternative electrocatalysts for the electrochemical HER.Based on this universal synthesis strategy, the exploration will expand to encompass additional combinations with various other metal elements to enhance energy production. Additionally, future research will focus on investigating the mechanisms involving structural interactions among these elements or the interfaces within unique metal structures to comprehend their impact. We will delve into researching an intriguing approach to multi-component material design by actively controlling the atomic charge transfer, short-range distortions, chemical bonding properties, and other functional characteristics within high entropy-stabilized alloy structures. We believe this work will not only position the HEA electrocatalyst group as a strong competitor in entering the hydrogen production market but also provide the impetus for a deeper understanding of their fundamentals.
Author contributions
S.J. carried out the conceptualization, data collection and analysis, and wrote the manuscript. A. J. B., S.W.B., and C.S.S. collected data and wrote the manuscript. M.B.R. designed the systems, analyzed the data, and wrote the manuscript. All authors commented on the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We are grateful for the assistance from Daniel P. Olds in collecting WAXS data. This work relates to Department of Navy award N00014-22-1-2654, issued by the Office of Naval Research. This research used beamline 28-ID-1 of the National Synchrotron Light Source II, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under contract no. DESC0012704. We are grateful to the UMass Lowell Core Research Facilities and the MIT.Nano facility.References
- Z. Zhou, Z. Pei, L. Wei, S. Zhao, X. Jian and Y. Chen, Energy Environ. Sci., 2020, 13, 3185–3206 RSC.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- S. Anantharaj, S. R. Ede, K. Karthick, S. Sam Sankar, K. Sangeetha, P. E. Karthik and S. Kundu, Energy Environ. Sci., 2018, 11, 744–771 RSC.
- S. Jeong, H. D. Mai, T. K. Nguyen, J. S. Youn, K. H. Nam, C. M. Park and K. J. Jeon, Appl. Catal., B, 2021, 293, 120227 CrossRef CAS.
- X. Liu, Y. Jiang, J. Huang, W. Zhong, B. He, P. Jin and Y. Chen, Carbon Energy, 2023, 5, 367–378 CrossRef CAS.
- Z. X. Ge, Y. Ding, T. J. Wang, F. Shi, P. J. Jin, P. Chen, B. He, S. B. Yin and Y. Chen, J. Energy Chem., 2023, 77, 209–216 CrossRef CAS.
- W. Ma, X. Zhang, W. Li, M. Jiao, L. Zhang, R. Ma and Z. Zhou, Nanoscale, 2023, 15, 11759–11776 RSC.
- Q. Xue, Z. Wang, Y. Ding, F. Li and Y. Chen, Chin. J. Catal., 2023, 45, 6–16 CrossRef CAS.
- J. N. Hansen, H. Prats, K. K. Toudahl, N. M. Secher, K. Chan, J. Kibsgaard and I. Chorkendorff, ACS Energy Lett., 2021, 6, 1175–1180 CrossRef CAS PubMed.
- Y. Jiang, H. Fu, Z. Liang, Q. Zhang and Y. Du, Chem. Soc. Rev., 2023, 53, 714–763 RSC.
- S. Zhang, S. E. Saji, Z. Yin, H. Zhang, Y. Du and C. Yan, Adv. Mater., 2021, 33, 2005988 CrossRef CAS PubMed.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- C. G. Morales-Guio, L. A. Stern and X. Hu, Chem. Soc. Rev., 2014, 43, 6555–6569 RSC.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, 4998 CrossRef PubMed.
- J. Shi, S. Xiang, D. Su, X. Liu, W. Zhang and L. Zhao, ChemSusChem, 2021, 14, 4525–4535 CrossRef CAS PubMed.
- X. Li, S. Takano and T. Tsukuda, J. Phys. Chem. C, 2021, 125, 23226–23230 CrossRef CAS.
- L. A. Kibler, J. M. Hermann, A. Abdelrahman, A. A. El-Aziz and T. Jacob, Curr. Opin. Electrochem., 2018, 9, 265–270 CrossRef CAS.
- Y. Jiang, Z. Liang, H. Fu, M. Sun, S. Wang, B. Huang and Y. Du, J. Am. Chem. Soc., 2024, 146, 9012–9025 CrossRef CAS PubMed.
- H. Lv, Z. Xi, Z. Chen, S. Guo, Y. Yu, W. Zhu, Q. Li, X. Zhang, M. Pan, G. Lu, S. Mu and S. Sun, J. Am. Chem. Soc., 2015, 137, 5859–5862 CrossRef CAS PubMed.
- H. Wang, S. Jiao, S. Liu, S. Wang, T. Zhou, Y. Xu, X. Li, Z. Wang and L. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 30479–30485 CrossRef CAS PubMed.
- X. Zhu, Q. Guo, Y. Sun, S. Chen, J. Q. Wang, M. Wu, W. Fu, Y. Tang, X. Duan, D. Chen and Y. Wan, Nat. Commun., 2019, 10, 1–11 CrossRef PubMed.
- M. V. Fonseca Guzman, M. E. King, N. L. Mason, C. S. Sullivan, S. Jeong and M. B. Ross, Matter, 2023, 6, 838–854 CrossRef CAS.
- A. Devi, H. Minhas, L. Sahoo, N. Rashi, S. Gratious, A. Das, S. Mandal, B. Pathak and A. Patra, Nanoscale, 2023, 16, 1758–1769 RSC.
- J. Hao, H. Zhu, Y. Li, P. Liu, S. Lu, F. Duan, W. Dong, Y. Lu, T. Liu and M. Du, Chem. Eng. J., 2021, 404, 126523 CrossRef CAS.
- J. Shen, Z. Hu, K. Chen, C. Chen, Y. Zhu and C. Li, Mater. Today Nano, 2023, 21, 100282 CrossRef CAS.
- X. Xu, Y. Guo, B. P. Bloom, J. Wei, H. Li, H. Li, Y. Du, Z. Zeng, L. Li and D. H. Waldeck, ACS Nano, 2020, 14, 17704–17712 CrossRef CAS PubMed.
- J. Hao, Z. Zhuang, K. Cao, G. Gao, C. Wang, F. Lai, S. Lu, P. Ma, W. Dong, T. Liu, M. Du and H. Zhu, Nat. Commun., 2022, 13, 1–13 Search PubMed.
- D. Wu, K. Kusada, T. Yamamoto, T. Toriyama, S. Matsumura, S. Kawaguchi, Y. Kubota and H. Kitagawa, J. Am. Chem. Soc., 2020, 142, 13833–13838 CrossRef CAS PubMed.
- H. Fu, Y. Jiang, M. Zhang, Z. Zhong, Z. Liang, S. Wang, Y. Du and C. Yan, Chem. Soc. Rev., 2024, 53, 2211–2247 RSC.
- H. Liu, L. Syama, L. Zhang, C. Lee, C. Liu, Z. Dai and Q. Yan, SusMat, 2021, 1, 482–505 CrossRef CAS.
- A. Sivanantham, H. Lee, S. W. Hwang, B. Ahn and I. S. Cho, J. Mater. Chem. A, 2021, 9, 16841–16851 RSC.
- M. Slobodyan, E. Pesterev and A. Markov, Mater. Today Commun., 2023, 36, 106422 CrossRef CAS.
- D. B. Miracle and O. N. Senkov, Acta Mater., 2017, 122, 448–511 CrossRef CAS.
- S. Jiang, L. Zhu, Z. Yang and Y. Wang, Int. J. Hydrogen Energy, 2021, 46, 36731–36741 CrossRef CAS.
- D. Wu, K. Kusada, Y. Nanba, M. Koyama, T. Yamamoto, T. Toriyama, S. Matsumura, O. Seo, I. Gueye, J. Kim, L. S. R. Kumara, O. Sakata, S. Kawaguchi, Y. Kubota and H. Kitagawa, J. Am. Chem. Soc., 2022, 144, 3365–3369 CrossRef CAS PubMed.
- G. Feng, F. Ning, J. Song, H. Shang, K. Zhang, Z. Ding, P. Gao, W. Chu and D. Xia, J. Am. Chem. Soc., 2021, 143, 17117–17127 CrossRef CAS PubMed.
- B. Lay, Y. M. Sabri, S. J. Ippolito and S. K. Bhargava, Phys. Chem. Chem. Phys., 2014, 16, 19522–19529 RSC.
- M. Khawaji and D. Chadwick, Catal. Today, 2019, 334, 122–130 CrossRef CAS.
- Y. Lou, C. Dai, W. Chang, H. Qian, L. Huang, C. Du and D. Zhang, Corros. Sci., 2020, 165, 108390 CrossRef CAS.
- P. Li, Y. Xuan, B. Jiang, S. Zhang and C. Xia, Electrochem. Commun., 2022, 134, 107188 CrossRef CAS.
- X. Cui, W. Li, K. Junge, Z. Fei, M. Beller and P. J. Dyson, Angew. Chem., Int. Ed., 2020, 59, 7501–7507 CrossRef CAS PubMed.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 1–21 Search PubMed.
- W. Sheng, Z. Zhuang, M. Gao, J. Zheng, J. G. Chen and Y. Yan, Nat. Commun., 2015, 6, 6–11 Search PubMed.
- T. Shinagawa and K. Takanabe, Phys. Chem. Chem. Phys., 2015, 17, 15111–15114 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01538j |
| This journal is © The Royal Society of Chemistry 2024 |