
Synergistic effect of ROS-generating polydopamine on drug-induced bone tissue regeneration†
Hyeju
Han‡
ab,
Bongkyun
Kang‡
b,
Shazid Md.
Sharker
 c,
Tabassum Binte
Kashem
c,
Yuejin
Kim
ab,
Jeehee
Lee
d,
Minok
Park
e,
Eunjeong
Kim
bf,
YunJae
Jung
g,
Jinkyu
Lim
*h,
Seungwon
Ryu
*g and
Kyueui
Lee
*abi
c,
Tabassum Binte
Kashem
c,
Yuejin
Kim
ab,
Jeehee
Lee
d,
Minok
Park
e,
Eunjeong
Kim
bf,
YunJae
Jung
g,
Jinkyu
Lim
*h,
Seungwon
Ryu
*g and
Kyueui
Lee
*abi
aDepartment of Chemistry, Kyungpook National University, Daegu 41566, South Korea. E-mail: kyueui@knu.ac.kr
bKNU Institute of Basic Sciences and KNU G-LAMP Project Group, Kyungpook National University, Daegu 41566, South Korea
cDepartment of Pharmaceutical Sciences, North South University, Dhaka 1229, Bangladesh
dDepartment of Orthopaedic Surgery, Stanford University School of Medicine, Stanford, CA 94305, USA
eEnergy Technologies Area, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
fBK21 FOUR KNU Creative BioResearch Group, Department of Biology, College of Natural Sciences, Kyungpook National University, Daegu 41566, Republic of Korea
gDepartment of Microbiology, Gachon University College of Medicine, Incheon 21999, South Korea
hDepartment of Energy and Environmental Engineering, The Catholic University of Korea, Bucheon 14662, South Korea
iBiomedical Research Institute, Kyungpook National University Hospital, Daegu 41940, South Korea
First published on 10th October 2024
Abstract
A PHD (prolyl hydroxylase) inhibitor, 1,4-dihydrophenonthrolin-4-one-3-carboxylic acid (1,4-DPCA), is a drug that can artificially promote tissue regeneration by enhancing metabolic activity through the upregulation of hypoxia inducible factor 1 subunit alpha (Hif-1α) under normoxic conditions. This study presents a novel design methodology for a drug delivery system to maximize the regenerative effect of 1,4-DPCA. Specifically, by encapsulating 1,4-DPCA in polydopamine (PDA) that generates reactive oxygen species (ROS), the combined effects of Hif-1α upregulation and the induction of cellular antioxidant defense mechanisms by localized ROS can significantly enhance tissue regeneration. The study confirmed that each material (PDA and 1,4-DPCA) triggers a positive synergistic effect on the regenerative mechanisms. As a result, the use of a PDA drug delivery system loaded with 1,4-DPCA showed approximately six times greater bone regeneration compared to the control (no treatment) in a mouse calvarial defect model.
 Kyueui Lee | Kyueui Lee earned his B.S. and Ph.D. degrees in Chemistry from KAIST. He later served as a visiting scholar and postdoctoral researcher in the Department of Bioengineering at UC Berkeley. He is currently an assistant professor in the Department of Chemistry at Kyungpook National University. His research focuses on the synthesis of functional biomedical polymers, understanding the mechanisms of their biological responses, and conducting translational research based on these findings. |
Introduction
Bone regeneration is crucial for restoring the structural integrity and functionality of damaged bone tissue. Traditionally, bone regeneration has relied on the use of autografts, allografts, and various biomaterials to replace or repair defective bone.1–4 However, these methods have resulted in significant challenges, including donor site morbidity, limited graft availability, immune rejection, and a heightened risk of infection.5 Furthermore, these approaches often fall short of fully replicating the natural bone healing process, leading to less-than-ideal outcomes in terms of integration and mechanical stability. In response to these limitations, there is increasing interest in drug-induced therapeutic strategies that specifically enhance bone regeneration at the targeted site.The compound 1,4-dihydrophenonthrolin-4-one-3-carboxylic acid (1,4-DPCA) is a small molecule that promotes tissue regeneration by acting as a proline hydroxylase (PHD) inhibitor.6 The detailed mechanisms are as follows: 1,4-DPCA inactivates PHD, leading to the upregulation of Hif-1α instead of its spontaneous degradation. This upregulation promotes erythropoiesis, metabolism, and angiogenesis, thereby inducing tissue regeneration.7,8 Notably, several studies have reported that the regenerative effect of 1,4-DPCA caused by increased Hif-1α protein expression is particularly effective in bone regeneration. For example, 1,4-DPCA injected into the injured area of a mouse has been shown to promote cartilage tissue regeneration,6,9 and its delivery has facilitated alveolar bone regeneration in a periodontal disease mouse model.10 Furthermore, it was revealed that this bone regenerative effect is accelerated by the inherent properties of Hif-1α, including its inhibitory effect on the inflammatory response11 and its ability to regulate osteogenic differentiation.12 However, despite the effective regenerative capabilities of 1,4-DPCA, several issues need to be addressed. For example, due to its inherent hydrophobicity, it has low solubility in water, making it difficult to use independently. Additionally, there are concerns about cytotoxicity when using high concentrations of 1,4-DPCA. A hydrogel-based drug delivery system (DDS) that chemically binds to 1,4-DPCA and releases it via ester hydrolysis has been developed to overcome this;6,9 however, rapid decomposition caused by enzymatic reactions in vivo limits control over drug release. Taken together, the development of a new DDS for 1,4-DPCA is essential to maximize its tissue regeneration effect.
A representative synthetic polyphenol, polydopamine (PDA), is highly valued for its role as a versatile drug carrier.13 This capability stems from PDA's unique chemical structure, which allows it to encapsulate a wide range of chemical drugs efficiently. The catechol groups in PDA facilitate diverse non-covalent interactions, including hydrogen bonding, electrostatic interactions, and π–π stacking.14,15 These interactions are not only pivotal for stabilizing the encapsulated drugs but also enhance the loading capacity of PDA, enabling it to control release the various types of therapeutic agents effectively.16,17 This multifunctionality makes PDA an advantageous candidates in the design of targeted drug delivery systems, offering potential improvements in drug solubility, stability, and controlled release profiles.
The efficacy of PDA extends beyond its role as a versatile drug carrier; it is also recognized for its potential to enhance bone regeneration.18 PDA can produce ROS and induce localized oxidative stress, similar to natural polyphenols.19,20 The generated ROS can activate the expression of antioxidant genes by regulating the Nuclear factor erythroid-2-related factor 2 (Nrf2) signaling pathway,21–23 which is crucial for maintaining cellular homeostasis within bone tissue.24,25 This regulation plays a significant role in mitigating inflammation,26 and promoting the proliferation and differentiation of osteoblasts, which are essential for bone formation.27,28 Therefore, maintaining an appropriate level of H2O2 production can support the bone healing process.29–31 In this context, a ROS-generating PDA-based drug carrier could be particularly advantageous for enhancing bone regeneration, addressing the specific needs of bone repair and recovery.
Based on these considerations, we hypothesized that loading 1,4-DPCA onto PDA and delivering it as a single DDS could maximize bone regeneration. This is because the regenerative ability of 1,4-DPCA, combined with the promoted antioxidant gene expression from ROS delivery by the PDA nanoparticle-based drug carrier, can achieve a synergistic effect. Additionally, the drug release kinetics of 1,4-DPCA loaded onto PDA can be controlled by non-covalent interactions (e.g., hydrogen bonding) between the drug and the drug carrier, which we expect to address the inherent cytotoxicity problem of 1,4-DPCA associated with its burst release.
Herein, we demonstrated that encapsulating the regenerative drug 1,4-DPCA in PDA drug carriers and delivering it to bone defect sites can maximize bone regeneration efficacy. Scheme 1 illustrates the detailed mechanism by which the developed DDS (PDA w/1,4-DPCA NPs) promotes bone tissue regeneration. In the DDS, PDA NPs induce ROS generation, which activates Nrf2, thereby promoting the expression of antioxidant genes, reducing inflammation, and enhancing cell proliferation. Simultaneously, 1,4-DPCA inhibits prolyl hydroxylase (PHD), stabilizing Hif-1α, which promotes erythropoiesis, metabolism, and angiogenesis. The ROS-generating capability of the developed DDS was confirmed through a peroxide assay. Subsequently, RT-qPCR and ELISA analyses demonstrated that the released ROS and 1,4-DPCA each promote the expression of antioxidant genes (heme oxygenase 1 (Hmox1) and Glutathione S-transferase alpha 1 (Gsta1)) and Hif-1α, respectively. As a result of the synergistic effects on tissue regeneration, bone tissue regeneration was maximized, as confirmed by in vitro ALP analysis and an in vivo mouse cranial defect model. Considering the versatility of the tissue regeneration mechanism of the developed DDS, it is expected that its use can be extended beyond bone defect-related diseases to other conditions requiring accelerated tissue regeneration.
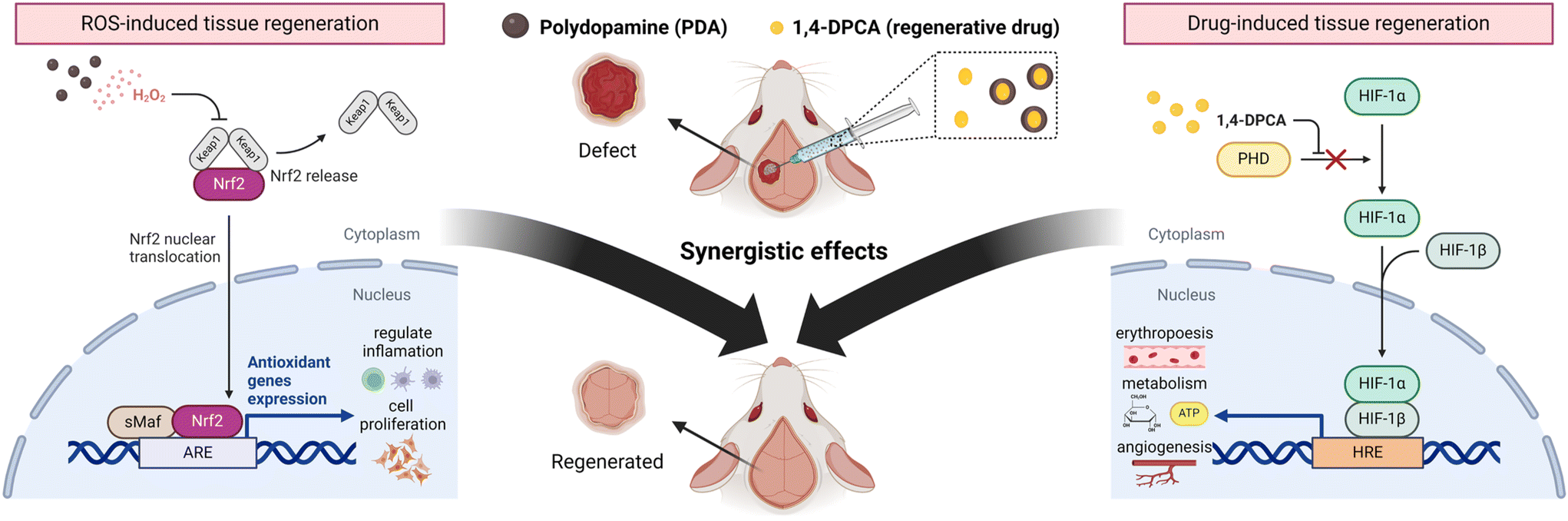 | ||
| Scheme 1 The combination of PDA NPs and 1,4-DPCA leads to a synergistic effect that enhances bone tissue regeneration in vivo. | ||
Experimental section
Chemicals and materials
Dopamine hydrochloride was purchased from Thermo Scientific (Massachusetts, MA, USA), and sodium chloride (NaCl) was purchased from SAMCHUN (Pyeongtaek, South Korea). Ammonium hydroxide, hydrochloric acid, and hexane were purchased from OCI (Seoul, South Korea). All other chemical reagents were purchased from Sigma-Aldrich (St Louis, MO, USA). L929 murine fibroblast and MC3T3-E1 murine preosteoblast cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Minimum Essential Medium Alpha Modification (Alpha MEM) was purchased from WelGene (Gyeongsan, South Korea). Dulbecco's Modified Eagle Medium (DMEM, high glucose), fetal bovine serum (FBS), penicillin–streptomycin, trypsin 0.25% solution, and Dulbecco's phosphate-buffered saline (DPBS) were purchased from Cytiva (Marlborough, MA, USA). The Cell Counting Kit-8 (CCK-8) for cytotoxicity test was purchased from Dojindo (Kumamoto, Japan).Synthesis and characterization of 1,4-DPCA
The drug 1,4-DPCA was synthesized following the previous paper.6,9 In detail, the synthesis of 1,4-DPCA was performed by the following procedures: 8-aminoquinoline (1.6 g, 11.1 mmol) and diethyl ethoxymethylenemalonate (2.36 mL, 11.7 mmol) were heated to 100 °C for 1 h. Then, diphenyl ether (33 mL) was added, and the mixture was refluxed at 250 °C for 2 h before cooling to room temperature. The precipitate product (1) was separated by centrifugation, washed with 10 mL of diethyl ether, and subsequently washed twice with 5 mL of diethyl ether. The product (1) (2.00 g, 7.46 mmol) was combined with 40 mL of 10% (w/v) KOH, refluxed (110 °C) for 1 h, allowed to cool to room temperature, and the residual diphenyl ether was extracted using 28 mL of petroleum ether (boiling point 80–110 °C). The product was precipitated with 40 mL of 10% (w/v) HCl, filtered, washed with deionized water, and dried under high vacuum conditions overnight. The synthesized 1,4-DPCA was confirmed using proton nuclear magnetic resonance (1H NMR) spectroscopy (AVANCE III 500, Bruker, Billerica, MA, USA).Synthesis of the PDA NPs
A cosolvent system consisting of deionized water (8 mL), ethanol (4 mL), and 0.2 mL of 25% NH4OH was used as the reaction solution. Dopamine hydrochloride (0.05 g) dispersed in deionized water (1 mL) was gradually injected into the mixture solution. After the mixture was allowed to react with constant stirring (200 rpm) for 24 h, the PDA NPs were collected by centrifugation at 8000 rpm for 5 min. The process of washing with deionized water and centrifugation at 8000 rpm for 5 min was performed three times. The prepared PDA NPs were lyophilized and frozen before use.Synthesis and characterization of 1,4-DPCA-loaded PDA NPs
The drug 1,4-DPCA (0.01 g) was dispersed in a cosolvent composed of ethanol (4 mL) and deionized water (8 mL) with 0.2 mL of 25% ammonium hydroxide (NH4OH). Subsequently, 0.05 g of dopamine hydrochloride was dispersed in 1 mL of deionized water and was injected into the 1,4-DPCA mixture. By varying the reaction time while maintaining constant stirring at 200 rpm, 1,4-DPCA-loaded PDA NPs (PDA w/DPCA NPs) of different average size of 282 nm were produced over stirring durations of 24 h. The nanoparticles were collected by centrifugation at 8000 rpm for 5 min. The process of washing with deionized water, followed by centrifugation, was repeated three times. The gathered nanoparticles were then lyophilized and frozen for later use. The average hydrodynamic diameter of the nanoparticles was determined by dynamic light scattering (ELSZ-2000ZS, Otsuka Electronics, Japan).Morphological analysis of PDA w/DPCA NPs
The morphology (i.e., surface and nanostructure properties) of PDA w/DPCA NPs was observed using scanning electron microscopy (SEM) (SU8220 & SU8230, Hitachi, Tokyo, Japan) and transmission electron microscopy (TEM) (Tecnai 12, Phillips, Amsterdam, Netherlands).X-ray photoelectron spectroscopy analysis
The element composition of the synthesized NPs was analyzed by X-ray photoelectron spectroscopy (XPS) (Thermo Fisher, Nexsa, MA, USA). For sample preparation, copper double-sided tape was attached to a silicon wafer, and the sample was securely positioned and dried with nitrogen. The deconvoluted spectra were obtained using Avantage software (version 4.0).Calculation of the drug loading efficiency
To calculate the drug loading efficiency, we forcefully released the total amount of drug that was loaded into the carrier. Briefly, 1 mg of lyophilized PDA w/DPCA NPs was dispersed in 50 mL of dimethyl sulfoxide (DMSO). The solution was subjected to tip sonication using a pulsed power system (Sonics & Materials, Inc.) for 100 h, employing a cycle of 30 s on/30 s off at 30 Amp% and 500 W. After drug release, centrifugation was performed to collect the supernatant for high-performance liquid chromatography (HPLC) analysis using a Nexera X2 system (Shimadzu, Kyoto, Japan). The mobile phase, consisting of acetonitrile and water (30/70; v/v), was used as the eluent at a constant flow rate of 1 mL min−1 for HPLC analysis. The fully released drug was quantified using the standard curve of 1,4-DPCA.In vitro drug release test
To assess the in vitro drug release rate, 3 mg of lyophilized PDA w/DPCA NPs with a particle size of 282 nm was introduced into a conical tube containing 3 mL of 1× phosphate-buffered saline (1× PBS) at pH 7.4. The samples were allowed to release the 1,4-DPCA over periods ranging from 1 h to 31 days. Following each release interval, centrifugation was performed to separate the supernatant for HPLC analysis. The HPLC analyses used a mobile phase of acetonitrile and water (30/70; v/v) as the eluent, maintaining a constant flow rate of 1 mL min−1. The concentration of the drug released at each time point was quantified against the standard curve of 1,4-DPCA.Quantification of ROS from PDA w/DPCA NPs
The concentrations of ROS (i.e., hydrogen peroxide) generated from PDA w/DPCA NPs and its control PDA were quantified according to the instructions of the Quantitative Peroxide Assay Kit (Thermo Fisher Scientific, MA, USA). Briefly, samples were prepared at different concentrations (10, 25, and 50 μg mL−1) in 1× PBS solution (pH 7.4). Each solution mixture was incubated for 20 minutes at room temperature, and the absorbance was measured at 595 nm using a microplate reader (Infinite 200 Pro, Tecan, Männedorf, Switzerland). A standard curve for the calculation was generated by measuring the absorbance at the same wavelength (595 nm) with a 0.001 to 1 mM H2O2 solution.Enzyme-linked immunosorbent assay (ELISA)
MC3T3-E1 cells were thawed, cultured in T-75 flasks through three cycles for adaptation, then dissociated with 0.25% trypsin-EDTA (Thermo Fisher Scientific, MA, USA), and counted using 0.4% trypan blue (Sigma-Aldrich, MO, USA). The cells were seeded at 2.2 × 106 cells per well in 100 mm dishes and incubated for 2 days in Alpha MEM medium supplemented with 10% FBS and 1% penicillin–streptomycin antibiotics in an incubator with 5% CO2 at 37 °C. Then, to induce osteogenic differentiation, a special medium containing 50 μg mL−1 of L-ascorbic acid 2-phosphate and 10 mM β-glycerophosphate was prepared and PDA NPs, PDA w/DPCA NPs, and 1,4-DPCA were added at a concentration of 10 μg ml−1. In addition, equal amounts of DMSO, which was used to dissolve 1,4-DPCA, were added to all samples and the cells were incubated for 3 days. The differentiated cells were lysed in RIPA buffer (ELPIS Biotech, Daejeon, South Korea) to extract cellular proteins, which were then quantified using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, MA, USA). Quantification of Hif-1α in MC3T3-E1 cells was performed using the human/mouse total HIF-1α/HIF1A Kit (R&D Systems, MN, USA), with 100 μg of total protein used per well. Absorbance was measured at 450 nm with a correction at 540 nm using a microplate reader (Infinite 200 Pro, Tecan, Männedorf, Switzerland).Western blot analysis
MC3T3-E1 cells were thawed, grown in T-75 flasks for three cycles for adaptation, then dissociated with 0.25% trypsin-EDTA (Thermo Fisher Scientific, MA, USA) and counted using 0.4% trypan blue (Sigma-Aldrich, MO, USA). Cells were seeded at 2.2 × 106 cells per well in 100 mm dishes and incubated for 2 days in Alpha-MEM medium supplemented with 10% FBS and 1% penicillin–streptomycin in an incubator with 5% CO2 at 37 °C. Then, to induce osteogenic differentiation, a specialized medium containing 50 μg ml−1L-ascorbic acid 2-phosphate and 10 mM β-glycerophosphate was prepared and PDA NPs, PDA w/DPCA NPs and 1,4-DPCA were added at a concentration of 10 μg ml−1. In addition, equal amounts of DMSO, which was used to dissolve 1,4-DPCA, were added to all samples and the cells were incubated for 3 days. The differentiated cells were lysed in RIPA buffer (ELPIS Biotech, Daejeon, South Korea) to extract cellular proteins, which were then quantified using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, MA, USA). The resulting cell lysates were denatured and separated on sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE), followed by transfer to nitrocellulose membranes (GE Healthcare, IL, USA). The membranes were then blocked in 5% non-fat milk in Tris-buffered saline supplemented with Tween 20 (TBST) for 1 hour at room temperature, followed by immunoblotting with primary antibodies overnight at 4 °C. The membranes were then washed twice with TBST for 20 minutes each and immunoblotted with secondary antibodies for 2 hours at room temperature, followed by two washes with TBST for 20 minutes each. Protein bands were detected with SuperSignal™ West Femto maximum sensitivity substrate (Thermo Fisher Scientific, MA, USA) on a ChemiDoc™ XRS + system (Bio-rad, CA, USA) using Hif-1α (abcam, Cambridge, UK) and β-actin (Cell Signalling Technology, MA, USA) antibodies. Bands were quantified using ImageJ software.Flow cytometric analysis of Hif-1α protein expression
To evaluate Hif-1α protein expression in MC3T3 cells, treated cells, as in the ELISA and western blot experiments, were pre-washed with sterile PBS and harvested by trypsin treatment. The cells were then filtered through 70 μm strainers and blocked with anti-mouse CD16/CD32 (TruStain FcX antibody, Biolegend, CA, USA) for 10 minutes on ice. After blocking, the cells were fixed and permeabilized using the FoxP3/Transcription Factor Fixation/Permeabilization Kit (Invitrogen, CA, USA) for 30 minutes. Next, the cells were stained with anti-human/mouse Hif-1α monoclonal antibody (clone 241812, APC-conjugated, R&D Systems, MN, USA) for 1 hour on ice. Hif-1α expression was detected using a FACSymphony A5 flow cytometer (BD Biosciences, CA, USA) and analyzed using FlowJo software (version 10, BD Biosciences, CA, USA).RNA purification and real-time quantitative PCR
The MC3T3-E1 cells were cultured for 2 days in Alpha MEM medium supplemented with 10% FBS and 1% penicillin–streptomycin antibiotics in an incubator with 5% CO2 at 37 °C. Then, to induce osteogenic differentiation, a special medium containing 50 μg mL−1 of L-ascorbic acid 2-phosphate and 10 mM β-glycerophosphate was prepared and PDA NPs, PDA w/DPCA NPs, and 1,4-DPCA were added at a concentration of 10 μg ml−1. In addition, equal amounts of DMSO, which was used to dissolve 1,4-DPCA, were added to all samples and the cells were incubated for 3 days. Total RNA was isolated from the cells using TRIzol™ Reagent (Invitrogen, MA, USA) and prepared for complementary DNA synthesis using the PrimeScript 1st strand cDNA Synthesis Kit (Takara Bio, Shiga, Japan). Gene expression was determined by RT-qPCR analysis using AccuPower® 2X GreenStar™ qPCR Master Mix (BIONEER, Daejeon, South Korea) on CFX Duet Real-time PCR system (Bio-rad, CA, USA). Gene expression levels were normalized using the β-actin housekeeping gene.Cytotoxicity test
The cytotoxicity of the synthesized 1,4-DPCA, PDA NPs, and PDA w/DPCA NPs with a particle size of 282 nm was evaluated using a CCK-8 assay. Briefly, mouse fibroblast L929 cells were seeded into a 96-well cell culture plate (Costar, USA) at a density of 1 × 104 cells per 100 μL per well. They were cultured in DMEM supplemented with 10% FBS and 1% penicillin–streptomycin and incubated in a 5% CO2 environment at 37 °C. After 24 h of culture, a range of compounds (10, 25, and 50 μg mL−1) were added to the cells. Samples treated with only DMEM and DMSO were used as controls. After 24 h of incubation, 10 μL of CCK-8 solution was added to each well and further incubated for 3 h. Subsequently, absorbance was measured at 450 nm using a microplate reader (Infinite 200 Pro, Tecan, Männedorf, Switzerland). Cell viability was calculated according to the following equation:
Hemolysis assay
The hemolytic activity of the PDA w/DPCA NPs with a particle size of 282 nm was investigated according to the reference.32 EDTA-stabilized single-donor human whole blood was purchased from Innovative Research (Novi, MI, USA) and centrifuged at 500g for 5 min. The pellet was washed with 150 mM NaCl solution and pH 7.4 1× PBS buffer, then resuspended in a series of buffers representing a pH range from physiological (pH 7.4) to late endolysosomes (pH 6.2). A volume of 190 μL of red blood cell (RBC) suspension was seeded in each well of a 96-well plate. Subsequently, 10 μL of various concentrations of the DDS, prepared in DPBS, were added to each well. The plate was then incubated at 37 °C for 1 h. Afterward, the 96-well plate was centrifuged at 500g for 5 min to pellet the RBCs. The absorbance of hemoglobin in the supernatants was measured at 590 nm using a microplate reader (Infinite 200 Pro M Plex, Tecan, Männedorf, Switzerland). Considering that, in addition to hemoglobin, other cytoplasmic components derived from erythrocytes, including proteins and carbohydrates, may contain small amounts of signal that interfere with the spectrophotometric measurements, 100% hemolysis was calibrated with Triton X-100-treated erythrocyte lysates. Hemolysis percentages of the RBCs was calculated using the following formula: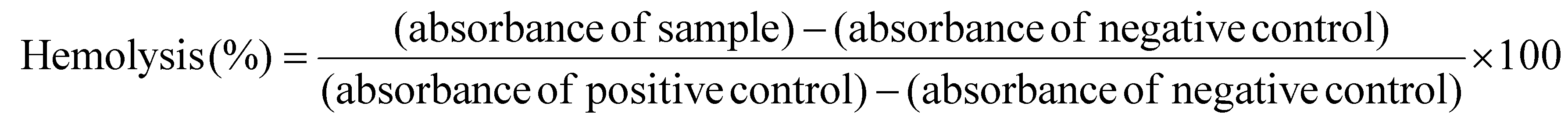
A hemolysis percent value <2% indicates that the test sample is not hemolytic; between 2% and 5% indicates that the test sample is slightly hemolytic; and >5% indicates that the test sample is hemolytic.
Alkaline phosphatase activity test
MC3T3-E1 cells were seeded into a 96-well cell culture plate at a density of 1 × 104 cells per well. After 24 h, the culture medium supplemented with 10% FBS and 1% penicillin–streptomycin antibiotics in Alpha MEM was replaced with an osteogenic medium (OSM). The OSM was prepared by adding 10 mM β-glycerol phosphate and 50 μg mL−1L-ascorbic acid 2-phosphate to the culture medium. PDA w/DPCA NPs were added to the medium at a concentration of 10 μg mL−1 and mixed with OSM. For 1, 3, and 7 days, the cells were cultured while replacing the medium with either OSM (control) or OSM containing PDA w/DPCA NPs. After 1, 3, and 7 days, the cells were washed with DPBS and lysed with lysis buffer. The absorbance was measured at a wavelength of 405 nm using a microplate reader (Infinite 200 Pro M Plex, Tecan, Männedorf, Switzerland) with an ALP kit (Sigma Aldrich, St Louis, MO, USA) according to the following equation:where ODT50 = OD value at 405 nm of sample at 50 min, ODT0 = OD value at 405 nm of sample at 0 min, ODCal = OD value at 405 nm of the calibrator solution included in the kit, ODBlank = OD value at 405 nm of deionized water, RxnVol = total reaction volume (200 μL), T = reaction time (50 min), and SmplVol = amount of sample (50 μL) used in the reaction.

In vivo experiment using a mouse calvarial defect model
The animals were housed under standard laboratory conditions (relative humidity 55–65%, room temperature 23 ± 2 °C, and 12 h/12 h light/dark cycle). Adult Swiss albino mice 7–8 weeks of age with a weight of 30–35 g were obtained from the animal house of the Department of Pharmaceutical Sciences, North South University (NSU), Bangladesh. The animals were fed a standard diet with ad libitum access to water. All experiments were performed with approval by the NSU Institutional Animal Care and Use Committee (2023/OR-NSU/IACUC/1101). Mice were housed in separate cages, and allowed to acclimate to the animal housing conditions for 7 days before surgery. The mice were anesthetized with an intraperitoneal injection of ketamine (75 mg per kg body weight).33 A sagittal skin incision was made over the scalp, from the frontal bone to the occipital bone, and the skin flap included the periosteum. Full-thickness circular defects (5 mm in diameter) were created in the left parietal bone using a 5 mm diamond-coated trephine bur (Cogoogon, China) with a slow dental handpiece (Marathon-N3, SMT, China).34 Untreated, empty calvarial defects were used as negative controls. The experimental group included groups treated with PDA NPs or PDA w/DPCA NPs with a particle size of 282 nm. The drug loading concentration was 10 μg mL−1. To minimize the risk of the drug spreading to other areas, the solution was applied drop by drop on the calvarial defect side and allowed to accumulate in the damaged area. Given that PDA NPs readily aggregate, the solution was used after bath sonication before use. The incisions were closed with 5–0 catgut sutures (Ethicon), and a warming lamp was used to keep the mice warm until they achieved full recovery. The animals were returned to their cages and monitored daily for complications or abnormal behavior.Histological evaluation based on hematoxylin and eosin and Masson's trichrome staining
Bone sections were collected after 4 weeks of treatment. Twelve samples were demineralized with 10% EDTA, and embedded in paraffin. The sections were stained with hematoxylin and eosin (H&E) and Masson's trichrome (MT). All tissue sections were imaged using an optical microscope (Euromex, BB.1153-PLi, Holland). Bone regeneration and the area containing osteoblasts were analyzed using ImageJ version 1.40 (National Institutes of Health, Bethesda, MD, USA).35Micro X-ray analysis
Four weeks post-surgery, the skulls (n = 5) were fixed overnight in 10% neutral buffered formalin at room temperature, followed by storage in 1× PBS at 4 °C until they were ready for micro X-ray imaging. Imaging was performed using a Triup Mobile X-ray Machine (TRP-100, Triup International, China). The scans were conducted according to the calibration parameters, with standardized reconstructions achieved at settings of 40 kV, 10 mAs, and an exposure duration of 0.1 s.Statistical analysis
The reported values are presented as the mean ± standard error (SE). For comparisons involving three or more groups or non-normally distributed data, one-way ANOVA or the Kruskal–Wallis test, a non-parametric method, were utilized to assess statistical differences. All statistical analyses were performed using OriginPro 9. A p-value of less than 0.05 was considered statistically significant.Results and discussion
Fabrication of the DDS (PDA NPs loaded with 1,4-DPCA)
The preparation of the DDS begins by dispersing the regenerative drug, 1,4-DPCA, in a mixture of ethanol and deionized water. The 1,4-DPCA used for DDS formation was synthesized based on previous literature,9 and the successful synthesis was confirmed using NMR spectroscopy (Fig. S1 and S2†). In the reaction solution, 1,4-DPCA exhibited immediate aggregation due to its inherent hydrophobicity, as demonstrated by the Tyndall effect (light scattering effect) (Fig. S3†) and dynamic light scattering (DLS) analysis confirmed that the size of the drug aggregates was 33 nm ± 5.6 (Fig. S4A†). Subsequently, dopamine hydrochloride and a base (ammonia) were added to the reaction solution to induce the formation of polydopamine. During this process, various catecholic building blocks, such as dopamine quinone, leukodopamine-chrome, dopachrome, and 5,6-dihydroxyindole—oxidative derivatives of dopamine—are generated and polymerized into polydopamine through either chemical conjugation or physical assembly (Fig. 1).36–38 The resulting polydopamine, due to its inherent molecular adhesiveness, coats the surface of the drug aggregates, forming a core (drug)–shell (PDA) DDS structure. TEM analysis revealed that the morphology of the formed DDS was spherical, similar to conventional PDA nanoparticles (Fig. 2). However, while the size of PDA nanoparticles synthesized under the same reaction conditions was 222 nm ± 6.2 nm (Fig. S4B†), the average diameter of the DDS was larger, measuring 282 nm ± 26.2 nm (Fig. S4C†). This size difference further confirms the effective loading of 1,4-DPCA (the drug) onto the DDS. Additionally, by subtracting the diameter of the drug aggregates from the total DDS diameter and dividing by two, the thickness of the polydopamine coating on the drug aggregates was calculated to be approximately 124.5 nm.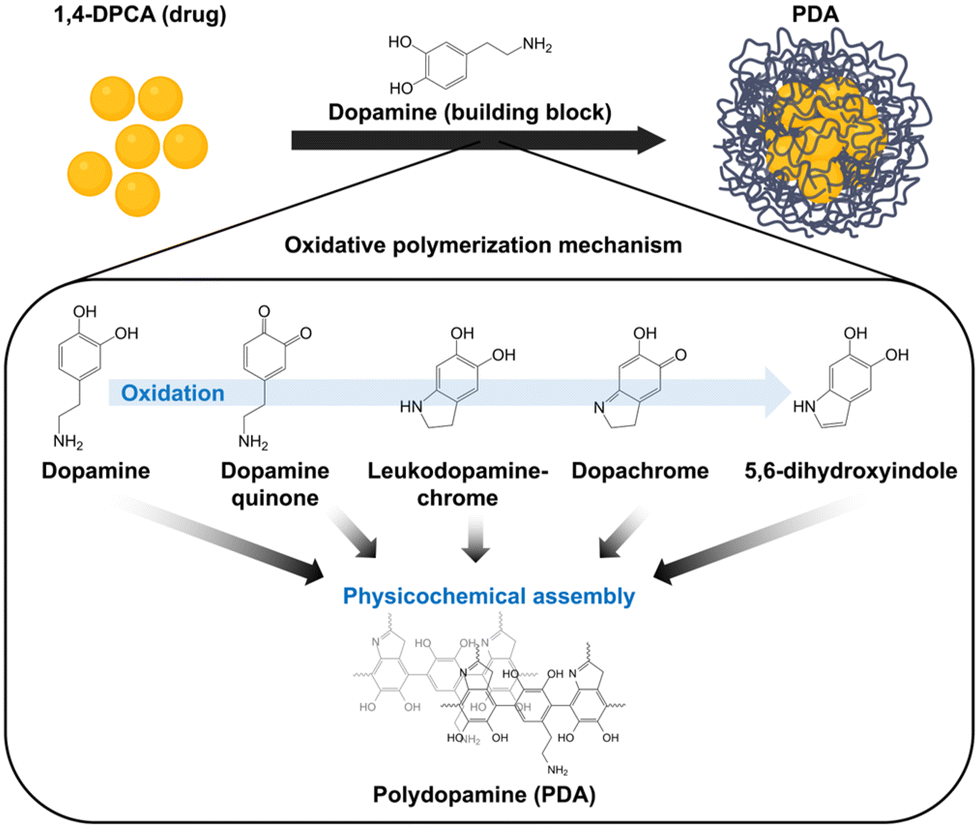 | ||
| Fig. 1 Schematic representation of the preparation of the drug delivery system (1,4-DPCA-loaded polydopamine nanoparticles). | ||
 | ||
| Fig. 2 Transmission electron microscopy images of (A) polydopamine nanoparticles (PDA NPs) and (B) polydopamine nanoparticles with 1,4-DPCA (PDA w/DPCA NPs). | ||
Surface analysis of DDS to demonstrate the location of the loaded drug
To confirm whether 1,4-DPCA was loaded at the core level of the PDA NPs, as illustrated in Fig. 1, XPS surface analysis was performed. Given that 1,4-DPCA has different proportions of chemical bonds (N1s and O1s) compared with PDA (Fig. 3), the XPS spectra of the PDA NPs with randomly incorporated 1,4-DPCA would show distinct differences from those of pure PDA NPs. However, according to the XPS results, the proportion of each chemical bond in PDA w/DPCA NPs was comparable to that of pure PDA NPs. For example, in the high-resolution N1s XPS spectrum, the proportions (%) of the –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–, –NH–, and –NH2 peaks for PDA NPs and PDA w/DPCA NPs were 23%, 58%, and 20%, and 25%, 57%, and 18%, respectively. By contrast, the proportions (%) of the –NC–, –NH–, and –NH2 peaks for 1,4-DPCA were 53%, 25%, and 22%, respectively. Similarly, in the high-resolution O1s XPS spectrum, the proportions (%) of the OC and O–C peaks for PDA NPs and PDA w/DPCA NPs were 92% and 8%, and 93% and 7%, respectively. For 1,4-DPCA, the proportions (%) of the OC and O–C peaks were 59% and 41%, respectively. Because XPS analysis usually examines up to tens of nanometers beneath the surface39 and does not reach the core of the PDA NPs, we can deduce that 1,4-DPCA is mainly located within the core of the nanoparticles.
C–, –NH–, and –NH2 peaks for PDA NPs and PDA w/DPCA NPs were 23%, 58%, and 20%, and 25%, 57%, and 18%, respectively. By contrast, the proportions (%) of the –NC–, –NH–, and –NH2 peaks for 1,4-DPCA were 53%, 25%, and 22%, respectively. Similarly, in the high-resolution O1s XPS spectrum, the proportions (%) of the OC and O–C peaks for PDA NPs and PDA w/DPCA NPs were 92% and 8%, and 93% and 7%, respectively. For 1,4-DPCA, the proportions (%) of the OC and O–C peaks were 59% and 41%, respectively. Because XPS analysis usually examines up to tens of nanometers beneath the surface39 and does not reach the core of the PDA NPs, we can deduce that 1,4-DPCA is mainly located within the core of the nanoparticles.
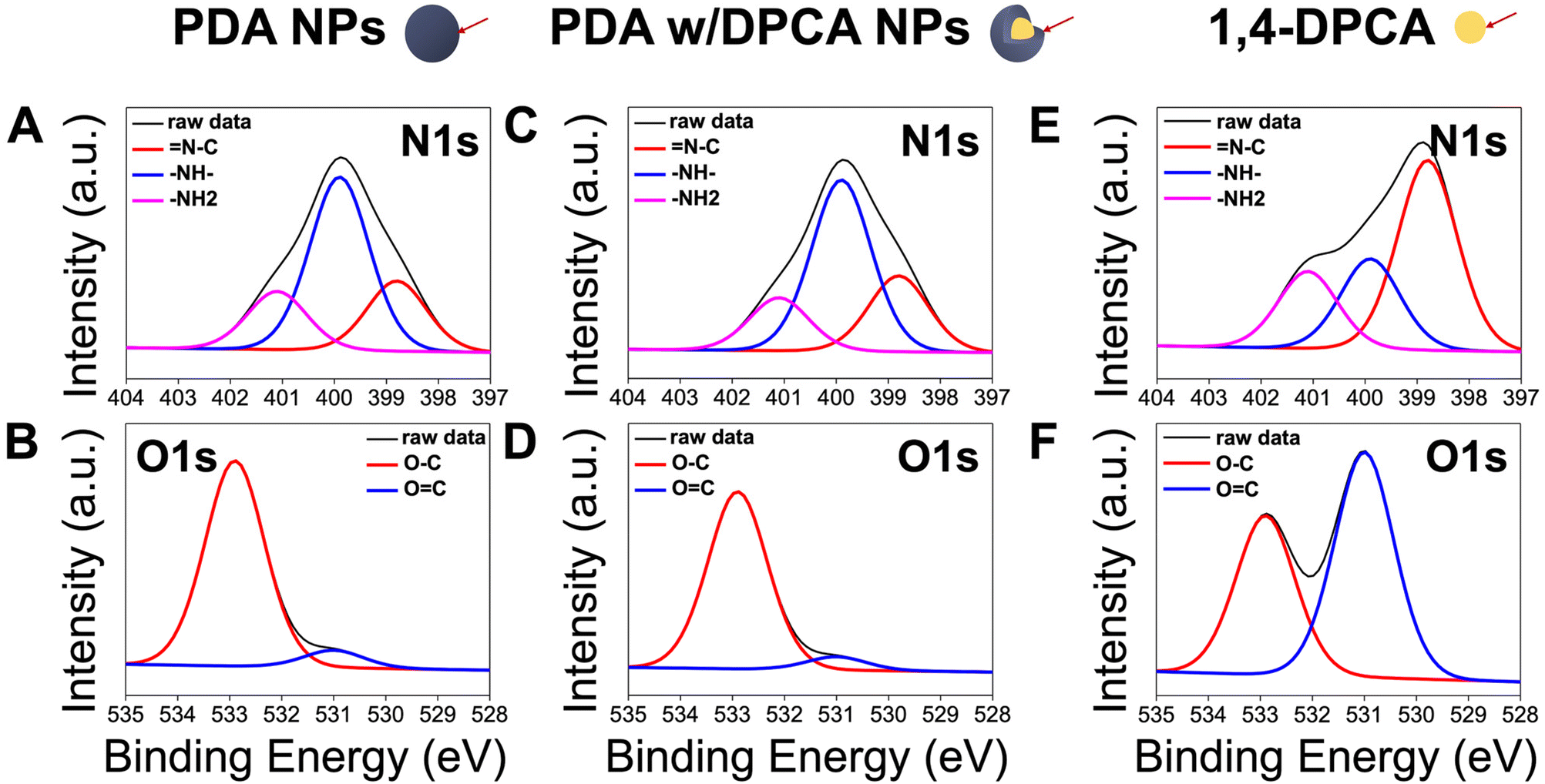 | ||
| Fig. 3 High-resolution N1s and O1s XPS spectra of (A and B) PDA NPs, (C and D) PDA NPs with DPCA, and (E and F) 1,4-DPCA. | ||
To further validate this conclusion, we conducted theoretical calculations based on two hypotheses: (1) the drug is randomly distributed within the PDA nanoparticles, and (2) the drug is concentrated in the core. These calculations were based on the atomic percentage (at%) values obtained from high-resolution XPS data, focusing particularly on the peaks showing the largest differences between 1,4-DPCA and PDA, namely the O–C bond in the O1s peak (Table S1†) and the –NH– bond in the N1s peak (Table S2†). The calculations were also based on the assumption that the drug loading efficiency is approximately 10 wt% (please refer to the Experimental section for detailed calculation methods). As a result, the case where the drug aggregates are located in the core of the PDA nanoparticles yielded values closer to the actual experimental results compared to the randomly distributed case (Tables S1 and S2†). Therefore, combining the XPS analysis and DLS data, we conclude that the developed DDS has a core–shell structure.
Controlled drug release from the DDS in vitro
We first demonstrated through HPLC whether the drug was successfully loaded into the synthesized DDS. For this, we released 1,4-DPCA from the PDA w/DPCA NPs at a concentration of 50 μg mL−1 in a 1× PBS solution (pH 7.4) for 1 hour and analyzed the obtained supernatant by HPLC. The peak appeared at the same retention time as that of 1,4-DPCA, which confirms that the regenerative drug was successfully loaded into the PDA NPs (Fig. 4A). We further validated the drug release profile of PDA w/DPCA NPs via HPLC analysis using the standard curve of 1,4-DPCA (Fig. 4B). To achieve this, we first measured the experimental drug loading efficiency of the PDA w/DPCA NPs, which was found to be approximately 10 wt%. Based on this, we calculate the quantity of drug released from the total amount of drug loaded into the NPs. As a result, PDA w/DPCA NPs steadily released 9.8 wt% of the loaded drug within 7 days (168 hours, Fig. 4C). The sustained drug release behavior can be attributed to the strong chemical interactions between PDA and 1,4-DPCA since the encapsulated drug must traverse the PDA layers during release. To confirm the influence of the chemical interaction between 1,4-DPCA and PDA on the drug release kinetics, we performed column chromatography by comparing the drug release rates of a column packed with PDA-coated silica beads with those of a column packed with only silica beads. We observed a significant decrease in the drug release rate of the column packed with PDA-coated silica beads (Fig. S5†). This result confirms our initial hypothesis, indicating that interaction with the PDA coating slows the release rate of the drug. This controlled release is especially advantageous for enhancing drug delivery efficacy of 1,4-DPCA, as it is highly toxic when released in bursts. | ||
| Fig. 4 (A) Confirmation of drug release from PDA w/DPCA NPs, (B) standard curve for the calculation of drug release using various concentrations of 1,4-DPCA. (C) In vitro drug release profile of PDA w/DPCA NPs. a.u. denotes arbitrary unit. | ||
ROS generation and activation of cellular antioxidant response by the DDS
ROS, including hydrogen peroxide (H2O2), can be generated from the drug carrier polydopamine as a byproduct through the autoxidation process of the catechol moiety.40 To demonstrate that polydopamine generates H2O2, we performed a peroxide assay to quantify the H2O2 produced (Fig. 5A). The assay was conducted by dissolving synthesized PDA NPs and PDA w/DPCA NPs at various concentrations (10, 25, 50 μg mL−1) in 1× PBS solution (pH 7.4) and measuring H2O2 concentration after 20 minutes of incubation at room temperature. Results showed that PDA NPs produced 9.3, 9.0, and 10.6 μM of H2O2 at 10, 25, and 50 μg mL−1, respectively (Fig. 5B). In contrast, PDA w/DPCA NPs generated approximately two-fold higher amounts of H2O2: 21.1, 26.3, and 29.5 μM at the same concentrations (Fig. 5C). This can be attributed to the electron-donating amine groups in 1,4-DPCA promoting the oxidation of the catechol moiety in polydopamine.41 Excessive ROS can oxidize cellular components, leading to cell death and triggering inflammatory responses. However, inflammation is typically induced when ROS levels reach around 300–500 μM.42 In our study, the amount of ROS generated by PDA NPs or PDA w/DPCA NPs at concentrations of 10–50 μg mL−1 remained well below this threshold, with a maximum of 29.5 μM. This indicates that at these concentrations, PDA nanoparticles release minimal ROS and are unlikely to induce an inflammatory response. Furthermore, previous studies have shown that PDA NPs help modulate oxidative stress without causing inflammation, further supporting the biocompatibility and safety of the developed DDS.43,44 | ||
| Fig. 5 (A) Schematic of peroxide assay. Quantified amount of hydrogen peroxide generated with (B) PDA NPs and (C) PDA w/DPCA NPs in pH 7.4 PBS solution (n = 7). (D) Relative gene expression of Hmox1, Gsta1, and Nrf2 in MC3T3-E1 cells treated with PDA NPs, PDA w/DPCA NPs, and 1,4-DPCA (n = 3). Asterisk symbolizes the statistical significance (p-values): **** indicate p ≤ 0.0001, n.s. = statistically not significant. | ||
The minimal ROS generated by polydopamine induces appropriate levels of oxidative stress at the targeted site, activating the Nrf2/Heme oxygenase 1 (HO-1) signaling pathway. This leads to the dissociation of Nrf2 from Kelch-like ECH associated protein 1 (Keap1), its translocation to the nucleus, binding to small Maf (musculoaponeurotic fibrosarcoma) proteins (sMaf) proteins, and activation of the antioxidant responsive element (ARE), thereby promoting the expression of antioxidant genes such as Hmox1 and Gsta1 (Scheme 1). To verify this, we analyzed the expression of Nrf2, Hmox1, Gsta1 genes in each state using qPCR. When MC3T3-E1 cells were treated with PDA NPs, PDA w/DPCA NPs, and 1,4-DPCA, there was no significant difference in the mRNA levels of Nrf2 (Fig. 5D), indicating that none of the components of the DDS affected the basal Nrf2 gene expression. On the other hand, the mRNA levels of Nrf2 target genes, Hmox1 and Gsta1, showed a significant increase in the groups treated with PDA NPs and PDA w/DPCA NPs. Specifically, Hmox1 mRNA levels increased by 4.5-fold in the PDA NPs-treated group and by 7.4-fold in the PDA w/DPCA NPs-treated group, compared to a 1.1-fold increase in the 1,4-DPCA-treated control group (Fig. 5D). Similarly, Gsta1 mRNA levels rose by 1.5-fold in the PDA NPs-treated group and by 5.2-fold in the PDA w/DPCA NPs-treated group, while the 1,4-DPCA-treated group showed a decrease to 0.63-fold (Fig. 5D). These results suggest that treatment with PDA NPs and PDA w/DPCA NPs successfully enhances the expression of Nrf2 target genes Hmox1 and Gsta1, which are involved in the antioxidant response. This implies that PDA NPs and PDA w/DPCA NPs induce ROS generation, thereby activating the Nrf2 pathway and enhancing cellular antioxidant defense mechanisms. Considering the superior ROS generation and the resultant upregulation of antioxidant gene expression observed in PDA w/DPCA NPs compared to PDA NPs, it suggests that the combination of PDA and DPCA more effectively activates the Nrf2 pathway and accelerates tissue regeneration.
Demonstration of Hif-1α upregulation at the mRNA and protein levels
To investigate the contribution of 1,4-DPCA released from PDA NPs to the upregulation of Hif-1α protein and mRNA levels (Scheme 1), we treated MC3T3-E1 cells with PDA w/DPCA NPs and conducted ELISA, western blot, FACS, and RT-qPCR analyses. ELISA results showed that PDA NPs and PDA w/DPCA NPs treatment significantly increased Hif-1α protein levels by 1.6- and 1.8-fold, respectively, while 1,4-DPCA treatment led to only a 1.2-fold increase (Fig. 6A). Similarly, western blot results confirmed that Hif-1α protein levels increased significantly by 1.2- and 2.3-fold with PDA NPs and PDA w/DPCA NPs treatment, respectively, while 1,4-DPCA treatment resulted in only a 1.7-fold increase (Fig. 6B and S6†). FACS analysis also demonstrated at the single-cell level that PDA w/DPCA NPs treatment led to a significantly greater increase in Hif-1α expression compared to the control (PDA NPs treatment alone) (Fig. 6C). These results consistently indicate that PDA w/DPCA NPs induced the greatest increase in Hif-1α protein levels. Interestingly, despite the theoretical reduction of DPCA loaded onto the PDA w/DPCA NPs to about one-tenth compared to the control (1,4-DPCA only), the effect on Hif-1α protein levels was significantly greater. These findings emphasize the effective drug delivery capability of our DDS on Hif-1α expression. Additionally, it is noteworthy that PDA itself significantly increased HIF-1α protein expression compared to the control. This could be attributed to the ROS (e.g., H2O2) generated by PDA, which mediates the dimerization of PHD via disulfide bonding, leading to the stabilization of HIF-1α.45 This suggests that the synergistic effect related to ROS and 1,4-DPCA's PHD inhibition resulted in the highest overexpression of HIF-1α protein in the PDA w/DPCA NPs treated samples. RT-qPCR results showed no statistically significant difference in Hif-1α mRNA expression levels with PDA NPs or PDA w/DPCA NPs treatment compared to the control, whereas 1,4-DPCA treatment significantly increased Hif-1α mRNA expression by 1.6-fold (Fig. S7†). This suggests that while PDA NPs and PDA w/DPCA NPs may initially promote a transient upregulation of Hif-1α mRNA, rapid mRNA degradation may prevent detection at the three-day time point.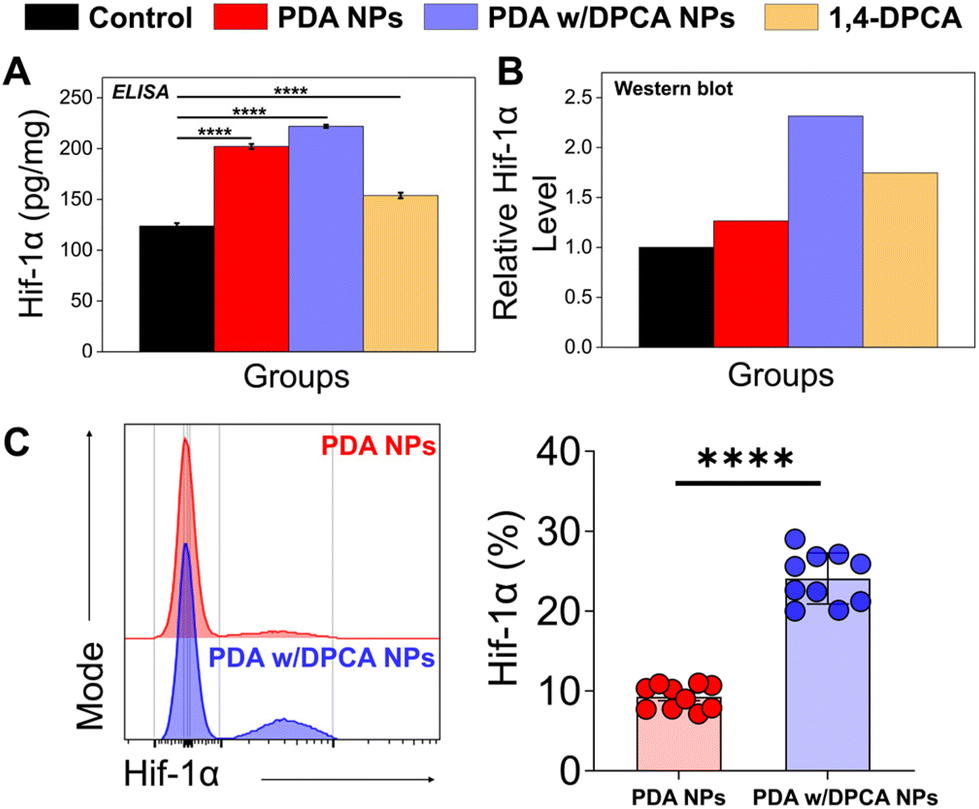 | ||
| Fig. 6 (A) Relative Hif-1α protein levels in MC3T3-E1 cells treated with PDA NPs, PDA w/DPCA NPs, and 1,4-DPCA were assessed using ELISA, and (B) western blot analysis (n = 6). (C) Histogram of Hif-1α flow cytometric data showing the frequency of Hif-1α-expressing cells (n = 10). Asterisks indicate statistical significance (p-values): **** represents p ≤ 0.0001, n.s. = not statistically significant. | ||
In vitro biocompatibility tests
To evaluate the biocompatibility of the DDS (PDA w/DPCA NPs) and establish the optimal concentration for application, cytotoxicity tests were conducted using CCK-8 and hemolysis assays at concentrations of 10, 25, and 50 μg mL−1. The CCK-8 assay results revealed that, following treatment with PDA w/DPCA NPs or PDA NPs, all sample groups at concentrations below 25 μg mL−1 showed cell viability above 80% compared to the untreated control group. According to ISO 10993-5 guidelines, cell viability above 80% indicates non-cytotoxicity, meaning that DDS concentrations of 25 μg mL−1 or lower are biocompatible (Fig. 7A). However, at the higher concentration of 50 μg mL−1, a mild cytotoxic response was observed. In the hemolysis assay, as the concentration of PDA w/DPCA NPs increased, the hemolysis rate also rose, with a slight increase observed in the pH range of late endolysosomes (pH 6.2). At the highest concentration of 50 μg mL−1, the hemolysis rate was approximately 1.9% across all pH ranges (physiological, early endosomal, and late endo–lysosomal). At concentrations of 10 and 20 μg mL−1, the DDS showed a hemolysis rate of approximately 1% across all pH ranges (Fig. 7B). According to ASTM E2524-08 standards, a hemolysis rate greater than 5% indicates that the test substance damages erythrocytes. Therefore, the safety of the DDS was demonstrated at all concentrations in the hemolysis assay. Based on the CCK-8 assay and hemolysis results, 10 μg mL−1 was established as the optimal DDS delivery concentration, and subsequent experiments were conducted at this concentration.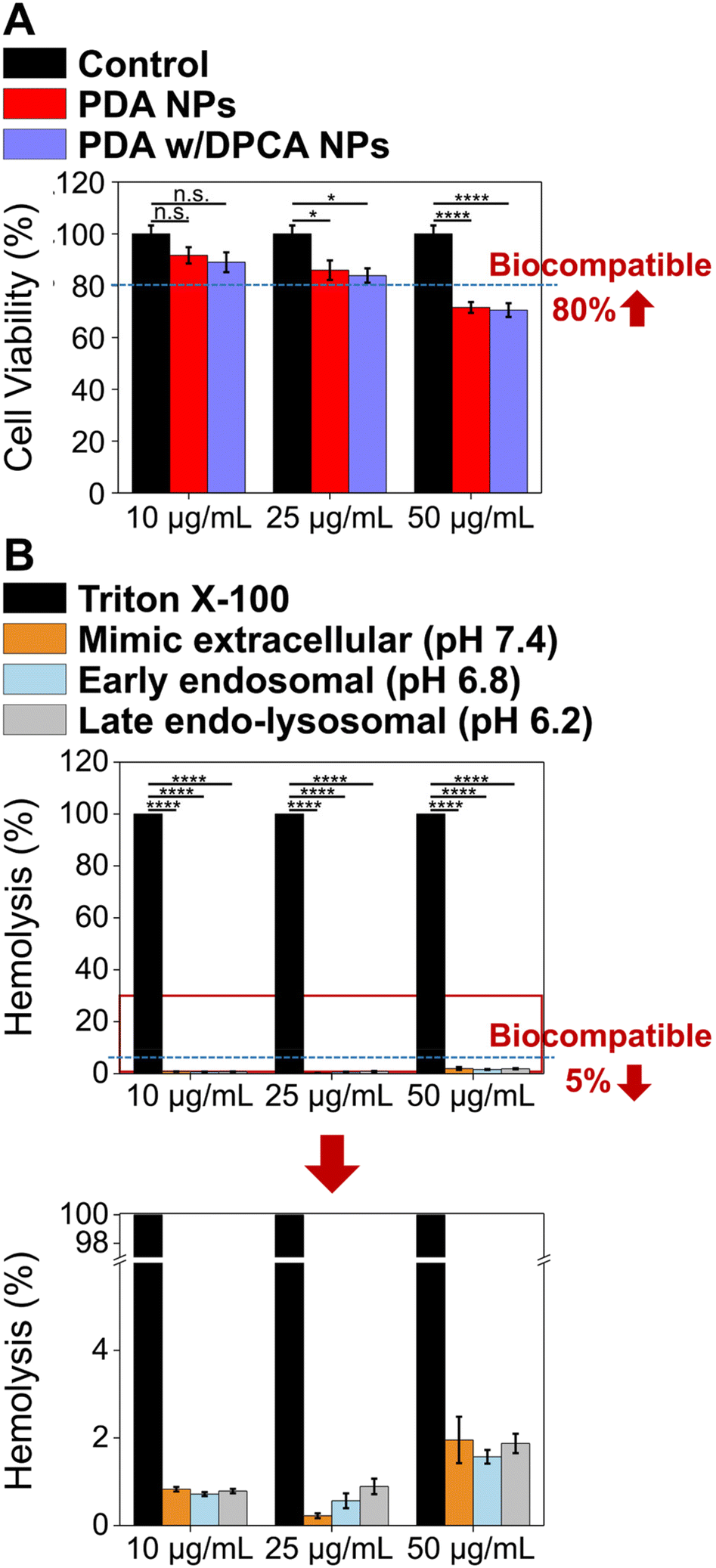 | ||
| Fig. 7 (A) In vitro cell cytotoxicity evaluated by CCK-8 assay. The bar graph shows the cell viability of cells treated with polydopamine nanoparticles (PDA NPs) and PDA NPs with 1,4-DPCA (PDA w/DPCA NPs) (n = 5). (B) In vitro hemolysis test results of PDA w/DPCA NPs (n = 4). Asterisk symbolizes the statistical significance (p-values): *, **** indicate p ≤ 0.05, p ≤ 0.0001, n.s. = not significant. | ||
In vitro and in vivo demonstration of bone regeneration using the DDS
To demonstrate the in vitro bone regeneration effect of the developed DDS, we evaluated the ALP activity of MC3T3-E1 cells treated with PDA w/DPCA NPs. ALP activity is a crucial indicator of early osteoblast differentiation and mineralization for bone development. Thus, ALP activity can confirm active bone formation in osteocytes and/or osteoblasts.46 Here, we tested the ALP activity of MC3T3-E1 cells treated with PDA w/DPCA NPs at a concentration of 10 μg mL−1. Specifically, ALP activity was monitored over 1, 3, and 7 days (Fig. S8†). Upon evaluating the changes in ALP activity in MC3T3-E1 cells following treatment with PDA w/DPCA NPs and their subsequent differentiation, it was observed that all groups exhibited enhanced bone activity compared to the untreated control group at different differentiation durations. Specifically, ALP activity increased to 1.2-fold over control at 1 day, 1.8-fold at 3 days, and 1.1-fold at 7 days. These findings suggest that the designed drug carrier facilitates bone formation and osteoblast differentiation in MC3T3-E1 cells.To evaluate the in vivo bone regeneration efficacy of the optimized DDS, we applied PDA w/DPCA NPs to a mouse calvarial defect model and compared the results with those of the control groups. The experimental timeline for evaluating the in vivo bone regeneration efficacy using a mouse calvarial defect model is depicted in Fig. 8A. Overall, treatment with PDA w/DPCA NPs led to the defect being fully filled with new bone, resulting in higher density, uniform appearance, and increased stability, closely resembling the surrounding old bone. In contrast, the control groups showed disordered fibrous connective tissue at the defect margin. The quantitative results (Fig. 8B and C) calculated from MT-stained images (Fig. S9A†) revealed the near absence of bone tissue and collagen in the untreated group. However, the group treated with PDA NPs displayed improved bone regeneration, attributed to the upregulated antioxidant gene expression as confirmed in Fig. 4D, which induced an anti-inflammatory effect.43 This was further enhanced in the group treated with PDA w/DPCA NPs, which exhibited the highest new bone formation ratio at the calvarial defect site, confirmed by both MT and H&E staining (Fig. S9A and S9B†). The PDA w/DPCA NPs treated group showed an approximately 6-fold increase in bone formation compared to the untreated group, indicating a synergistic effect of regenerative drug (1,4-DPCA) with ROS-generating drug carrier (PDA NPs) in significantly improving bone formation.
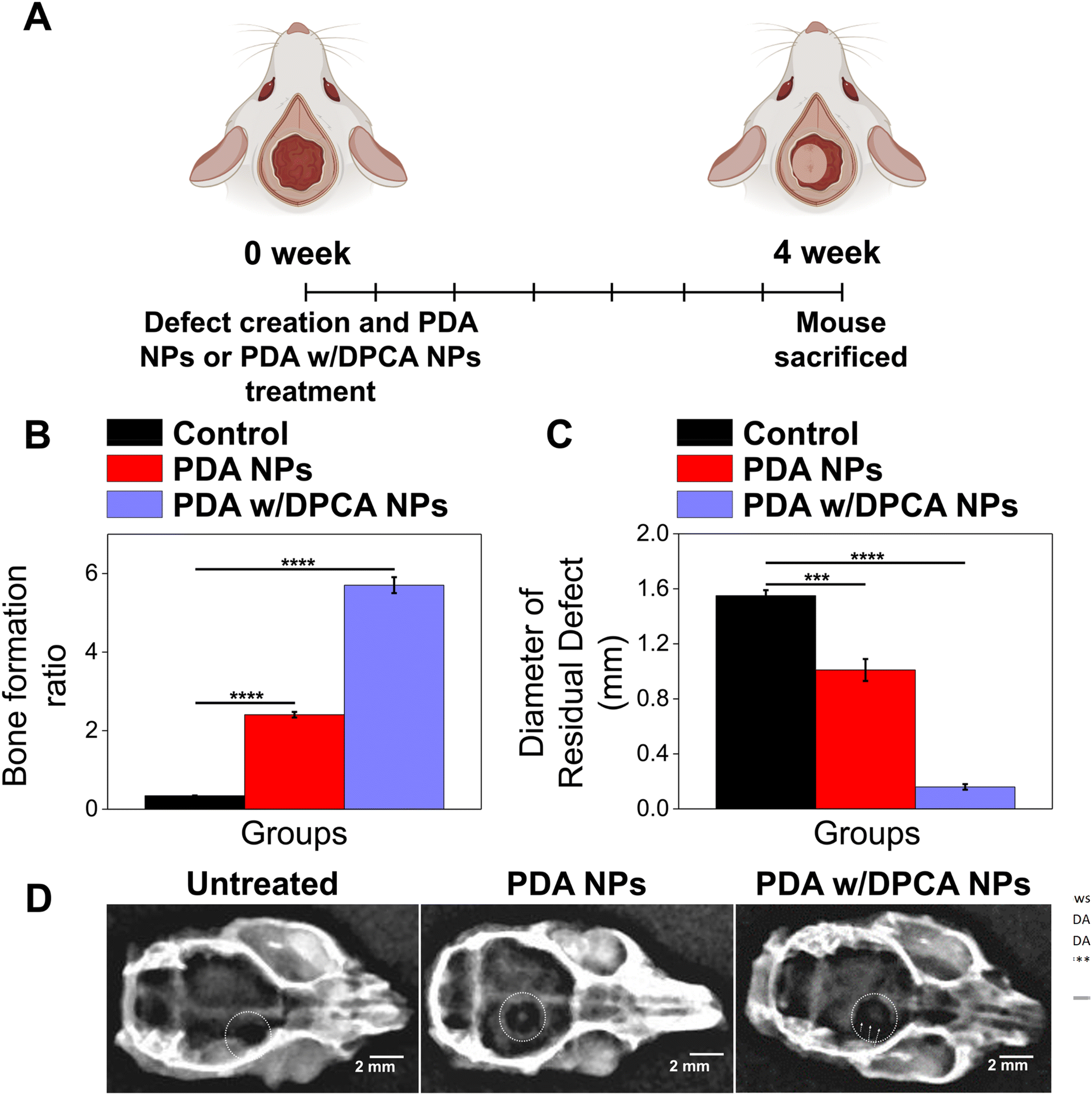 | ||
| Fig. 8 (A) Experimental timeline for evaluating in vivo bone regeneration efficacy using a mouse calvarial defect model. (B) The proportion of new bone matrix within the calvarial defect site: reported values were calculated by using Masson's trichrome (MT)-stained images. (C) The average diameter of the residual defect at the site four weeks post-surgery was measured for the untreated group, the PDA NPs-treated group, and the PDA w/DPCA NPs-treated group; the initial defect area was 2 mm on the mouse calvarial bone. (D) Three-dimensional reconstructed micro-X-ray images of calvarial bone defect sites at 4 weeks post-surgery for the following groups: the untreated group, the PDA NPs-treated group, and the PDA w/DPCA NPs-treated group. The dotted lines highlight the areas of bone defects and regeneration. Data are presented as the mean ± SD (n = 4). The regions of tissue regeneration are indicated by a black dotted line. Asterisk symbolizes the statistical significance (p-values): ***, **** indicate p ≤ 0.001, p ≤ 0.0001. | ||
Furthermore, using 3D reconstructed micro-X-ray imaging, we analyzed the entire skull 4 weeks post-surgery to confirm the induction of new bone formation at the calvarial bone defect site (Fig. 8D). No bone formation was observed in the untreated control group. In contrast, new bone formation was detected at the center of the defect in the PDA NPs-treated group. The group treated with PDA w/DPCA NPs exhibited more extensive bone formation, visibly greater across a larger area of the defect center compared to the PDA NPs-treated group. This suggests that newly formed bone cells are gradually assembling to close the defect site. This observation aligns with the results from the MT and H&E staining (Fig. S9A and S9B†). The above results clearly demonstrate the efficacy of our designed DDS for bone tissue regeneration.
Conclusion
In this study, we designed a novel drug delivery system incorporating the regenerative drug 1,4-DPCA into a polydopamine (PDA)-based nanocarrier. This delivery system leverages polydopamine's ROS generation to activate the cellular antioxidant defense system and stimulates tissue regeneration through 1,4-DPCA's upregulation of Hif-1α, thereby maximizing tissue regeneration at the target site through their synergistic effects. In this process, 1,4-DPCA enhances PDA's ROS generation, further boosting antioxidant gene expression, while PDA also aids in increasing Hif-1α protein expression by 1,4-DPCA. This indicates that each material helps amplify the other's regenerative mechanisms. Additionally, the inherent sustained release capability of the drug delivery system addresses the potential drug toxicity issues associated with burst release of 1,4-DPCA. Moreover, even with a small loading of about 10% of the drug, bone regeneration capacity was shown to be 6-fold greater than that of the untreated control, as verified by the mouse calvarial defect model. The results provide valuable insights into how the synergistic interaction between PDA and 1,4-DPCA can maximize tissue regeneration. Furthermore, CCK-8 and hemolysis analyses indicated that the developed drug delivery system can be easily applied in vivo in an injectable form. Given that the synergetic regenerative mechanisms could be applicable to various tissues, we believe that the potential application of our developed drug delivery system could extend beyond bone regeneration to other disease models.Author contributions
Conceptualization, H. H., B. K., K. L.; methodology, H. H., B. K., S. M. S., T. B. K., Y. K., S. R., K. L.; investigation H. H., B. K., S. M. S., T. B. K., Y. K., J. L., M. P., E. K., Y. J., J. L., S. R., K. L.; data curation, H. H., B. K., S. R., K. L.; formal analysis, H. H., B. K., S. M. S., T. B. K., Y. K., J. L., M. P., E. K., Y. J., J. L., S. R., K. L.; validation, J. L., S. R., K. L.; visualization, H. H., B. K., S. M. S., T. B. K., Y. K., S. R.; writing – original draft, H. H., B. K., S. M. S., T. B. K., Y. K., S. R., K. L.; writing – review and editing, H. H., B. K., S. M. S., T. B. K., Y. K., J. L., M. P., E. K., Y. J., J. L., S. R., K. L.; funding acquisition, E. K., Y. J., K. L.; project administration, J. L., S. R., K. L.; resources, S. M. S., Y. J., S. R., K. L.; supervision, K. L. All authors have read and agreed to the submitted version of the manuscript.Data availability
The authors state that all data supporting the results of this study can be found within the paper and its ESI.† If raw data in different formats are required, they can be provided by the corresponding author upon reasonable request.Conflicts of interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: H. H. and K. L. have a patent pending to Kyungpook National University.Acknowledgements
This research was supported by grants of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HR22C1832), the National Research Foundation of Korea (NRF) grants funded by the Korea government (MSIT) (No. RS-2023-00212797 and No. NRF-2021R1A5A2030333), and Global – Learning & Academic research institution for Master's PhD students, and Postdocs (LAMP) Program of the National Research Foundation of Korea (NRF) grant funded by the Ministry of Education (No. RS-2023-00301914). This work was also supported by the Korea Institute of Energy Technology Evaluation and Planning (KETEP) and the Ministry of Trade, Industry & Energy (MOTIE) of the Republic of Korea (No. 20224000000150). We would like to acknowledge BioRender.com for providing the tools to create the illustrations used in this publication.References
- W. C. Broaddus, K. L. Holloway, C. J. Winters, M. R. Bullock, R. S. Graham, B. E. Mathern, J. D. Ward and H. F. Young, J. Neurosurg., 2002, 96, 244–247 Search PubMed.
- S. Farah, D. G. Anderson and R. Langer, Adv. Drug Delivery Rev., 2016, 107, 367–392 CrossRef CAS PubMed.
- Z. Lv, Y. Ji, G. Wen, X. Liang, K. Zhang and W. Zhang, Burns Trauma, 2024, 12, tkae036 CrossRef PubMed.
- R. Jiao, X. Lin, J. Wang, C. Zhu, J. Hu, H. Gao and K. Zhang, J. Pharm. Anal., 2023 DOI:10.1016/j.jpha.2023.12.015.
- M. J. Imola, D. D. Hamlar, W. Shao, K. Chowdhury and S. Tatum, Arch. Facial Plast. Surg., 2001, 3, 79–90 CrossRef CAS PubMed.
- Y. Zhang, I. Strehin, K. Bedelbaeva, D. Gourevitch, L. Clark, J. Leferovich, P. B. Messersmith and E. Heber-Katz, Sci. Transl. Med., 2015, 7, 290ra92 Search PubMed.
- H. Choudhry and A. L. Harris, Cell Metab., 2018, 27, 281–298 CrossRef CAS PubMed.
- S. Chen and N. Sang, J. Cell. Biochem., 2016, 117, 267–278 CrossRef CAS PubMed.
- J. Cheng, D. Amin, J. Latona, E. Heber-Katz and P. B. Messersmith, ACS Nano, 2019, 13, 5493–5501 CrossRef CAS PubMed.
- E. Zebrowitz, A. Aslanukov, T. Kajikawa, K. Bedelbaeva, S. Bollinger, Y. Zhang, D. Sarfatti, J. Cheng, P. B. Messersmith, G. Hajishengallis and E. Heber-Katz, Front. Dent. Med., 2022, 3, 992722 CrossRef PubMed.
- K. Nagai, H. Ideguchi, T. Kajikawa, X. Li, T. Chavakis, J. Cheng, P. B. Messersmith, E. Heber-Katz and G. Hajishengallis, FASEB J., 2020, 34, 13726–13740 CrossRef CAS PubMed.
- Y. Zhuang, Z. Zhao, M. Cheng, M. Li, J. Si, K. Lin and H. Yu, Front. Cell Dev. Biol., 2022, 10, 836285 CrossRef PubMed.
- A. Menichetti, D. Mordini and M. Montalti, Nanomaterials, 2024, 14, 303 CrossRef CAS PubMed.
- H. Lee, N. F. Scherer and P. B. Messersmith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12999–13003 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- R. S. Ambekar and B. Kandasubramanian, Biomater. Sci., 2019, 7, 1776–1793 RSC.
- Z. Wang, Y. Duan and Y. Duan, J. Controlled Release, 2018, 290, 56–74 CrossRef CAS PubMed.
- X. Zhang, Z. Li, P. Yang, G. Duan, X. Liu, Z. Gu and Y. Li, Mater. Horiz., 2021, 8, 145–167 RSC.
- F. Razaviamri, S. Singh, J. Manuel, Z. Zhang, L. M. Manchester, C. L. Heldt and B. P. Lee, ACS Appl. Mater. Interfaces, 2024, 16, 26998–27010 CrossRef CAS PubMed.
- Y. Jeong and K. Lee, Appl. Sci., 2022, 12, 8710 CrossRef CAS.
- L. Xing, H. Zhang, R. Qi, R. Tsao and Y. Mine, J. Agric. Food Chem., 2019, 67, 1029–1043 CrossRef CAS PubMed.
- D. Wang, Y. Wang, X. Wan, C. S. Yang and J. Zhang, Toxicol. Appl. Pharmacol., 2015, 283, 65–74 CrossRef CAS PubMed.
- X. Gao, Z. Xu, G. Liu and J. Wu, Acta Biomater., 2021, 119, 57–74 CrossRef CAS PubMed.
- Q. Li, Z. Qiu, Y. Wang, C. Guo, X. Cai, Y. Zhang, L. Liu, H. Xue and J. Tang, Front. Dent. Med., 2021, 22, 1473 CAS.
- D. H. Choi, K. E. Lee, S. Y. Oh, S. M. Lee, B. S. Jo, J. Y. Lee, J. C. Park, Y. J. Park, K. D. Park, I. Jo and Y. S. Park, Biomaterials, 2021, 278, 121156 CrossRef CAS PubMed.
- T. Xu, J. Hu, C. Fang, T. Luo, J. Liu and K. Zhang, ACS Mater. Lett., 2023, 5, 1892–1901 CrossRef CAS.
- Q. Ma, Annu. Rev. Pharmacol. Toxicol., 2013, 53, 401–426 CrossRef CAS PubMed.
- T. W. Kensler, N. Wakabayashi and S. Biswal, Annu. Rev. Pharmacol. Toxicol., 2007, 47, 89–116 CrossRef CAS PubMed.
- Y. Choe, J. Y. Yu, Y. O. Son, S. M. Park, J. G. Kim, X. Shi and J. C. Lee, J. Cell. Biochem., 2012, 113, 1426–1436 CrossRef CAS PubMed.
- M. Schäfer and S. Werner, Pharmacol. Res., 2008, 58, 165–171 CrossRef PubMed.
- L. Zheng, S. Liu, X. Cheng, Z. Qin, Z. Lu, K. Zhang and J. Zhao, Adv. Sci., 2019, 6, 1900099 CrossRef PubMed.
- B. C. Evans, C. E. Nelson, S. S. Yu, K. R. Beavers, A. J. Kim, H. Li, H. M. Nelson, T. D. Giorgio and C. L. Duvall, J. Visualized Exp., 2013, e50166 Search PubMed.
- T. Histing, D. Stenger, S. Kuntz, C. Scheuer, A. Tami, P. Garcia, J. H. Holstein, M. Klein, T. Pohlemann and M. D. Menger, J. Surg. Res., 2012, 175, 271–277 CrossRef PubMed.
- O. O. Aalami, R. P. Nacamuli, K. A. Lenton, C. M. Cowan, T. D. Fang, K. D. Fong, Y. Y. Shi, H. M. Song, D. E. Sahar and M. T. Longaker, Plast. Reconstr. Surg., 2004, 114, 713–720 CrossRef PubMed.
- Y. R. Hong, T.-H. Kim, K.-H. Park, J. Kang, K. Lee, E. K. Park, T.-G. Kwon, J. O. Lim and C.-W. Oh, Tissue Eng. Regener. Med., 2023, 20, 69–81 CrossRef CAS PubMed.
- H. Hemmatpour, O. De Luca, D. Crestani, M. C. A. Stuart, A. Lasorsa, P. C. A. van der Wel, K. Loos, T. Giousis, V. Haddadi-Asl and P. Rudolf, Nat. Commun., 2023, 14, 664 CrossRef CAS PubMed.
- Y. Kim, K.-I. Min, S. Jeong and K. Lee, Bull. Korean Chem. Soc., 2023, 44, 147–152 CrossRef CAS.
- K. Lee, M. Park, K. G. Malollari, J. Shin, S. M. Winkler, Y. Zheng, J. H. Park, C. P. Grigoropoulos and P. B. Messersmith, Nat. Commun., 2020, 11, 4848 CrossRef CAS PubMed.
- F. A. Stevie and C. L. Donley, J. Vac. Sci. Technol., A, 2020, 38, 063204 CrossRef CAS.
- H. Meng, Y. Liu and B. P. Lee, Acta Biomater., 2017, 48, 144–156 CrossRef CAS PubMed.
- G. P. Maier, C. M. Bernt and A. Butler, Biomater. Sci., 2018, 6, 332–339 RSC.
- W. H. Park, Oncol. Rep., 2018, 40, 1787–1794 CAS.
- H. Zhao, Z. Zeng, L. Liu, J. Chen, H. Zhou, L. Huang, J. Huang, H. Xu, Y. Xu, Z. Chen, Y. Wu, W. Guo, J. H. Wang, J. Wang and Z. Liu, Nanoscale, 2018, 10, 6981–6991 RSC.
- S. Jia, S. Dong, H. Liu, H. Yu, Z. Chen, S. Wang, W. Li, R. Peng, F. Li, Q. Jiang and J. Liu, Biomater. Sci., 2022, 10, 3309–3322 RSC.
- G. Lee, H.-S. Won, Y.-M. Lee, J.-W. Choi, T.-I. Oh, J.-H. Jang, D.-K. Choi, B.-O. Lim, Y. J. Kim, J.-W. Park, P. Puigserver and J.-H. Lim, Sci. Rep., 2016, 6, 18928 CrossRef CAS PubMed.
- V. Bradaschia-Correa, A. M. Josephson, A. J. Egol, M. M. Mizrahi, K. Leclerc, J. Huo, B. N. Cronstein and P. Leucht, Tissue Cell, 2017, 49, 545–551 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02887b |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2024 |