
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multigram-scale chemoenzymatic synthesis of diverse aminopolycarboxylic acids as potential metallo-β-lactamase inhibitors†
Mohammad Faizan
Bhat
,
Alejandro
Prats Luján
 ,
Mohammad
Saifuddin
,
Peter
Fodran
and
Gerrit J.
Poelarends
*
,
Mohammad
Saifuddin
,
Peter
Fodran
and
Gerrit J.
Poelarends
*
Department of Chemical and Pharmaceutical Biology, Groningen Research Institute of Pharmacy, University of Groningen, Antonius Deusinglaan 1, 9713 AV Groningen, The Netherlands. E-mail: g.j.poelarends@rug.nl; Tel: +31-50-3633354 Web: https://www.rug.nl/staff/g.j.poelarends/ Web: https://twitter.com/gpoelarends
First published on 12th December 2023
Abstract
Toxin A, a precursor to naturally occurring aspergillomarasmine A, aspergillomarasmine B, lycomarasmine and related aminopolycarboxylic acids, was synthesized as the desired (2S,2′S)-diastereomer on a multigram-scale (>99% conversion, 82% isolated yield, dr > 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) from commercially available starting materials using the enzyme ethylenediamine-N,N′-disuccinic acid lyase. A single-step protection route of this chiral synthon was developed to aid N-sulfonylation/-alkylation and reductive amination at the terminal primary amine for easy derivatization, followed by global deprotection to give the corresponding toxin A derivatives, including lycomarasmine, in moderate to good yields (23–66%) and with high stereopurity (dr > 95:5). Furthermore, a chemoenzymatic route was developed to introduce a click handle on toxin A (yield 72%, dr > 95:5) and its cyclized congener for further analogue design. Finally, a chemoenzymatic route towards the synthesis of photocaged aspergillomarasmine B (yield 8%, dr > 95:5) was established, prompting further steps into smart prodrug design and precision delivery. These new synthetic methodologies have the prospective of facilitating research into the finding of more selective and potent metallo-β-lactamase (MBL) inhibitors, which are urgently needed to combat MBL-based infections.
:5) from commercially available starting materials using the enzyme ethylenediamine-N,N′-disuccinic acid lyase. A single-step protection route of this chiral synthon was developed to aid N-sulfonylation/-alkylation and reductive amination at the terminal primary amine for easy derivatization, followed by global deprotection to give the corresponding toxin A derivatives, including lycomarasmine, in moderate to good yields (23–66%) and with high stereopurity (dr > 95:5). Furthermore, a chemoenzymatic route was developed to introduce a click handle on toxin A (yield 72%, dr > 95:5) and its cyclized congener for further analogue design. Finally, a chemoenzymatic route towards the synthesis of photocaged aspergillomarasmine B (yield 8%, dr > 95:5) was established, prompting further steps into smart prodrug design and precision delivery. These new synthetic methodologies have the prospective of facilitating research into the finding of more selective and potent metallo-β-lactamase (MBL) inhibitors, which are urgently needed to combat MBL-based infections.
Antibacterial resistance remains one of the biggest public health challenges, especially because of the rate and magnitude at which it is acquired. While β-lactam containing drugs continue to be the most effective and widely used class of antibiotics,1 exhaustive and improper use has led to the rapid development of resistance among microorganisms.2 The mechanism of resistance mainly involves two enzyme classes, serine-β-lactamases (SBLs) and metallo-β-lactamases (MBLs), that cleave the β-lactam ring of the antibiotics and render them ineffective.3 While combination therapies with co-drugs (clavulanic acid, sulbactam, and tazobactam) are available to fight SBL producing bacteria, there is no clinically approved treatment for the highly resistant MBL producing bacteria.4 In particular, since its discovery in 2008, bacteria expressing New Delhi metallo-β-lactamase-1 (NDM-1) have emerged as one of the most clinically relevant challenges.5
Promisingly, the aminopolycarboxylic acid aspergillomarasmine A (AMA, Fig. 1) was discovered from a screen of 500 fungal natural products and identified as a selective and potent inhibitor of NDM-1 with an IC50 in the low-micromolar range.6 Interest in AMA as a scaffold for further development of drug candidates led to several total synthesis reports for AMA and analogues, based on a late-stage oxidation strategy (14 steps, 4% yield),7 an approach employing o-nosyl aziridine as a key intermediate (9 steps, 1% yield),8 a sulfamidate approach (6 steps, 19% yield),9 a Mitsunobu approach (6 steps, 28% yield),10 and an attractive biocatalytic strategy using AMA synthase (Fig. 1).11 Interestingly, biocatalytic approaches gain momentum in the synthesis of natural products.12–15 We have previously developed a chemoenzymatic route to AMA (Fig. 1), aspergillomarasmine B (AMB), toxin A (the natural precursor to AMA and AMB) and analogous compounds.16 This synthetic route benefits from a highly regio- and enantioselective carbon–nitrogen bond-forming step catalyzed by ethylenediamine-N,N′-disuccinic acid lyase (EDDS lyase). However, several limitations such as difficult purification and over-alkylation, resulting in relatively low product yields, inspired us to explore alternative routes towards the synthesis of diverse toxin A derivatives.
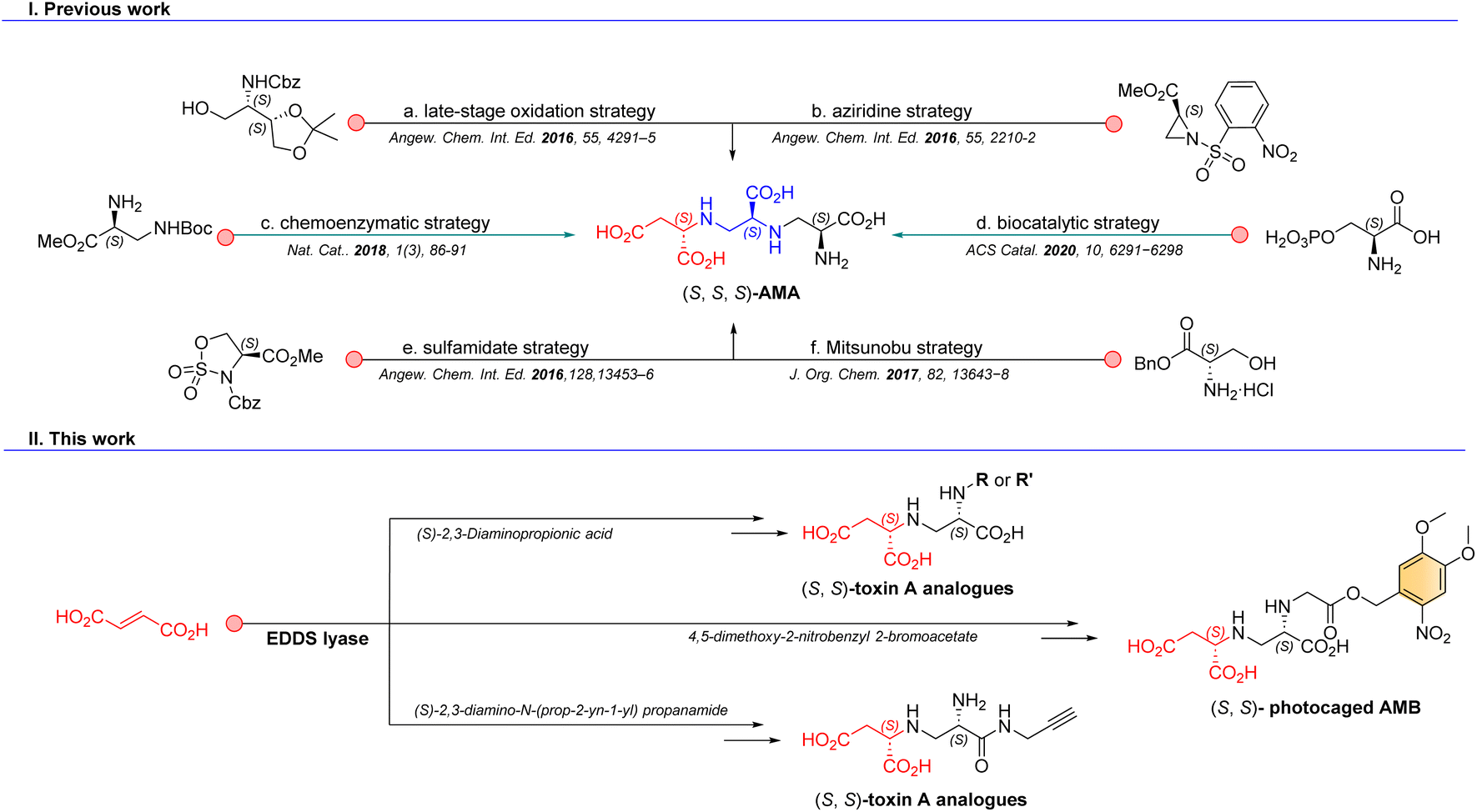 | ||
| Fig. 1 Synthesis of complex aminopolycarboxylic acids. (I) Previous chemical, chemoenzymatic and biocatalytic synthesis strategies towards the aminopolycarboxylic acid AMA and (II) our chemoenzymatic synthesis strategies towards related aminopolycarboxylic acids (i.e., toxin A derivatives). | ||
Along these lines, we envisaged that protection of the carboxylic groups of toxin A (compound 2 in Scheme 1) would lend the primary amine free for the synthesis of broad range of aminopolycarboxylic acids. Furthermore, to facilitate further derivatization of the toxin A pharmacophore, a new retrosynthetically designed chemoenzymatic route with inclusion of a propargylamine click handle was also anticipated. In contrast to our previous work,16,17 where an excess of chiral amine 1 (Scheme 1) was used to push the equilibrium of the enzymatic reaction, we decided to use an excess of fumarate in our new method to achieve multigram-scale synthesis of toxin A. We reasoned that the use of an excess of the chiral amine is not economically feasible, and furthermore it demands the use of a time and resource consuming purification procedure, which is based on a two-steps ion-exchange chromatography protocol, that eventually results in a significant loss of product yield.
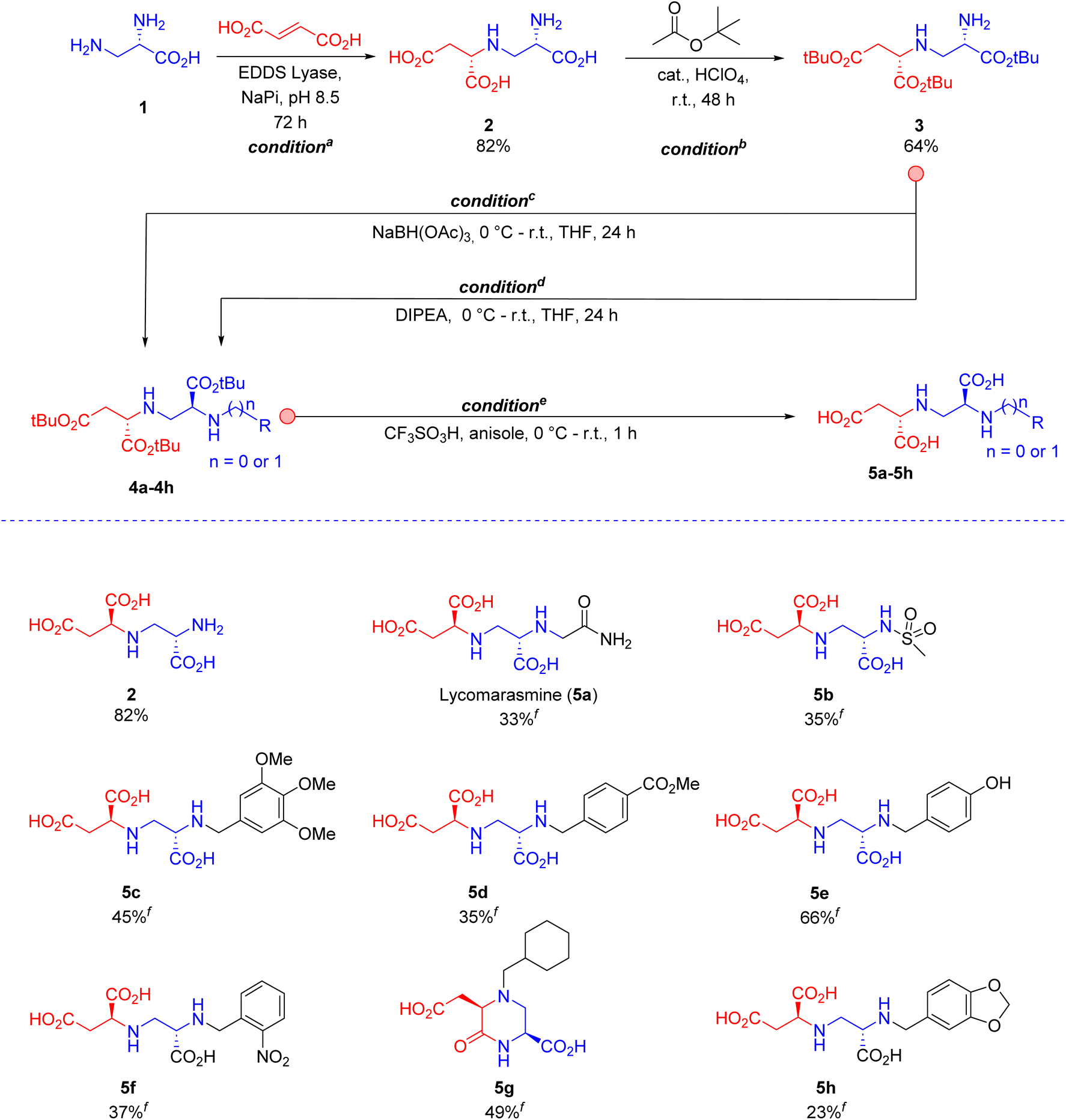 | ||
| Scheme 1 (Chemo)enzymatic synthesis of toxin A (2) and derivatives 5a–5h. Conditions and reagents: aDiamine substrate (1, 5.0 g, 35.6 mmol), fumaric acid (20.6 g, 177.9 mmol) and purified EDDS lyase (0.005 mol% based on diamine substrate 1) in buffer (200 mL, 50 mM Na2HPO4, pH 8.5), at room temperature for 72 h. b200 mL tBuOAc, 2 mL 70% HClO4, at room temperature for 48 h. cCorresponding aromatic aldehyde precursors for products 5c–5h, NaBH(OAc)3 (2 equiv.), in THF, 0 °C to room temperature for 24 h. dCorresponding bromo or sulfonyl derivatives for products 5a and 5b, respectively; DIPEA (2 equiv.), in THF, 0 °C to room temperature for 24 h. eTrifluoromethanesulfonic acid (5 equiv.), anisole (5 equiv.) in DCM, 0 °C to room temperature for 1 h. A slightly different protocol for workup was used for the synthesis of 5g (ESI†). fIsolated yield of final products 5a–5h from intermediates 4a–4h after full deprotection and ion-exchange chromatography. The absolute configuration of toxin A (2) was assigned by 1H NMR spectroscopy using an authentic standard with known (2S,2′S) configuration.16 With both stereogenic centres being derived from (2S,2′S)-2, products 5a–5h have the correct absolute configuration. The dr of 5a–h was determined by 1H NMR to be >95:5 (ESI Fig. S1–S26†), indicating that no noteworthy epimerization occurred during the derivatization of 2. | ||
Accordingly, we started with 1 g of amine 1, a 4-fold molar excess of fumarate, and 0.1 mol% of EDDS lyase in 200 mL of aqueous buffer (pH = 8.5, NaPi). Under these reaction conditions, 92% conversion of amine 1 into toxin A (2) was observed after 48 h (analyzed by 1H NMR). Although these results were noteworthy, a two-step purification procedure, consisting of an anion-exchange step to remove the remaining amine and a cation-exchange step to remove the remaining fumarate, was still demanded. After optimization of the reaction conditions (conditiona in Scheme 1), we found that a higher concentration of the two substrates in 200 mL buffer (pH 8.5, NaPi), using a 5-fold molar excess of fumarate over amine 1 and a prolonged reaction time (72 h), allowed >99% conversion of the amine into toxin A even with a substantial reduction in biocatalyst loading (0.005 mol%). Gratifyingly, a single cation-exchange purification step afforded 7 g of toxin A (isolated yield of 82%) as the desired (2S,2′S)-diastereomer (diastereomeric ratio (dr) > 95:5 based on 1H NMR), with the (2S)-stereogenic centre being set by EDDS lyase and the (2′S)-stereogenic centre derived from the starting substrate 1 (Scheme 1).16 This method (see ESI† for details) was repeated several times to obtain a total of ∼100 g of toxin A (2, Scheme 1) for the follow-up chemistry.
Next, several methods were attempted for the protection of the carboxylic acid groups of compound 2, such as esterification (benzyl alcohol, methanol, ethanol) under acidic conditions (HCl, H2SO4, SOCl2, and p-TsOH), but unfortunately these conditions either gave a low yield or the cyclization into a previously reported inactive toxin A congener.8 In addition, a three-step protection–deprotection sequence (cbz protection of amine, tBu protection of carboxylic acids, and cbz deprotection) also proved difficult. Finally, a catalytic amount of perchloric acid (conditionb in Scheme 1; see ESI† for details) and the use of tert-butyl acetate both as a solvent and tBu protecting agent provided an adequate solution, leading to fully protected compound 3 in 64% yield, without the need to purify this intermediate for the next step.
To demonstrate the synthetic usefulness of intermediate 3, reductive amination (conditionc, Scheme 1) and N-alkylation/N-sulfonylation (conditiond) were employed for the efficient synthesis of intermediates 4a–4h (see ESI† for details) in moderate to good yield (24–85%). Finally, the previously published method by Koteva et al.8 was used to fully deprotect the isolated intermediates (conditione, ESI†) and give the corresponding substituted aminopolycarboxylic acid products 5a–5h (Scheme 1) in modest to good yield (23–66%) and with high dr values of >95:5.8 With both stereogenic centres being derived from (2S,2′S)-2, products 5a–5h have the correct absolute configuration. Notably, the natural product lycomarasmine 5a, that was previously inaccessible with our one-pot synthesis strategy,16 could now be synthesized with 13% overall yield.
Next, we reasoned that inclusion of a click-handle into the toxin A scaffold would promote further derivatization. Towards this end, we performed amidation of the starting (S)-2,3-diaminopropionic acid (1) with propargylamine, after a protection–deprotection sequence (Scheme 2) to give the click-handle installed precursor 8. To our delight, the synthesized intermediate 8 was readily accepted by EDDS lyase as a non-native substrate for the hydroamination reaction (conversion >99%, 72 h) with 0.1 mol% biocatalyst loading to yield the desired compound 9 in multigram amount (5.1 g, yield 72%, dr > 95:5). Although, this intermediate in itself is an important building block for click-chemistry, we went further and synthesized a tBu protected open chain and methyl protected cyclic congener (10 and 11, respectively, Scheme 2). Hence, this method can be used in the future for further development of important cyclic and acyclic chiral synthons.
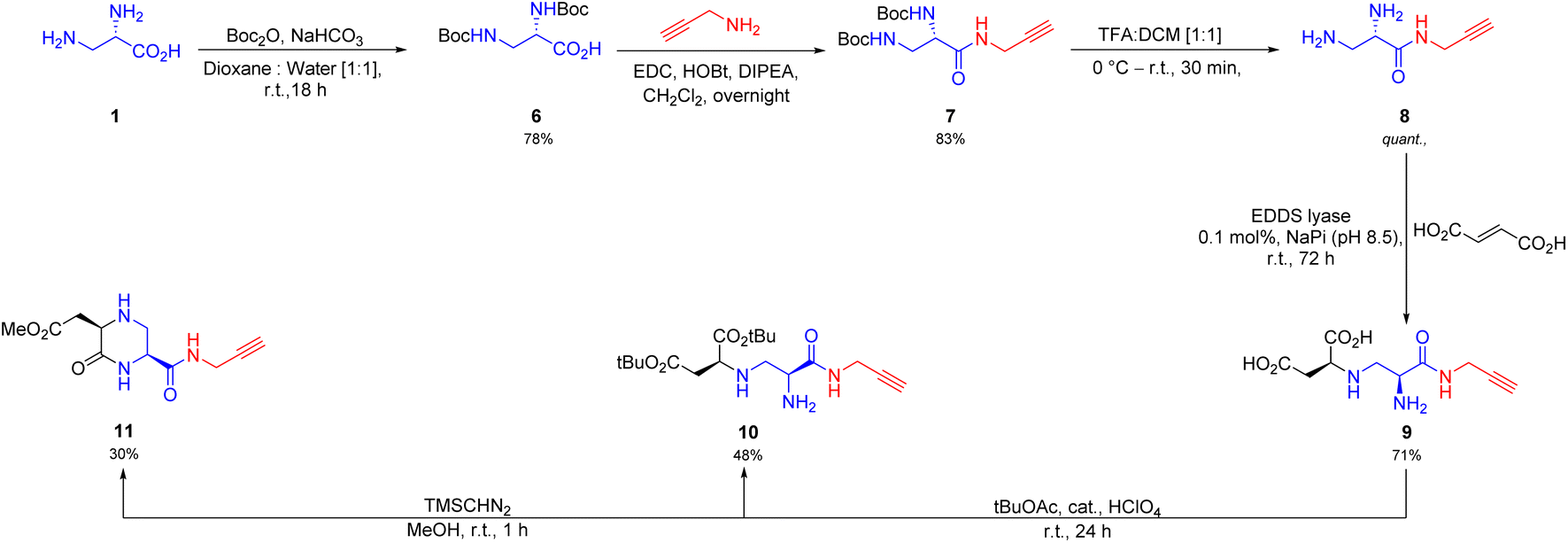 | ||
| Scheme 2 Chemoenzymatic synthetic route towards click-handle installed toxin A derivatives. The dr of 9 was determined to be >95:5 by 1H NMR and the absolute configuration was tentatively assigned as (S,S) based on analogy. | ||
Finally, inspired by the recent advances in photocleavable prodrug design for precision therapy18 and safety,18–21 we embarked upon retrosynthetic analysis towards a photocleavable AMB (Scheme 3). We chose AMB for photocaging over AMA owing to its relatively easy synthesis but comparable inhibitory activity.17 Towards this end, N-alkylation of fully protected toxin A intermediate 3 with freshly synthesized photocaged-bromoacetic acid 12 (Scheme 3, see ESI† for details) was performed following our newly established synthetic method (conditiond, see ESI† for details) to give 13 in 58% isolated yield. Finally, a standard deprotection procedure (conditione), gave the desired photocaged AMB product 14 (overall yield 8%; dr > 95:5 based on 1H NMR, with both stereogenic centres being derived from (2S,2′S)-2).
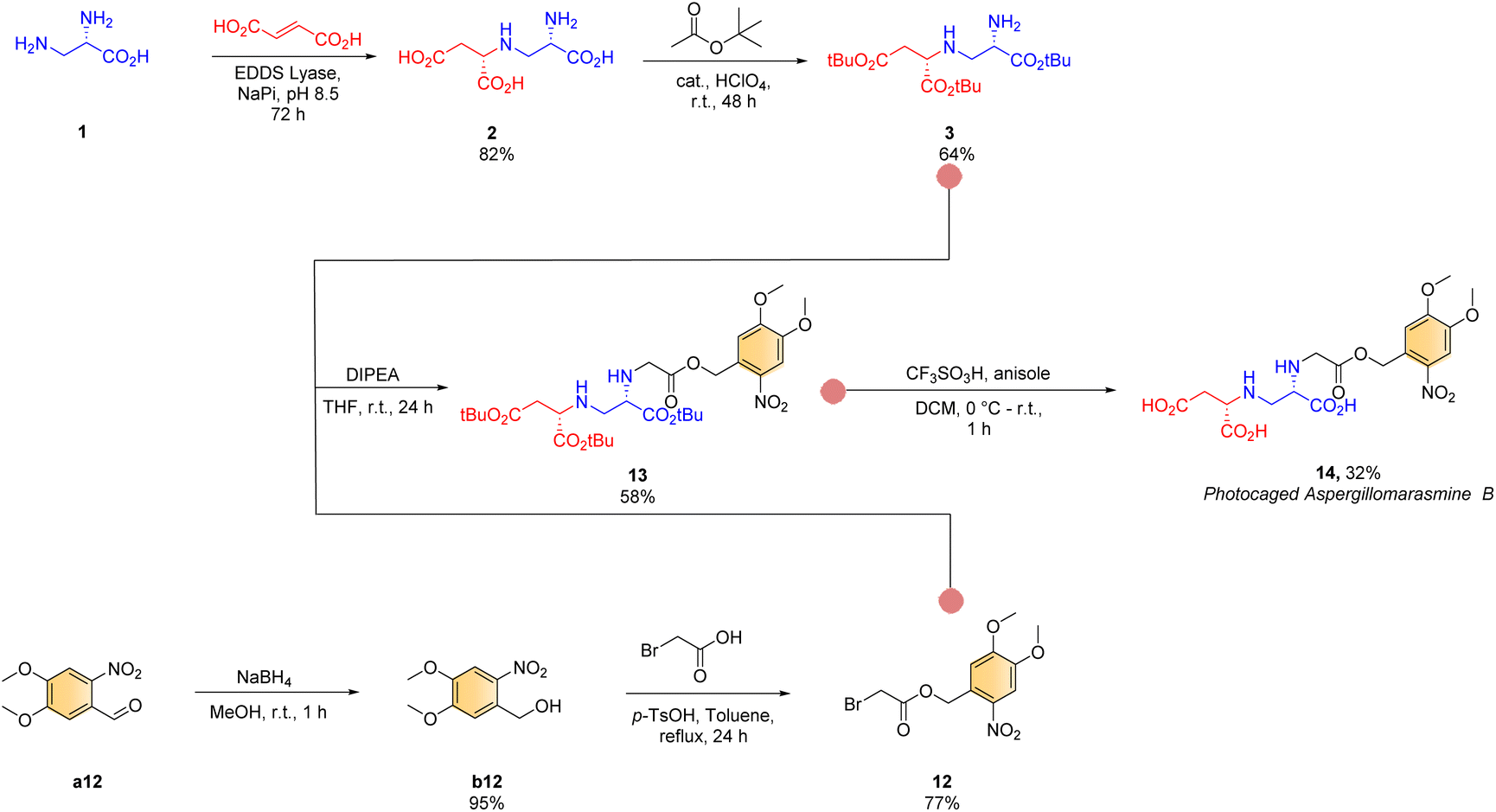 | ||
| Scheme 3 Chemoenzymatic route towards the synthesis of photocaged aspergillomarasmine B. | ||
In conclusion, we have developed efficient chemoenzymatic routes for the facile and stereoselective synthesis of various complex aminopolycarboxylic acids at milligram- to gram-scale. We first optimized and scaled-up a biocatalytic methodology to prepare multigram amounts of (2S,2′S)-toxin A, with the (2S)-stereogenic centre being selectively installed by EDDS lyase and the (2′S)-stereogenic centre derived from the starting amine substrate. Subsequent chemical N-functionalization of this key chiral building block gives access to a series of toxin A derivatives with retention of configuration. Indeed, no significant epimerization has been observed during the N-functionalization procedures, providing the final products with high diastereomeric purity (dr > 95:5). We have also installed a click-handle into toxin A to further facilitate derivatization, enabling the preparation of large compound libraries for MBL inhibitor screening. Finally, inspired by recent developments in photocleavable prodrug design, we have used a retrosynthetic approach to chemoenzymatically prepare photocaged AMB. In future work, we aim to characterize this compound for its efficiency of uncaging and usefulness as MBL inhibitor in precision treatment of infections using model systems. Taken together, the new methodologies and compounds reported herein have the potential to facilitate research into the discovery of more potent and more selective MBL inhibitors, which are urgently needed to battle MBL-based infections.
Experimental section
For detailed experimental procedures and characterization of compounds, see ESI.†Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no. 754425, and from the Netherlands Organization of Scientific Research (NWO-VICI grant 724.016.002).References
- S. C. Abeylath and E. Turos, Expert Opin. Drug Delivery, 2008, 5, 931–949 CrossRef CAS PubMed.
- D. Brown, Nat. Rev. Drug Discovery, 2015, 14, 821–832 CrossRef CAS PubMed.
- A. Bergstrom, A. Katko, Z. Adkins, J. Hill, Z. Cheng, M. Burnett, H. Yang, M. Aitha, M. R. Mehaffey and J. S. Brodbelt, ACS Infect. Dis., 2018, 4, 135–145 CrossRef CAS PubMed.
- D. Y. Wang, M. I. Abboud, M. S. Markoulides, J. Brem and C. J. Schofield, Future Med. Chem., 2016, 8, 1063–1084 CrossRef CAS PubMed.
- T. R. Walsh, J. Weeks, D. M. Livermore and M. A. Toleman, Lancet Infect. Dis., 2011, 11, 355–362 CrossRef PubMed.
- A. M. King, S. A. Reid-Yu, W. Wang, D. T. King, G. De Pascale, N. C. Strynadka, T. R. Walsh, B. K. Coombes and G. D. Wright, Nature, 2014, 510, 503 CrossRef CAS PubMed.
- D. Liao, S. Yang, J. Wang, J. Zhang, B. Hong, F. Wu and X. Lei, Angew. Chem., Int. Ed., 2016, 55, 4291–4295 CrossRef CAS PubMed.
- K. Koteva, A. M. King, A. Capretta and G. D. Wright, Angew. Chem., Int. Ed., 2016, 55, 2210–2212 CrossRef CAS PubMed.
- S. A. Albu, K. Koteva, A. M. King, S. Al-Karmi, G. D. Wright and A. Capretta, Angew. Chem., 2016, 128, 13453–13456 CrossRef.
- J. Zhang, S. Wang, Y. Bai, Q. Guo, J. Zhou and X. Lei, J. Org. Chem., 2017, 82, 13643–13648 CrossRef CAS PubMed.
- Q. Guo, D. Wu, L. Gao, Y. Bai, Y. Liu, N. Guo, X. Du, J. Yang, X. Wang and X. Lei, ACS Catal., 2020, 10, 6291–6298 CrossRef CAS.
- E. Cigan, B. Eggbauer, J. H. Schrittwieser and W. Kroutil, RSC Adv., 2021, 11, 28223–28270 RSC.
- T. Hudlicky, ACS Omega, 2018, 3, 17326–17340 CrossRef CAS PubMed.
- J. H. Schrittwieser and V. Resch, RSC Adv., 2013, 3, 17602–17632 RSC.
- T. Sugai, S. Higashibayashi and K. Hanaya, Tetrahedron, 2018, 74, 3469–3487 CrossRef CAS.
- H. Fu, J. Zhang, M. Saifuddin, G. Cruiming, P. G. Tepper and G. J. Poelarends, Nat. Catal., 2018, 1, 186–191 CrossRef CAS.
- K. H. Tehrani, H. Fu, N. C. Brüchle, V. Mashayekhi, A. P. Luján, M. J. van Haren, G. J. Poelarends and N. I. Martin, Chem. Commun., 2020, 56, 3047–3049 RSC.
- J. M. Silva, E. Silva and R. L. Reis, J. Controlled Release, 2019, 298, 154–176 CrossRef CAS PubMed.
- A. V. Cheng and W. M. Wuest, ACS Infect. Dis., 2019, 5, 816–828 CrossRef PubMed.
- I. S. Shchelik, A. Tomio and K. Gademann, ACS Infect. Dis., 2021, 7, 681–692 CrossRef CAS PubMed.
- P. T. Wong, S. Tang, J. Mukherjee, K. Tang, K. Gam, D. Isham, C. Murat, R. Sun, J. R. Baker and S. K. Choi, Chem. Commun., 2016, 52, 10357–10360 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob01405c |
| This journal is © The Royal Society of Chemistry 2024 |