
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSymmetry breaking enhances the catalytic and electrocatalytic performance of core/shell tetrametallic porous nanoparticles†
Apoko S.
Omondi
 ab,
Dávid
Kovács
ab,
György Z.
Radnóczi
a,
Zsolt E.
Horváth
a,
István
Tolnai
a,
András
Deák
a and
Dániel
Zámbó
*a
ab,
Dávid
Kovács
ab,
György Z.
Radnóczi
a,
Zsolt E.
Horváth
a,
István
Tolnai
a,
András
Deák
a and
Dániel
Zámbó
*a
aHUN-REN Centre for Energy Research, Konkoly-Thege M. út 29-33., H-1121 Budapest, Hungary. E-mail: daniel.zambo@ek.hun-ren.hu
bDepartment of Physical Chemistry and Materials Science, Faculty of Chemical Technology and Biotechnology, Budapest University of Technology and Economics, Műegyetem rkp. 3., H-1111 Budapest, Hungary
First published on 5th November 2024
Abstract
The performance of functional nanocatalysts can be extended by integrating multiple types of metals into well-designed nanoparticles. A porous multimetallic shell grown around high-quality monometallic seeds significantly enhances the availability of active sites. Here, tetrametallic core/shell nanoparticles (Au@mPdPtIr) featuring micro- and mesoporous shells are synthesized with strict control over the overall particle morphology. To reveal the impact of the core nanoparticle morphology on the optical, structural and electrocatalytic properties, tetrametallic particles are prepared using gold cores with different shapes but identical volumes and surface chemistry. Our general synthetic approach ensures the successful and reliable synthesis of porous trimetallic shells around the cores, keeping the final atomic composition of the different multimetallic particles identical. The results clearly highlight the significance of the core morphology in the catalytic performance and the superior activity of symmetry-broken core/shell particles in heterogeneous as well as electrocatalytic oxidation reactions. These can be attributed to the fine structural details of the deposited trimetallic shells and their influence on the charge carrier transport between the multimetallic particles and the organic test molecules. While all nanocatalysts show excellent morphological robustness, the optimal morphology also depends on the reaction type and conditions of the specific reaction.
1. Introduction
Shape-controlled colloidal synthesis of metallic nanoparticles has been explored to utilize noble metal nanoparticles in a number of applications including the key areas of sensing,1,2 surface-enhanced Raman scattering (SERS),3–5 plasmonics,6–8 drug delivery,9,10 and heterogeneous catalysis11–14 in recent decades. The well-developed synthetic pathways over the years have allowed the preparation of particles of various shapes and sizes with good shape purity and size monodispersity, aspects that are of key importance in advanced applications of mono-, bi-, tri-, and multimetallic nanoparticles. Noble metals are of particular interest in optics and catalysis, including heterogeneous catalysis and electrocatalysis. Although platinum is considered as a benchmark material in catalysis,15 the presence of other noble metals (e.g. Pd, Au, and Ir) can alter its electronic structure and adsorption/desorption processes further enhancing its catalytic performance.16 Thus, the combination of more than one noble metal can result in synergistic physicochemical properties which can be further improved upon reducing the particle size to the nanoscale.17 It has been previously reported that noble metal bimetallic nanoparticles possess superior catalytic activity compared to their pure noble monometallic counterparts.18–21 Multimetallic synergy itself has already been utilized in various intermetallic, core/shell22 and alloy23 nanoparticles showing outstanding catalytic performance and multifunctionality.24 Upon combining the compositional variety with meso- and microporosity, the resulting high surface area and improved mass transport enhance the availability of catalytically active surface sites which make them even more promising nanocatalysts compared to commercial catalysts, including Pt on carbon supports (Pt/C).16,25 The synergistic effects arise due to ligand and steric effects, leading to the manipulation of the d-band electronic structure.16,21,26–28 These features have a profound influence on the catalytic performance by enhancing the binding strength of the catalyst and the adsorbate materials to promote the electrocatalytic efficiency.29Hence, tetrametallic noble metal mesoporous core/shell nanoparticles provide multiple benefits including enhanced electrocatalytic performance and stability. Moreover, the mesoporous core/shell framework offers virtually unlimited possibilities for enhancing catalytic stability and activity, extending the utilization efficiency of the scarce and costly metals while simultaneously accelerating mass and charge transfer.30,31 When gold is used as the core metal, it has been found that its inherent stability and resistance to electrochemical leaching grant long-term stability to the shell metals and further enhance their surface features (e.g., analyte adsorption and product desorption).18
The effect of the nanoparticle morphology on the catalytic activity has already been reported for monometallic gold nanoparticles. For instance, when comparing the degradation rate of methylene blue in the presence of gold NPs, nanoprisms were found to be the most effective catalysts being superior to other morphologies (sphere, cube, rod), or even bimetallic (AuR/Pd or AuR/Pt) particles.32 Nevertheless, the presence of a metallic shell improved the catalytic activity compared to the monometallic nanorods exceeding the achievable turnover by using Au spheres (AuNSs) and nanorods (AuRs). It has also been demonstrated earlier for bimetallic core/shell particles33,34 that there is a correlation between the shape of the gold core in Au@Pt particles and their activity towards the oxygen reduction reaction: on comparing cubic, octahedral and rod-like core morphologies, Auoct@Pt core/shell particles were found to possess the highest electrocatalytic activity, indicating that the catalytic performance is in correlation with the tailored core's shape.
Core/shell bi- and trimetallic noble metal nanoparticles have previously been synthesized through in situ homogeneous nucleation (one-pot synthesis) including seed-mediated co-reduction,35 and a two-step synthesis where nucleation and formation of the core material are separated from the shell growth.36 It is clear that the utilization efficiencies of these core/shell multimetallic nanoparticles when used as electrocatalysts or heterocatalysts do not depend only on the size and composition.14 The shape of the core also plays a fundamental role in determining the features that make them explicitly more attractive.28 This is attributed to the interaction between the core and the shell metal(s) that leads to the modification of their shell's electronic structure through charge transfer effects at the core/shell interfaces partially governed by the electronegativities of the metallic elements involved.37 The charge transfer effect results in a shift in the d-band centers of the shell metals making attachment and detachment of adsorbates more facile. Furthermore, Zhang and Lu investigated the effect of core shape on the catalytic effectiveness of Au@Pt by synthesizing a coreless Pt dendrite, Au@Pt with different cores (octahedral, rods and cubes). It was reported that the order of catalytic effectiveness followed Auoct@Pt > Aurod@Pt > Aucube > Ptdend.28,33 In their work, Kang et al. reported the existence of the effective charge transfer effect to be superior when octahedral gold cores were used in trimetallic Au@PdPt as opposed to spherical gold cores of the same noble metal combination. This led to a superior catalytic activity of Auoct@PdPt compared to AusphPdPt in the ORR.14 Introduction of Ir can further enhance the catalytic activity of Pt in a wire-like Au@dendritic-PtIr structure, where d electron transfer from Ir to Pt weakens the adsorption of intermediates, especially the poisonous COad.38 These findings underscore the fact that the core morphology has a profound impact on the performance of noble metal multimetallic nanoparticles in the applications of heterogeneous catalysis, photocatalysis and electrocatalysis as well. The morphology of the core in seed-mediated multimetallic core/shell nanoparticle synthesis also determines the shape of the multicomponent core/shell NPs.
In this work, we sought to investigate the effect of core morphology in core/shell porous Au@mPdPtIr tetrametallic nanoparticles on the (electro)catalytic properties by applying symmetry breaking during the core NP synthesis. A trimetallic shell with well-defined micro- and mesoporosity was grown around the gold core with different morphologies and excellent shape and size uniformity. The porous nature of these nanocrystals offers a high surface-to-volume ratio and access to high-index facets around curvatures, tips and branches. It is clear that a nanostructured interface has an impact on the activation energy in electron-transfer reactions,11 since the shape determines the fraction of atoms located on edges and corners.31 However, little or no attempt has been made to systematically investigate the impact of core morphology on electrocatalytic and catalytic performance when the concentration of gold in these cores of equal volume is kept similar in the nanomaterials. Therefore, we synthesized five different gold core nanoparticle types (nanospheres, nanorods, nanoprisms, nanooctahedra and nanobipyramids) with equal gold (Au0) concentrations and volumes by wet chemical methods followed by shell growth resulting in trimetallic palladium, platinum and iridium shells. Our approach ensures that the overall synthesis is uniform and versatile regardless of the shape and size of the cores. After in-depth structural and optical characterization, their catalytic performance was investigated in relevant test reactions, such as degradation of p-nitrophenol, alcohol electrooxidation (EOR and MOR) in an alkaline medium and the formic acid electrooxidation reaction (FAOR) in an acidic medium. Due to the strict control over the synthesis parameters and electrode fabrication process, it was guaranteed that the volume of all gold cores, the elemental composition of the tetrametallic NPs and the catalyst loading were identical, which enabled the direct comparison and evaluation of the impact of core morphology on the catalytic performance.
2. Results and discussion
2.1. Synthesis and structural characterization of the nanoparticles
The synthesis of the tetrametallic core/shell nanoparticles was performed in a two-step process. First, monodisperse gold nanoparticles of different morphologies (spheres, rods, prisms, octahedra and bipyramids) with the target sizes and particle volumes were synthesized (see the Experimental section for details) and used as core particles. A trimetallic shell consisting of Pd, Pt and Ir was deposited around the core particles in a controlled manner.39 The volume and dimensions of the Au nanorods were used as a reference and the synthesis conditions were carefully tuned to obtain the desired dimensions (corresponding to nearly identical particle volumes) for the other core particle types (Fig. S1†). The synthesis of gold nanorods was performed in the presence of a binary surfactant mixture (CTAB, sodium oleate). Gold nanospheres were prepared by slow addition of Au3+ and sodium citrate solution to smaller Au nanoparticles used as seeds and their ligands were replaced by CTAB in a separate step. Nanoprisms and nanooctahedra were synthesized by a similar approach, where Au seed particles were grown in the presence of CTAC and iodide in a two-step protocol. The preparation of gold bipyramids includes two main steps: (i) the synthesis of pentatwinned gold seeds and (ii) their overgrowth into bipyramids in an acidic solution containing Ag+ and CTAB. The dimensions of all particle types were first precisely fine-tuned in smaller batches and then upscaled to provide NP stock solutions with higher Au0 concentrations for the growth of the trimetallic shell. In cases where shape impurities were present (nanoprisms, nanobipyramids), careful purification steps were conducted by surfactant-mediated flocculation depletion (using CTAC or BDAC) to achieve excellent shape and size purity (discussed below). The careful preparation resulted in almost 100% shape purity for all core particles as shown in Fig. 1a (and Fig. S2†). It has to be noted that the quality of nanoprisms and nanobipyramids could be dramatically increased by the applied post-synthetic treatments which manifests in their sharp resonance peaks (Fig. 1b). The purification using BDAC to selectively flocculate the bipyramids is a key tool that simplified the separation of the small spherical nanoparticles into a pinkish-red supernatant that was carefully decanted. Similarly, the gold nanoprisms were also successfully purified by a two-step depletion process as described in the Experimental section. Thus, the core particles exhibited appropriate size and shape monodispersity (Fig. S1 and Table S1†). | ||
| Fig. 1 Physical appearance of the aqueous solution of core particles and their corresponding SEM images (a). Extinction spectra of the core particles (b). Scale bars are 100 nm. | ||
The trimetallic microporous and mesoporous shell was grown over each core morphology in two steps but in one pot. The ions of the metal precursors (Pd2+, Pt2+ and Pt4+) were reduced by ascorbic acid in the presence of HCl and F127 block copolymer micelles. These micelles act as a template for the micro- and mesoporosity of the trimetallic shell. The reaction was carried out at a temperature of 70 °C for 40 minutes under stirring. To decorate the deposited Pd/Pt shell with Ir, the temperature was raised to 80 °C and the reduction of Ir3+ was achieved by using ascorbic acid and NaBH4 in a reaction time of 20 minutes. The concentration and volume of the precursor shell materials, the gold core concentration and the reaction conditions were kept identical in all the five model systems ensuring that the final composition of the tetrametallic particles remained the same. Upon performing precise analysis of the elemental composition by ICP-OES, the concentrations of the tetrametallic nanoparticle solutions were set identical to guarantee the same particle loading for further catalytic experiments.
2.2 Structural and optical characterization and surface chemistry
After purification and washing, the ultimate core nanoparticle solutions were concentrated in 50 mM CTAB or CTAC to ensure the uniformity of their surface chemistry. Fig. 1a shows the physical appearance of the core particles’ diluted aqueous solutions as well as the corresponding SEM images, demonstrating the excellent quality of the NPs. All the synthesized nanoparticles were analyzed by UV-Vis spectroscopy and their extinction spectra are presented in Fig. 1b. The extinction spectra feature well-defined and narrow localized surface plasmon resonance (LSPR) peaks due to the light induced collective oscillation of the conduction band electrons. While nanospheres show a single extinction peak, additional peaks beside the dominant dipolar plasmon resonance mode appear for symmetry-broken particle types, dictated by the geometry of the particles.40 It has to be mentioned though, that in the present work the impact of localized surface plasmons on charge-carrier dynamics can be ignored as the assessment of the particles’ functional properties does not rely on photogenerated charge carriers. In contrast, the main role of the gold core particle is that a large variety of particle morphologies becomes synthetically accessible and – as shown later – it can act as an electron source during intraparticle charge separation.During the synthesis of the core particles, positively charged CTA+ ligands were used as stabilizers except for Au nanospheres which first have citrate on their surface. Consequently, these as-synthesized gold nanospheres have a negative zeta potential (−42.9 ± 0.9 mV) and electrophoretic mobility (−3.2 ± 0.05 μm cm V−1 s−1). To ensure uniformity in surface charge and chemistry in all the nanoparticle morphologies, a surfactant exchange to CTAB was conducted and a reversal in the zeta potential (+52.9 ± 2.8 mV) and electrophoretic mobility (+4.1 ± 0.2 μm cm V−1 s−1) depicted a successful surfactant exchange (see Fig. S4† for the electrophoretic mobilities of the particles).
Fig. 2 summarizes the structural properties of the particles after growing the micro- and mesoporous trimetallic shells around the Au cores. Due to the thick shell and the dominance of Pt, the nanoparticle solutions became black. Both the elemental distribution maps and the SEM images confirm the successful synthesis of the core/shell particles possessing significant porosity and outstanding shape and size purity (see Fig. S4 and S5† for more detailed TEM and SEM characterization). This porous nature endows the particles with enhanced surface area and improved availability of the reactive surface sites. Similarly to the AuR@mPdPtIr particles in our previous study,39 a well-defined Pd and Pt layer can be identified around the Au cores (Fig. S6†). The average thicknesses of these layers are nearly identical for all the synthesized model systems (Fig. S7†). However, due to the spectral overlapping of Pt and Ir during elemental mapping, separate detection of Ir cannot be reliably performed at the applied low Ir contents. Nevertheless, the exact composition of the tetrametallic NPs was determined by ICP-OES, which shows not only the presence of each applied element but their exact concentration as well (Fig. 3a) and confirms the nearly identical composition of the five different core/shell particle types.
 | ||
| Fig. 2 Photographs of the nanoparticle solutions, TEM-EDS elemental maps and SEM images of the multimetallic nanoparticles containing a gold core with spherical, rod-shaped, prism-shaped, octahedral, and bipyramidal morphology (left to right). The spatial distribution of the elements is color coded. Scale bars are 20 nm and 100 nm for TEM and SEM images, respectively. | ||
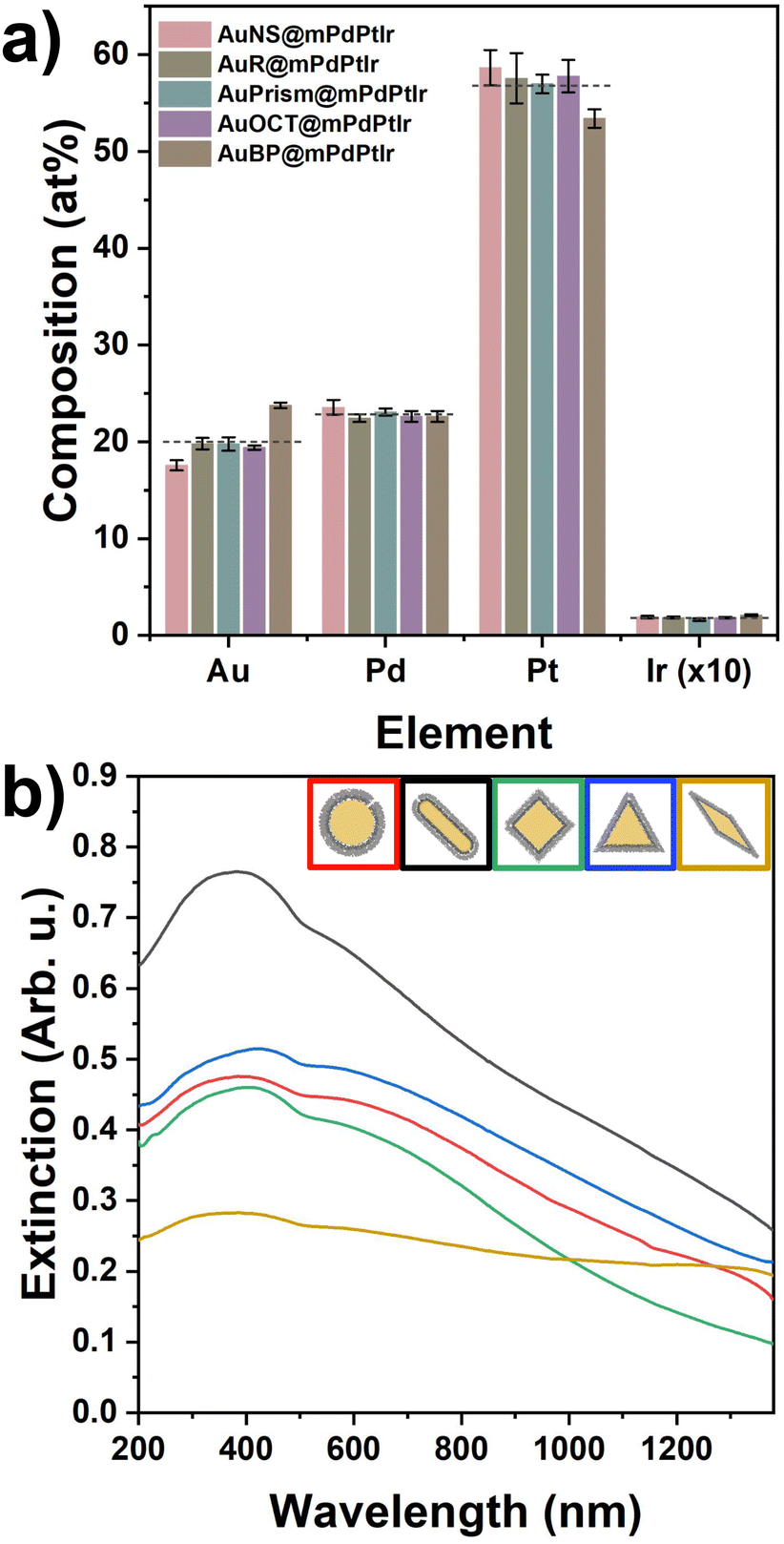 | ||
| Fig. 3 Elemental composition (in atomic%) of the different tetrametallic nanoparticles measured by ICP-OES (a). Dashed lines represent the mean value of the corresponding elements in the five different nanoparticle model systems. UV-Vis-NIR extinction spectra of the tetrametallic nanoparticles (b). | ||
Upon shell growth, the extinction spectra of all the core/shell tetrametallic nanoparticles changed dramatically due to the thick, porous trimetallic shell, showing extensive extinction in the UV to NIR region (Fig. 3b). Moreover, inversion of the originally positive electrophoretic mobilities can be observed for all particle types after the shell growth (electrophoretic mobilities before and after the shell growth are shown in Fig. S3†). The resulting negatively charged surface sites imply an intraparticle charge transfer in the multielemental system. The interaction between the elements results in the negative charging of the particle surface and is based most likely on the realignment of the Fermi levels upon equilibrating the contact potentials. To rule out the possible impact of any charged molecules attached to the nanoparticle surface on the observed negative electrophoretic mobilities during the synthesis, a control experiment was conducted with spherical Au cores, ascorbic acid and sodium borohydride following the same steps and conditions applied in the tetrametallic nanoparticle preparation, however, without introducing the precursors of Pd, Pt and Ir metals (Fig. S8†). The electrophoretic mobility of the AuNSs becomes virtually 0 μm cm V−1 s−1 after the addition of ascorbic acid at 70 °C. Importantly, this value does not change upon introducing the reducing agents (ascorbic acid and NaBH4) in the next step at 80 °C, which confirms that the negative charges of the tetrametallic particles do not simply originate from residual ascorbate, or borohydride anions. In contrast, extensively negatively charged particle surfaces can be obtained upon performing the deposition of the trimetallic shell.
X-ray diffractograms of the core particle samples (Fig. 4) were recorded in the range of 2θ = 30°–90°. Peaks of the face-centered cubic (fcc) Au phase are situated in their correct positions as denoted by the orange reference lines. The variation of Au peak intensity ratios can be attributed to texture effects: diffraction of close-packed particles of different shapes self-assembled in blocks (see Fig. 1a) obviously produces different line intensities. In the range of 2θ = 60°–70° for some of the XRD curves, the peaks are attributed to the (400) line and the satellites of the single crystalline Si substrate. XRD plots of the multimetallic particles recorded in the range of 2θ = 35°–50° show the presence of the (111) and (200) lines of Au, and, separately from them, overlapping (111) and (200) lines of Pd, Pt and Ir. The position of Au lines corresponds again to the reference values, indicating a negligible extent of Pd, Pt or Ir diffusion into the Au matrix.
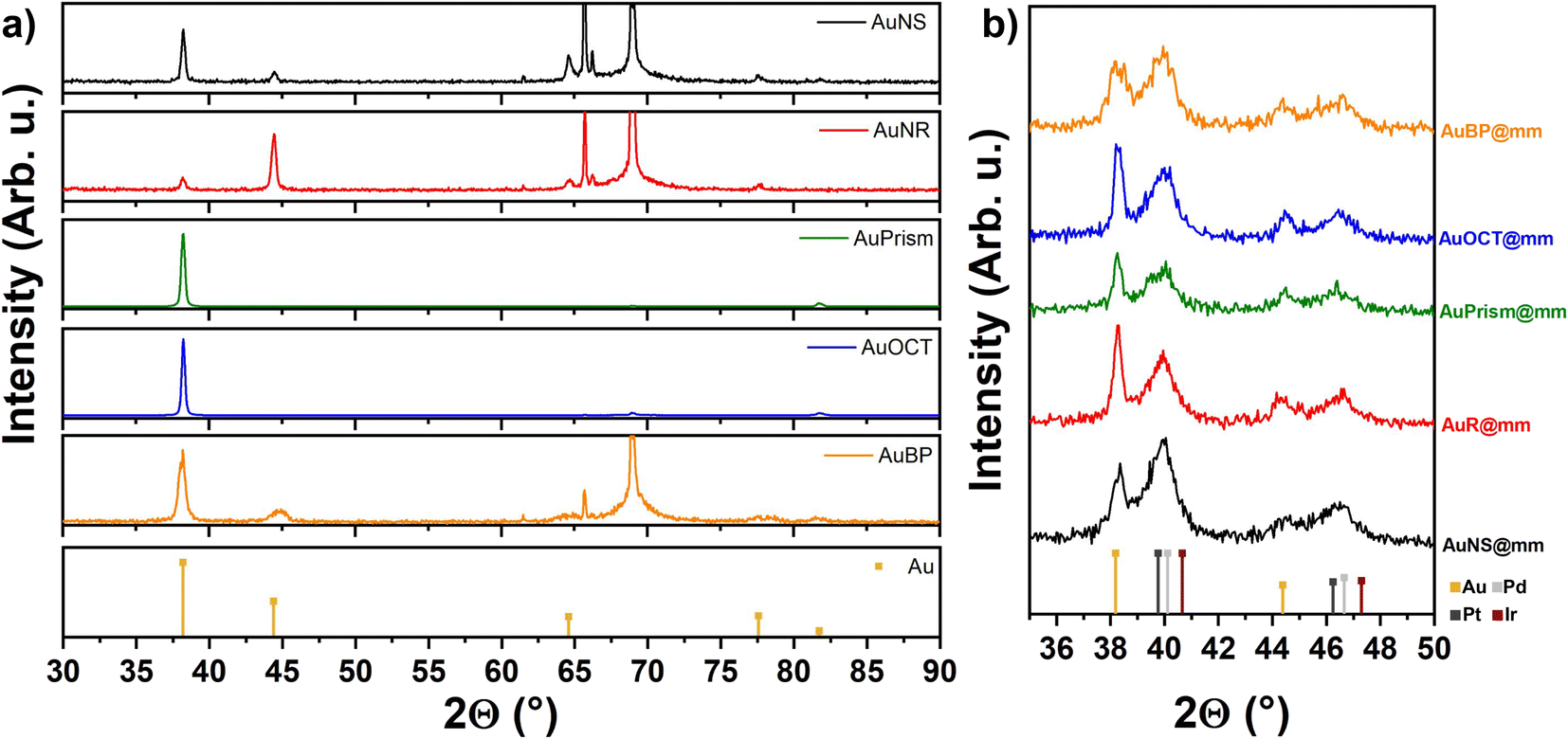 | ||
| Fig. 4 XRD profiles of the core (a) and multimetallic particles (b) including the reference lines of the metals. | ||
2.3. Heterogeneous catalysis: reduction of p-nitrophenol
The heterogeneous catalytic reduction of p-nitrophenol (pNP) to 4-aminophenol (pAP) was studied under aqueous conditions using NaBH4 as a hydride source, with the tetrametallic catalysts being the electron transfer agents. The hydrolysis reaction of borohydride raises the pH causing the deprotonation of pNP into the 4-nitrophenolate anion that has an intense yellow color and an absorption peak centered at 400 nm. This enables easy monitoring of its degradation by UV-Vis spectrophotometry.41,42 Due to the presence of the tetrametallic particles in the solution during the reduction reaction, the recorded extinction spectra were corrected with the extinction of the particles measured separately under the same conditions (but without adding pNP). After this background correction, the time-resolved absorbance data at 400 nm were plotted (with 42 s temporal resolution) against time for each of the five catalysts. While pNP remains stable during the time course of the experiment in the absence of the catalyst (also see Fig. S9†), it is clear that core/shell nanoparticles and their specific symmetry breaking (spherical vs. other particle morphology) have a profound impact on the catalytic performance of the as-synthesized catalysts (Fig. 5).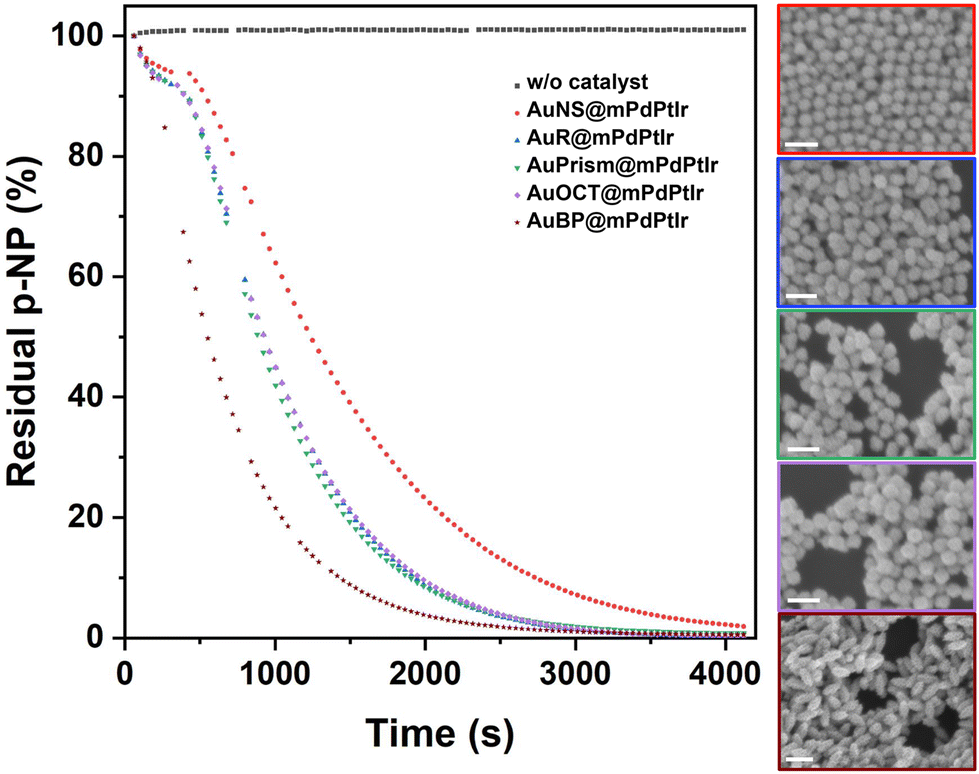 | ||
| Fig. 5 Residual p-nitrophenol in % as a function of time for the investigated model systems calculated from the measured absorbance at 400 nm over time. All decay curves represent the average of at least three consecutive measurements. Insets show the SEM images of the particles (color coded to the absorbance data) collected after the catalytic reaction by centrifugation (scale bars are 200 nm). | ||
Tetrametallic NPs with spherical cores were found to be the least active in the pNP–pAP conversion, while tetrametallic nanobipyramids exhibited the highest rate constant. Despite the similar fine structure of the shell for all particle types (Fig. S10†), particles with nanorod, nanoprism or nanooctahedron cores show nearly identical catalytic performance, but in general all symmetry-broken particles outperform the activity of the spherical multimetallic NPs (see the fitted pseudo-first order kinetics and the rate constants in Fig. S11†). It has to be noted, however, that all types of tetrametallic particles retain their original morphology as well as porosity after the catalytic reaction (Fig. 5 insets), which underlines the excellent stability of their meso- and microstructure, enabling their recyclability.39
Control experiments performed with core-only Au nanoparticles (AuNSs, AuPrisms, AuBPs) highlight the role of the trimetallic shell in the overall activity (Fig. S12†). Gold cores with different morphologies (even though their concentrations are identical to that of multimetallic particles) show no activity (AuNS, AuPrism), or much lower activity in the pNP degradation reaction (AuBP) compared to their tetrametallic counterparts.
2.4. Morphology-dependent electrochemically active surface areas
The electrochemically active surface areas (ECSAs) of all the electrocatalysts were determined by first calculating the double layer capacitance of working electrodes that were fabricated by depositing the multimetallic particles on a glassy carbon electrode (GCE). Briefly, the differences in anodic and cathodic currents were measured in a potential window with non-faradaic processes at different scan rates (Fig. 6). By plotting and fitting the current differences as a function of the scan rate, the slopes provide the double layer capacitances (cDL). The ECSA values were obtained by calculating the ratio of cDL/cS (where cS is the specific capacitance in 1 M KOH; cS = 0.022 mF cm−2)43 and the values were normalized to the catalyst loading on the GCE surface (Fig. 6f). For the five different model systems, the ECSA values were found to be 118.59, 86.84, 74.68, 70.54 and 16.12 m2 g−1 for AuBP@mPdPtIr, AuR@mPdPtIr, AuOCT@mPdPtIr, AuPrism@mPdPtIr and AuNS@mPdPtIr, respectively. This depicts that symmetry breaking itself has a significant effect on the availability of active sites and the nanoparticles with symmetry broken cores are inherently electrochemically more active than the nanoparticles with a spherical core morphology.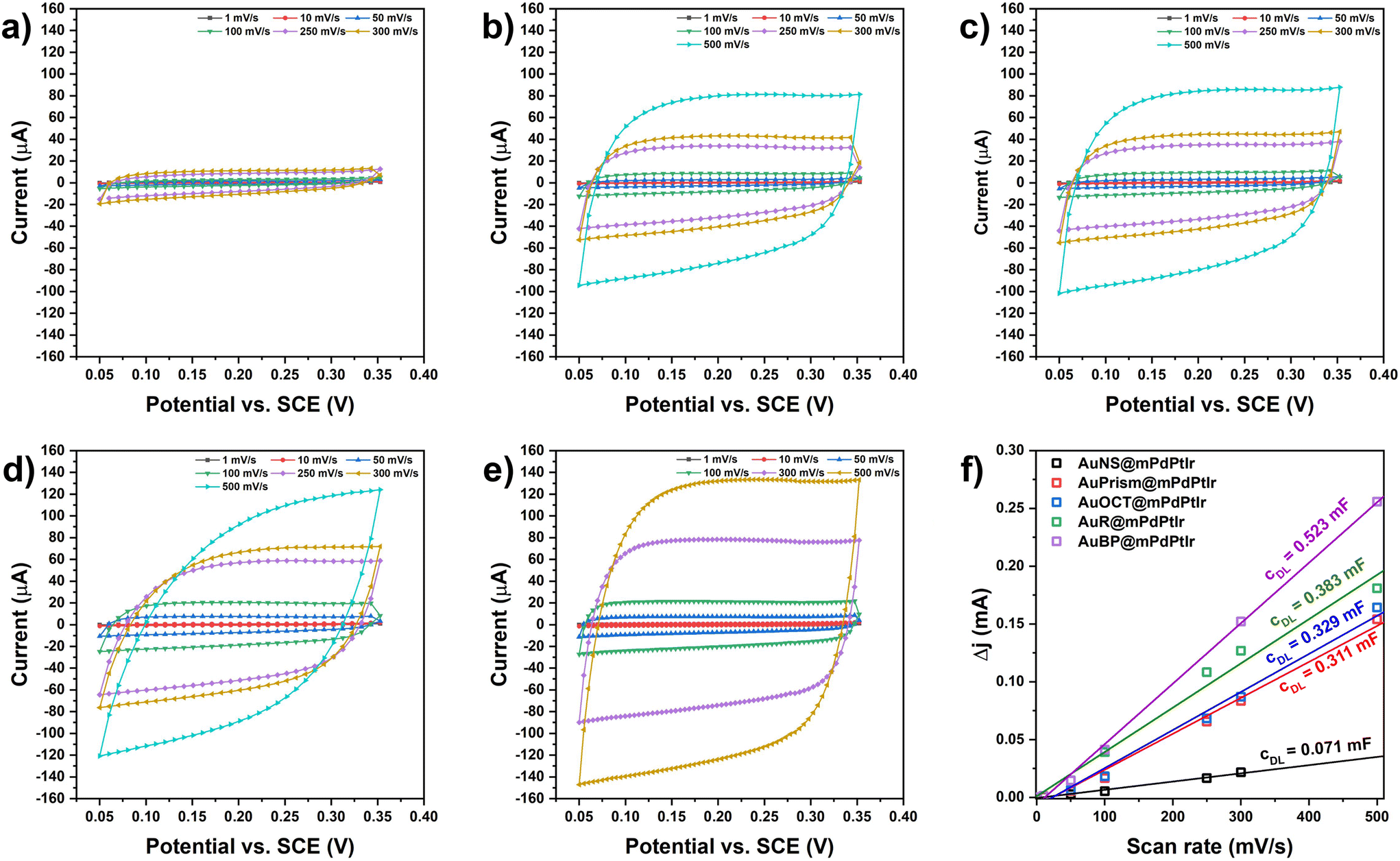 | ||
| Fig. 6 Scan rate dependent CV curves of GCE electrodes modified by multimetallic nanoparticles with spherical (a), prism-shaped (b), octahedral (c), rod-shaped (d), and bipyramidal (e) cores in 1 M KOH electrolyte. Double layer capacitances for the different types of multimetallic nanoparticle electrodes (f). | ||
2.5. Application of tetrametallic nanoparticles in electrocatalytic oxidation reactions
Direct alcohol fuel cells have been in the spotlight towards generating sustainable and clean energy. Given their electrochemical activity towards alcohol oxidation reactions, noble metal nanoparticles are promising candidates in this electrochemical conversion of alcohols to generate energy and other useful chemicals that have a lower impact on the environment. To this end, the electrocatalytic activity of various tetrametallic electrocatalysts was investigated in the electrooxidation reaction of methanol (MOR) and ethanol (EOR) in alkaline media, and formic acid (FAOR) in an acidic medium. The electrochemical measurements were conducted in a typical three electrode electrochemical cell consisting of a GCE with the tetrametallic NPs deposited and dried on its surface as the working electrode, a coiled Pt wire auxiliary electrode as the counter electrode and Ag/AgCl (3.5 M KCl) as the reference electrode. Importantly, the electrode preparation does not require any binding agent (e.g., Nafion) and stable particle coatings can be realized by fine-tuning the concentration of the NP stocks (ctotal metal = 10 g L−1). Moreover, external heat or light sources are not used in the test reactions performed at room temperature. Cyclic voltammetry measurements for the EOR and MOR of all the electrocatalysts were conducted in argon saturated electrolytes containing 1 M KOH and 1 M EtOH or MeOH. The investigated bias potential range was −0.7 to 0.5 V for both the EOR and MOR. A scan rate of 10 mV s−1 was used to record the voltammograms of all the nanoparticle morphologies. Since a plain GCE shows virtually no current in the presence of alcohols (Fig. S13†), the activity toward the EOR and MOR can be directly attributed to the tetrametallic nanoparticles on the GCE and as such, the currents were normalized with respect to the catalyst loading on the GCE (Fig. 7a and b). The voltammograms show solely oxidative peaks both in the anodic and cathodic scan, indicating the sufficient conversion of the alcohols. The lowest onset potential can be assigned to the AuBP@mPdPtIr particles (Eonset (MOR) = −0.65 V; Eonset (EOR) = −0.68 V); however, all other morphologies show similarly low onset potentials (−0.6 V). Tetrametallic nanoparticles with prism-shaped cores are the most active catalysts for the MOR and EOR, demonstrating high mass activity (480 mA mg−1 and 350 mA mg−1 for the MOR and EOR, respectively). Focusing on the methanol oxidation reaction, the shape of the anodic oxidation peak is different for AuPrism@mPdPtIr. In contrast to other morphologies, where a shoulder appears next to the main peak, prisms show a distinct, sharper single oxidation peak. This indicates a different oxidation mechanism and the coupled adsorption/desorption processes for prism-shaped particles. Complete oxidation of methanol to CO2 involves the transfer of 6 electrons, during which different byproducts and intermediates, such as COad, HCHOad, COHad and H(3−x)COad, are generated.44,45 Although COad was identified as the main poison of the Pt-based catalyst in the recent decade,46,47 Rus et al. highlighted the important role of OHad and its supply in the oxidation process.45 Hydroxide anions oxidize the poisoning COad to CO2 and control the surface pH.48 During methanol oxidation, the pH of the catalyst surface decreases and is governed by the OH− concentration involving the mass transport and bulk OH− concentration in the overall process. By using sufficient OH− concentration in the electrolyte, however, the reaction rate is governed by the most sluggish side process, making it more likely to be transport limited.45 For Pd nanoparticles, it was found that the oxidation of methanol can proceed through multiple intermediate steps manifesting in 2 or more oxidation peaks in the forward scan.49 Moreover, the structure and morphology of the nanocatalyst also influence the velocity of adsorption and desorption processes: porous Pd nanoflowers49 as well as dendritic PtRu nanocatalyst involve multiple intermediate steps from which the rate limiting step can be the adsorption and dehydrogenation of MeOH.50 Thus, it can be concluded that the porosity dramatically enhances the availability of active sites (and thus the mass activity); at the same time, however, the mass transport throughout the pore system might generate a broader oxidation peak of methanol in the anodic scan. Introduction of gold in general reduces the binding energy of CO;51 hence the planar 〈111〉 faces of Au nanoprisms might be responsible for the different shape of the MOR oxidation peak. Additionally, nanoprisms have the smallest dimension in the Z direction (the thickness of the prisms is only 18 nm); therefore the pore system can also be more permeable for the analyte and its intermediates further accelerating the mass transport. This is also valid for the EOR, where the AuPrism@mPdPtIr particles are found to be superior in terms of peak width and mass activity. Nevertheless, symmetry-breaking itself has a profound effect on the overall activity in general, greatly enhancing the performance of the nanocatalysts (low onset and overpotential can be achieved). These findings imply that the shape of the tetrametallic catalyst essentially alters the electrocatalytic activity of the particles even if the catalyst loading, the composition and the volume of the cores are identical.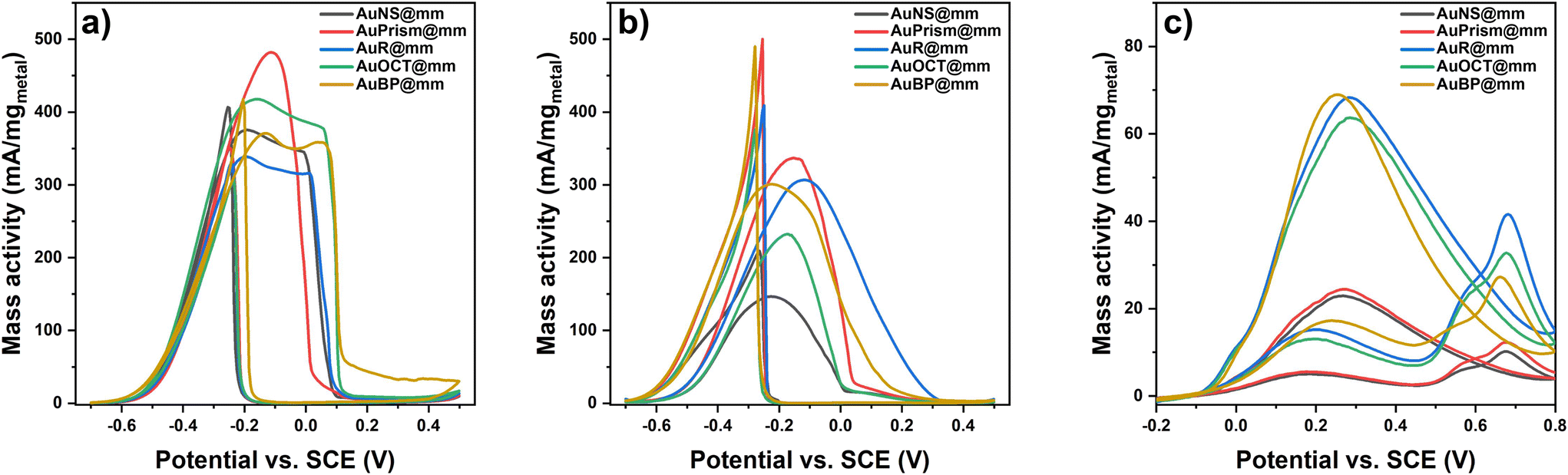 | ||
| Fig. 7 Mass activity of different types of multimetallic nanoparticle electrodes in the MOR (a), EOR (b) and FAOR (c). The total metal concentration was kept constant during the preparation of all electrodes. | ||
To test the tetrametallic nanoparticles in another reaction, the formic acid oxidation reaction (FAOR) was performed in an argon-saturated solution of 1 M formic acid in 0.5 M sulfuric acid supporting electrolyte in the potential window of −0.2 to 0.8 V. Acidic conditions were chosen since the production of formate (being the active species) is more favorable at acidic pH.52Fig. 7c shows the voltammograms for the FAOR in the presence of the nanoparticles with different morphologies. The shape of the CV curves is similar for all model systems and includes two main oxidation events in the forward scan: (i) the bulk electrooxidation of formic acid following the direct pathway and (ii) the oxidation of CO in an indirect pathway at higher potentials.53 The highest oxidation peak in the cathodic scan belongs to the reaction of the formate anion with the COad-free catalyst surface and is usually referred to as the ‘reactivation’ peak.54,55 The separation of the two main peaks in the anodic scan indicates that the formation of COad is suppressed in the FAOR (compared to the MOR and EOR) and offers a way to directly oxidize formic acid at low overpotentials (0.2 V), which is in line with the literature.56 While particles with AuNS and AuPrism exhibit low performance in the FAOR, other symmetry-broken particles have comparably higher mass activity which is ca. 3.5 times higher than that of spherical NPs. It was reported earlier that the synergy between Au, Pd and Pt can enhance the catalytic activity in the FAOR compared to pure Pt. Palladium endows the multimetallic nanoparticles with an advantageous property in formic acid oxidation reaction, namely that almost no CO can be detected on its surface in contrast to Pt.57,58 It has to be noted that Pd does not tolerate acidic conditions and shows significant degradation, while Pt is prone to poisoning by CO due to its higher binding energy.29 Nevertheless, incorporating Au into Pd and Pt structures significantly suppresses the instability of Pd and lowers the CO binding energy on Pt; hence, synergistic effects make the AuPdPt multicomponent particles significantly more active in the FAOR.29 Our results show that multielemental synergy accompanied by symmetry-breaking provides a high-performance electrocatalyst for formic acid oxidation in an acidic medium with a mass activity as high as 70 mA mgmetal−1.
On performing the EOR, MOR and FAOR on core-only Au nanoparticles (that is spheres, prisms and bipyramids), a much lower activity and a higher overpotential in all electrooxidation reactions are observed (Fig. S14†). The porous tetrametallic nanoparticles outperform the mass activity of the plain Au cores with even 2–3 orders of magnitude underlining the advantageous effect of the porous trimetallic shell in all tested reactions.
It can be concluded that the nanocatalyst's performance in pNP degradation and electrooxidation of alcohols and formic acid can be significantly improved via exploiting the novel structural and synergistic properties of the prepared multimetallic catalysts which possess high porosity and enhanced availability of the active sites. Calculating the surface-to-volume ratio of the synthesized core particles indicates that the observed trend of the activities in the test reactions are not governed by the – otherwise slight – surface area differences of the cores. AuBP@mPdPtIr particles show the highest rate constant for pNP degradation although the prisms’ surface-to-volume ratio exceeds that of AuBPs (Fig. 8a). Moreover, tetrametallic particles with octahedral cores outperform the electrocatalytic activity of AuRs and AuBPs in the MOR, even if the trend in the surface area suggested a different order of reactivity (Fig. 8b).
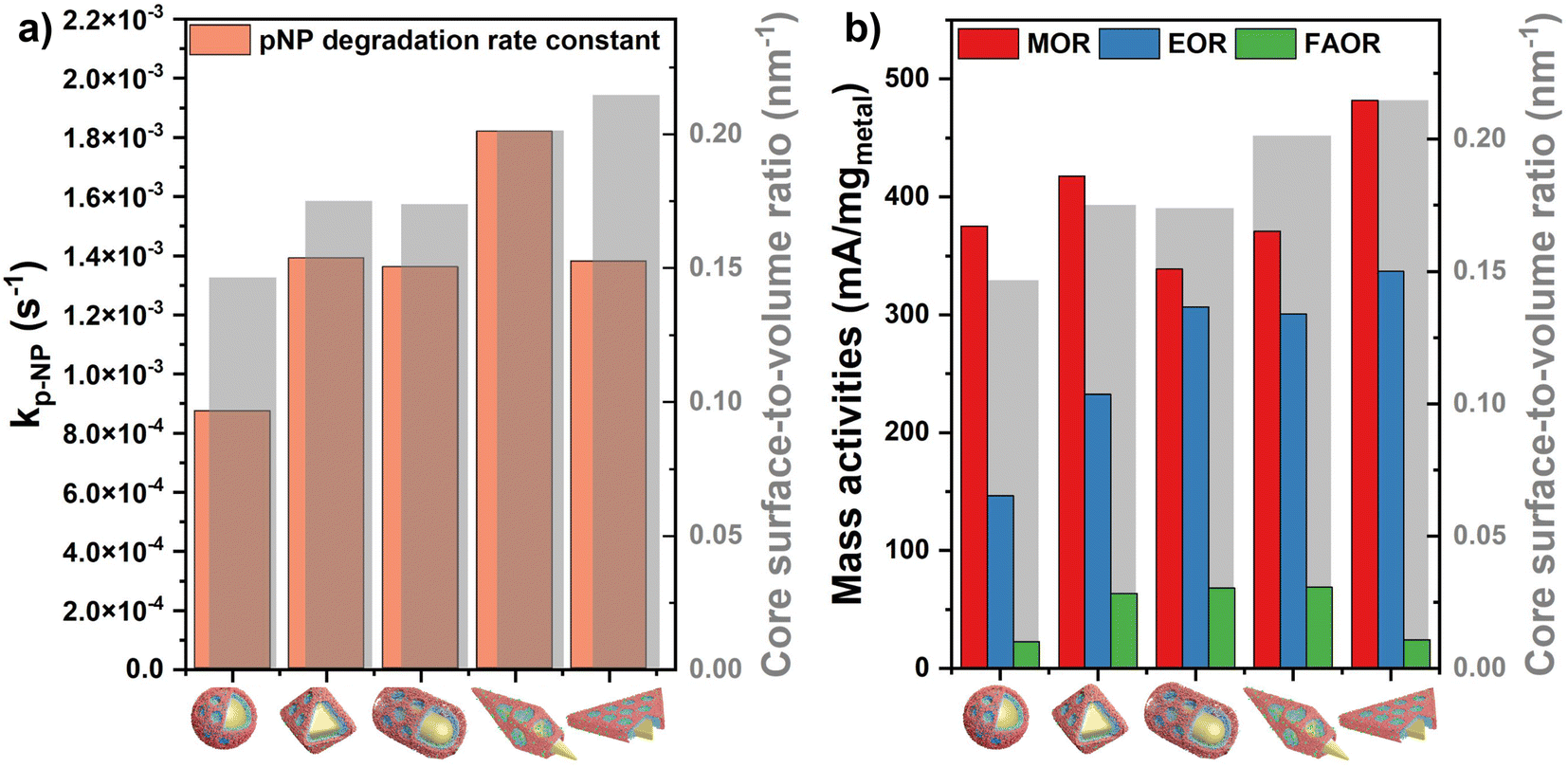 | ||
| Fig. 8 Comparison of the catalytic performance of the tetrametallic nanoparticles with different core morphologies in the degradation reaction of p-nitrophenol (a) and electrooxidation reactions (b). Gray bars represent the surface-to-volume ratios of the corresponding Au core particles. | ||
On comparing the activity of the different nanoparticle types investigated, symmetry-broken nanocatalysts were found to clearly outperform the ones with symmetrical cores both in heterogeneous and electrocatalytic reactions. For heterogeneous pNP degradation reactions, the observed trend of the rate constants correlates well with the ECSA values measured with the electrode-deposited NPs, indicating the central role of the active site availability during electron transfer reactions for the case of homogeneously dispersed particles as well. Nevertheless, in electrocatalytic reactions, the outstanding performance of the nanoprism-shaped particles might be correlated with the structure of the trimetallic shell. Based on the elemental maps in Fig. 2 and Fig. S6,† AuPrism@PdPtIr particles show the most compact Pd adlayer most likely due to the planar faces dominating the prism shaped cores’ surface. It can be anticipated that the electron transfer towards the catalytically active Pt sites is more efficient in this arrangement, which might be due to the better contact between the Pd and Pt domains. Moreover, the planar faces of prism-shaped particles might further facilitate the overall electron transfer towards the active sites upon applying a driving bias potential. Hence, the shape of the cores (and thus, of the multimetallic particles as well) is of importance for designing catalysts for heterogeneous and electrocatalytic reactions and symmetry-breaking opens up a new route towards more active and durable nanocatalysts.
3. Conclusion
The effect of the core morphology in tetrametallic Au@PdPtIr core/shell nanoparticles on their catalytic and electrocatalytic properties has been revealed by carefully controlling the size, composition and shape of the particles while keeping all parameters identical. Gold core particles having the same volume were synthesized by wet-chemical seeded-growth methods and subsequent purification approaches to obtain particles with high shape and size purity. After depositing a porous, trimetallic shell (PdPtIr) onto the cores via a universal synthetic approach, multimetallic NPs with highly accessible surface sites were characterized and their elemental composition was analyzed. An identical total metal concentration was set in all particle solutions to ensure the same catalyst loading on the glassy carbon electrode used in the evaluation of their electrocatalytic performance. It was shown that symmetry breaking of the core in general improves the catalytic activity of the porous tetrametallic NPs in different test reactions: heterogeneous pNP to pAP conversion, alcohol electrooxidation and formic acid electrooxidation reactions. Although all particle types show pronounced activity due to the multielemental synergy and the anticipated intraparticle charge transport, particles with bipyramidal Au cores exhibit the highest rate constant in pNP degradation, while other symmetry broken NPs also surpass the spherical ones. Similar conclusions can be drawn from the electrocatalytic oxidation reactions, where anisometric cores (especially AuPrisms and AuBPs) were found to be superior underlining the importance of the shape of the core particles (and the overall tetrametallic NPs) in application-related reactions. All investigated reactions were performed under ambient conditions and do not require a binding agent for electrode preparation. Since catalytic performance and activity can change from reaction to reaction, fine-tuned and tailored nanoparticles are required for particular applications. This work highlights the important role of the particle morphology in the (electro)catalytic properties and offers a novel method to control the shape, size and composition of multimetallic particles to design nanocatalysts with enhanced performance for targeted application.4. Experimental section/methods
4.1. Chemicals
Hydrogen tetrachloroaurate (HAuCl4), silver nitrate (AgNO3), potassium tetrachloroplatinate (K2PtCl4), hexachloroplatinic acid (H2PtCl6), sodium tetrachloropalladate (Na2PdCl4), sodium hexachloroiridate (Na3IrCl6), cetyltrimethylammonium chloride solution (CTAC, 25wt%), benzyldimethylhexadecylammonium chloride (BDAC), sodium iodide (NaI), potassium iodide (KI), Pluronic F-127, ascorbic acid (AA), sodium borohydride (NaBH4), potassium hydroxide (KOH), p-nitrophenol (pNP), hydrochloric acid (HCl), nitric acid (HNO3), formic acid (HCOOH) and sulfuric acid (H2SO4, 99.999%) were purchased from Merck. Cetyltrimethylammonium bromide (CTAB) and sodium oleate (NaOL) were supplied by Thermo Fisher Scientific and TCI Chemicals, respectively. Methanol, ethanol, and 2-propanol were supplied by VWR Avantor. Ultrapure water (>18.2 MΩ cm) was produced using an Adrona B300 purification system. All glassware and stirring bars were cleaned with aqua regia (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 V/V ratio of cc. HCl and HNO3), rinsed thoroughly with ultrapure water, and dried at 70 °C. Metal precursors, NaBH4, NaOL, NaI and KI were stored in a glove-box and their solutions were prepared freshly.
:1 V/V ratio of cc. HCl and HNO3), rinsed thoroughly with ultrapure water, and dried at 70 °C. Metal precursors, NaBH4, NaOL, NaI and KI were stored in a glove-box and their solutions were prepared freshly.
4.2. Synthesis of gold nanorods
Gold nanorods with dimensions of 76 × 26 nm were synthesized following a seed mediated growth based on the work of Ye et al. with slight modifications.59,60 Briefly, seed solutions were initially prepared in a 22 mL glass vial by sequentially adding 2.5 mL of 0.5 mM HAuCl4 to 2.5 mL of 200 mM CTAB under mild stirring to homogenize for approximately 2 minutes. While the contents were under fast stirring at 1100 rpm, 0.5 mL of 6 mM NaBH4 was added to the vial in one quick shot. The stirring was continued for 1 minute and then the stirring bar was removed. The seed solutions were then aged for 30 minutes in a heat block maintained at 30 °C. Three parallel seed solutions were prepared and the one with an appropriate spectrum and Au0 concentration was used for the growth.The nanorods were grown in a 250 mL Schott glass bottle. 1.8 g of CTAB and 0.3086 g of sodium oleate were mixed in the vial and 50 mL of MilliQ water was added. Under stirring in a water bath, the mixture temperature was brought to 60 °C to dissolve the CTAB/oleate mixture. The mixture was then cooled down to 30 °C under tap water flow and kept at 30 °C during the consecutive addition of the other reagents in a water bath. First, 2.0 mL of 4 mM AgNO3 was added under mild stirring and stirred for 15 minutes (250 rpm). The stirring speed was increased to 700 rpm followed by the addition of 50 mL of 1 mM HAuCl4 solution for 90 minutes until the solution turned colorless. Afterwards, 420 μL of cc. HCl was added to adjust the pH under stirring at 400 rpm for 15 minutes followed by the addition of 250 μL of 64 mM ascorbic acid. The mixture was vigorously stirred for 30 seconds. Finally, 80 μL of seed solution was injected under stirring and shaking to further homogenize. The stirring bar was removed and the vial was placed in a water bath set at 28 °C overnight.
To stop the growth, the as-synthesized sample was centrifuged in 50 mL Falcon tubes at 6500 rcf for 20 minutes. The supernatant was carefully removed, and the precipitate was collected and redispersed in 50 mM CTAB to an approximate total volume of 12 mL. The concentrated sample was diluted with a factor of 20 in a cuvette and the extinction at 400 nm was measured to calculate the Au0 concentration.61 According to this, the concentrated NR stock solution has a gold concentration of 1.07 mM (0.21 g L−1). This solution shows excellent temporal stability for up to 2 years if stored in a water bath set to 28 °C.
4.3. Synthesis of gold nanoprisms
The gold nanoprisms were synthesized through a seed mediated growth that also involves an intermediate growth to obtain the final nanoprisms. The synthetic method by Scarabelli et al.62 was adopted and upscaled. Three parallel seed solutions were prepared following the method of Wu et al.63 In a 22 mL vial, 4.7 mL of 100 mM CTAC was added followed by 25 μL of 50 mM HAuCl4 under mild stirring (300 rpm). The rpm was increased to 1100 rpm and then 300 μL of 10 mM NaBH4 solution was quickly injected in one shot and stirred for 2 minutes. The seed solutions were aged for 90 minutes in a heat block maintained at 30 °C without the stirring bars.The seed solution for the intermediate growth was diluted by adding 500 μL of seed solution to 4.5 mL of 100 mM CTAC.
Two solutions for the intermediate growth were prepared in a tube labeled A and an Erlenmeyer flask labeled B. 8 mL and 40 mL of MilliQ water were added to A and B, respectively. 1.6 mL and 40 mL of 100 mM CTAC, 40 μL and 1000 μL of 50 mM HAuCl4, and 15 μL and 600 μL of 10 mM NaI were added in this order. The contents of B were continuously stirred to homogenize, while A was homogenized by shaking.
To perform the intermediate growth, 40 μL and 800 μL of 100 mM ascorbic acid were respectively added to A and B, while B was under stirring. 100 μL of the diluted seed was then injected into solution A which was then quickly shaken to homogenize (ca. 2 seconds). Then, 6.4 mL of A was rapidly injected into B while under vigorous stirring at 500 rpm for 2 minutes. The stirring speed was afterwards reduced to 250 rpm for 80 minutes. Two growth solutions were prepared in this manner. Their UV-VIS extinction profiles were identical and as such they were merged into a total volume of 176 mL.
4.4. Depletion and concentration of Au nanoprisms
The as-synthesized gold nanotriangles were purified by surfactant-mediated depletion using CTAC based on the work of Scarabelli et al.62 Briefly, 35 mL of the solution were put in 5 × 50 mL Falcon tubes and to this, 5.11 mL of 25% CTAC was added to each tube to set the CTAC concentration to 0.14 M. The solutions were left undisturbed overnight. 37 mL of the pink supernatant was carefully decanted from each tube followed by the addition of 5.61 mL of MilliQ water to set back the CTAC concentration to 0.05 M. To initiate the second depletion, the solutions were each topped up to 35 mL with 0.05 M CTAC followed by the addition of 4.16 mL of 25% CTAC to set the CTAC concentration to 0.125 M. After overnight depletion, the supernatant was carefully decanted and the precipitate was redispersed in a total volume of 77 mL (50 mM CTAC) in a glass vial. The UV-VIS spectrum of the purified sample was obtained and used to determine the Au0 concentration (0.415 mM). This final AuPrism stock solution can be stored at room temperature for further use.4.5. Synthesis of gold nanospheres
Gold nanospheres were synthesized by the seed-mediated growth of the Turkevich seeds.64 44.4 mL of MilliQ water was measured into a blue-capped vial and 1.2 mL of 0.01 M HAuCl4 was added. The mixture was brought to boil and then 1.2 mL of sodium citrate (0.171 g/15 mL MilliQ water) was added. The mixture was boiled for a further 15 minutes to obtain a ruby red solution of approximately 18 nm sized gold spherical particles. For growth, two solutions A and B were prepared in Falcon tubes as follows: 101.4 μL of HAuCl4 was added to 9.899 mL of MilliQ water to make solution A. Solution B contained 9.25 mL of MilliQ water, 250 μL of 1% sodium citrate and 500 μL of 1% ascorbic acid. In a 100 mL three-neck flask, mixture of 17 mL of MilliQ water and 3 mL of the pre-synthesized seeds was prepared. Solutions A and B were carefully introduced dropwise into the flask through a peristaltic pump (Ismatec Masterflex) at a rate of 250 μL min−1 while the contents were moderately stirred at 280 rpm at ambient temperature until all of solutions A and B were completely added. To upscale the synthesis, 68 mL of MilliQ water and 12 mL of seeds were introduced into a 250 mL three-neck flask under stirring at 280 rpm. To this, 50 mL of solution A (49.495 mL of MilliQ water and 507 μL of HAuCl4) and 50 mL of solution B (46.25 mL of MilliQ water, 1.25 mL of 1% sodium citrate and 2.50 mL of 1% ascorbic acid) were added at a rate of 250 μL min−1 and stirred steadily until the addition was complete. Surfactant exchange from citrate to CTAB was accomplished by adding 100 mL of 200 mM CTAB to 100 mL of the as-synthesized gold nanosphere stock. The mixture was gently shaken to mix and kept in a water bath at 28 °C overnight. Finally, the gold nanospheres were concentrated by centrifugation at 4500 rcf for 30 minutes. The supernatant was carefully decanted, and the precipitate was redispersed in ca. 8 mL of 50 mM CTAB. Samples were prepared for zeta potential, electrophoretic mobility, and UV-VIS measurements in both surfactants. The concentration of Au0 in the AuNS stock solution was 4.58 mM.4.6. Synthesis of gold octahedra
Gold octahedra were synthesized by the seed-mediated growth process. The seeds were prepared in triplicate to ensure reproducibility by the method of Wu et al.63 and the growth typically similar to that of gold nanoprisms was adopted from Chang et al.65 In a 22 mL glass vial, 5 mL of 0.5 mM HAuCl4 was added to 5 mL of 0.2 M CTAC and the mixture was gently stirred to homogenize. While the stirring speed was increased to 1100 rpm, 0.45 mL of 20 mM NaBH4 at room temperature was swiftly injected. The stirring continued for about one minute and the stirring bars were then removed. The seed solutions were aged for 60 minutes in a heat block maintained at 30 °C.Two growth solutions were prepared and labeled as A and B. For solution A, 9.5 mL of 105.26 mM CTAC, 250 μL of 0.01M HAuCl4, 5 μL of 0.01 M KI and 220 μL of 0.04 M ascorbic acid were sequentially added in a 22 mL glass vial. For solution B, 95 mL of 105.26 mM CTAC, 2.5 mL of 0.01M HAuCl4, 50 μL of 0.01 M KI and 2.2 mL of 0.04 M ascorbic acid were sequentially added in a 250 mL Schott glass bottle under stirring. Afterwards, 70 μL of seed solution was injected into vial A and the solution was shaken to homogenize. After 6 seconds, 700 μL from A was quickly added to solution B under vigorous stirring. The contents of the flask were then moderately stirred for another 15 minutes at room temperature.
The as-synthesized gold octahedra were divided into 4 Falcon tubes (25 mL each) and centrifuged at 3000 rcf for 20 minutes; the supernatant was decanted and the precipitate was redispersed in 50 mM CTAC to a total volume of 8 mL. The Au octahedra stock solution has an Au0 concentration of 2.36 mM.
4.7. Synthesis of gold bipyramids
The synthesis of gold bipyramids was preceded by the preparation of pentatwinned seeds. The whole synthesis procedure is described in the paper of Zámbó et al.66 In a 40 mL glass vial, 16.5 mL of 60.6 mM CTAC was introduced and stirred in a 30 °C water bath. To this, 200 μL of 25 mM HAuCl4 and 2 mL of 50 mM sodium citrate were sequentially added under stirring and the solution was stirred for 10 minutes at 30 °C. Finally, 1.2 mL of 10 mM NaBH4 at room temperature was swiftly injected while the vial contents were stirred at 1100 rpm. After 1 minute of stirring, the stirring bar was removed, and the seeds were aged undisturbed in an oven (Memmert UN 55) maintained at 40 °C for five days. The formation of the pentatwinned seeds was accompanied by a color change (from brown to red) as well as the appearance of an LSPR peak at 515 nm.The growth was achieved by adding different seed volumes to fine tune the size of the bipyramids in the desired size range. The dimensions of the bipyramids were found to be suitable in the case of using 35 μL of seeds; thus, an upscaled synthesis was performed via increasing the total volume of the solution with a factor of 10.
Briefly, 200 mL of 100 mM CTAB was added to a 250 mL Schott glass bottle followed by 4 mL of 25 mM HAuCl4. The mixture was mildly stirred to homogenize, and under stirring, 2 mL of 10 mM AgNO3 was added. Afterwards, 4 mL of 1 M HCl and 1.6 mL of 100 mM ascorbic acid were added sequentially. The solution turned colorless. Finally, 350 μL of seed solution was injected under stirring for 2 minutes; thereafter the stirring bar was removed and the glass was placed in a water bath (30 °C) for 4 hours and then in a 28 °C water bath overnight.
The as-synthesized bipyramid solution was concentrated by centrifugation (6 × 36 mL) at 3000 rcf for 20 minutes. The pinkish supernatants were decanted, and the precipitates were redispersed in 10–10 mL of 10 mM CTAB and centrifuged again. The subsequent precipitates were then collected and redispersed in 1 mM CTAB to a total volume of 15 mL.
4.8. Purification of Au bipyramids
The as-synthesized AuBP solution contains a remarkable amount of spherical impurities. The concentrated bipyramid stock in 1 mM CTAB was purified to enhance the shape purity. To purify the sample, BDAC-mediated flocculation depletion was employed following the method by Lee et al.67 To 3 mL of concentrated bipyramid solution in 1 mM CTAB in 5 Falcon tubes, 3 mL of MilliQ water and 4 mL of 0.5 mM BDAC were added, and the tubes were carefully secured in a rack and placed in an oven set at 30 °C for 5 hours. Finally, the pink supernatant (containing the spherical impurities) was decanted, and the residual bipyramid precipitate was washed to eliminate BDAC by redispersing in 10 mL of 1 mM CTAB and centrifuged twice at 3000 rcf for 20 minutes. Finally, the precipitate was collected to a total volume of 6 mL. The concentration of CTAB and Au0 in the final stock was 50 mM and 10.138 mM, respectively.4.9. Synthesis of tetrametallic nanoparticles
The syntheses were performed according to our previous work.39 Stock solutions of the Au core particles (rods, spheres, octahedra, prisms and bipyramids) were further concentrated in two consecutive centrifugation–redispersion cycles.For AuNR, AuNS, AuOCT, AuPrism and AuBP, 11.258 mL, 2.616 mL, 5.084 mL, 28.892 mL and 1.183 mL of solution were centrifuged, respectively. The precipitate was redispersed in 600 μL of 3 wt% F-127. This solution was centrifuged again, and the precipitate was redispersed in 50 μL of 3 wt% F-127 solution to obtain the concentrated AuNP stock solutions (cAu0 = 239.8 mM). In a 40 mL screw cap (with septum) vial, 1.419 mL of water, 0.501 mL of 10 wt% F127 solution and 60 μL of 6 M HCl were mixed and heated to 70 °C in a temperature-controlled heat block. Under stirring (200 rpm), 120 μL of 0.1 M Na2PdCl4, 120 μL of 0.1 M K2PtCl4, and 180 μL of 0.1 M H2PtCl6 were subsequently added. Afterwards, the concentrated AuNP stock (50 μL) was added followed by the rapid injection of 3.0 mL of 0.1 M AA. The solution was stirred for 40 minutes at 70 °C. At this stage, the concentrations of F-127 and the core Au particles were 1 wt% and 2.2 mM, respectively.
Then, the temperature was increased to 80 °C and 60 μL of 0.1 M Na3IrCl6 solution was injected. After adding 807 μL of 0.1 M AA, the growth of iridium was initiated by injecting 284 μL of 25 mg mL−1 NaBH4 solution through the septum of the cap. This highly concentrated borohydride solution at 80 °C decomposes quickly forming extensive foam and pressure inside the vial. Hence, the pressure build-up must be avoided by spiking an additional needle through the septum. Moreover, using a taller vial (even if the total volume of the solution would not justify it) helps avoid the foam flowing out. The solution was stirred at 80 °C for 20 minutes and then cooled to room temperature naturally.
The tetrametallic nanoparticles were purified and concentrated in 50 mL Falcon tubes. The as-synthesized solution was divided into 3 Falcon tubes and 20 mL of MilliQ water was added to each followed by centrifugation at 3500 rcf for 15 minutes. After removing the supernatant, 15 mL of MilliQ water was added to each tube and a second round of centrifugation was performed. After removing the supernatant, the total volume of the final stock solution was set to ca. 3 mL. The total metal concentration of the tetrametallic nanoparticle stocks was determined by ICP-OES and then set to 2.154 g L−1 in terms of total metal concentration in all nanoparticle solutions.
4.10. Optical characterization
Extinction spectra of the core and core/shell particles were recorded using a Shimadzu UV 3600i UV-Vis-NIR spectrophotometer in the wavelength range of 200–1350 nm. The stock solutions were diluted in a QS quartz macro cuvette.4.11. Electron microscopy
Stock solutions (100 μL) were diluted (5×) and purified further by centrifugation. The precipitates were redispersed in 200 μL of MilliQ water and 1.5 μL was drop-cast onto a Si (SEM) or carbon-coated Cu grid (TEM). SEM secondary electron images were taken with a Zeiss LEO 1540-XB operating at 5 keV equipped with an Oxford Instruments UltimMax 40 detector for energy-dispersive X-ray investigations. TEM, STEM images and TEM-EDX maps were taken with a JEOL 3010 microscope at 300 keV and a Thermo Fisher THEMIS 200 spherical aberration-corrected transmission electron microscope at 200 keV, respectively.4.12. XRD sample preparation and measurements
Nanoparticle stock solutions were concentrated to the value of ca. 30 mM in terms of metal concentration via centrifugation. 10–20 μL solution was drop-cast onto a 5 × 5 mm Si substrate and dried under ambient conditions. Measurements were performed using a Bruker AXS D8 Discover X-ray diffractometer equipped with a Göbel-mirror and scintillation detector with Cu Kα (λ = 1.5406 Å) radiation. The X-ray beam dimensions were 1 mm × 5 mm, with a 2Θ step size of 0.02° and a scan speed of 0.2° min−1. While the range of 30–90° was recorded with lower resolution, higher resolution scans were performed in the relevant 2Θ range for the elements (35–50°). We used the Diffrac.EVA program and the ICDD PDF database for phase identification. Powder diffraction file (PDF) cards 04-0784 (gold), 04-0802 (platinum), 46-1043 (palladium) and 46-1044 (iridium) were used to identify the XRD peaks.4.13. Zeta potential and electrophoretic mobility
For the measurement of size, zeta-potential and electrophoretic mobility distributions, folded capillary cells (DTS-1070, filled with diluted nanoparticle solutions) were used in a Malvern ZetaSizer Nano ZS (λ = 635 nm).4.14. ICP-OES
A set of calibration solutions (0 ppm, 0.01 ppm, 0.1 ppm, 1 ppm, 10 ppm) were prepared freshly from Merck standard solutions (metal concentration of 1000 ppm) prior to the measurement. A portion of the mesoporous tetrametallic stock solutions (100 μL) was digested in aqua regia (675 μL of cc. HCl + 225 μL of cc. HNO3) for 15 minutes. After the dissolution, the solutions were topped up to 10 mL by adding 8.9 mL of MilliQ water and 100 μL of Y standard solution (100 ppm Y in 20% HNO3). Hence, the total dilution of the stock solutions was 100×. The concentration of the metals was determined using a PerkinElmer Avio 200 ICP-OES instrument by considering the wavelength of 242.795 nm for Au, 340.458 nm and 324.270 nm for Pd, 214.423 nm and 265.945 nm for Pt and 224.268 nm and 212.681 nm for Ir to avoid spectral overlapping. Due to the relatively low Ir loading of the particles, Ir was determined in the diluted stock solution at 40×, but all other measurement parameters were kept the same.4.15. Heterogeneous catalysis (pNP–pAP conversion) using solution-dispersed nanoparticles
The reduction of p-nitrophenol (pNP) was conducted in triplicate for each morphology of tetrametallic nanoparticles in PMMA disposable cuvettes. 976 μL of MilliQ water, 6.66 μL of 12 mM pNP, 20 μL of 100 mM NaBH4 and 3 μL of nanoparticle solution (2.154 g L−1) were consecutively introduced into a PMMA disposable cuvette and thoroughly homogenized. The time between the borohydride addition and the catalyst injection in all cases was kept constant at 30 seconds. Time-resolved UV-Vis measurements were done in a Shimadzu UV 3600i UV-Vis-NIR spectrophotometer in the wavelength range of 240–600 nm and the decay of absorption at 400 nm attributed to the absorption maximum of the p-nitrophenolate anion was monitored for 100 cycles (the duration of each cycle was 42 s). After the measurement, the solution was centrifuged and the precipitate was redispersed in 100 μL of MilliQ water. A 1 μL sample of the solution was drop-cast onto a silicon wafer for SEM to check the morphology of the particles after heterogeneous pNP degradation in 90 minutes.4.16. Electrocatalytic alcohol and formic acid oxidation reactions
EOR, MOR and FAOR experiments were carried out in a conventional three-electrode electrochemical cell setup consisting of an Ag/AgCl (3M KCl, ASL Japan) reference electrode, a coiled Pt wire auxiliary electrode (BASi) and a glassy carbon electrode (GCE, 3 mm in diameter, ASL Japan) as the working electrode. Prior to any electrochemical measurements, the GCE was polished using a microfibre pad and 1μm diamond polishing paste, rinsing with MilliQ water and ultrasonication in MilliQ water for 3 minutes. These steps were repeated twice more and then GCE was dried under nitrogen. Tetrametallic nanoparticles (20 μL) were centrifuged and concentrated to achieve a total metal concentration of 10 g L−1. The concentrated mNP solutions (2 μL) were deposited and dried on the surface of GCE electrodes under ambient conditions. Alcohols (EtOH, MeOH) were added to 1 M KOH solution to set the alcohol concentration to 1 M. Also, formic acid was added to 0.5 M sulfuric acid to set its concentration to 1 M, and all the electrolytes were bubbled with Ar for 30 minutes. The electrodes were immersed in a glass cell (12 mL in total) equipped with a Teflon cap and the cell was air tightly closed. Cyclic voltammetry measurements were performed using a PalmSens EmStat4X HR potentiostat in the bias potential range of −0.7 to 0.5 V (EOR and MOR) and −0.2 to 0.8 V (FAOR) applying automatic current ranging.Author contributions
D. Z. conceived the concept. A. S. O., D. K. and D. Z. carried out the synthesis and optical and structural characterization of the nanoparticles. A. S. O. and D. Z. performed the catalytic experiments. A. S. O. and D. Z. analyzed and visualized the data. G. Z. R and D. Z. performed the TEM and SEM investigations. Z. E. H. carried out the XRD measurements. I. T. performed the ICP-OES elemental analyses. D. Z. took the lead in writing the manuscript, funding acquisition and supervision. A. D. contributed to the writing, editing of the paper and the discussion of the data. The final manuscript was written with the contributions of all authors. All authors have given approval for the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
A.S.O. and D.K. acknowledge the support of Pro Progressio and the József Varga Foundation. The work was financially supported by the NRDI Fund of Hungary under the grant OTKA FK 142148. Project no. TKP2021-NKTA-05 has been implemented with the support provided by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund (NRDI), financed under the TKP2021 funding scheme. The support of VEKOP-2.3.3-15-2016-00002 is also acknowledged.References
- T. K. Sau, A. L. Rogach, F. Jäckel, T. A. Klar and J. Feldmann, Adv. Mater., 2010, 22, 1805–1825 CrossRef CAS.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS.
- W. Lu, A. K. Singh, S. A. Khan, D. Senapati, H. Yu and P. C. Ray, J. Am. Chem. Soc., 2010, 132, 18103–18114 CrossRef CAS.
- X. Qian, J. Li and S. Nie, J. Am. Chem. Soc., 2009, 131, 7540–7541 CAS.
- J. W. Hong, S.-U. Lee, Y. W. Lee and S. W. Han, J. Am. Chem. Soc., 2012, 134, 4565–4568 CAS.
- M. B. Cortie and A. M. McDonagh, Chem. Rev., 2011, 111, 3713–3735 CAS.
- M. Rycenga, C. M. Cobley, J. Zeng, W. Li, C. H. Moran, Q. Zhang, D. Qin and Y. Xia, Chem. Rev., 2011, 111, 3669–3712 CAS.
- M. Kim, Y. W. Lee, D. Kim, S. Lee, S.-R. Ryoo, D.-H. Min, S. B. Lee and S. W. Han, ACS Appl. Mater. Interfaces, 2012, 4, 5038–5043 CAS.
- P. Ghosh, G. Han, M. De, C. Kim and V. Rotello, Adv. Drug Delivery Rev., 2008, 60, 1307–1315 CAS.
- H. Jang, S.-R. Ryoo, K. Kostarelos, S. W. Han and D.-H. Min, Biomaterials, 2013, 34, 3503–3510 CAS.
- B. Kumar Jena and C. Retna Raj, Electrochem. Commun., 2008, 10, 951–954 CAS.
- L. Yaqoob, T. Noor and N. Iqbal, RSC Adv., 2021, 11, 16768–16804 RSC.
- M. Hosseini, T. Barakat, R. Cousin, A. Aboukaïs, B.-L. Su, G. De Weireld and S. Siffert, Appl. Catal., B, 2012, 111–112, 218–224 CrossRef CAS.
- S. W. Kang, Y. W. Lee, Y. Park, B.-S. Choi, J. W. Hong, K.-H. Park and S. W. Han, ACS Nano, 2013, 7, 7945–7955 CrossRef CAS.
- J. N. Hansen, H. Prats, K. K. Toudahl, N. Mørch Secher, K. Chan, J. Kibsgaard and I. Chorkendorff, ACS Energy Lett., 2021, 6, 1175–1180 CrossRef CAS.
- L. Sun, H. Lv, J. Feng, O. Guselnikova, Y. Wang, Y. Yamauchi and B. Liu, Adv. Mater., 2022, 34, 2201954 CrossRef CAS PubMed.
- K. Loza, M. Heggen and M. Epple, Adv. Funct. Mater., 2020, 30, 1909260 CrossRef CAS.
- B. A. Yusuf, W. Yaseen, J. Xie, A. A. Babangida, A. I. Muhammad, M. Xie and Y. Xu, Nano Energy, 2022, 104, 107959 CrossRef CAS.
- K. D. Gilroy, A. Ruditskiy, H.-C. Peng, D. Qin and Y. Xia, Chem. Rev., 2016, 116, 10414–10472 CrossRef CAS.
- C. Xie, Z. Niu, D. Kim, M. Li and P. Yang, Chem. Rev., 2020, 120, 1184–1249 CrossRef CAS.
- D. Wang, A. Villa, F. Porta, L. Prati and D. Su, J. Phys. Chem. C, 2008, 112, 8617–8622 CrossRef CAS.
- Y. Yang, H. Jia, N. Hu, M. Zhao, J. Li, W. Ni and C. Zhang, J. Am. Chem. Soc., 2024, 146, 7734–7742 CrossRef CAS.
- L. Hao, N. Hu, L. Wang, H. Jia and W. Ni, ACS Appl. Nano Mater., 2024, 7, 23028–23036 CrossRef CAS.
- H. Kim, T. Y. Yoo, M. S. Bootharaju, J. H. Kim, D. Y. Chung and T. Hyeon, Adv. Sci., 2022, 9, 2104054 CrossRef CAS PubMed.
- A. K. Ipadeola, K. Eid, A. K. Lebechi, A. M. Abdullah and K. I. Ozoemena, Electrochem. Commun., 2022, 140, 107330 CrossRef CAS.
- K. Sasaki, H. Naohara, Y. Choi, Y. Cai, W.-F. Chen, P. Liu and R. R. Adzic, Nat. Commun., 2012, 3, 1115 CrossRef.
- B. D. Adams, G. Wu, S. Nigro and A. Chen, J. Am. Chem. Soc., 2009, 131, 6930–6931 CrossRef CAS.
- W. Zhang and X. Lu, Nanotechnol. Rev., 2013, 2, 487–514 CAS.
- C. Li, O. J. H. Chai, Q. Yao, Z. Liu, L. Wang, H. Wang and J. Xie, Mater. Horiz., 2021, 8, 1657–1682 RSC.
- H. M. Song, D. H. Anjum and N. M. Khashab, RSC Adv., 2012, 2, 3621 RSC.
- R. Narayanan and M. A. El-Sayed, Nano Lett., 2004, 4, 1343–1348 CrossRef CAS.
- H. L. K. S. Stolle, J. J. Kluitmann, A. Csáki, J. M. Köhler and W. Fritzsche, Catalysts, 2021, 11, 1442 CrossRef CAS.
- Y. Kim, J. W. Hong, Y. W. Lee, M. Kim, D. Kim, W. S. Yun and S. W. Han, Angew. Chem., Int. Ed., 2010, 49, 10197–10201 CrossRef CAS PubMed.
- R. Jin, Nanotechnol. Rev., 2012, 1, 31–56 CAS.
- R. G. Weiner, M. R. Kunz and S. E. Skrabalak, Acc. Chem. Res., 2015, 48, 2688–2695 CrossRef CAS.
- H. Jung, M. E. King and M. L. Personick, Curr. Opin. Colloid Interface Sci., 2019, 40, 104–117 CrossRef CAS.
- G. Li, W. Zhang, N. Luo, Z. Xue, Q. Hu, W. Zeng and J. Xu, Nanomaterials, 2021, 11, 1926 CrossRef CAS.
- X. Wang, J. Qi, X. Luo, Z. Yang, Y. Fan, Z. Jiang, C. Liu, J. Yang and W. Chen, Int. J. Hydrogen Energy, 2021, 46, 36771–36780 Search PubMed.
- D. Zámbó, D. Kovács, G. Z. Radnóczi, Z. E. Horváth, A. Sulyok, I. Tolnai and A. Deák, Small, 2024, 20, 2400421 CrossRef.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 Search PubMed.
- R. D. Neal, Y. Inoue, R. A. Hughes and S. Neretina, J. Phys. Chem. C, 2019, 123, 12894–12901 CrossRef CAS.
- N. K. Reddy Bogireddy, Y. R. Mejia, T. M. Aminabhavi, V. Barba, R. H. Becerra, A. D. Ariza Flores and V. Agarwal, J. Environ. Manage., 2022, 316, 115292 CrossRef CAS PubMed.
- N. K. Shrestha, S. A. Patil, J. Han, S. Cho, A. I. Inamdar, H. Kim and H. Im, J. Mater. Chem. A, 2022, 10, 8989–9000 RSC.
- L. Lai, G. Yang, Q. Zhang, H. Yu and F. Peng, J. Power Sources, 2021, 509, 230397 Search PubMed.
- E. D. Rus, R. H. Wakabayashi, H. Wang and H. D. Abruña, ChemPhysChem, 2021, 22, 1397–1406 CrossRef CAS.
- E. N. El Sawy and V. I. Birss, ACS Appl. Mater. Interfaces, 2018, 10, 3459–3469 CrossRef CAS PubMed.
- L. Huang, X. Zhang, Y. Han, Q. Wang, Y. Fang and S. Dong, Chem. Mater., 2017, 29, 4557–4562 CrossRef CAS.
- D. Y. Chung, K.-J. Lee and Y.-E. Sung, J. Phys. Chem. C, 2016, 120, 9028–9035 CrossRef CAS.
- Z. Yin, H. Zheng, D. Ma and X. Bao, J. Phys. Chem. C, 2009, 113, 1001–1005 CrossRef CAS.
- X. Teng, S. Maksimuk, S. Frommer and H. Yang, Chem. Mater., 2007, 19, 36–41 CrossRef CAS.
- J. Suntivich, Z. Xu, C. E. Carlton, J. Kim, B. Han, S. W. Lee, N. Bonnet, N. Marzari, L. F. Allard, H. A. Gasteiger, K. Hamad-Schifferli and Y. Shao-Horn, J. Am. Chem. Soc., 2013, 135, 7985–7991 CrossRef CAS PubMed.
- C. Busó-Rogero, A. Ferre-Vilaplana, E. Herrero and J. M. Feliu, Electrochem. Commun., 2019, 98, 10–14 CrossRef.
- H. Fan, M. Cheng, L. Wang, Y. Song, Y. Cui and R. Wang, Nano Energy, 2018, 48, 1–9 CrossRef CAS.
- N. Perini, M. V. F. Delmonde, C. Ranjan and H. Varela, J. Solid State Electrochem., 2020, 24, 1811–1818 CrossRef CAS.
- J. Joo, T. Uchida, A. Cuesta, M. T. M. Koper and M. Osawa, J. Am. Chem. Soc., 2013, 135, 9991–9994 CrossRef CAS PubMed.
- H. Okamoto, W. Kon and Y. Mukouyama, J. Phys. Chem. B, 2005, 109, 15659–15666 CrossRef CAS PubMed.
- M. Arenz, V. Stamenkovic, T. J. Schmidt, K. Wandelt, P. N. Ross and N. M. Markovic, Phys. Chem. Chem. Phys., 2003, 5, 4242–4251 CAS.
- H. Miyake, T. Okada, G. Samjeské and M. Osawa, Phys. Chem. Chem. Phys., 2008, 10, 3662 RSC.
- X. Ye, C. Zheng, J. Chen, Y. Gao and C. B. Murray, Nano Lett., 2013, 13, 765–771 CrossRef CAS.
- D. Kovács, A. Deák, G. Z. Radnóczi, Z. E. Horváth, A. Sulyok, R. Schiller, O. Czömpöly and D. Zámbó, J. Mater. Chem. C, 2023, 11, 8796–8807 RSC.
- L. Scarabelli, A. Sánchez-Iglesias, J. Pérez-Juste and L. M. Liz-Marzán, J. Phys. Chem. Lett., 2015, 6, 4270–4279 CrossRef CAS.
- L. Scarabelli and L. M. Liz-Marzán, ACS Nano, 2021, 15, 18600–18607 CrossRef CAS.
- H.-L. Wu, C.-H. Kuo and M. H. Huang, Langmuir, 2010, 26, 12307–12313 CrossRef CAS.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55 RSC.
- C.-W. Chang, S.-P. Chu and W.-L. Tseng, J. Chromatogr. A, 2010, 1217, 7800–7806 CrossRef CAS PubMed.
- D. Zámbó, P. Rusch, F. Lübkemann and N. C. Bigall, ACS Appl. Mater. Interfaces, 2021, 13, 57774–57785 CrossRef PubMed.
- J.-H. Lee, K. J. Gibson, G. Chen and Y. Weizmann, Nat. Commun., 2015, 6, 7571 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr03589e |
| This journal is © The Royal Society of Chemistry 2025 |