
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructurally and surficially activated TiO2 nanomaterials for photochemical reactions
Si Yin
Tee
 *a,
Junhua
Kong
a,
Justin Junqiang
Koh
a,
Choon Peng
Teng
a,
Xizu
Wang
a,
Xiaobai
Wang
a,
Siew Lang
Teo
a,
Warintorn
Thitsartarn
a,
Ming-Yong
Han
*b and
Zhi Wei
Seh
*a
*a,
Junhua
Kong
a,
Justin Junqiang
Koh
a,
Choon Peng
Teng
a,
Xizu
Wang
a,
Xiaobai
Wang
a,
Siew Lang
Teo
a,
Warintorn
Thitsartarn
a,
Ming-Yong
Han
*b and
Zhi Wei
Seh
*a
aInstitute of Materials Research and Engineering (IMRE), Agency for Science, Technology and Research (A*STAR), 2 Fusionopolis Way, Innovis #08-03, Singapore 138634, Republic of Singapore. E-mail: teesyi@imre.a-star.edu.sg; sehzw@imre.a-star.edu.sg
bInstitute of Molecular Plus, Tianjin University, Tianjin 300072, China. E-mail: han_mingyong@tju.edu.cn
First published on 9th September 2024
Abstract
Renewable fuels and environmental remediation are of paramount importance in today's world due to escalating concerns about climate change, pollution, and the finite nature of fossil fuels. Transitioning to sustainable energy sources and addressing environmental pollution has become an urgent necessity. Photocatalysis, particularly harnessing solar energy to drive chemical reactions for environmental remediation and clean fuel production, holds significant promise among emerging technologies. As a benchmark semiconductor in photocatalysis, TiO2 photocatalyst offers an excellent solution for environmental remediation and serves as a key tool in energy conversion and chemical synthesis. Despite its status as the default photocatalyst, TiO2 suffers from drawbacks such as a high recombination rate of charge carriers, low electrical conductivity, and limited absorption in the visible light spectrum. This review provides an in-depth exploration of the fundamental principles of photocatalytic reactions and presents recent advancements in the development of TiO2 photocatalysts. It specifically focuses on strategic approaches aimed at enhancing the performance of TiO2 photocatalysts, including improving visible light absorption for efficient solar energy harvesting, enhancing charge separation and transportation efficiency, and ensuring stability for robust photocatalysis. Additionally, the review delves into the application of photodegradation and photocatalysis, particularly in critical processes such as water splitting, carbon dioxide reduction, nitrogen fixation, hydrogen peroxide generation, and alcohol oxidation. It also highlights the novel use of TiO2 in plastic polymerization and degradation, showcasing its potential for converting plastic waste into valuable chemicals and fuels, thereby offering sustainable waste management solutions. By addressing these essential areas, the review offers valuable insights into the potential of TiO2 photocatalysis for addressing pressing environmental and energy challenges. Furthermore, the review encompasses the application of TiO2 photochromic systems, expanding its scope to include other innovative research and applications. Finally, it addresses the underlying challenges and provides perspectives on the future development of TiO2 photocatalysts. Through addressing these issues and implementing innovative strategies, TiO2 photocatalysis can continue to evolve and play a pivotal role in sustainable energy and environmental applications.
 Si Yin Tee | Si Yin Tee obtained her PhD in Biomedical Engineering from National University of Singapore. Currently, she is working as a research scientist at the Institute of Materials Research and Engineering, A*STAR. Her research focuses on the development of functional metal and semiconductor nanostructures for biomedical, environmental and energy applications. |
 Ming-Yong Han | Ming-Yong Han has served at National University of Singapore, Institute of Materials Research and Engineering, and Tianjin University. Throughout his career, Ming-Yong Han has passionately tackled interdisciplinary challenges at the intersections of nanoscience, nanotechnology, biotechnology, and energy/biomedical applications. His contributions are extensive, encompassing the publication of ∼300 papers with ∼30 |
 Zhi Wei Seh | Zhi Wei Seh is a Senior Principal Scientist at the Institute of Materials Research and Engineering, A*STAR. He received his BS and PhD degrees in Materials Science and Engineering from Cornell University and Stanford University, respectively. His research interests lie in the design of new battery materials beyond lithium-ion technology. As a Highly Cited Researcher, he is widely recognized for designing the first yolk–shell nanostructure in lithium–sulfur batteries, which is a licensed technology. He pioneered the first study of sodium dendrites using cryogenic TEM, and the first anode-free magnesium battery with five times the energy density of standard magnesium batteries. |
1 Introduction
Photocatalysis stands as a promising solution for addressing a wide array of environmental and energy-related challenges by facilitating photochemical catalytic reactions. This process harnesses the abundant and renewable energy of sunlight to drive chemical transformations without requiring additional energy input, thereby positioning it as an attractive avenue for diverse applications. Typically, photocatalysis employs a semiconductor photocatalyst that absorbs photons, generating active electron–hole pairs upon exposure to light. This photoexcitation leads to the generation of electrons in the conduction band (CB) and electronic vacancies or holes in the valence band (VB). Among the numerous photocatalysts derived from a variety of semiconducting materials, including metal oxides,1 oxysulfides,2 metal sulfides,3 oxynitrides,4 and their composites,5 metal oxides hold particular importance due to their favorable band gap and band edge positions. Notably, TiO2 has emerged as one extensively studied semiconductor in photocatalysis due to its exceptional optical and electronic properties, as well as its unique chemical and physical characteristics facilitating a broad spectrum of reactions under solar irradiation. Since its pioneering role in photo-assisted water splitting in the early 1970s,6 TiO2 has garnered significant attention as a photocatalyst, leading to extensive research into the fabrication, structure, and applications of nanostructured TiO2-based photocatalysts.7,8 These photocatalysts have found widespread consideration for numerous processes, such as environmental purification,9–12 water splitting,13,14 carbon dioxide reduction,15,16 nitrogen fixation,17,18 hydrogen peroxide generation,19,20 alcohol oxidation,21,22 and many more.However, despite its remarkable properties, the widespread utilization of TiO2 in photocatalytic applications faces inherent limitations. A primary challenge arises from its relatively large band gap of 3.2 eV, necessitating the use of high-energy photons for activation, primarily in the ultraviolet (UV) region below 400 nm, which represents only a small fraction of the total solar spectrum. Consequently, TiO2 photocatalysts exhibit limited efficiency under natural sunlight conditions, with a substantial portion of solar energy remaining untapped. Moreover, TiO2 is constrained by rapid recombination of electron–hole pairs, resulting in lifetimes typically ranging from 10−12 to 10−11 seconds, further diminishing its photocatalytic performance. To overcome these challenges, substantial efforts have been directed towards extending the working spectrum of TiO2 photocatalysts into the visible light range, which accounts for 45% of total solar energy, and enhancing their charge carrier separation capabilities. Various approaches have thus been explored, aiming to unlock the full potential of TiO2 photocatalysts for solar-driven processes, including bandgap engineering, surface modification, defect engineering, nanostructuring, and hybridization.
(I) Band gap engineering involves modifying the electronic structure of TiO2 to shift its band gap, typically achieved through doping with metal or non-metal ions. This introduces donor or acceptor states below or above the conduction or valence bands, respectively, enabling electronic transitions with lower energy compared to pure TiO2. As a result, visible light absorption and photocatalytic activity are enhanced.23 (II) Surface modification of TiO2, through functionalization or nanoparticle deposition, creates localized energy states within the band gap.24 For instance, deposition of noble metals like Pt or Au can trap visible light and enhance charge separation, thereby improving photocatalytic activity under visible light irradiation.24 (III) Defect engineering through introducing hydrogen to TiO2 can create oxygen vacancies and modify its electronic structure, narrowing the band gap and improving visible light absorption.25 (IV) Nanostructuring TiO2 at the nanoscale can create quantum confinement effects or surface defects, modifying its electronic properties and band gap.26 With the large surface-to-volume ratio of nanomaterials, abundant surface reaction sites are offered, potentially modulating the catalytic activity of surface atoms and leading to improved catalytic performance.27–30 (V) Hybridization techniques for TiO2 involve forming composites with various materials such as graphitic carbon nitride, MXenes, metal–organic frameworks, and aerogels. These materials are integrated with TiO2 to enhance its photocatalytic performance by leveraging their unique properties. For instance, graphitic carbon nitride can improve visible light absorption,31 MXenes can enhance conductivity and charge separation,32 metal–organic frameworks can provide high surface area and tunable porosity,33 and aerogels can offer lightweight structures with high surface area.34 By combining TiO2 with these materials, hybrid composites can achieve superior light absorption, increased charge carrier separation, and improved catalytic activity, addressing the limitations of pure TiO2.
In light of the significant research advancements in the field of TiO2, a brief search of the literature in the Web of Science database for “TiO2” returns over 260![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 results, demonstrating the extensive interest and investigation in this material. When specifically searching for “TiO2 photocatalyst”, the results narrow to over 30000, indicating the focused research on its photocatalytic properties. However, this number may still underrepresent the true scope, as many relevant studies might not explicitly label TiO2 as a photocatalyst. This extensive body of research highlights the critical importance of TiO2 in the field of photocatalysis, underlining its significant role and impact in advancing environmental remediation and energy conversion technologies over several decades.
000 results, demonstrating the extensive interest and investigation in this material. When specifically searching for “TiO2 photocatalyst”, the results narrow to over 30000, indicating the focused research on its photocatalytic properties. However, this number may still underrepresent the true scope, as many relevant studies might not explicitly label TiO2 as a photocatalyst. This extensive body of research highlights the critical importance of TiO2 in the field of photocatalysis, underlining its significant role and impact in advancing environmental remediation and energy conversion technologies over several decades.
For a quick overview of the research history, we present a brief timeline of milestones in the development of TiO2 photocatalysts for various applications (Fig. 1). The first investigation of TiO2 as a photocatalyst began in the 1970s when it was identified as a semiconductor capable of splitting water under UV light. During the 1980s and 1990s, significant advancements in doping techniques enhanced its photocatalytic efficiency under visible light. The early 2000s introduced engineered nanostructures and facets, which further improved TiO2's performance. In the 2010s, research focused on defect engineering and developing heterojunctions and composite/hybrid materials, incorporating TiO2 with other semiconductors and noble metals to boost its photocatalytic activity. Recent years have seen progress in single-atom co-catalysts, with atomically dispersed metal atoms on TiO2 tuning active sites, selectivity, and stability. Moving forward, 3D printing technology has enabled the creation of complex TiO2 nanostructures with favorable properties for photochemical reactions. These advancements have expanded TiO2's applications in environmental remediation and energy conversion, with photoreforming waste polymers into sustainable hydrogen fuel and chemical feedstock marking a significant leap in waste-to-energy technology.
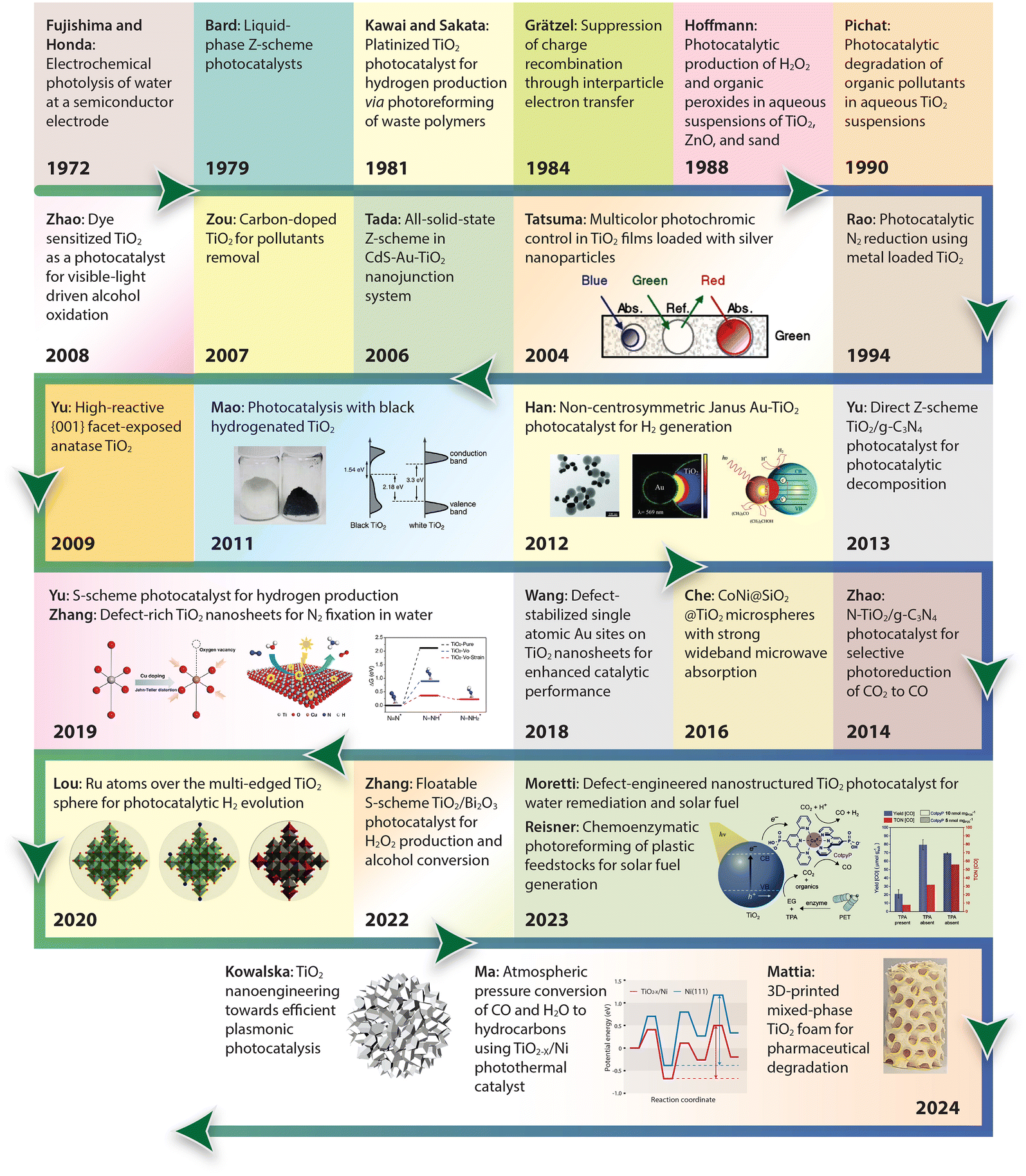 | ||
| Fig. 1 Timeline of milestones in TiO2 nanomaterials development, highlighting continuous advancements and innovations in the field. 1972, Fujishima and Honda: Electrochemical photolysis of water at a semiconductor electrode.35 1979, Bard: Liquid-phase Z-scheme photocatalysts.36 1981, Kawai and Sakata: Platinized TiO2 photocatalyst for hydrogen production via photoreforming of waste polymers.37 1984, Grätzel: Suppression of charge recombination through interparticle electron transfer.38 1988, Hoffmann: Photocatalytic production of H2O2 and organic peroxides in aqueous suspensions of TiO2, ZnO, and sand.39 1990, Pichat: Photocatalytic degradation of organic pollutants in aqueous TiO2 suspensions.40 1994, Rao: Photocatalytic N2 reduction using metal loaded TiO2.41 2004, Tatsuma: Multicolor photochromic control in TiO2 films loaded with silver nanoparticles. Reproduced with permission from ref. 42. Copyright 2004, American Chemical Society. 2006, Tada: All-solid-state Z-scheme in CdS-Au-TiO2 nanojunction system.43 2007, Zou: Carbon-doped TiO2 for pollutants removal.44 2008, Zhao: Dye sensitized TiO2 as a photocatalyst for visible-light driven alcohol oxidation.45 2009, Yu: High-reactive (001) facet-exposed anatase TiO2.46 2011, Mao: Photocatalysis with black hydrogenated TiO2. Reproduced with permission from ref. 25. Copyright 2011, American Association for the Advancement of Science. 2012, Han: Non-centrosymmetric Janus Au-TiO2 photocatalyst for H2 generation Reproduced with permission from ref. 13. Copyright 2012, Wiley-VCH. 2013, Yu: Direct Z-scheme TiO2/g-C3N4 photocatalyst for photocatalytic decomposition.47 2014, Zhao: N-TiO2/g-C3N4 photocatalyst for selective photoreduction of CO2 to CO.48 2016, Che: CoNi@SiO2 @TiO2 microspheres with strong wideband microwave absorption.49 2018, Wang: Defect-stabilized single atomic Au sites on TiO2.50 2019, Yu: S-scheme photocatalyst for hydrogen production.51 2019, Zhang: Defect-rich TiO2 nanosheets for N2 fixation in water. Reproduced with permission from ref. 52. Copyright 2019, Wiley-VCH. 2020, Lou: Ru atoms over the multi-edged TiO2 sphere for photocatalytic H2 evolution. Reproduced with permission from ref. 53. Copyright 2020, American Association for the Advancement of Science. 2022, Zhang: Floatable S-scheme TiO2/Bi2O3 photocatalyst for H2O2 Production and alcohol conversion.54 2023, Moretti: Defect-engineered nanostructured TiO2 photocatalyst for water remediation and solar fuel.55 2023, Reisner: Chemoenzymatic photoreforming of plastic feedstocks for solar fuel generation. Reproduced with permission from ref. 56. Copyright 2023, Wiley-VCH. 2024, Mattia: 3D-printed mixed-phase TiO2 foam for pharmaceutical degradation. Reproduced with permission from ref. 57. Copyright 2024, Royal Society of Chemistry. 2024, Ma: Atmospheric pressure conversion of CO and H2O to hydrocarbons using TiO2−x/Ni photothermal catalyst.58 2024, Kowalska: TiO2 nanoengineering towards efficient plasmonic photocatalysis. Reproduced with permission from ref. 59. Copyright 2024, Elsevier. | ||
This review provides a comprehensive and interdisciplinary perspective on the advancements in TiO2 photocatalysts, bridging materials science, chemistry, environmental science, and engineering to offer a holistic understanding of their diverse applications. Going beyond general advancements, this review offers a detailed analysis of specific techniques such as doping, metal nanoparticle incorporation for surface plasmon resonance, morphological control, and hybridization. These approaches target three main steps: improving visible light absorption for efficient solar energy harvesting, enhancing charge separation and transportation efficiency, and maximizing charge utilization while ensuring good stability for robust photocatalysis. Recent progress includes the development of single-atom co-catalysts, advanced heterojunctions, and sophisticated 3D printing technologies for creating complex TiO2 nanostructures.
These developments have significantly broadened TiO2's applications. In the realm of environmental remediation, TiO2 photocatalysts have shown significant promise in breaking down pollutants under visible light, making them viable for water and air purification processes. Their antibacterial properties also present opportunities for public health improvements. For sustainable energy production, TiO2 has been explored extensively for water splitting, which generates hydrogen as a clean fuel. In addition, it is used for carbon dioxide reduction, converting carbon dioxide into valuable fuels, and nitrogen fixation, which is critical for producing ammonia for fertilizers.
Emerging applications such as hydrogen peroxide generation and alcohol oxidation are also discussed, illustrating the broadening scope of TiO2 photocatalysis. The review delves into the novel application of TiO2 in plastic polymerization and degradation processes. Here, TiO2 photocatalysts facilitate the breakdown of plastic waste into valuable chemicals and fuels, offering a sustainable approach to waste management and resource recovery. Recent research highlights the potential of photocatalytic depolymerization of various plastics and the conversion of plastic-derived intermediates into useful products, addressing plastic pollution and promoting circular economy principles (Fig. 2).
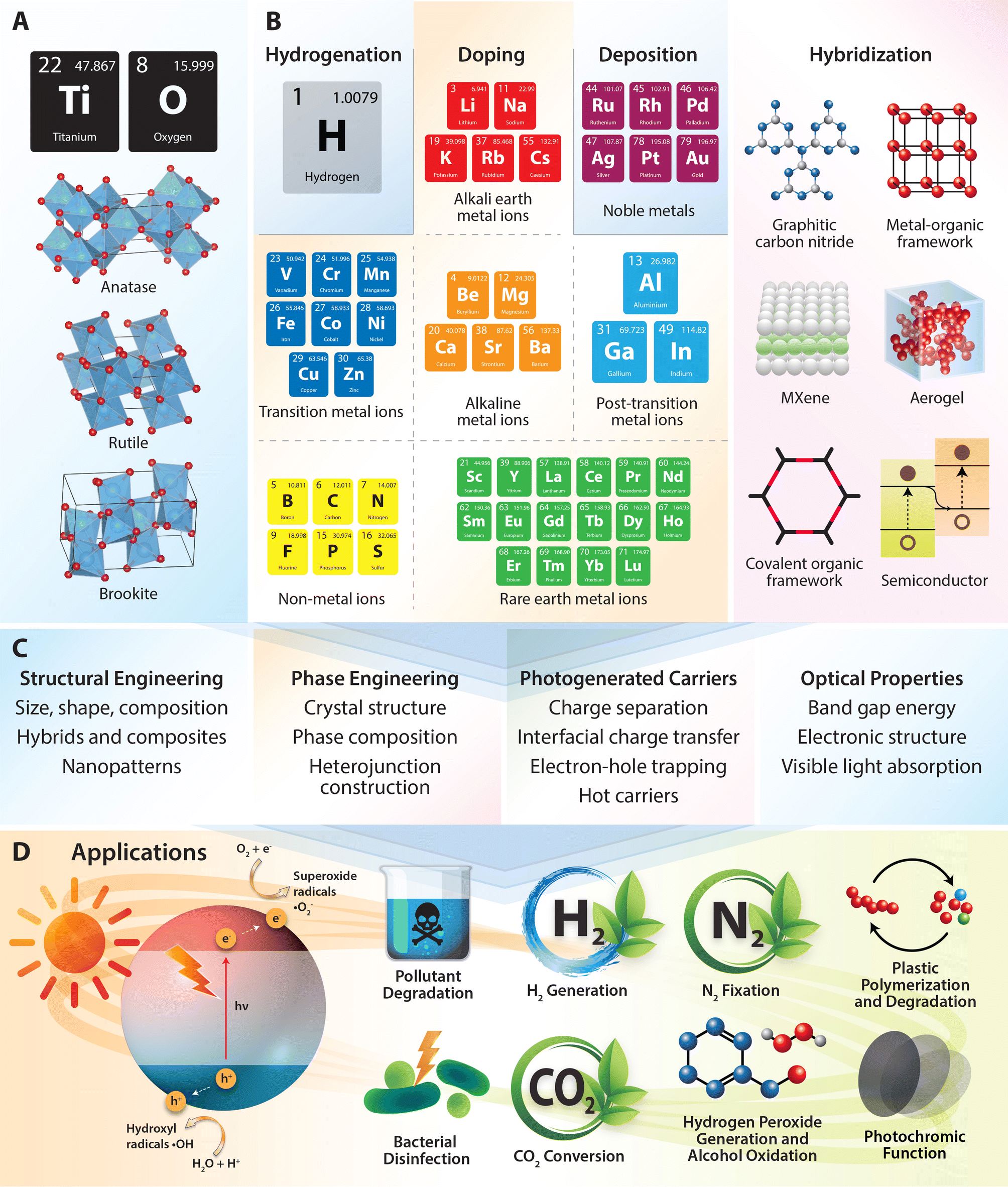 | ||
| Fig. 2 An overview on modification strategies of TiO2 for photocatalytic applications. (A) Crystal structures of TiO2 in anatase, rutile, and brookite phases. (B and C) Modification strategies used to optimize structural, electronic, and surface properties of TiO2 semiconductor. Key techniques include hydrogenation (defect engineering), ion-doping (metal/non-metal), noble metal deposition, and hybridization/compositing with other materials. (D) Schematic illustration of the mechanism of TiO2 photocatalysis for applications in environmental remediation, bacterial disinfection, hydrogen generation, carbon dioxide reduction, nitrogen fixation, hydrogen peroxide generation, alcohol oxidation, photochromic functions, plastic polymerization and degradation. | ||
Finally, the review presents a summary and outlook on the major challenges and promising perspectives for future research in TiO2 photocatalysts. Overall, the advancements in TiO2-based photocatalysts are anticipated to provide robust solutions to some of the most pressing energy and environmental challenges facing society today. The review highlights the transformative potential of TiO2 photocatalysts in addressing these global issues and sets a clear roadmap for future research directions aimed at maximizing their efficiency and practical applicability.
2 Photochemical reactions of TiO2 nanomaterials
TiO2 occurs in nature in three distinct polymorphs: rutile, anatase, and brookite, with rutile being the most abundant followed by anatase and then brookite. In all three forms of TiO2, the TiO6 octahedra exhibit distortion, with titanium ions coordinated to six oxygen ions. In anatase, the TiO6 octahedra are arranged in a tetragonal structure where each octahedron shares four edges with neighboring octahedra but lacks corners. This unique configuration results in a lattice with specific characteristics such as high surface area and excellent photocatalytic activity. Rutile also features a tetragonal structure, albeit with a different alignment of the TiO6 octahedra. In rutile, each octahedron shares two opposing edges to create linear chains in the (001) direction, which are interconnected at their corners, leading to a denser crystal lattice compared to anatase. Brookite's orthorhombic crystal structure is distinguished by octahedra sharing three edges and three corners, resulting in a more intricate lattice structure compared to anatase and rutile. Among these polymorphs, rutile is the most thermodynamically stable phase of TiO2, while anatase and brookite are metastable phases that can transition into rutile at elevated temperatures.60,61 The varied properties and functionalities of these TiO2 polymorphs establish them as essential materials in photochemical reactions (refer to Table 1).| Photocatalysts | Synthesis methods | Pollutants | Degradation efficiency | Ref. |
|---|---|---|---|---|
| TiO2 nanosheets | Solvothermal | Rhodamine B | >90% in 15 min under UV light | 250 |
| TiO2 nanorods | Hydrothermal | p-Cresol | >90% in 75 min under UV-visible light | 267 |
| TiO2 nanostructures | Hydrothermal | Rhodamine B | >90% in 150 min under UV light | 251 |
| TiO2 nanotubes | Hydrothermal | Orange II | 89.46% in 2000 min under UV light | 392 |
| TiO2 nanotubes | Electrochemical anodization | Volatile organic compounds | 72.1% in 30 min under UV-LED | 393 |
| TiO2 nanotubes | Electrochemical anodization | β-Blocker metoprolol | 87.09% (milli-water) and 62.05% (tap water) in 120 min under UV-LED | 394 |
| TiO2 nanotubes | Hydrothermal | H2S | 10 ppm to 1.0 ppb in 3 h under UV light | 395 |
| Mesoporous TiO2 nanoshell@polyimide nanofibers | In situ complexation-hydrolysis | Methylene blue | 95% in 40 min under UV light | 396 |
| Nano flower-like rutile TiO2 | Hydrothermal | Methylene blue | 98.95% in 180 min under solar light | 252 |
| Raschig rings-supported TiO2 | Sol–gel | Rhodamine 6G | 91% in 30 min under UV light | 397 |
| TiO2 films | Magnetron sputtering | (i) Methylene blue, (ii) E. coli bacteria | (i) 45% in 8 h under UVB light, (ii) >90% in 45 min under UVB light | 280 |
| TiO2 sheets | Chemical synthesis/hydrogenation | Rhodamine B | 99% in 75 min under simulated solar light | 398 |
| TiO2 hollow spheres | Hard template-based approach | Ciprofloxacin | 82% in 6 h under simulated solar light with AM1.5G filter, 100 W Xe arc lamp | 55 |
| TiO2 particles | Ultrasound assisted sol–gel | Metformin | 75.4% in 120 min under UV light | 61 |
TiO2 photocatalytic reactions rely on the fundamental generation of electron–hole pairs through photoexcitation. When TiO2 is exposed to UV light, electron–hole pairs are generated between the valence band (VB) and conduction band (CB). These pairs subsequently migrate to the surface of the TiO2, where they either undergo recombination and release the energy as heat, become trapped in metastable surface states, or interact with adsorbed electron donors/acceptors. In photodegradation reactions, the holes can directly react with organic compounds or oxidize water to produce hydroxyl radicals (˙OH), initiating an oxidative process that leads to the breakdown of organic compounds. Meanwhile, the electrons can react with organic compounds to form reduction products or with molecular oxygen to generate superoxides (˙O2−), which can trigger the formation of various reactive oxygen species (e.g., ˙O2−, ˙OH, H2O2, etc.).62 These photogenerated reactive oxidative species play a crucial role not only in degrading organic compounds but also in the photocatalytic inactivation of bacteria by oxidizing their cell walls and inducing cell lysis. In aerobic conditions, bacterial cells are inactivated through oxidation, whereas in anaerobic conditions, bacterial cells are inactivated through reduction, with the cells capable of trapping the electrons to prevent charge recombination.63
When a photocatalyst is utilized for water splitting to generate hydrogen, it is essential that the conduction band (CB) level is more negative than the reduction potential of H+ to H2 (0 V vs. NHE at pH 0), and the valence band (VB) should be more positive than the oxidation potential of H2O to O2 (1.23 V vs. NHE). This requirement indicates that a minimum photon energy of 1.23 eV is needed to facilitate photocatalytic water splitting, corresponding to a wavelength of approximately 1000 nm. However, an activation barrier exists in the charge transfer process between the solid photocatalyst and water molecules, necessitating a photon energy exceeding the band gap of the photocatalyst to effectively split pure water at a reasonable rate.14
In the realm of carbon dioxide reduction, the process involves converting carbon dioxide into valuable chemical products like hydrocarbons or synthetic fuels by leveraging the reactivity of electron–hole pairs generated by the photocatalyst. The photocatalytic reduction of carbon dioxide with TiO2 entails a series of steps facilitated by photoexcited charge carriers. Upon light exposure, TiO2 absorbs photons, creating electron–hole pairs that migrate to the semiconductor's surface. Carbon dioxide molecules adsorb onto the TiO2 surface, where photoexcited electrons can reduce them to form various carbon-based products such as carbon monoxide (CO), methane (CH4), or formic acid (HCOOH). This reduction process involves multiple proton–electron transfer steps, followed by the release of the newly formed carbon-based products from the TiO2 surface into the environment.64
Similarly, in nitrogen fixation, electron–hole pairs play a pivotal role in catalyzing the reduction of nitrogen molecules to produce ammonia, a critical component in fertilizers and various chemical processes. The thermodynamically non-spontaneous reaction (N2 + 3H2O → 2NH3 + 1.5O2), which combines water splitting and nitrogen fixation, can be accomplished with solar energy input. The photocatalytic process of nitrogen fixation can be delineated into multiple stages. Initially, photogenerated electrons are promoted to the CB, creating vacancies (or holes) in the VB. Subsequently, some of these electrons and holes may recombine, while others migrate to the catalyst surface to participate in redox reactions. Specifically, water can be oxidized to oxygen by the holes, whereas nitrogen is reduced to form ammonia through successive transfers of photogenerated electrons and protons sourced from water.17
3 Modification of electronic structures by doping
Despite significant advancements in the design and improvement of TiO2 photocatalysts, challenges persist when using pure TiO2 due to its low quantum efficiency and limited utilization of visible light. To address these challenges, considerable efforts have been directed towards modifying the electronic structure of TiO2 to shift its absorption into the visible range and enhance its photocatalytic performance. One approach involves doping, which introduces permissible electronic states between the CB and VB by incorporating metal ion (Table 2) and non-metal ion (Table 3) dopants. It is well-known that metal ions incorporated into the TiO2 lattice typically introduce donor states below the conduction band, while non-metal ions contribute to the formation of acceptor states above the valence band (Fig. 3A). These donor and acceptor states play crucial roles in altering the electronic structure of TiO2, affecting its band gap and, consequently, its optical and photocatalytic properties.62,65 In this section, we outline recent progress in fabricating various TiO2 semiconductors to enhance photoconversion efficiency in the visible light region, employing strategies such as doping with metal ions and non-metal ions.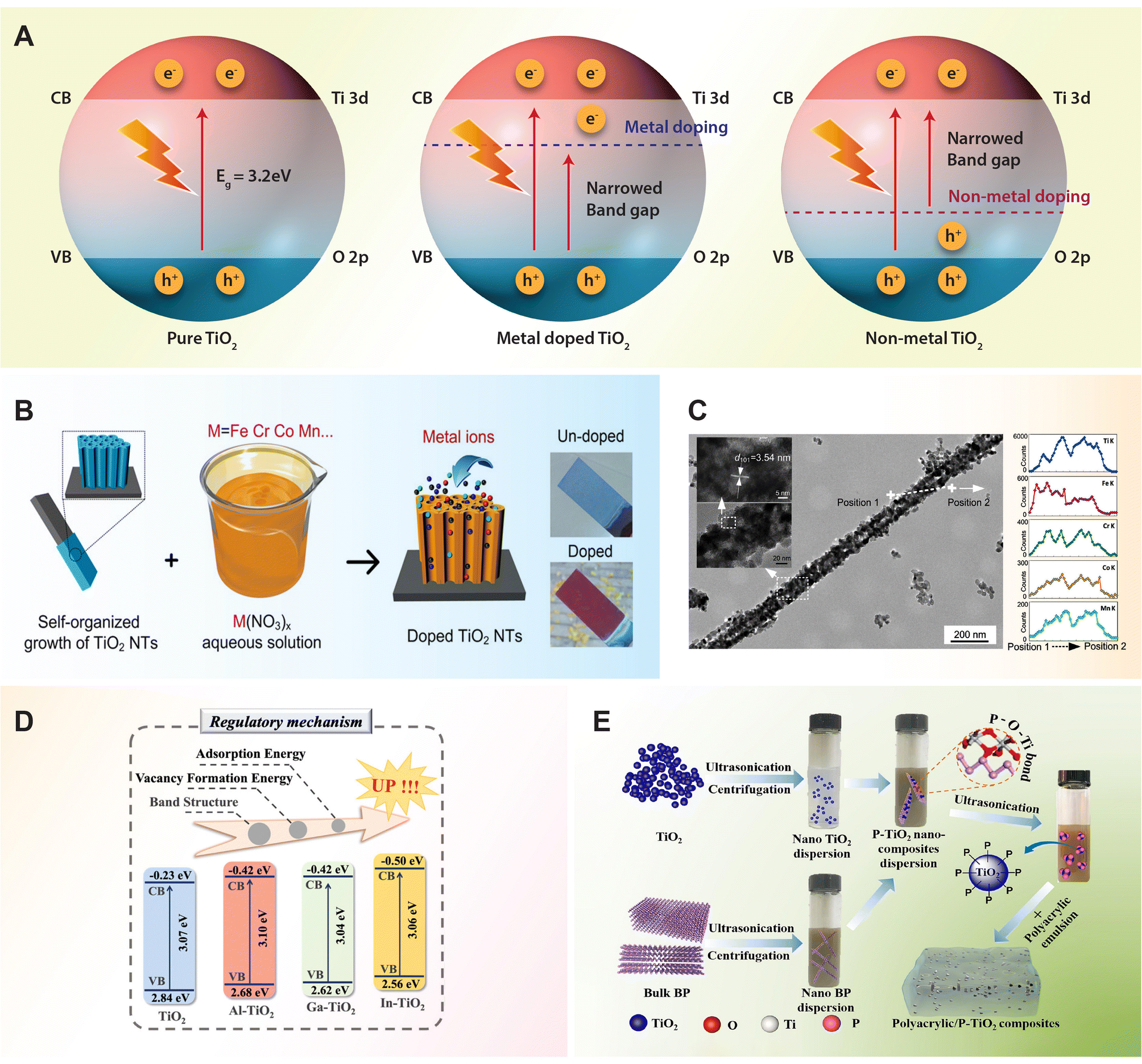 | ||
| Fig. 3 (A) Schematic diagram of photocatalytic mechanisms of pure, metal and non-metal doped TiO2 semiconductor. (B) Metal ions doping of transition metal ions (Cr, Co, Fe, Mn) doped TiO2 nanotubes through anodization in an aqueous solution. (C) Transmission electron microscopy (TEM) image of transition metal ion doped TiO2 nanotube with corresponding elemental composition line scans. Reproduced with permission from ref. 84. Copyright 2019, American Chemical Society. (D) Band structure position of trivalent metal ions (Al, Ga, In)-doped TiO2. Reproduced with permission from ref. 85. Copyright 2024, American Chemical Society. (E) Schematic illustration of fabricating phosphorus doped TiO2/polyacrylic composites by using black phosphorous as the phosphate source. Reproduced with permission from ref. 90. Copyright 2024, Elsevier. | ||
| Photocatalysts | Synthesis methods | Pollutants | Degradation efficiency | Ref. |
|---|---|---|---|---|
| Na–TiO2 nano-powder | Sol–gel | Methylene blue | 92.5% in 60 min under UV light, peak λmax = 365 nm | 72 |
| Na–TiO2 nanotubes | Electrochemical anodization | Methylene blue | 97.3% in 180 min under solar light | 73 |
| Rb–TiO2 nanoparticles | Sol–gel | Methylene blue | 97% in 60 min under UV light | 77 |
| Mg–TiO2 nanoparticles | Sol–gel | Methyl orange | 47.82% in 120 min under UV light | 80 |
| Ca–TiO2 nanofibers | Sol–gel electrospinning | Rhodamine B | 95% in 100 min under UV light | 399 |
| Ba–TiO2−x (x = 5%) | Template assisted synthesis | Rhodamine B | 99.4% in 30 min under simulated solar light | 400 |
| Ba–TiO2 quantum dots | Co-precipitation | Methylene blue | 99.5% in 120 min under visible light | 81 |
| Fe–TiO2 nanoparticles | Sol–gel | Methyl orange | 98% in 60 min under visible light | 88 |
| Fe–TiO2 nanoparticles | Electrospray-assisted flame spray pyrolysis | Escherichia coli | 99.4% in 5 h under UV light | 87 |
| In–TiO2 nanoparticles | Sol–gel | H2S | 100% in 120 min under UV light | 85 |
| Mo–TiO2 particles | Sol–gel/underwater plasma | Rhodamine B | 96% in 60 min under visible light | 256 |
| W–TiO2 particles | Sol–gel/underwater plasma | Rhodamine B | 96% in 60 min under visible light | 256 |
| Tb–TiO2 coating | Plasma electrolytic oxidation | Methyl orange | 75% in 12 h under simulated sunlight | 92 |
| Nd–TiO2 nanoparticles | Sol–gel | Methyl orange | 96.5% in 120 min under simulated sunlight | 91 |
| Eu–TiO2 nanorods | Hydrothermal | Methyl orange | 100% in 20 min under simulated sunlight | 89 |
| Er–Ce co-doped TiO2 nanoparticles | Sol–gel | (i) Staphylococcus aureus, (ii) E. coli | (i) 91.23% and (ii) 92.8% in 20 min under simulated double solar radiation | 281 |
| Photocatalysts | Synthesis methods | Pollutants | Degradation efficiency | Ref. |
|---|---|---|---|---|
| B–TiO2 particles | Sol–gel | (i) Diuron, (ii) o-phenylphenol, (iii) 2-methyl-4-chlorophenoxy-acetic acid, (iv) terbuthylazine | (i) and (ii) 70–80% in 120 min, (iii) >90% in 45 min, (iv) 50% in 120 min under solar light | 271 |
| B–TiO2 nanoparticles | Solvothermal | (i) 2,4-Dichloro-phenol, (ii) bisphenol-A, (iii) ibuprofen & flurbiprofen | (i) 75.1% in 5 h, (ii) 97.7% in 4 h, (iii) 81–85% in 5 h under visible light | 275 |
| B–TiO2 nanocrystals | Electrochemical anodization/hydrothermal | Rhodamine B | >90% in 60 min under visible light | 401 |
| B–TiO2 nanoparticles | Co-precipitation | Rhodamine B | >90% in 90 min under visible light | 99 |
| C–TiO2 particles | Controlled hydrolysis | Caffeic acid | >90% in 120 min under visible light | 402 |
| C–TiO2 core–shell nanostructures (TiO2@C) | In situ polymer, Encapsulation–graphitization | Methylene orange | >90% in 30 min under UV light | 104 |
| C–TiO2 single-crystal nanorods | Hydrothermal | (i) Methylene blue, (ii) Rhodamine B, (iii) p-Nitrophenol | (i) 98.3% in 60 min, (ii) 99.4% in 20 min, (iii) 63% in 80 min under visible light | 403 |
| C–TiO2 nanoparticles | Vapour-assisted solvothermal | (i) Phenol, (ii) Methyl orange | (i) 94.6% in 75 min, (ii) >90% in 75 min under visible light | 404 |
| C–TiO2 core–shell nanostructures | Sol–gel | Methylene blue | 90.1% in 120 min under visible light | 107 |
| C–TiO2 nanoparticles | Hydrothermal | Rhodamine B | >90% in 120 min under UV light | 109 |
| Biochar–TiO2 particles | Hydrolysis | Methyl orange | 83.23% in 150 min under UV light | 405 |
| C/C-doped TiO2 hollow microsphere | Sol–gel | Rhodamine B | 96% in 140 min under UV light | 406 |
| C–nanohorns–TiO2 nanoflowers | Solvothermal | Methylene blue and methyl orange | 90% in 4 h under solar light | 407 |
| N–TiO2 nanoparticles | Sol–gel | Rhodamine B | 90% in 40 min under visible light | 116 |
| N–TiO2 nanoparticles | Sol–gel | Methyl orange | 90% in 200 min under UV-vis light | 257 |
| N–TiO2 nanoparticles | Graft polymerization | Methyl orange | 65% in 60 min under visible light | 408 |
| N–TiO2 nanoparticles | Plasma-assisted electrolysis | Methyl orange | 91% in 300 min under visible light | 409 |
| N–TiO2 nanoparticles | Co-precipitation | Rhodamine B | 99.2% in 540 min under visible light | 410 |
| N–TiO2 nanoparticles | Solvothermal | Methylene blue | 92% in 500 min under visible light | 119 |
| P–TiO2 particles | Post-phosphation | Bisphenol A | 92% in 67 min under sunlight | 411 |
| P–TiO2 powders | Microwave-hydrothermal | Methylene blue | >90% in 100 min under visible light | 258 |
| P–TiO2 nano-powders | Emulsion-based sol–gel | Methylene blue | >90% in 30 min under simulated solar light | 412 |
| P–TiO2 nanoparticles | Solvothermal/heat treatment | Ciprofloxacin | >90% in 60 min under visible light | 277 |
| Mesoporous P–TiO2 | Microwave-assisted sol–gel | Sulfamethazine | >90% in 300 min under visible light | 413 |
| P–TiO2/polyacrylic composite | Sol–gel | Formaldehyde | 100% in 60 min under visible light | 90 |
| S–TiO2 nanoparticles | Solvothermal | Phenol | 85.4% in 10 h under visible light | 139 |
| S–TiO2 nanorods | Oxidant peroxide method assisted hydrothermal | Methyl orange and phenol | 80% in 120 min under visible light | 138 |
| S–TiO2 nanoparticles | Hydrothermal | Rhodamine B | 80% in 60 min under visible light | 134 |
| S–TiO2 particles | Flame spray pyrolysis | Acetaldehyde | 75% in 300 min under visible light | 137 |
| S–TiO2 nanoparticles | Sol–gel | Diclofenac | 93% in 4 h under visible light | 136 |
| Meso-macroporous S–TiO2 nanoparticles | Ball-milling | (i) Methylene blue, (ii) Methyl orange, (iii) 4-Nitro-phenol | (i) 98% in 100 min, (ii) 60% in 100 min, (iii) 50% in 80 min under visible light | 135 |
| F–TiO2 nanorods | Hydrolysis/refluxing process | Methylene blue | 90% in 130 min under visible light | 414 |
| F–TiO2 hollow spiny nanocubes | Hydrothermal/photoreduction | Phenol | 58.6% in 60 min under visible light | 143 |
| F–TiO2 nanoparticles | Hydrothermal | 4-Methoxybenzyl alcohol | 56.4% in 240 min under visible light | 142 |
| F–TiO2/exfoliated bentonite | Sol–gel | Toluene | 11% in 80 min under visible light | 144 |
3.1 Metal ions
Metal-ion doping of TiO2 involves the introduction of metal ions into the TiO2 lattice structure to modify its properties. This helps reduce carrier recombination by creating electron trap centers, ultimately enhancing its photocatalytic activity.66 Various metal ions such as alkali, alkaline earth, transition, and rare-earth can be incorporate into the TiO2 matrix to alter its electronic structure and tailor its photocatalytic activity.For the larger alkali ions, potassium doping is considered effective for stabilizing the anatase phase of TiO2, as alkaline elements promote the formation of anatase crystalline phase. Doping with potassium can control the agglomeration of TiO2 particles, improve photocatalytic efficiency, enhance the adsorption of reactants on the catalyst, reduce the electron–hole recombination rate, and increase the specific surface area of TiO2.74 For instance, K-doped TiO2 thin films with a concentration of 10 wt% resulted in a reduction in the optical band gap energy of TiO2 from 3.5 to 3.0 eV, thereby enhancing light absorption capabilities.75 Furthermore, the presence of hydroxyl groups on the surface of K-doped TiO2 is likely to enhance its photocatalytic activity and hydrophilic properties.76 This makes it suitable for various applications such as synthesizing water-dispersible coatings and achieving optimal photovoltaic performance. For rubidium doping, it can induce distortion in the TiO2 lattice, promoting the formation of oxygen vacancies. These vacancies act as traps for capturing photo-generated electron–hole pairs, reducing their recombination rate. Furthermore, rubidium doping converts Ti4+ into Ti3+ through charge compensation, potentially forming impurity levels in the titania lattice that restrain carrier recombination and extend light absorption. Meanwhile, rubidium oxide on the TiO2 surface can transfer and capture electrons, thereby reducing the recombination rate of photo-generated carriers.77 Metal dopants with larger ionic radii may not substitute Ti4+ in the TiO2 lattice easily, leading them to diffuse on the surface and inhibit the growth of TiO2 grains, ultimately enhancing its photocatalytic properties.78
The roles of transition metal ions in charge trapping, recombination, interfacial transfer, and photocatalytic activity were systematically studied by doping six types of transition metal ions, including V, Mn, Fe, Cu, Ce, and W, into the surface lattice of TiO2 powders. Among these ions, Fe and Cu were found to enhance interfacial charge transfer by inhibiting defect-mediated annihilation, promoting d–d transitions, and inducing thermally induced de-trapping. Conversely, Mn ions introduced states in the mid-band-gap region, leading to the trapping of charge carriers and significant consumption via intra-atomic relaxation. Additionally, Ce and W ions formed strong bonds with O2− radicals, thereby limiting charge utilization and photocatalytic performance.86 Particularly, Fe metal ions are widely studied for doping into the TiO2 lattice due to their notable effects. However, the efficacy of Fe-doped TiO2 has been hindered by the formation of an amorphous contamination layer, primarily composed of iron oxide, on the nanoparticle surface.87 The removal of the contamination layer through acid treatment has proven to be effective in enhancing the photocatalytic activity from 24% to 98%.88
Doping TiO2 with 1 mol% neodymium results in modified TiO2 with solely the anatase phase, demonstrating superior photocatalytic activity.91 This enhancement is attributed to the formation of bonds between the rare earth element and TiO2, which not only increases photocatalytic reactivity but also restrains the generation of the rutile phase within the TiO2 structure. The exclusive formation of the anatase phase and the facilitation of bonding interactions with the rare earth element led to improved performance in the modified TiO2 composition. The study of Tb-doped TiO2 photocatalytic activity revealed that the substitution of Ti4+ ions with Tb3+ ions induce the creation of oxygen vacancies within the TiO2 lattice, due to a large mismatch in ionic radii and charge imbalance.92 As the Tb3+ content increases, the absorption edge of the material shifts gradually towards higher wavelengths. This observed red shift is attributed to charge-transfer transitions between the f electrons of Tb3+ ions and either the conduction or valence band of TiO2. The study of different rare earth ions, including Pr3+, Eu3+, Er3+, Y3+, Ho3+, Yb3+, and Nd3+, reveals that TiO2 doping with these ions leads to enhanced photocatalytic activity.93–95 Among these ions, Ho3+ stands out for its significant absorption in the visible light region, which can contribute to improved photocatalytic performance under visible light irradiation.96 This finding highlights the importance of selecting appropriate dopants with favorable electronic structures to maximize the utilization of visible light for photocatalysis.
3.2 Non-metal ions
The development of metal-incorporated TiO2 visible light photocatalysts often encounters challenges such as low thermal stability, photo-corrosion, and increased likelihood of serving as recombination centers due to the localized d-states of the dopants deep in the band gap of TiO2.97,98 To address these drawbacks associated with metal-incorporated photocatalysts, extensive research has focused on modifying TiO2 with non-metal elements to achieve stable visible-light-active TiO2. Non-metal-incorporated TiO2 crystals have shown greater success compared to metal doping because they can introduce mid-gap states acting as electron donors or acceptors within the band gap of TiO2. These mid-gap states effectively reduce the band gap energy and enhance the photoconversion efficiency of TiO2 in the visible light region.In many cases, doping techniques are utilized to enhance visible light absorption in photocatalytic materials, but they often result in the formation of defect sites, which can compromise photocatalytic activity. One strategy to mitigate the probability of charge recombination is to encapsulate C-doped TiO2 particles with nano-sized graphene.111 This approach aims to facilitate the effective transfer of photogenerated electrons to surface active sites by reducing the interfacial charge-transfer resistance between C-doped TiO2 and reactants. As a result, the prolonged lifetime of photogenerated charges over the C-doped TiO2 nanoparticles enables the generation of a large number of hydroxyl radicals with high oxidizing power for photodegradation purposes.
Another approach to modifying TiO2 with carbon involves the formation of core–shell nanostructure photocatalysts. In this method, carbon-modified TiO2 core–shell nanostructures were fabricated using an acidified sol–gel system with titanium-n-butoxide and a regenerated cellulose membrane.104 This approach has been shown to enhance the visible light photocatalytic performance. The synergistic effect of the carbon shell and TiO2 promotes the formation of a large number of hydroxyl radicals due to the carbon's photosensitizer behavior, thus supporting higher photocatalytic activity. The enhanced visible light absorption capability is primarily attributed to the incorporation of carbon dopants at interstitial positions in the TiO2 lattice, forming O–Ti–C or Ti–O–C bonds. In addition, the formation of the carbon core–shell nanostructure occurs through a carbonaceous layer grafted onto the surface of TiO2via Ti–O–C and Ti–OCO bonds. In this study, the calcination temperature is identified as an important parameter for controlling the thickness of the carbon shell coating on TiO2, as well as its crystallinity, surface area, and optical properties. Specifically, increasing the calcination temperature from 75 to 500 °C results in a narrowing of the band gap from 2.95 to 2.80 eV, and an increase in the thickness of the carbon shell from 0.40 to 1.20 nm, respectively. However, excessive carbon thickness is found to be detrimental to photocatalysis because it can shield the passage of light and hinder reactant adsorption on the TiO2 surface.
Early studies on phosphorus-modified TiO2 primarily focused on improving thermal stability and surface area.127,128 Surface modification of TiO2 with phosphoric acid is a common technique for preparing P-doped TiO2 photocatalysts. Phosphate anions from phosphoric acid adsorb strongly on the surface of TiO2, significantly influencing the interfacial and surface chemistry of TiO2.129 The incorporation of phosphorus into the TiO2 lattice using phosphoric acid as the phosphorus source via a sol–gel method resulted in a redshift in the absorption edge due to the formation of Ti–O–P bonds in the crystal lattice.130 Furthermore, phosphorus doping affects the crystallization of TiO2 by inhibiting crystal growth and suppressing the phase transformation from anatase to rutile. Apart from these observed merits, the enhanced visible light activity is primarily attributed to increased hole signal intensity, indicating efficient trapping of photogenerated holes.
Apart from post-treating TiO2 with phosphoric acid, other methods of introducing phosphorus into TiO2 include using elemental red phosphorus via a ball milling process131 or decomposing black phosphorus via high-intensity ultrasonication.90 In the case of black phosphorus, it can absorb onto the TiO2 surface through electrostatic interactions, leading to the formation of uniformly dispersed P–TiO2 composites. These P–TiO2 composites can then be blended with a polyacrylic matrix to prepare functional coatings with photocatalytic properties (Fig. 3E). During the decomposition process of black phosphorus, P5+ ions are produced and incorporated into the crystal lattice of TiO2 to form P–O–Ti bonds. This incorporation leads to the generation of more defects in the P–TiO2 crystals, which helps trap electrons and suppress the recombination of electron–hole pairs. Consequently, the photocatalytic ability of P–TiO2 is expected to be superior to both TiO2 and black phosphorus.
The early studies on sulfur doping of TiO2 have shown that the band gap modification is achieved by substituting lattice oxygen with S2− ions to form Ti–S bonds, representing anionic substitution.133 In this work, S-doped TiO2 was synthesized through the oxidative annealing of titanium disulfide. This doping of sulfur induces a significant shift in the absorption edge towards lower energy by mixing the S 3p states with the VB.134 The use of titanium disulfide as starting material was later extended in hydrothermal oxidation reaction to prepare anionic S-doped TiO2.134 Subsequent to thermal annealing, various chemical processes have been developed for synthesizing S-doped TiO2, including ball milling,135 sol gel,136 flame spray pyrolysis,137 oxidant peroxide method,138 hydrothermal reaction,134 and solvothermal reactions.139 The choice of preparation method and sulfur sources significantly influences the ionic form of sulfur doping in TiO2. Studies have shown that thiourea and carbon disulfide as sulfur sources favor the formation of anionic sulfur doping in the TiO2 crystal lattice. When TiS2 or CS2 is used, most of the sulfur from TiS2 or CS2 is oxidized, leaving residual S as S2−, which replaces oxygen atoms in the O–Ti–O framework. Conversely, using thiourea as the sulfur source promotes the cationic doping of S atoms into the TiO2 crystal lattice.138,140
To attain optimal photocatalytic performance, numerous studies on F-doped TiO2 have focused on controlling fluorine doping concentration during synthesis. For instance, in a hydrothermal synthesis of F-doped rutile single crystalline TiO2, the optical band gap of the photocatalyst was effectively adjusted from 3.05 to 2.58 eV by varying the fluorine doping concentration.141 Besides, the concentration of fluorine doping has been identified as a crucial factor in shaping the morphology of TiO2. Using TiCl4 as a precursor, a range of TiO2 hierarchical nanostructures, including pompon-like and football-like microspheres composed of aligned rutile and anatase nanoparticles, have been synthesized using a hydrothermal method in the presence of NaF.151 An incremental increase in the concentration of NaF leads to a sequential red shift of the absorption band edge of F-doped TiO2 compared to pure TiO2. Consequently, the band gap of F-doped TiO2 can be tuned from 3.0 eV to 2.2 eV, with the degree of tuning dependent on the fluorine doping concentration.
4 Noble metals-supported TiO2 hybrids
The integration of noble metals onto the surface of TiO2 introduces new functionalities by leveraging the intrinsic properties of metal nanoparticles, significantly enhancing their performance as photocatalysts. Noble metal nanoparticles, such as silver (Ag) and gold (Au), exhibit strong absorption across the UV to near-infrared (NIR) spectrum due to their surface plasmon resonance, which can be tailored by controlling their size, shape, and surrounding medium.29 Despite Ag's susceptibility to oxidation, it remains a top candidate for plasmonic applications due to its low optical loss in the visible and NIR spectral regions. On the other hand, Au offers superior stability alongside excellent performance in the visible and NIR ranges. While aluminium and copper are alternative plasmonic materials, their poor chemical stability limits their applicability. Palladium (Pd) and platinum (Pt) are catalytically active metals, but both do not support strong plasmonic resonances at visible wavelengths. In hybrid systems formed by depositing noble metals onto TiO2, these metal nanoparticles serve as antennae, efficiently absorbing visible light and generating energetic electrons and holes in the semiconductor.152,153 This synergistic effect between TiO2 and noble metals enhances photoreactivity across a broad range of the solar spectrum, overcoming the inherent limitations of wide band gap TiO2 semiconductors.In contrast to conventional semiconductor photocatalysis, plasmonic photocatalysis exhibits two distinct features: the Schottky junction and localized surface plasmon resonance (LSPR).154 In semiconductor-noble metal hybrid systems, when illuminated with UV light, the excited electrons of the semiconductor are transferred from the CB to the Fermi level of noble metals, leading to the separation of electron–hole pairs. This metal–semiconductor interface, known as the Schottky junction, promotes charge separation and transfer at the interface. Meanwhile, LSPR plays a crucial role in plasmonic photocatalysis by inducing the collective oscillation of conduction electrons at the interface of metallic structures upon excitation by incident electromagnetic radiation of the same frequency. When noble metals absorb visible light through LSPR, charge carriers in the noble metals are directly injected from excited plasmonic metal nanostructures into the semiconductor, contributing to strong visible light absorption and the generation of active charge carriers.155 Both the Schottky junction and LSPR significantly enhance the photocatalysis process and the overall photocatalytic efficiency of TiO2 semiconductor (Table 3).
4.1 Ag on TiO2
Functionalizing Ag nanoparticles presents challenges related to stability, aggregation, and size control, compounded by their susceptibility to oxidation over time, which can degrade their surface properties. However, the work of Awazu et al. represents a significant advancement in plasmonic photocatalysis by combining TiO2 with optically active Ag nanoparticles.156 In this approach, the excitation of surface plasmons on the Ag nanoparticle surface amplifies the near-field amplitude at specific wavelengths in the near UV region, leading to higher concentrations of charge carriers in TiO2 and thereby increasing the efficiency of photocatalysis. The important feature to enable plasmonic photocatalysis in this study is to deposit TiO2 on Ag nanoparticles coated with thin protective SiO2 shell to prevent oxidation. This protective shell ensures that the photodegradation rate by the TiO2 photocatalyst modified with protected Ag nanoparticles is 7 times faster than that of pure TiO2 under UV irradiation. Alternatively, a polymer shell can be used to protect Ag nanoparticles from oxidation, forming a core–shell structure with an Ag core and a polymer shell. For instance, polyacrylic acid is employed for polymer encapsulation on the Ag nanoparticles using Layer-by-Layer synthesis, enabling precise control of the polymer shell thickness at the nanoscale without compromising the plasmon-induced enhancement of the near-field.157 The TiO2 photocatalyst modified with protected Ag core–shell structures demonstrates a 15% enhancement in their photocatalytic activity in air compared to pure TiO2. Notably, this enhanced activity is retained over time, even upon aging in air, whereas TiO2 modified with bare Ag nanoparticles loses its plasmonic properties gradually. This phenomenon can be attributed to the progressive oxidation of the unprotected Ag nanoparticles to Ag2O at the surface, causing the detrimental loss of their plasmonic properties.The performance of the plasmonic photocatalyst Ag/TiO2 is a strong function of the size and shape of Ag nanostructures.158 Tailoring the size and shape of Ag nanostructures enables the control of the properties of Ag surface plasmons, in turn tuning their impact on the photocatalytic activity of TiO2. The enhanced photochemical activity of Ag/TiO2 composite systems is largely dependent on the plasmon-mediated transfer of energy from Ag nanoparticles to TiO2 to increase the concentration of electron–hole pairs in the composite. In this case, the Ag nanocubes exhibit superior amplifications in the photochemical reactivity relative to that of the Ag nanospheres and nanowires of similar size for identical Ag mass (volume). The enhancement reactivity of nanocubes can be explained by their large extinction cross-section, more specifically, a higher scattering efficiency.
4.2 Au on TiO2
The photocatalytic activity of Au/TiO2 is influenced by various factors including the Au loading, phase composition, particle size, shape, surface area, and spatial structuring.24,159–161 Studies have shown that smaller Au nanoparticles tend to favor enhanced photocatalytic performance.162–165 This size effect is attributed to the shift of the Fermi energy to a more negative potential for smaller nanoparticles, reducing the potential difference between the CB of TiO2 and the Fermi level of the metal nanoparticles. As a result, electron accumulation in the metal nanoparticles increases, leading to an upward shift of the Fermi level to the CB of TiO2, promoting rapid electron transfer from TiO2 to metal nanoparticles for improved photooxidation reactions by photoexcited holes. Conversely, chemically aggregated nanospheres or nanorods may not be as beneficial to the photocatalytic activity of TiO2 due to differences in their co-catalytic efficiency, influenced by factors such as the percentage of surface-active atoms and Fermi energy shifts resulting from changes in size, shape, and surface-to-volume ratio. Nanorods, for instance, might reduce substrate adsorption and block light penetration on the oxide surface, leading to lower photogeneration of charged species and reduced photoactivity. To control the dispersibility of the co-catalyst nanoparticles, Tatsuma et al. prepared Au/TiO2 composites using the electron trap-mediated deposition method, leveraging the electron traps present in TiO2.166 This method offered higher dispersibility of the Au co-catalyst compared to the photodeposition method whereby the amount of deposited Au could be controlled and preventing overloading.Numerous studies have demonstrated that plasmonic metals, when integrated with TiO2 semiconductor in structurally optimized configurations, can significantly boost light absorption.167,168 Li et al. developed spinous TiO2-based octahedral nanocages through a template-assisted approach, resulting in enhanced photocatalytic performance compared to spinousless nanocages.169 Zhao et al. designed an oxygen vacancy-rich 2D Au/TiO2 hybrid nanosheet derived from 2D Mxene, achieved via in situ growth of Au nanoparticles on preformed TiO2 nanosheets.170 The synergistic interplay between Au active species and abundant oxygen vacancies from TiO2 significantly lowered the reaction barrier and improved catalytic reactions. Guo et al. fabricated a hierarchical forest-like plasmonic superstructure consisting of vertically printed macro-sized TiO2 pillar arrays as tree trunks, dense TiO2 nanorod arrays as branches, and self-assembled Au nanoparticles as leaves. This plasmonic superstructure effectively absorbs light through surface plasmon resonance effects and multiple scattering, offering high light absorption capacity and interconnect mass transfer channels (Fig. 4).
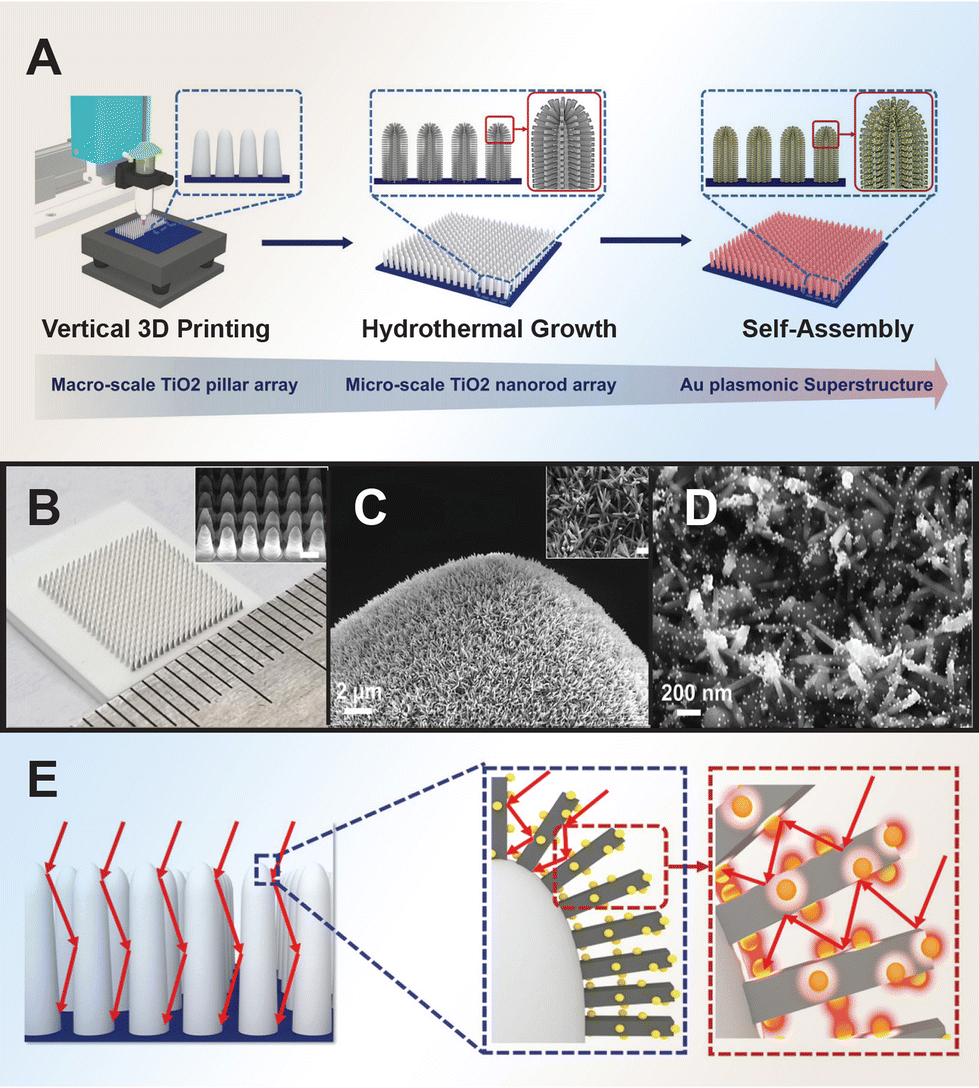 | ||
| Fig. 4 Fabrication of forest-inspired hierarchical Au plasmonic superstructure. (A) Printing of TiO2 pillar arrays and hydrothermal grown layer of dense TiO2 nanorod arrays, followed by self-assembling Au nanoparticles on the hierarchical TiO2 structure. (B) Optical image of a printed TiO2 pillar array. (C) Scanning electron microscopy (SEM) images of the TiO2 nanorods growing on the top surface of a 3D printed TiO2 pillar. (D) SEM image of Au nanoparticles assembled on TiO2 nanorods. (E) Schematic illustration of the light-trapping approaches of Au plasmonic superstructure through the multiple light scattering of the vertically aligned pillars and nanorods in multiscale and LSPR effects of Au nanoparticles. Reproduced with permission from ref. 185. Copyright 2021, Wiley-VCH. | ||
4.3 Multimetals on TiO2
Noble metals like Ag, Au, Pt, and Pd serve as potent co-catalysts in TiO2, offering numerous benefits to the photocatalytic process. They essentially act as electron traps, assisting in the separation and transfer of charges on the semiconductor surface. This facilitates the creation and movement of electron–hole pairs, thereby improving catalytic reactions. Furthermore, they contribute to modifying the energy levels and band structures of TiO2, broadening its light absorption spectrum to include visible light, and ultimately enhancing its overall photocatalytic performance.13,171,172 On the other hand, multi-metallic nanoparticles of noble metals represent a novel class of nanomaterials with distinct properties compared to monometallic nanoparticles.173–176 They offer enhanced versatility and technological utility due to their unique properties and the ability to tune their LSPR properties through configurational and elemental modifications.176–178 These multi-metallic nanoparticles exhibit multi-functionality, wide tunability, and multiple plasmon bands, making them ideal components for TiO2 photocatalysts.179,180 Bimetallic combinations like Au–Ag,181,182 Au–Pd,183 and Au–Pt184 loaded onto TiO2 have demonstrated efficient photocatalytic activity, due to their ability to efficiently separate electron–hole pairs at the alloy/TiO2 junction. Moreover, controlling the morphology and structure of plasmonic nanoparticles supported on TiO2 can significantly influence product selectivity in photocatalytic reactions.Studies on visible-light-responsive Au–Ag alloy nanoparticles loaded onto TiO2 have demonstrated successful control over the apparent photocatalytic activity by altering the alloy composition. The photocatalytic reactions were found to be based on plasmon-induced charge separation.179,186 For instance, colloidal suspensions of AuxAg1−x alloys with composition x ranging from 0.2 to 1 exhibit an intense SPR band in the broad visible light range of 420 to 520 nm. When Au0.3Ag0.7 alloy nanoparticles are deposited on TiO2, this plasmonic photocatalyst generates SPR at 490 nm, corresponding to the maximum intensity of solar light.179 The TiO2 photocatalyst containing 1% of each metal (Ag, Au, Pt, Pd) in an alloy structure exhibited a remarkable increase in photoactivity compared to monometallic nanoparticles.
5 Defective TiO2 nanomaterials
Defect engineering in TiO2 involves intentionally introducing imperfections or vacancies into the crystal lattice to tailor its properties for specific applications. The prevalence of these defects has been extensively investigated through theoretical calculations and experimental analyses due to its ability to modify the electronic structure, charge carrier dynamics, and surface reactivity of TiO2.55,187 Oxygen vacancies and Ti3+ interstitials are two commonly studied defects in TiO2, which are known to influence the electronic properties of TiO2 by generating intermediate bands, hence reducing its band gap.188,189 Moreover, oxygen vacancies can enhance the donor density and facilitate the separation and mobility of photogenerated electron–hole pairs.190 By strategically controlling the types and concentrations of defects, defect engineering enables the development of TiO2-based materials with enhanced performance.Oxygen vacancies are introduced into the TiO2 lattice through various methods, including annealing in an oxygen-deficient atmosphere or as a byproduct of doping processes involving anions or cations.125,191 These vacancies can create intermediate energy states within the band gap of TiO2, effectively extending its light absorption into the NIR region. As a result, TiO2 materials with oxygen vacancies exhibit enhanced photoactivity, particularly under visible light irradiation. Black TiO2, rich in oxygen vacancies, has been shown to significantly improve the photocatalytic performance compared to white (stoichiometric) TiO2. Additionally, colored TiO2 materials such as blue or gray variants can be obtained through hydrogenation, resulting in sub-stoichiometric oxide. These sub-stoichiometric oxides possess altered chemical compositions and structures, characterized by features like surface disordered shells, oxygen vacancies, Ti3+ centers, and surface hydroxyl groups or Ti–H bonds.188
In 2011, Chen et al. introduced black TiO2 with a narrowed band gap of 1.5 eV to enhance full spectrum sunlight absorption by subjecting TiO2 nanoparticles to hydrogen thermal treatment.25 This process induced disorder layers on the TiO2 surface, resulting in the formation of defective black TiO2−x nanoparticles and giving rise to an increase in photocatalytic activity. Following this, Hu et al. developed black TiO2 hollow spheres with a narrow bandgap using a template-free solvothermal approach.192 The TiO2 hollow frameworks, stabilized by encircled protectors like amine molecules, maintained high structural integrity and improved crystallinity of the anatase phase during high-temperature hydrogenation. The thermal hydrogenation process created a disordered shell layer over the crystalline TiO2 core, with dispersed Ti3+ within the hollow structure frameworks. The resulting mesoporous black TiO2 hollow spheres exhibited a high photo-response in visible-light absorption and significantly improved photocatalytic activity, attributed to their high crystallinity, hollow structure, Ti3+ content in the frameworks, and surface disorderliness.
