
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Immunomodulatory nanoparticles activate cytotoxic T cells for enhancement of the effect of cancer immunotherapy
Kory
Wells
 ab,
Tongrui
Liu
a,
Lei
Zhu
a and
Lily
Yang
*ab
ab,
Tongrui
Liu
a,
Lei
Zhu
a and
Lily
Yang
*ab
aDepartment of Surgery, Emory University School of Medicine, Winship Cancer Institute, Clinic C, Room 4088, 1365 C Clifton Road, NE, Atlanta, GA 30322, USA. E-mail: Lyang02@emory.edu; Tel: +1 4040778429
bWinship Cancer Institute, Emory University School of Medicine, Atlanta, GA 30322, USA
First published on 6th September 2024
Abstract
Cancer immunotherapy represents a promising targeted treatment by leveraging the patient's immune system or adoptive transfer of active immune cells to selectively eliminate cancer cells. Despite notable clinical successes, conventional immunotherapies face significant challenges stemming from the poor infiltration of endogenous or adoptively transferred cytotoxic T cells in tumors, immunosuppressive tumor microenvironment and the immune evasion capability of cancer cells, leading to limited efficacy in many types of solid tumors. Overcoming these hurdles is essential to broaden the applicability of immunotherapies. Recent advances in nanotherapeutics have emerged as an innovative tool to overcome these challenges and enhance the therapeutic potential of tumor immunotherapy. The unique biochemical and biophysical properties of nanomaterials offer advantages in activation of immune cells in vitro for cell therapy, targeted delivery, and controlled release of immunomodulatory agents in vivo. Nanoparticles are excellent carriers for tumor associated antigens or neoantigen peptides for tumor vaccine, empowering activation of tumor specific T cell responses. By precisely delivering immunomodulatory agents to the tumor site, immunoactivating nanoparticles can promote tumor infiltration of endogenous T cells or adoptively transferred T cells into tumors, to overcoming delivery and biological barriers in the tumor microenvironment, augmenting the immune system's ability to recognize and eliminate cancer cells. This review provides an overview of the current advances in immunotherapeutic approaches utilizing nanotechnology. With a focus on discussions concerning strategies to enhance activity and efficacy of cytotoxic T cells and explore the intersection of engineering nanoparticles and immunomodulation aimed at bolstering T cell-mediated immune responses, we introduce various nanoparticle formulations designed to deliver therapeutic payloads, tumor antigens and immunomodulatory agents for T cell activation. Diverse mechanisms through which nanoparticle-based approaches influence T cell responses by improving antigen presentation, promoting immune cell trafficking, and reprogramming immunosuppressive tumor microenvironments to potentiate anti-tumor immunity are examined. Additionally, the synergistic potential of combining nanotherapeutics with existing immunotherapies, such as immune checkpoint inhibitors and adoptive T cell therapies is explored. In conclusion, this review highlights emerging research advances on activation of cytotoxic T cells using nanoparticle agents to support the promises and potential applications of nanoparticle-based immunomodulatory agents for cancer immunotherapy.
Introduction
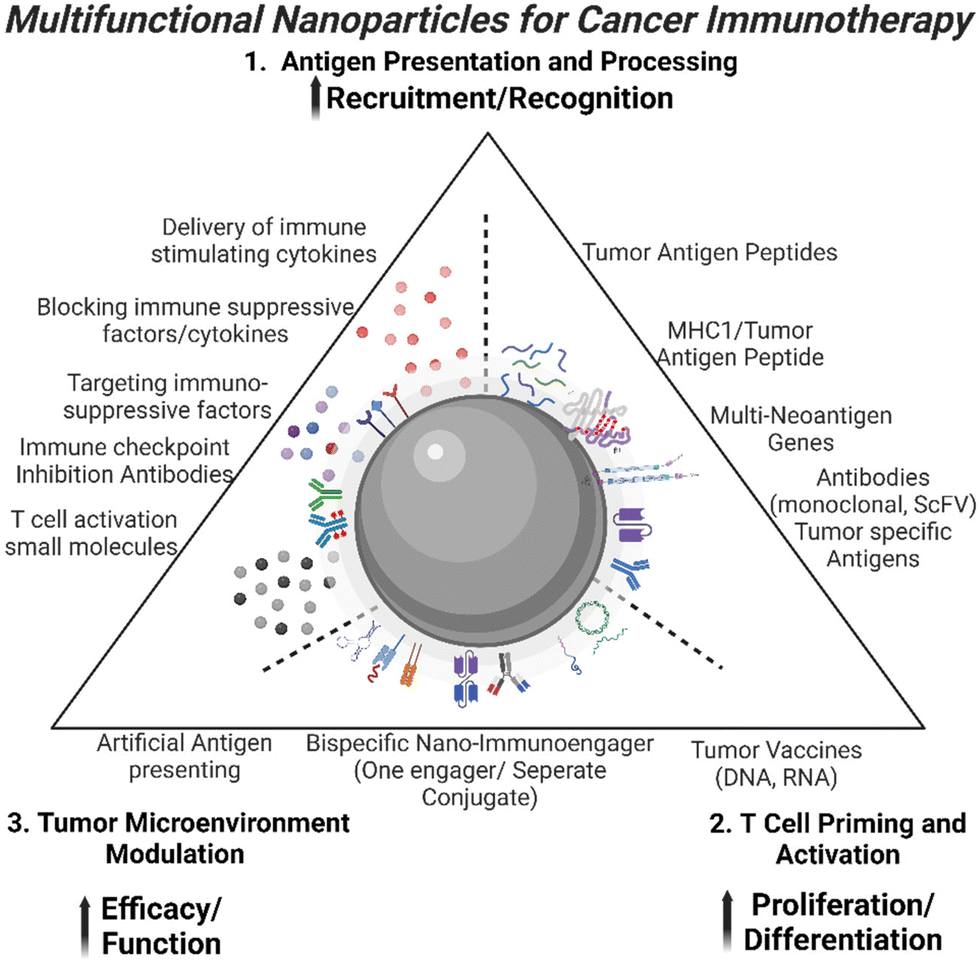
T cell-mediated immune responses against tumors form the backbone of cancer immunotherapy, offering a potent avenue to combat malignancies.1 Efforts to activate endogenous T-cell responses within the tumor microenvironment (TME) have gained prominence. Strategies involve the blockade of immune checkpoints, such as anti-PD-1 or CTLA-4 monoclonal antibodies, unleashing the potential of cytotoxic T cells to recognize and eliminate cancer cells.2–4 These groundbreaking approaches underscore the dynamic interplay between T cells and tumors, highlighting the versatility of immunotherapeutic strategies in redirecting the immune system's inherent power to combat cancer. Clinical translation of T cell immunotherapy in solid cancers faces substantial challenges that impede its widespread efficacy, including tumor heterogeneity, immune evasion, immunosuppressive TME, and limited T cell infiltration.5 In order to address these challenges, current strategies include improving T cell trafficking, modifying the immunosuppressive TME, enhancing T cell's specificity and activity, overcoming tumor heterogeneity by targeting multiple antigens, and utilizing combination therapies.6 It is likely that combinatory approaches are necessary to tackle different barriers faced by cancer immunotherapy simultaneously to achieve significant improvement in therapeutic responses. Recent developments in multifunctional nanoparticles (NPs) for the enhancement of therapeutic response to cancer immunotherapy have shown promises in applications of NP-mediated immunotherapeutic for the treatment of solid tumors.7 Here, we provide an overview of the development of various NP platforms or delivery systems with the ability of enhancement of antigen presentation, activation, and stimulation of proliferative cytotoxicity T cells, and inhibition of immunosuppressive TME.NPs offer a versatile platform to enhance responses to immunotherapy and T cell-based therapies. With their small size and unique physicochemical properties, NPs have emerged as powerful tools in the realm of cancer immunotherapy. They can efficiently deliver tumor antigens for presentation to T cells, serve as carriers for immunostimulatory molecules, genetic material delivery, and carry immunomodulatory agents and adjuvants to create a favorable microenvironment.8 NPs also improve targeted drug delivery, sustain release of immunotherapeutic agents, and possess multifunctionality for optimized treatment responses.9 The integration of NPs extending beyond conventional therapies provides a dynamic landscape for advancing T cell activation in a hostile solid TME. The focus on the development of various NP approaches for T cell activation stems from the pivotal role these NPs play in optimizing and enhancing immunotherapeutic strategies. This exemplifies the critical interplay between NPs and T cells, underscoring their significance in modulating immune responses. With the recent push for personalized medicine and success of immunotherapy, many treatment modalities have shifted its focus from the tumor itself to the host immune system, mobilizing immune stimulatory cells and impairing immunosuppression.10–14 Thus recent advances in applications of nanotechnology in immunotherapy have shown the potential of the novel design and function of NP-based agents in targeting and improving the cancer immunity cycle, i.e. activation of tumor specific immune responses, modulation of the immunosuppressive TME.15–18 Here, we provide an overview of the development of various NP platforms or delivery systems with the ability of enhancement of antigen presentation, activation, and stimulation of proliferation of cytotoxic T cells, and inhibition of immunosuppression in tumors (Fig. 1). Additionally, information on mechanisms of those NP immunotherapeutic agents on regulation of tumor immune response and activation of tumor specific cytotoxic T cells should shed light on the promising areas of research to accelerate development and refinement of personalized cancer immunotherapy using novel nanotechnology.
 | ||
| Fig. 1 Nanoparticle-mediated immunoactivating and immunomodulating strategies for activation of T cell immune response. Figure created with Biorender. | ||
Emerging roles of nanotechnology in cancer treatment
Nanotechnology and its recent advancements have become an innovative platform in biomedical research that has the potential to reshape the landscape of cancer therapy, imaging, and drug delivery. NPs are nanometer-sized particles with the ability to encapsulate and/or conjugate therapeutic agents within the particle through functional modifications onto its surface. In addition to the capacity for surface engineering, the dimensions of NPs are similar to macromolecules or biomolecules that can improve drug stability and targeted efficacy while also being able to be used as tools in imaging-guided cancer therapy.19–21 Their unique physiochemical properties, including size, surface charge and composition allow versatility that makes them an ideal candidate for creating multifunctional and personalized nanoformulations for a range of biomedical applications.19–21 One of the key modifiable aspects of NPs is their nanomaterials, the composition can range from organic polymers to inorganic metals each providing distinct properties.19,21–23 For the development of cancer therapeutic agents that require large doses and repeated administrations, it is critical to use nanomaterials that are biocompatible and biodegradable.The design of these NPs takes into account the functional or anatomical changes expected by exposure to the NP (e.g., mechanism of action) and their specific biochemical interactions (e.g., TME targets). Chemical functionalization with ligands can be used to design delivery systems with dual, multi-targeting NPs that have targeting and pharmacological activities.21,24,25 Due to NP's large surface-to-volume ratio, they not only allow high-capacity drug loading but also the attachment of targeting ligands that facilitate their movement through tissues, binding to their targets, and intracellular trafficking into cancer cells.21,24,25 Therefore, with the addition of ligands, such as peptides, antibodies (fragments), proteins, carbohydrates, polypeptides, DNA/RNA and small molecules, these targeting agents not only increase targeted particle uptake into the tumor but also improve particle distribution in the tumor and within its cells. In addition to material composition, wide varieties of payload can be incorporated into NPs, which range from chemotherapy or small molecule drugs, proteins/peptides, nucleic acids and/or imaging agents.19–21,25 As a result, therapeutic cargo can be delivered to the site of interest efficiently, promoting precise and controlled release. NP delivery into tumors is mediated by two main mechanisms: active and passive targeting. Passive targeting takes advantage of the characteristics of tumors, being able to transport through leaky tumor vasculatures, also known as enhanced permeability and retention effect (EPR).26 Nanotherapeutics tend to accumulate in the tumor interstitial space after moving across the leaky vasculature with gap openings. In contrast, active targeting relies on the use of specific ligands to bind to a highly expressed target in tumor tissues.26 However, many NPs engineered to target biomarkers expressed on tumor cells only delivered into the tumor interstitial space by passive targeting before reaching their tumor cell targets. Thus, targeting tumor endothelial cells as well as tumor cells represents truly an actively targeted delivery that brings the NP across tumor vessels into the tumor interstitial space and then tumor cells.25,27,28
Various organic and inorganic materials have been used to develop NPs with unique biochemical and biophysical properties. NPs produced from lipids, polymers, dendrimers, and hyaluronic acid are commonly used as drug and nuclear acid delivery carriers.19,23,25 Lipid nanoparticles (LNPs) and liposomes are made of lipid bilayers with a function of encapsulating hydrophobic or hydrophilic drugs to provide controlled release. Currently FDA approved NP/drugs for solid cancer therapy are mostly based on liposome or LNPs, such as liposome doxorubicin (Doxil)29 and liposomal irinotecan (Onivyde).30 Recent successes in application of LNPs for the delivery of mRNAs for the COVID-19 vaccine have accelerate the translational process of LNPs carrying mRNAs for cancer vaccine and therapy.31,32 Self-assembling polymeric NPs have been investigated extensively as drug delivery systems.33 Bioactive and biomimetic hyaluronic acid NPs are a promising class of drug carriers with tumor targeting and TME modulating activities.23 Cationic polymers can be used to improve delivery of DNA/RNA and penetrate cell membranes by the action of charges resulting in the escape of degradation via endosomes.34 Polymeric micelles carrying paclitaxel have also been approved for the treatment of breast and ovarian cancer.35 Proteins and peptides are excellent biocompatible and biodegradable materials for the development of nanotherapeutics. Human albumin NPs encapsulated paclitaxel (Abraxane) is commonly used for the treatment of cancer patients with advanced diseases.36 Inorganic NPs, such as gold, silver, iron oxide, and hafnium oxide NPs, carbon nanotubes, and quantum dots have been developed as theragnostic agents for optical and magnetic resonance imaging (MRI) tumor imaging, drug delivery, photodynamic therapy, magnetic hyperthermia therapy and radiation therapy.19,22,37 Superparamagnetic iron oxide NPs have been approved by the FDA for tumor imaging, assessing permeability of tumors for drug delivery by non-invasive MRI, and for the treatment of severe anemia.19,38 Extensive preclinical studies demonstrated the ability of magnetic iron oxide NPs as MRI contrasts of tumor imaging, which has the potential for clinical translation for tumor detection, monitoring drug delivery and therapeutic response, and mediating magnetic hyperthermal therapy.19 Hafnium oxide NPs have been approved by the FDA and European Medicines Agency (EMA) for local administrations to enhance responses to radiotherapy in tumors.39 Iron oxide NPs (NanoTherm) have been approved by the EMA for thermal ablation of glioblastoma, prostate, and pancreatic cancer.38 Gold NPs have optical, photothermal and radioenhancement activities.40 Many types of NPs with hybrid nanomaterials that confer multi-functionality have also been developed, such as biomimetic nanocarriers engineered with synthesized NP cores and coated with biological membranes derived from various cells.41 DNA or RNA coated or encapsulated NPs for gene delivery, biomarker targeting, tumor vaccine, and biosensor for biomarker detections.42 At present, extensive preclinical and clinical studies are ongoing for translation of various types of nanotherapeutics for cancer treatment and detection.
Immunomodulatory properties of NPs and nanotherapeutics in solid tumors
The immunomodulatory properties of NPs have profound effects on the immune response, influencing diverse stages of the immune activation cascade. NPs can dynamically interact with immune cells, shaping their responses in a manner that spans from cytokine production to the recruitment of immune cells.43,44 This multifaceted impact is particularly crucial given the intricate adaptation of immune responses at various stages. They play a role in immune cell recruitment, influencing the types and quantities of immune cells participating in the response.45 This modulation extends across the distinct phases of immune activation, from the initial recognition of antigens to the effector functions that eliminate target cells.46–50 Understanding and harnessing these immunomodulatory properties of NPs open avenues for finely tuning immune responses, potentially enhancing the efficacy of immunotherapies at multiple levels and providing a refined approach to cancer treatment. Increasing evidence supports the effect of NPs on modulating tumor immune microenvironment. NPs, upon entering the body, involve uptake processes like pinocytosis, phagocytosis, and endocytosis.51 The protein corona formed on NPs’ surfaces is recognized by immune receptors, including Toll-like receptor (TLR) and complement receptors, and influences NP clearance by being taken up by macrophages.52 These NPs are able to circulate through the venous and lymphatic drainage and increase antigen presentation, triggering a cascade of immune responses.53 As engineered particles, NP agents can interact with phagocytic cells, such as macrophages of the innate immune cells, to be taken up nonspecifically by macrophages. Internalization of NPs into macrophages leads to the activation of macrophages and production of inflammatory cytokines and factors that promote infiltration and activation of immune effector cells, including CD4+ and CD8+ T cells.54 It has been shown that systemic delivery of 100 nm hydroxyethyl starch-coated iron oxide NPs without payload drug into mice bearing orthotopic mammary tumors altered immune microenvironment in tumors and induced tumor infiltration of CD8+ T cells. This immune activation effect was independent from the conjugation of an antibody targeting ligand or the level of intratumoral delivery of the NP.55 Metal NPs (i.e. iron oxide and titanium dioxide) have been shown to promote the activation of immune cells and cytokine production, inducing potent humoral and cellular immune responses.56,57 In addition, they induce exosomes that have been shown to target antigen-presenting cells to initiate Th1-type immune activation, demonstrating their role in activation and modulation of immune responses.58 This interaction between NPs and immune cells can be used in the design of nanotherapies, as it can influence the efficacy and safety of these systems in various biomedical applications.In the realm of immunotherapy, T cells have become the focal point for innovative strategies. However, tumors present barriers including the immunosuppressive TME, heterogeneity in antigen expression, and limitations in T cell persistence and function within solid tumors.59 The heterogeneous nature of solid tumors poses challenges for effective recognition and activation of cytotoxic T cells. Furthermore, TME hurdles effector T cell infiltrating and trafficking into tumors.60 The physical barriers include inadequate vasculature and dense stromal barriers.61 For the low level of cytotoxic T cells that enters into tumor tissue, they are trapped in the immunosuppressive TME in solid tumors, limiting their cytotoxic effect on tumor cells. These obstacles necessitate a deeper understanding and innovative strategies to unlock the full potential of T cells in fighting solid tumors. The ability of NPs to modulate these immune responses can be harnessed to address these challenges and advance the efficacy and widespread applicability of T cell-based immunotherapies.
Leveraging NPs to enhance antigen presentation and T cell activation
Central to tumor specific T cell response are antigen-presenting cells (APCs), including dendritic cells (DCs) and macrophages that capture, process, and present tumor antigens to T lymphocytes. This multifaceted response involves two key T cell subsets: helper T cells and cytotoxic T cells. DCs, pivotal players in antigen presentation, capture these antigens and process it into antigenic peptides to form a complex with an MHC II molecule for presentation to CD8+ cytotoxic and CD4+ T helper cells to activate tumor-specific T cells, initiating a cascade of events leading to the activation of adaptive immunity.62 However, some limitations can hinder the activation of immune effector cells by APCs, such as weak immunogenicity of tumor associated antigens, low level of neoantigen on tumor cells, immune tolerance acquired by tumor cells, and upregulation of immune regulatory factors.63 Inability of adequately presenting tumor antigens to activate T and B lymphocytes can lead to the immune evasion of tumor cells. A promising strategy of immunotherapy is to increase antigen presentation to immune effector cells, stimulating the expansion of tumor-specific T lymphocytes, by therapeutic tumor vaccine using tumor antigen peptides on immunoadjuvant carriers that enhance immunogenicity of tumor antigens.64 One of the most commonly used adjuvants is poly ICLC, a synthetic double stranded-RNA mimic that stimulates innate immune response via toll-like receptor 3 (TLR3) and cytosolic melanoma differentiation-associated protein 5 to activate the production of cytokines and chemokines.65 Despite of extensive preclinical studies using various neoantigens and adjuvant carriers, delivered as mRNAs, peptides, and proteins, have demonstrated tumor specific T cell responses and tumor growth inhibition in mouse tumor models, results of clinical trials in human cancer patients did not show significant therapeutic efficacy on tumor growth, although the activation and expansion of tumor antigen specific T cells were found in the patients.66–68 It is clear that the combination of tumor vaccine with other cancer therapeutic approaches are necessary to enhance overall therapeutic efficacy in cancer patients. Supporting this notion, results of a recent clinical trial (KEYNOTE-942) showed that intramuscular injection of a single synthetic mRNA coding for up to 34 neoantigens encapsulated in LNPs in combination with an anti-PD1 antibody (KEYTRUDA) in melanoma patients after surgery significantly improved recurrence-free survival of the patients.69 Since mRNA and peptide-based tumor vaccine have a poor stability and delivery efficiency into APCs.70,71 a promising approach to overcome these limitations is designing NP delivery platforms that can carry diverse types of tumor antigens, such as proteins, peptides, mRNAs, or DNA. Following local or systemic delivery, a high level of NPs has a tendency to accumulate in lymphoid organs, such as the draining lymph node and spleen, where antigen presentation and activation of T cells take place.72–75 Study results also showed that NP size affected its accumulation in the draining lymph nodes. Intradermal delivery of 25 nm polymeric NPs led to significantly higher efficiency entering into lymphatic capillaries and lymph nodes to activate dendritic cells than that of 100 nm NPs.76 However, smaller NPs (<10 nm) can rapidly pass through lymph nodes to enter the blood circulation and thus, have a low level of lymph node accumulation. Furthermore, NP shape is an important factor in determining lymphatic transportation as well as interaction with and internalization by immune cells. Previous reports showed that rod-shaped NPs had a higher lymphatic uptake than spherical shaped NPs.77–79 Increasing evidence also supports the effect of NP's surface modifications on lymph node accumulation. For example, polyethylene glycol (PEG) coating increased lymphatic permeability and lymph node accumulation.80 Intradermal delivery of PEG-coated magnetic iron oxide NPs (20 nm) showed higher levels of uptake by dendritic cells and lymph node homing compared with amphiphilic polymer coated NP.81 It has been shown that the form of antigens (i.e., peptides, mRNAs, and plasmids) administered in vaccination can dramatically impact localization in lymphoid tissues and provide a new rationale for the enhanced immune responses observed following immunization with immune complexes or NPs.82 Polymeric NPs (30 nm) incorporated with immune adjuvant CpG targeted to the draining lymph nodes to induce DC maturation and increase the level of activated CD8+ T cells.83 Therefore, strategically designed NPs carrying tumor antigens have the potential to increase targeted delivery into lymph nodes and interaction with APCs to activate immune response.NPs as carriers for tumor antigen peptides and protein
The T-cell receptor (TCR) of CD8+ T cells are capable of recognizing the antigen peptide-MHC I complex to produce cytotoxic response against antigen expressing target cells.1 Therefore, a critical factor is the selection of proper tumor antigens for therapeutic cancer vaccines to exert specific cytotoxicity against tumor cells. Tumor antigens can be classified into tumor-associated antigens (TAA) and tumor-specific antigens (TSA).63,66 Despite TAAs being expressed in both normal cells and tumor cells, tumor cells overexpress those antigens but their levels in normal cells are very low. On the other hand, TSAs are derived from genetic mutations that are specific to tumor cells. Currently, many TAAs and TSAs have been identified as targets for protein or peptide-based therapeutic cancer vaccines.66 However, peptide and protein vaccines have historically shown limited efficacy in cancer treatment.84 Recent advancements in nanotechnology enabled encapsulation or conjugation of a variety of peptides and proteins and significantly improved their therapeutic effect in effect in several cancer types.85–89As nanocarriers for tumor antigens, NPs not only increase stability and bioavailability of tumor antigen proteins or peptides, and uptake by APCs, but also have the capability of carrying multiple immunomodulatory agents, such as inhibitors of immune checkpoint proteins and small molecule agents, to induce a synergistic effect. A liposome delivery platform (DepoVax) has been developed as a therapeutic vaccine for breast or ovarian cancer. HLA-A*0201/H2Dd transgenic mice were treated with naturally processed peptides bound to HLA-A2 molecules that were isolated from breast and ovarian tumor cell lines, as antigens in DepoVax (DPX-0907) along with adjuvant and a T helper peptide epitope, that bound to and activated CD4+ and CD8+ T cells.85 Immunization with those liposomes elicited tumor specific immune response, represented with increased frequency of IFN-γ secreting cells in the lymph nodes and reduced regulatory T cells in the spleen. Furthermore, results of a phase I clinical trial after subcutaneous (SC) injection of DPX-0907 carrying 7 MHC class I-presented peptides that were isolated from HLA-A2+ breast, ovarian and prostate cancer cells induced rapid and sustained immune responses to multiple antigens in several patients while generating multi-functional T cells and antigen-specific T cell responses.86
NP-based vaccines were further optimized to deliver multiple peptides and proteins for promoting their recognition and activation of both CD8+ T and CD4+ T cells by presenting multiple epitopes to bind to MHC class I and II molecules in APCs. A cholesteryl pullulan (CHP) nanocomplex carrying truncated HER2 protein (CHP-HER2 vaccine) was developed.87 BALB/c mice immunized with CHP-HER2 activated HER2 specific CD8+ and CD4+ T-cell clones, resulting in rejection of HER2-expressing murine sarcomas in a CD8+ T-cell-dependent manner.87 Further investigations in a Phase I trial demonstrated induction of HER2-specific CD8+ and/or CD4+ T-cell immune response in HLA-A2402-positive patients with therapy-refractory HER2-expressing cancers.88 By complexing tumor antigen protein and carbohydrates with cholesteryl moieties, the CHP-HER2 was not only tolerable but multiple treatments and boosters increased the proportion of HER2-specific T cells.
NPs have been used to improve the delivery of peptides and proteins by enabling precise targeting and controlled release. Building on this progress, researchers are exploring novel methods where the vaccine carrier is constructed from the peptides or proteins themselves. This approach, along with various vaccine generation techniques like self-assembling peptides,89,90 protein-based NPs91,92 and nucleic acid nanomaterials31,93 enables the combination of different peptides or proteins with chemotherapy or other agents, enhancing their therapeutic potential. Taking advantage of the known properties of DNA/RNA and proteins/peptides, increasing attention has been devoted to design NP vaccines for tumor immunotherapy using combinatorial approaches. These optimized NPs are constructed using different biomolecular building blocks to self-assemble into hybrid nanostructures based on their highly specific interaction patterns of the secondary/tertiary structures.94,95 Those genetically engineered nanocarriers enable precise control over the stereochemistry, structure, and self-assembly behavior of NPs. Additionally, molecular modifications produce NPs with controlled properties, such as surface charge, environmental responsiveness, drug encapsulation, stability, and ligand display, which allow for distinct morphologies of nanostructures, tailored functionalization with multiple functional groups for conjugation of targeting moieties and imaging agents to improve vaccine design or drug delivery.
For example, self-assembling peptides (Ac-AAVVLLLW-COOH) were engineered onto the N-terminal of peptide epitopes from a linear cytotoxic T-cell epitope derived from MAGE-A3 (KVAELVHFL) and pan HLA-DR epitope PADRE (AKFVAAWTLKA), which lead to the production of a peptide-based tumor vaccine NP.89 The self-assembled NPs had increased stability, showed good biocompatibility to human red blood cells. The peptide NPs have strong effect on activation of DCs and induction of antigen specific CD4+ and CD8+ T cells in vitro compared to soluble peptides.89 Subcutaneous immunization into healthy C57BL/6 mice increased uptake and presentation to APCs and activated specific T cells that secreted IFN-γ.89 Similarly, a trivalent peptide-hydrogel that co-deliver antigen epitopes was produced by co-assembling three antigenic epitopes (gp100, Tyr369, and MART-126) with the self-assembling peptide FEFEFKFK (F peptide). Such a design greatly promoted antigen presentation to DCs and their subsequent homing to the draining lymph nodes, eliciting antitumor CD8+ T cell response in the absence of additional immunoadjuvants, resulting in significant inhibition of the B16 melanoma tumor growth in C57BL/6 mice.90 T cell targeted NPs were developed, in combination with multiple therapies to provide additional functions that better meet the needs of clinical application. For example, a co-delivery nanocomplex with a universal nitrated T helper cell epitope, neoantigen epitope (Adpgk mutation), and adjuvant was developed. By incorporating CD4+ T cell epitopes into DNA-coupled nitrated T helper cell epitope NP, this nanovaccine could promote simultaneous delivery of adjuvant and neoantigen to lymph nodes and DCs, improving the antigen uptake and presentation, leading to the production of neoantigen-specific cytotoxic T cells and memory T cells, and tumor growth inhibition and resistance to tumor cell re-challenging in the MC38 mouse colon cancer model.96 Therefore, the assembly of antigenic epitope-conjugated peptides offers a simple, customizable approach for the development of cancer vaccines with remarkable therapeutic efficacy, thereby providing a highly versatile platform for the application of personalized multivalent tumor vaccines.
This concept is further supported by study results in a preclinical murine model where neoantigens were identified by whole-exome sequencing and each neoantigen peptide was conjugated to amphiphilic lipids (DSPE-PEG2000-NHS) to synthesize NPs. The nine-tumor antigen peptide assembled NPs were then mixed to generate a personalized neoantigen nanovaccine (PNVAC). By creating these amphiphilic DSPE-PEG2000-peptides that self-assembled into a core/shell structure, these PNVAC triggered superior protective efficacy against tumor recurrence and promoted a longer survival than neoantigen peptide alone, especially when combined with anti-PD-1 treatment in a murine gastric tumor mode.97 Given the biological and functional significance of PNVAC, the approach has been translated into a phase I clinical trial (ChiCTR1800017319), where a PNVAC platform for adjuvant cancer immunotherapy is generated for patients with a high-risk gastric/gastroesophageal junction cancer after surgery and adjuvant chemotherapy. Consistent with the preclinical studies, the PNVAC carrying patients’ specific neoantigen peptides not only induced significant CD4+ and CD8+ T cell-mediated immune responses, but also showed clearly prolonged disease-free survival in the patients. Induction of T cell responses against 77.8% of immunized neoepitopes in the enrolled twenty-nine patients were detected. Importantly, the levels of antigen-experienced memory T cells were increased, resulted in persistent immune response for over one year.97
Immunomodulatory factors added to NP-based peptide vaccines
In addition to nanostructures and their compositions, delivery of tumor antigens can be readily modified to incorporate a variety of molecules, such as chemical compounds, peptides/proteins, and oligonucleotides, as well as pathogen-associated molecular patterns that function as adjuvants to enable the formation of predefined nanovaccine platforms that are uniform in size, highly stable and consistent in peptide loading. By anchoring different molecules on the backbone of NPs, and/or controlling the shape/morphology, antigens encapsulated into NPs release slowly and enhance immune responses, while antigens adsorbed onto NPs with quick release also improve immune responses.98 For example, synthetic long peptides (SLP) were used to produce a SLP-containing cationic lipoplexes (SLP-Lpxs) that improve delivery of peptides to the spleen and lymphatics. Using a KRASG12D mutation as a neoantigen, vaccination of mice with SLP-Lpx conjugated with the KRAS mutation peptides elicited potent CD4+ and CD8+ T cell responses that inhibited the tumor growth in three KRAS gene transfected mouse colon and lung cancer models. Results also showed that the composition of the liposome is more critical than the overall charge when it comes to site-specific delivery. These SLP-Lpxs increased targeting to the spleen and lymph nodes resulting in increased numbers of hematopoietic and T cell infiltration into the tumor. Furthermore, addition of CpG (ODN 1826) to the peptide–liposomes not only enhanced stability of the lipoplexes (Lpx) but they exhibited increased targeting and uptake by myeloid cells, specifically CD11b+CD11c− macrophages, CD11b+ DCs and consistently elevated antigen-specific CD8+ T cell responses while maintaining CD4+ T cell responses.99Nanoformulations provide unique properties for tumor vaccine in the areas of targeted delivery into desired organs, prolonged epitope persistence in vivo, retaining structures for immunogenicity and recognition, enhanced co-stimulation signals, and increased local inflammation to trigger non-specific proliferation of lymphocytes that enhanced overall efficacy of peptide-based vaccines.100,101 These formulations, in addition to acting as a vaccine delivery system, can be used as immune stimulation adjuvants. Therefore, strategically designed NPs have the potential for translational development of tumor specific peptide-based NP vaccines for cancer immunotherapy. Immune stimulating adjuvants, such as agonists for TLR and stimulator of interferon genes (STING) could enhance humoral immune response by inducing IFN-γ secretion from T helper cells and regulating MHC-II class antigen expression in APCs for the production of tumor antigen-specific cytotoxic T cells.100,101
For instance, a vaccine platform (SNP-7/8a) based on charge-modified long peptide with tumor antigen epitope-TLR-7/8a conjugates has been developed. The design of charge modifications on the peptides chemically programmed to self-assemble into micelle NPs of uniform size (∼20 nm) irrespective of the peptide antigen composition.102 This approach provided precise loading of diverse peptide neoantigens derived from the MC38 mouse colon tumor cells known to bind MHC-I (Aatf, Adpgk, Cpne1, Dpagt, Irgq, Med12 and Reps1) in combination with adjuvant TLR-7/8a in NPs102 (Fig. 2A). Results showed that the treatment with such tumor vaccine NPs increased uptake by and activation of APCs and promoted T-cell immunity. SNP-7/8a induced higher magnitude of T cells by both local (SC) and systemic intravenous (IV) routes of vaccination in comparison with that of native long peptide microparticles/aggregates (MP-7/8a).102 In addition, delivery of nonimmunogenic neoantigens (M01, M07, M21 and M39) from the B16-F10 melanoma tumor cell line in SNP-7/8a also limited tumor growth. SC or IV delivery of neoantigen peptides containing SNP-7/8a and adjuvant polyICLC increased DC recruitment, antigen uptake and activated CD8 T-cell responses in the lymph node of the mouse tumor model. The combination treatment of SNP-7/8a carrying tumor antigen peptides, Trp 1 or M39, with immune checkpoint inhibitors, an anti-PD-L1 antibody, resulted in significant tumor growth inhibition, with IV delivery having a stronger effect than SC delivery102 (Fig. 2A). Thus, the peptide-based vaccine platforms, such as SNP-7/8a, have the potential for priming tumor specific T cells and as combination therapy with other immunomodulatory agents, such as immune checkpoint inhibitors.
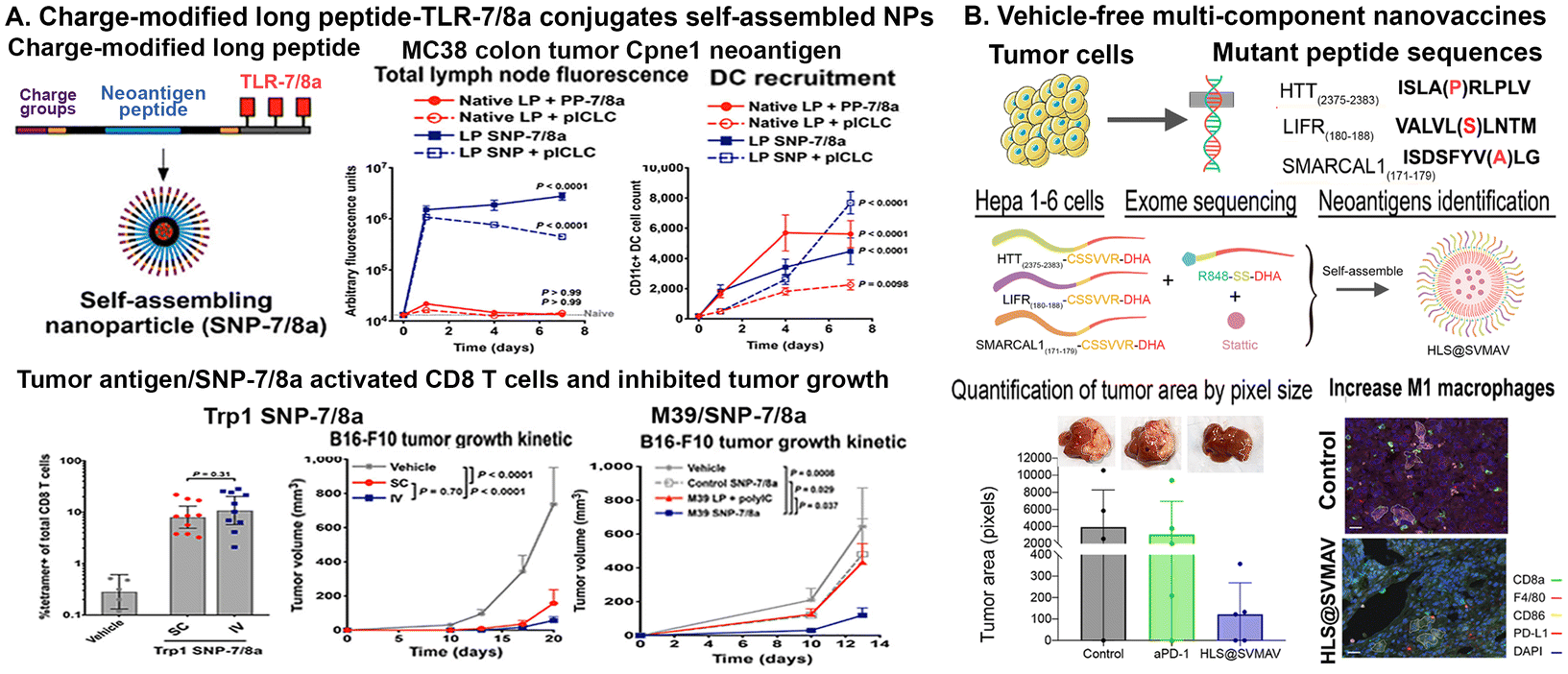 | ||
| Fig. 2 Self-assembling nanoparticles carrying tumor antigen peptides and immune modulatory agents for tumor vaccine. (A) A vaccine platform, SNP-7/8a, based on charge-modified long peptide with tumor antigen epitope-TLR-7/8a conjugates that are chemically programmed to self-assemble into nanoparticles of uniform size (∼20 nm). SNP-7/8a carrying Cpne1 neoantigen derived from a mouse colon cancer cell line (MC38) selectively accumulated in lymph nodes and increased DC recruitment and CD8+ T cell response after SC delivery into C57BL/6 mice (Upper panel). Tumor bearing mice received Trp 1 neoantigen in SNP-7/8a by the SC or IV injection, in combination with i.p. delivery of anti-PD-L1 antibody activated CD8 T cells and significantly inhibited the growth of B16-F10 mouse melanoma tumors. By i.v. injection of another neoantigen, M39/SNP-7/8a, anti-tumor growth effect was further demonstrated (Lower panel). Reproduced from Lynn, et al., Nat. Biotechnol., 2020, 38, 320–332, with permission from Springer Nature. (B) Self-assembling vehicle-free multi-component antitumor nanovaccines (SVMAV) carrying tumor specific neoantigen inhibited tumor growth and activated immune responses in an orthotopic mouse hepatocellular carcinoma model. Three neoantigens with mutations identified by exome sequencing of hepatoma cells (Hepa 1–6) were conjugated to DHA and self-assembled into nanoparticles. Liver tumor volumes were quantified by pixel counting. Immunofluorescence labeling of HCC tissues showed increased the F4/80+CD86+ M1 macrophages (dashed circles). Reprint from Zhang et al., J. Immunother. Cancer, 2021,9(8), e003132, published by BMJ, with permitted under CC BY 4.0. | ||
NPs have been engineered for the modular incorporation of neoantigens and adjuvants to promote activation and antigen cross-presentation of APCs and cross-priming of neoantigen-specific CD8+ T cells.103 The pool of vaccine primed T cells could be expanded to higher numbers in vivo using heterologous prime-boost immunization with vaccines and cytokines, such as IL-2, which promote T-cell expansion.104 Recently researchers have been investigating new approaches to improve uptake and interaction with DCs. Although targeting the activation of the innate immune system has been shown to increase antigen presentation, other methods can be used to improve presentation of tumor antigens to T cells, such as targeting the immunosuppressive environment or intrinsic immunosuppressive signaling. For example, silencing gene expression of an immunosuppressive factor, STAT3, in combination with nanovaccine delivery have been shown to decrease immunosuppressive cells and increase DC maturation.105,106 Therefore, by tailoring nanovaccine design, researchers could not only target neoantigens but also pathways involved in immune activation and antigen presentation. For example, a self-assembling vehicle-free multi-component antitumor nanovaccine (SVMAV) has been developed using an unsaturated fatty acid docosahexaenoic acid (DHA)-conjugated antigen and R848 (a TLR 7/8 agonist) encapsulated with stattic, a STAT3 inhibitor107 (Fig. 2B). To determine whether the platform could be used for both neoantigen vaccines and personalized vaccines, the SVMAV encapsulated with predicted mouse hepatoma Hepa1–6 cell line-specific neoantigens were mixed with R848-SS-DHA and stattic and then assembled into the personalized SVMAV loaded with mutant antigens, including Htt, Lifr, and Smarcal1107 (Fig. 2B). In an orthotopic hepatocellular carcinoma model, they showed that SVMAV efficiently migrated into Lymph nodes, promoted the antigen uptake by DCs, stimulated DC maturation, and enhanced antigen presentation, priming CD8+ T cells for exert neoantigen-specific killing. SVMAV did not only yield a strong antitumor effect for primary melanoma allografts, but also exhibited a protective effect for lung metastases. Moreover, the combination treatment of SVMAV and anti-PD-1 antibody showed synergistic antitumor activity and extended the survival duration of melanoma-bearing mice107 (Fig. 2B). Furthermore, new peptide NPs produced with extracellular matrix mimicking peptides (RLDI and RQDT) could self-assemble into ordered nanofibers, nanotubes, and nanovesicles.108 This platform increased diversity in peptide bond formation and enhanced stability due to the presence of two cysteine residues at their ends that resulted in the ability to recruit DCs, promote their maturation, target lymph nodes efficiently, which addressed the challenges associated with insufficient numbers, immature states, and the low homing efficiency of DCs. In the MC38-ovalbumin (OVA) colon cancer model, OVA peptide antigen or peptides from tumor cell lysate were slowly and continuously released from the hydrogel. This peptide hydrogel and released antigens enhance the activation of CD8+ and CD4+ T cells and differentiate CD8+ T cells into cytotoxic T lymphocytes, resulting in killing tumor cells. When combined with the tumor vaccine and anti-PD-1 treatment, RLDI and RQDT effectively boosted the immune activation, further enhancing the antitumor efficacy of the vaccine without causing noticeable side effects.108 Therefore, by combining peptide/protein-based vaccines with other peptides or agents that act as adjuvants or immune modulators, it is possible to further enhance the vaccine's ability to induce a strong and durable anti-tumor immune response.
Antigen presenting cell mimetic NPs
NPs have been rationally designed to carry TAAs or TSAs with costimulatory molecules and adjuvants to facilitate activating tumor specific T cell responses. These NPs are called artificial antigen presenting cells (aAPCs) due to their ability to directly activate the TCRs of the antigen epitope specific T cells64 (Fig. 3A). The activation of CD4+ and CD8+ T cells begins with the presentation of TAA or TSA antigen peptides together with MHC molecules by antigen-presenting cells (APCs) to T cells. The cytotoxic effect of a CD8+ T cell requires its TCRs to recognize a specific antigen peptide presented by MHC I molecule on tumor cells. The binding of the TCRs of the CD8+ T cells to the peptide/MHC 1 complex led to activate CD8+ T cells to initiate tumor cell killing.109 It has been shown that the binding of multiple TCRs of a T cell with antigen/MHC complexes (>20 complexes) is necessary to activate CD8+ T cells.110 However, a low level of tumor antigen expression and lack of strong immunogenic epitopes as well as downregulation of MHC molecules in APC (MHC II) and tumor cells (MHC I) due to immune evasion limit the ability of activation of CD4+ helper and CD8+ cytotoxicity T cells.1,6 To address these challenges, novel strategies have been developed to enhance T cell activation and overcome immunosuppression within the TME.12 One promising approach involves the design of engineered NP-based APCs conjugated with a relatively high density of tumor antigen/MHC complexes that enable simultaneously engaging multiple TCRs to active antigen specific T cells in a more potent and targeted manner64,111 (Fig. 3A and B). NPs have a large surface area and can be functionalized for controlled conjugation of tumor antigen peptide/MHC I/β 2-microglobulin complexes at a desired density for the TCR binding and T cell activation. Shapes, sizes, and nanomaterials can be optimized to mimic the physiological interactions among T cells and APCs in the artificial membrane to cluster the TCR–MHC molecules, and formation of immune synapse to achieve a maximal effect of T cell activation.112–114 Importantly, additional costimulatory factors can also be conjugated to the same NP to provide necessary secondary signals for T cell activation.115 To build upon the concept of a high density of ligands affecting stimulation, a study compared two systems of aAPC, one in which monosialoganglioside (GM1) and cholera toxin B MHC–peptide complexes were uniformly distributed on the surface of the liposomal aAPC and another in which the same MHC–peptide complexes were clustered in microdomains on the aAPC surface. Using stimulation assays comparing the two types of aAPC with natural APCs, they were able to induce ex vivo activation of human polyclonal antigen-specific CD4+ T cells while also showing the advantages of mimicking the naturally patchy distribution of T cell ligands on APC membranes to efficiently manipulate T cells.116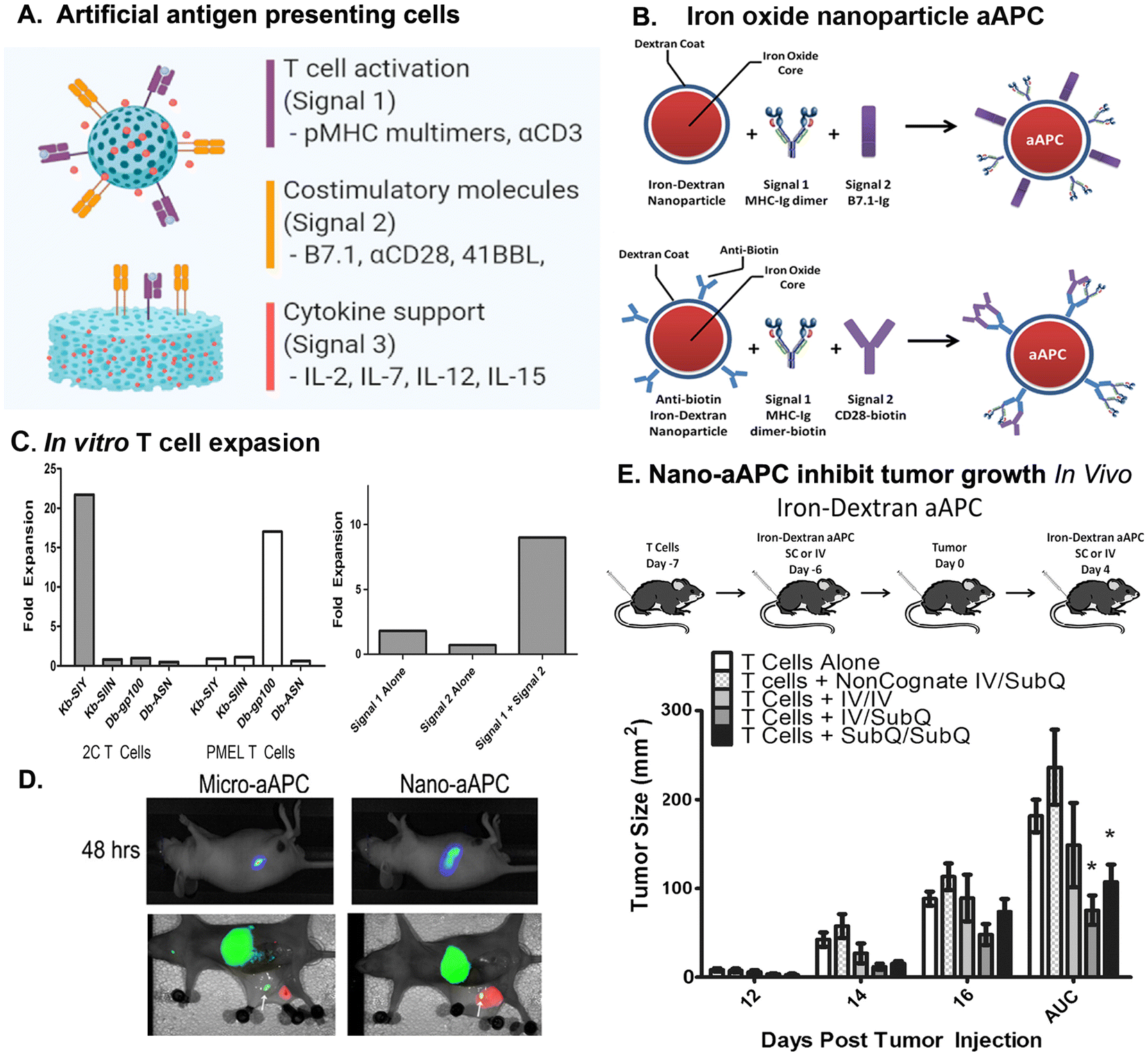 | ||
| Fig. 3 Design of aAPCs and the effect of iron oxide nanoparticle based aAPC on T cell expansion and tumor growth inhibition in the B16 mouse melanoma tumor model. (A) A nanoparticle aAPC consists of a nanoparticle core conjugated with signal molecules, including Signal 1 to activate T cells (pMHC molecule or an αCD3 antibody for antigen-specific or polyclonal T cell activation) and Signal 2 as costimulatory molecule (agonistic αCD28 antibodies or B7.1 molecules). Additionally, cytokines (IL-2, IL-7, IL-12, or IL-15) are important for T cells activation that serve as Signal 3. Reprinted from Isser, et al., Biomaterials, 2021, 268, 120584, with permission from Elsevier. (B) Synthesis and characterization of iron-dextran nano-aAPC. Nano-aAPC (size 50–100 nm) were synthesized in one of two ways: (1) direct chemical coupling of soluble MHC-Ig dimer (signal 1) and B7.1-Ig (signal 2) to the surface of iron oxide, dextran-coated particles. (2) Binding of biotinylated MHC-Ig dimer (signal 1) and biotinylated anti-CD28 (signal 2) to anti-biotin coated particles. (C) Nano aAPC induced T cell expansion is antigen-specific and depends on both signal 1 and signal 2. (D) Optical imaging shows that Nano-aAPCs induce a high level of lymph node drainage following s.c. injection. Simultaneous NIR images of biodistribution for pMEL T cells (green) and aAPC (red) revealed that IV delivered T cells accumulated in the lymph nodes and spleen. SC injected nano-aAPCs reached inguinal lymph node. (E) Iron-dextran aAPCs, injected IV or SC, activate naive pMEL T cells and inhibited the growth of mouse melanoma tumors. (B, C, D, &E) Reproduced from Perica, et al., Nanomedicine, 2014, 10, 119–129, with permission from Elsevier. | ||
Since the percentage of tumor antigen specific CD8+ cells are extremely low in vivo, ex vivo expansion of those tumor specific CD8+ T cells and then infusion back to the patients have the potential to increase therapeutic efficacy. Antigen specific aAPCs have been used ex vivo to activate and induce proliferation of tumor antigen specific T cells, as well as their potential for controlled delivery to the tumor site for in vivo stimulation of cytotoxic T cells. The earlier aAPCs used microbeads to produce aAPC by coupling HLA-A2 Ig and anti-CD28 human monoclonal antibody onto Dynabeads.117 These NPs successfully generated Mart-1- and CMV-specific cytotoxic T cells from human peripheral blood T lymphocytes and showed robust expansion and functional activity ex vivo118 and in vivo in a human/SCID mouse melanoma model.117 It has been shown that 7–30 TCRs form protein clusters with a dimension of 35–70 nm in radius and 300 nm at the longest length scale on the cellular membrane.119 The peptide/MHC complexes on APCs also form 25–125 complex cluster with sizes about 70 to 600 nm.120 A previous study showed that activation of T cells requires a minimal density of 90–140 stimulating MHCII molecules per μm2 of membrane area.121 It has been shown that CD4+ T cells are insensitive to activation when the density of peptide/MHC is very low and a maximum linear distance of 60–70 nm is required for CD4+ T cell activation. Using 50, 300, 600, and 4500 nm size of superparamagnetic iron oxide NPs conjugated with a T cell specific SIY tumor antigen peptide (SIYRYYGL)/MHC-Ig complex and anti-CD28 antibody, ex vivo stimulation study showed that aAPCs could bind to antigen specific TCRs on CD4+ or CD8+ T cells to induce TCR clustering and T cell activation in a size dependent manner.122 The aAPCs that are larger than 300 nm activate T cells more efficiently than smaller 50 nm aAPCs. The 50 nm aAPCs require saturating doses or require artificial magnetic clustering to activate T cells. Thus, there is a need to engage local islands of peptide/MHC complexes and co-stimulatory aCD28 antibody with a greater than 50 nm in diameter for effective CD8+ T cell stimulation. In another study, nano-aAPCs were produced by direct chemical conjugation of MHC-Ig dimers loaded with GP100 peptide to iron oxide NPs (50–100 nm in diameter)111 (Fig. 3B). Tumor antigen peptide(GP100)/MHCI (Signal 1) and anti-CD28 antibody (signal 2) dependent expansion of tumor antigen specific T cells using the nanoaAPCs were demonstrated in vitro in cultured mouse splenocytes derived from pMEL (pMEL TCR/Thy1a Rag−/−) transgenic mice, whose T cells recognize GP100 peptide and MHC Class I H2-Db111 (Fig. 3C). In comparison with large microparticle aAPCs in several μm size, nano-aAPCs induced a higher level of lymph node drainage following SC injection than Micro-aAPCs (Fig. 3D). The iron-dextra aAPCs, injected either IV or SC, activated adoptive transferred pMEL T cells, leading to tumor rejection in the mouse melanoma model (Fig. 3E). The activation of cytotoxic T cells by the nano-aAPCs was further supported by the detection of the production of critical effector cytokines, such as IFNγ, secreting cytotoxic granules, and surface expression of the degranulation marker CD107a.111 On the other hand, the aAPCs produced from iron-dextran NPs coated with murine MHC II or human counterpart HLA II loaded with GP61–80 peptide/MHC II complexes and co-stimulatory proteins could activate and expand rare subsets of endogenous murine and human CD4+ T cells ex vivo. These MHC II aAPC expanded murine CD4+ T cells displayed high levels of effector cytokine production and demonstrated lytic capacity in vitro and in vivo in the B16 melanoma mice model, which, in turn enhanced cytokine production and memory formation in CD8+ T cells, while also specifically expanding initially undetectable antigen-specific murine and human CD4+ T cells from endogenous T cell repertoires.123 By targeting specific MHC/peptide complexes and costimulatory molecules, NPs have the capability of enriching, expanding and modulating the effector and helper functions of CD4+ T cells. The ability of aAPCs to deliver to lymphoid organs and tumors to activate tumor antigen specific T cells should have a great impact on cancer immunotherapy. Therefore, aAPCs resemble nanometer-scale structures of signaling molecules of APCs that should be more efficient in interaction with TCRs on the effector T cells to activate their function. By providing peptide loaded MHC and costimulatory factors, these NPs can be used in vitro for ex vivo stimulation to expand antigen-specific T cells as well as in vivo to induce specific/rare endogenous subsets of antigen specific T cells.
The feasibility of the application of aAPC in cancer patients was examined in a clinical trial. Results showed that aAPCs produced by conjugating HLA-A*0201 molecules coupled with tumor antigen MART-1 or gp100 class I-restricted peptides and costimulatory anti-CD28 antibody to iron-dextran NPs rapidly expanded the antigen-specific CD8+ T cells from the peripheral blood obtained from melanoma patients without or with received anti-PD-1 antibody treatment.124 The expanded CD8+ T cells have high avidity and potent lytic function. CD8+ T cells had a predominantly memory stem cell phenotype (CD45RA+/CD62L+/CD95+) that expressed ICOS, PD-1, Tim3, and LAG3; and lacked CD28. CD8+ T cells from patients with melanoma were polyfunctional, which had highly diverse T-cell receptor V beta repertoire, expressed IL-2, IFNγ, and TNFα, and exhibited cytolytic activity against tumor cell lines.124 As an emerging technology, aAPCs have the potential to induce therapeutic cellular immunity without the need for autologous antigen-presenting cells (APCs). However, some of the limitations of aAPCs include only selected MHC specific antigen peptides can be used to stimulate specific T cell populations and difficulties in completely engineering an immune costimulatory microenvironment that resembles complicated in vivo immune responses. Therefore, researchers have been looking to identify optimal formulations to further mimic APCs by adding cytokines to manipulate the response. These aAPC NPs present a high density of adaptor elements for attaching both recognition ligands and co-stimulatory ligands to a biodegradable core encapsulating the different cytokines implicated in T cell differentiation and function, including expansion, survival, effector function, and memory of stimulated T cells.125 By providing a local and sustained release of cytokines, coupled with the ease of attaching multiple ligands, it is feasible to produce a durable, artificial antigen-presenting system for T-cell expansion.
Enhancing the immune response with NP delivery of mRNAs encoding tumor antigens as tumor vaccines
In the intricate realm of antitumor immunity, T cells take center stage as the orchestrators of the anticancer immune response, closely collaborating with the rest of the immune system. mRNA can act as a pivotal link between innate and adaptive immunity, encoding antigens that trigger antigen recognition, costimulation, and cytokine secretion to drive this process. mRNA vaccines are developed using mRNAs encoding known tumor antigen or newly identified immunogenic tumor antigen gene sequences from genetic and proteomic analyses of tumor samples.126,127 mRNAs were transcribed from engineered mRNA expressing plasmids and purified in vitro, and these mRNAs are then used for in vivo delivery and uptake by APCs for translating and processing into antigenic peptides for presentation.126,127 However, there are several barriers to success of mRNA vaccines, including (1) extracellular barrier leading to degradation, (2) endocytic barriers prevent uptake, (3) endosomal and cytosolic barriers preventing translation, and leading to (4) inefficient immune stimulation via limited MHC presentation and production.128 Strategies to address these challenges have been developed to increase stability and translation of mRNAs, such as incorporating modified nucleosides to increase stability and optimizing untranslated regions in mRNAs to improve translation and expression126 (Fig. 4A). To further increase stability and delivery, lipid nanoparticles (LNPs) have been used for the development of mRNA-based therapeutics. Encapsulation of mRNAs in LNPs increases cell uptake by endocytosis that leads to cytoplasmic translocation and protein translation and processing to finally form a complex with MHC I/II and β 2-microglobulin for antigen presentation.129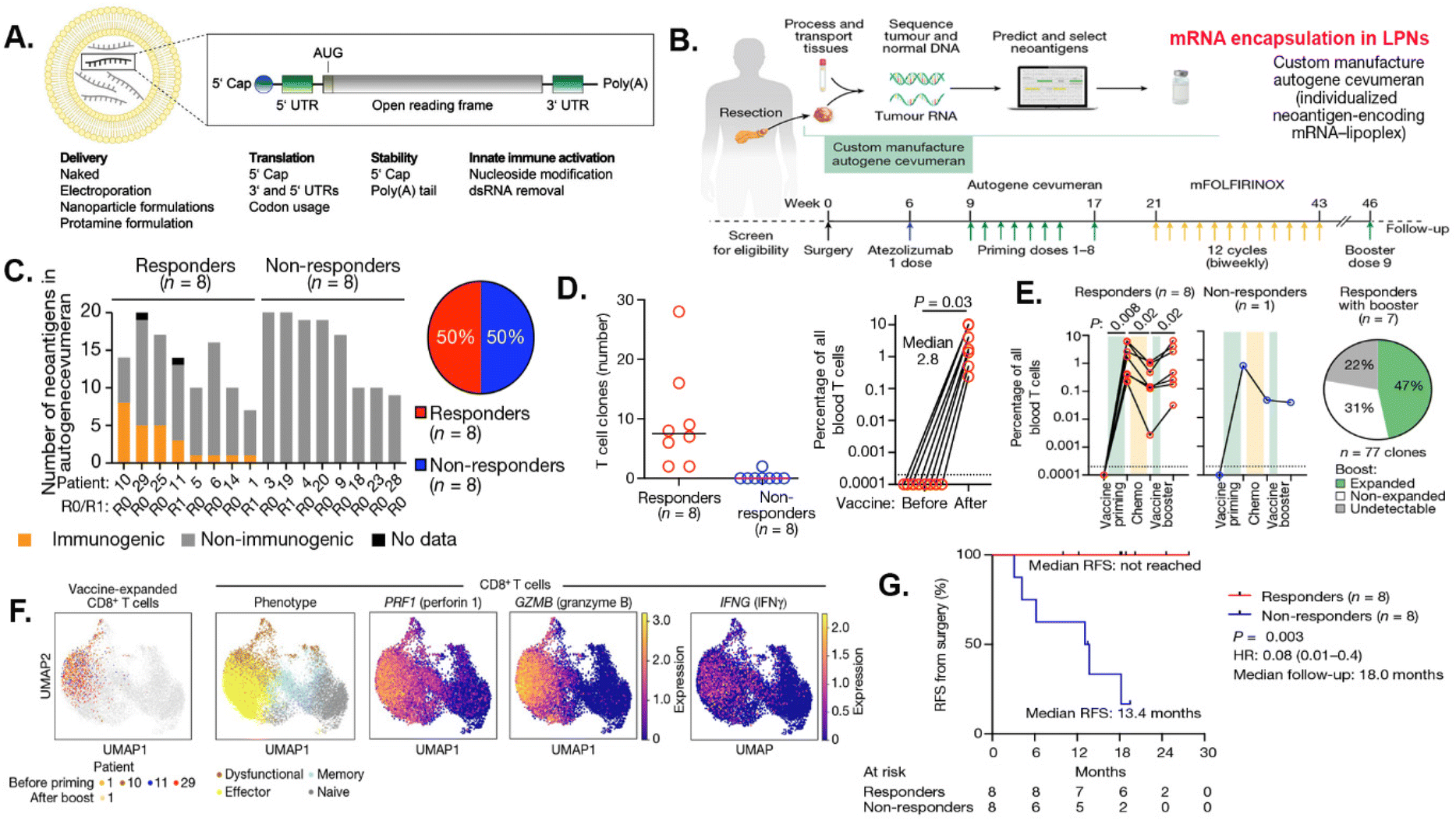 | ||
| Fig. 4 Lipid nanoparticle mRNA carriers for personalized RNA neoantigen vaccines that activated tumor specific T cell responses in pancreatic cancer patients. (A) Illustration of design and structure of a lipid-based mRNA nanoparticle and synthetic mRNA. Reprint from Beck et al., Mol. Cancer, 2021, 20, 69, with permission from Springer Nature. (B) A clinical trial design for identification of neoantigens, mRNA production and encapsulation, and immunization and treatment protocols in pancreatic cancer patients. An individualized mRNA neoantigen vaccine contained up to 20 MHC I and MHC II restricted neoantigens in LPNs was delivered IV into the patients. (C) PBMCs collected from pancreatic patients after atezolizumab, vaccine priming, and mFOLFIRINOX were analyzed for IFN-γ+ T cells specific to all individual vaccine neoantigens by ELISpot. Proportion of vaccine responders and non-responders is shown. (D) Number of unique antigen specific T cells clones before vaccine (left) and peak expansion aggregate percentage (right). (E) Aggregate percentage of vaccine-expanded T cell clones with priming, chemotherapy and booster in peripheral blood and percentage of primed clones that re-expand with booster (right). (F) Single-cell phenotypes of vaccine-expanded CD8+ T cells. Dots indicate blood CD8+ T cells. Colored dots (far left) indicate vaccine-expanded clones. (G) mRNA vaccine response correlates with delayed PDAC recurrence in pancreatic cancer patients. Recurrence-free survival (RFS) from surgery or from the date of the last vaccine priming dose is shown. (B, C. D, E, F and G) Reproduced from Beck et al., Nature, 2023, 618, 144–150, with permission from Springer Nature. | ||
Lipid nanoparticle as mRNA delivery carrier
RNA-lipoplexes or lipid NPs (LNPs) encapsulated with mRNAs have shown promise in protecting RNAs from ribonucleases in vivo and efficiently delivering encoded antigens to DCs and macrophages in various lymphoid compartments.126,129 This delivery strategy addresses challenges, such as nucleic acid degradation, low cellular uptake, and increase the blood half-life of mRNAs after systemic administration.126,129 LNPs play a crucial role in the delivery of mRNAs for therapeutic applications, such as vaccines and gene therapies. These NPs are composed of lipids with both hydrophilic and hydrophobic components, allowing them to self-assemble into structures like vesicles or micelles, forming the core structure of LNPs.126,129 The process involves formulating LNPs with cationic lipids like DOTAP or DOTMA, helper lipids such as cholesterol, and polyethylene glycol (PEG) for stability. Cholesterol can also improve intracellular delivery of mRNAs. The negatively charged mRNA molecules then interact with cationic lipid and cholesterol to be encapsulated within LNPs, protecting them from degradation.129 LNPs are designed to interact efficiently with cell membranes to facilitate cellular uptake. After uptake by APCs, LNPs escape endosomes to release mRNAs into the cytoplasm and bind to ribosomes for translation of antigen proteins or peptides, which are then processed into antigenic peptides to form a complex with MHC II for antigen presentation. Extensive research into the application of LNPs for mRNA delivery has been ongoing prior to the COVID-19 pandemic. However, the remarkable success of mRNA-based COVID-19 vaccines has significantly accelerated both the progress and public interest in this field.130 The demonstrated efficacy and safety of these vaccines have propelled mRNA technology into the spotlight, garnering increased attention and resources for further advancements and applications beyond infectious diseases.By screening different lipids and LNP formulations, researchers found that addition of chemical modifications, like esters/disulfide bonds, thiols and amines, modification of LNP structures, incorporating hydrophobic tails or changes in the lipid membrane components of LNPs, leading to the selective delivery into lymphoid organs, or precise delivery into the tumor for in situ tumor vaccine.131–134 Those modifications endowed LNPs with less inner hydrophobicity and fewer surface charges with proper stability and balanced hydrophobicity that benefit the cellular uptake of LNPs and the intracellular trafficking of mRNAs, thus resulted in improved delivery efficiency of mRNAs. LNP-mRNA vaccines effectively activate T cell immunity and produce anti-tumor effect. In preclinical studies, LNP-carrying mRNAs encoding for Trp-2 TAA induced strong CD8+ T cell activation and decreased tumor volume with an increased animal survival in the highly aggressive B16-F10 murine melanoma model and reduced the incidence of tumor metastatic nodules in the lung.135 In a different study, lymph node-targeting LNP carrying OVA mRNAs increased the expression OVA antigens in the lymph nodes compared with LNP formulated with ALC-0315, a synthetic lipid used in the COVID-19 vaccine.136 Results also showed that the chemical structures influenced mRNA expression and the optimal formulation contained shorter length lipids, ester linkers, and methyl groups of the amine head led to efficient delivery into lymph nodes. A recent report from a phase 1 clinical trial (NCT02410733) using a RNA-lipoplex vaccine (RNA-LPX) encoding four melanoma TAA (NY-ESO-1, MAGE-A3, tyrosinase, and TPTE) in combination with the immune checkpoint therapy showed the ability of mRNA-LNPs to induce a durable and strong CD4+ and CD8+ T cell immunity against the above tumor antigens in cancer patients with unresectable melanoma.137
Personalized neoantigen mRNAs for tumor vaccines
Tumor vaccines using known TAA or TSA epitopes have the ability to induce immune responses to tumor antigens that present in cancers without the challenges of identifying a patient's unique individual TAAs or TSAs for synthesizing a custom vaccine strategy. Immunogenicity or the ability to trigger an immune response, is closely correlated with mutations shown by tumors with higher mutational loads are generally more immunogenic. Personalized tumor vaccines are essential due to the inherent diversity in the genetic makeup of tumors among individual patients. Novel LNP-mRNA vaccines and treatments hold tremendous potential for future development of tumor vaccines, with improved targeting strategies, and the generation of individualized, multiepitope neoantigens. Currently, about 70 clinical trials using LNPs/mRNAs for cancer vaccine are ongoing.138,139 Therefore, the next step for the future cancer vaccines includes the development of personalized medicine using tumor specific neoantigens, as well as exploring the combination with other immunotherapeutics.To improve the effect of tumor vaccine in highly heterogeneous human tumors, mRNA vaccines containing multiple neoantigens identified from tumor tissues by the whole genome sequencing have been developed.140,141 In a phase I/II trial (NCT03480152), tumor tissues were analyzed by exome sequencing to identify mutations and predicate neoepitopes for constructing a single mRNA encoding up to 20 different antigens, including HLA class I candidate neoantigens with patients’ specific mutations in TP53, KRAS, or PIK3CA gene. Results of this trial showed encapsulating these mRNA into LNPs were safe and elicited mutation-specific T cell responses against predicted neoepitopes.140 In another phase 1 clinical trial, genetic mutations in surgically resected tumors in pancreatic cancer patients were identified by whole genome sequencing. Immunogenic neoantigen peptides were selected to engineer a multiplexed mRNA consisting of 20 immunogenic neoantigen epitopes (autogene cevumeran) (Fig. 4B). Results showed that a combination of adjuvant personalized tumor vaccine, with immune checkpoint therapy, atezolizumab (anti-PD-L1 antibody), and a modified version of a four-drug chemotherapy regimen (mFOLFIRINOX, comprising folinic acid, fluorouracil, irinotecan and oxaliplatin), significantly increased recurrence-free and overall survival of pancreatic cancer patients141 (Fig. 4C). The above treatment led to the activation of T cells producing IFNγ and expressing lytic markers such as perforin 1 and granzyme B. The combination therapy was tolerable and induced de novo expansion of neoantigen-specific T cells, with half of them targeting more than one vaccine neoantigen. In addition, patients with vaccine-expanded T cells (responders) had a longer median recurrence-free survival compared with patients without vaccine-expanded T cells141 (Fig. 4D–G). Novel LNP-mRNA vaccines and treatments hold tremendous potential for future development of tumor vaccines, with improved targeting strategies, and the generation of individualized, multiepitope neoantigens.
Immunomodulatory effect of optimized LNPs with mRNA vaccines
To further improve the effect of mRNA tumor vaccine and broaden the vaccine applicability, adjuvants to enhance antigen presentation and boost the immune response have been incorporated in the LNPs, leading to stronger and longer immunity. Multifunctional NPs that are able to deliver both tumor antigens and adjuvants have the potential to enhance immune responses. These NPs can be engineered to target specific sites and cells (i.e. tumor, draining lymph nodes,93 DCs,142 macrophages143), and be modified with additional ligands to improve circulation, infiltration, and interactions. Increasing efforts has been devoted toward the development of LNP with self-adjuvant effects by optimization of lipid components of NP to activate TLR or STING to promote APC maturation, antigen process and presentation, triggering enhanced antitumor immunity and effective inhibition of the tumor growth. TLRs are innate immune receptors that recognize pathogen-associated molecular patterns to activate immune responses by triggering a signaling cascade that leads to upregulation of co-stimulatory molecules and secretion of pro-inflammatory cytokines, leading to the activation and maturation of APCs and promoting effective antigen presentation to T cells.144 STING is a key adaptor protein in the cytosolic DNA sensing pathway that induces the production of type I interferons and proinflammatory cytokines. It has been shown that LNPs produced with lipids with cyclic amino head groups activated STING pathway and induced APC maturation. Treatment with the cyclic lipid LNPs carrying mRNAs encoding a tumor-associated antigen (Trp2) inhibited tumor growth and prolonged mice survival in the B16F10 melanoma model.145 Furthermore, a combination of the LNP delivery of a viral oncogenic (papillomavirus E7 protein) mRNA with anti-PD-1 antibody therapy induces a strong anti-tumor response and increased CD8+ T cell infiltration and activity in tumors in the TC-1 mouse lung cancer model.145 Additionally, screening ionizable lipids of LNP formulations using human primary APCs led to the identification of a L17-F05 mRNA delivery LNP with six-membered cyclic amine heads with a high mRNA loading. Subcutaneous delivery of L17-F05 mRNA vaccines led to selective accumulation in APCs of the lymph nodes activated STING pathway in macrophages, enhanced innate immune responses, upregulated proinflammatory genes, including IFNb1, IL-6, and TNF, and IFN-I production, which promoted maturation of APCs. L17-F05 mRNA vaccines encoding OVA, Gp100 and Trp2 inhibited tumor growth and prolonged the survival of mice bearing B16F10 melanoma.133,146 Furthermore, agonists for TLR and STING have been used in multifunctional LNPs to either co-deliver both antigen and molecular adjuvants as one vaccine or to act as adjuvant therapies to mRNA vaccines.145,147 LNP-delivered cGAMP (cGAMP-NP) effectively activated STING pathway and induced innate and adaptive host immune responses to preexisting tumors in a mouse triple negative mammary tumor model (C3(1)Tag). cGAMP-NP treatment induced M2 to M1 macrophage transition, increased intratumoral infiltration of IFN-γ-producing CD8+ T cells and further prevented the development of secondary tumors.148 Systemic administration of stearic acid lipid nanoparticles with ovalbumin (OVA)-coding mRNA and TLR4 agonists (MPLA) led to the selective delivery in the spleen and induced strong antigen-specific cytotoxic T cell response and persistent immune memory, preventing the growth of EG.7-OVA tumors.149 Taken together, by designing NPs for efficient co-delivery of both neoantigens and synergistic adjuvants, the engineered nanovaccines can elicit potent neoantigen-specific immune responses. By targeting sites of the lymphoid system or TME, and stimulating the innate and adaptive immune response, these nanocarriers with multiple agents can promote DC maturation and antigen presentation, and ultimately abrogate immunosuppression in TME.NP-mediated modulation of costimulatory molecules and signals regulating CD8+ T cell function
An effective T-cell response involves a coordinated effort of various immune cells. This collaborative and orchestrated response from macrophages, DCs, B cells, NK cells, and neutrophils ensures the elimination of threats and the establishment of immune memory for future protection while conversely impacting T cell function through costimulatory molecules and cytokine production, ultimately shaping the inflammatory milieu. Cytotoxic T cell activity is tightly regulated by functional status of CD8+ T cells, cytokines and immune checkpoint factors.1 Costimulatory molecules play a pivotal role in regulating immune responses. They ensure T cells receive adequate signals before initiating an immune response. Nevertheless, there are two types of signals crucial in manipulating response. They can provide signals that either enhance (immunostimulatory) or dampen (immunoinhibitory) the activation of immune cells, particularly T cells. Cytokines such as IL-2, IL-12, IL-21, IL-15 and IL-18, have been shown to be involved in the activation of T cell function.150 Immunostimulatory molecules like CD28, CD40, and ICOS provide signals that promote the activation, proliferation, and effector functions of T cells2,3 whereas immunoinhibitory molecules like PD1, LAG3, and CTLA-4 provide signals that suppress T cell activation, preventing excessive immune responses. Additionally, the release of immunosuppressive soluble factors, TGF-β, and cytokine IL-6, and IL-10, further contributes to the hostile tumor environment acting to downregulate T cell function, impairing their ability to recognize and eliminate cancer cells.2,150,151 The balance between immunostimulatory and immunoinhibitory signals is critical for a functional immune system and maintaining immune homeostasis. Therapeutically modulating these signals can have overwhelming results for treating cancer, from enhancing immune responses against cancer.NPs are ideal carriers for delivery multiple immunostimulant agents
NPs can be used to target different cellular receptors that have been implicated in controlling T cell homeostasis. Incorporating biomolecules with T cell activation function, such as small molecules, nucleic acids, peptides, and proteins in the NPs can enhance T cell activity via direct interaction or uptake by cells. NPs are used as a combined immunotherapy to enhance immune activation and reverse immunosuppression by efficiently delivery into immune organs to activate T cells,152 influencing costimulatory receptors on T cells, such as 4-1BB and OX40, or delivering stimulating agents, i.e. antibodies,153 checkpoint inhibitors154 or the combination of both.155 It has been shown that conjugating anti-CD40 antibody and CpG to the surface of PEGylated liposomes, restricted the immunostimulatory effect to the treated tumor and lymph nodes, resulting in tumor growth inhibition while minimizing the inflammatory side effects associated with systemic exposure to these agonists.156 An “Immunoswitch particle” consisting of iron-dextran NPs conjugated with an agonistic antibody against 4-1BB (a co-stimulatory receptor on effector T cells) and antagonistic antibodies against PD-L1 was developed, which enabled the simultaneous blockade of the inhibitory checkpoint PD-L1 signal and stimulation of T cells via the 4-1BB co-stimulatory pathway.157 In mouse melanoma and colon cancer models, co-delivery of those immunomodulatory agents led to a stronger anti-tumor effect compared to that of the single antibody treatment. Activation of tumor-specific T cells was evident by increased IFN-γ production, CD107a expression, and detection of polyclonal T cell repertoires against various tumor antigens.157 In another approach, phospholipid-derived NP, PL1, was developed to deliver OX40 mRNA, a costimulatory receptor for activating CD8 and CD4 T cells. Results showed that PL1-OX40 mRNA NPs induced the OX40 expression in both T cells and DCs in vitro and in vivo. Furthermore, a combination of PL1-OX mRNA delivery with an agonistic anti-OX40 antibody exhibited significantly improved antitumor activity compared to anti-OX40 antibody alone in multiple mouse tumor models.158 In both A20 lymphoma and B16F10 tumor mouse models, the above therapy was further combined with anti-PD-1 and anti-CTLA-4 antibodies, resulted in enhancement in the immune response, reduced lung metastasis and protected from B16F10 tumor cell re-challenging.158In addition, NPs act as carrier systems targeting prominent immune checkpoint receptors and their interactions with cognate ligands to maintain T cell activation over an extended duration. NPs have the ability to carry different immunostimulatory agents to block or silence specific genes in order to improve the responsiveness of cancer immune checkpoint therapy to activate cytotoxic effect of CD8+ T cells.159,160 To overcome immune exhaustion, a PD-1 gene engineered exosome together with an immune adjuvant imiquimod (PD1-Imi Exo) was used to block the PD1/PDL1 while releasing imiquimod to promote the maturation of dendritic cells. The treatment reversed the effect on the immune exhaustion through activating and restoring function of CD8+ T cells while increasing the memory T cells in the spleens.161 Results showed that PD1-Imi Exo had a strong binding with both cancer cell and DCs, and demonstrated a remarkable therapeutic efficacy in the melanoma tumor-bearing mice as well as in the mammary tumor-bearing mice.161 By combining multiple stimulatory agents targeting the various mechanisms involved in t cell activation, it was possible to reverse CD8+ T cell exhaustion and increase CD8+ T cell cytotoxic activity.
NPs overcome the limitations and risk of cytokine therapy
Cytokines are small proteins that play crucial roles in immune responses, acting as signaling molecules that regulate immune cells via proliferation and survival, immunomodulation, and differentiation. They are produced by a variety of cells, including immune cells such as macrophages, T cells, and B cells, as well as non-immune cells like fibroblasts and endothelial cells.150,162 Delivery of immunostimulatory cytokines to activate immune cells, especially CD8+ T cells, showed promises in cancer immunotherapy, however the pluripotent effects of cytokines and systemic side effects limit their clinical applications.163 Using NP to deliver cytokines involved in the differentiation and activation of T cells, such as IL-2 and IL-12, blocking cytokines/agonists involved in immunosuppression like IL-15 and GMCSF or a combination of different cytokines, the effects of significant tumor growth inhibition and reduction in systemic toxicity have been demonstrated in various mouse tumor models.164–171 For example, IL-2 is a potent T cell activator for immunotherapy, but has severe systemic toxicity. Thus, IL-2 was loaded into porous silica NPs (degradable nanoBALL), which improved intratumoral delivery of IL-2 following intravenous injection, resulting in a sustained and targeted release in the tumors in the B16F10 mouse tumor model164 Such an approach also showed significant improvement in pharmacokinetics in healthy non-human primates following both SC and/or IV injection.164 The sustained and targeted release kinetics stimulated the proliferation of CD8+ T cells without affecting the development of regulatory T cells, a common side effect associated with excessive IL-2. NP delivery of IL-2 not only accelerated the recruitment of NK, DCs and T cells in vivo at the tumor site and secondary lymphoid organs but also showed no notable exhaustion or immunosuppressive effects on functional T cells, including CD8+ and CD4+ T cells.164 The treatment resulted in stronger anti-tumor efficacy at lower doses, thereby reducing systemic toxicity compared to systemic IL-2 in both subcutaneous and orthotopic mouse melanoma models.The ability of nanodelivery systems to generate long-lasting, tumor-specific immune responses without the toxicity, was further shown to enable the combination of multiple therapeutic approaches. Notably, preclinical studies demonstrated efficacy of NP-mediated delivery of IL-12 alone and in combination with other treatments like chemotherapy in several cancer types.167,172–174 These data led to the development of EGEN-001 (GEN-1), which is a novel immunotherapeutic agent comprising a human IL-12 expressing plasmid encapsulated within a synthetic polyethyleneglycol–polyethyleneimine–cholesterol (PPC) designed to facilitate plasmid delivery in vivo. In ovarian tumor bearing mice, this NP was able to shift the immune response towards a Th1 state, improving mouse survival after treatment with GEN-1 alone or with taxol/paraplatin chemotherapy.175 Results from a clinical trial in ovarian cancer patients with recurrent or persistent epithelial ovarian cancers (EOC) showed that the combination of GEN-1 with pegylated liposomal doxorubicin induced partial responses with a clinical benefit of 57.1% in the patients. Intraperitoneal delivery led to NP/drug accumulation in peritoneal cavity and increased the levels of IFN-γ and TNF-α, demonstrating activation of immune pathways that are important to T cell activation and function.176 NPs have also been used to deliver cytokines and/or agonists that block immunosuppressive factors within the TME while simultaneously stimulating key immune effector cells. For example, a biodegradable core–shell NP that combines features of PEGylated liposomes and polymers enabled sustained and simultaneous release of both hydrophobic drugs and hydrophilic cytokines.155 Nanoscale liposomal polymeric gels (nanolipogels; nLGs) encapsulated with IL-2 and TGF-β receptor-I inhibitor, SB505124, was developed.156 Treatment with nLGs releasing TGF-β inhibitor and IL-2 significantly delayed tumor growth and increased survival in B16/B6 melanoma-bearing mice.156 It has been shown that the antitumor effect was induced by increasing in the activity of NK cells and intratumoral infiltration of activated CD8+ T-cells while reducing the accumulation of regulatory T cells after intratumoral or systemic administration. Additionally, LNPs encapsulated with mRNAs encoding cytokines, including IL-12, IL-27 and GM-CSF, have been developed. By engineering ionizable lipids containing di-amino and various head groups, two or more mRNAs were encapsulated in the NPs while also influencing the particles’ interactions with cellular and endosome membranes. Successful delivery of three types of cytokine mRNAs into tumors and tumor growth inhibition have been demonstrated in a melanoma tumor model.177 Intratumoral injection of the NP/cytokine mRNAs led to a marked infiltration of immune effector cells, including IFN-γ and TNF-α producing NK and CD8+ T cells in tumors.177 NP-mediated delivery of multiple cytokines has the potential to orchestrate a coordinated and potent immune response against cancer cells by boosting or prolonging immune responses.
T cell activation by modulation of the TME using combinatorial approaches
The TME plays a pivotal role in regulating T cell activity, impacting therapeutic response to immunotherapy. Within the TME, various cells and factors interact to create a complex milieu that can either promote or suppress the function of cytotoxic T cells. Tumor-infiltrating lymphocytes, including CD8+ cytotoxic and CD4+ helper T cells, are central to mounting an effective anti-tumor immune response. However, the presence of immune suppressive cells such as regulatory T cells, MDSCs, and tumor-associated macrophages (TAMs), and cancer-associated fibroblasts (CAFs) can hinder activation and function of effector T cells. Additionally, factors secreted by the tumor and stromal cells, such as cytokines (e.g., TGF-β, and IL-10) and metabolites (e.g., adenosine), contribute to the immunosuppressive environment within the TME, further dampening T cell responses. In addition to the TME composition, tumors present a variety of physical barriers that limit the activity of T cells within the TME, including the dense extracellular matrix, which can physically impede T cell infiltration in the tumor. Additionally, tumors often exhibit abnormal vasculature and high interstitial fluid pressure, further hindering T cell migration and function within the TME. Therefore, NPs with the ability of targeting and overcoming these barriers to modulate the TME into a favorable setting for T cell activation can enhance the effect of immunotherapy.NPs transforming the physiological landscape in the TME
NPs have been engineered to target specific molecules or functional status of immune cells, such as macrophages, DCs, or T cells, in the TME.178–183 For example, LNPs produced from sugar-alcohol-derived ionizable lipids encapsulated with mRNAs encoded with either CD40 ligand or CD40 were developed as a CATCH approach to enhance immune response by modulating an immunosuppressive TME.180 Delivery of LNPs containing CD40 ligand mRNA induced immunogenic cell death in tumors to release of tumor-associated antigens as well as increased CD40 ligand expression. Adoptively transferred dendritic cells containing lipid nanoparticle encapsulating CD40 mRNA were then activated by the CD40 ligand molecules expressed in tumor cells, which led to the production of cytokines and chemokines, and the upregulation of co-stimulatory molecules on dendritic cells.180 The combined treatment reprogrammed the TME and primed the T-cell responses, which resulted in the reduction of the established tumors, suppresses the development of distal metastatic lesions, and protection from tumor cell re-challenging in mouse melanoma and colon cancer models.180 Taking advantage of the upregulation of specific biomarkers implicated in a poor response in pancreatic ductal adenocarcinoma, such as CXCR4 and PD-L1, PEG-modified mesoporous Cu2MoS4 NPs loaded with PD-L1 inhibitor (BMS-1) and CXCR4 inhibitor (Plerixafor) were developed.184 Treatment with the NP/drug actively remodeled the TME, resulting in increased production of immunostimulatory cytokines, INF-γ and IL-12, and decreased levels of immunosuppressive cytokines, IL-6, IL-10 and IFN-α, which then led to increased infiltration and activation of CD4+ and CD8+ T cells in a mouse pancreatic tumor model. The effect of tumor growth inhibition was demonstrated in the mouse tumor model.184 This evidence supports an alternative approach of targeting costimulatory or modulatory molecules on the T cell surface and expanding the focus to the TME that is also capable of activating effector T cell function.184Alternatively, NPs can be used to reduce the effect of the negative regulatory cells on function and differentiation of APCs and effector T cells, by targeting MDSCs, regulatory T cells, TAMs or cancer-associated fibroblasts (CAFs).185–188 For example, a combination of liposomes encapsulating phosphoinositide 3-kinase gamma (PI3Kγ) inhibitor (IPI-549) and photosensitizer chlorin e6 with photodynamic therapy showed an enhanced therapeutic response in the CT 26 mouse colon cancer model through photodynamic therapy induced immunogenic tumor cell death and PI3Kγ inhibition in the myeloid-derived suppressive cells (MDSCs).185 Moreover, M2 macrophage binding peptide (M2pep) conjugated hollow copper sulfide NPs (CuS NPs) loaded with an immunomodulatory agent, imiquimod, could self-assemble into supramolecular aggregates following targeted delivery into M2 macrophages to efficiently drive reprogramming them into M1 macrophages in tumors. In combination with the hyperthermia property of CuS NPs, the treatment induced immunogenic cell death of tumor cells, increased tumor infiltration of APCs, and CD4+ and CD8+ T cells in the 4T1 mouse mammary tumor model, leading to the tumor growth inhibition and improvement of mouse survival in the above tumor model.186
In another approach, a TME responsive PEG-S-S-PLA NP carrying IL-12 gene expressing plasmids and PLX3397, an inhibitor for colony-stimulating factor-1 receptor (CSF-1R) (pIL-12 + PLX@cR-PssPD) disintegrated following delivery into a highly reducing TME to release of the CSF-1R and IL-12 expressing plasmid DNA.189 Intraperitoneal delivery of pIL-12 + PLX@cR-PssPD inhibited the growth of primary tumor and reduced peritoneal metastases in the CT26 mouse colon cancer model. The treatment led to the activation of CD4+ and CD8+ T cells, promotion of the repolarization of TAMs, reduction of MDSCs and regulatory T cells, and increases in DC maturation and secretion of anti-tumor cytokines.189 To advance further, another study used tunable components to generate a NP with the ability to induce tertiary lymphoid structures (TLSs) in tumors. One of the key components for a response to immunotherapy is the formation of TLSs since this site is where the adaptive immune response initiates, including processing tumor-associated antigens by APCs, generating a large number of B cells within the TLSs, and activating specific T cells to locally enhance antitumor immunity. A nanovaccine consisting of Epstein–Barr virus nuclear antigen 1 (EBNA1) and a bi-adjuvant of Mn2+ and CpG formulated with tannic acid was developed190 (Fig. 5A and B). In a nasopharyngeal carcinoma mouse model, this nanovaccine was not only able to significantly foster TLSs’ formation but also enhance local immune responses, leading to a delay in tumor outgrowth, and prolonging the median survival time of the mice.190 These NPs were able to remodel the TME by acting on DCs’ membrane lymphotoxins-α and -β pathways, subsequently enhancing the expression of downstream chemokines, CCL19/CCL21, CXCL10 and CXCL13, in the TME, enhancing the normalization of blood vessels, lymphatic vessels, and high endothelial venules in tumors, which were more conducive to reducing the interstitial pressure of tumors and facilitating the migration or homing of peripheral lymphocytes to tumors190 (Fig. 5C and D). Therefore, NPs can be used to induce substantial changes in the TME, especially in solid tumors to provide necessary conditions for motivating multifunctioning T cells in the tumors.
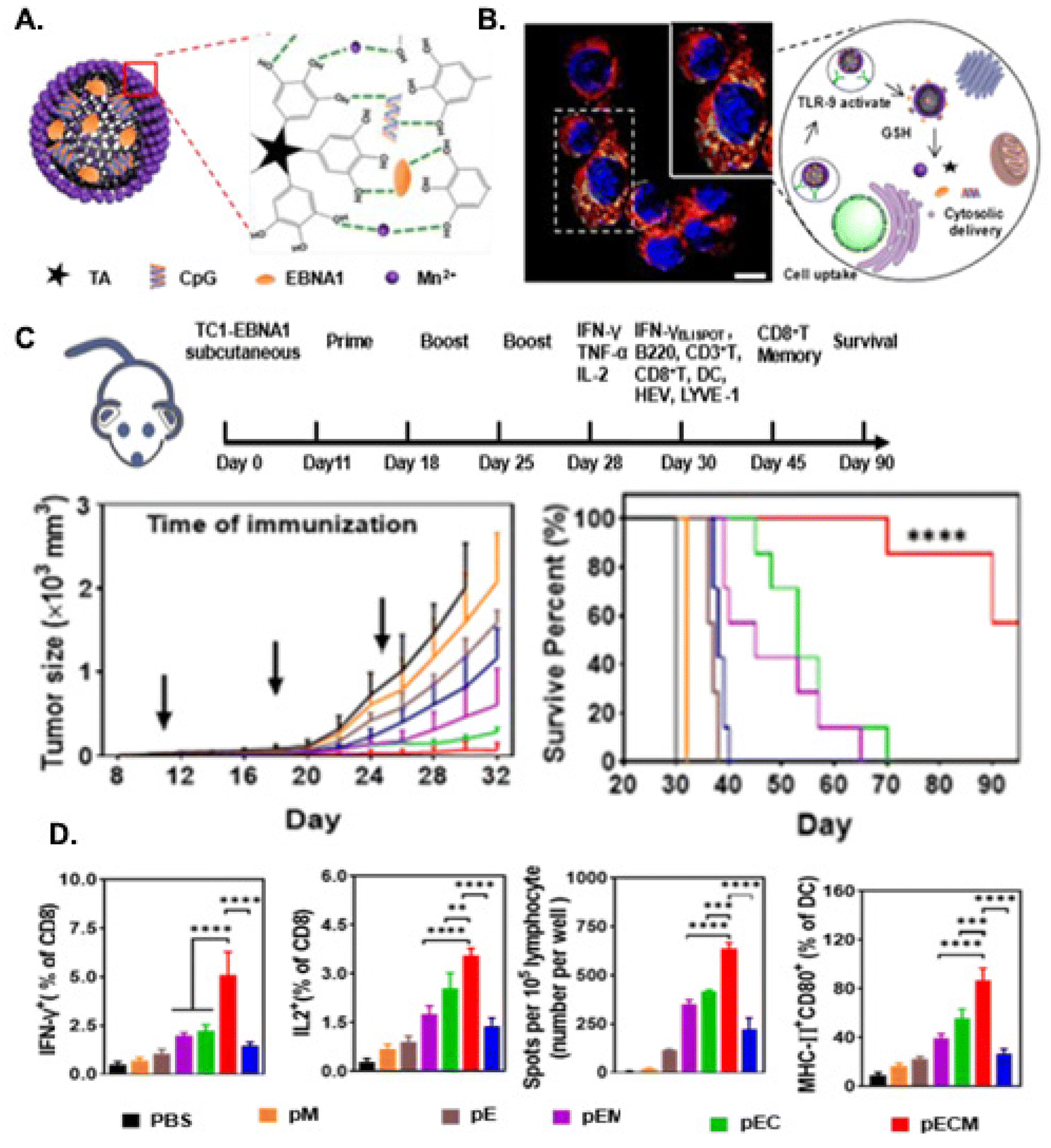 | ||
| Fig. 5 Effect of tertiary lymphoid mimicking nanoparticle on TME modulation. (A) Nanovaccine consisting of Epstein–Barr virus nuclear antigen 1 (EBNA1) and a bi-adjuvant of Mn2+ and CpG formulated with tannic acid (pECM) with different components formulated by hydrogen bond interaction. (B) Confocal microscopy photograph of localization of pECM in Raw264.7. Blue: DAPI; green: LysoTracker Green; red: Cy5-labeled EBNA1Δ93-236. (C) Timeline of vaccination and evaluation of the mouse injected with NPC cells. (Left) Tumor growth curves of different treatment groups. (Right) Survival curves of different treatment groups. (D) Cytokine levels by flow cytometry and ELISPOT. Reproduced from Wen et al., ACS Nano, 2023, 17, 7194–7206, with permission from American Chemical Society. | ||
Achieving therapeutic synergy with novel NP formulations
By strategically designing, loading and manufacturing NPs, researchers can create simultaneous and/or sequential immune stimulatory/modulatory therapeutic effects that can be used in combination with other therapies. For example, to activate immune responses in immune ‘cold tumor’, such as pancreatic cancer, a lipid-bilayer coated mesoporous silica NP carrying dual oxaliplatin and IDO inhibitor (OX/IND-MSNP) has been developed191 (Fig. 6A). Chemotherapy drug, Oxaliplatin (OX), can induce immunogenic cell death in tumor cells. Indoleamine 2,3-dioxy-genase (IDO) controls immune surveillance by converting L-tryptophan to L-kynurenine (Kyn), which interferes in the development of cytotoxic T cells and induces regulatory T cells. Systemic delivery of OX/IND-MSNP led to selective accumulation in orthotopic pancreatic tumors and significant inhibition of tumor growth. The treatment also increased recruiting cytotoxic T lymphocytes into tumors and reduced the level of Foxp3+ regulatory T cells191 (Fig. 6B–E). Additionally, researchers have begun integrating multiple manufacturing approaches to develop a one-step formulation of NPs designed with immune potentiating, and T cells-priming activities. Using controllable self-assembling into desired nanostructures by rationally designed oligonucleotide sequences, DNA has been used as a biomaterial to produce size and shape defined nanostructures.192,193 Using DNA nanocomplexes as scaffolds, various metal ions can bind to DNAs via coordination interactions and assemble into metal–DNA nanostructures. Results of a recent study showed that intratumoral administrations of metal–DNA NPs produced by incorporating an immunostimulatory CpG oligonucleotide formed nanostructures with a high level of gold ions enhanced the photothermal effect in the CT26 mouse colon cancer model. CpG-gold NP (CpG-GDS) treatment in combination with a laser irradiation not only abolished primary SC tumors but also prevented the growth of lung metastatic lesions following the tumor cell re-challenging. Importantly, the combination therapy induced high levels of inflammation cytokines, IL-6, IL-12, and TNF-α, in the serum of mice, activated DCs and produced strong immune responses to inhibit tumor growth after tumor cell re-challenging.192 Additionally, a metal “X” Framework consisting of CpG DNA oligodeoxynucleotides and metal iron Hf4+ (Hf-CpG) has been shown to enhance the therapeutic effect of radiotherapy, resulting in elimination of primary tumors, inhibition of tumor metastasis and prolonged survival in the CT-26 mouse colon cancer model following intratumoral delivery of Hf-CpG and radiation therapy. Increased therapeutic response was found to be associated with activation of DCs and CD8+ and CD4+ T cells as well as the percentage of effector memory T cells. Higher levels of TNF-α and IFN-γ were also detected in mice following the combination therapy.193 Taken together, these delivery platforms can be engineered with specific immunomodulatory function(s) such as T cells priming, activation, proliferation, expansion, and memory that can be used alone or in conjunction with current treatment modalities creating novel promising nano-mediated therapeutic approaches. | ||
| Fig. 6 Effect of a lipid-bilayer coated mesoporous silica nanoparticle carrying dual oxaliplatin and indoleamine 2,3-dioxy-genase (IDO) inhibitor agents on tumor growth inhibition and activation of T cell response in a mouse pancreatic cancer model derived from a KPC transgenic mouse tumor cells. (A). Schematic of the structure of an OX/IND-MSNP. A lipid bilayer (LB) containing lipid conjugated IDO inhibitor, indoximod (IND) and stable entrapment of oxaliplatin (OX) in the pores that is sealed by a coating. (B) Systemic delivery of OX plus IND-NV by MSNP induced effective tumor growth inhibition in the KPC mouse pancreatic cancer model. Representative IVIS imaging of tumor volumes shown as intensity of bioluminescence signals on days 10, 18, 27, and 36. (C) OX/IND-MSNP treatment significantly increased survival of tumor bearing mice. (D & E) The dual agent treatment markedly increased the effector CD8+ T cells (CD8+/Treg ratio, D) and intratumoral infiltrating CD8+ T cells determined by PET imaging using a radiolabeled anti-CD8 probe (E). Reproduced from Lu et al., Nat. Commun., 2017, 8, 1811, with permission from Springer Nature. | ||
Conclusions
Summarizing the potential of nanotechnology in T cell activation
NPs offer a promising avenue for addressing numerous challenges encountered by cancer immunotherapy. With a focus on T cell activation, the adaptability of NPs facilitates the design for specific functions. These rationally engineered NPs carry immunoactivating and modulating agents, either as single or combination, to act upon critical steps in the induction of a tumor specific T cell response. Recognizing their potential to influence various facets of T cell activation, such as serving as artificial antigen-presenting cells (aAPCs) and acting as nanocarriers for tumor antigens, diverse costimulatory molecules and cytokine signaling. NPs can strategically target different stages of the T cell activation process. This targeted approach enables NPs to enhance the key signals required for the optimal activation and functioning of T cells. Considering the diverse nature of NPs, they can function as synthetic immune cells, transporting a spectrum of small molecules, peptides, proteins, antibodies, and DNAs/RNAs. These NPs can be equipped to carry specific antigen peptides for TCR/MHC recognition, costimulatory molecules to enhance or prolong the response, and deliver cytokines/chemokines crucial for the differentiation and development of immune effector cells that are necessary for an effective anti-tumor response. By engineering multifunctional NPs, it becomes possible not only to overcome challenges in tumors but also to synergize with different therapies and remodel the TME to foster potent efficacy. Studies have demonstrated that NPs can activate both endogenous and adoptively transferred cells, highlighting their capacity to influence the immune response. The versatility of NPs is evident in their ability to act on T cells in vitro, ex vivo, and in vivo, as demonstrated in various research studies.194–196 Given the growing emphasis on T cell-based immunotherapies, researchers are directing their efforts towards identifying the optimal T cell subset and modifications to the TME that can support this ideal T cell subset. Therefore, comprehending the T cell activation cascade and applying that knowledge to NPs holds the potential to contribute to the development of optimal therapies. The mechanisms of action of these NPs vary depending on the modifications made to the NPs, therefore they can be used for a personalized approach by essentially combining releasing immune brakes (inhibiting immune suppressors) while simultaneously stepping on the gas (activating antitumor immune cells). The promising aspect of this design is its ability to precondition the tumor milieu into a favorable environment for proper T cell function, both for endogenous and adoptively transferred T cells.Future prospects and developments
Identifying promising candidates that target various mechanisms of T cell activation is crucial for advancing the development of nano-based immunotherapies. By targeting the various critical steps required for effective immunity against cancer cells or the cancer-immunity cycle, including release of cancer cell antigens, antigen presentation, priming and activation of T cells in secondary lymphoid organs, trafficking of activated T cells to tumors, infiltration of T cells into tumors, recognition of cancer cells by tumor-infiltrating T cells, and T cell-mediated selective targeting and killing of cancer cells,14 NPs can be combined with multiple therapies and functions in order to improve current treatment modalities. NPs have been designed to not only target specific cells and increase infiltration of T cells in tumors, but also encapsulate different agents (i.e., metabolic, tumor-responsive, immunotherapy, and/or chemotherapy) to work synergistically to overcome the physical barriers presented by solid tumors. These NPs can target multiple cell death and survival mechanisms of tumor cells (i.e., autophagy, immunogenic cell death, and apoptosis), leading to sustained and precise “on tumor” activation of T cells and therapeutic efficacy. We anticipate that those immunotherapy approaches have the potential for adjuvant therapy following surgery to remove primary tumors or the combination therapy for surgically unresectable, locally advanced, and metastatic cancers. By identifying novel antigens/receptors, optimizing cellular fitness, enhancing cell–cell interactions, and activating key signal pathways for developing multifunctional NPs with specific immunological function, it is possible to create artificial immune cells that mimic and mediate immune response to overcome immune evasion in tumors. These NPs can supply many essential signals required for T cell activation, and their formulations can be fine-tuned to achieve outcomes that are crucial for a robust immune response. By delivering these necessary signals, the NPs can activate the immune effector cells and enhance immune response in an immunosuppressive TME. Moreover, these NPs can be tailored to individual patients, addressing specific deficiencies and requirements to enhance each patient's immune response to personalized therapeutic strategies. Through the conjugation of ligands to the NP's surface and the encapsulation of diverse payloads, these NPs can be intricately designed to meet specific therapeutic objectives for effective inhibition of tumor growth, activation of tumor specific immune response and reduction of systemic toxicity associated with cancer therapeutic agents. Therefore, advances in understanding mechanisms and signal molecules in regulation of activation of tumor specific T cells should allow designing more effective immunotherapy NPs that carry multiple immune stimulatory/modulatory agents for efficient delivery into tumors with controlled simultaneous or sequential release of those agents for cancer immunotherapy.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
Dr Lily Yang is the President of MIGRA-Therapeutics, LLC. Dr Lei Zhu is the Chief Operating Officer of MIGRA-Therapeutics, LLC.Acknowledgements
Kory Wells is supported by a NIH grant award (R01CA261251, Yang). This research was supported by NIH R41CA247165 (Yang and Zhu).References
- H. Raskov, A. Orhan, J. P. Christensen and I. Gogenur, Br. J. Cancer, 2021, 124, 359–367 CrossRef CAS PubMed.
- M. Kuske, M. Haist, T. Jung, S. Grabbe and M. Bros, Cancers, 2022, 14(7), 1710 CrossRef CAS PubMed.
- A. Chow, K. Perica, C. A. Klebanoff and J. D. Wolchok, Nat. Rev. Clin. Oncol., 2022, 19, 775–790 CrossRef PubMed.
- E. Goleva, T. Lyubchenko, L. Kraehenbuehl, M. E. Lacouture, D. Y. M. Leung and J. A. Kern, Ann. Allergy, Asthma, Immunol., 2021, 126, 630–638 CrossRef CAS PubMed.
- M. Morotti, A. Albukhari, A. Alsaadi, M. Artibani, J. D. Brenton, S. M. Curbishley, T. Dong, M. L. Dustin, Z. Hu, N. McGranahan, M. L. Miller, L. Santana-Gonzalez, L. W. Seymour, T. Shi, P. Van Loo, C. Yau, H. White, N. Wietek, D. N. Church, D. C. Wedge and A. A. Ahmed, Br. J. Cancer, 2021, 124, 1759–1776 CrossRef PubMed.
- F. Fuchsl and A. M. Krackhardt, Cancers, 2022, 14(14), 4192 CrossRef PubMed.
- V. Gowd, A. Ahmad, M. Tarique, M. Suhail, T. A. Zughaibi, S. Tabrez and R. Khan, Semin. Cancer Biol., 2022, 86, 624–644 CrossRef CAS PubMed.
- T. A. Debele, C. F. Yeh and W. P. Su, Cancers, 2020, 12(12), 3773 CrossRef CAS PubMed.
- X. Feng, W. Xu, Z. Li, W. Song, J. Ding and X. Chen, Adv. Sci., 2019, 6, 1900101 CrossRef PubMed.
- Y. P. Liao, D. Schaue and W. H. McBride, Front. Biosci., 2007, 12, 3576–3600 CrossRef CAS PubMed.
- D. S. Vinay, E. P. Ryan, G. Pawelec, W. H. Talib, J. Stagg, E. Elkord, T. Lichtor, W. K. Decker, R. L. Whelan, H. Kumara, E. Signori, K. Honoki, A. G. Georgakilas, A. Amin, W. G. Helferich, C. S. Boosani, G. Guha, M. R. Ciriolo, S. Chen, S. I. Mohammed, A. S. Azmi, W. N. Keith, A. Bilsland, D. Bhakta, D. Halicka, H. Fujii, K. Aquilano, S. S. Ashraf, S. Nowsheen, X. Yang, B. K. Choi and B. S. Kwon, Semin. Cancer Biol., 2015, 35(Suppl), S185–S198 CrossRef PubMed.
- P. Wu, J. Han, Y. Gong, C. Liu, H. Yu and N. Xie, Pharmaceutics, 2022, 14(10), 1990 CrossRef CAS PubMed.
- S. Lim, J. Park, M. K. Shim, W. Um, H. Y. Yoon, J. H. Ryu, D. K. Lim and K. Kim, Theranostics, 2019, 9, 7906–7923 CrossRef CAS PubMed.
- I. Mellman, D. S. Chen, T. Powles and S. J. Turley, Immunity, 2023, 56, 2188–2205 CrossRef CAS PubMed.
- Q. Liu, Y. Duo, J. Fu, M. Qiu, Z. Sun, D. Adah, J. Kang, Z. Xie, T. Fan, S. Bao, H. Zhang, L.-P. Liu and Y. Cao, Nano Today, 2021, 36, 101023 CrossRef CAS.
- Q. Li, Y. Liu, Z. Huang, Y. Guo and Q. Li, Front. Bioeng. Biotechnol., 2022, 10, 878524 CrossRef PubMed.
- K. Shi, M. Haynes and L. Huang, Front. Chem. Sci. Eng., 2017, 11, 676–684 CrossRef CAS.
- S. Gao, D. Yang, Y. Fang, X. Lin, X. Jin, Q. Wang, X. Wang, L. Ke and K. Shi, Theranostics, 2019, 9, 126–151 CrossRef CAS PubMed.
- W. Fan, B. Yung, P. Huang and X. Chen, Chem. Rev., 2017, 117, 13566–13638 CrossRef CAS PubMed.
- P. P. Adiseshaiah, R. M. Crist, S. S. Hook and S. E. McNeil, Nat. Rev. Clin. Oncol., 2016, 13, 750–765 CrossRef CAS PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- L. Zhu, H. Mao and L. Yang, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2022, 14, e1793 CAS.
- J. H. Kim, M. J. Moon, D. Y. Kim, S. H. Heo and Y. Y. Jeong, Polymers, 2018, 10(10), 1133 CrossRef PubMed.
- R. Bajracharya, J. G. Song, B. R. Patil, S. H. Lee, H. M. Noh, D. H. Kim, G. L. Kim, S. H. Seo, J. W. Park, S. H. Jeong, C. H. Lee and H. K. Han, Drug Delivery, 2022, 29, 1959–1970 CrossRef CAS PubMed.
- K. Ulbrich, K. Hola, V. Subr, A. Bakandritsos, J. Tucek and R. Zboril, Chem. Rev., 2016, 116, 5338–5431 CrossRef CAS PubMed.
- N. Bertrand, J. Wu, X. Xu, N. Kamaly and O. C. Farokhzad, Adv. Drug Delivery Rev., 2014, 66, 2–25 CrossRef CAS PubMed.
- M. Izci, C. Maksoudian, B. B. Manshian and S. J. Soenen, Chem. Rev., 2021, 121, 1746–1803 CrossRef CAS PubMed.
- W. C. Chen, A. X. Zhang and S.-D. Li, Eur. J. Nanomed., 2012, 4, 89–93 CrossRef.
- Y. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS PubMed.
- H. Zhang, OncoTargets Ther., 2016, 9, 3001–3007 CrossRef CAS PubMed.
- L. Schoenmaker, D. Witzigmann, J. A. Kulkarni, R. Verbeke, G. Kersten, W. Jiskoot and D. J. A. Crommelin, Int. J. Pharm., 2021, 601, 120586 CrossRef CAS PubMed.
- L. Milane and M. Amiji, Drug Delivery Transl. Res., 2021, 11, 1309–1315 CrossRef CAS PubMed.
- N. Kamaly, B. Yameen, J. Wu and O. C. Farokhzad, Chem. Rev., 2016, 116, 2602–2663 CrossRef CAS PubMed.
- I. M. S. Degors, C. Wang, Z. U. Rehman and I. S. Zuhorn, Acc. Chem. Res., 2019, 52, 1750–1760 CrossRef CAS PubMed.
- Z. He, X. Wan, A. Schulz, H. Bludau, M. A. Dobrovolskaia, S. T. Stern, S. A. Montgomery, H. Yuan, Z. Li, D. Alakhova, M. Sokolsky, D. B. Darr, C. M. Perou, R. Jordan, R. Luxenhofer and A. V. Kabanov, Biomaterials, 2016, 101, 296–309 CrossRef CAS PubMed.
- A. Sparreboom, C. D. Scripture, V. Trieu, P. J. Williams, T. De, A. Yang, B. Beals, W. D. Figg, M. Hawkins and N. Desai, Clin. Cancer Res., 2005, 11, 4136–4143 CrossRef CAS PubMed.
- J. Verma, S. Lal and C. J. F. V. Noorden, Eur. J. Nanomed., 2015, 7, 271–287 Search PubMed.
- K. Mahmoudi, A. Bouras, D. Bozec, R. Ivkov and C. Hadjipanayis, Int. J. Hyperthermia, 2018, 34, 1316–1328 CrossRef PubMed.
- S. Bonvalot, P. L. Rutkowski, J. Thariat, S. Carrere, A. Ducassou, M. P. Sunyach, P. Agoston, A. Hong, A. Mervoyer, M. Rastrelli, V. Moreno, R. K. Li, B. Tiangco, A. C. Herraez, A. Gronchi, L. Mangel, T. Sy-Ortin, P. Hohenberger, T. de Baere, A. Le Cesne, S. Helfre, E. Saada-Bouzid, A. Borkowska, R. Anghel, A. Co, M. Gebhart, G. Kantor, A. Montero, H. H. Loong, R. Verges, L. Lapeire, S. Dema, G. Kacso, L. Austen, L. Moureau-Zabotto, V. Servois, E. Wardelmann, P. Terrier, A. J. Lazar, J. Bovee, C. Le Pechoux and Z. Papai, Lancet Oncol., 2019, 20, 1148–1159 CrossRef CAS PubMed.
- J. F. Dorsey, L. Sun, D. Y. Joh, A. Witztum, G. D. Kao, M. Alonso-Basanta, S. Avery, S. M. Hahn, A. Al Zaki and A. Tsourkas, Transl. Cancer Res., 2013, 2, 280–291 CAS.
- P. Dash, A. M. Piras and M. Dash, J. Controlled Release, 2020, 327, 546–570 CrossRef CAS PubMed.
- A. J. Sinegra, M. Evangelopoulos, J. Park, Z. Huang and C. A. Mirkin, Nano Lett., 2021, 21, 6584–6591 CrossRef CAS PubMed.
- M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2013, 42, 5552–5576 RSC.
- S. Singha, K. Shao, K. K. Ellestad, Y. Yang and P. Santamaria, ACS Nano, 2018, 12, 10621–10635 CrossRef CAS PubMed.
- N. K. Lee, S.-N. Kim and C. G. Park, Biomater. Res., 2021, 25, 44 CrossRef CAS PubMed.
- A. L. Silva, C. Peres, J. Conniot, A. I. Matos, L. Moura, B. Carreira, V. Sainz, A. Scomparin, R. Satchi-Fainaro, V. Preat and H. F. Florindo, Semin. Immunol., 2017, 34, 3–24 CrossRef CAS PubMed.
- R. H. Fang and L. Zhang, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 305–326 CrossRef PubMed.
- D. R. Getts, L. D. Shea, S. D. Miller and N. J. King, Trends Immunol., 2015, 36, 419–427 CrossRef CAS PubMed.
- Q. Muhammad, Y. Jang, S. H. Kang, J. Moon, W. J. Kim and H. Park, Biomater. Sci., 2020, 8, 1490–1501 RSC.
- Y. Liu, J. Hardie, X. Zhang and V. M. Rotello, Semin. Immunol., 2017, 34, 25–32 CrossRef CAS PubMed.
- P. Foroozandeh and A. A. Aziz, Nanoscale Res. Lett., 2018, 13, 339 CrossRef PubMed.
- P. Ray, N. Haideri, I. Haque, O. Mohammed, S. Chakraborty, S. Banerjee, M. Quadir, A. E. Brinker and S. K. Banerjee, J. Immunol. Sci., 2021, 5(1), 19–33 CrossRef.
- S. Panico, S. Capolla, S. Bozzer, G. Toffoli, M. Dal Bo and P. Macor, Pharmaceutics, 2022, 14(12), 2605 CrossRef CAS PubMed.
- D. Boraschi, P. Italiani, R. Palomba, P. Decuzzi, A. Duschl, B. Fadeel and S. M. Moghimi, Semin. Immunol., 2017, 34, 33–51 CrossRef CAS PubMed.
- P. Korangath, J. D. Barnett, A. Sharma, E. T. Henderson, J. Stewart, S. H. Yu, S. K. Kandala, C. T. Yang, J. S. Caserto, M. Hedayati, T. D. Armstrong, E. Jaffee, C. Gruettner, X. C. Zhou, W. Fu, C. Hu, S. Sukumar, B. W. Simons and R. Ivkov, Sci. Adv., 2020, 6, eaay1601 CrossRef CAS PubMed.
- L. Liu, R. Sha, L. Yang, X. Zhao, Y. Zhu, J. Gao, Y. Zhang and L. P. Wen, ACS Appl. Mater. Interfaces, 2018, 10, 41197–41206 CrossRef CAS PubMed.
- F. Zhao, C. Wang, Q. Yang, S. Han, Q. Hu and Z. Fu, Life Sci., 2018, 202, 44–51 CrossRef CAS PubMed.
- M. Zhu, X. Tian, X. Song, Y. Li, Y. Tian, Y. Zhao and G. Nie, Small, 2012, 8, 2841–2848 CrossRef CAS PubMed.
- M. Binnewies, E. W. Roberts, K. Kersten, V. Chan, D. F. Fearon, M. Merad, L. M. Coussens, D. I. Gabrilovich, S. Ostrand-Rosenberg, C. C. Hedrick, R. H. Vonderheide, M. J. Pittet, R. K. Jain, W. Zou, T. K. Howcroft, E. C. Woodhouse, R. A. Weinberg and M. F. Krummel, Nat. Med., 2018, 24, 541–550 CrossRef CAS PubMed.
- A. Labani-Motlagh, M. Ashja-Mahdavi and A. Loskog, Front. Immunol., 2020, 11, 940 CrossRef CAS PubMed.
- S. K. Sriraman, B. Aryasomayajula and V. P. Torchilin, Tissue Barriers, 2014, 2, e29528 CrossRef PubMed.
- L. J. Appleman and V. A. Boussiotis, Immunol. Rev., 2003, 192, 161–180 CrossRef CAS PubMed.
- R. G. Gupta, F. Li, J. Roszik and G. Lizee, Cancer Discovery, 2021, 11, 1024–1039 CrossRef CAS PubMed.
- A. Isser, N. K. Livingston and J. P. Schneck, Biomaterials, 2021, 268, 120584 CrossRef CAS PubMed.
- H. Sultan, A. M. Salazar and E. Celis, Semin. Immunol., 2020, 49, 101414 CrossRef CAS PubMed.
- N. Biswas, S. Chakrabarti, V. Padul, L. D. Jones and S. Ashili, Front. Immunol., 2023, 14, 1105420 CrossRef CAS PubMed.
- S. C. Pao, M. T. Chu and S. I. Hung, Pharmaceutics, 2022, 14(4), 867 CrossRef CAS PubMed.
- P. D. Katsikis, K. J. Ishii and C. Schliehe, Nat. Rev. Immunol., 2024, 24, 213–227 CrossRef CAS PubMed.
- J. S. Weber, M. S. Carlino, A. Khattak, T. Meniawy, G. Ansstas, M. H. Taylor, K. B. Kim, M. McKean, G. V. Long, R. J. Sullivan, M. Faries, T. T. Tran, C. L. Cowey, A. Pecora, M. Shaheen, J. Segar, T. Medina, V. Atkinson, G. T. Gibney, J. J. Luke, S. Thomas, E. I. Buchbinder, J. A. Healy, M. Huang, M. Morrissey, I. Feldman, V. Sehgal, C. Robert-Tissot, P. Hou, L. Zhu, M. Brown, P. Aanur, R. S. Meehan and T. Zaks, Lancet, 2024, 403, 632–644 CrossRef CAS PubMed.
- Q. Wu, S. G. Medina, G. Kushawah, M. L. DeVore, L. A. Castellano, J. M. Hand, M. Wright and A. A. Bazzini, eLife, 2019, 8, e45396 CrossRef PubMed.
- J. F. Yao, H. Yang, Y. Z. Zhao and M. Xue, Curr. Drug Metab., 2018, 19, 892–901 CrossRef CAS PubMed.
- Y. N. Zhang, J. Lazarovits, W. Poon, B. Ouyang, L. N. M. Nguyen, B. R. Kingston and W. C. W. Chan, Nano Lett., 2019, 19, 7226–7235 CrossRef CAS PubMed.
- J. Lee, S. Kang, H. Park, J. G. Sun, E. C. Kim and G. Shim, Pharmaceutics, 2023, 15(2), 565 CrossRef CAS PubMed.
- J. P. Almeida, A. L. Chen, A. Foster and R. Drezek, Nanomedicine, 2011, 6, 815–835 CrossRef CAS PubMed.
- K. H. Chen, D. J. Lundy, E. K. Toh, C. H. Chen, C. Shih, P. Chen, H. C. Chang, J. J. Lai, P. S. Stayton, A. S. Hoffman and P. C. Hsieh, Nanoscale, 2015, 7, 15863–15872 RSC.
- S. T. Reddy, A. J. van der Vlies, E. Simeoni, V. Angeli, G. J. Randolph, C. P. O'Neil, L. K. Lee, M. A. Swartz and J. A. Hubbell, Nat. Biotechnol., 2007, 25, 1159–1164 CrossRef CAS PubMed.
- W. Zhang, R. Taheri-Ledari, F. Ganjali, S. S. Mirmohammadi, F. S. Qazi, M. Saeidirad, A. KashtiAray, S. Zarei-Shokat, Y. Tian and A. Maleki, RSC Adv., 2022, 13, 80–114 RSC.
- N. Kapate, J. R. Clegg and S. Mitragotri, Adv. Drug Delivery Rev., 2021, 177, 113807 CrossRef CAS PubMed.
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS PubMed.
- J. McCright, C. Skeen, J. Yarmovsky and K. Maisel, Acta Biomater., 2022, 145, 146–158 CrossRef CAS PubMed.
- E. Yang, W. Qian, Z. Cao, L. Wang, E. N. Bozeman, C. Ward, B. Yang, P. Selvaraj, M. Lipowska, Y. A. Wang, H. Mao and L. Yang, Theranostics, 2015, 5, 43–61 CrossRef PubMed.
- J. T. Martin, C. A. Cottrell, A. Antanasijevic, D. G. Carnathan, B. J. Cossette, C. A. Enemuo, E. H. Gebru, Y. Choe, F. Viviano, S. Fischinger, T. Tokatlian, K. M. Cirelli, G. Ueda, J. Copps, T. Schiffner, S. Menis, G. Alter, W. R. Schief, S. Crotty, N. P. King, D. Baker, G. Silvestri, A. B. Ward and D. J. Irvine, npj Vaccines, 2020, 5, 72 CrossRef CAS PubMed.
- S. N. Thomas, E. Vokali, A. W. Lund, J. A. Hubbell and M. A. Swartz, Biomaterials, 2014, 35, 814–824 CrossRef CAS PubMed.
- R. A. Fenstermaker, M. J. Ciesielski, J. Qiu, N. Yang, C. L. Frank, K. P. Lee, L. R. Mechtler, A. Belal, M. S. Ahluwalia and A. D. Hutson, Cancer Immunol. Immunother., 2016, 65, 1339–1352 CrossRef CAS PubMed.
- M. Karkada, G. M. Weir, T. Quinton, L. Sammatur, L. D. MacDonald, A. Grant, R. Liwski, R. Juskevicius, G. Sinnathamby, R. Philip and M. Mansour, J. Immunother., 2010, 33, 250–261 CrossRef PubMed.
- N. L. Berinstein, M. Karkada, M. A. Morse, J. J. Nemunaitis, G. Chatta, H. Kaufman, K. Odunsi, R. Nigam, L. Sammatur, L. D. MacDonald, G. M. Weir, M. M. Stanford and M. Mansour, J. Transl. Med., 2012, 10, 156 CrossRef CAS PubMed.
- X.-G. Gu, M. Schmitt, A. Hiasa, Y. Nagata, H. Ikeda, Y. Sasaki, K. Akiyoshi, J. Sunamoto, H. Nakamura, K. Kuribayashi and H. Shiku, Cancer Res., 1998, 58, 3385–3390 CAS.
- S. Kitano, S. Kageyama, Y. Nagata, Y. Miyahara, A. Hiasa, H. Naota, S. Okumura, H. Imai, T. Shiraishi, M. Masuya, M. Nishikawa, J. Sunamoto, K. Akiyoshi, T. Kanematsu, A. M. Scott, R. Murphy, E. W. Hoffman, L. J. Old and H. Shiku, Clin. Cancer Res., 2006, 12, 7397–7405 CrossRef CAS PubMed.
- S. Khazaei, R. Varela-Calviño, M. Rad-Malekshahi, F. Quattrini, S. Jokar, N. Rezaei, S. Balalaie, I. Haririan, N. Csaba and M. Garcia-Fuentes, Drug Delivery Transl. Res., 2024, 14, 455–473 CrossRef CAS PubMed.
- H. Song, Q. Su, Y. Nie, C. Zhang, P. Huang, S. Shi, Q. Liu and W. Wang, Acta Biomater., 2023, 158, 535–546 CrossRef CAS PubMed.
- X. Wang, Y. Liu, C. Xue, Y. Hu, Y. Zhao, K. Cai, M. Li and Z. Luo, Nat. Commun., 2022, 13, 5685 CrossRef CAS PubMed.
- Y. Shen, M. Wang, H. Wang, J. Zhou and J. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 19907–19917 CrossRef CAS PubMed.
- Y. Xiang, M. Tian, J. Huang, Y. Li, G. Li, X. Li, Z. Jiang, X. Song and X. Ma, J. Nanobiotechnol., 2023, 21, 324 CrossRef CAS PubMed.
- A. Shaban, J. Telegdi and G. Vastag, in Fundamentals of Bionanomaterials, ed. A. Barhoum, J. Jeevanandam and M. K. Danquah, Elsevier, 2022, pp. 111–138, DOI:10.1016/B978-0-12-824147-9.00005-4.
- L. P. Datta, S. Manchineella and T. Govindaraju, Biomaterials, 2020, 230, 119633 CrossRef CAS PubMed.
- Y. Kang, W. Zhang, Q. Yu, L. Gao, J. Quan, F. Gu, Y. Wu, Y. Tian, Z. Wu, S. Shao, H. Zhou, S. Duan, Y. Zhou, L. Zhang, X. Gao, H. Tian and W. Yao, Cancer Immunol. Immunother., 2023, 72, 2741–2755 CrossRef CAS PubMed.
- Q. Liu, Y. Chu, J. Shao, H. Qian, J. Yang, H. Sha, L. Cen, M. Tian, Q. Xu, F. Chen, Y. Yang, W. Wang, K. Wang, L. Yu, J. Wei and B. Liu, Adv. Sci., 2022, 10, e2203298 CrossRef PubMed.
- P. Gu, A. Wusiman, Y. Zhang, Z. Liu, R. Bo, Y. Hu, J. Liu and D. Wang, Mol. Pharm., 2019, 16, 5000–5012 CrossRef CAS PubMed.
- C. A. Arbelaez, J. Estrada, M. A. Gessner, C. Glaus, A. B. Morales, D. Mohn, H. Phee, J. R. Lipford and J. A. Johnston, npj Vaccines, 2020, 5, 106 CrossRef CAS PubMed.
- Z. Xu, S. Ramishetti, Y. C. Tseng, S. Guo, Y. Wang and L. Huang, J. Controlled Release, 2013, 172, 259–265 CrossRef CAS PubMed.
- J. J. Baljon, A. J. Kwiatkowski, H. M. Pagendarm, P. T. Stone, A. Kumar, V. Bharti, J. A. Schulman, K. W. Becker, E. W. Roth, P. P. Christov, S. Joyce and J. T. Wilson, ACS Nano, 2024, 18, 6845–6862 CrossRef CAS PubMed.
- G. M. Lynn, C. Sedlik, F. Baharom, Y. Zhu, R. A. Ramirez-Valdez, V. L. Coble, K. Tobin, S. R. Nichols, Y. Itzkowitz, N. Zaidi, J. M. Gammon, N. J. Blobel, J. Denizeau, P. de la Rochere, B. J. Francica, B. Decker, M. Maciejewski, J. Cheung, H. Yamane, M. G. Smelkinson, J. R. Francica, R. Laga, J. D. Bernstock, L. W. Seymour, C. G. Drake, C. M. Jewell, O. Lantz, E. Piaggio, A. S. Ishizuka and R. A. Seder, Nat. Biotechnol., 2020, 38, 320–332 CrossRef CAS PubMed.
- J. Nam, S. Son, K. S. Park and J. J. Moon, Adv. Sci., 2021, 8, 2002577 CrossRef CAS PubMed.
- J. R. Vasconcelos, M. R. Dominguez, A. F. Araujo, J. Ersching, C. A. Tararam, O. Bruna-Romero and M. M. Rodrigues, Front. Immunol., 2012, 3, 358 Search PubMed.
- G. Zhu, L. Mei, H. D. Vishwasrao, O. Jacobson, Z. Wang, Y. Liu, B. C. Yung, X. Fu, A. Jin, G. Niu, Q. Wang, F. Zhang, H. Shroff and X. Chen, Nat. Commun., 2017, 8, 1482 CrossRef PubMed.
- Z. Liu, Q. Wang, Y. Feng, L. Zhao, N. Dong, Y. Zhang, T. Yin, H. He, X. Tang, J. Gou and L. Yang, Chem. Eng. J., 2023, 475, 146474 CrossRef CAS.
- L. Zhang, J. Huang, X. Chen, C. Pan, Y. He, R. Su, D. Guo, S. Yin, S. Wang, L. Zhou, J. Chen, S. Zheng and Y. Qiao, J. Immunother. Cancer, 2021, 9, e003132 CrossRef PubMed.
- R. Luo, Y. Wan, G. Liu, J. Chen, X. Luo, Z. Li, D. Su, N. Lu and Z. Luo, Biomacromolecules, 2024, 25, 1408–1428 CrossRef CAS PubMed.
- E. Riquelme, L. J. Carreno, P. A. Gonzalez and A. M. Kalergis, Eur. J. Immunol., 2009, 39, 2259–2269 CrossRef CAS PubMed.
- G. F. Gao, Z. Rao and J. I. Bell, Trends Immunol., 2002, 23, 408–413 CrossRef CAS PubMed.
- K. Perica, A. De Leon Medero, M. Durai, Y. L. Chiu, J. G. Bieler, L. Sibener, M. Niemoller, M. Assenmacher, A. Richter, M. Edidin, M. Oelke and J. Schneck, Nanomedicine, 2014, 10, 119–129 CrossRef CAS PubMed.
- E. Ben-Akiva, J. W. Hickey, R. A. Meyer, A. Isser, S. R. Shannon, N. K. Livingston, K. R. Rhodes, A. K. Kosmides, T. R. Warren, S. Y. Tzeng, J. P. Schneck and J. J. Green, Acta Biomater., 2023, 160, 187–197 CrossRef CAS PubMed.
- B. Prakken, M. Wauben, D. Genini, R. Samodal, J. Barnett, A. Mendivil, L. Leoni and S. Albani, Nat. Med., 2000, 6, 1406–1410 CrossRef CAS PubMed.
- A. C. Wauters, J. F. Scheerstra, I. G. Vermeijlen, R. Hammink, M. Schluck, L. Woythe, H. Wu, L. Albertazzi, C. G. Figdor, J. Tel, L. Abdelmohsen and J. C. M. van Hest, ACS Nano, 2022, 16, 15072–15085 CrossRef CAS PubMed.
- S. Walter, L. Herrgen, O. Schoor, G. Jung, D. Wernet, H. J. Buhring, H. G. Rammensee and S. Stevanovic, J. Immunol., 2003, 171, 4974–4978 CrossRef CAS PubMed.
- F. Giannoni, J. Barnett, K. Bi, R. Samodal, P. Lanza, P. Marchese, R. Billetta, R. Vita, M. R. Klein, B. Prakken, W. W. Kwok, E. Sercarz, A. Altman and S. Albani, J. Immunol., 2005, 174, 3204–3211 CrossRef CAS PubMed.
- M. Durai, C. Krueger, Z. Ye, L. Cheng, A. Mackensen, M. Oelke and J. P. Schneck, Cancer Immunol. Immunother., 2009, 58, 209–220 CrossRef CAS PubMed.
- M. Oelke, M. V. Maus, D. Didiano, C. H. June, A. Mackensen and J. P. Schneck, Nat. Med., 2003, 9, 619–624 CrossRef CAS PubMed.
- B. F. Lillemeier, M. A. Mortelmaier, M. B. Forstner, J. B. Huppa, J. T. Groves and M. M. Davis, Nat. Immunol., 2010, 11, 90–96 CrossRef CAS PubMed.
- M. F. S. Lindenbergh and W. Stoorvogel, Annu. Rev. Immunol., 2018, 36, 435–459 CrossRef CAS PubMed.
- J. Deeg, M. Axmann, J. Matic, A. Liapis, D. Depoil, J. Afrose, S. Curado, M. L. Dustin and J. P. Spatz, Nano Lett., 2013, 13, 5619–5626 CrossRef CAS PubMed.
- J. W. Hickey, F. P. Vicente, G. P. Howard, H. Q. Mao and J. P. Schneck, Nano Lett., 2017, 17, 7045–7054 CrossRef CAS PubMed.
- A. Isser, A. B. Silver, H. C. Pruitt, M. Mass, E. H. Elias, G. Aihara, S. S. Kang, N. Bachmann, Y. Y. Chen, E. K. Leonard, J. G. Bieler, W. Chaisawangwong, J. Choy, S. R. Shannon, S. Gerecht, J. S. Weber, J. B. Spangler and J. P. Schneck, Nat. Commun., 2022, 13, 6086 CrossRef CAS PubMed.
- J. Ichikawa, T. Yoshida, A. Isser, A. S. Laino, M. Vassallo, D. Woods, S. Kim, M. Oelke, K. Jones, J. P. Schneck and J. S. Weber, Clin. Cancer Res., 2020, 26, 3384–3396 CrossRef CAS PubMed.
- K. Perica, A. K. Kosmides and J. P. Schneck, Biochim. Biophys. Acta, 2015, 1853, 781–790 CrossRef CAS PubMed.
- J. D. Beck, D. Reidenbach, N. Salomon, U. Sahin, O. Tureci, M. Vormehr and L. M. Kranz, Mol. Cancer, 2021, 20, 69 CrossRef CAS PubMed.
- C. L. Lorentzen, J. B. Haanen, O. Met and I. M. Svane, Lancet Oncol., 2022, 23, e450–e458 CrossRef CAS PubMed.
- C. Pollard, S. De Koker, X. Saelens, G. Vanham and J. Grooten, Trends Mol. Med., 2013, 19, 705–713 CrossRef CAS PubMed.
- X. Hou, T. Zaks, R. Langer and Y. Dong, Nat. Rev. Mater., 2021, 6, 1078–1094 CrossRef CAS PubMed.
- H. Parhiz, E. N. Atochina-Vasserman and D. Weissman, Lancet, 2024, 403, 1192–1204 CrossRef CAS PubMed.
- Z. Chen, Y. Tian, J. Yang, F. Wu, S. Liu, W. Cao, W. Xu, T. Hu, D. J. Siegwart and H. Xiong, J. Am. Chem. Soc., 2023, 145, 24302–24314 CrossRef CAS PubMed.
- K. C. Choi, D. H. Lee, J. W. Lee, J. S. Lee, Y. K. Lee, M. J. Choi, H. Y. Jeong, M. W. Kim, C. G. Lee and Y. S. Park, Int. J. Mol. Sci., 2024, 25(3), 1388 CrossRef CAS PubMed.
- K. Kubara, K. Yamazaki, T. Miyazaki, K. Kondo, D. Kurotaki, T. Tamura and Y. Suzuki, Mol. Ther., 2024, 32, 704–721 CrossRef CAS PubMed.
- Y. Yan, X. Liu, L. Wang, C. Wu, Q. Shuai, Y. Zhang and S. Liu, Biomaterials, 2023, 301, 122279 CrossRef CAS PubMed.
- Y. Wang, L. Zhang, Z. Xu, L. Miao and L. Huang, Mol. Ther., 2018, 26, 420–434 CrossRef CAS PubMed.
- J. Chen, Z. Ye, C. Huang, M. Qiu, D. Song, Y. Li and Q. Xu, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2207841119 CrossRef CAS PubMed.
- U. Sahin, P. Oehm, E. Derhovanessian, R. A. Jabulowsky, M. Vormehr, M. Gold, D. Maurus, D. Schwarck-Kokarakis, A. N. Kuhn, T. Omokoko, L. M. Kranz, M. Diken, S. Kreiter, H. Haas, S. Attig, R. Rae, K. Cuk, A. Kemmer-Bruck, A. Breitkreuz, C. Tolliver, J. Caspar, J. Quinkhardt, L. Hebich, M. Stein, A. Hohberger, I. Vogler, I. Liebig, S. Renken, J. Sikorski, M. Leierer, V. Muller, H. Mitzel-Rink, M. Miederer, C. Huber, S. Grabbe, J. Utikal, A. Pinter, R. Kaufmann, J. C. Hassel, C. Loquai and O. Tureci, Nature, 2020, 585, 107–112 CrossRef CAS PubMed.
- E. Kon, N. Ad-El, I. Hazan-Halevy, L. Stotsky-Oterin and D. Peer, Nat. Rev. Clin. Oncol., 2023, 20, 739–754 CrossRef CAS PubMed.
- M. May, Nat. Med., 2024, 8, 2097–2098 CrossRef PubMed.
- G. Cafri, J. J. Gartner, T. Zaks, K. Hopson, N. Levin, B. C. Paria, M. R. Parkhurst, R. Yossef, F. J. Lowery, M. S. Jafferji, T. D. Prickett, S. L. Goff, C. T. McGowan, S. Seitter, M. L. Shindorf, A. Parikh, P. D. Chatani, P. F. Robbins and S. A. Rosenberg, J. Clin. Invest., 2020, 130, 5976–5988 CrossRef CAS PubMed.
- L. A. Rojas, Z. Sethna, K. C. Soares, C. Olcese, N. Pang, E. Patterson, J. Lihm, N. Ceglia, P. Guasp, A. Chu, R. Yu, A. K. Chandra, T. Waters, J. Ruan, M. Amisaki, A. Zebboudj, Z. Odgerel, G. Payne, E. Derhovanessian, F. Muller, I. Rhee, M. Yadav, A. Dobrin, M. Sadelain, M. Luksza, N. Cohen, L. Tang, O. Basturk, M. Gonen, S. Katz, R. K. Do, A. S. Epstein, P. Momtaz, W. Park, R. Sugarman, A. M. Varghese, E. Won, A. Desai, A. C. Wei, M. I. D'Angelica, T. P. Kingham, I. Mellman, T. Merghoub, J. D. Wolchok, U. Sahin, O. Tureci, B. D. Greenbaum, W. R. Jarnagin, J. Drebin, E. M. O'Reilly and V. P. Balachandran, Nature, 2023, 618, 144–150 CrossRef CAS PubMed.
- E. Ben-Akiva, J. Karlsson, S. Hemmati, H. Yu, S. Y. Tzeng, D. M. Pardoll and J. J. Green, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2301606120 CrossRef PubMed.
- H. Peng, B. Chen, W. Huang, Y. Tang, Y. Jiang, W. Zhang and Y. Huang, Nano Lett., 2017, 17, 7684–7690 CrossRef CAS PubMed.
- S. Adams, Immunotherapy, 2009, 1, 949–964 CrossRef CAS PubMed.
- L. Miao, L. Li, Y. Huang, D. Delcassian, J. Chahal, J. Han, Y. Shi, K. Sadtler, W. Gao, J. Lin, J. C. Doloff, R. Langer and D. G. Anderson, Nat. Biotechnol., 2019, 37, 1174–1185 CrossRef CAS PubMed.
- D. Guerini, Cells, 2022, 11(7), 1159 CrossRef CAS PubMed.
- Y. Wu, X. Liang, C. Mao and Y. Jiang, ACS Nano, 2023, 17, 21782–21798 CrossRef PubMed.
- N. Cheng, R. Watkins-Schulz, R. D. Junkins, C. N. David, B. M. Johnson, S. A. Montgomery, K. J. Peine, D. B. Darr, H. Yuan, K. P. McKinnon, Q. Liu, L. Miao, L. Huang, E. M. Bachelder, K. M. Ainslie and J. P. Ting, JCI Insight, 2018, 3(22), e120638 CrossRef PubMed.
- L. Pan, L. Zhang, W. Deng, J. Lou, X. Gao, X. Lou, Y. Liu, X. Yao, Y. Sheng, Y. Yan, C. Ni, M. Wang, C. Tian, F. Wang and Z. Qin, J. Controlled Release, 2023, 357, 133–148 CrossRef CAS PubMed.
- D. J. Propper and F. R. Balkwill, Nat. Rev. Clin. Oncol., 2022, 19, 237–253 CrossRef CAS PubMed.
- K. E. de Visser and L. M. Coussens, Cancer Immunol. Immunother., 2005, 54, 1143–1152 CrossRef CAS PubMed.
- J. Ma, F. Liu, W. C. Sheu, Z. Meng, Y. Xie, H. Xu, M. Li, A. T. Chen, J. Liu, Y. Bao, X. Zhang, S. Zhang, L. Zhang, Z. Zou, H. Wu, H. Wang, Y. Zhu and J. Zhou, Nano Lett., 2020, 20, 4084–4094 CrossRef CAS PubMed.
- Y. Mi, C. C. Smith, F. Yang, Y. Qi, K. C. Roche, J. S. Serody, B. G. Vincent and A. Z. Wang, Adv. Mater., 2018, 30, e1706098 CrossRef PubMed.
- S.-Y. Li, Y. Liu, C.-F. Xu, S. Shen, R. Sun, X.-J. Du, J.-X. Xia, Y.-H. Zhu and J. Wang, J. Controlled Release, 2016, 231, 17–28 CrossRef CAS PubMed.
- Y. Hu, X. Liu, M. Ran, T. Yang, T. Li, Y. Wu, Y. Lin, Z. Qian and X. Gao, Mater. Today Nano, 2022, 17, 100151 CrossRef CAS.
- B. Kwong, H. Liu and D. J. Irvine, Biomaterials, 2011, 32, 5134–5147 CrossRef CAS PubMed.
- A. K. Kosmides, J. W. Sidhom, A. Fraser, C. A. Bessell and J. P. Schneck, ACS Nano, 2017, 11, 5417–5429 CrossRef CAS PubMed.
- W. Li, X. Zhang, C. Zhang, J. Yan, X. Hou, S. Du, C. Zeng, W. Zhao, B. Deng, D. W. McComb, Y. Zhang, D. D. Kang, J. Li, W. E. Carson, 3rd and Y. Dong, Nat. Commun., 2021, 12, 7264 CrossRef CAS PubMed.
- G. Kwak, D. Kim, G. H. Nam, S. Y. Wang, I. S. Kim, S. H. Kim, I. C. Kwon and Y. Yeo, ACS Nano, 2017, 11, 10135–10146 CrossRef CAS PubMed.
- Y. Wu, W. Gu, J. Li, C. Chen and Z. P. Xu, Nanomedicine, 2019, 14, 955–967 CrossRef CAS PubMed.
- P. Li, Y. Xie, J. Wang, C. Bao, J. Duan, Y. Liu, Q. Luo, J. Xu, Y. Ren and M. Jiang, Bioact. Mater., 2024, 34, 466–481 CAS.
- M. J. Smyth, E. Cretney, M. H. Kershaw and Y. Hayakawa, Immunol. Rev., 2004, 202, 275–293 CrossRef CAS PubMed.
- P. Rider, Y. Carmi and I. Cohen, Int. J. Cell Biol., 2016, 2016, 9259646 Search PubMed.
- J. Kim, S. Kang, K. W. Kim, M.-G. Heo, D.-I. Park, J.-H. Lee, N. J. Lim, D.-H. Min and C. Won, Biomaterials, 2022, 280, 121257 CrossRef CAS PubMed.
- J. Park, S. H. Wrzesinski, E. Stern, M. Look, J. Criscione, R. Ragheb, S. M. Jay, S. L. Demento, A. Agawu, P. Licona Limon, A. F. Ferrandino, D. Gonzalez, A. Habermann, R. A. Flavell and T. M. Fahmy, Nat. Mater., 2012, 11, 895–905 CrossRef CAS PubMed.
- A. E. Barberio, S. G. Smith, S. Correa, C. Nguyen, B. Nhan, M. Melo, T. Tokatlian, H. Suh, D. J. Irvine and P. T. Hammond, ACS Nano, 2020, 14, 11238–11253 CrossRef CAS PubMed.
- T. Li, Z. Liu, X. Fu, Y. Chen, S. Zhu and J. Zhang, Eur. J. Pharm. Biopharm., 2022, 177, 175–183 CrossRef CAS PubMed.
- Y. Li, Z. Su, W. Zhao, X. Zhang, N. Momin, C. Zhang, K. D. Wittrup, Y. Dong, D. J. Irvine and R. Weiss, Nat. Cancer, 2020, 1, 882–893 CrossRef CAS PubMed.
- A. E. Barberio, S. G. Smith, I. S. Pires, S. Iyer, F. Reinhardt, M. B. Melo, H. Suh, R. A. Weinberg, D. J. Irvine and P. T. Hammond, Bioeng. Transl. Med., 2023, 8, e10453 CrossRef CAS PubMed.
- L. Tang, Y. Zheng, M. B. Melo, L. Mabardi, A. P. Castano, Y. Q. Xie, N. Li, S. B. Kudchodkar, H. C. Wong, E. K. Jeng, M. V. Maus and D. J. Irvine, Nat. Biotechnol., 2018, 36, 707–716 CrossRef CAS PubMed.
- M. Wang, J. Zhang, J. Tang, X. Cai, R. Dou, C. Guo and Y. Hu, JCIS Open, 2023, 9, 100081 CrossRef.
- Q. Hu, L. Shang, M. Wang, K. Tu, M. Hu, Y. Yu, M. Xu, L. Kong, Y. Guo and Z. Zhang, Adv. Healthcare Mater., 2020, 9, e1901858 CrossRef PubMed.
- Y. Sun, L. Liu, L. Zhou, S. Yu, Y. Lan, Q. Liang, J. Liu, A. Cao and Y. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 45873–45890 CrossRef CAS PubMed.
- M. S. Sabel, J. Skitzki, L. Stoolman, N. K. Egilmez, E. Mathiowitz, N. Bailey, W. J. Chang and A. E. Chang, Ann. Surg. Oncol., 2004, 11, 147–156 CrossRef PubMed.
- J. G. Fewell, M. Matar, G. Slobodkin, S.-O. Han, J. Rice, B. Hovanes, D. H. Lewis and K. Anwer, J. Controlled Release, 2005, 109, 288–298 CrossRef CAS PubMed.
- P. H. Thaker, W. E. Brady, H. A. Lankes, K. Odunsi, W. H. Bradley, K. N. Moore, C. Y. Muller, K. Anwer, R. J. Schilder and R. D. Alvarez, Gynecol. Oncol., 2017, 147, 283–290 CrossRef CAS PubMed.
- J.-Q. Liu, C. Zhang, X. Zhang, J. Yan, C. Zeng, F. Talebian, K. Lynch, W. Zhao, X. Hou and S. Du, J. Controlled Release, 2022, 345, 306–313 CrossRef CAS PubMed.
- E. J. Comparetti, P. M. P. Lins, J. Quitiba and V. Zucolotto, J. Biomed. Mater. Res., Part A, 2022, 110, 1499–1511 CrossRef CAS PubMed.
- M. Tang, B. Chen, H. Xia, M. Pan, R. Zhao, J. Zhou, Q. Yin, F. Wan, Y. Yan, C. Fu, L. Zhong, Q. Zhang and Y. Wang, Nat. Commun., 2023, 14, 5888 CrossRef CAS PubMed.
- Y. Zhang, X. Hou, S. Du, Y. Xue, J. Yan, D. D. Kang, Y. Zhong, C. Wang, B. Deng, D. W. McComb and Y. Dong, Nat. Nanotechnol., 2023, 18, 1364–1374 CrossRef CAS PubMed.
- Y. Li, K. Tang, X. Zhang, W. Pan, N. Li and B. Tang, Chem. Sci., 2021, 13, 105–110 RSC.
- I. Tombacz, D. Laczko, H. Shahnawaz, H. Muramatsu, A. Natesan, A. Yadegari, T. E. Papp, M. G. Alameh, V. Shuvaev, B. L. Mui, Y. K. Tam, V. Muzykantov, N. Pardi, D. Weissman and H. Parhiz, Mol. Ther., 2021, 29, 3293–3304 CrossRef CAS PubMed.
- D. Wang, T. Wang, H. Yu, B. Feng, L. Zhou, F. Zhou, B. Hou, H. Zhang, M. Luo and Y. Li, Sci. Immunol., 2019, 4, eaau6584 CrossRef CAS PubMed.
- Z. Yao, C. Qi, F. Zhang, H. Yao, C. Wang, X. Cao, C. Zhao, Z. Wang, M. Qi and C. Yao, Acta Biomater., 2024, 173, 365–377 CrossRef CAS PubMed.
- D. Ding, H. Zhong, R. Liang, T. Lan, X. Zhu, S. Huang, Y. Wang, J. Shao, X. Shuai and B. Wei, Adv. Sci., 2021, 8, e2100712 CrossRef PubMed.
- J. Li, Y.-F. Ding, Z. Wang, Q. Cheng, J. Wei, Z. Yang and R. Wang, Nano Today, 2024, 54, 102104 CrossRef CAS.
- S. Zhou, Z. Zhen, A. V. Paschall, L. Xue, X. Yang, A. G. Bebin-Blackwell, Z. Cao, W. Zhang, M. Wang, Y. Teng, G. Zhou, Z. Li, F. Y. Avci, W. Tang and J. Xie, Adv. Funct. Mater., 2021, 31(7), 2007017 CrossRef CAS PubMed.
- M. Liu, W. Song and L. Huang, Cancer Lett., 2019, 448, 31–39 CrossRef CAS PubMed.
- Y. Hu, W. Nie, L. Lyu, X. Zhang, W. Wang, Y. Zhang, S. He, A. Guo, F. Liu, B. Wang, Z. Qian and X. Gao, ACS Nano, 2024, 18, 3295–3312 CrossRef CAS PubMed.
- Z. Wen, H. Liu, D. Qiao, H. Chen, L. Li, Z. Yang, C. Zhu, Z. Zeng, Y. Chen and L. Liu, ACS Nano, 2023, 17, 7194–7206 CrossRef CAS PubMed.
- J. Lu, X. Liu, Y. P. Liao, F. Salazar, B. Sun, W. Jiang, C. H. Chang, J. Jiang, X. Wang, A. M. Wu, H. Meng and A. E. Nel, Nat. Commun., 2017, 8, 1811 CrossRef PubMed.
- D. Kim, S. J. Kim, J. Jeong, S. Han, H. Kim, S. Lee, I. Choi, J. Hong, J. O. Jin and J. B. Lee, ACS Nano, 2024, 18, 1744–1755 CrossRef CAS PubMed.
- Y. Yang, B. Liu, Y. Liu, J. Chen, Y. Sun, X. Pan, J. Xu, S. Xu, Z. Liu and W. Tan, Nano Lett., 2022, 22, 2826–2834 CrossRef CAS PubMed.
- K. Perica, A. Tu, A. Richter, J. G. Bieler, M. Edidin and J. P. Schneck, ACS Nano, 2014, 8, 2252–2260 CrossRef CAS PubMed.
- S. Y. Li, Y. Liu, C. F. Xu, S. Shen, R. Sun, X. J. Du, J. X. Xia, Y. H. Zhu and J. Wang, J. Controlled Release, 2016, 231, 17–28 CrossRef CAS PubMed.
- P. M. Cevaal, A. Ali, E. Czuba-Wojnilowicz, J. Symons, S. R. Lewin, C. Cortez-Jugo and F. Caruso, ACS Nano, 2021, 15, 3736–3753 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |