
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence“Sergeants-and-soldiers” principle in the synthesis of intrinsically chiral Au13 clusters†
Katsuya
Mutoh
 ,
Sonomi
Kawakita
,
Teppei
Yahagi
and
Takuya
Nakashima
*
,
Sonomi
Kawakita
,
Teppei
Yahagi
and
Takuya
Nakashima
*
Department of Chemistry, Graduate School of Science, Osaka Metropolitan University, 3-3-138 Sugimoto, Sumiyoshi, Osaka 558-8585, Japan. E-mail: takuya.nakashima@omu.ac.jp
First published on 28th October 2024
Abstract
Surface ligand modification on atomically precise metal clusters is one of the important strategies not only to stabilize monodisperse clusters, but also to derive unique optical and electronic properties. The chiral coordination of surface ligands on clusters controls the global chirality in clusters giving rise to unambiguous optical activity including circular dichroism (CD) and circularly polarized luminescence (CPL). Stereoselective synthesis of intrinsically chiral clusters has been achieved by the use of chiral ligands stabilizing one-handed clusters selectively. Herein, we investigate the effect of chiral ligand coordination on the deracemization of intrinsically chiral Au13 superatom clusters. The mixture of achiral bis(diphenylphosphino)ethane (DPPE) and chiral (R,R)-1,2-bis[(2-methoxyphenyl)phenylphosphino]ethane (R-DIPAMP) led to the formation of a mixture of [Au13(R-DIPAMP)x(DPPE)5−xCl2]3+ clusters with varied x-values (x = 0–5). Reverse phase HPLC successfully fractionalized the mixture into solutions composed of single component Au13 clusters depending on the x-values. The separated Au13 clusters afforded similar optical activity to that of [Au13(R-DIPAMP)5Cl2]3+ in the CD study regardless of x-values (x = 1–4). The DFT calculation supports that the coordination of a single DIPAM ligand is enough to bias the twisting direction in the Au13 superatom with dictating the tortional orientation of other four DPPE ligands. The emergence of the “sergeants-and-soldiers principle”, wherein a small number of chiral ligands determine the chiral orientation of other achiral ligands, is thus demonstrated in the synthesis of intrinsically chiral Au13 clusters.
Introduction
Chiral amplification phenomena have received significant attention in several research areas in chemical sciences, not only for the development of asymmetric catalysts and nonlinear optical materials,1–6 but also for seeking the origin of homochirality evolution in nature.7–10 Effective communications among chiral/achiral building units in helical polymers and supramolecular assemblies have been investigated in detail to clarify the specific stereochemical interaction for chiral amplification.11,12 The “Sergeants-and-soldiers principle” is one of the representative chiral amplification expressions.13,14 An intrinsically chiral system, such as a helical polymer, built by achiral monomers usually consists of a racemic mixture of left- and right-hand ones. The introduction of a small amount of chiral monomers with a similar structure to achiral ones effectively deracemizes the system by selectively stabilizing one-handed helical polymers. The achiral monomer units obey the orientation given by the chiral ones aligning in a one-hand direction together in a helical polymer. Chirality could thus be significantly amplified by a slight chiral input as a result of strong cooperative interactions among monomers and the entire polymer, producing chiral polymers with a large helical sense excess.Ligand-protected metal clusters have been extensively investigated in recent years owing to their unique electronic properties derived from an unambiguous atomically precise structure revealed by a single crystal X-ray diffraction (SCXRD) study.15–20 Chiral metal clusters have constituted an important class of chiral nanomaterials due to their fascinating chiroptical properties.21–25 Some clusters possess intrinsic chirality in their asymmetric structures and their deracemization has been achieved by employing chiral ligands,26 chiral HPLC separation27 and spontaneous resolution.28,29 Recent works have focused on the dynamic control of chirality in intrinsically chiral metal clusters through ligand engineering.30–32
The icosahedral Au13 structure is one of the commonly observed superatomic fundamental units in which 12 gold atoms occupy the vertices of the icosahedron and one remaining gold is situated in the center. While the stable icosahedral Au13 superatom alone is achiral and highly symmetric, the intrinsic chirality or chiral distortion can be induced mostly by the coordination of bidentate ligands such as bis(diphenylphosphino)ethane (DPPE, Fig. 1).33,34 The detailed investigation on the crystal structure of the [Au13(DPPE)5Cl2]3+ cluster demonstrated the distortion between the two gold pentagons (blue and red in Fig. 1 bottom) arranging five bridging DPPE ligands in a helical propeller pattern, indicating the existence of intrinsic chirality in the Au13 superatom.34,35 The deracemization of the Au13-based cluster has been demonstrated by the enantioselective synthesis with chiral ligands,36–40 spontaneous resolution41 and enantioseparation with a chiral column.42 It is of interest to note that the heteroatom doping41,43 in the center of Au13 with Ir3+ successfully leads to the spontaneous resolution of the [IrAu12(DPPE)5Cl2]+ cluster suppressing the rapid racemization upon crystallization. Given the inherent energy barrier for the racemization of the [Au13(DPPE)5Cl2]3+ cluster is not so small, a slight chiral input may enhance the activation energy biasing the formation of the Au13 cluster with a unidirectional twist. In this study, we investigate the effect of the coordination of a chiral ligand on the chirality induction in the Au13 superatom cluster. We prepared Au13 clusters by using a mixed ligand system of achiral DPPE and chiral (R,R)-1,2-bis[(2-methoxyphenyl)phenylphosphino]-ethane (R-DIPAMP)36 with a changing DPPE/R-DIPAMP (A/C) ratio (Fig. 1). The as-prepared samples displayed optical activity depending on the A/C ratio, while they were composed of a mixture of Au13 clusters with different ligand compositions ([Au13(R-DIPAMP)x(DPPE)5−xCl2]3+, x = 0–5). The as-prepared samples were therefore separated by means of reverse phase HPLC depending on the x-value. Circular dichroism (CD) measurement for the fractionalized eluents clearly suggested the non-additive effect of the chiral ligand on the chirality induction, demonstrating the “sergeants-and-soldiers principle” in the synthesis of metal clusters for the first time. The entire twist in the Au13 superatom is dictated by a single DIPAMP ligand in the [Au13(R-DIPAMP)1(DPPE)4Cl2]3+ cluster. The twist in Au13 propagates to the helical torsion and arrangement of other four DPPE ligands.
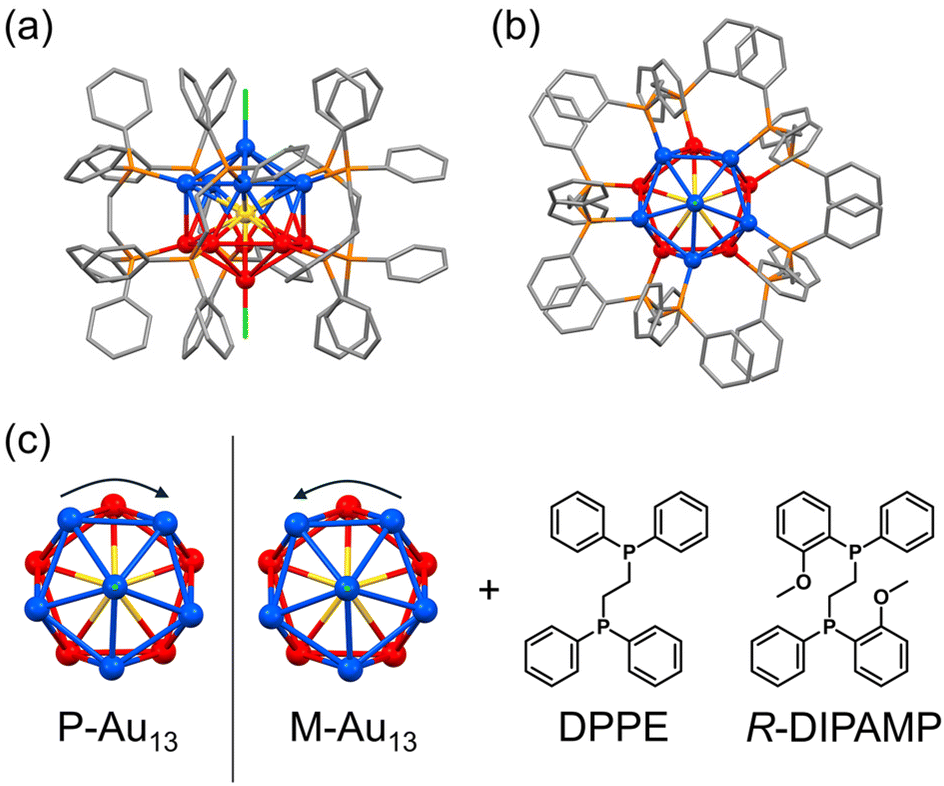 | ||
| Fig. 1 Structural models of (a and b) [Au13(DPPE)5Cl5]3+ cluster, and (c) the top views of right- and left-handed helically distorted Au13 cores and chemical structures of achiral (DPPE) and chiral (R-DIPAMP) ligands discussed in this study. | ||
Results and discussion
DFT calculations for Au13 clusters protected by DPPE and R-DIPAMP ligands ([Au13(R-DIPAMP)x(DPPE)5−xCl2]3+, called R0–R5: x = 0–5, respectively) were first carried out to predict the geometries with left- and right-hand twists and their stabilities. Fig. 2 shows the energy diagrams of the P- and M-[Au13(R-DIPAMP)x(DPPE)5−xCl2]3+ clusters for the right- and left-hand twisted structures (Fig. 1), respectively. The chiral arrangement of the ligands results in chiroptical properties originating from the helical right- and left-hand structures (global chirality) of Au13 clusters.34,36 It should be noted that the calculation based on the [Au13(PMe2Ph)10Cl2]3+ structure resulted in an achiral Au13 superatom core (θ2/θ1 = 1, Fig. 2).34 The chiral distortion is therefore introduced by the bridging nature of DPPE coordination with a certain P–P distance. When the Au13 superatom core is protected by the DPPE ligand, the energy difference between P- and M-Au13 clusters is zero because they are enantiomers to each other (Fig. 2). The enantioseparation of them with a chiral column was reported to be unsuccessful probably due to the rapid racemization.35 In contrast, the protection by R-DIPAMP ligands gives diastereomeric isomers with different energies between P- and M-clusters. The DFT calculations for R0–R5 reveal that the M-type conformation is destabilized by increasing the number of chiral ligands. While R2 and R3 should have two structural isomers in terms of ligand sequence for each, these isomers were estimated to have negligible energy difference (∼0.2 kJ mol−1, Fig. S1†). The ratio of averaged torsion angles (θ2/θ1) between the two Au-pentagons (Fig. 2a) slightly increases in the order of R1–R5 (Table 1), whereas the differences are negligible (<2%) compared to the differences in the energy. The optimized structures for P- and M-conformations of R5 (Fig. 2b) show the distinctive different arrangement of methoxy groups in R-DIPAMPs on the exterior shell. The methoxy groups are present in the latitudinal direction of the clusters in P-Au13, whereas, in M-Au13, they are located in the equatorial plane of the clusters, leading to a more crowded stereostructure. This conformational difference in R-DIPAMP can be considered as the primary origin of the pronounced energy differences between P-Au13 and M-Au13.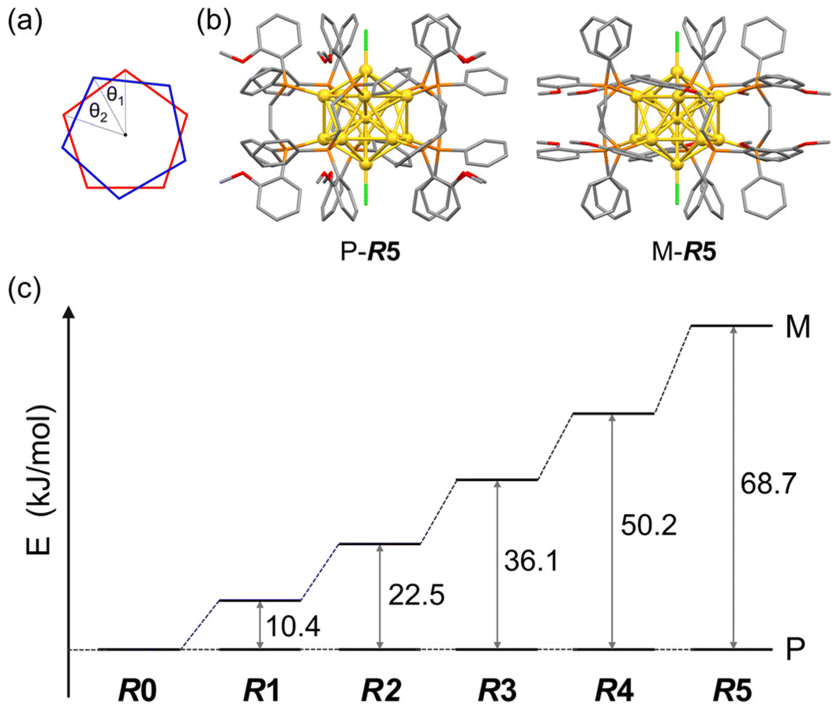 | ||
| Fig. 2 (a) The torsion angles between the two equatorial Au5 pentagons in Fig. 1c. (b) The optimized structures of P- and M-[Au13(R-DIPAMP)5Cl2]3+ clusters by DFT calculation. (c) The energy differences between the P- and M-Au13 clusters for R0–R5. | ||
| R1 | R2 | R3 | R4 | R5 | |
|---|---|---|---|---|---|
| K | 1.48 × 10−2 | 1.15 × 10−4 | 4.69 × 10−7 | 1.56 × 10−9 | 9.12 × 10−13 |
| θ 2/θ1 | 1.200 | 1.207 | 1.212 | 1.217 | 1.220 |
The DFT-optimized structures for P-Au13 and M-Au13 also disclosed the characteristic conformational differences in the diphosphine ligands. The diphosphine ligands coordinate on the surface by tilting to the right (P-R1) or left (M-R1) with respect to the central axis of the icosahedron (Cl–Cl axis), determining the global chirality of Au13 clusters. Fig. 3 shows the Newman projections along the ethylene C–C axis for DPPE and R-DIPAMP ligands in R1. The entire ethylene unit adopts an eclipsed conformation, rather than a staggered one. The dihedral angles between the two H–C bonds (φ, Fig. 3) are calculated to be ca. +4° (clockwise) for DPPE and R-DIPAMP in the stable P-R1, indicating that DPPE and R-DIPAMP coordinate to the Au13 core with similar geometries regardless of the presence of methoxy groups in P-R1. In contrast, the difference in the conformation of the ethylene unit was observed in M-R1. While the ethylene units of DPPE in M-R1 have a mirror-image form of those in P-R1 (anticlockwise, φ ∼−4°), that of R-DIPAMP keeps the clockwise direction (φ ∼+5.6°). A similar tendency of the coordination of R-DIMPAMP is also confirmed in the optimized structure of P- and M-R5. This result suggests that the R-DIPAMP ligand prefers clockwise conformation (in terms of φ) against the global chirality of M-Au13. Thus, a single R-DIPAMP ligand plays the role of “sergeant” transferring its chiral coordination structure to other four DPPE on the surface of Au13 in P-R1 while its role is invalid in M-R1.
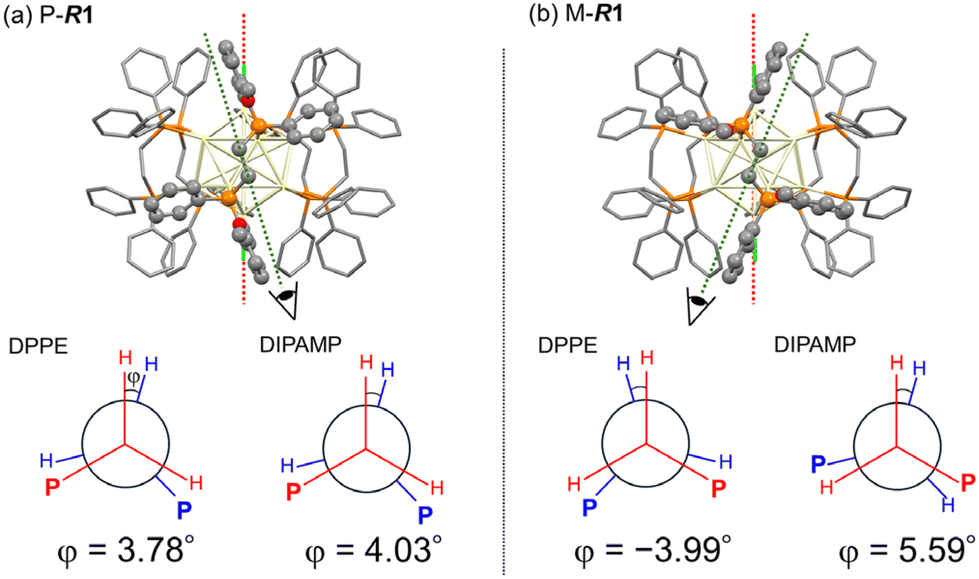 | ||
| Fig. 3 The optimized structures and Newman projections along the ethylene C–C axis for DPPE and R-DIPAMP ligands of (a) P-R1 and (b) M-R1. R-DIPAMP ligands are highlighted with a ball and stick model. The dihedral angle for the DPPE ligand was calculated as an averaged value of four DPPE ligands. | ||
Given that the P- and M-Au13 isomers are assumed to be in equilibrium, the equilibrium constants (K) for R1–R5 at 298 K can be estimated approximating the calculated energy differences as ΔG (Table 1). The population ratio of unstable M-R1 at 298 K was calculated to be 1.46% and that for the other R2–R5 was approximately zero. While the activation barrier of twist inversion could not be calculated easily, the estimated energy difference suggests that the formation of the P-Au13 cluster can be significantly biased by the introduction of just one R-DIPAMP ligand.
The synthesis of a series of R0–R5 clusters was performed through a previously reported method by changing the ratio of the gold complexes, Au2(DPPE)Cl2/Au2(R-DIPAMP)Cl2, Achiral/Chiral (A/C) ratios = 5/0, 4/1, 3/2, 2/3, 1/4 and 0/5, in the reduction step with NaBH4.33,36 The reduction of the mixed solution of gold complexes, followed by acid (HCl) etching yielded a mixture of Au13 clusters at room temperature. The as-prepared samples were treated by reverse phase column chromatography (Wakosil 100C18, Wako-Fuji Film) using a methanol solution containing 0.5 vol% trifluoroacetic acid (TFA) and 0.5 vol% diethylamine (DEA) as an eluent to remove larger agglomerates. The treated samples afforded almost identical absorption spectra indicating the successful formation of Au13 clusters regardless of the A/C ratio (Fig. S2†). Meanwhile, they exhibited optical activity dependent on the A/C ratio for the preparation condition. The A/C = 5/0 and 0/5 samples exclusively contain R0 and R5 with no and the largest optical activity, respectively, while other ratios should give the mixtures of R0–R5. To compare the optical activity, CD spectra were translated to g-spectra (g = Δε/ε) with the equation of g = CD[mdeg]/(32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 980·Abs) (Fig. 4a). The plot of anisotropy value (g-value) at 550 nm clearly demonstrated the dependence of optical activity on the A/C ratio for the preparative condition. The non-linear relationship between the g-value and C/(A + C) ratio suggested the emergence of chiral amplification or the non-additive effect of the chiral ligand on the entire optical activity of Au13 clusters. However, the unseparated samples contain R0–R5 clusters with a certain distribution. The ESI-MS spectrum for the A/C = 1/4 sample indeed demonstrated that the solution contains all possible compositions (Fig. 5). Furthermore, the result of DFT calculation motivated us to isolate R0–R5 species to investigate the effect of single chiral ligand coordination on the optical activity of Au13 clusters.
980·Abs) (Fig. 4a). The plot of anisotropy value (g-value) at 550 nm clearly demonstrated the dependence of optical activity on the A/C ratio for the preparative condition. The non-linear relationship between the g-value and C/(A + C) ratio suggested the emergence of chiral amplification or the non-additive effect of the chiral ligand on the entire optical activity of Au13 clusters. However, the unseparated samples contain R0–R5 clusters with a certain distribution. The ESI-MS spectrum for the A/C = 1/4 sample indeed demonstrated that the solution contains all possible compositions (Fig. 5). Furthermore, the result of DFT calculation motivated us to isolate R0–R5 species to investigate the effect of single chiral ligand coordination on the optical activity of Au13 clusters.
 | ||
| Fig. 4 (a) g-Spectra of the sample prepared with A/C = 5/0–0/5 ratios and (b) the plot of g-values at 550 nm as a function of C/(A + C) ratio in the synthetic condition. | ||
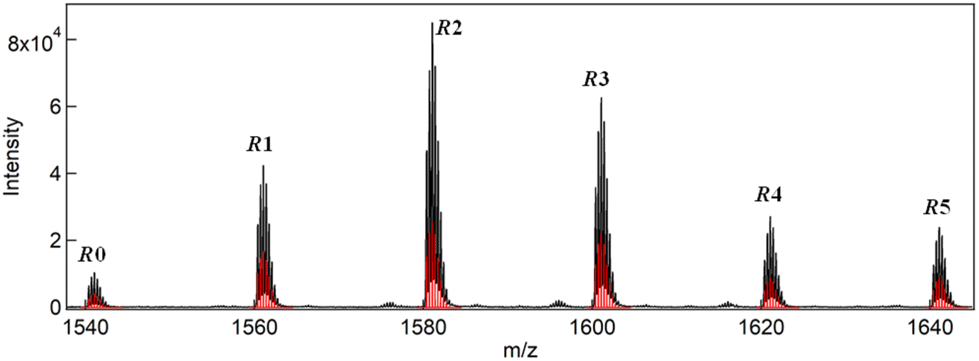 | ||
| Fig. 5 ESI-MS spectrum of the sample prepared with A/C = 1/4 ratio together with simulated isotope peak patterns (red bars) for R0–R5. | ||
Negishi and coworkers demonstrated the precise HPLC separation of nonionic PdAu24(SR)18−n(SR′)n clusters by gradient elution using a reverse phase column.44,45 On the other hand, a tricationic [Au13(bisNHC)5Cl2]3+ (NHC: N-heterocyclic carbene) cluster was subjected to a chiral column using methanol as an eluent with TFA/DEA additives.42 In the present study, a preparative reverse phase column (InertSustain C30, GL Science) was employed and a gradient elution condition was considered. Finally, the successful separation was achieved by using the mixture of methanol containing TFA (0.2 vol%) and DEA (0.2 vol%) and water (see the ESI† for details). Fig. 6 shows the result of the HPLC separation. The A/C = 5/0 sample gave a single peak at 52–53 min collected as F0 (fraction 0, Fig. 6), which was confirmed as the R0 cluster with ESI-MS (Fig. S3†). Meanwhile, the A/C = 0/5 sample afforded a peak at the shorter retention time assigned to R5, which was collected as F5. The retention times in the HPLC charts for A/C = 4/1–1/4 samples were adjusted by using R0 peaks as a standard. The A/C = 1/4 sample, ESI-MS data of which demonstrated the presence of all species R0–R5 (Fig. 5), displayed 4 more peaks in-between the peaks assigned to R0 and R5, indicating the separation of all the species. To confirm the separation of R1–R4, the eluate was fractionalized into F1–F4 depending on the peak positions (Fig. 6). The ESI-MS measurement demonstrated that F1–F4 exclusively contain R1–R4, respectively (Fig. S3†). While two isomers with different ligand sequences for R2 and R3 could not be discriminated, successful separation was achieved in terms of ligand compositions.
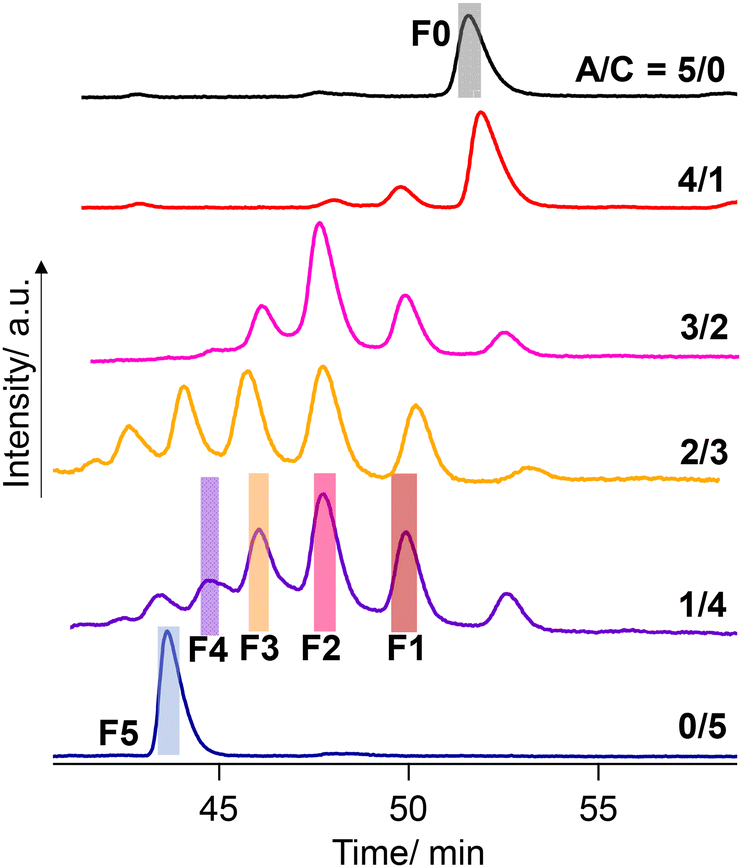 | ||
| Fig. 6 HPLC chromatograms for the mixture solutions of [Au13(R-DIPAMP)x(DPPE)5−xCl2]3+ clusters synthesized with different A/C ratios. | ||
The peak intensity ratio in the ESI-MS spectrum for the A/C = 1/4 sample (Fig. 5) well accorded with that in the elution profile (Fig. 6) corresponding to the abundance ratio of R0–R5. However, this distribution was far different from the theoretical one assuming the random incorporation of the ligand based on the given A/C ratio (Fig. S4†). The A/C = 1/4 ratio should give R4 and R5 as the major and second major products with 45 and 30% probabilities and there is a negligible chance for the R0 formation. The experimental result suggests the less reactivity of the DIPAMP ligand for the coordination on the Au13 cluster than DPPE. The A/C = 4/1 condition highlights the less reactivity of DIPAMP by the formation of R0 as the major product with faint formation of R1 and R2 (Fig. 6). The two-step reaction mechanism for the efficient synthesis of Au13 clusters has been discussed by Shichibu and Konishi.33 The first reduction process of Au2(diphosphine)Cl2 gives a mixture of polydispersed gold clusters with various compositions. These polydispersed clusters converged to [Au13(diphosphine)5Cl2]3+ upon the addition of HCl aq. Most of the kinetically formed metastable clusters were subjected to etching being consumed as precursors forming the most stable one. While both the enantiomers of [Au13(DPPE)5Cl2]3+ have identical stability, the M-Au13 clusters containing R-DIPAMP ligands should have less stability than P-ones (Fig. 2), being subject to etching. Furthermore, achiral DPPE is considered to preferentially participate in the coordination on the preformed M-Au13 superatom over R-DIPAMP with adopting the torsional conformation appropriate for the left-hand twisted cluster (Fig. 3). The formation of R0 thus preferentially proceeds in the condition of a small chiral ligand content.
When the content of chiral ligand changed from A/C = 4/1 to 3/2, the major product drastically changed from R0 to R2. While the incorporation of R-DIPAMP in the Au13 cluster formation seems less probable than DPPE, the yield of R2 is higher than that of R1. The coordination of the first R-DIPAMP ligand might facilitate the introduction of the second R-DIPAMP in a similar manner to the emergence of the allosteric effect in the chiral guest incorporation by prochiral double decker porphyrins.46,47 More specifically, if an Au13 superatom with P-type twist induced by the single R-DIPAMP coordination had a vacant coordination site, R-DIPAMP with a geometry preorganized (Fig. 3) for the coordination on the P-type cluster could be more feasible than DPPE to fill that site. However, the inherent less reactivity with sterically less flexible nature compared to DPPE may result in the less formation probabilities of R3, R4 and R5 for the conditions of A/C = 3/2, 2/3 and 1/4. Another possibility includes the selective consumption of the chiral DIPAMP ligand for the formation of cluster species other than Au13, resulting in the decrease of practical chiral ligand ratio to form Au13 clusters.
The absorption and CD measurements were carried out for the fractions of F1–F5. As described above, the ESI-MS measurement for F1–F5 demonstrated the successful isolation of Au13 clusters into a single composition of R1–R5, respectively. At the very least, no trace of R0 was contained in those fractions because of its well-separated peak position in the HPLC charts (Fig. 6). All F1–F5 fractions containing R1–R5, respectively, exhibited almost identical absorption and CD profiles (Fig. S5 and S6†). Furthermore, the g-spectra of R1–R4 showed similar profiles to that of R5 with very similar g-values at 550 nm (Fig. 7). The contamination of R0 in the solutions should result in a lower optical activity of the unseparated samples than that of R5 (Fig. 4). The result thus confirmed that the single coordination of the R-DIPAMP ligand is enough to induce a unidirectional helical twist in the Au13 superatom as predicted by the DFT study. Meanwhile, other four achiral DPPE ligands adopt the helical torsional conformation following the one-hand twist in the Au13 superatom (Fig. 3).
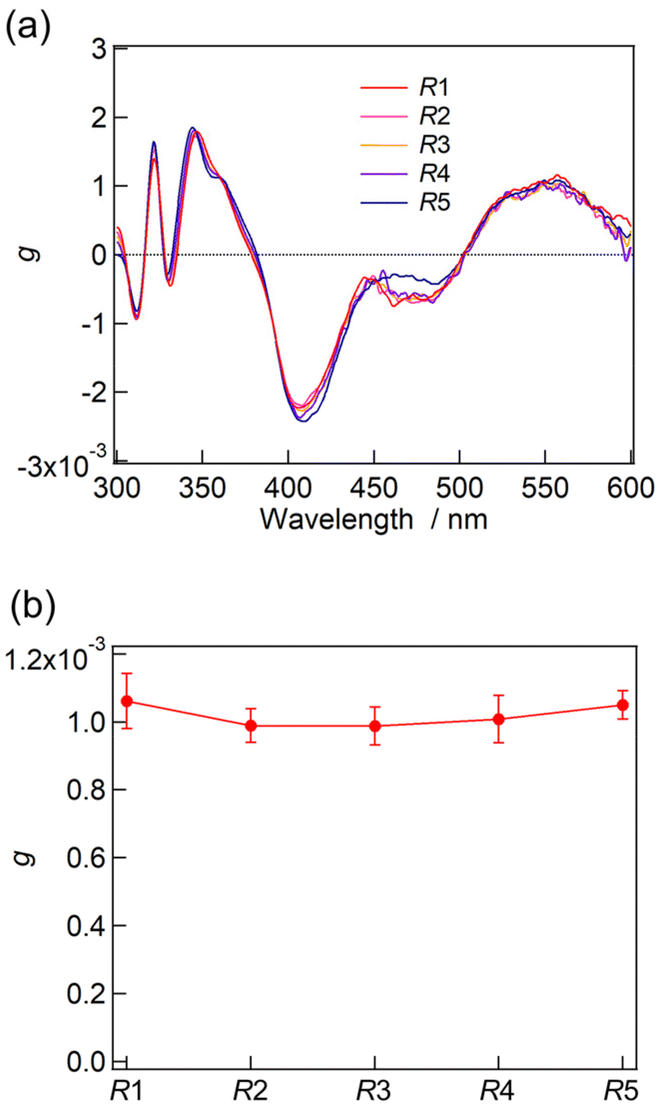 | ||
| Fig. 7 (a) g-Spectra of R1–R5 separated by HPLC. (b) The plot of g-values at 550 nm for R1–R5. | ||
Bürgi and coworkers have extensively utilized the HPLC technique to monitor the ligand exchange reaction of the intrinsically chiral Au38(SR)24 clusters and to isolate the ligand-exchanged products.30,48–51 The intrinsically chiral Au38 enantiomer demonstrated the diastereoselective ligand exchange with chiral ligands, suppression of racemization by the introduction of a chiral ligand and modulation of optical activity by the ligand exchange. In the present study, the HPLC separation of chiral–achiral mixed-ligand-protected clusters disclosed the clear composition–chiroptical property relationship with a one-to-one correspondence.
The CD spectra of R1–R5 were well reproduced by DFT calculations (Fig. S7†). While the compositions in the ligand shell are different, R1–R5 gave very similar CD spectra which also supports that the first Cotton effect originates from the internal chirality in the helical superatom core (Au13). The average torsion angles calculated from the optimized structures of R1–R5 slightly increased by increasing the number of R-DIPAMP as discussed above (Table 1). Although the nearly similar torsion angles of R1–R5 may have minimal effect on the intensity of the CD signal, these experimental results support the argument that the chiroptical properties arise from the differences in the angle between the two Au-pentagons.34
Conclusion
The effect of a chiral ligand on the chirality induction in [Au13(diphosphine)5Cl2]3+ clusters has been investigated. The DFT calculation indicated that the coordination of the single chiral ligand DIPAMP could greatly stabilize the one-handed twist structure in an icosahedral Au13 superatom core with 98.5% occupancy over the opposite-twist regardless of the presence of other four achiral DPPE ligands. While the synthesis in a mixed ligand system of DPPE and R-DIPAMP led to the formation of a mixture of [Au13(R-DIPAMP)x(DPPE)5−xCl2]3+ clusters, they have been successfully separated into individual compositions in terms of x-values (x = 0–5) by reverse-phase HPLC. The fractionalized HPLC eluents including R1–R5, free from [Au13(DPPE)5Cl2]3+ (R0), afforded identical optical activity clearly supporting the result of DFT calculation. Thus the non-additive effect of the chiral ligand on the chirality induction in the Au13 cluster has been demonstrated in theoretical and experimental manners. In other words, the chirality of the DIPAMP ligand could be successfully communicated into a chiral conformation of DPPE.52 The HPLC separation technique53,54 should provide the opportunity of clear role division between different ligands for mixed-ligand clusters, further expanding the usage of ligands as toolboxes in functional clusters.55Author contributions
K. M.: investigation, funding acquisition, visualization, writing – original draft, and writing – review & editing. S. K.: investigation. T. Y.: investigation. T. N.: conceptualization, investigation, funding acquisition, resources, supervision, and writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by JST CREST (Grant Number: JPMJCR20B2), JSPS KAKENHI grant numbers JP22H05134 (TN) for Transformative Research (A) “Revolution of Chiral Materials Science using Helical Light”, JP23H01938 (TN) and JP24K01458 (KM) for Scientific Research (B), and by Tokyo Ohka Foundation for The Promotion of Science and Technology for K. M. A part of this work was conducted in the Institute for Molecular Science, supported by “Advanced Research Infrastructure for Materials and Nanotechnology in Japan (ARIM)” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT). Proposal Number JPMXP1224MS0001.References
- R. Noyori and M. Kitamura, Angew. Chem., Int. Ed. Engl., 1991, 30, 49–69 CrossRef.
- H. B. Kagan, Synlett, 2001, 888–899 CrossRef CAS.
- T. Satyanarayana, S. Abraham and H. B. Kagan, Angew. Chem., Int. Ed., 2009, 48, 456–494 CrossRef CAS PubMed.
- S. Sioncke, T. Verbiest and A. Persoons, Mater. Sci. Eng., R, 2003, 42, 115–155 CrossRef.
- X. Yan, Q. Wang, X. Chen and Y.-B. Jiang, Adv. Mater., 2020, 32, 1905667 CrossRef PubMed.
- M. Rok, A. Miniewicz, M. Zdończyk, B. Zarychta, J. W. Mikurenda, S. Bartkiewicz, M. Wiśniewska-Bełej, J. Cybińska and A. Piecha-Bisiorek, J. Phys. Chem. Lett., 2024, 15, 5276–5287 CrossRef CAS PubMed.
- J. S. Siegel, Chirality, 1998, 10, 24–27 CrossRef CAS.
- D. G. Blackmond and M. Klussmann, Chem. Commun., 2007, 3990–3996 RSC.
- K. K. Konstantinov and A. F. Konstantinova, Origins Life Evol. Biospheres, 2018, 48, 93–122 CrossRef CAS PubMed.
- N. Suzuki and Y. Itabashi, Symmetry, 2019, 11, 966 CrossRef CAS.
- A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2007, 46, 8948–8968 CrossRef CAS PubMed.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed.
- M. M. Green and P. Ridy, J. Am. Chem. Soc., 1989, 111, 6452–6454 CrossRef.
- A. R. A. Palmans, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Angew. Chem., Int. Ed. Engl., 1997, 36, 2648–2651 CrossRef CAS.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed.
- S. Takano and T. Tsukuda, J. Am. Chem. Soc., 2021, 143, 1683–1698 CrossRef CAS PubMed.
- Y. Saito, C. Murata, M. Sugiuchi, Y. Shichibu and K. Konishi, Coord. Chem. Rev., 2022, 470, 214713 CrossRef CAS.
- W. Du, S. Jin, L. Xiong, M. Chen, J. Zhang, X. Zou, Y. Pei, S. Wang and M. Zhu, J. Am. Chem. Soc., 2017, 139, 1618–1624 CrossRef CAS PubMed.
- S. Chen, S. Wang, J. Zhong, Y. Song, J. Zhang, H. Sheng, Y. Pei and M. Zhu, Angew. Chem., Int. Ed., 2015, 54, 3145–3149 CrossRef CAS PubMed.
- Y. Li, T. Higaki, X. Du and R. Jin, Adv. Mater., 2020, 32, e1905488 CrossRef PubMed.
- Y. Zhu, J. Guo, X. Qiu, S. Zhao and Z. Tang, Acc. Mater. Res., 2020, 2, 21–35 CrossRef.
- Y. Shichibu and K. Konishi, ChemNanoMat, 2022, 8, e202200194 CrossRef CAS.
- J.-H. Huang, X.-Y. Dong, Y.-J. Wang and S.-Q. Zang, Coord. Chem. Rev., 2022, 470, 214729 CrossRef CAS.
- K. K. Ramankutty, Nanoscale, 2024, 16, 11914–11927 RSC.
- J. Kumar, T. Kawai and T. Nakashima, Chem. Commun., 2017, 53, 1269–1272 RSC.
- I. Dolamic, S. Knoppe, A. Dass and T. Bürgi, Nat. Commun., 2012, 3, 798 CrossRef PubMed.
- J. H. Huang, Z. Y. Wang, S.-Q. Zang and T. C. W. Mak, ACS Cent. Sci., 2020, 6, 1971–1976 CrossRef CAS PubMed.
- S. Chen, W. Du, C. Qin, D. Liu, L. Tang, Y. Liu, S. Wang and M. Zhu, Angew. Chem., Int. Ed., 2020, 59, 7542–7547 CrossRef CAS PubMed.
- Y. Wang, B. Nieto-Ortega and T. Bürgi, Nat. Commun., 2020, 11, 4562 CrossRef CAS PubMed.
- H. Yoshida, M. Kuzuhara, R. Tanibe, T. Kawai and T. Nakashima, J. Phys. Chem. C, 2021, 125, 27009–27015 CrossRef CAS.
- T. Nakashima, R. Tanibe, H. Yoshida, M. Ehara, M. Kuzuhara and T. Nakashima, Angew. Chem., Int. Ed., 2022, 61, e202208273 CrossRef CAS PubMed.
- Y. Shichibu and K. Konishi, Small, 2010, 6, 1216–1220 CrossRef CAS PubMed.
- Y. Shichibu, Y. Ogawa, M. Sugiuchi and K. Konishi, Nanoscale Adv., 2021, 3, 1005–1011 RSC.
- J. Zhang, Y. Zhou, K. Zheng, H. Abroshan, D. R. Kauffman, J. Sun and G. Li, Nano Res., 2018, 11, 5787–5798 CrossRef CAS.
- Y. Yang, Q. Zhang, Z.-J. Guan, Z.-A. Nan, J.-Q. Wang, T. Jia and W.-W. Zhan, Inorg. Chem., 2019, 58, 3670–3675 CrossRef CAS PubMed.
- P. Luo, X.-J. Zhai, S. Bai, Y.-B. Si, X.-Y. Dong, Y.-F. Han and S.-Q. Zang, Angew. Chem., Int. Ed., 2023, 62, e202219017 CrossRef CAS PubMed.
- H. Hirai, T. Nakashima, S. Takano, Y. Shichibu, K. Konishi, T. Kawai and T. Tsukuda, J. Mater. Chem., 2023, 11, 3095–3100 CAS.
- Y. Sato, M. Mitani and H. Yao, J. Phys. Chem. C, 2020, 124, 25547–25556 CrossRef CAS.
- E. L. Albright, S. Malola, S. I. Jacob, H. Yi, S. Takano, K. Mimura, T. Tsukuda, H. Häkkinen, M. Nambo and C. M. Crudden, Chem. Mater., 2024, 36, 1279–1289 CrossRef CAS.
- H. Hirai, S. Takano, T. Nakamura and T. Tsukuda, Inorg. Chem., 2020, 59, 17889–17895 CrossRef CAS PubMed.
- H. Yi, K. M. Osten, T. I. Levchenko, A. J. Veinot, Y. Aramaki, T. Ooi, M. Nambo and C. M. Crudden, Chem. Sci., 2021, 12, 10436–10440 RSC.
- H. Hirai, S. Takano, T. Nakashima, T. Iwasa, T. Taketsugu and T. Tsukuda, Angew. Chem., Int. Ed., 2022, 61, e202207290 CrossRef CAS PubMed.
- Y. Niihori, M. Matsuzaki, T. Pradeep and Y. Negishi, J. Am. Chem. Soc., 2013, 135, 4946–4949 CrossRef CAS PubMed.
- Y. Niihori, M. Matsuzaki, C. Uchida and Y. Negishi, Nanoscale, 2014, 6, 7889–7896 RSC.
- S. Shinkai, M. Ikeda, A. Sugasaki and M. Takeuchi, Acc. Chem. Res., 2001, 34, 494–503 CrossRef CAS PubMed.
- T. Ikeda, K. Sada, S. Shinkai and M. Takechi, Supramol. Chem., 2011, 23, 59–64 CrossRef CAS.
- S. Knoppe, R. Azoulay, A. Dass and T. Bürgi, J. Am. Chem. Soc., 2012, 134, 20302–20305 CrossRef CAS PubMed.
- S. Knoppe, S. Michalet and T. Bürgi, J. Phys. Chem. C, 2013, 117, 15354–15361 CrossRef CAS.
- A. Baghdasaryan, K. Martin, L. M. L. Daku, M. M. Talamo, N. Avarvari and T. Bürgi, Nanoscale, 2020, 12, 18160–18170 RSC.
- Y. Wang, Ee. Makkonen, X. Chen and T. Bürgi, Chem. Sci., 2021, 12, 9413–9419 RSC.
- I. Dolamic, B. Varnholt and T. Bürgi, Nat. Commun., 2015, 6, 7117 CrossRef CAS PubMed.
- Y. Niihori, C. Uchida, W. Kurashige and Y. Negishi, Phys. Chem. Chem. Phys., 2016, 18, 4251–4265 RSC.
- Y. Niihori, D. Shima, K. Yoshida, K. Hamada, L. V. Nair, S. Hossain, W. Kurashige and Y. Negishi, Nanoscale, 2018, 10, 1641–1649 RSC.
- K. Konishi, M. Iwasaki, M. Sugiuchi and Y. Shichibu, J. Phys. Chem. Lett., 2016, 7, 4267–4274 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and supplemental simulation and spectral data. See DOI: https://doi.org/10.1039/d4nr03810j |
| This journal is © The Royal Society of Chemistry 2024 |