To date, various synthetic techniques beyond hydrogenation have been explored to synthesize black TiO2 with broad spectrum absorption. These methods include metal reduction, plasma-assisted processes, NaBH4 reduction, electrochemical reduction, laser ablation in liquid, and oxidation approaches.193 In the case of black TiO2 hollow spheres synthesized via aluminum reduction, the formation of oxygen vacancy defects generates mid-gap states, facilitating electron excitation at lower energies.194 This electron transition from the valence band to the oxygen vacancy mid-levels, or vice versa, enables absorption of visible and infrared light. However, despite these optical enhancements, photogenerated electrons residing at the energy levels of oxygen vacancies are unable to participate in hydrogen production (H+ → H2) as their energy levels are situated below the reduction potential of H2O/H2. Consequently, this absence of photocatalytic activity under visible light illumination highlights a limitation in utilizing these oxygen vacancies for hydrogen production.
Sub-stoichiometric titania (TiO2−x), also known as colored titania (gray, blue, brown, or black), exhibits efficient light absorption across the UV to the IR region of the solar spectrum. Numerous studies have highlighted its enhanced photoactivity in various oxidation reactions under visible light compared to ordinary white (stoichiometric) TiO2.188,195 The synthesis of these materials typically involves thermal treatment of TiO2 in different reducing atmospheres, including vacuum, Ar, H2/Ar, and pure H2.196 Generally, increasing the reduction level results in a higher defect density, such as oxygen vacancies and Ti3+ centers, within the TiO2 lattice, leading to darker-colored TiO2 powder.
6 Composites of TiO2 nanomaterials
Composite photocatalysts have emerged as promising materials for overcoming the intrinsic limitations of individual semiconductor photocatalysts. The tailored properties of composite photocatalysts play an essential role in maximizing their photocatalytic performance by improving light absorption, charge separation, redox capability, and chemical reactivity.197,198 For instance, the inclusion of carbon-based materials such as graphene or carbon nanotubes in TiO2-based nanocomposites has been particularly effective. When combining TiO2 with graphene, excited electrons from the conduction band of TiO2 can transfer to graphene through a percolation mechanism. This transfer results in the formation of a heterojunction at the interface which effectively separates the photoinduced electron–hole pairs, thereby suppressing charge recombination.199 Hybridization with carbon quantum dots results in the formation of new electronic structures and an increase in the photo-response.200 Combining TiO2 with various materials such as carbon nitride, metal–organic frameworks, Mxene, and aerogels can lead to the development of advanced composite photocatalysts with enhanced properties and functionalities.6.1 TiO2/C3N4
Carbon nitride (g-C3N4) is a 2D polymer composed of carbon and nitrogen atoms arranged in a graphitic structure. Due to its polymeric nature, the surface chemistry of g-C3N4 can be readily modulated through surface engineering at the molecular level.201 In comparison with TiO2, g-C3N4 has a moderate band gap ranging from 2.7 to 2.8 eV, allowing it to absorb visible light with an onset around 450 to 460 nm.202 Since the metal-free semiconductor was reported to generate hydrogen from water by Wang et al. under visible-light irradiation,203 g-C3N4 has been widely employed as a guest semiconductor to modify TiO2. The favorable alignment of band positions between g-C3N4 and TiO2 confers a greater driving force for charge transfer across the heterointerface in constructed TiO2/g-C3N4 heterostructures. This alignment promotes spatial separation of photogenerated electron–hole pairs, consequently enhancing the overall photoconversion efficiency of the system.204–2066.2 TiO2/MXene
Titanium carbide (Ti3C2, a member of the ‘MXene’ family) has emerged as a new class of promising 2D materials because of their unique properties including high conductivity, high structural, chemical stability, and tailored surface chemistry.207,208 Moreover, Ti3C2 has a large proportion of Ti, which can be readily transformed into TiO2.209 The Schottky junction formed at the interface between Ti3C2 and TiO2 could promote the photoinduced charge separation. By a hydrothermal oxidation of layered Ti3C2, the Ti atoms on Ti3C2 afforded the Ti source and nucleating sites for the in situ growth of highly active (001) facets TiO2 nanosheets on Ti3C2, and it also forms an interfacial heterojunction between 2D Ti3C2 and TiO2 to prevent the defect-induced recombination. This is related to the large gap in work function of the two phases between –OH terminated Ti3C2 (1.8 eV) and TiO2 (001) surface (4.924 eV), thus hindering the electron transfer from TiO2 to Ti3C2, instead allowing the flow of photogenerated holes. As such, the low function –OH terminated Ti3C2 serves as a reservoir of holes through the hole trapping by the Schottky-junction, yielding the spatial separation of photogenerated electrons and holes.198,2106.3 TiO2/MOF
Metal organic frameworks (MOFs) are a new generation of organic-inorganic hybrid porous materials, making up by metal ions/clusters connected by organic ligands. These structures provide a large surface area and tunable pore sizes, making them excellent candidates for hosting catalytic metal clusters within their frameworks. The metal clusters can function as active sites for catalytic reactions. TiO2 can be synergistically coupled with the catalytic properties of metal clusters within the MOFs structure. This synergy allows for efficient utilization of photoexcited electrons generated by TiO2 in driving catalytic reactions facilitated by the metal clusters. By adjusting parameters such as pore size, surface area, and TiO2 loading, the photocatalytic activity and selectivity of the composite material can be optimized for specific catalytic applications. To give an illustration, combining TiO2 nanosheets with exposed (001) facets and NH2-UiO-66 (MOF) via an in situ growth strategy offers a promising approach for producing bifunctional materials capable of both capturing and photocatalytically reducing cardon dioxide (CO2) under UV-vis light irradiation. This strategy leverages the strengths of TiO2 nanosheets and NH2-UiO-66 MOF while allowing intimate contact between the two components, with the formation of a heterojunction for effective charge transfer.211 Photogenerated electrons from TiO2 can readily transfer to the catalytic sites within MOFs, facilitating gaseous adsorption and subsequent catalytic reactions.6.4 TiO2/COF
Covalent organic frameworks (COFs) are a novel class of crystalline organic polymers constructed from light elements such as carbon, hydrogen, nitrogen, oxygen, and sulfur.212–214 These frameworks are linked via strong covalent bonds, which bestow them with remarkable stability and robustness. COFs are characterized by unique properties including low density, large surface area, tunable pore size and structure, and easily customizable functionality.215–217 Despite these advantages, the photocatalytic performance of pure COFs is limited by high recombination rates of photoinduced electron–hole pairs. To address this limitation, COFs are often integrated with TiO2 to form composites. TiO2, with its band gap of approximately 3.2 eV, is highly effective in visible light-assisted photocatalysis, making it an ideal candidate for integration with COFs.218–220 This combination aids in the formation of heterostructures, significantly enhancing the photocatalytic functionality. TiO2/COF composites utilize COFs as efficient photosensors, improving light absorption and overall applicability. The formation of heterojunctions and band gap narrowing in TiO2/COF composites optimizes the interaction between TiO2 and COFs, which have a band gap between 2 and 2.8 eV. This optimization reduces the recombination rate of photoinduced electron–hole pairs, thereby enhancing the production of active species and overall photocatalytic activity.21,2216.5 TiO2/aerogel
Aerogels are a fascinating class of highly porous materials composed of a network of interconnected nanostructures which exhibit high surface areas, open pores, low densities, and unique physical properties.222,223 Aerogel can be prepared using sol–gel process followed by supercritical drying, allowing for the production of aerogel in various shapes and sizes that suitable for different applications.224,225 Particularly, TiO2 aerogels consisting of a network of interconnected TiO2 nanoparticles have emerged as a highly favorable architecture, offering ample active sites for photocatalytic reactions and long diffusion pathways for photogenerated electrons. Additionally, TiO2 aerogels have been observed to possess surface trap states where large amounts of photogenerated electrons can be stored upon illumination, which enhances their photocatalytic activity by promoting efficient charge separation and utilization in redox reactions. It was reported that the as-synthesized aerogel can store 1.7 times more electrons relative to commercial anatase nanoparticles.2266.6 TiO2/other semiconductor composites
Heterojunction photocatalysts are engineered by combining two distinct semiconductor materials. This coupling takes advantage of the unique electronic properties of each semiconductor, creating an interface where efficient charge separation can occur. When these heterojunctions are exposed to light, they generate electron–hole pairs in both semiconductor components.227–229 The type-II heterojunction, one of the most studied types, features staggered band structures where electrons accumulate in one semiconductor (photocatalyst II) and holes in the other (photocatalyst I), promoting efficient charge separation for photocatalysis.38,230,231 While the type-II heterojunction configuration enhances charge separation, it also reduces redox ability, which is not ideal for photocatalysis.Traditional Z-scheme photocatalysts, proposed to address the limitations of type-II heterojunctions, improve charge-separation efficiency while maintaining strong redox abilities.36 This system consists of two semiconductors with suitable intermediate couples, such as Fe3+/Fe2+, IO3−/I−, and I3−/I−, arranged in staggered band structures.232 In operation, photogenerated holes in the VB of photocatalyst I react with electron donors (D), creating electron acceptors (A). Simultaneously, photogenerated electrons in the CB of photocatalyst II react with A, forming D. The retained electrons in the CB of photocatalyst I and holes in the VB of photocatalyst II are then available for reduction and oxidation reactions, respectively.233 This charge-transfer mode endows the system with strong redox ability and spatially separated redox reaction sites, enhancing photocatalytic performance by facilitating more efficient and selective redox reactions. However, this system is limited to the solution phase, faces side reactions, light shielding issues, and pH sensitivity.234
The all-solid-state Z-scheme was introduced to overcome the limitations of traditional Z-scheme photocatalysts by replacing shuttle redox ion pairs with a solid conductor. In a three-component heterojunction (CdS–Au–TiO2), both the holes in CdS and electrons in TiO2 are injected into Au.43 This scheme utilizes a solid conductor instead of shuttle redox ion pairs, making it suitable for both liquid and gas applications and significantly shortening the charge-transfer length, thus accelerating charge transfer.235 In a typical all-solid-state Z-scheme, the CB electrons of photocatalyst II migrate to a solid conductor upon light irradiation and then to the VB of photocatalyst I. This innovation allows for applications in both liquid and gas phases and accelerates charge transfer, significantly enhancing the efficiency of the photocatalytic process. Despite these advancements, it faces challenges such as Schottky barriers, preferential electron transfer leading to neutralization at the conductor, and difficulties in synthesis and assembly.
Direct Z-scheme heterojunctions, evolving from traditional and all-solid-state Z-scheme heterojunctions, refine the concept by eliminating intermediate redox couples or conductors.236,237 Instead, they couple an oxidative and a reductive photocatalyst based on their VB and CB positions. This setup optimizes charge transfer and enhances photocatalytic activity by ensuring that electrons from the low VB photocatalyst are injected into the VB of the high CB photocatalyst.47,238 TiO2, known for its low VB position, is frequently used as an oxidative photocatalyst and is often coupled with high CB photocatalysts such as CdS,237 ZnIn2S4,239 and Cu2O.240,241 In this scheme, electrons excited to the CB of the low VB photocatalyst are injected into the VB of the high CB photocatalyst, optimizing charge transfer and enhancing photocatalytic activity.
Despite their advantages, Z-scheme heterojunctions face confusion and theoretical challenges. The S-scheme heterojunction, a recent advancement, addresses the limitations of Z-scheme heterojunctions by improving charge separation and maintaining strong redox abilities.51,234 It comprises an oxidation photocatalyst (OP) and a reduction photocatalyst (RP), where the RP has more negative CB and Fermi levels, and the OP has more positive VB. Electron transfer from the RP to the OP due to Fermi level differences generates an internal electric field and band bending, enhancing photo-induced carrier transfer. Under illumination, electrons in the OP's CB migrate to the RP's VB and combine with holes, differing from the type-II mechanism. This carrier transfer resembles a “step” at a macroscopic level and an “N” shape at a microscopic level, optimizing photocatalytic performance.242 A recent study reveals that the TiO2/perovskite (CsPbBr3) S-scheme heterojunction demonstrates electron transfer from CsPbBr3 quantum dots to TiO2, resulting in an internal electric field directed from CsPbBr3 to TiO2 upon hybridization.243 Upon light irradiation, this internal electric field drives the photoexcited electrons in TiO2 to CsPbBr3, forming an S-scheme heterojunction in the TiO2/CsPbBr3 nanohybrids that significantly enhances electron–hole pair separation. However, the S-scheme is still in its early stages and requires further research to overcome significant conceptual and practical challenges.
7 Applications of TiO2 nanomaterials
The objective of engineering functional TiO2 materials with tailored electronic properties through morphological control, crystal phase manipulation, doping, and hybridization, is to optimize their performance in photocatalytic applications. Extensive efforts have been dedicated to leveraging TiO2 for a wide range of photooxidation reactions such as photodegradation of organic pollutants and photocatalytic disinfection (Tables 1–5). Furthermore, TiO2 has shown promise in photoreduction reactions including hydrogen generation through water splitting (Table 6), carbon dioxide reduction (Table 7), nitrogen fixation (Table 8), hydrogen peroxide generation (Table 9), alcohol oxidation (Table 10) and even in photochromic applications. These advancements in TiO2 photocatalysis aim to unlock the full potential of TiO2 in addressing environmental challenges in water and air pollution as well as advancing sustainable energy solutions.| Photocatalysts | Synthesis methods (TiO2/noble metals) | Pollutants | Degradation efficiency | Ref. |
|---|---|---|---|---|
| Ag nanoparticles/mesoporous TiO2 | Pyrolysis/photodeposition | (i) Methylene blue, (ii) Phenol | (i) >90% in 300 min, (ii) 85% in 210 min under visible light | 260 |
| Ag/TiO2 particles | Commercial/electrochemical method | 2-Chlorophenol | 94% in 6 h under UV light | 415 |
| Amine-adsorbed Ag/TiO2 particles | Sol–gel | Phthalic acid | >90% in 210 min under visible light | 416 |
| Ag/TiO2 nanoparticles | Chemical synthesis | Methylene blue | 89.2% in 60 min under solar light | 417 |
| Biochar-coupled Ag and TiO2 particles | Hydrolysis/photodeposition | Methyl orange | 85.38% in 60 min under UV light | 418 |
| Ag/TiO2 particles | Laser pyrolysis/wet impregnation and chemical reduction | Methyl orange | 90% in 120 min under visible light | 419 |
| Ag/β-cyclodextrin TiO2 membrane | Hydrothermal method/electrospinning | Dimethyl-hydrazine | 96.8% in 80 min under visible light | 270 |
| Au/TiO2 nanotubes | Electro-spinning/deposition–precipitation | Methylene blue | >80% in 140 min under visible light | 420 |
| Mesoporous Au/TiO2 | Sol gel/deposition–precipitation | Safranin-O | 97% in 60 min under UV light and 87% in 90 min under solar light | 421 |
| One-dimensional Au/TiO2 nanoforests | Chemical synthesis/photoreduction | (i) Rhodamine B, (ii) p-Nitrophenol, (iii) Phenol | (i) >90% in 60 min under UV and >80% in 60 min under visible light, (ii) >40% in 60 min under UV light, (iii) >45% in 60 min under UV light | 422 |
| Au/TiO2 nanofibers | Electro-spinning | (i) Methylene blue, (ii) Rhodamine B | (i) 88% in 3 h under solar light, (ii) >90% in 3 h min under solar light | 12 |
| Au/TiO2 film | Commercial/sputtering | Methylene blue | 60% in 60 min under 532 nm laser | 423 |
| Au/TiO2 nanostructures | Chemical synthesis | Methylene blue | 97% in 150 min under visible LED light | 168 |
| Au/TiO2 nanorod | Hydrothermal/wet impregnation | Bisphenol A | 40% in 120 min under visible light (λmax = 520 nm) | 164 |
| Pt/TiO2 particles | Commercial/Photo-deposition | (i) Acetaminophen, (ii) Panadol | (i) 99% and (ii) 83% in 180 min under simulated solar light | 424 |
| Pt/TiO2@polymeric matrix | Commercial/chemical-reduction | Methylene blue | 83% in 130 min under UV and 94% in 400 min under sunlight | 262 |
| Pd/TiO2 particles | Chemical synthesis/photo-deposition | Amoxicillin | 97.5% in 5 h under visible light | 425 |
| Pd/TiO2 particles | Chemical synthesis/incipient wetness impregnation | Methyl violet | 95% in 20 min under UV light | 426 |
| Pd/TiO2 particles | Sol–gel | (i) Methylene blue, (ii) Methylene orange | (i) 99.4% and (ii) 92.6% in 120 min under UV light | 263 |
| Pd/TiO2 films | Sol–gel | Phenol | 80% in 5 h under UV light and 23% in 5 h under visible light | 427 |
| Au/Pt–TiO2 nanopillar arrays | Glancing angle deposition/successive ion layer adsorption and reaction | Methyl orange | 40% in 120 min under UV light | 184 |
| Ag@Au/TiO2 nanotubes | Electro-chemical anodization/displacement reaction | (i) Methyl orange, (ii) Cr(IV) ions | (i) 98.1% and (ii) 70.2% in 120 min under solar light | 428 |
| Au0.4Ag0.6/TiO2 | Chemical synthesis | Methylene blue | 99% in 120 min under visible light | 261 |
| Ag0.1Au0.1Pt0.1Pd0.1/TiO2 | Sol gel/ions reduction | Toluene | 86% in 60 min under LED light (λmax = 465 nm) | 429 |
| Photocatalysts | Synthesis methods | Pollutants | Degradation efficiency | Ref. |
|---|---|---|---|---|
| TiO2/Ti3C2Tx composite | Hydrothermal | Carbamazepine | 98.67% in 240 min under UV light and 55.83% in 8 h under solar light | 210 |
| BiOBr/TiO2 | Solvothermal | Rhodamine B | 99.9% in 10 min under visible light | 265 |
| In2S3/TiO2 | Hydrothermal | Rhodamine B | 98% in 10 min under simulated solar light | 430 |
| rGO/TiO2-B/W18O49 | Solvothermal | Rhodamine B | 100% in 15 min under full solar-spectrum | 264 |
| C-MoS2/TiO2 | Hydrothermal | Methylene blue | 99% in 60 min under simulated solar light | 266 |
| CdO/TiO2 | Sol–gel | Imazapyr herbicide | 100% in 180 min under visible light | 272 |
| Pt/Nb2O5/TiO2 | Photodeposition | (i) Diclofenac, (ii) Ketoprofen | (i) 100% in 20 min under UV light, (ii) 100% in 60 min under UV light | 431 |
| Bi2O3/rGO/TiO2 | Hydrothermal | Tetracycline | 94.3% in 90 min under visible light | 278 |
| Bi2O3/Ti3+–TiO2 | Hydrothermal | Tetracycline | 100% in 200 min under visible light (λ > 420 nm) | 432 |
| TiO2/g-C3N4 | Hydrothermal | Ciprofloxacin | 93.4% in 60 min under simulated solar light | 279 |
| g-C3N4@C-TiO2 | Hydrothermal | (i) Rhodamine B, (ii) Phenol | (i) 97% in 90 min under visible light, (ii) 92% in 60 min under visible light | 433 |
| Cu–Ni/TiO2 | Chemical method | Gaseous acetaldehyde | 88% in 180 min under UV light and 56% in 180 min under visible light | 282 |
| TiO2-RGO/LDHs | Hydrothermal | Gaseous: (i) toluene, (ii) methanol, (iii) ethyl acetate | (i) 69.9%, (ii) 91.6%, (iii) 99.9% in 60 min under simulated sunlight (λ ≥ 350 nm) | 283 |
| Photocatalysts | Synthesis methods | Light source | H2 generation rate | Quantum efficiencies | Ref. |
|---|---|---|---|---|---|
| TiO2 hollow spheres | Solvothermal/chemical reduction | 300 W Xe lamp | 0.182 mmol g−1 h−1 | — | 292 |
| TiO2 hollow sphere | Al reduction | 300 W Xe lamp | 56.7 mmol h−1 g−1 | 90.6% (365 nm) | 194 |
| TiO2 microspheres | Confinement reduction route | Visible light (λ > 400 nm) | 42.6 μmol h−1 | 12.7% (420 nm) and 2.8% (520 nm) | 189 |
| Sr-TiO2−x (x = 0.3%) + Pt co-catalyst | Template assisted synthesis | 300 W Xe lamp | 19442 μmol g−1 h−1 |
— | 400 |
| Rh-loaded TiO2 | Impregnation/calcination/hydrogenation | Solar simulator with AM 1.5G filter | 7.27 mmol g−1 h−1 | 0.0231% (420 nm) | 434 |
| Carbonate-doped phase-junction TiO2 nanotubes | Emulsion electrospinning method | Solar simulator with AM 1.5G filter | 6108 μmol h−1 g−1 | 83% (365 nm) | 290 |
| Anatase/rutile TiO2 with hydrogenated heterophase interface structures | Hydrothermal/hydrogenation | UV-vis light, 100 mW cm−2 | 136.5 μmol cm−2 h−1 | — | 289 |
| Anatase/rutile TiO2 | MOF-mediated synthesis | UV-vis light, 300 W Xe lamp | 1394 μmol g−1 h−1 | — | 294 |
| Ti3C2/TiO2 nanoflowers | Oxidation and alkalization/ion exchange and calcination | 300 W Xe arc lamp | 526 μmol g−1 h−1 | 5.86% (350 nm) | 208 |
| TiO2/Ti-MOF + Pt co-catalyst | Solvothermal | 300 W Xe lamp (420–760 nm) | 12.4 mmol g−1 h−1 | 19.17% (420 nm) | 295 |
| PdAu–TiO2–aerogel | Wet chemistry synthesis/supercritical drying | Solar simulator, 100 mW cm−2 | 22 μmol g−1 h−1 | — | 222 |
| Pd–TiO2–aerogel | Microwave synthesis/supercritical drying | Visible light (400–800 nm) | 117.5 mmol g−1 h−1 | 30.9% (430 nm) | 302 |
| N–TiO2–aerogel | Plasma-enhanced chemical vapor deposition | Visible-light irradiation | 3.1 mmol g−1 h−1 | 5.6% (450 nm) | 300 |
| TiO2/g-C3N4 core–shell fibers | Electrospinning | 300 W Xe lamp (λ ≥ 420 nm) | 436 μmol g−1 h−1 | — | 435 |
| TiO2 microflowers/g-C3N4 | Hydrothermal | 350 W Xe lamp | 4128 μmol g−1 h−1 | — | 436 |
| CdS/TiO2 nanofibers | Electrospinning | 350 W Xe lamp | 2.32 mmol g−1 h−1 | 10.14% | 437 |
| ZnIn2S4/TiO2 heterostructure | Hydrothermal | 300 W Xe arc lamp | 6.03 mmol g−1 h−1 | 10.49% (365 nm) | 438 |
| TiO2/NiCo2S4 core–shell structure | Solvothermal | 300 W Xe lamp | 8.55 mmol g−1 h−1 | — | 439 |
| Bi2WO6/TiO2 heterostructure | Solvothermal | 300W Xe lamp | 12.9 mmol g−1 h−1 | — | 440 |
| TiO2/ZnTe/Au nanocorncob | Hydrothermal | 300W Xe lamp | 3344.0 μmol g−1 h−1 | — | 441 |
| Multi-edged TiO2@Ru atoms | Chemical synthesis | 300W Xe lamp | 323.2 μmol h−1 per 50 mg | — | 53 |
| Ru single atoms-RuO2/TiO2 | Solvothermal/impregnation-adsorption | 300 W Xe lamp, 200 mW cm−2 | 2.91 mmol g−1 h−1 | 2.24% in benzyl-alcohol and 30.84% in methanol (365 nm) | 297 |
| Photocatalysts | Synthesis methods | Light source | Gas generation rate | Quantum efficiencies | Ref. |
|---|---|---|---|---|---|
| Eu–TiO2 nanoparticles | Sol–gel | 300 W Xe arc lamp | 65.53 μmol g−1 (CH4) and 42.91 μmol g−1 per 9 h (CO) | — | 442 |
| Ag/TiO2 nanoparticles | Chemical synthesis | 300 W Xe lamp with AM 1.5G filter | 46 mmol g−1 h−1 (CH4) | — | 308 |
| Cu0.8Au0.2/TiO2 | Photodeposition | 300 W Xe lamp (500 mW cm−2) with AM 1.5G filter | 3578.9 μmol g−1 h−1 (CH4) and 369.8 μmol g−1 h−1 for C2H4 | — | 312 |
| N-doped carbon dots decorated TiO2 hollow spheres | Chemical synthesis | 300 W Xe arc lamp | 26.8 μmol h−1 g−1 (CH4) | 0.87% (365 nm) | 443 |
| TiO2/NH2-UiO-66 (MOF) | Hydrothermal/microwave | UV-vis light (λ > 325 nm, 300 W) | 1.8 μmol g−1 h−1 (CO) | — | 309 |
| TiO2/MIL-101-Cr-NO2 (MOF) | Chemical synthesis | 300 W Xe lamp | 12 mmol g−1 h−1 (CO and CH4) | 11.3% (350 nm) | 319 |
| Cu/TiO2-aerogel | Hydrothermal/supercritical drying | UV-A/vis light (320–500 nm) | 28.2 μmol g−1 h−1 (CO) | — | 314 |
| Pd-porphyrin-based polymers coated hollow TiO2 | Chemical synthesis | 300 W Xe lamp (325–780 nm) | 48 μmol g−1 h−1 (CH4) and 34.0 μmol g−1 h−1 (CO) | — | 311 |
| TiO2@polydopamine hollow spheres | Chemical synthesis | 350 W Xenon lamp | 1.50 μmol g−1 h−1 (CH4) | — | 444 |
| (001)TiO2-g-C3N4/BiVO4 nanosheet | Solvothermal | 300 W Xe lamp with 420 nm cut-off filter | 65 μmol g−1 per 4 h (CH4) | — | 320 |
| ZnIn2S4 nanosheets/TiO2 nanobelts | Solvothermal | 300W Xe lamp | 1.135 μmol g−1 h−1 (CH4) | — | 239 |
| C-TiO2/β-Bi2O3 | Chemical synthesis | 300DUV Xe lamp with AM 1.5G filter | 31.07 μmol g−1 h−1 (CO) | — | 445 |
| TiO2/CsPbBr3 nanofibers | Electrospinning/colloidal synthesis | 300 W Xe arc lamp | 9.02 μmol g−1 h−1 (CO) | — | 243 |
| Photocatalysts | Synthesis methods | Light source | NH3 generation rate | Quantum efficiencies | Ref. |
|---|---|---|---|---|---|
| Defective TiO2 nanoparticles | Sol–gel | 300 W Xe lamp (full spectrum) | 324.86 mmol g−1 h−1 | 1.1% (365 nm) | 325 |
| Defective TiO2 particles | Hydrothermal | 300 W Xe lamp (full spectrum) | 64.82 μmol g−1 h−1 | — | 446 |
| Defective TiO2 nanotubes | Hydrothermal | 300 W Xe lamp (full spectrum) | 1.2 mmol L−1 h−1 | — | 330 |
| Defective Cu–TiO2 nanosheets | Hydrothermal | 300 W Xe lamp (full spectrum) | 78.9 μmol g−1 h−1 | 0.08% (600 nm) and 0.05% (700 nm) | 52 |
| Ru atom decorated TiO2 nanosheets | Hydrothermal | 300 W Xe lamp | 56.3 μg g−1 h−1 | — | 328 |
| Defective C-TiO2 | Calcination | 300 W Xe lamp (200–800 nm) | 84 μmol g−1 h−1 | 0.04% (400 nm) and 0.01% (420 nm) | 326 |
| Mo2C/TiO2 | Sintering | 300 W Xe lamp (UV) | 432 μg g−1 h−1 | 0.1% (365 nm) | 329 |
| TiO2@C/g-C3N4 | Calcination | 300 W Xe lamp with 420 nm cutoff filter | 250.6 mmol g−1 h−1 | 0.14% (420 nm) | 327 |
| Ce/S co-doped TiO2 | Chemical synthesis | 300 W Xe lamp with 420 nm cutoff filter | 382.4 mmol g−1 h−1 | 3.32% (420 nm) | 447 |
| N-TiO2 hollow microspheres | Hydrothermal | 300 W Xe lamp (λ > 400 nm) | 80.09 μmol g−1 h−1 | 0.07% (375 nm) | 114 |
| N-TiO2/Ti3C2 | Hydrothermal | 500 W Xe lamp (100 W cm−2) | 415.6 μmol g−1 h−1 | — | 32 |
| Photocatalysts | Synthesis methods | Light source | H2O2 generation rate | Quantum efficiencies | Ref. |
|---|---|---|---|---|---|
| TiO2−x/C3N5 | Hydrothermal/polymerization and solvent exfoliation | 300 W Xe lamp | 2.93 μmol L−1 min−1 | — | 339 |
| Au/Bi2O3–TiO2 | Chemical synthesis/deposition | 300 W Xe lamp | 11.2 mM per 12 h | — | 448 |
| TiO2/ZnIn2S4 | Chemical synthesis/solvothermal | Simulated natural light source (400 nm ≤ λ ≤ 760 nm, 100 mW cm−2) | 1530.59 μmol g−1 h−1 | 10.39% (400 nm) | 19 |
| TiO2/Au/MXene | Hydrothermal, photodeposition | UV light (360 nm < λ < 380 nm, 1 mW cm−2) | 6.80 mg per L per 4 h | — | 341 |
| TiO2@BTTA (COF) | Electrospinning/chemical synthesis | 300 W Xe lamp (λ = 350–780 nm, 0.64W cm−2) | 740 μmol L−1 h−1 | 5.48% (365 nm) | 342 |
| TiO2/Bi2O3 on polystyrene spheres | Hydrothermal/photodeposition | 300 W Xenon arc lamp (λ = 350–780 nm) | 1.15 mM h−1 | 1.25% (365 nm) | 54 |
| S-doped g-C3N4/TiO2 | Chemical synthesis/electrostatic self-assembly | 300 W Xe lamp | 2128 μmol h−1 g−1 | 0.61% (365 nm) | 340 |
| Photocatalysts | Synthesis methods | Light source | Alcohol | Product | Solvent | Conv. % | Sel. % | Ref. |
|---|---|---|---|---|---|---|---|---|
| Conv. %: conversion % and Sel %: selectivity %. | ||||||||
| Gd-TiO2/poly(o-phenylenediamine) nanowires | Hydrothermal/photopolymerization | Simulated solar light | Benzyl alcohol | Benzaldehyde | Acetonitrile | 96.0 | 97.5 | 346 |
| TiO2/Ti3C2 | Hydrothermal | 300 W Xe lamp (385–740 nm) | Furfuryl alcohol | Furfural | Acetonitrile | >99 | >99 | 22 |
| TiO2/Ti3C2 | Chemical synthesis/calcination oxidation | 300 W Xe lamp | Benzyl alcohol | Benzaldehyde | n-Hexane | 97 | 98 | 343 |
| TiO2@COF | Hydrothermal/chemical synthesis | White LED (5 W, λ = 420–780 nm, 150 mW cm−2) | Benzyl alcohol | Benzaldehyde | Acetonitrile | 92.5 | 99.9 | 21 |
| COF@TiO2 core–shell heterojunction | Chemical synthesis | 300 W Xe lamp (λ ≥ 420 nm) | Benzyl alcohol | Benzaldehyde | Benzotri-fluoride | 84 | 93 | 218 |
7.1 Photodegradation for environmental remediation
The rapid growing global populations, urbanization and industrialization have caused severe environmental impact such as global warming, climate change, and pollution. Water pollution is a major global environmental issue with detrimental effects on human health and the ecosystem, resulting from discharge of industrial effluent wastes, pharmaceutical wastes and leaching high content of organic substances such as chemical fertilizers and pesticides in aqueous environments.244–246 According to World Health Organization, an estimated 3.4 million deaths annually are attributable to water-related diseases. Moving forward, half of the world's population will be living in water-stressed areas by 2025. Therefore, re-use of wastewater and to recover water is becoming an important strategy, particularly the world's supply of fresh water is scarce. Over the last decades, Advanced Oxidation Processes (AOPs) have been regarded as effective methods in water purification and wastewater treatment. These processes are light-induced (UV or near-UV) and based on the generation of hydroxyl radicals to oxidize the harmful organic pollutants.247 Examples of the AOPs processes include ozonation, electrochemical processes, direct decomposition of water and photocatalysis (Fig. 5A).248,249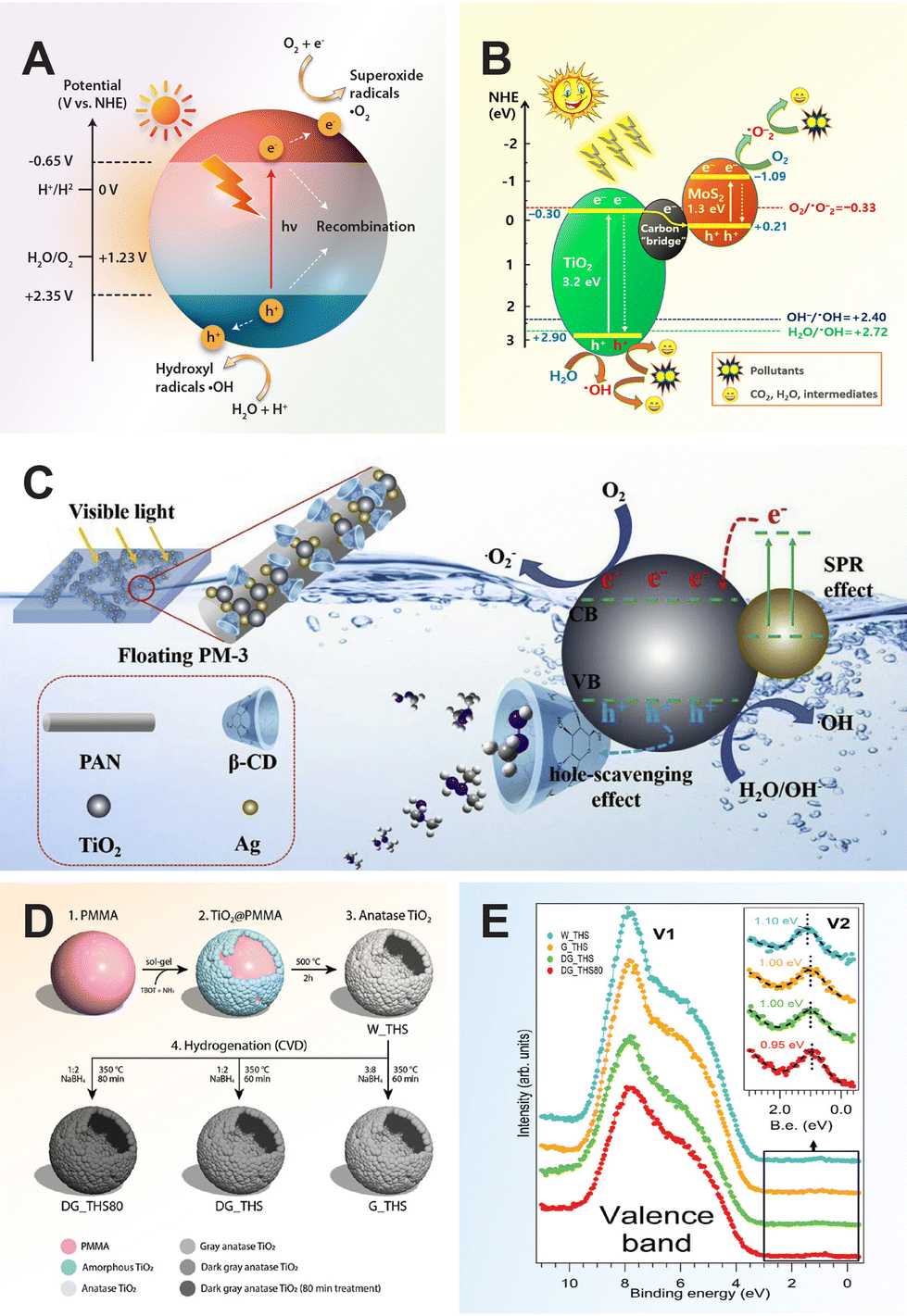 | ||
| Fig. 5 (A) Schematic illustration of a general photodegradation mechanism of TiO2 semiconductor. (B) Mechanism of Z-scheme heterojunction C-MoS/TiO2 photocatalyst for organic pollutant degradation under solar light. Reproduced with permission from ref. 266. Copyright 2022, Elsevier. (C) Photodegradation with Ag/β-cyclodextrin co-doped TiO2 floating photocatalytic membrane. Reproduced with permission from ref. 270. Copyright 2020, Elsevier. (D) Synthesis of TiO2 hollow spheres by a hard template-based method, followed by chemical reduction under controlled conditions. (E) X-ray photoelectron spectroscopy characterization showing the valence band spectra and a zoom of the fundamental gap region of the colored TiO2 hollow spheres, where defect states (V2) are detected. Reproduced with permission from ref. 55. Copyright 2023, Wiley-VCH. | ||
Photocatalysis stands out as one of the most successful and thoroughly investigated AOP. Its application offers a sustainable and environmentally friendly solution to address pollution challenges by utilizing light energy to initiate chemical reactions without producing additional pollutants. This technology finds widespread use in various photodegradation applications, encompassing the elimination of organic pollutants, dyes, and harmful chemicals from both air and water sources. Through photocatalysis, harmful contaminants can be efficiently degraded and transformed into harmless byproducts, contributing to cleaner and healthier environments. The photodegradation of various organic molecules/dyes and photocatalytic disinfection of microbial contaminants in the presence of TiO2 photocatalyst were reviewed and summarized in Table 1.
In recent studies on TiO2 facet engineering, a series of anatase TiO2 nanoparticles with varying percentages of (001) and (101) facets were synthesized via the hydrothermal method.251 By adjusting the hydrofluoric acid concentration, the (001) facet percentage was increased to nearly 100%. However, excessive hydrofluoric acid dissolved small crystal seeds, disrupting crystal growth balance. Optimal photocatalytic efficiency for Rhodamine B decomposition was achieved with 73% exposed (001) facets. Additionally, nanoflower-like rutile TiO2 was synthesized and immobilized on basil seeds for methylene blue photodegradation.252 This structure, with small particle size and large surface area, enhanced adsorption and degradation, achieving a 98.95% removal efficiency under solar light within 180 minutes.
Metal ion doping in TiO2 significantly enhances its photocatalytic properties, particularly for photodegradation applications.66,253,254 Li-doped TiO2 shows up to 2.2 times higher efficiency than undoped TiO267 and up to 5 times higher efficiency than commercial P25 under visible light.255 Specifically, doping TiO2 with 1.0 mol% Li+ lowers the anatase-to-rutile transformation temperature and creates a mixed-phase composition of 27.1% rutile and 72.9% anatase at 550 °C, significantly improving the photocatalytic degradation rate of methyl orange.67 In addition, transition metal ion doping with Al, Cu, Mo, and W also boosts photocatalytic performance for Rhodamine B dye degradation.256 Al and Cu doping increased activity to 70%, while Mo and W doping achieved 96% under visible light irradiation for 60 minutes. Surface analysis shows that Mo and W doping introduces surface hydroxyl groups essential for photodegradation and increases surface acidity, particularly in W-doped TiO2, which enhances its affinity for chemical particles with unpaired electrons.
The source of dopant and the doping process significantly influence nitrogen incorporation in N-doped TiO2 nanoparticles. Acidic doping using HNO3 incorporates nitrogen into substitutional positions, while doping with urea results in interstitial positions, both enhancing photocatalytic activity in the visible range.257 N-doped TiO2 from the acidic process demonstrates superior photodegradation of methyl orange due to better UV-visible light absorption. In P-doped TiO2, phosphorus doping in the form of P5+ inhibits crystalline growth, increases surface area, and narrows band gap energies, leading to effective methylene blue degradation.258 Cationic S-doped TiO2, with sulfur atoms as S4+ substituting lattice Ti atoms, shifts the absorption edge to a lower energy level, enabling visible light photocatalytic degradation of methylene blue and 2-propanol at longer wavelengths.259
Incorporating Ag nanoparticles into TiO2 enhances photocatalytic activity due to plasmonic effects.260 Ag/TiO2 composites with 50–100 nm Ag nanoparticles show improved degradation of methyl blue and phenol under simulated sunlight, due to better charge carrier separation and plasmon-enhanced light absorption. However, excessive Ag loading can reduce efficiency by covering active sites and promoting electron–hole recombination. Similarly, TiO2/Au nanofiber composites achieve high photocatalytic performance, with 88% degradation of methylene blue and nearly complete degradation of Rhodamine B under solar light.12 This is attributed to plasmonic hot electrons from Au nanoparticles and efficient charge separation at the Au–TiO2 interface. Bimetallic Au–Ag/TiO2 composites further enhance photocatalytic activity. The presence of Au–Ag alloy nanoparticles shifts the light absorption from UV to visible spectrum and improves plasmonic effects, resulting in up to 99% degradation of methylene blue under visible light.261
Encapsulating TiO2 within a polymer matrix and coating it with Pt nanoparticles enhances photocatalytic degradation of methylene blue.262 The Pt/TiO2 composite achieved 83% degradation under UV light in 130 minutes and 94% under direct sunlight in 400 minutes. Similarly, a Pd/TiO2 photocatalyst degraded binary dyes, achieving 83.4% mineralization of methylene blue and 75.3% of methyl orange under UV light in 180 minutes, with methylene blue degrading faster due to its thiazine structure compared to the azo bond in methyl orange.263 Au/Pt-modified TiO2 nanopillar arrays further improved photocatalytic efficiency. Depositing ∼4 nm Au/Pt nanoparticles onto the arrays enhanced UV and visible light absorption, leading to 21- and 13-times higher photocatalytic efficiency under UV-vis and visible light, respectively.184 This demonstrates the potential of combining noble metal nanoparticles with TiO2 to harness the entire solar spectrum effectively through plasmonic and electron sink effects.
Constructing heterojunctions by coupling two semiconductors together is one of the most efficient ways for achieving high pollutant degradation efficiency. For example, a Z-scheme reduced graphene oxide (rGO)/TiO2-bronze (TiO2-B)/W18O49 photocatalyst, composed of rGO, ultra-thin TiO2-B nanosheets, and W18O49 nanofibers, can absorb sunlight from ultraviolet to near-infrared regions.264 This broad absorption spectrum enables the efficient photocatalytic degradation of Rhodamine B by making full use of available light.
A 1D BiOBr/TiO2 nanorod heterojunction composite was designed through molecular and interface engineering for efficient removal of organic dye.265 This composite enhances photocatalytic activity by promoting charge migration and separation of photogenerated electron–hole pairs. The heterojunction acts as a nanochannel, facilitating the rapid transfer of photogenerated holes from the VB of BiOBr to the VB of TiO2 nanorods, leveraging BiOBr's higher VB (+3.04 eV) compared to TiO2 nanorods (+2.6 eV). This results in superior photodegradation of Rhodamine B, with an apparent rate constant of 0.49 min−1 and an 88.5% total organic carbon removal ratio. The efficient separation and extended lifetime of charge carriers allow the holes on the VB of TiO2 nanorods to oxidize Rhodamine B into CO2 and H2O.
A Z-scheme photocatalyst was constructed using a hydrothermal method, combining carbon-modified MoS2 (C-MoS2) sheets with octahedral anatase TiO2 nanocrystals, achieving 99% methylene blue degradation with a low catalyst loading (0.2 g L−1) under simulated solar light in 60 minutes.266 The C-MoS2 acts as an electron mediator, facilitating efficient electron–hole separation. The electron-rich (101)-faceted TiO2 supports the Z-scheme recombination of electrons from TiO2's CB and holes from MoS2's VB. The coupling of (101)-exposed TiO2 and 2H-MoS2, along with solid-state electron mediators 1T-MoS2 and carbon, enhances light absorption and accelerates charge transfer at the interface, significantly boosting photocatalytic activity compared to P25, MoS2/TiO2, and C-MoS2 alone (Fig. 5B). The effective separation of electron–hole pairs prolongs their lifetime, facilitating oxidation and reduction reactions in the degradation process.
Non-metal ion doping with sulfur in TiO2 photocatalysts enhances visible light absorption and reduces the band gap to 2.78 eV due to sulfur-induced structural defects.137 The sulfur is primarily present as cationic S6+/S4+ species, which enhance photocatalytic oxidation of acetaldehyde by capturing electrons and improving electrical conductivity. Comparative studies reveal that cationic S-doping (S6+ substituting Ti4+) reduces TiO2 grain size due to the smaller ionic radius of S6+, while anionic S-doping (S2− substituting O2−) increases grain size.138,140 Cationic S-doping leads to enhanced visible light absorption and photocatalytic activity through chemisorbed hydroxyls and photoinduced holes, while anionic S-doping contributes equally through electrons and holes.
The presence of bimetallic alloy nanoparticles, such as Au–Pd, significantly influences the photocatalytic reaction of phenol decomposition.269 These nanoparticles act as mediators in undesired redox reactions that would otherwise consume photogenerated radicals inefficiently. As a result, they enhance the photo-oxidation efficiency of toxic aromatic compounds like phenol. Furthermore, the TiO2 photocatalyst supported by Au–Pd demonstrated superior long-term photoactivity, achieving approximately 90% phenol decomposition under UV irradiation.
A floating photocatalytic membrane composed of Ag and β-cyclodextrin co-doped TiO2 has been developed for the dynamic adsorption and degradation of dimethylhydrazine under visible light.270 This membrane combines the photocatalytic properties of TiO2 with enhanced adsorption capabilities due to β-cyclodextrin, while Ag doping extends the light absorption into the visible spectrum, resulting in efficient degradation of the contaminant. When placed on the surface of shallow water, the membrane exhibits dual functionality through adsorption and photoactivity under visible light and oxygen. The unique cone-shaped structure of β-cyclodextrin, with its hydrophilic outer surface and lipophilic inner cavity, enhances the adsorptive capacity, making it effective in entrapping target pollutants like unsymmetrical dimethylhydrazine. Besides, Ag nanoparticles enhance light absorption via surface plasmon resonance, while TiO2 nanoparticles improve photo-response performance across UV and visible light regions. Upon excitation, β-cyclodextrin acts as a hole-scavenger, effectively suppressing electron–hole pair recombination (Fig. 5C).
To effectively degrade Imazapyr herbicide, mesoporous CdO–TiO2 nanocomposites were synthesized using a sol–gel method. These nanocomposites demonstrated photodegradation rates that were 12.2 and 24.5 times higher than those of TiO2 and P25, respectively.272 To selectively degrade 2,4-dichlorophenoxyacetic acid herbicide and imidacloprid insecticide from water, the combination of molecular imprinting and photocatalysis was investigated. These common agricultural pesticides were used as templates during synthesis and removed through calcination. The synthesized imprinted TiO2 material selectively interacted with the herbicide and insecticide, demonstrating significantly enhanced photocatalytic activity compared to bare TiO2.273,274
Non-metal ion doping in TiO2 has proven effective for degrading various pharmaceutical compounds in wastewater. A 8 wt% B-doped TiO2 achieved over 75% degradation efficiency for compounds like 2,4-dichlorophenol, bisphenol-A, ibuprofen, and flurbiprofen under visible light.275 A 5 wt% B-doped TiO2 showed 70% removal efficiency of metoprolol under simulated sunlight, compared to 48% with pure TiO2.276 The improved performance is due to increased visible light absorption, smaller crystal and particle sizes, mesoporous anatase-rutile structures, and interstitial boron positions reducing electron–hole recombination. Likewise, P-doped TiO2 with surface oxygen vacancies effectively degrades ciprofloxacin under visible light by narrowing the TiO2 band gap and enhancing charge separation.277 Surface oxygen vacancies act as charge traps and adsorption sites, facilitating efficient photogenerated charge transfer and additional reaction sites.
The effectiveness of nanostructured photocatalysts, particularly TiO2 hollow spheres made of self-assembled nanoparticles, is driven by their tailored chemical structure and morphology. These hollow spheres leverage a combination of factors such as high surface area and strong light scattering, which concentrate light within a confined volume, increasing absorption probabilities. Hydrogenated or colored TiO2 hollow spheres, consisting of hierarchically assembled nanoparticles (Fig. 5D), expand solar spectrum absorption up to 1200 nm.55 This engineered surface boosts charge photogeneration, leading to significant photocatalytic efficacy, achieving 82% degradation of ciprofloxacin after 6 hours under simulated sunlight. Valence band analysis shows prominent O 2p-related states between 3 and 10 eV (V1) and a less intense Ti 3d-derived state at around 1 eV (V2), as highlighted in the inset (Fig. 5E). The binding energy of V2 shifts from 1.10 to 0.95 eV as the TiO2 hollow spheres transition from white to dark gray, moving closer to the conduction band minimum. The V2/V1 ratio, increasing from 1.7 to 3.0, indicates a higher concentration of oxygen vacancies in the dark gray samples. This analysis highlights the defect characteristics of TiO2 hollow spheres and their implications for efficient photocatalysis.
The heterostructural TiO2/Ti3C2 nanosheet-based composite have been studied for the photocatalytic degradation of pharmaceuticals compounds. The TiO2 (0 0 1) facet-decorated Ti3C2Tx MXene was synthesized by a hydrothermal process and demonstrated a photocatalytic degradation of 98.67% of the antiepileptic drug carbamazepine, under UV light irradiation.210 The significant degradation enhancement from 60% for the pristine Ti3C2 MXene could be attributed to the extra holes and electrons generated by (001) facets of TiO2 embedded in Ti3C2 sheets, together with Schottky junctions formed between TiO2–MXene interfaces. Findings has revealed that pH was found to have a noteworthy effect on the carbamazepine degradation kinetics, with lower pH values of 3.0–5.0 are more favourable due to the nanocomposite surface being positively charged from H+ ions. Combined with a strong oxidation ability of TiO2/Ti3C2 photocatalyst, it would ultimately degrade the carbamazepine into CO2 and H2O by the end of the reaction.
A ternary heterojunction composite of Bi2O3, TiO2, and rGO was produced via a one-step hydrothermal process.278 This Bi2O3/rGO/TiO2 composite exhibited strong visible-light responsiveness and high separation efficiency of photogenerated carriers due to the Bi–Ti heterojunction, resulting in good photocatalytic activity towards tetracycline under visible-light irradiation. Besides, a Z-scheme nanocomposite of 1D/2D TiO2 nanorods and g-C3N4 nanosheets was successfully fabricated, achieving 93.4% degradation of ciprofloxacin in 60 minutes.279 Under simulated sunlight irradiation, the nanocomposite's photodegradation rate was 2.3 times higher than that of commercial TiO2 powder and 7.5 times higher than that of g-C3N4 nanosheets alone.
Recently, photocatalytic foams are emerging as an effective alternative to traditional slurry and supported catalysts due to their unique structural advantages. Their hierarchical porosity, encompassing both macro and micro levels, provides expansive surface areas akin to slurries. This structure facilitates better interaction between pollutants and the photocatalyst surface, overcoming the diffusion limitations typically associated with supported photocatalysts. The development of photocatalytic foams has been significantly advanced by 3D printing technologies. These technologies offer precise control over the design and fabrication process, allowing for the creation of complex structures with tailored porosity and flow characteristics. By building objects layer by layer from digital designs, 3D printing can produce foams with optimized pore sizes and shapes, enhancing pollutant flow and contact with the photocatalyst surface. Mattia and co-workers introduced 3D printed TiO2 foams that are nanoparticle-free, mechanically robust, and photoactive. These foams offer a promising alternative to slurry photocatalysts for the degradation of pharmaceuticals. In their study, the foams were tested using carbamazepine, a common pharmaceutical pollutant in waterways, within a recirculating flow reactor. The results revealed a quantum yield of 7.6 × 10−3 and an electrical energy per order of 67.6 kW h m−3. These figures indicate that the 3D printed TiO2 foams outperformed traditional TiO2 nanoparticle slurries in terms of efficiency and energy consumption.57
Transition metal ion doping in TiO2, such as Fe-doped TiO2, significantly enhances antibacterial properties. Under 365 nm UV light, Fe-doped TiO2 samples (1% to 10%) demonstrated inhibition rates of 67.5% to 99.4% against bacterial growth.87 This improvement is due to the formation of oxygen vacancies and a reduced optical gap in TiO2, leading to better light absorption and reactive oxygen species generation, which damage bacterial cells. Fe doping proves effective even against antibiotic-resistant strains like E. coli by generating reactive oxygen species, causing DNA damage, and peroxidizing membrane phospholipids, thus inhibiting respiration. Co-doping with cerium (Ce) and erbium (Er) also enhances antibacterial efficacy.281 Ce doping reduces the band gap, allowing absorption of both UV and visible light, while Er doping shifts NIR light into the visible range, increasing light absorption. The combination of Ce and Er co-doping inhibits the recombination of photogenerated charge carriers, resulting in antibacterial efficiencies of 91.23% against Staphylococcus aureus and 92.8% against E. coli.
Acetaldehyde, a common volatile organic compound in the environment, was used to assess the photodegradation efficiency of various TiO2 nanocomposites. In this study, Cu–Ni bimetallic nanowires were incorporated into a TiO2 matrix via a one-step hydrolysis process to form a new heterostructured photocatalyst.282 This composite achieved photodegradation efficiencies of 88% under UV light and 56% under visible light for flowing acetaldehyde gas. The enhanced performance is attributed to one-dimensional electron pathways, surface plasmon resonance effects, and an improved bimetallic Schottky barrier. This study provides insights into photon-generated carrier separation and transmission in metal–semiconductor networks and presents an effective method for developing bimetal-based heterostructured photocatalysts.
Combining rGO and layered double hydroxides (LDHs) with TiO2 synthesizes highly efficient sunlight-driven photocatalysts for degrading volatile organic pollutants such as toluene, methanol, and ethyl acetate.283 The TiO2-rGO/LDHs nanocomposite demonstrates superior photodegradation activity compared to pure TiO2 and TiO2-rGO samples. The enhanced performance is due to graphene's expanded light response range and inhibition of electron–hole pair recombination, while LDHs provide more hydroxide ions to accelerate oxidation reactions, resulting in increased radicals and improved pollutant degradation.
7.2 Photocatalytic water splitting for hydrogen production
Fossil fuel combustion is a primary contributor to greenhouse gas emissions and climate pollutants, causing climate change. The urgent need to transition to zero-emission energy sources drives the quest for alternatives to fossil fuels. Hydrogen emerges as a promising candidate, serving as a versatile and clean energy carrier. Through photocatalytic water splitting, hydrogen production becomes feasible using only water and solar light. This process initiates through photoelectric conversion within a semiconducting material, analogous to photovoltaic power generation. Unlike photovoltaic cells requiring external electrocatalysts,284 photocatalytic systems integrate both photoelectric conversion and catalytic functions within a single particle or composite. This integration allows for the direct conversion of solar energy into chemical energy, enabling the splitting of water molecules into hydrogen and oxygen without the need for additional components.14,285Water splitting is an uphill reaction, requiring an external energy input of at least 1.23 V to overcome the thermodynamic barrier associated with breaking the O–H bonds in water molecules and generating hydrogen and oxygen. Photocatalytic process aiming at water splitting is required to provide this minimum energy input to drive the reaction forward. TiO2 stands as the predominant semiconductor in photocatalytic water splitting. The combination of light absorption, charge separation, and surface redox reactions enables the photocatalytic generation of hydrogen from water using TiO2 as the catalyst. When TiO2 photocatalyst is exposed to light, at the catalyst surface, the photo-generated electrons react with water molecules, reducing them to hydrogen ions (H+). Simultaneously, the holes react with water molecules, oxidizing them to oxygen gas (O2) or hydroxyl radicals (˙OH). The accumulated H+ from the reduction reaction combine with the electrons at the catalyst surface, forming hydrogen gas (H2). Finally, the products of the redox reactions are desorbed from the catalyst surface, completing the catalytic cycle (Fig. 6A). Backus et al. studied the photocatalytic water dissociation at the TiO2–water interface with bulk water, revealing that the process begins with hole-assisted deprotonation of near-surface water molecules and the attachment of hydroxyl groups to the surface.286 These interfacial processes follow a biexponential model with time constants of 3 and 16 picoseconds. Understanding these timescales is crucial for optimizing the photocatalytic system by enhancing the desired reaction pathway and mitigating competing processes.
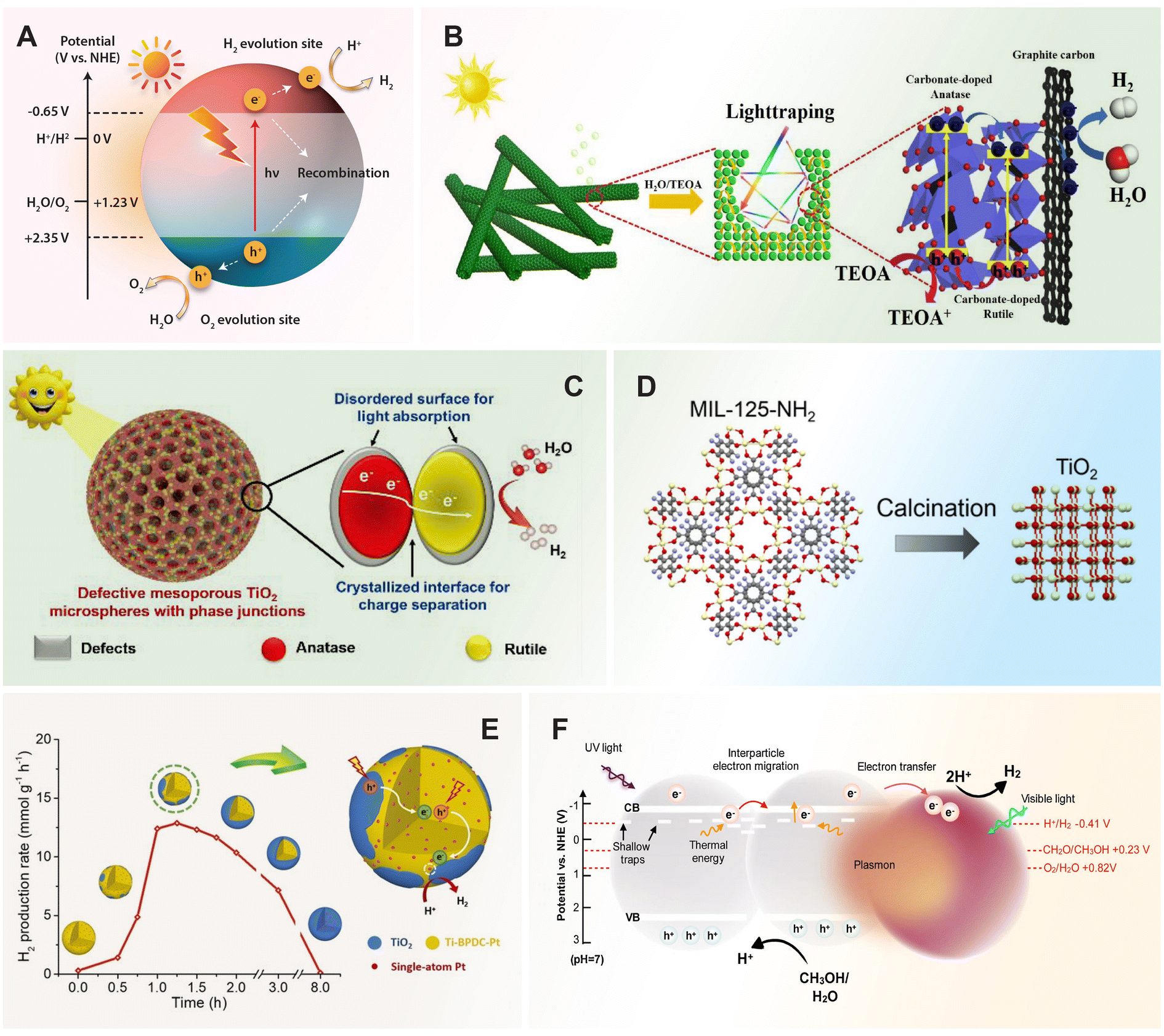 | ||
| Fig. 6 (A) Schematic illustration of the photocatalytic water splitting mechanism of TiO2 semiconductor. (B) Photocatalytic hydrogen evolution mechanism of mesoporous carbonate-doped phase-junction TiO2 nanotubes. Reproduced with permission from ref. 290. Copyright 2018, Elsevier. (C) Defective mesoporous TiO2 microspheres with phase junctions for visible-light driven water splitting. Reproduced with permission from ref. 189. Copyright 2019, Elsevier. (D) Preparation of MIL-125-NH2-derived TiO2. Reproduced with permission from ref. 294. Copyright 2018, American Chemical Society. (E) The progress of hydrogen evolution reaction activities with phase composition of TiO2/Ti-BPDC-Pt, finely tuned by varying pyrolysis duration. Reproduced with permission from ref. 295. Copyright 2023, Wiley-VCH. (F) Schematic illustration of the electron migration process in metal-containing TiO2 aerogel monoliths for the photocatalytic hydrogen evolution reaction. Reproduced with permission from ref. 222. Copyright 2020, Elsevier. | ||
All three polymorphs of TiO2 (anatase, rutile, brookite) have been studied for their photocatalytic activity. Anatase and rutile are often considered for this application while brookite TiO2 is less common in this context, with their photocatalytic potential less explored compared to other two. The photocatalytic performance of heterophase junction structures combining anatase and rutile TiO2 has garnered attention due to the distinct band gaps and aligned band-edge positions of these two phases. This configuration facilitates improved separation of photogenerated electron–hole pairs when exposed to solar light irradiation. The differing band gaps and matched band-edge positions create favorable conditions for efficient charge transfer across the heterojunction interface, enhancing the overall photocatalytic activity. As a result, these heterophase junction structures hold significant promise for applications in solar-driven photocatalysis, offering enhanced efficiency and performance compared to individual phases of TiO2.287,288
Combining a hydrogenation treatment with heterophase junctions further enhances their performance by introducing a disordered layer with oxygen vacancies and Ti3+ ions. This augmentation improves optical absorption, electrical conductivity, carrier transport, and separation efficiency. For instance, Hu et al. engineered a TiO2 photoelectrode composed of rutile TiO2 nanorods and anatase TiO2 branches with hydrogenated heterophase interfaces through hydrothermal synthesis, hydrogenation, and branching growth processes.289 The resulting structure exhibited anatase crystallographic planes in the TiO2 branches, along with a disordered layer within the heterophase interface. This optimized photoelectrode significantly enhanced the hydrogen evolution rate for photoelectrochemical water splitting, achieving a rate 20 times higher than that of unhydrogenated TiO2 nanorod arrays. Particularly, the hydrogenated interfaces between anatase branches and rutile nanorods play a crucial role by introducing oxygen vacancies and Ti3+ species. These modifications create new energy levels associated with oxygen vacancies and Ti–OH groups, situated below the band edge positions of the CB and VB of rutile TiO2 nanorods, respectively. The alignment of these energy levels with those of anatase branches and hydrogenated rutile nanorods mitigates the recombination of photogenerated carriers, thereby enhancing the overall photoelectrochemical (PEC) performance. This improved carrier separation and reduced recombination contribute to the enhanced efficiency of the heterophase junction structure in facilitating photocatalytic water splitting.
By leveraging morphology, electronic, crystal, and textural structures, electrospun mesoporous carbonate-doped phase-junction TiO2 nanotubes demonstrated exceptional photocatalytic hydrogen evolution activity, achieving 6108 μmol h−1 g−1, which is nearly 6 times higher than that of commercially available P25. The porous nanotube architecture extended the optical path through multiple reflections, enhancing light harvesting efficiency. Carbon doping reduced the bandgap of TiO2, significantly enhancing visible light photocatalytic activity. Moreover, the phase junction interface improved charge separation and transfer, resulting in photocurrent densities 2 and 18 times higher than those of pristine anatase and rutile phase samples, respectively (Fig. 6B).290 These synergistic effects led to the exceptional photocatalytic performance of the electrospun mesoporous carbonate-doped phase-junction TiO2 nanotubes. Apart from 1D TiO2 nanostructures with phase junctions and porous structures, TiO2 hollow spheres have been widely investigated in the field of photocatalytic water splitting due to their unique structural characteristics and enhanced light harvesting capabilities.291 The TiO2 spheres composed of a crystalline core and an amorphous shell structured nanocrystallites, exhibited a hydrogen production rate of 0.182 mmol g−1 h−1, which was twice that of pristine TiO2.292
Zhang et al. engineered defective mesoporous TiO2 microspheres with phase junctions featuring controllable defect locations using a confinement reduction method.189 This method exploits on the confinement decomposition effect, which allows for efficient defect production under mild conditions without compromising the mesostructures and phase junctions of the pristine mesoporous microspheres. Furthermore, by adjusting the reduction temperature, defects can be relocated from the nanocrystalline-exposed surfaces to the phase junction interfaces, enabling precise tuning of defect locations (Fig. 6C). The defect formation altered the energy band structure and mediated the visible light adsorption of the TiO2 microspheres. The well-retained phase junction structure facilitated effective photo-induced charge separation. This engineered photocatalyst exhibited a hydrogen production rate of 42.6 μmol h−1 (based on 50 mg of catalyst) under visible-light irradiation (λ > 400 nm). Moreover, the apparent quantum efficiencies were measured at 12.7% and 2.8% at wavelengths of 420 nm and 520 nm, respectively.
In the realm of MOFs, several studies have explored the combination of TiO2 with MOFs to enhance the efficiency of photocatalytic systems for hydrogen evolution reaction under visible light irradiation.293 The 3D porous structure of TiIV-based MOFs provides a structured environment for the controlled growth and arrangement of TiO2 crystals. Specifically, the templating effect of MIL-125-NH2 enables the formation of well-defined TiO2 nanoparticles with customized size, morphology, and crystallinity, leading to optimized photocatalytic performance. Studies have demonstrated that starting with MIL-125-NH2 as a MOF precursor, TiO2 particles can be synthesized at various temperatures while retaining the well-defined crystal shape of the parent MOF and controlled phase composition (Fig. 6D).294 For instance, a mixed TiO2 phase comprising 66% anatase and 34% rutile exhibited a remarkable hydrogen evolution rate of 1394 μmol g−1 h−1, surpassing both commercial Degussa P25 TiO2 and conventionally synthesized TiH4O4- and MIL-167-derived TiO2. This superior performance is credited to the unique templating effect of MIL-125-NH2, which promotes the formation of nanosized anatase and rutile nanoparticles in effective contact, thereby enhancing electron–hole separation and overall photocatalytic efficiency.
A recently developed TiO2/Ti-BPDC-Pt photocatalyst, featuring a TiO2/Ti-MOF heterojunction with high-density Pt single-atomic co-catalysts, has shown promise for photocatalytic hydrogen evolution (Fig. 6E).295 Single-atom co-catalysts have emerged as an efficient and cost-effective approach in heterogeneous photocatalysis due to their high atomic utilization efficiency and excellent catalytic activity. These isolated metal heteroatoms can enhance semiconductor band structures, improving light absorption and facilitating electron collection to boost surface charge separation and transfer.296,297 In this study, a TiO2/Ti-BPDC heterojunction is tailored through a surface pyrolytic reconstruction approach, resulting in an intimate interface between TiO2 and the Ti-based MOF, which facilitates efficient charge separation and migration. The strategic placement of Pt within the electron-enriched domain of the heterojunction further enhances the utilization of separated electrons for the reduction of protons, thus promoting hydrogen production. This engineered TiO2/Ti-BPDC-Pt catalyst exhibits superior activity, achieving a hydrogen evolution rate of 12.4 mmol g−1 h−1, surpassing other TiO2- or MOF-based catalysts.
Among the Pt-group metals, Ru stands out as a competitive alternative to Pt due to its cost-effectiveness and comparable hydrogen evolution performance.298 The coexistence of two forms of Ru species such as nanoparticles and single atoms, supported on MOF-derived N-doped TiO2/C hybrids exhibits superior photocatalytic hydrogen evolution reaction.299 This enhanced performance results from the synergistic coupling of Ru nanoparticles and Ru single atoms. Similarly, atomically dispersed Ru atoms on multi-edged TiO2 spheres significantly enhance hydrogen evolution by effectively transferring photogenerated electrons to isolated Ru atoms and facilitating charge separation and transport through the multi-edged TiO2 structure.53 The in situ X-ray absorption fine structure technique was used to examine the dynamic changes of isolated sites during the catalytic process. Upon light irradiation, the Ru species experience gradual changes in valence and configuration, facilitating the photo-splitting of water into solar fuels.
When Ti3C2 MXene was converted into 3D porous frameworks of Ti3C2–TiO2 nanoflowers, the in situ growth of TiO2 on the surface of Ti3C2 offers intimate interaction between TiO2 and Ti3C2 for photocatalytic overall water splitting.208 The photogenerated electrons can transfer from the CB of TiO2 to Ti3C2, where Ti3C2 serving as an electron sink. The formation of a possible Schottky junction at the interface between Ti3C2 and TiO2 enhances the separation of photogenerated charge carriers, effectively suppressing recombination. As a result, more electrons participate in the photoreduction process for hydrogen evolution, while more holes engage in the photooxidation process for oxygen evolution. This mechanism contributes to the enhanced efficiency of photocatalytic water splitting facilitated by Ti3C2–TiO2 nanoflowers.
TiO2 aerogel monoliths hold significant promise as efficient and sustainable photocatalysts for hydrogen generation due to the synergetic effect between their building blocks and their 3D macroscopic structure. The porous structure of TiO2 aerogels promotes light trapping and diffusion within the monolith. Upon light entering the aerogel, it undergoes multiple scattering events, resulting to prolonged interaction lengths and increased absorption probabilities. This improves the efficiency of light harvesting by ensuring that a larger fraction of incident photons is absorbed by the TiO2 matrix. Likewise, the interconnected pores and tortuous pathways within the aerogel matrix enable light to travel over millimeter length scales through the monolith, hence increasing the probability of photon absorption by TiO2 nanoparticles.222 To preserve the fragile structure of the 3D TiO2 network while enabling it sensitive to visible light, monolithic aerogels composed Pd modified TiO2 nanoparticle was doped with nitrogen in a gas-phase nitridation process using plasma-enhanced chemical vapor deposition at low temperature.300 The nitridation-induced nitrogen doping and defect engineering in TiO2 aerogels, coupled with Pd nanoparticle loading achieved the desired enhancement in optical absorption and charge separation efficiency, and hence outperformed the undoped material in visible light-driven photocatalysis for hydrogen production.
Besides doping, the assembly of non-doped TiO2 aerogel composite with noble metal nanoparticles (Au, Pd, PdAu) has been shown to exhibit superior visible light-induced photocatalytic hydrogen production compared to their corresponding powders.222 By leveraging the plasmonic properties of noble metal nanoparticles and the catalytic activity of TiO2, the formation of inter-particle contacts during nanoparticle assembly lead to the creation of shallow traps within the composite material, resulting to an absorption band around 400–500 nm in the visible range of the electromagnetic spectrum. The combination of shallow traps absorption in the visible range and the LSPR of the metal nanoparticles promoted the hydrogen evolution through the near-field electromagnetic mechanism. Further, thermal energy generated by the LSPR of the plasmonic particles promotes the shallow-trap electron migration process (Fig. 6F). Overall, the observed 3.5 times increase in hydrogen generation underlines the importance of the both the aerogel 3D structure and the type of metal nanoparticle on the photocatalytic activity of the aerogels in enabling light-harvesting and efficient mass transport of reactants to the surface-active sites.
Among the noble metal nanoparticles, Pd has been recognized as a particularly effective photocatalyst under visible light irradiation. One of the key reasons for the high photocatalytic activity of Pd is attributed to the generation of hot electrons from its 4d orbital under visible light irradiation.301 By using a microwave-assisted non-aqueous sol–gel method, Pd-modified TiO2 nanoparticles can be synthesized with simultaneous incorporation of Pd ions in the TiO2 lattice and formation of Pd metal nanoparticles on the surface of the TiO2 nanoparticles.302 The subsequent assembly of Pd-modified TiO2 nanoparticles into macroscopic aerogels results in photocatalyst with a narrow band gap, primarily due to the formation of Pd 4d energy levels, oxygen vacancies, and Ti3+ centers within the TiO2 lattice. While Pd doping and Pd nanoparticle loading can enhance visible-light absorption in TiO2 aerogels, excessive Pd accumulation may hinder charge generation and separation due to shadowing effects and high Schottky barriers.
7.3 Chemical transformations of CO2 for sustainable future
Carbon dioxide (CO2) emissions are the primary driver of global warming and climate change. Limiting global warming to 1.5 °C requires rapid and sustained reductions in CO2 emissions and reaching net-zero emissions in the energy sector by 2050.303 To mitigate this crisis, turning CO2 into valuable chemicals or fuels has the potential to mitigate global warming by treating hundreds of millions of tons of CO2 annually. Among various CO2 conversion approaches, photocatalytic CO2 reductions is regarded as one of the most ideal approaches by mimicking natural photosynthesis. Generally, CO2 can be photocatalytically reduced into several carbonaceous molecules including CO, HCOOH, HCHO, CH3OH, and CH4 (Fig. 7A). In this regard, numerous visible-light-driven photocatalysts for the CO2 conversion have been developed, particularly to optimize the structure and composition of semiconductor photocatalysts to improve their visible light absorption and charge separation efficiency, e.g., creating heterojunctions, forming surface defects, incorporating metal co-catalysts, and engineering exposed crystal facets etc.16,48,216,304–307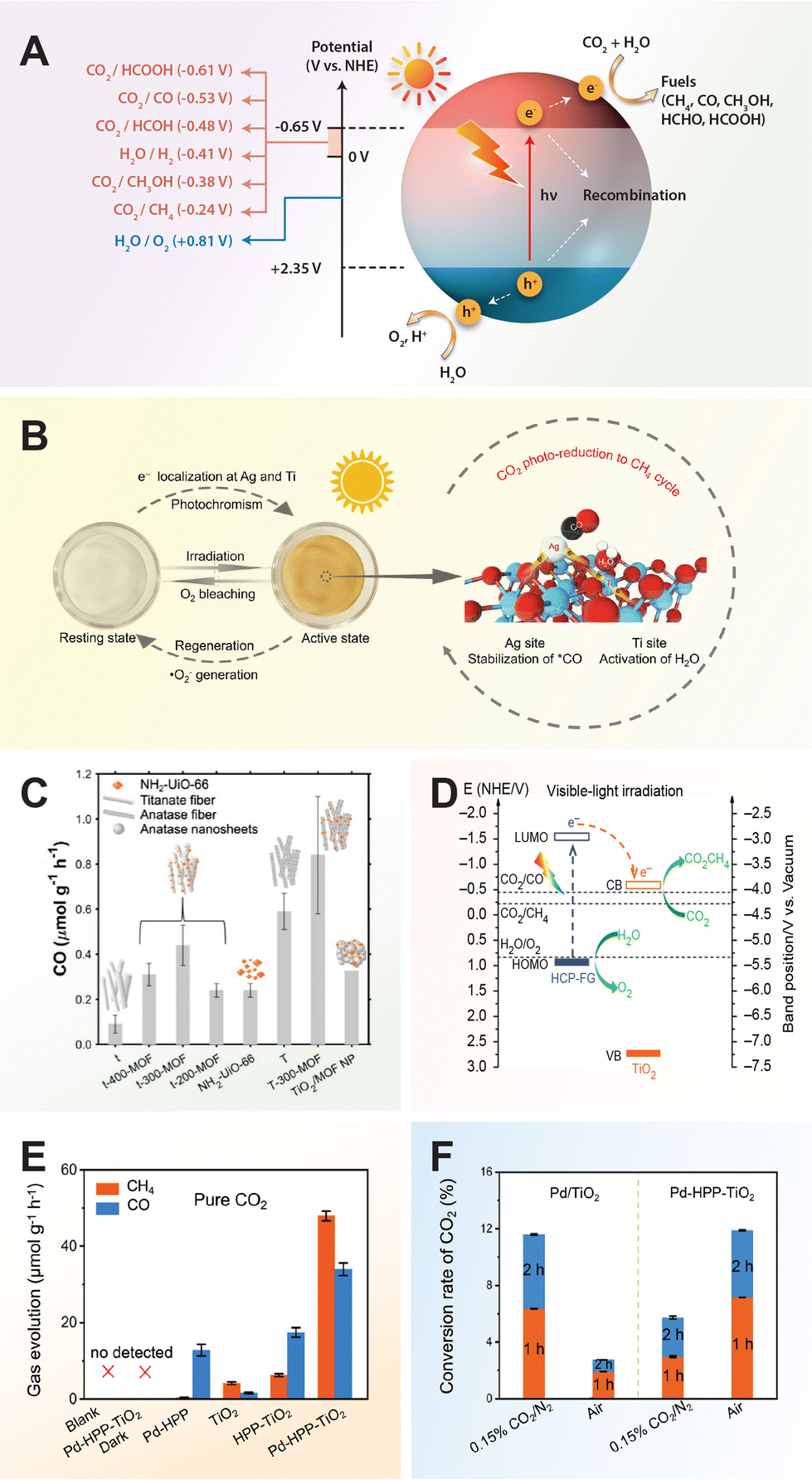 | ||
| Fig. 7 (A) Schematic illustration of the CO2 photoreduction mechanism of TiO2 semiconductor. (B) Photochromic cycle of Ag/TiO2 (left) and the photocatalytic CO2 to CH4 conversion cycle over the Ag–Ti active site on Ag/TiO2 (right). Reproduced with permission from ref. 308. Copyright 2024, Royal Society of Chemistry. (C) Photocatalytic CO evolution rates of TiO2/NH2-UiO-66 nanocomposite compared to titanate fiber. Reproduced with permission from ref. 309. Copyright 2019, Wiley-VCH. (D) Proposed mechanism of charge separation and transfer within the TiO2/graphene composite under visible-light (λ ≥ 420 nm) irradiation. Reproduced with permission from ref. 310. Copyright 2019, Springer Nature Group. (E) The evolution rates of CH4 and CO in photocatalytic CO2 reduction and (F) comparison of the conversion yield of CO2 in Pd/TiO2 and Pd-HPP-TiO2. Reproduced with permission from ref. 311. Copyright 2022, Springer Nature Group. | ||
Recently, a TiO2 catalyst was engineered by anchoring single Ag atoms onto the surface of the anatase TiO2 nanoparticles for photocatalytic CO2 to CH4 conversion. The Ag/TiO2 catalyst exhibited photochromism which was attributed to the trapping of photogenerated electrons.308 The resulting active state of the photochromic catalyst effectively facilitates the separation and migration of photogenerated charge carriers. In addition, the isolated Ag atoms and adjacent Ti sites play complementary roles in catalyzing the conversion of CO2 to CH4. The isolated Ag atoms serve to stabilize two key intermediates (*CO and *CHO) formed during the reaction process, while the adjacent Ti sites are responsible for activating water molecules to generate more protons, hence both simultaneously affording active sites to increase the production of CH4 rather than CO (Fig. 7B). Ultimately, the Ag/TiO2 catalyst achieved high activity and selectivity of 46.0 mmol g−1 h−1 and 91% respectively, for photocatalytic CO2 methanation.
Besides Ag atoms, Cu single atoms and Au–Cu alloy nanoparticles were co-loaded on TiO2 by photodeposition for the photocatalytic production of solar fuels from CO2 and H2O.312 The optimized photocatalyst achieved high formation rates of 3578.9 μmol g−1 h−1 for CH4 and 369.8 μmol g−1 h−1 for C2H4. The synergy between Cu single atoms and Au–Cu alloy nanoparticles enhanced the adsorption and activation of CO2 and H2O and lowered the activation energy barrier for CH4 and C2H4 formation, enabling highly efficient and stable production of these solar fuels.
Typically, the scope of CO2 conversion work is often limited to the low adsorption of CO2 on the surface of photocatalysts owing to the low specific surface area and the lack of matched pores. The efficiency of CO2 conversion heavily relies on the availability of CO2 molecules near the active sites of the photocatalyst, where they can interact with photoexcited electrons and participate in catalytic reactions. Facing this challenge, combining a CO2 capture material with semiconductor photocatalysts has been proposed to provide an attractive means for increasing CO2 conversion efficiency. Xu et al. designed photocathode comprised of a layered hybrid heterojunction of TiO2 on 2D Ti3C2 MXene nanosheets, functionalized with imine ligands and Pd nanoparticles (Pd/N-TiO2/Ti3C2) for CO2 conversion.313 The photocathode of Pd/N-TiO2/Ti3C2 exhibited an evolution rate of total hydrocarbon (formate, methanol, ethanol) 5-fold higher than that of Pd-Ti3C2 photocathode. The electrode was stable for 16 hours without a change in its efficiency. The high efficiency of Pd/N-TiO2/Ti3C2 is ascribed to the high surface area and narrow band gap (2.1 eV) TiO2/Ti3C2 heterojunction materials which provide abundant active sites in favor of the adsorption of CO2 molecules and promote strong visible light absorption, respectively as well as the plasmonic resonance effect of Pd nanoparticles. Kreft et al. synthesized Cu/TiO2-aerogel composite featuring Cu(II)-nanoparticles on the surface of a highly porous TiO2 aerogel for aqueous CO2 reduction to CO without the need for external sacrificial reagents.314 Notably, the presence of O2 in the reaction environment enhances CO productivity while suppressing H2 generation.
Combining TiO2 photocatalyst with MOFs allows for the synergy between the light-absorption and electron-generation capabilities of TiO2 with a high concentration of open active sites in the framework of MOFs for CO2 capture and conversion processes.315,316 In addition, optimizing the morphology of the heterojunction components and engineering the interface between the two materials are critical for achieving a close interaction and maximizing photocatalytic performance. This can involve controlling the size, shape, and distribution of TiO2 nanoparticles on the MOF surface. Studies have shown that improved CO2 photoreduction was achieved through synthesizing TiO2 nanoparticles onto various preformed MOFs, producing TiO2/HKUST-1,317 TiO2/Co-ZIF-9,318 and TiO2/NH2-UiO-66 composites.211 The superior photocatalytic activity is ascribed to the development of an intimate interaction between TiO2 and MOFs forming a heterojunction, while retaining the high CO2 uptake and porosity of MOFs. The TiO2/MOF composites exhibited better durability and significantly more efficient in reducing CO2 to CO compared to their individual components. Crake et al. synthesized TiO2/NH2-UiO-66 nanocomposites with superior photocatalytic activity in CO2 photoreduction, specifically focusing on the role of heterojunctions, highlights the importance of both crystalline phase and morphology control in enhancing charge transfer and overall photocatalytic performance.309 Forming anatase phase nanofibers and growing MOF particles on their surface allows for precise control of composite morphology, which in turn maximizes charge transfer efficiency. Accordingly, the electrons transfer from TiO2 into the MOF and holes from the MOF into TiO2, accompanied by strong band bending in TiO2 induced by the MOF, leading to an improved charge separation and facilitating efficient charge transfer in the heterojunction structure. Therefore, the TiO2/NH2-UiO-66 nanocomposite produced 9 times more CO when compared to titanate under UV-vis light irradiation, confirming the synergistic effect of forming a composite (Fig. 7C).
Wang et al. incorporated TiO2 units within the pores of a chromium terephthalate-based MOF (MIL-101) and its derivatives, forming “molecular compartments” where photocatalytic reactions can occur in a confined environment.319 These compartments facilitate the close proximity of TiO2 units and catalytic metal clusters, promoting efficient charge transfer and catalytic activity for CO2 reduction. The observed apparent quantum efficiency for CO2 photoreduction of 11.3% at 350 nm in the composite consisting of 42% TiO2 in a MIL-101 derivative (42%-TiO2-in-MIL-101-Cr-NO2) demonstrates the effectiveness of this composite for photocatalytic CO2 conversion. Furthermore, TiO2 units in one type of compartment in this composite are 44-fold more active than those in the other type, emphasizing the importance of precise positioning of TiO2 within the composite. Simultaneously, the photocatalytic process generates molecular oxygen as a byproduct, contributing to the overall sustainability of the CO2 conversion process.
Wang et al. developed a porous composite structure by integrating anatase TiO2 crystals with reactive (001) facets on graphene surface and subsequently encapsulated in hypercrosslinked polymer layers by in situ knitting strategy.310 Given abundant adsorptive sites of the porous capture materials for efficient CO2 uptake, the photoreduction of CO2 of modified TiO2 photocatalyst proceeded under modest conditions without sacrificial reagents and co-catalysts, yielding 27.62 μmol g−1 h−1 for CH4 production under visible-light irradiation. In comparison, the CO2 conversion products were hardly identified over commercial TiO2 (P25), and pristine TiO2 with reactive (001) facets because of their weak visible light-responsive ability.
With the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels lying more negative than the VB and CB levels of TiO2, respectively, the hypercrosslinked polymer-graphene can act both as CO2 adsorbent and photosensitizer, in which their photoinduced electron–hole pairs are migrated and separated at the interface with TiO2via their interfacial interaction. Therefore, the CO2 reduction is prone to happen at the catalytic sites on TiO2 instead on hypercrosslinked polymer-graphene, giving rise to better CO2 conversion efficiency (Fig. 7D). Moreover, a selectivity of 83.7% for CH4 production is achieved during the photocatalytic reaction, suggesting negligible side reaction of hydrogen evolution under visible-light irradiation.
Recently, Ma et al. reported a novel approach for the preparation of a composite photocatalyst by hyper-crosslinking porphyrin-based polymers (HPP) on the surface of hollow TiO2, followed by coordination with Pd(II).311 In a pure CO2 environment, this composite photocatalyst, Pd-HPP TiO2 exhibited remarkable CO2 reduction efficiency, achieving high evolution rates of 48.0 and 34.0 μmol g−1 h−1 for CH4 and CO, respectively (Fig. 7E). In the presence of 5 vol% O2, the CO2 reduction over a catalyst without hyper-crosslinked porphyrin-based polymer (Pd/TiO2) drastically decreases to only 6% of that observed in pure CO2. On the other hand, the inhibition from O2 is significantly mitigated over composite photocatalyst (Pd-HPP TiO2), which maintains 46% of the CH4 evolution rate observed in pure CO2 conditions. The composite photocatalyst, Pd-HPP-TiO2 demonstrated notable photocatalytic activity even in the presence of air, with a CO2 conversion yield of 12% and CH4 production of 24.3 μmol g−1 after 2 hours of UV-visible light irradiation (Fig. 6F). This performance is 4.5 times higher than that observed over Pd/TiO2. The hyper-crosslinked porphyrin-based polymer layer effectively enriches CO2 at Pd(II) sites, thereby mitigating the reduction of O2. Moreover, water adsorbed on TiO2 undergoes oxidation by the holes present in the valence band of TiO2. This process serves to reduce charge recombination, thereby enhancing CO2 conversion efficiency.
Bian et al. developed a cascade Z-Scheme photocatalytic system using 2D g-C3N4 for the reduction half-reaction and 2D BiVO4 nanosheets for the oxidation half-reaction, combined with an energy platform of (001)TiO2.320 This (001)TiO2-g-C3N4/BiVO4 nanosheet heterojunction exhibited exceptional photocatalytic activity for CO2 photoreduction and water splitting without cocatalysts, achieving a 19-fold improvement in photoactivity for CO2 reduction to CO under visible-light irradiation compared to BiVO4. This performance surpasses other reported Z-Scheme systems, even those using noble metals as mediators.
7.4. Nitrogen fixation for ammonia production
Since the initial discovery in 1977 that nitrogen (N2) could be reduced to ammonia (NH3) on the surface of TiO2,321 the pursuit of achieving N2 fixation using sunlight has become a significant research focus. N2 fixation is the conversion of atmospheric N2 into NH3 or other N2-containing compounds, which is an essential process for the production of fertilizers and various industrial applications.322 As is known, N2 is a highly stable compound with a bond dissociation energy of N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N of approximately 941 kJ mol−1, making it thermodynamically challenging to convert it into NH3 or other N-containing compounds. The Haber–Bosch process, which is the most widely used method for industrial NH3 production, achieves the industrial-scale NH3 synthesis through high temperatures (300–500 °C) and pressures (200–300 atm), which leads to huge energy consumption and high CO2 emission.323 Therefore, a less energy consuming alternative would be highly desirable.
N of approximately 941 kJ mol−1, making it thermodynamically challenging to convert it into NH3 or other N-containing compounds. The Haber–Bosch process, which is the most widely used method for industrial NH3 production, achieves the industrial-scale NH3 synthesis through high temperatures (300–500 °C) and pressures (200–300 atm), which leads to huge energy consumption and high CO2 emission.323 Therefore, a less energy consuming alternative would be highly desirable.
Photocatalytic conversion of N2 to NH3 is a green alternative for the Haber–Bosch process. It typically involves using light energy to generate electrons and holes within a semiconductor and combine with water protons to reduce N2 to NH3 (Fig. 8A).17,324 The current strategies for developing efficient TiO2 photocatalysts for N2 fixation prioritize the creation of active sites through the introduction of defects in TiO2. These defects, such as oxygen vacancies, aim to weaken the NN triple bond of adsorbed N2 molecules by facilitating the transfer of electrons into the antibonding orbital of N2. This activation process enables their subsequent reaction with photogenerated electrons for N2 reduction reactions. In addition, defects in the TiO2 structure enable efficient charge separation and accelerate charge carrier transfer from photocatalysts to the adsorbed reactants.18 It was demonstrated that by fine-tuning the concentration of oxygen vacancies, TiO2 can achieve a 3-fold increase in charge separation efficiency compared to pristine TiO2.325 The oxygen vacancy defect structures coordinate both the charge separation efficiency and the dissociative adsorption capacity of N2, leading to a normalized N2 photofixation rates of 324.86 mmol g−1 h−1 (under full spectrum illumination), with corresponding apparent quantum yields of 1.1% under 365 nm illumination.
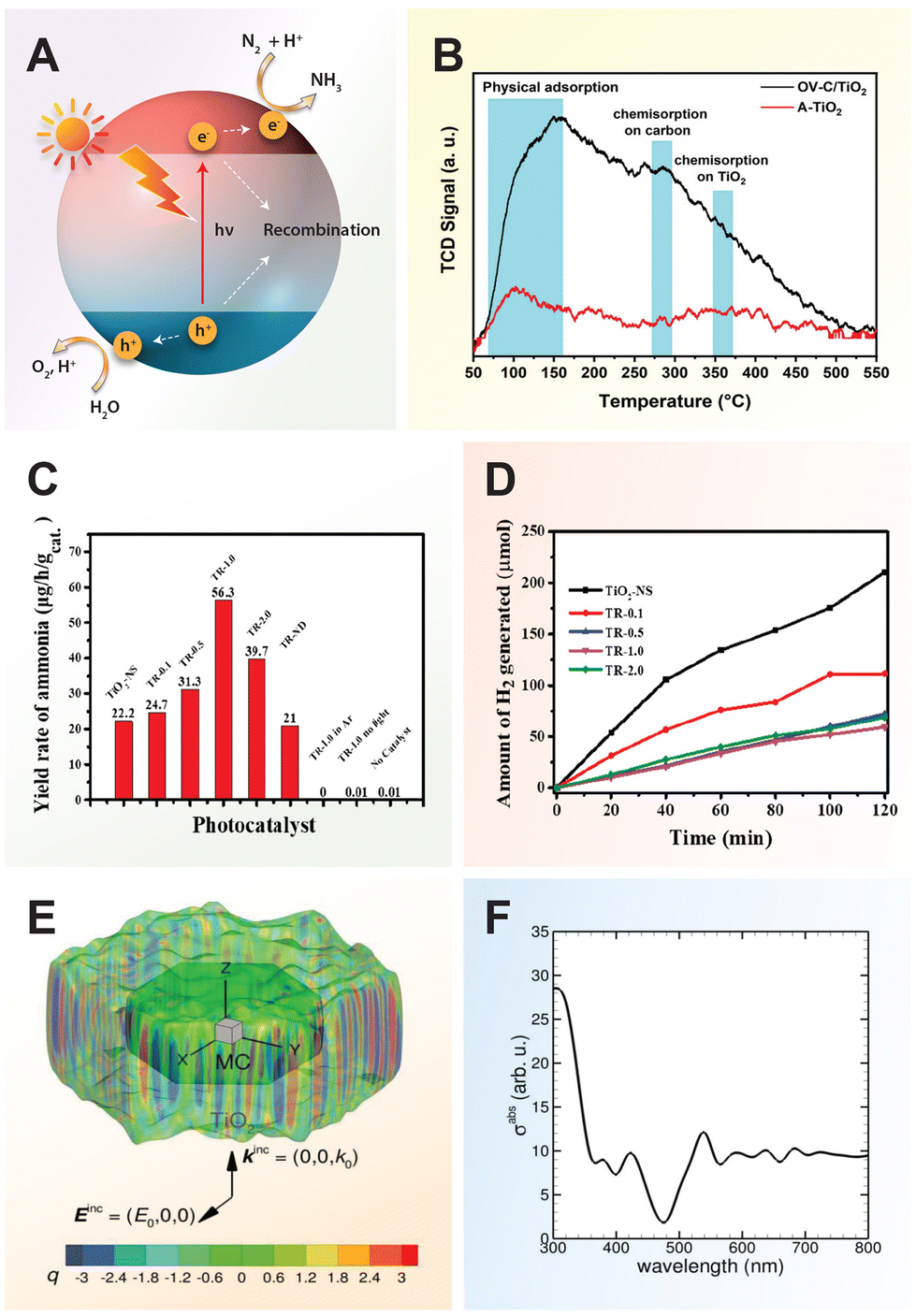 | ||
| Fig. 8 (A) Schematic illustration of the N2 photoreduction mechanism of TiO2 semiconductor. (B) N2-temperature programmed desorption profiles of the commercial anatase TiO2 and oxygen vacancy-rich C-TiO2. Reproduced with permission from ref. 326. Copyright 2022, Wiley-VCH. (C) The yield rate of photocatalytic fixation of N2 to NH3 and (D) the quantity of H2 generated in water by single Ru atom decorated TiO2 nanosheet photocatalysts. Reproduced with permission from ref. 328. Copyright 2019, American Chemical Society. (E) Surface charge distribution of a carbon coated hexagonal Mo2C and TiO2 particle with core–shell structure under the illumination of sunlight and (F) corresponding absorption cross sections across the spectrum of sunlight. Reproduced with permission from ref. 329. Copyright 2023, Elsevier. | ||
The oxygen-rich TiO2, prepared by calcination of Ti3C2 MXene, exhibited superior photocatalytic N2 fixation performance compared to both P25 and commercial anatase TiO2.326 Specifically, it achieved a N2 fixation rate of 84 μmol g−1 h−1, utilizing CH3OH as the proton source. Typically, chemisorption of N2 on carbon occurs at around 280 °C, while on TiO2, it occurs at approximately 360 °C. In contrast, physical adsorption of N2 takes place at a lower temperature of about 120 °C.327 The significantly higher temperature programmed desorption signal observed for oxygen vacancy-rich C-TiO2 compared to commercial anatase TiO2 indicates stronger chemisorption of N2 on the former (Fig. 8B). This enhanced chemisorption capability of oxygen vacancy-rich C-TiO2 is crucial for the activation of N2, highlighting its potential for efficient N2 reduction in photocatalytic processes.
Controlling defects in TiO2 nanotubes can be achieved through an amine-assisted remedying strategy using urea, dicyandiamide, and cyanamide as precursors.330 This approach involves the preparation of hydrogen-treated TiO2 nanotubes, which are engineered to possess oxygen vacancies. By using urea, dicyandiamide, or cyanamide as precursors, the process can be optimized to introduce and regulate the concentration of oxygen vacancies in the TiO2 nanotubes. The resulting defect-rich nanotubes extended the visible light absorption and suppressed the recombination of photogenerated electron–hole pairs, which led to improved photocatalytic performance in N2 fixation reactions. The NH3 production rate, reaching 1.2 mmol L−1 h−1 under full spectrum light irradiation, represents a significant enhancement compared to pristine TiO2, with an approximate 8.6-fold increase in efficiency.
While another effective way of increasing NH3 production yield in photocatalytic N2 fixation is through doping TiO2 with metal heteroatoms.52 Copper, as a dopant, can effectively regulate the concentration of oxygen vacancies and introduce substantial compressive strain in ultrathin TiO2 nanosheets. Particularly, defect-rich TiO2 nanosheets containing 6 mol% copper demonstrated stable performance for the photocatalytic reduction of N2 to NH3 in water, showcasing superior photoactivity even up to 700 nm. Modifications with oxygen vacancies and strain effects in TiO2 nanosheets enable strong chemisorption and activation of molecular N2 and water, leading to high rates of NH3 evolution under visible-light irradiation. Analysis using diffuse reflectance infrared Fourier transformation spectroscopy provides compelling evidence that NN triple bonds can be activated on the defect-rich TiO2 nanosheets containing 6 mol% copper, forming NH4+ species under light irradiation. The observed rates of O2 (59.1 μmol g−1 h−1) and NH3 (78.9 μmol g−1 h−1) evolution during the tests closely match the theoretical ratio of 3:4 for the reaction N2 + 3H2O → 2NH3 + 1.5 O2, with no detectable N2H4 byproduct observed. This suggests high selectivity and efficiency of the photocatalytic N2 fixation process on the defect-rich TiO2 nanosheets.
The recent development of single atom decorated TiO2 semiconductor with engineered oxygen vacancies has demonstrated remarkable catalytic activity in photocatalytic N2 fixation. Single atom metals dispersed on supports offer homogeneous catalytically active sites, a low-coordination environment for metal atoms, and maximum metal utilization efficiency, resulting in enhanced catalytic activity, stability, and selectivity across various processes.53,328,331 The composite catalyst containing 1 wt% of Ru exhibited a significantly improved ammonia generation rate of 56.3 μg g−1 h−1, more than doubling that of pure TiO2 nanosheets (Fig. 8C).328 Upon decoration with Ru, the photocatalytic activity of TiO2 nanosheets for the competing reaction, such as the hydrogen evolution reaction decreased. The hydrogen evolution activity was suppressed, reaching an almost saturated value of 30 μmol h−1 at 0.5 wt% Ru, compared to 105.3 μmol h−1 in pure TiO2 nanosheets (Fig. 8D). This decrease in hydrogen evolution reaction activity with the introduction of Ru species is attributed to the interference with the transportation of photoelectrons from TiO2 to H+ by combining with the oxygen vacancies, thus inhibiting hydrogen evolution reaction.
In addition to metal doping, non-metal introduction into the TiO2 lattice has been investigated as a strategy in modifying the electronic structure of TiO2 and facilitating the separation of photogenerated charge carriers. Specifically, N-doping in Ti3C2–TiO2 was found to enhance the carrier separation and improve the ammonia production yield to 415.6 μmol g−1 h−1, which represents an 8-fold increase compared to pure TiO2.32 Likewise, N-doping TiO2 hollow microspheres along with the creation of oxygen vacancies displayed an ammonia yield of 80.09 μmol g−1 h−1.114 This enhancement is associated with the synergistic effect between the N-doping, oxygen vacancies, and hollow microsphere structure that collectively contributes to the boosted efficiency of photocatalytic N2 fixation. Firstly, the incorporation of nitrogen dopants narrows the bandgap of TiO2 from 3.18 eV to 2.83 eV, leading to improved absorption of visible light. Further, the creation of oxygen vacancies assists to impede the recombination of photo-generated carriers, hence increasing the efficiency of charge separation. Besides, the hollow microspheres structure of the catalysts provides a large surface area and promotes light absorption and utilization. These findings highlight the potential of tailored nanostructures and dopants for enhancing the performance of photocatalysts in nitrogen fixation applications.
Recently, a composite material consisting of carbon coated hexagonal Mo2C and TiO2 was developed for photocatalytic N2 fixation.329 In contrast to pure TiO2, this composite photocatalyst effectively separates the photogenerated electrons and holes, facilitating the efficient conversion of atmospheric N2 into NH3 directly from air. The carbon-coated hexagonal photocatalyst demonstrated a remarkable enhancement in photocatalytic N2 fixation performance, achieving a 16-fold increase compared to pure TiO2 under room temperature and ambient pressure conditions. In the proposed heterojunction formed by TiO2 and Mo2C, an important aspect is the alignment of their conduction band potentials. Specifically, the conduction band potential of Mo2C is strategically positioned to facilitate the conversion of N2 to NH3. Relative to TiO2, the conduction band potential of Mo2C is lower, thereby creating a favorable energy gradient that facilitates the transfer of photoelectrons from TiO2 to Mo2C. Moreover, the graphitized carbon layer structure enhances the concentration of photoelectrons on Mo2C, owing to the highly conductive carbon layer. N2 molecules readily adsorb onto the carbon vacancies of the molybdenum atoms, where they receive photoelectrons. Subsequently, the activated nitrogen species combine with protons in water, ultimately undergoing reduction to form ammonia. The hydroxyl radicals generated by photocatalytic water splitting of the catalyst, undergo oxidation by TiO2 holes, leading to the formation of O2. The validation from numerical simulations further supports the consistency between the trend of absorption cross-sections of the core–shell structured carbon-coated hexagonal Mo2C/TiO2 across the sunlight spectrum in simulations and the absorption measured in experiments (Fig. 8E and F). This agreement between simulation and experimental results provides confidence in the accuracy of the proposed model and supports further exploration of the material's photocatalytic properties and potential applications.332
7.5. Photocatalytic hydrogen peroxide production
Hydrogen peroxide (H2O2) is a potent oxidising agent that decomposes into water and oxygen. It is widely used in chemical manufacturing, paper making, textile bleaching and water treatment industries. Currently, H2O2 is primarily produced industrially through the anthraquinone oxidation, which is energy intensive process that generates large amounts of hazardous by-products.333,334 One alternative method is via the direct synthesis from hydrogen gas and oxygen gas in the presence of noble metal or alloy catalysts but the inherent risk of explosions from the combustible hydrogen-oxygen mixture severely limits its use on a larger industrial scale.335–337 A safer and greener method of H2O2 production is to utilise TiO2 as a photocatalyst for reducing water and oxygen into H2O2. However, pristine TiO2 has significant limitations, such as poor light absorption and low H2O2 yield, due to the combination of synthesized H2O2 with hydroxyl groups forming peroxide complexes that decompose H2O2.337,338 To address these issues, various modification strategies have been employed in TiO2-based photocatalytic systems to enhance H2O2 production.For example, Gan et al. synthesized ultrathin C3N5 nanosheets and assembled them on oxygen-deficient TiO2 arrays.339 This coupling created a type-II heterojunction with an internal electric field to drive carrier separation and charge transfer, leading to efficient photocatalytic H2O2 production. The optimal heterojunction achieved the highest H2O2 formation rate of 2.93 μmol L−1 min−1 in a 90% water/10% ethanol mixture, which is about 4.1 times higher than that of the TiO2−x arrays. Similarly, a S-scheme heterojunction photocatalyst was created by coupling TiO2 with three-dimensionally ordered macroporous sulfur-doped graphitic carbon nitride using electrostatic self-assembly.340 This photocatalyst achieved high photocatalytic H2O2 production activity, yielding 2128 μmol h−1 g−1 without the need for hole scavengers.
Another composite material uses Au and MXene as co-catalysts to explore the relationship between the photocatalytic activity of the brookite TiO2/Au/MXene composite under UV light for the formation of hydrogen peroxide.341 This combination effectively improved the electron–hole pair separation and charge transfer rate of brookite TiO2. Under UV light exposure, brookite TiO2 forms electron–hole pairs, with electrons transferring to the conduction bands of Au and MXene. Concurrently, holes oxidize 2-propanol to create protons, while electrons react with O2 to form superoxide anion radicals, ultimately producing H2O2. The composite with 0.4% gold and 15% MXene content was optimal, generating 6.80 mg L−1 H2O2 at pH 3 after 4 hours of UV photo-irradiation. This H2O2 production was 10 times greater than that of brookite TiO2 alone.
Hu et al. synthesized a 2D/1D hierarchical layered ZnIn2S4 modified TiO2 photocatalyst for H2O2 production, which was rapidly activated into hydroxyl radicals for wastewater purification in a Fenton-like cascade reaction.19 This process is driven by a Z-scheme heterojunction mechanism, where photoexcited electrons from TiO2 recombine with holes from ZnIn2S4, leading to the reduction of O2 to H2O2. The ZnIn2S4/TiO2 catalyst achieved an H2O2 evolution rate of 1530.59 μmol g−1 h−1 with a 10.39% external quantum efficiency under 400 nm irradiation. The produced H2O2 was activated by unsaturated sulfur atoms in ZnIn2S4, degrading 90% of tetracycline antibiotics (50 ppm) in wastewater within 1 hour (Fig. 9A).
 | ||
| Fig. 9 (A) Schematic illustration of the preparation of the ZnIn2S4/TiO2 heterojunction for in situ H2O2 production and its activation for water remediation under visible light irradiation. Reproduced with permission from ref. 19. Copyright 2022, Elsevier. (B) Charge carrier transfer mechanism of S-scheme TiO2@BTTA photocatalysts. Reproduced with permission from ref. 342. Copyright 2023, Elsevier. (C) Schematic illustration comparing traditional solid photocatalyst with floatable TiO2/Bi2O3 photocatalyst, highlighting redox reactions occur at the distinct gas-liquid-solid tri-phase interface. Reproduced with permission from ref. 54. Copyright 2022, Wiley-VCH. | ||
Yang et al. developed a composite system comprising TiO2 nanofibers hybridized with porous 4,4,4-(benzene-1,3,5-triyltris(ethyne-2,1-diyl))tribenzaldehyde (BTTA) COF, serving as a dual-function photocatalyst for simultaneous hydrogen peroxide production and selective furfuryl alcohol oxidation. This composite system offers several advantages, including a large surface area, superior light absorption, efficient carrier separation, and enhanced redox power through the S-scheme heterojunction (Fig. 9B). Specifically, the TiO2-BTTA composite with a 6:1 weight ratio exhibited the highest H2O2 production activity at 740 μmol L−1 h−1 and achieved approximately 92% oxidation of furfuryl alcohol.
Going beyond traditional powder photocatalysts, which suffer from severe agglomeration, limited light absorption, poor gas reactant accessibility, and difficulties in reuse, the development of floatable composite photocatalysts presents a promising alternative. These novel floatable composite photocatalysts were synthesized by immobilizing hydrophobic TiO2 and Bi2O3 on lightweight polystyrene spheres using hydrothermal and photodeposition methods.54 The photocatalysts are solar transparent and improve contact between reactants and the photocatalyst. Floatable photocatalysts offer significant advantages, such as the rapid diffusion of oxygen to the solid-water interface, which enhances H2O2 evolution (Fig. 9C). This rapid oxygen delivery facilitates electron consumption, thereby minimizing electron–hole recombination. By combining the benefits of floatable and S-scheme photocatalysts, the TiO2/Bi2O3 composite achieves a significantly enhanced H2O2 yield of 1.15 mM h−1 and an furoic acid formation rate of 0.45 mM h−1. The innovative design maximizes photocatalytic reaction kinetics and provides a new route for efficient heterogeneous catalysis.
7.6. Photocatalytic selective oxidation of alcohol
The selective oxidation of alcohols to produce aldehydes, carboxylic acids, or ketones is an important chemical process in industry.218,343,344 However, the industrial alcohol oxidation processes use strong oxidants which are toxic with hazardous by-products and require energy intensive conditions such as high temperature and pressure. Hence, there is a pressing need to develop environmentally friendly and sustainable processes for alcohol oxidation under ambient conditions. One promising strategy for oxidation of alcohols is through photocatalysis which can be performed under room temperature and atmospheric pressure and taps on renewable solar energy. TiO2 has garnered significant attention as a photocatalyst for alcohol oxidation due to its abundance, low cost, and non-toxicity. However, its large bandgap limits its activity to the UV region, resulting in lower solar energy utilization.Courtois et al. investigated the photoreforming of tertiary alcohols on Pt-loaded rutile TiO2(110).345 The process involves hole-mediated disproportionation, which results in the formation of an alkane and the corresponding ketone. Wolde and co-workers synthesized gadolinium-doped TiO2 nanorods decorated with poly(o-phenylenediamine) nanowires to enhance photocatalytic performance.346 Gadolinium doping increases adsorption capacity, introduces half-filled f-orbitals, and causes an optical redshift. Poly(o-phenylenediamine), a conducting polymer, improves photocatalytic activity in the visible region by acting as an electron donor and hole transporter. The composite was tested for photocatalytic benzyl alcohol oxidation coupled with p-dinitrobenzene reduction under simulated solar light. The best performance was achieved with 10% gadolinium doping, resulting in a 90.6% yield of benzaldehyde from benzyl alcohol and an 89.1% yield of p-nitroaniline from p-dinitrobenzene in acetonitrile, while in water, the yields were 70.5% for benzaldehyde and 85.0% for p-phenylenediamine.
When Ti3C2 MXene is combined with TiO2, it significantly enhances the photocatalytic selective oxidation of alcohols, such as benzyl alcohol. Accordingly, the selective oxidation of benzyl alcohol to benzaldehyde over TiO2 relies on the synergy between electrons and holes. Ti3+ species in TiO2 are crucial for generating alcohol cation radicals and activating molecular O2, which enhances the conversion efficiency of benzyl alcohol.347,348 However, the VB potential of TiO2 (2.9 V vs. RHE) is more positive than the benzaldehyde/oxidized benzaldehyde redox potential (2.5 V vs. RHE), leading to further oxidation of benzaldehyde to byproducts like benzoic acid and CO2, reducing selectivity. Therefore, creating TiO2 with a high concentration of Ti3+ species and a less positive VB could improve both conversion efficiency and selectivity for benzaldehyde during the photocatalytic oxidation of benzyl alcohol. The TiO2/Ti3C2 composite enhances the photocatalytic selective oxidation of alcohols by stabilizing oxygen vacancies and Ti3+ species. This stabilization boosts the production of active intermediates for the conversion of benzyl alcohol to benzaldehyde. The up-shifted valence band of TiO2 in the composite prevents further oxidation of benzaldehyde, increasing selectivity and efficiency.343
Recently, Ti3C2/TiO2 nanocomposite was used for photocatalytic oxidation of biomass-derived alcohols, and other aromatic alcohols to corresponding aldehydes.22 The enhanced photocatalytic activity is attributed to the abundant functional groups on the Ti3C2 MXene and the in situ formation of TiO2 nanoparticles on the MXene sheets. The proposed reaction mechanism involves TiO2 nanoparticles forming a close interface with MXene nanosheets. Under full-spectrum illumination, electrons are excited from the valence band to the conduction band of TiO2, then quickly transferred to the Ti3C2 nanosheets, aiding the separation and prolonging the lifespan of photogenerated carriers. The system facilitates oxidation through photogenerated holes and tert-butoxy radicals, driven by the reduction of adsorbed oxygen. The Ti3C2/TiO2 composite shows higher photocatalytic conversion efficiency and selectivity for furfuryl alcohol oxidation than pristine TiO2, with the optimized photocatalyst achieving a 99% yield of furfural.
Besides MXene, Lu et al. developed core–shell composites composed of TiO2 nanobelts as the core and COFs of varying thicknesses as the shell, which enhanced the reactivity, selectivity, and stability for benzyl alcohol oxidation.21 The COFs, with their narrow bandgap, absorbed visible light and generated electrons and holes in their conduction and valence bands, respectively. Electrons migrated from the COF's conduction band to TiO2's conduction band, while holes facilitated the oxidation of benzyl alcohol to form carbon-centered radicals. These radicals reacted with superoxide radicals, produced by the accumulated electrons activating O2, to form benzaldehyde. The TiO2@COF composite with a 15 nm COF shell demonstrated the highest benzyl alcohol conversion (92.5%) with a maximum rate constant of 6.73 × 10−2 h−1 under visible light (>420 nm), performing 10.1 times better than TiO2 alone and 12.9 times better than COF alone.
7.7. Photochromic systems for electronic devices
A TiO2-based color switching system typically involves the reversible color displaying capability of TiO2, which allow the material to change its color in response to an external stimulus, such as an applied voltage or light irradiation.349–351 A 2D TiO2 layer has been used as the electrochromic layer combining with a MXene layer (e.g., Ti3C2Tx) for flexible transparent electrodes in electrochromic devices.352 To attain the desired TiO2 phase, the anatase TiO2 phase was derived from Ti3C2Tx through annealing in air, which demonstrated with good electrochromic properties. The Ti3C2Tx and TiO2 2D nanosheets were self-assemble into Ti3C2Tx and TiO2 films through liquid/liquid interfacial self-assembly to give a uniform and high coverage layer and transferred consecutively onto a polyethylene terephthalate substrate to form TiO2/Ti3C2Tx heterostructures. The heterostructures demonstrated outstanding electrochromic performance (e.g., fast coloration speed and high coloration efficiency) because of the 2D nature of the TiO2 flakes as well as the well-balanced porosity and loosely networked structures that promote efficient ion diffusion and electron transport. In addition, the 2D network structure also assists in redistributing the induced strain uniformly, enhancing both the mechanical strength and the flexibility of the film, which is important for the application in flexible electrochromic devices.Several reports have demonstrated the photoreversible color-switching systems based on photocatalytic TiO2 nanocrystals to induce a reversible redox reaction of chromic dyes, resulting in changes in color.353,354 Later developments in TiO2-based photoreversible color switching systems focused on improving cyclability and coloring duration through doping and surface engineering.355–357 For instance, binding organic sacrificial electron donors to the surface of TiO2 is beneficial for the stabilization of nanocrystals and removal of photogenerated holes, thereby enabling cycling of the color-switching system until the surface-bound sacrificial donors are exhausted or depleted. On the other hand, the doping-induced lattice distortion in anatase TiO2 nanocrystals promotes efficient charge separation and migration, altering the kinetics of redox reactions, and tuning the material's optical properties. These enhancements enable TiO2 nanocrystals to undergo rapid and reversible changes in color upon exposure to light.355,358
By leveraging on the photocatalytic property and color changing capability of TiO2 nanocrystals under redox reaction (Ti3+ and Ti4+), a photoreversible color-switching paper based on these TiO2 nanocrystals can repeatedly write and erase content in response to UV irradiation and oxygen exposure (Fig. 10A).359 To promote good coloration response in atmospheric conditions, N-doping was introduced in creating large quantity of defects (oxygen vacancies) on the TiO2 surface to improve the chemisorption of polyol groups for scavenging photo-generated holes. In addition, N-doping also induces substantial physisorption of polyol molecules that acts as a protective layer on the surface of TiO2 nanocrystals by limiting the diffusion of ambient oxygen molecules, thereby reducing the rate of oxidation of Ti3+ species back to Ti4+ (Fig. 10B). When exposed to UV irradiation, the TiO2 nanocrystals undergo photoexcitation, generating electrons that induce reduction of Ti4+ to Ti3+ ions in the nanocrystal lattice. This led to a color change from white to black or dark blue at the irradiated regions, allowing for visible writing or marking on the paper surface (Fig. 10C). To erase the writing or markings, the rewritable paper is exposed to oxygen or air, which serves as an oxidizing agent. Oxygen molecules react with the reduced Ti3+ ions in the TiO2 nanocrystals, oxidizing them back to Ti4+ ions and restoring the original white color of the paper. This color-switching systems process is typically rapid, enabling multiple cycles of writing and erasing without degradation of the paper substrate.
 | ||
| Fig. 10 Photochromic function of TiO2 semiconductor. (A) Schematic illustration of the writing process on the rewritable paper using a laser pen and (B) light-printed pattern on rewritable film using a UV lamp. (C) Photoreversible color-switching mechanism of N-doped TiO2 nanocrystals. Reproduced with permission from ref. 359. Copyright 2022, Wiley-VCH. (D) Color-switching process and the corresponding color change of the film under continuous UV irradiation. Reproduced with permission from ref. 356. Copyright 2019, Elsevier. | ||
Combining oxygen vacant TiO2 and reduced graphene oxide enables the realization of four distinct types of photochromic devices, each exhibiting unique properties and functionalities (i.e., multi-wavelength photochromism, intensity-dependent photochromism, time-dependent photochromism, dual-response hydro-/photochromism).356 All of the mentioned photochromic devices can exhibit rapid and reversible color changes through precise control of applied stimuli and demonstrate distinct multi-color states. By carefully engineering the composition and structure of the composite material, the multi-wavelength photochromic device can selectively respond to specific regions of the electromagnetic spectrum. The intensity-dependent photochromic device responds sensitively to variations in the intensity of incident light. In the time-dependent photochromic device, color changes evolve gradually over time in response to continuous light illumination (Fig. 10D). The dual-response hydro-/photochromic device exhibits reversible color changes in response to both light irradiation and variations in humidity. The success of these designs hinge on the development of a highly reductive catalyst as well as the manipulation of the film structure and the dye reduction kinetics to exhibit distinctive and customizable responses to various stimuli, such as light, humidity, or temperature changes. This approach enables the development of versatile and functional photochromic materials with applications across diverse fields, including optoelectronics, advanced sensors, displays, and rewritable media.
7.8. Photocatalytic reactions for plastics: polymerization, degradation and conversion
The ability of semiconductors to generate free radicals upon photoexcitation, allows them to be employed for initiating heterogenous free radical polymerization, which is a commonly used method for synthesizing polymers from vinyl monomers. The radicals are generated via the photodegradation of surrounding organic compounds360 or water when they react with the holes in the VB of TiO2 (refer to section 1). These compounds containing radicals would then serve as the initiators for free radical polymerization to take place. The first photocatalytic surface-Initiated polymerization TiO2 photoinitiated polymerization were reported by Kraeutler et al., for the bulk polymerization methyl methacrylate (MMA).361 Other semiconductors, such as ZnO and CdS, shows similar photoinitiation ability for free radical polymerization.362–364 Apart from PMMA,365–368 polymerization of other vinyl monomers have also been reported including homopolymers such as poly(styrene),362,364 and co-polymers consisting of different repeat units.369 Interestingly, such heterogeneous surface-initiated polymerization technique opens the pathway to fabricate core–shell structures that have a polymer shell encapsulating the inorganic semiconducting particles. For instance, Kong et al. synthesized TiO2 particles embedded within antibacterial poly[2-(tert-butylamino)ethyl methacrylateco-ethylene glycol dimethacrylate] shell.369 Wang et al. synthesized core–shell nanospheres with TiO2 core embedded within PMMA, poly(styrene) or Poly(N-isopropylacrylamide) (PNIPAM) shell, along with tunable spherical structures that can be manipulated through the compatibility between the polymers and TiO2 particles.370 For other non-photoactive particles (e.g., SiO2), a thin layer of TiO2 coating would allow them to achieve similar photocatalytic polymerization ability.370,371On the other hand, the generation of radical by TiO2 can also lead to depolymerization, which reduces the molecular weight of polymers, and hence, resulting in degradation to the physical properties of plastic materials. Polyolefins, the largest class of polymers produced, can be subjected to accelerated degradation by radicals generated by TiO2, despite being amongst the most chemically resistant among plastics.372,373 For instance, the hydroxyl radicals generated by TiO2 can lead to hydrogen abstraction of polyolefins, forming alkyl radicals that causes a series of reaction including chain scissions. Zapata et al. reported about 57–60% reduction in molecular weight of polypropylene with TiO2 nanotubes embedded within as compared to 37% reduction for pure polypropylene, after subjecting to 0.55 W m−2 of 340 nm irradiation for 10 days.374 Similarly, Day reported significant reduction in embrittlement times (defined as 50% reduction in elongation as break) of 100 μm thick polyethylene films embedded with different forms (e.g. polymorphs, coated/uncoated) of TiO2.375 Uncoated anatase appears to the most active, with 20000 mg kg−1 incorporated embedded in polyethylene, a ten times reduction in embrittlement time is observed as compared to the pristine polyethylene. Zhao et al. also reported on enhanced photocatalytic degradation of polyethylene using copper phthalocyanine modified TiO2, leading to faster photodegradation rates.449 On the other hand, TiO2 particles have commonly been incorporated into polyvinyl chloride to prevent UV degradation.16 Their ability for UV absorption can compete with direct UV-induced degradation associated with vinyl polymers, given the bonds energy of C–C (375 kJ mol−1) and C–H (420 kJ mol−1) falls within the range of UV light energies.372 However, they will also induce photocatalytic oxidative degradation with radical generation, leading to the loss of gloss on the polyvinyl chloride surface and the degradation of mechanical properties. Nevertheless, it has been reported that the penetration of UV light reduces by 90% at a depth of 20 μm,376,377 thereby limiting the degradation to a shallow surface, while the bulk of polyvinyl chloride remains largely protected.
Photocatalytic depolymerization also opens up a pathway for plastic recycling, as monomeric building blocks can be recovered for re-polymerization. Such chemical recycling techniques are often less energy-intensive than conventional mechanical method.378–380 In comparison, mechanical recycling methods causes material degradation leading to lower quality and reduced properties of the recycled materials, thereby often being labelled as a “downcycling” process. This is unlike chemical recycling that deconstructs the material at a molecular level, allowing a renewed production of the pristine material. For example, Daraboina and Madras employed TiO2 synthesized by combustion synthesis to perform photocatalytic degradation of PMMA, poly(butyl methacrylate) (PBMA) and their co-polymers, yielding their respective monomers.381 These recovered monomers can serve as the feedstock for the repolymerization of these poly (alkyl methacrylates).382
Similarly, photocatalysis can also be an effective tool to convert plastic materials to other chemical species beyond their monomeric/oligomeric form, allowing them to be repurposed upon reaching their end of life.383–386 Recently, Nguyen and Edalati employed TiO2 for the photoconversion of poly(ethylene terephthalate) (PET) to terephthalate and acetic acid, and the latter is widely used in chemical, plastic and food production.387 They also found brookite polymorph of TiO2 to be most active in this photocoversion process, followed by rutile, and finally anatase. Other than such direct photoconversion of plastics, the polymers also can undergo an initial step of degradation/depolymerization to smaller chemical compound prior to photocatalytic conversion into other useful molecules. For example, Han et al. reported photocatalytic conversion of ethylene glycol derived from alkaline hydrolyzed PET, into glycolic acid, glycolaldehyde, and ethanol, using photocatalytic carbonized polymer dots graphitic carbon nitride.388 Similarly, for TiO2, Bhattacharjee et al. performed enzymatic pretreatments on polyesters plastic prior to photo conversion of PET and polycaprolactone using Pt-loaded TiO2, resulting in the formation of pentanal and formic acid products.56 In cases where the chemical products obtained are of higher value than the initial plastic, such processes would be referred to as an “upcycling” process. For example, using TiO2 modified with potassium stearate or N,N-diethyl-3-(trimethoxysilyl)propan-1-amine, Peng et al. reported the successful photoconversion of poly(styrene) wastes into benzoic acid with 18–44.2 mol% yields under mild conditions.389 Such aromatic compounds would find applications food, cosmetic and pharmaceutical products.
In addition, many of the photoconversion process of plastic materials described above can be conducted with a simultaneous production of hydrogen fuel. This process commonly known as photoreforming first described by Kawai and Sakata in 1981, using Xe-lamp irradiation on platinized TiO2 photocatalyst in the presence of water and chlorine or nitrogen containing organic molecules.37 The difference between photoforming and typical photocatalytic water splitting is the participation of the organics (i.e., polymer or plastic-derived chemicals) as a feedstock for hydrogen production. In photoreforming, the oxidation of the organic compound replaces oxygen evolution reaction for hole consumption, thereby acting as a hole scavenger that supresses the electron–hole recombination, resulting in the increase rate of H+/H2O reduction boosting H2O production as compared to typical photocatalytic water splitting.384,390,391 More importantly, the additional production of clean H2 fuel further adds value to the photocatalytic conversion of plastic materials.
8. Conclusions and perspectives
Photocatalysis is recognized as a promising sustainable technology for solving environmental and energy problems by harnessing solar light. Numerous research attempts in the field of photocatalysis have clearly shown that TiO2 is one of the most extensively studied photocatalysts due to its excellent photocatalytic properties. The continuous progress in TiO2 photocatalyst research is driven by the material's inherent advantages such as chemical stability, non-toxicity, cost-effectiveness, and strong oxidative power under UV light. However, the challenges of narrow light absorption and fast charge recombination have prompted extensive research into TiO2 material engineering. In view of these developments, the emergence of TiO2 photocatalysis and the modification methods of TiO2-based photocatalysts were comprehensively reviewed in this paper.Advancements in engineering TiO2 through doping have extended its activity into the visible light range. However, metal doping faces challenges such as thermal instability, high fabrication costs, and increased charge-carrier recombination, which reduce efficiency. Conversely, non-metal doping is generally more cost-effective and stable but requires careful control to avoid disrupting the lattice structure. By integrating techniques like morphological control, crystal phase manipulation, doping, and hybridization, researchers have improved charge carrier separation and extended the applicability of TiO2 in a wide range of photooxidation and photoreduction reactions. These applications include organic pollutant degradation, photocatalytic disinfection, hydrogen generation, CO2 reduction, nitrogen fixation, hydrogen peroxide generation, alcohol oxidation, plastic polymerization and degradation, and photochromic applications.
Future research in TiO2 photocatalysis is poised to focus on several key areas, particularly the modification of TiO2 materials to enhance their performance. Advances in 3D printing technologies have significantly impacted the development of these materials. 3D printing allows for precise control over the design and fabrication process, enabling the creation of complex structures with tailored porosity and flow characteristics. By building objects layer by layer from digital designs, 3D printing can produce photocatalytic materials with optimized pore sizes and shapes, enhancing the flow of pollutants and improving contact with the photocatalyst surface. This precision in design is expected to lead to significant improvements in the efficiency and effectiveness of photocatalytic materials.
Another promising area of future research is the development of single-atom catalysts. These catalysts represent the ultimate small-size limit for metal particles, containing isolated metal atoms singly dispersed on supports. Single-atom catalysts maximize the efficiency of metal atom use, which is particularly important for supported noble metal catalysts. The well-defined and uniform dispersion of single atoms offers great potential for achieving high catalytic activity and selectivity. By utilizing each metal atom to its fullest potential, single-atom catalysts can provide exceptional performance in various catalytic applications, including photocatalysis.
Photocatalytic upcycling and depolymerization of polymers have emerged as promising strategies to address plastic pollution and advance a circular economy. Recycling polymers with backbone structures primarily composed of C–C bonds is particularly challenging due to the lack of functional groups, making these materials difficult to break down and reuse. Consequently, the valorization of common commodity plastics such as polyethylene (PE) and polypropylene (PP), which are major contributors to the plastic pollution problem, is a crucial next step.
Traditional studies of TiO2 photocatalysis have used static ensemble-averaged methods, providing valuable but limited insights into fundamental processes. Future research will utilize advanced in situ and time-resolved techniques to capture real-time dynamic processes, such as ultrafast electron dynamics and charge carrier lifetimes, revealing key mechanisms like charge separation and transfer. For example, in situ extended X-ray absorption fine structure has been used to study the dynamic changes in chemical valence and coordination environment of isolated metal centers in a multi-edged TiO2-supported single-atom Ru photocatalyst. Additionally, femtosecond time-resolved, surface-specific vibrational sum frequency generation spectroscopy has investigated the photo-induced reaction at the TiO2–water interface, observing the interfacial water molecule reactions with high temporal precision. In situ mass spectrometry will further aid in analyzing gaseous products and intermediates, elucidating reaction pathways and mechanisms.
Future research in TiO2 photocatalysis will increasingly rely on theoretical calculations, such as density functional theory, molecular dynamics simulations, and kinetic modeling, to complement experimental techniques. These computational methods will model electronic structures, predict reactivity, and develop comprehensive reaction models, providing a holistic understanding of photocatalytic mechanisms. This integration will enable the optimization and development of more efficient and tailored photocatalysts for various applications. Currently, research on TiO2 photocatalysts largely depends on experimental work, which can be complex and costly. Moving forward, the incorporation of machine learning, big data, and artificial intelligence holds significant potential to generate high-performance models that predict material properties and elucidate property–structure relationships. These advanced computational tools can streamline the research process through high-throughput screening, reduce experimental complexity, and lower costs. This approach will not only enhance our understanding of TiO2-based photocatalysts but also drive the development of more effective and efficient materials, advancing their practical applications on an industrial scale.
Author contributions
Conceptualization and writing – original draft: S. Y. Tee, J. Kong, J. J. Koh, C. P. Teng, X. Wang, X. Wang, S. L. Teo. Conceptualization, supervision and writing – review & editing: W. Thitsartarn, M-Y Han, and Z. W. Seh. All authors have read and agreed to the published version of the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is supported by the National Research Foundation, Singapore, and Agency for Science, Technology and Research (A*STAR), under its low carbon energy research (LCER) phase 2 HETFI Directed Hydrogen programme (Grant No.: U2307D4001), the A*STAR SERC MTC YIRG (Grant No.: M21K3c0125), and the A*STAR IMRE–SCG Chemicals Advanced Composite Joint Lab (IAF-ICP Project No.: I1801E0024).References
- P. Riente and T. Noël, Catal. Sci. Technol., 2019, 9, 5186–5232 RSC.
- C. Larquet and S. Carenco, Front. Chem., 2020, 8, 179 CrossRef CAS PubMed.
- H. Hao and X. Lang, ChemCatChem, 2019, 11, 1378–1393 CrossRef CAS.
- M. Ahmed and G. Xinxin, Inorg. Chem. Front., 2016, 3, 578–590 RSC.
- D. Sudha and P. Sivakumar, Chem. Eng. Process.: Process Intensif., 2015, 97, 112–133 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- Y. Tanaka, S. L. Lim, W. P. Goh, C. Jiang, S. Y. Tee, T. Ye, X. Li, K. H. Nguyen, C. J. J. Lee, N. Ding, Z. Liu, J. Wu, J. Zhang and M. Y. Han, ChemNanoMat, 2018, 4, 394–400 CrossRef CAS.
- C. P. Teng, M. Y. Tan, J. P. W. Toh, Q. F. Lim, X. Wang, D. Ponsford, E. M. J. Lin, W. Thitsartarn and S. Y. Tee, Materials, 2023, 16, 3856 CrossRef CAS PubMed.
- S. Banerjee, D. D. Dionysiou and S. C. Pillai, Appl. Catal., B, 2015, 176–177, 396–428 CrossRef CAS.
- P. V. Laxma Reddy, B. Kavitha, P. A. Kumar Reddy and K. H. Kim, Environ. Res., 2017, 154, 296–303 CrossRef CAS PubMed.
- S. Zhang, J. Zhang, J. Sun and Z. Tang, Chem. Eng. Process.: Process Intensif., 2020, 147, 107746 CrossRef CAS.
- K. Y. Tang, J. X. Chen, E. D. R. Legaspi, C. Owh, M. Lin, I. S. Y. Tee, D. Kai, X. J. Loh, Z. Li, M. D. Regulacio and E. Ye, Chemosphere, 2021, 265, 129114 CrossRef CAS PubMed.
- Z. W. Seh, S. Liu, M. Low, S. Y. Zhang, Z. Liu, A. Mlayah and M. Y. Han, Adv. Mater., 2012, 24, 2310–2314 CrossRef CAS PubMed.
- S. Y. Tee, K. Y. Win, W. S. Teo, L. D. Koh, S. Liu, C. P. Teng and M. Y. Han, Adv. Sci., 2017, 4, 1600337 CrossRef PubMed.
- S. Jiang, K. Zhao, M. Al-Mamun, Y. L. Zhong, P. Liu, H. Yin, L. Jiang, S. Lowe, J. Qi, R. Yu, D. Wang and H. Zhao, Inorg. Chem. Front., 2019, 6, 1667–1674 RSC.
- J. Wang, R. T. Guo, Z. X. Bi, X. Chen, X. Hu and W. G. Pan, Nanoscale, 2022, 14, 11512–11528 RSC.
- X. Chen, N. Li, Z. Kong, W.-J. Ong and X. Zhao, Mater. Horiz., 2018, 5, 9–27 RSC.
- M. Cheng, C. Xiao and Y. Xie, J. Mater. Chem. A, 2019, 7, 19616–19633 RSC.
- J. Hu, T. Yang, J. Chen, X. Yang, J. Qu and Y. Cai, J. Chem. Eng., 2022, 430, 133039 CrossRef CAS.
- Z. Chen, D. Yao, C. Chu and S. Mao, Chem. Eng. J., 2023, 451, 138489 CrossRef CAS.
- G. Lu, X. Huang, Z. Wu, Y. Li, L. Xing, H. Gao, W. Dong and G. Wang, Appl. Surf. Sci., 2019, 493, 551–560 CrossRef CAS.
- I. R. Warkad, R. Paul, S. Parthiban and M. B. Gawande, J. Environ. Chem. Eng., 2024, 12, 113128 CrossRef CAS.
- W. J. Ong, L. L. Tan, S. P. Chai, S. T. Yong and A. R. Mohamed, Nanoscale, 2014, 6, 1946–2008 RSC.
- A. Bumajdad and M. Madkour, Phys. Chem. Chem. Phys., 2014, 16, 7146–7158 RSC.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- E. Roduner, Chem. Soc. Rev., 2006, 35, 583–592 RSC.
- M. Valden, Science, 1998, 281, 1647–1650 CrossRef CAS PubMed.
- X. Wang, Z. Li, J. Shi and Y. Yu, Chem. Rev., 2014, 114, 9346–9384 CrossRef CAS PubMed.
- S. Y. Tee and E. Ye, Mater. Adv., 2021, 2, 1507–1529 RSC.
- J. Wang, Z. Wang, W. Wang, Y. Wang, X. Hu, J. Liu, X. Gong, W. Miao, L. Ding, X. Li and J. Tang, Nanoscale, 2022, 14, 6709–6734 RSC.
- X.-X. Fang, L.-B. Ma, K. Liang, S.-J. Zhao, Y.-F. Jiang, C. Ling, T. Zhao, T.-Y. Cheang and A.-W. Xu, J. Mater. Chem. A, 2019, 7, 11506–11512 RSC.
- Z. Ding, M. Sun, W. Liu, W. Sun, X. Meng and Y. Zheng, Sep. Purif. Technol., 2021, 276, 119287 CrossRef CAS.
- D. Yu, Q. Shao, Q. Song, J. Cui, Y. Zhang, B. Wu, L. Ge, Y. Wang, Y. Zhang, Y. Qin, R. Vajtai, P. M. Ajayan, H. Wang, T. Xu and Y. Wu, Nat. Commun., 2020, 11, 927 CrossRef CAS PubMed.
- E. Abraham, V. Cherpak, B. Senyuk, J. B. ten Hove, T. Lee, Q. Liu and I. I. Smalyukh, Nat. Energy, 2023, 8, 381–396 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- A. J. Bard, J. Photochem., 1979, 10, 59–75 CrossRef CAS.
- T. Kawai and T. Sakata, Chem. Lett., 1981, 10, 81–84 CrossRef.
- N. Serpone, E. Borgarello and M. Grätzel, J. Chem. Soc., Chem. Commun., 1984, 342–344 RSC.
- C. Kormann, D. W. Bahnemann and M. R. Hoffmann, Environ. Sci. Technol., 1988, 22, 798–806 CrossRef CAS PubMed.
- J. C. D'Oliveira, G. Al-Sayyed and P. Pichat, Environ. Sci. Technol., 2002, 24, 990–996 CrossRef.
- N. N. Rao, S. Dube, Manjubala and P. Natarajan, Appl. Catal., B, 1994, 5, 33–42 CrossRef CAS.
- K. Naoi, Y. Ohko and T. Tatsuma, J. Am. Chem. Soc., 2004, 126, 3664–3668 CrossRef CAS PubMed.
- H. Tada, T. Mitsui, T. Kiyonaga, T. Akita and K. Tanaka, Nat. Mater., 2006, 5, 782–786 CrossRef CAS PubMed.
- W. Ren, Z. Ai, F. Jia, L. Zhang, X. Fan and Z. Zou, Appl. Catal., B, 2007, 69, 138–144 CrossRef CAS.
- M. Zhang, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2008, 47, 9730–9733 CrossRef CAS PubMed.
- D. Zhang, G. Li, X. Yang and J. C. Yu, Chem. Commun., 2009, 4381–4383 RSC.
- J. Yu, S. Wang, J. Low and W. Xiao, Phys. Chem. Chem. Phys., 2013, 15, 16883–16890 RSC.
- S. Zhou, Y. Liu, J. Li, Y. Wang, G. Jiang, Z. Zhao, D. Wang, A. Duan, J. Liu and Y. Wei, Appl. Catal., B, 2014, 158–159, 20–29 CrossRef CAS.
- Q. Liu, Q. Cao, H. Bi, C. Liang, K. Yuan, W. She, Y. Yang and R. Che, Adv. Mater., 2016, 28, 486–490 CrossRef CAS PubMed.
- J. Wan, W. Chen, C. Jia, L. Zheng, J. Dong, X. Zheng, Y. Wang, W. Yan, C. Chen, Q. Peng, D. Wang and Y. Li, Adv. Mater., 2018, 30, 1705369 CrossRef PubMed.
- J. Fu, Q. Xu, J. Low, C. Jiang and J. Yu, Appl. Catal., B, 2019, 243, 556–565 CrossRef CAS.
- Y. Zhao, Y. Zhao, R. Shi, B. Wang, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2019, 31, e1806482 CrossRef PubMed.
- H. Zhang, S. Zuo, M. Qiu, S. Wang, Y. Zhang, J. Zhang and X. W. D. Lou, Sci. Adv., 2020, 6, eabb9823 CrossRef CAS PubMed.
- B. He, Z. Wang, P. Xiao, T. Chen, J. Yu and L. Zhang, Adv. Mater., 2022, 34, e2203225 CrossRef PubMed.
- L. Liccardo, M. Bordin, P. M. Sheverdyaeva, M. Belli, P. Moras, A. Vomiero and E. Moretti, Adv. Funct. Mater., 2023, 33, 2212486 CrossRef CAS.
- S. Bhattacharjee, C. Guo, E. Lam, J. M. Holstein, M. Rangel Pereira, C. M. Pichler, C. Pornrungroj, M. Rahaman, T. Uekert, F. Hollfelder and E. Reisner, J. Am. Chem. Soc., 2023, 145, 20355–20364 CrossRef CAS PubMed.
- Z. Warren, T. T. Guaraldo, I. Barisic, G. A. Zoumpouli, J. Wenk and D. Mattia, J. Mater. Chem. A, 2024, 12, 10913–10922 RSC.
- X. Qin, M. Xu, J. Guan, L. Feng, Y. Xu, L. Zheng, M. Wang, J.-W. Zhao, J.-L. Chen, J. Zhang, J. Xie, Z. Yu, R. Zhang, X. Li, X. Liu, J.-X. Liu, J. Zheng and D. Ma, Nat. Energy, 2024, 9, 154–162 CrossRef CAS.
- Z. Wei, L. Wu, X. Yue, H. Mu, Z. Li, Y. Chang, M. Janczarek, S. Juodkazis and E. Kowalska, Appl. Catal., B, 2024, 345, 123654 CrossRef CAS.
- D. A. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2010, 46, 855–874 CrossRef.
- D. R. Eddy, G. A. Nur Sheha, M. D. Permana, N. Saito, T. Takei, N. Kumada, Irkham, I. Rahayu, I. Abe, Y. Sekine, T. Oyumi and Y. Izumi, Chemosphere, 2024, 351, 141206 CrossRef CAS PubMed.
- C. Chen, W. Ma and J. Zhao, Chem. Soc. Rev., 2010, 39, 4206–4219 RSC.
- W. Wang, J. C. Yu, D. Xia, P. K. Wong and Y. Li, Environ. Sci. Technol., 2013, 47, 8724–8732 CrossRef CAS PubMed.
- J. Ran, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed.
- P. S. Basavarajappa, S. B. Patil, N. Ganganagappa, K. R. Reddy, A. V. Raghu and C. V. Reddy, Int. J. Hydrogen Energy, 2020, 45, 7764–7778 CrossRef CAS.
- M. Sultana, A. Mondal, S. Islam, M. A. Khatun, M. H. Rahaman, A. K. Chakraborty, M. S. Rahman, M. M. Rahman and A. S. M. Nur, Curr. Res. Green Sustainable Chem., 2023, 7, 100383 CrossRef CAS.
- F. Zou, J. Hu, W. Miao, Y. Shen, J. Ding and X. Jing, ACS Omega, 2020, 5, 28510–28516 CrossRef CAS PubMed.
- M. E. Kurtoglu, T. Longenbach and Y. Gogotsi, Int. J. Appl. Glass Sci., 2011, 2, 108–116 CrossRef CAS.
- E. Aubry, M. N. Ghazzal, V. Demange, N. Chaoui, D. Robert and A. Billard, Surf. Coat. Technol., 2007, 201, 7706–7712 CrossRef CAS.
- Y. Bessekhouad, D. Robert, J. V. Weber and N. Chaoui, J. Photochem. Photobiol., A, 2004, 167, 49–57 CrossRef CAS.
- G. Yang, Z. Yan, T. Xiao and B. Yang, J. Alloys Compd., 2013, 580, 15–22 CrossRef CAS.
- I. Singh and B. Birajdar, RSC Adv., 2017, 7, 54053–54062 RSC.
- H. Rahman, A. Norbert, P. S. Nair, J. A. Joseph, S. Shaji, U. Deshpande, J. Naduvath, A. Shanu and R. R. Philip, Opt. Mater., 2022, 134, 113172 CrossRef CAS.
- E. Shin, S. Jin, J. Kim, S.-J. Chang, B.-H. Jun, K.-W. Park and J. Hong, Appl. Surf. Sci., 2016, 379, 33–38 CrossRef CAS.
- Z. N. Kayani, A. Iqbal, Z. Bashir, S. Riaz and S. Naseem, Inorg. Chem. Commun., 2023, 151, 110560 CrossRef CAS.
- Z. Ma, X. Ma, L. Yang, X. Liu, Z. Zhong and B. Hou, Appl. Surf. Sci., 2022, 580, 152274 CrossRef CAS.
- L. Jia, L.-M. Yang, W. Wang, S.-T. Huang and Z. Xu, Rare Met., 2019, 43, 555–561 CrossRef.
- B. Roose, S. Pathak and U. Steiner, Chem. Soc. Rev., 2015, 44, 8326–8349 RSC.
- D. Fang, F. He and J. Xie, J. Energy Inst., 2019, 92, 319–331 CrossRef CAS.
- K. Athira, K. T. Merin, T. Raguram and K. S. Rajni, Mater. Today: Proc., 2020, 33, 2321–2327 CAS.
- M. Ikram, M. A. Ul Haq, A. Haider, J. Haider, A. Ul-Hamid, I. Shahzadi, M. A. Bari, S. Ali, S. Goumri-Said and M. B. Kanoun, Nanoscale Adv., 2022, 4, 3996–4008 RSC.
- S. P. Keerthana, R. Yuvakkumar, G. Ravi, M. Thambidurai, H. D. Nguyen and D. Velauthapillai, RSC Adv., 2023, 13, 18779–18787 RSC.
- E. Santos, A. C. Catto, A. F. Peterline and W. Avansi Jr, Appl. Surf. Sci., 2022, 579, 152146 CrossRef CAS.
- D. Zhang, J. Chen, Q. Xiang, Y. Li, M. Liu and Y. Liao, Inorg. Chem., 2019, 58, 12511–12515 CrossRef CAS PubMed.
- L. Huang, G. He, Y. Yuan, T. C. Zhang, Y. Wang and S. Yuan, Ind. Eng. Chem. Res., 2024, 63, 7154–7165 CAS.
- S.-m. Chang and W.-s. Liu, Appl. Catal., B, 2014, 156–157, 466–475 CrossRef CAS.
- J. Poostforooshan, S. Belbekhouche, V. Olszok, M. F. B. Stodt, M. Simmler, M. Bierwirth, H. Nirschl, J. Kiefer, U. Fritsching and A. P. Weber, ACS Appl. Nano Mater., 2023, 6, 22660–22672 CrossRef CAS.
- V. Moradi, M. B. G. Jun, A. Blackburn and R. A. Herring, Appl. Surf. Sci., 2018, 427, 791–799 CrossRef CAS.
- L. Song, X. Zhao, L. Cao, J. W. Moon, B. Gu and W. Wang, Nanoscale, 2015, 7, 16695–16703 RSC.
- R. Zhu, H. Jiang, Y. Xie, D. Xu, Y. Duo, Y. Zhu, Y. Mei and D. Xie, Prog. Org. Coat., 2024, 189, 108272 CrossRef CAS.
- J. Liang, J. Wang, K. Song, X. Wang, K. Yu and C. Liang, J. Rare Earths, 2020, 38, 148–156 CrossRef CAS.
- S. Stojadinović, N. Tadić, N. Radić, B. Grbić and R. Vasilić, Surf. Coat. Technol., 2018, 337, 279–289 CrossRef.
- J. Reszczyńska, T. Grzyb, J. W. Sobczak, W. Lisowski, M. Gazda, B. Ohtani and A. Zaleska, Appl. Surf. Sci., 2014, 307, 333–345 CrossRef.
- J. Reszczyńska, T. Grzyb, J. W. Sobczak, W. Lisowski, M. Gazda, B. Ohtani and A. Zaleska, Appl. Catal., B, 2015, 163, 40–49 CrossRef.
- J. Reszczyńska, T. Grzyb, Z. Wei, M. Klein, E. Kowalska, B. Ohtani and A. Zaleska-Medynska, Appl. Catal., B, 2016, 181, 825–837 CrossRef.
- V. Đorđević, B. Milićević and M. D. Dramićanin, Rare Earth-Doped Anatase TiO2 Nanoparticles, IntechOpen Inc., 2017 Search PubMed.
- H. Dong, G. Zeng, L. Tang, C. Fan, C. Zhang, X. He and Y. He, Water Res., 2015, 79, 128–146 CrossRef CAS PubMed.
- J. P. Jeon, D. H. Kweon, B. J. Jang, M. J. Ju and J. B. Baek, Adv. Sustainable Syst., 2020, 4, 2000197 CrossRef CAS.
- P. Niu, G. Wu, P. Chen, H. Zheng, Q. Cao and H. Jiang, Front. Chem., 2020, 8, 172 CrossRef CAS PubMed.
- A. Zaleska, E. Grabowska, J. W. Sobczak, M. Gazda and J. Hupka, Appl. Catal., B, 2009, 89, 469–475 CrossRef CAS.
- M. Quesada-Gonzalez, N. D. Boscher, C. J. Carmalt and I. P. Parkin, ACS Appl. Mater. Interfaces, 2016, 8, 25024–25029 CrossRef CAS PubMed.
- Y. Cong, X. Li, Y. Qin, Z. Dong, G. Yuan, Z. Cui and X. Lai, Appl. Catal., B, 2011, 107, 128–134 CrossRef CAS.
- C. Liu, H. Chen, K. Dai, A. Xue, H. Chen and Q. Huang, Mater. Res. Bull., 2013, 48, 1499–1505 CrossRef CAS.
- J. Zhang, M. Vasei, Y. Sang, H. Liu and J. P. Claverie, ACS Appl. Mater. Interfaces, 2016, 8, 1903–1912 CrossRef CAS PubMed.
- A. Trapalis, N. Todorova, T. Giannakopoulou, N. Boukos, T. Speliotis, D. Dimotikali and J. Yu, Appl. Catal., B, 2016, 180, 637–647 CrossRef CAS.
- D. He, Y. Li, I. Wang, J. Wu, Y. Yang and Q. An, Appl. Surf. Sci., 2017, 391, 318–325 CrossRef CAS.
- M. A. Mohamed, W. N. Wan Salleh, J. Jaafar, M. S. Rosmi, Z. A. Mohd Hir, M. Abd Mutalib, A. F. Ismail and M. Tanemura, Appl. Surf. Sci., 2017, 393, 46–59 CrossRef CAS.
- B. Tang, H. Chen, H. Peng, Z. Wang and W. Huang, Nanomaterials, 2018, 8, 105 CrossRef PubMed.
- Y. Duan, X. Chen, X. Zhang, W. Xiang and C. Wu, Solid State Sci., 2018, 86, 12–18 CrossRef CAS.
- Y.-T. Lin, C.-H. Weng, Y.-H. Lin, C.-C. Shiesh and F.-Y. Chen, Sep. Purif. Technol., 2013, 116, 114–123 CrossRef CAS.
- S. Yu, H. J. Yun, Y. H. Kim and J. Yi, Appl. Catal., B, 2014, 144, 893–899 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- S. A. Ansari, M. M. Khan, M. O. Ansari and M. H. Cho, New J. Chem., 2016, 40, 3000–3009 RSC.
- C. Li, M. Gu, M. Gao, K. Liu, X. Zhao, N. Cao, J. Feng, Y. Ren, T. Wei and M. Zhang, J. Colloid Interface Sci., 2022, 609, 341–352 CrossRef CAS PubMed.
- L. Hu, J. Wang, J. Zhang, Q. Zhang and Z. Liu, RSC Adv., 2014, 4, 420–427 RSC.
- L. Huang, W. Fu, X. Fu, B. Zong, H. Liu, H. Bala, X. Wang, G. Sun, J. Cao and Z. Zhang, Mater. Lett., 2017, 209, 585–588 CrossRef CAS.
- P. H. Le, L. T. Hieu, T. N. Lam, N. T. N. Hang, N. V. Truong, L. T. C. Tuyen, P. T. Phong and J. Leu, Micromachines, 2018, 9, 618 CrossRef PubMed.
- J. Wang, C. Fan, Z. Ren, X. Fu, G. Qian and Z. Wang, Dalton Trans., 2014, 43, 13783–13791 RSC.
- S. Samangsri, S. Chiarakorn and T. Areerob, IOP Conf. Ser.: Mater. Sci. Eng., 2019, 576, 012033 CAS.
- H. Wu, D. Yang, X. Zhu, P. Gu, H. Sun, P. Wangyang, J. Li, X. He and L. Fan, CrystEngComm, 2018, 20, 4133–4140 RSC.
- Y.-C. Tang, X.-H. Huang, H.-Q. Yu and L.-H. Tang, Int. J. Photoenergy, 2012, 2012, 1–10 Search PubMed.
- R. Quesada-Cabrera, C. Sotelo-Vázquez, M. Quesada-González, E. P. Melián, N. Chadwick and I. P. Parkin, J. Photochem. Photobiol., A, 2017, 333, 49–55 CrossRef CAS.
- C. W. Dunnill and I. P. Parkin, Dalton Trans., 2011, 40, 1635–1640 RSC.
- F. Xiao, W. Zhou, B. Sun, H. Li, P. Qiao, L. Ren, X. Zhao and H. Fu, Sci. China Mater., 2018, 61, 822–830 CrossRef CAS.
- C. Foo, Y. Li, K. Lebedev, T. Chen, S. Day, C. Tang and S. C. E. Tsang, Nat. Commun., 2021, 12, 661 CrossRef CAS PubMed.
- K. Wang, J. Yu, L. Liu, L. Hou and F. Jin, Ceram. Int., 2016, 42, 16405–16411 CrossRef CAS.
- L. Körösi, S. Papp, I. Bertóti and I. Dékány, Chem. Mater., 2007, 19, 4811–4819 CrossRef.
- X. Fan, T. Yu, Y. Wang, J. Zheng, L. Gao, Z. Li, J. Ye and Z. Zou, Appl. Surf. Sci., 2008, 254, 5191–5198 CrossRef CAS.
- A. V. Akimov, A. J. Neukirch and O. V. Prezhdo, Chem. Rev., 2013, 113, 4496–4565 CrossRef CAS PubMed.
- N. O. Gopal, H.-H. Lo, T.-F. Ke, C.-H. Lee, C.-C. Chou, J.-D. Wu, S.-C. Sheu and S.-C. Ke, J. Phys. Chem. C, 2012, 116, 16191–16197 CrossRef CAS.
- S. A. Ansari and M. H. Cho, Sci. Rep., 2016, 6, 25405 CrossRef CAS PubMed.
- T. Ohno, M. Akiyoshi, T. Umebayashi, K. Asai, T. Mitsui and M. Matsumura, Appl. Catal., A, 2004, 265, 115–121 CrossRef CAS.
- T. Umebayashi, T. Yamaki, H. Itoh and K. Asai, Appl. Phys. Lett., 2002, 81, 454–456 CrossRef CAS.
- F. Wang, F. Li, L. Zhang, H. Zeng, Y. Sun, S. Zhang and X. Xu, Mater. Res. Bull., 2017, 87, 20–26 CrossRef CAS.
- V. V. Pillai, S. P. Lonkar and S. M. Alhassan, ACS Omega, 2020, 5, 7969–7978 CrossRef CAS PubMed.
- C. Yi, Q. Liao, W. Deng, Y. Huang, J. Mao, B. Zhang and G. Wu, Sci. Total Environ., 2019, 684, 527–536 CrossRef CAS PubMed.
- T. Boningari, S. N. R. Inturi, M. Suidan and P. G. Smirniotis, J. Chem. Eng., 2018, 339, 249–258 CrossRef CAS.
- S. A. Bakar and C. Ribeiro, J. Mol. Catal. A: Chem., 2016, 421, 1–15 CrossRef.
- Y. Niu, M. Xing, B. Tian and J. Zhang, Appl. Catal., B, 2012, 115–116, 253–260 CrossRef CAS.
- D. Ma, Y. Xin, M. Gao and J. Wu, Appl. Catal., B, 2014, 147, 49–57 CrossRef CAS.
- W. Q. Fang, X. L. Wang, H. Zhang, Y. Jia, Z. Huo, Z. Li, H. Zhao, H. G. Yang and X. Yao, J. Mater. Chem. A, 2014, 2, 3513 RSC.
- M. Bellardita, C. Garlisi, A. M. Venezia, G. Palmisano and L. Palmisano, Catal. Sci. Technol., 2018, 8, 1606–1620 RSC.
- X. Kang, X. Z. Song, Y. Han, J. Cao and Z. Tan, Sci. Rep., 2018, 8, 5904 CrossRef PubMed.
- Q. Wang, B. Rhimi, H. Wang and C. Wang, Appl. Surf. Sci., 2020, 530, 147286 CrossRef CAS.
- F. Zuo, L. Wang, T. Wu, Z. Zhang, D. Borchardt and P. Feng, J. Am. Chem. Soc., 2010, 132, 11856–11857 CrossRef CAS PubMed.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed.
- A. S. Ichimura, B. Mack, S. M. Usmani and D. Mars, Chem. Mater., 2012, 24, 2324–2329 CrossRef CAS PubMed.
- X. Liu, G. Du and M. Li, ACS Omega, 2019, 4, 14902–14912 CrossRef CAS PubMed.
- G. Liu, H. G. Yang, J. Pan, Y. Q. Yang, G. Q. Lu and H. M. Cheng, Chem. Rev., 2014, 114, 9559–9612 CrossRef CAS PubMed.
- Y. Cao, Q. Li, C. Li, J. Li and J. Yang, Appl. Catal., B, 2016, 198, 378–388 CrossRef CAS.
- Y. Dong, M. Kapilashrami, Y. Zhang and J. Guo, CrystEngComm, 2013, 15, 10657 RSC.
- S. Sarina, E. R. Waclawik and H. Zhu, Green Chem., 2013, 15, 1814 RSC.
- X. C. Ma, Y. Dai, L. Yu and B. B. Huang, Light: Sci. Appl., 2016, 5, e16017 CrossRef CAS PubMed.
- X. Zhang, Y. L. Chen, R. S. Liu and D. P. Tsai, Rep. Prog. Phys., 2013, 76, 046401 CrossRef PubMed.
- S. T. Kochuveedu, Y. H. Jang and D. H. Kim, Chem. Soc. Rev., 2013, 42, 8467–8493 RSC.
- K. Awazu, M. Fujimaki, C. Rockstuhl, J. Tominaga, H. Murakami, Y. Ohki, N. Yoshida and T. Watanabe, J. Am. Chem. Soc., 2008, 130, 1676–1680 CrossRef CAS PubMed.
- R. Asapu, N. Claes, S. Bals, S. Denys, C. Detavernier, S. Lenaerts and S. W. Verbruggen, Appl. Catal., B, 2017, 200, 31–38 CrossRef CAS.
- H. Chakhtouna, H. Benzeid, N. Zari, A. E. K. Qaiss and R. Bouhfid, Environ. Sci. Pollut. Res. Int., 2021, 28, 44638–44666 CrossRef CAS PubMed.
- Z. Wei, L. Rosa, K. Wang, M. Endo, S. Juodkazis, B. Ohtani and E. Kowalska, Appl. Catal., B, 2017, 206, 393–405 CrossRef CAS PubMed.
- R. Asapu, N. Claes, R.-G. Ciocarlan, M. Minjauw, C. Detavernier, P. Cool, S. Bals and S. W. Verbruggen, ACS Appl. Nano Mater., 2019, 2, 4067–4074 CrossRef CAS.
- S. Liu, M. D. Regulacio, S. Y. Tee, Y. W. Khin, C. P. Teng, L. D. Koh, G. Guan and M. Y. Han, Chem. Rec., 2016, 16, 1965–1990 CrossRef CAS PubMed.
- R. Kaur and B. Pal, J. Mol. Catal. A: Chem., 2012, 355, 39–43 CrossRef CAS.
- D. Tsukamoto, Y. Shiraishi, Y. Sugano, S. Ichikawa, S. Tanaka and T. Hirai, J. Am. Chem. Soc., 2012, 134, 6309–6315 CrossRef CAS PubMed.
- Š. Slapničar, G. Žerjav, J. Zavašnik, M. Finšgar and A. Pintar, J. Environ. Chem. Eng., 2023, 11, 109835 CrossRef.
- Z. Guo, G. Yu, Z. Zhang, Y. Han, G. Guan, W. Yang and M. Y. Han, Adv. Mater., 2023, 35, e2206700 CrossRef PubMed.
- T. Akiyama, H. Nagakawa and T. Tatsuma, Phys. Chem. Chem. Phys., 2023, 25, 9031–9035 RSC.
- M. Gao, L. Zhu, W. L. Ong, J. Wang and G. W. Ho, Catal. Sci. Technol., 2015, 5, 4703–4726 RSC.
- N. Celebi, M. Y. Aydin, F. Soysal, Y. O. Ciftci and K. Salimi, J. Alloys Compd., 2021, 860, 157908 CrossRef CAS.
- J. Li, L. Zu, Y. Li, C. Jin, Y. Qin, D. Shi and J. Yang, J. Colloid Interface Sci., 2014, 426, 90–98 CrossRef CAS PubMed.
- S. Zhao, H.-X. Liu, Y. Qiu, S.-Q. Liu, J.-X. Diao, C.-R. Chang, R. Si and X.-H. Guo, J. Mater. Chem. A, 2020, 8, 6586–6596 RSC.
- Z. W. Seh, S. Liu, S. Y. Zhang, M. S. Bharathi, H. Ramanarayan, M. Low, K. W. Shah, Y. W. Zhang and M. Y. Han, Angew. Chem., Int. Ed., 2011, 50, 10140–10143 CrossRef CAS PubMed.
- A. Zada, P. Muhammad, W. Ahmad, Z. Hussain, S. Ali, M. Khan, Q. Khan and M. Maqbool, Adv. Funct. Mater., 2019, 30, 1906744 CrossRef.
- J. Yang, X. Chen, X. Yang and J. Y. Ying, Energy Environ. Sci., 2012, 5, 8976 RSC.
- G. Guisbiers, S. Mejia-Rosales, S. Khanal, F. Ruiz-Zepeda, R. L. Whetten and M. Jose-Yacaman, Nano Lett., 2014, 14, 6718–6726 CrossRef CAS PubMed.
- G. Darabdhara, B. Sharma, M. R. Das, R. Boukherroub and S. Szunerits, Sens. Actuators, B, 2017, 238, 842–851 CrossRef CAS.
- S. Kunwar, P. Pandey and J. Lee, ACS Omega, 2019, 4, 17340–17351 CrossRef CAS PubMed.
- M. Sui, S. Kunwar, P. Pandey and J. Lee, Sci. Rep., 2019, 9, 16582 CrossRef PubMed.
- H. Yu, Y. Peng, Y. Yang and Z.-Y. Li, npj Comput. Mater., 2019, 5, 45 CrossRef.
- S. W. Verbruggen, M. Keulemans, M. Filippousi, D. Flahaut, G. Van Tendeloo, S. Lacombe, J. A. Martens and S. Lenaerts, Appl. Catal., B, 2014, 156–157, 116–121 CrossRef CAS.
- M. Zhu, Y. Wang, Y. H. Deng, X. Peng, X. Wang, H. Yuan, Z. J. Yang, Y. Wang and H. Wang, Nanoscale, 2020, 12, 7035–7044 RSC.
- P. Reñones, L. Collado, A. Iglesias-Juez, F. E. Oropeza, F. Fresno and V. A. de la Peña O'Shea, Ind. Eng. Chem. Res., 2020, 59, 9440–9450 CrossRef.
- D. Tsukamoto, A. Shiro, Y. Shiraishi, Y. Sugano, S. Ichikawa, S. Tanaka and T. Hirai, ACS Catal., 2012, 2, 599–603 CrossRef CAS.
- R. Su, L. Kesavan, M. M. Jensen, R. Tiruvalam, Q. He, N. Dimitratos, S. Wendt, M. Glasius, C. J. Kiely, G. J. Hutchings and F. Besenbacher, Chem. Commun., 2014, 50, 12612–12614 RSC.
- S. Shuang, R. Lv, Z. Xie and Z. Zhang, Sci. Rep., 2016, 6, 26670 CrossRef CAS PubMed.
- W. Guo, Y. Liu, Y. Sun, Y. Wang, W. Qin, B. Zhao, Z. Liang and L. Jiang, Adv. Funct. Mater., 2021, 31, 2100768 CrossRef CAS.
- H. Nishi, T. Torimoto and T. Tatsuma, Phys. Chem. Chem. Phys., 2015, 17, 4042–4046 RSC.
- S. Wang, L. Pan, J. J. Song, W. Mi, J. J. Zou, L. Wang and X. Zhang, J. Am. Chem. Soc., 2015, 137, 2975–2983 CrossRef CAS PubMed.
- X. Liu, G. Zhu, X. Wang, X. Yuan, T. Lin and F. Huang, Adv. Energy Mater., 2016, 6, 1600452 CrossRef.
- W. Zhang, H. He, Y. Tian, H. Li, K. Lan, L. Zu, Y. Xia, L. Duan, W. Li and D. Zhao, Nano Energy, 2019, 66, 104113 CrossRef CAS.
- R. Fernández-Climent, S. Giménez and M. García-Tecedor, Sustainable Energy Fuels, 2020, 4, 5916–5926 RSC.
- F. Yu, C. Wang, Y. Li, H. Ma, R. Wang, Y. Liu, N. Suzuki, C. Terashima, B. Ohtani, T. Ochiai, A. Fujishima and X. Zhang, Adv. Sci., 2020, 7, 2000204 CrossRef CAS PubMed.
- W. Hu, W. Zhou, K. Zhang, X. Zhang, L. Wang, B. Jiang, G. Tian, D. Zhao and H. Fu, J. Mater. Chem. A, 2016, 4, 7495–7502 RSC.
- T. S. Rajaraman, S. P. Parikh and V. G. Gandhi, J. Chem. Eng., 2020, 389, 123918 CrossRef CAS.
- H. Song, C. Li, Z. Lou, Z. Ye and L. Zhu, ACS Sustainable Chem. Eng., 2017, 5, 8982–8987 CrossRef CAS.
- L. Andronic and A. Enesca, Front. Chem., 2020, 8, 565489 CrossRef CAS PubMed.
- X. Chen, L. Liu and F. Huang, Chem. Soc. Rev., 2015, 44, 1861–1885 RSC.
- L. Gu, J. Wang, H. Cheng, Y. Zhao, L. Liu and X. Han, ACS Appl. Mater. Interfaces, 2013, 5, 3085–3093 CrossRef CAS PubMed.
- C. Peng, X. Yang, Y. Li, H. Yu, H. Wang and F. Peng, ACS Appl. Mater. Interfaces, 2016, 8, 6051–6060 CrossRef CAS PubMed.
- L. L. Tan, W. J. Ong, S. P. Chai and A. R. Mohamed, Nanoscale Res. Lett., 2013, 8, 465 CrossRef PubMed.
- I.-A. Baragau, J. Buckeridge, K. G. Nguyen, T. Heil, M. T. Sajjad, S. A. J. Thomson, A. Rennie, D. J. Morgan, N. P. Power, S. A. Nicolae, M.-M. Titirici, S. Dunn and S. Kellici, J. Mater. Chem. A, 2023, 11, 9791–9806 RSC.
- X. Zhou, Y. Fang, X. Cai, S. Zhang, S. Yang, H. Wang, X. Zhong and Y. Fang, ACS Appl. Mater. Interfaces, 2020, 12, 20579–20588 CrossRef CAS PubMed.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- D. Zeng, W.-J. Ong, H. Zheng, M. Wu, Y. Chen, D.-L. Peng and M.-Y. Han, J. Mater. Chem. A, 2017, 5, 16171–16178 RSC.
- D. Zeng, W. J. Ong, Y. Chen, S. Y. Tee, C. S. Chua, D. L. Peng and M. Y. Han, Part. Syst. Charact., 2017, 35, 1700251 CrossRef.
- T. Hong, S. Anwer, J. Wu, C. Deng and H. Qian, Front. Chem., 2022, 10, 1050046 CrossRef CAS PubMed.
- M. Ghidiu, M. R. Lukatskaya, M. Q. Zhao, Y. Gogotsi and M. W. Barsoum, Nature, 2014, 516, 78–81 CrossRef CAS PubMed.
- Y. Li, X. Deng, J. Tian, Z. Liang and H. Cui, Appl. Mater. Today, 2018, 13, 217–227 CrossRef.
- C. J. Zhang, S. Pinilla, N. McEvoy, C. P. Cullen, B. Anasori, E. Long, S.-H. Park, A. Seral-Ascaso, A. Shmeliov, D. Krishnan, C. Morant, X. Liu, G. S. Duesberg, Y. Gogotsi and V. Nicolosi, Chem. Mater., 2017, 29, 4848–4856 CrossRef CAS.
- A. Shahzad, K. Rasool, M. Nawaz, W. Miran, J. Jang, M. Moztahida, K. A. Mahmoud and D. S. Lee, Chem. Eng. J., 2018, 349, 748–755 CrossRef CAS.
- A. Crake, K. C. Christoforidis, A. Kafizas, S. Zafeiratos and C. Petit, Appl. Catal., B, 2017, 210, 131–140 CrossRef CAS.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- T. Banerjee, K. Gottschling, G. Savasci, C. Ochsenfeld and B. V. Lotsch, ACS Energy Lett., 2018, 3, 400–409 CrossRef CAS PubMed.
- Y. Cai, Y. Yu, J. Wu, J. Qu, J. Hu, D. Tian and J. Li, Nanoscale, 2024, 16, 961–977 RSC.
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- J. Qu, T. Yang, P. Zhang, F. Yang, Y. Cai, X. Yang, C. M. Li and J. Hu, Appl. Catal., B, 2024, 348, 123827 CrossRef CAS.
- J. Hao, Y. Tang, J. Qu, Y. Cai, X. Yang and J. Hu, Small, 2024, 2404139 CrossRef PubMed.
- H. Li, H. Liu, C. Li, J. Liu, J. Liu and Q. Yang, J. Mater. Chem. A, 2020, 8, 18745–18754 RSC.
- X. Han, W. Dong, L. Li and X. Zhou, Chem. Commun., 2023, 59, 11863–11866 RSC.
- A. Putta Rangappa, D. Praveen Kumar, K. H. Do, J. Wang, Y. Zhang and T. K. Kim, Adv. Sci., 2023, 10, e2300073 CrossRef PubMed.
- P. Sarkar, I. H. Chowdhury, A. Chakraborty, M. Goswami, M. K. Naskar, A. Khan and S. M. Islam, Ind. Eng. Chem. Res., 2024, 63, 5591–5607 CrossRef CAS.
- A. L. Luna, F. Matter, M. Schreck, J. Wohlwend, E. Tervoort, C. Colbeau-Justin and M. Niederberger, Appl. Catal., B, 2020, 267, 118660 CrossRef CAS.
- Y. Si, J. Yu, X. Tang, J. Ge and B. Ding, Nat. Commun., 2014, 5, 5802 CrossRef PubMed.
- W. Liu, A. K. Herrmann, N. C. Bigall, P. Rodriguez, D. Wen, M. Oezaslan, T. J. Schmidt, N. Gaponik and A. Eychmuller, Acc. Chem. Res., 2015, 48, 154–162 CrossRef CAS PubMed.
- P. Rusch, D. Zambo and N. C. Bigall, Acc. Chem. Res., 2020, 53, 2414–2424 CrossRef CAS PubMed.
- A. Rose, A. Hofmann, P. Voepel, B. Milow and R. Marschall, ACS Appl. Energy Mater., 2022, 5, 14966–14978 CrossRef CAS.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- Z. Liu, S. Y. Tee, G. Guan and M. Y. Han, Nanomicro Lett., 2024, 16, 95 CAS.
- J. Hu, D. Chen, Z. Mo, N. Li, Q. Xu, H. Li, J. He, H. Xu and J. Lu, Angew. Chem., Int. Ed., 2019, 58, 2073–2077 CrossRef CAS PubMed.
- N. Serpone, P. Maruthamuthu, P. Pichat, E. Pelizzetti and H. Hidaka, J. Photochem. Photobiol., A, 1995, 85, 247–255 CrossRef CAS.
- I. Bedja and P. V. Kamat, J. Phys. Chem., 2002, 99, 9182–9188 CrossRef.
- H. Li, W. Tu, Y. Zhou and Z. Zou, Adv. Sci., 2016, 3, 1500389 CrossRef PubMed.
- K. Maeda, ACS Catal., 2013, 3, 1486–1503 CrossRef CAS.
- Q. Xu, L. Zhang, B. Cheng, J. Fan and J. Yu, Chem, 2020, 6, 1543–1559 CAS.
- P. Zhou, J. Yu and M. Jaroniec, Adv. Mater., 2014, 26, 4920–4935 CrossRef CAS PubMed.
- W. Yu, S. Zhang, J. Chen, P. Xia, M. H. Richter, L. Chen, W. Xu, J. Jin, S. Chen and T. Peng, J. Mater. Chem. A, 2018, 6, 15668–15674 RSC.
- J. Low, B. Dai, T. Tong, C. Jiang and J. Yu, Adv. Mater., 2019, 31, e1802981 CrossRef PubMed.
- Q. Xu, L. Zhang, J. Yu, S. Wageh, A. A. Al-Ghamdi and M. Jaroniec, Mater. Today, 2018, 21, 1042–1063 CrossRef CAS.
- G. Yang, D. Chen, H. Ding, J. Feng, J. Z. Zhang, Y. Zhu, S. Hamid and D. W. Bahnemann, Appl. Catal., B, 2017, 219, 611–618 CrossRef CAS.
- U. S. Meda, K. Vora, Y. Athreya and U. A. Mandi, Process Saf. Environ. Prot., 2022, 161, 771–787 CrossRef CAS.
- M. E. Aguirre, R. Zhou, A. J. Eugene, M. I. Guzman and M. A. Grela, Appl. Catal., B, 2017, 217, 485–493 CrossRef CAS.
- Y. Li, Z. Xia, Q. Yang, L. Wang and Y. Xing, J. Mater. Sci. Technol., 2022, 125, 128–144 CrossRef CAS.
- F. Xu, K. Meng, B. Cheng, S. Wang, J. Xu and J. Yu, Nat. Commun., 2020, 11, 4613 CrossRef CAS PubMed.
- R. P. Schwarzenbach, T. Egli, T. B. Hofstetter, U. von Gunten and B. Wehrli, Annu. Rev. Environ. Resour., 2010, 35, 109–136 CrossRef.
- D. Antonio da Silva, R. P. Cavalcante, R. F. Cunha, A. J. Machulek and S. Cesar de Oliveira, Chemosphere, 2018, 207, 457–468 CrossRef CAS PubMed.
- M. Ahmed, M. O. Mavukkandy, A. Giwa, M. Elektorowicz, E. Katsou, O. Khelifi, V. Naddeo and S. W. Hasan, npj Clean Water, 2022, 5, 12 CrossRef CAS.
- J. Giménez, B. Bayarri, Ó. González, S. Malato, J. Peral and S. Esplugas, ACS Sustainable Chem. Eng., 2015, 3, 3188–3196 CrossRef.
- A. R. Khataee and M. Fathinia, Recent Advances in Photocatalytic Processes by Nanomaterials, 2013 Search PubMed.
- S. Foteinis and E. Chatzisymeon, Nanostruct. Photocatal., 2020, 75–97 CAS.
- C. Hu, X. Zhang, W. Li, Y. Yan, G. Xi, H. Yang, J. Li and H. Bai, J. Mater. Chem. A, 2014, 2, 2040 RSC.
- W. Wang, J. Fang, Y. Zhou, W. Zhang and C. Lu, RSC Adv., 2016, 6, 67556–67564 RSC.
- S. S. Alias, Z. Harun, F. H. Azhar, S. A. Ibrahim and B. Johar, J. Cleaner Prod., 2020, 251, 119448 CrossRef CAS.
- T. Rajaramanan, S. Shanmugaratnam, V. Gurunanthanan, S. Yohi, D. Velauthapillai, P. Ravirajan and M. Senthilnanthanan, Catalysts, 2021, 11, 690 CrossRef CAS.
- M. K. Hossain, M. M. Hossain and S. Akhtar, ACS Omega, 2023, 8, 1979–1988 CrossRef CAS PubMed.
- L. Elsellami, H. Lachheb and A. Houas, Mater. Sci. Semicond. Process., 2015, 36, 103–114 CrossRef CAS.
- A. Khlyustova, N. Sirotkin, T. Kusova, A. Kraev, V. Titov and A. Agafonov, Mater. Adv., 2020, 1, 1193–1201 RSC.
- L. Bergamonti, G. Predieri, Y. Paz, L. Fornasini, P. P. Lottici and F. Bondioli, Microchem. J., 2017, 133, 1–12 CrossRef CAS.
- J. Niu, P. Lu, M. Kang, K. Deng, B. Yao, X. Yu and Q. Zhang, Appl. Surf. Sci., 2014, 319, 99–106 CrossRef CAS.
- T. Ohno, M. Akiyoshi, T. Umebayashi, K. Asai, T. Mitsui and M. Matsumura, Appl. Catal., A, 2004, 265, 115–121 CrossRef CAS.
- T. Liu, B. Li, Y. Hao, F. Han, L. Zhang and L. Hu, Appl. Catal., B, 2015, 165, 378–388 CrossRef CAS.
- X. Yang, Y. Wang, L. Zhang, H. Fu, P. He, D. Han, T. Lawson and X. An, Catalysts, 2020, 10, 139 CrossRef CAS.
- E. D. Sultanova, I. R. Nizameev, K. V. Kholin, M. K. Kadirov, A. S. Ovsyannikov, V. A. Burilov, A. Y. Ziganshina and I. S. Antipin, New J. Chem., 2020, 44, 7169–7174 RSC.
- C. H. Nguyen, C.-C. Fu and R.-S. Juang, J. Cleaner Prod., 2018, 202, 413–427 CrossRef CAS.
- C. Jia, X. Zhang, K. Matras-Postolek, B. Huang and P. Yang, Carbon, 2018, 139, 415–426 CrossRef CAS.
- C. Xue, T. Zhang, S. Ding, J. Wei and G. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 16091–16102 CrossRef CAS PubMed.
- V. Q. Nguyen, A. H. Mady, M. A. Mahadadalkar, M. L. Baynosa, D. R. Kumar, A. M. Rabie, J. Lee, W. K. Kim and J. J. Shim, J. Colloid Interface Sci., 2022, 606, 337–352 CrossRef CAS PubMed.
- R. Khunphonoi and N. Grisdanurak, Chem. Eng. J., 2016, 296, 420–427 CrossRef CAS.
- A. M. Alotaibi, S. Sathasivam, B. A. D. Williamson, A. Kafizas, C. Sotelo-Vazquez, A. Taylor, D. O. Scanlon and I. P. Parkin, Chem. Mater., 2018, 30, 1353–1361 CrossRef CAS.
- R. Su, R. Tiruvalam, Q. He, N. Dimitratos, L. Kesavan, C. Hammond, J. A. Lopez-Sanchez, R. Bechstein, C. J. Kiely, G. J. Hutchings and F. Besenbacher, ACS Nano, 2012, 6, 6284–6292 CrossRef CAS PubMed.
- Y. Zhang, Q. Li, Q. Gao, S. Wan, P. Yao and X. Zhu, Appl. Catal., B, 2020, 267, 118715 CrossRef CAS.
- D. H. Quiñones, A. Rey, P. M. Álvarez, F. J. Beltrán and G. Li Puma, Appl. Catal., B, 2015, 178, 74–81 CrossRef.
- R. M. Mohamed and Z. I. Zaki, J. Environ. Chem. Eng., 2021, 9, 104732 CrossRef CAS.
- R. Fiorenza, A. Di Mauro, M. Cantarella, V. Privitera and G. Impellizzeri, J. Photochem. Photobiol., A, 2019, 380, 111872 CrossRef CAS.
- R. Fiorenza, A. Di Mauro, M. Cantarella, C. Iaria, E. M. Scalisi, M. V. Brundo, A. Gulino, L. Spitaleri, G. Nicotra, S. Dattilo, S. C. Carroccio, V. Privitera and G. Impellizzeri, J. Chem. Eng., 2020, 379, 122309 CrossRef CAS.
- E. Bilgin Simsek, Appl. Catal., B, 2017, 200, 309–322 CrossRef CAS.
- R. P. Cavalcante, R. F. Dantas, B. Bayarri, O. González, J. Giménez, S. Esplugas and A. Machulek, Catal. Today, 2015, 252, 27–34 CrossRef CAS.
- X. Feng, P. Wang, J. Hou, J. Qian, Y. Ao and C. Wang, J. Hazard. Mater., 2018, 351, 196–205 CrossRef CAS PubMed.
- R. Yu, Y. Yang, Z. Zhou, X. Li, J. Gao, N. Wang, J. Li and Y. Liu, Sep. Purif. Technol., 2022, 299, 121712 CrossRef CAS.
- K. Hu, R. Li, C. Ye, A. Wang, W. Wei, D. Hu, R. Qiu and K. Yan, J. Cleaner Prod., 2020, 253, 120055 CrossRef CAS.
- S. Varnagiris, M. Urbonavicius, S. Sakalauskaite, R. Daugelavicius, L. Pranevicius, M. Lelis and D. Milcius, Sci. Total Environ., 2020, 720, 137600 CrossRef CAS PubMed.
- Y. Ren, Y. Han, Z. Li, X. Liu, S. Zhu, Y. Liang, K. W. K. Yeung and S. Wu, Bioact. Mater., 2020, 5, 201–209 Search PubMed.
- S. Zhu, X. Xie, S.-C. Chen, S. Tong, G. Lu, D. Y. H. Pui and J. Sun, Appl. Surf. Sci., 2017, 408, 117–124 CrossRef CAS.
- X. Gao, K. Zheng, Q. Zhang, X. Cao, S. Wu and J. Su, Appl. Surf. Sci., 2022, 586, 152882 CrossRef CAS.
- S. Y. Tee, C. J. Lee, S. S. Dinachali, S. C. Lai, E. L. Williams, H. K. Luo, D. Chi, T. S. Andy Hor and M. Y. Han, Nanotechnology, 2015, 26, 415401 CrossRef PubMed.
- F. Yang, P. Wang, J. Hao, J. Qu, Y. Cai, X. Yang, C. M. Li and J. Hu, Nano Energy, 2023, 118, 108993 CrossRef CAS.
- E. H. G. Backus, S. Hosseinpour, C. Ramanan, S. Sun, S. J. Schlegel, M. Zelenka, X. Jia, M. Gebhard, A. Devi, H. I. Wang and M. Bonn, Angew. Chem., Int. Ed., 2024, 63, e202312123 CrossRef CAS PubMed.
- J. Low, J. Yu, M. Jaroniec, S. Wageh and A. A. Al-Ghamdi, Adv. Mater., 2017, 29, 1601694 CrossRef PubMed.
- M. Ge, Q. Li, C. Cao, J. Huang, S. Li, S. Zhang, Z. Chen, K. Zhang, S. S. Al-Deyab and Y. Lai, Adv. Sci., 2017, 4, 1600152 CrossRef PubMed.
- J. Hu, S. Zhang, Y. Cao, H. Wang, H. Yu and F. Peng, ACS Sustainable Chem. Eng., 2018, 6, 10823–10832 CrossRef CAS.
- P. Wang, Q. Zhou, Y. Xia, S. Zhan and Y. Li, Appl. Catal., B, 2018, 225, 433–444 CrossRef CAS.
- H. Li, Z. Bian, J. Zhu, D. Zhang, G. Li, Y. Huo, H. Li and Y. Lu, J. Am. Chem. Soc., 2007, 129, 8406–8407 CrossRef CAS PubMed.
- W. Cao, B. Wei, X. Fu, N. Ma, H. Gao and L. Xu, RSC Adv., 2016, 6, 108969–108973 RSC.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro, B. Ferrer and H. Garcia, Chem. Rev., 2023, 123, 445–490 CrossRef CAS PubMed.
- S. Kampouri, C. P. Ireland, B. Valizadeh, E. Oveisi, P. A. Schouwink, M. Mensi and K. C. Stylianou, ACS Appl. Energy Mater., 2018, 1, 6541–6548 CrossRef CAS.
- X. He, Y. Ding, Z. Huang, M. Liu, M. Chi, Z. Wu, C. U. Segre, C. Song, X. Wang and X. Guo, Angew. Chem., Int. Ed., 2023, 62, e202217439 CrossRef CAS PubMed.
- Z.-H. Xue, D. Luan, H. Zhang and X. W. Lou, Joule, 2022, 6, 92–133 CrossRef CAS.
- B. Xing, T. Wang, Z. Zheng, S. Liu, J. Mao, C. Li and B. Li, Chem. Eng. J., 2023, 461, 141871 CrossRef CAS.
- B. Lu, L. Guo, F. Wu, Y. Peng, J. E. Lu, T. J. Smart, N. Wang, Y. Z. Finfrock, D. Morris, P. Zhang, N. Li, P. Gao, Y. Ping and S. Chen, Nat. Commun., 2019, 10, 631 CrossRef CAS PubMed.
- B. Yan, D. Liu, X. Feng, M. Shao and Y. Zhang, Adv. Funct. Mater., 2020, 30, 2003007 CrossRef CAS.
- J. Kwon, K. Choi, M. Schreck, T. Liu, E. Tervoort and M. Niederberger, ACS Appl. Mater. Interfaces, 2021, 13, 53691–53701 CrossRef CAS PubMed.
- Z. Li and X. Meng, J. Alloys Compd., 2020, 830, 154669 CrossRef CAS.
- J. Kwon, K. Choi, E. Tervoort and M. Niederberger, J. Mater. Chem. A, 2022, 10, 18383–18395 RSC.
- G. Bersalli, T. Trondle and J. Lilliestam, Commun. Earth Environ., 2023, 4, 44 CrossRef PubMed.
- V. Kumaravel, J. Bartlett and S. C. Pillai, ACS Energy Lett., 2020, 5, 486–519 CrossRef CAS.
- S. A. Rawool, K. K. Yadav and V. Polshettiwar, Chem. Sci., 2021, 12, 4267–4299 RSC.
- Y. Wei, F. You, D. Zhao, J. Wan, L. Gu and D. Wang, Angew. Chem., Int. Ed., 2022, 61, e202212049 CrossRef CAS PubMed.
- W. Jiang, H. Loh, B. Q. L. Low, H. Zhu, J. Low, J. Z. X. Heng, K. Y. Tang, Z. Li, X. J. Loh, E. Ye and Y. Xiong, Appl. Catal., B, 2023, 321, 122079 CrossRef CAS.
- C. Ban, Y. Wang, Y. Feng, Z. Zhu, Y. Duan, J. Ma, X. Zhang, X. Liu, K. Zhou, H. Zou, D. Yu, X. Tao, L. Gan, G. Han and X. Zhou, Energy Environ. Sci., 2024, 17, 518–530 RSC.
- A. Crake, K. C. Christoforidis, A. Gregg, B. Moss, A. Kafizas and C. Petit, Small, 2019, 15, e1805473 CrossRef PubMed.
- S. Wang, M. Xu, T. Peng, C. Zhang, T. Li, I. Hussain, J. Wang and B. Tan, Nat. Commun., 2019, 10, 676 CrossRef CAS PubMed.
- Y. Ma, X. Yi, S. Wang, T. Li, B. Tan, C. Chen, T. Majima, E. R. Waclawik, H. Zhu and J. Wang, Nat. Commun., 2022, 13, 1400 CrossRef CAS PubMed.
- Y. Yu, X. Dong, P. Chen, Q. Geng, H. Wang, J. Li, Y. Zhou and F. Dong, ACS Nano, 2021, 15, 14453–14464 CrossRef CAS PubMed.
- Y. Xu, S. Wang, J. Yang, B. Han, R. Nie, J. Wang, J. Wang and H. Jing, Nano Energy, 2018, 51, 442–450 CrossRef CAS.
- S. Kreft, R. Schoch, J. Schneidewind, J. Rabeah, E. V. Kondratenko, V. A. Kondratenko, H. Junge, M. Bauer, S. Wohlrab and M. Beller, Chem, 2019, 5, 1818–1833 CAS.
- L. Liang, C. Liu, F. Jiang, Q. Chen, L. Zhang, H. Xue, H. L. Jiang, J. Qian, D. Yuan and M. Hong, Nat. Commun., 2017, 8, 1233 CrossRef PubMed.
- L. Liang, C. Liu, F. Jiang, Q. Chen, L. Zhang, H. Xue, H. L. Jiang, J. Qian, D. Yuan and M. Hong, Nat. Commun., 2017, 8, 1233 CrossRef PubMed.
- R. Li, J. Hu, M. Deng, H. Wang, X. Wang, Y. Hu, H. L. Jiang, J. Jiang, Q. Zhang, Y. Xie and Y. Xiong, Adv. Mater., 2014, 26, 4783–4788 CrossRef CAS PubMed.
- S. Yan, S. Ouyang, H. Xu, M. Zhao, X. Zhang and J. Ye, J. Mater. Chem. A, 2016, 4, 15126–15133 RSC.
- Z. Jiang, X. Xu, Y. Ma, H. S. Cho, D. Ding, C. Wang, J. Wu, P. Oleynikov, M. Jia, J. Cheng, Y. Zhou, O. Terasaki, T. Peng, L. Zan and H. Deng, Nature, 2020, 586, 549–554 CrossRef CAS PubMed.
- J. Bian, Z. Zhang, J. Feng, M. Thangamuthu, F. Yang, L. Sun, Z. Li, Y. Qu, D. Tang, Z. Lin, F. Bai, J. Tang and L. Jing, Angew. Chem., Int. Ed., 2021, 60, 20906–20914 CrossRef CAS PubMed.
- G. N. Schrauzer and T. D. Guth, J. Am. Chem. Soc., 1977, 99, 7189–7193 CrossRef CAS.
- P. W. Huang and M. C. Hatzell, Nat. Commun., 2022, 13, 7908 CrossRef CAS PubMed.
- C. J. van der Ham, M. T. Koper and D. G. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC.
- S. Chen, D. Liu and T. Peng, Sol. RRL, 2020, 5, 2000487 CrossRef.
- G. Zhang, X. Yang, C. He, P. Zhang and H. Mi, J. Mater. Chem. A, 2020, 8, 334–341 RSC.
- J. Qian, S. Zhao, W. Dang, Y. Liao, W. Zhang, H. Wang, L. Lv, L. Luo, H. Y. Jiang and J. Tang, Adv. Sustainable Syst., 2021, 5, 2000282 CrossRef CAS.
- Q. Liu, L. Ai and J. Jiang, J. Mater. Chem. A, 2018, 6, 4102–4110 RSC.
- S. Liu, Y. Wang, S. Wang, M. You, S. Hong, T.-S. Wu, Y.-L. Soo, Z. Zhao, G. Jiang, Q. Jieshan, B. Wang and Z. Sun, ACS Sustainable Chem. Eng., 2019, 7, 6813–6820 CrossRef CAS.
- L. Chen, J. Shou, Y. Chen, W. Han, X. Tu, L. Zhang, Q. Sun, J. Cao, Y. Chang and H. Zheng, Chem. Eng. J., 2023, 451, 138592 CrossRef CAS.
- J. Wang, W. Lin, Y. Ran, J. Cui, L. Wang, X. Yu and Y. Zhang, J. Phys. Chem. C, 2019, 124, 1253–1259 CrossRef.
- Y. Tang, C. Asokan, M. Xu, G. W. Graham, X. Pan, P. Christopher, J. Li and P. Sautet, Nat. Commun., 2019, 10, 4488 CrossRef PubMed.
- W. Li, A. Elzatahry, D. Aldhayan and D. Zhao, Chem. Soc. Rev., 2018, 47, 8203–8237 RSC.
- C. Samanta, Appl. Catal., A, 2008, 350, 133–149 CrossRef CAS.
- G. Gao, Y. Tian, X. Gong, Z. Pan, K. Yang and B. Zong, Chin. J. Catal., 2020, 41, 1039–1047 CrossRef CAS.
- J. Liu, Y. Zou, B. Jin, K. Zhang and J. H. Park, ACS Energy Lett., 2019, 4, 3018–3027 CrossRef CAS.
- J. S. J. Hargreaves, Y.-M. Chung, W.-S. Ahn, T. Hisatomi, K. Domen, M. C. Kung and H. H. Kung, Appl. Catal., A, 2020, 594, 117419 CrossRef CAS.
- H. Hou, X. Zeng and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 17356–17376 CrossRef CAS PubMed.
- Y. Zhao, Y. Kondo, Y. Kuwahara, K. Mori and H. Yamashita, Catal. Today, 2024, 425, 114350 CrossRef CAS.
- W. Gan, X. Fu, J. Jin, J. Guo, M. Zhang, R. Chen, C. Ding, Y. Lu, J. Li and Z. Sun, J. Colloid Interface Sci., 2024, 653, 1028–1039 CrossRef CAS PubMed.
- Z. Jiang, Q. Long, B. Cheng, R. He and L. Wang, J. Mater. Process. Technol., 2023, 162, 1–10 CAS.
- X. Sun, T. Wang, C. Wang and T. Ohno, Catal. Sci. Technol., 2023, 13, 6799–6811 RSC.
- Y. Yang, J. Liu, M. Gu, B. Cheng, L. Wang and J. Yu, Appl. Catal., B, 2023, 333, 122780 CrossRef CAS.
- X. Bao, H. Li, Z. Wang, F. Tong, M. Liu, Z. Zheng, P. Wang, H. Cheng, Y. Liu, Y. Dai, Y. Fan, Z. Li and B. Huang, Appl. Catal., B, 2021, 286, 119885 CrossRef CAS.
- Z. Shen, Y. Hu, B. Li, Y. Zou, S. Li, G. Wilma Busser, X. Wang, G. Zhao and M. Muhler, J. Energy Chem., 2021, 62, 338–350 CrossRef CAS.
- C. Courtois, M. Eder, K. Schnabl, C. A. Walenta, M. Tschurl and U. Heiz, Angew. Chem., Int. Ed., 2019, 58, 14255–14259 CrossRef CAS PubMed.
- G. S. Wolde, D.-H. Kuo, M. H. Urgesa and T. N. Gemeda, Chem. Eng. J., 2023, 469, 143916 CrossRef CAS.
- X. Lang, W. Ma, C. Chen, H. Ji and J. Zhao, Acc. Chem. Res., 2014, 47, 355–363 CrossRef CAS PubMed.
- Q. Wang, M. Zhang, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2010, 49, 7976–7979 CrossRef CAS PubMed.
- Y. Ohko, T. Tatsuma, T. Fujii, K. Naoi, C. Niwa, Y. Kubota and A. Fujishima, Nat. Mater., 2003, 2, 29–31 CrossRef CAS PubMed.
- M. Barawi, L. De Trizio, R. Giannuzzi, G. Veramonti, L. Manna and M. Manca, ACS Nano, 2017, 11, 3576–3584 CrossRef CAS PubMed.
- W. Wang, L. Liu, J. Feng and Y. Yin, Small Methods, 2017, 2, 1700273 CrossRef.
- R. Li, X. Ma, J. Li, J. Cao, H. Gao, T. Li, X. Zhang, L. Wang, Q. Zhang, G. Wang, C. Hou, Y. Li, T. Palacios, Y. Lin, H. Wang and X. Ling, Nat. Commun., 2021, 12, 1587 CrossRef CAS PubMed.
- W. Wang, M. Ye, L. He and Y. Yin, Nano Lett., 2014, 14, 1681–1686 CrossRef CAS PubMed.
- W. Wang, N. Xie, L. He and Y. Yin, Nat. Commun., 2014, 5, 5459 CrossRef CAS PubMed.
- K. Yang, X. Chen, Z. Zheng, J. Wan, M. Feng and Y. Yu, J. Mater. Chem. A, 2019, 7, 3863–3873 RSC.
- A. T. Smith, H. Ding, A. Gorski, M. Zhang, P. A. Gitman, C. Park, Z. Hao, Y. Jiang, B. L. Williams, S. Zeng, A. Kokkula, Q. Yu, G. Ding, H. Zeng and L. Sun, Matter, 2020, 2, 680–696 CrossRef.
- Y. Wei, B. Han, Z. Dong and W. Feng, J. Mater. Sci. Technol., 2019, 35, 1951–1958 CrossRef CAS.
- U. Joost, A. Šutka, M. Oja, K. Smits, N. Döbelin, A. Loot, M. Järvekülg, M. Hirsimäki, M. Valden and E. Nõmmiste, Chem. Mater., 2018, 30, 8968–8974 CrossRef CAS.
- R. Aleisa, J. Feng, Z. Ye and Y. Yin, Angew. Chem., Int. Ed., 2022, 61, e202203700 CrossRef CAS PubMed.
- B. Kraeutler, C. D. Jaeger and A. J. Bard, J. Am. Chem. Soc., 2002, 100, 4903–4905 CrossRef.
- B. Kraeutler, H. Reiche, A. J. Bard and R. G. Hocker, J. Polym. Sci., Polym. Lett. Ed., 1979, 17, 535–538 CrossRef CAS.
- A. J. Hoffman, H. Yee, G. Mills and M. R. Hoffmann, J. Phys. Chem., 1992, 96, 5540–5546 CrossRef CAS.
- C. Lü, Y. Cheng, Y. Liu, F. Liu and B. Yang, Adv. Mater., 2006, 18, 1188–1192 CrossRef.
- A. J. Hoffman, G. Mills, H. Yee and M. R. Hoffmann, J. Phys. Chem., 1992, 96, 5546–5552 CrossRef CAS.
- R. Ojah and S. K. Dolui, J. Photochem. Photobiol., A, 2005, 172, 121–125 CrossRef CAS.
- J. Wang and X. Ni, J. Appl. Polym. Sci., 2008, 108, 3552–3558 CrossRef CAS.
- C. Dong and X. Ni, J. Macromol. Sci., Part A: Pure Appl.Chem., 2004, 41, 547–563 CrossRef.
- Z. Y. Huang, T. Barber, G. Mills and M. B. Morris, J. Phys. Chem., 1994, 98, 12746–12752 CrossRef CAS.
- H. Kong, J. Song and J. Jang, Environ. Sci. Technol., 2010, 44, 5672–5676 CrossRef CAS PubMed.
- X. Wang, Q. Lu, X. Wang, J. Joo, M. Dahl, B. Liu, C. Gao and Y. Yin, ACS Appl. Mater Interfaces, 2016, 8, 538–546 CrossRef CAS PubMed.
- Q. Zhang, D. Q. Lima, I. Lee, F. Zaera, M. Chi and Y. Yin, Angew. Chem., Int. Ed., 2011, 50, 7088–7092 CrossRef CAS PubMed.
- X. L. García-Montelongo, A. Martínez-de la Cruz, S. Vázquez-Rodríguez and L. M. Torres-Martínez, Mater. Res. Bull., 2014, 51, 56–62 CrossRef.
- P. A. Kots, S. Liu, B. C. Vance, C. Wang, J. D. Sheehan and D. G. Vlachos, ACS Catal., 2021, 11, 8104–8115 CrossRef CAS.
- P. A. Zapata, A. Zenteno, N. Amigó, F. M. Rabagliati, F. Sepúlveda, F. Catalina and T. Corrales, Polym. Degrad. Stab., 2016, 133, 101–107 CrossRef CAS.
- R. E. Day, Polym. Degrad. Stab., 1990, 29, 73–92 CrossRef CAS.
- A. X. Gao, J. D. Bolt and A. A. Feng, Plast., Rubber Compos., 2008, 37, 397–402 CrossRef CAS.
- C. Anton-Prinet, G. Mur, M. Gay, L. Audouin and J. Verdu, Polym. Degrad. Stab., 1998, 61, 211–216 CrossRef CAS.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- T. El Darai, A. Ter-Halle, M. Blanzat, G. Despras, V. Sartor, G. Bordeau, A. Lattes, S. Franceschi, S. Cassel, N. Chouini-Lalanne, E. Perez, C. Déjugnat and J.-C. Garrigues, Green Chem., 2024, 26, 6857–6885 RSC.
- R. S. Braido, L. E. P. Borges and J. C. Pinto, J. Anal. Appl. Pyrolysis, 2018, 132, 47–55 CrossRef CAS.
- N. Daraboina and G. Madras, Ind. Eng. Chem. Res., 2008, 47, 6828–6834 CrossRef CAS.
- B.-S. Kang, S. G. Kim and J.-S. Kim, J. Anal. Appl. Pyrolysis, 2008, 81, 7–13 CrossRef CAS.
- L. Wimberger, G. Ng and C. Boyer, Nat. Commun., 2024, 15, 2510 CrossRef CAS PubMed.
- T. Uekert, C. M. Pichler, T. Schubert and E. Reisner, Nat. Sustainable, 2021, 4, 383–391 CrossRef.
- O. G. Mountanea, E. Skolia and C. G. Kokotos, Green Chem., 2024, 26, 8528–8549 RSC.
- R. Cao, D. Xiao, M. Wang, Y. Gao and D. Ma, Appl. Catal., B, 2024, 341, 123357 CrossRef CAS.
- T. T. Nguyen and K. Edalati, Chemosphere, 2024, 355, 141785 CrossRef CAS PubMed.
- M. Han, S. Zhu, C. Xia and B. Yang, Appl. Catal., B, 2022, 316, 121662 CrossRef CAS.
- Z. Peng, R. Chen and H. Li, ACS Sustainable Chem. Eng., 2023, 11, 10688–10697 CrossRef CAS.
- S. Feng, P. T. T. Nguyen, X. Ma and N. Yan, Angew. Chem., Int. Ed., 2024, e202408504, DOI:10.1002/anie.202408504.
- F. Eisenreich, Angew. Chem., Int. Ed., 2023, 62, e202301303 CrossRef CAS PubMed.
- M. Zulfiqar, S. Chowdhury, S. Sufian and A. A. Omar, J. Cleaner Prod., 2018, 203, 848–859 CrossRef CAS.
- S. Weon, E. Choi, H. Kim, J. Y. Kim, H. J. Park, S. M. Kim, W. Kim and W. Choi, Environ. Sci. Technol., 2018, 52, 9330–9340 CrossRef CAS PubMed.
- Y. Ye, Y. Feng, H. Bruning, D. Yntema and H. H. M. Rijnaarts, Appl. Catal., B, 2018, 220, 171–181 CrossRef CAS.
- Y. Uesugi, H. Nagakawa and M. Nagata, ACS Omega, 2022, 7, 11946–11955 CrossRef CAS PubMed.
- G. Dong, Y. Wang, H. Lei, G. Tian, S. Qi and D. Wu, J. Cleaner Prod., 2020, 253, 120021 CrossRef CAS.
- E. Pino, C. Calderon, F. Herrera, G. Cifuentes and G. Arteaga, Front. Chem., 2020, 8, 365 CrossRef CAS PubMed.
- L. Wang, D. Wu, Z. Guo, J. Yan, Y. Hu, Z. Chang, Q. Yuan, H. Ming and J. Wang, J. Alloys Compd., 2018, 745, 26–32 CrossRef CAS.
- L. Minchi, F. Cao, Z. Xinni, C. Youqiang and L. Xuhua, Chem. Phys. Lett., 2019, 736, 136807 CrossRef.
- C. Lv, X. Lan, L. Wang, Q. Yu, M. Zhang, H. Sun and J. Shi, Catal. Sci. Technol., 2019, 9, 6124–6135 RSC.
- C. Ni, Y. Tang, H. R. S. Abdellatif, X. Huang, D. Xie and J. Ni, J. Electrochem. Soc., 2020, 167, 126505 CrossRef CAS.
- F. Venditti, F. Cuomo, A. Ceglie, P. Avino, M. V. Russo and F. Lopez, Langmuir, 2015, 31, 3627–3634 CrossRef CAS PubMed.
- J. Shao, W. Sheng, M. Wang, S. Li, J. Chen, Y. Zhang and S. Cao, Appl. Catal., B, 2017, 209, 311–319 CrossRef CAS.
- Y. Ma, L. Han, H. Ma, J. Wang, J. Liu, L. Cheng, J. Yang and Q. Zhang, Catal. Commun., 2017, 95, 1–5 CrossRef.
- L. Lu, R. Shan, Y. Shi, S. Wang and H. Yuan, Chemosphere, 2019, 222, 391–398 CrossRef CAS PubMed.
- L. Ji, X. Liu, T. Xu, M. Gong and S. Zhou, J. Sol-Gel Sci. Technol., 2019, 93, 380–390 CrossRef.
- N. Ramesh Reddy, M. Mamatha Kumari, M. V. Shankar, K. Raghava Reddy, S. W. Joo and T. M. Aminabhavi, J. Environ. Manag., 2020, 277, 111433 CrossRef PubMed.
- J. T. Park, D. J. Kim, D. H. Kim and J. H. Kim, Mater. Lett., 2017, 202, 66–69 CrossRef CAS.
- T. H. Kim, G. M. Go, H. B. Cho, Y. Song, C. G. Lee and Y. H. Choa, Front. Chem., 2018, 6, 458 CrossRef CAS PubMed.
- A. Sanchez-Martinez, O. Ceballos-Sanchez, C. Koop-Santa, E. R. López-Mena, E. Orozco-Guareño and M. García-Guaderrama, Ceram. Int., 2018, 44, 5273–5283 CrossRef CAS.
- C.-Y. Kuo, C.-H. Wu, J.-T. Wu and Y.-R. Chen, React. Kinet., Mech. Catal., 2014, 114, 753–766 CrossRef.
- I. Ganesh, Appl. Surf. Sci., 2017, 414, 277–291 CrossRef CAS.
- S. Y. Mendiola-Alvarez, M. A. Hernandez-Ramirez, J. L. Guzman-Mar, L. L. Garza-Tovar and L. Hinojosa-Reyes, Environ. Sci. Pollut. Res. Int., 2019, 26, 4180–4191 CrossRef CAS PubMed.
- J. Zhao, W. Li, X. Li and X. Zhang, RSC Adv., 2017, 7, 21547–21555 RSC.
- N. F. Jaafar, A. A. Jalil, S. Triwahyono, J. Efendi, R. R. Mukti, R. Jusoh, N. W. C. Jusoh, A. H. Karim, N. F. M. Salleh and V. Suendo, Appl. Surf. Sci., 2015, 338, 75–84 CrossRef CAS.
- S. I. Mogal, D. O. Shah, T. Mukherjee, T. Shripathi and M. K. Mishra, ACS Omega, 2018, 3, 12802–12812 CrossRef CAS PubMed.
- J. Singh, N. Tripathi and S. Mohapatra, Nano-Struct. Nano-Objects, 2019, 18, 100266 CrossRef CAS.
- R. Shan, L. Lu, J. Gu, Y. Zhang, H. Yuan, Y. Chen and B. Luo, Mater. Sci. Semicond. Process., 2020, 114, 105088 CrossRef CAS.
- M. Scarisoreanu, A. G. Ilie, E. Goncearenco, A. M. Banici, I. P. Morjan, E. Dutu, E. Tanasa, I. Fort, M. Stan, C. N. Mihailescu and C. Fleaca, Appl. Surf. Sci., 2020, 509, 145217 CrossRef CAS.
- Y. Wen, B. Liu, W. Zeng and Y. Wang, Nanoscale, 2013, 5, 9739–9746 RSC.
- A. Bumajdad, M. Madkour, Y. Abdel-Moneam and M. El-Kemary, J. Mater. Sci., 2013, 49, 1743–1754 CrossRef.
- Y. Yu, W. Wen, X. Y. Qian, J. B. Liu and J. M. Wu, Sci. Rep., 2017, 7, 41253 CrossRef CAS PubMed.
- M. A. Ibrahem, B. G. Rasheed, R. I. Mahdi, T. M. Khazal, M. M. Omar and M. O'Neill, RSC Adv., 2020, 10, 22324–22330 RSC.
- O. Nasr, O. Mohamed, A.-S. Al-Shirbini and A.-M. Abdel-Wahab, J. Photochem. Photobiol., A, 2019, 374, 185–193 CrossRef CAS.
- K. H. Leong, H. Y. Chu, S. Ibrahim and P. Saravanan, Beilstein J. Nanotechnol., 2015, 6, 428–437 CrossRef PubMed.
- K. Saeed, I. Khan, T. Gul and M. Sadiq, Appl. Water Sci., 2017, 7, 3841–3848 CrossRef CAS.
- L. Rossi, P. I. Villabrille, S. Morales-Torres and J. A. Rosso, Mater. Chem. Phys., 2023, 302, 127740 CrossRef CAS.
- Q. Wang, X. Wang, M. Zhang, G. Li, S. Gao, M. Li and Y. Zhang, J. Colloid Interface Sci., 2016, 463, 308–316 CrossRef CAS PubMed.
- A. Malankowska, A. Mikołajczyk, J. Mędrzycka, I. Wysocka, G. Nowaczyk, M. Jarek, T. Puzyn and E. Mulkiewicz, Environ. Sci.: Nano, 2020, 7, 3557–3574 RSC.
- M. A. Mahadadalkar, G. Dhakal, S. Sahoo, D. Ranjith Kumar, M. L. Baynosa, V. Q. Nguyen, M. S. Sayed, A. M. Rabie, W. K. Kim and J.-J. Shim, J. Ind. Eng. Chem., 2023, 124, 402–411 CrossRef CAS.
- O. Sacco, J. J. Murcia, A. E. Lara, M. Hernández-Laverde, H. Rojas, J. A. Navío, M. C. Hidalgo and V. Vaiano, Mater. Sci. Semicond. Process., 2020, 107, 104839 CrossRef CAS.
- T. Tang, Z. Yin, J. Chen, S. Zhang, W. Sheng, W. Wei, Y. Xiao, Q. Shi and S. Cao, Chem. Eng. J., 2021, 417, 128058 CrossRef CAS.
- X. Li, J. Xiong, Y. Xu, Z. Feng and J. Huang, Chin. J. Catal., 2019, 40, 424–433 CrossRef CAS.
- S. Fang, Y. Liu, Z. Sun, J. Lang, C. Bao and Y. H. Hu, Appl. Catal., B, 2020, 278, 119316 CrossRef CAS.
- J. Liu, J. Zheng, G. Yue, H. Li, Z. Liu, Y. Zhao, N. Wang, C. Sun and Z. Cui, RSC Adv., 2022, 12, 10258–10266 RSC.
- J. Wang, G. Wang, X. Wang, Y. Wu, Y. Su and H. Tang, Carbon, 2019, 149, 618–626 CrossRef CAS.
- H. Ge, F. Xu, B. Cheng, J. Yu and W. Ho, ChemCatChem, 2019, 11, 6301–6309 CrossRef CAS.
- J. Li, C. Wu, J. Li, B. Dong, L. Zhao and S. Wang, Chin. J. Catal., 2022, 43, 339–349 CrossRef CAS.
- M.-J. Ran, M. Wang, Z.-Y. Hu, Y.-F. Huang, L.-D. Wang, L. Wu, M.-M. Yuan, J. Zhang, B. Li, G. Van Tendeloo, Y. Li and B.-L. Su, J. Mater. Sci. Technol., 2025, 212, 182–191 CrossRef.
- Y.-Q. Wang, C. Yang and L.-H. Gan, Int. J. Hydrogen Energy, 2023, 48, 19372–19384 CrossRef CAS.
- W. Zhang, Y. Hu, C. Yan, D. Hong, R. Chen, X. Xue, S. Yang, Y. Tian, Z. Tie and Z. Jin, Nanoscale, 2019, 11, 9053–9060 RSC.
- C.-y. Huang, R.-t. Guo, W.-g. Pan, J.-y. Tang, W.-g. Zhou, H. Qin, X.-y. Liu and P.-y. Jia, J. CO2 Util., 2018, 26, 487–495 CrossRef CAS.
- D.-E. Lee, D. J. Kim, S. Moru, M. G. Kim, W.-K. Jo and S. Tonda, Appl. Surf. Sci., 2021, 563, 150292 CrossRef CAS.
- A. Meng, B. Cheng, H. Tan, J. Fan, C. Su and J. Yu, Appl. Catal., B, 2021, 289, 120039 CrossRef CAS.
- C. Ban, Y. Wang, J. Ma, Y. Feng, J. Ding, Y. Duan, X. Liu, B. Zhang, J. Tang, X. Tao, L. Gan, S. Tan and X. Zhou, Sep. Purif. Technol., 2023, 326, 124745 CrossRef CAS.
- W. Ding, X. Li, S. Su, Z. Liu, Y. Cao, L. Meng, S. Yuan, W. Wei and M. Luo, Nanoscale, 2023, 15, 4014–4021 RSC.
- P. Zhang, L. Chen, D.-H. Kuo, B. Wu, Z. Su, D. Lu, Q. Wu, J. Li, J. Lin and X. Chen, J. Mater. Chem. A, 2024, 12, 7163–7177 RSC.
- L. Feng, B. Li, Y. Xiao, L. Li, Y. Zhang, Q. Zhao, G. Zuo, X. Meng and V. A. L. Roy, Catal. Commun., 2021, 155, 106315 CrossRef CAS.
- X. Zhao, Z. Li, Y. Chen, L. Shi and Y. Zhu, Appl. Surf. Sci., 2008, 254, 1825–1829 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |