
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Clemastine/tamoxifen hybrids as easily accessible antileishmanial drug leads†
V. S.
Agostino
 ad,
M. L.
Buerdsell
a,
S. R. B.
Uliana
b,
P. W.
Denny
c,
A. C.
Coelho
d and
P. G.
Steel
*a
ad,
M. L.
Buerdsell
a,
S. R. B.
Uliana
b,
P. W.
Denny
c,
A. C.
Coelho
d and
P. G.
Steel
*a
aDepartment of Chemistry, Durham University, UK. E-mail: p.g.steel@durham.ac.uk
bDepartment of Parasitology, Biomedical Sciences Institute, University of Sao Paulo, Brazil
cDepartment of Biosciences, Durham University, UK
dDepartment of Animal Biology, Institute of Biology, University of Campinas, Brazil
First published on 5th February 2024
Abstract
A library of hybrid molecules is developed based on the common chemical features shared by clemastine and tamoxifen both of which are well known for their antileishmanial activities. In the initial screening against Leishmania major and L. amazonensis promastigotes, as well as cytotoxicity assays using HepG2 cells, several hybrids showed submicromolar activity against the parasite and no toxicity against human cells. The compounds with an EC50 < 2 μM against promastigotes of both species and a selectivity index >10 were further characterized against intracellular amastigotes as well as promastigotes of species that cause both visceral and cutaneous leishmaniasis, such as L. infantum and L. braziliensis, respectively. These sequential screenings revealed the high pan-activity of this class of molecules against these species, with several compounds displaying an EC50 ≤ 2 μM against both promastigotes and intracellular amastigotes. Two of them were identified as the potential templates for lead optimization of this series having shown the highest activities against all species in both stages of parasite. The present findings can serve as a good starting point in the search for novel antileishmanial compounds that are easy to access and highly active.
1. Introduction
Leishmaniasis is a group of vector-borne neglected tropical diseases caused by approximately 20 species of the parasite of the genus Leishmania.1 According to the World Health Organization (WHO), more than one billion people are at risk of infection in over 90 countries where the leishmaniases are endemic with major implications for economic viability, demonstrated by the 1.6 million DALYs (disability-adjusted life years) lost due to the disease.2 The leishmaniases are caused by approximately 20 different parasite species leading to different clinical manifestations ranging from lesions in the skin to highly disfiguring destruction of facial mucosa and potentially lethal infections in the liver, spleen, and bone-marrow.3–5 The actual impact of the disease is challenged by limitation on diagnosis and the number of cases that are under reported. However, recent estimates by WHO suggest that there are at least 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 new cases of visceral leishmaniases (VL) leading approximately 20000 deaths each year.6,7 Cutaneous leishmaniasis (CL) is more prevalent with over 1 million new annual cases.7 Although generally non-fatal, CL is often associated with other potentially lethal secondary infections, as well as a social stigma that is correlated with longer term mental health issues leading to a collective adverse impact to over 40 million people worldwide.8–10 This situation is compounded by the fact that the current therapeutic arsenal is limited to a few drugs which have multiple shortcomings including severe side effects, unpleasant modes of administration, high cost, species dependent activity and the emergence of resistant parasites.11–13 These issues result in poor patient adherence, impairing the efficacious elimination of the disease. Collectively, this makes the search for new alternative cheap broad-spectrum treatments for leishmaniasis an urgent issue.
000 new cases of visceral leishmaniases (VL) leading approximately 20000 deaths each year.6,7 Cutaneous leishmaniasis (CL) is more prevalent with over 1 million new annual cases.7 Although generally non-fatal, CL is often associated with other potentially lethal secondary infections, as well as a social stigma that is correlated with longer term mental health issues leading to a collective adverse impact to over 40 million people worldwide.8–10 This situation is compounded by the fact that the current therapeutic arsenal is limited to a few drugs which have multiple shortcomings including severe side effects, unpleasant modes of administration, high cost, species dependent activity and the emergence of resistant parasites.11–13 These issues result in poor patient adherence, impairing the efficacious elimination of the disease. Collectively, this makes the search for new alternative cheap broad-spectrum treatments for leishmaniasis an urgent issue.
The cost of, and time required for, de novo drug discovery challenges the development of new chemotherapies and renders the adoption of a drug repurposing strategy attractive.14–16 Towards this end, we have previously reported that the breast cancer drug, tamoxifen,17,18 and the antihistamine clemastine fumarate19 have promising activity against several species of Leishmania, both in vitro and in vivo. Tamoxifen is a well-known selective estrogen receptor modulator (SERM) that has been used since the 1970s in the treatment and prevention of estrogen-dependent breast cancer.20,21 Even though estrogen receptors have not been identified in Leishmania,17 tamoxifen displayed micromolar activity against intramacrophage amastigotes of L. braziliensis (EC50 = 1.9 μM), L. amazonensis (EC50 = 4.5 μM) and L. infantum (EC50 = 2.4 μM), in addition to also clearing infections in vivo in mouse models of the disease when administered intraperitoneally.17,22–25 Clemastine fumarate is a first-generation H1 receptor antagonist26 with sub-micromolar activity against L. amazonensis (EC50 = 0.46 μM), and an in vivo efficacy comparable to that obtained with the clinically used meglumine antimoniate (Glucantime®) in a mouse model of L. amazonesis infection.19
Although both compounds exhibit polypharmacology, they share a common mode of action involving the inhibition of inositol phosphorylceramide synthase (IPCS), which is a key enzyme in the sphingolipid biosynthetic pathway of the parasite.19,27 Moreover, both share common structural elements, in particular an aminoalkoxy side chain (Fig. 1, head group, blue) and a diarylmethane (Fig. 1, scaffold, red), and it was therefore hypothesised that these features contribute to their common antileishmanial activity. Whilst a number of syntheses have been reported for each compound,28,29 both present challenges, particularly in the control of stereochemistry. In this report we describe the synthesis of hybrid compounds that build on this commonality, are simple to access and retain good levels of antileishmanial activity across a broad spectrum of parasite species.
 | ||
| Fig. 1 Clemastine and tamoxifen common structural features, an aminoalkoxy chain (blue) and a diaryl system (red). | ||
2. Results and discussion
2.1. Design and synthesis of clemastine/tamoxifen hybrid molecules
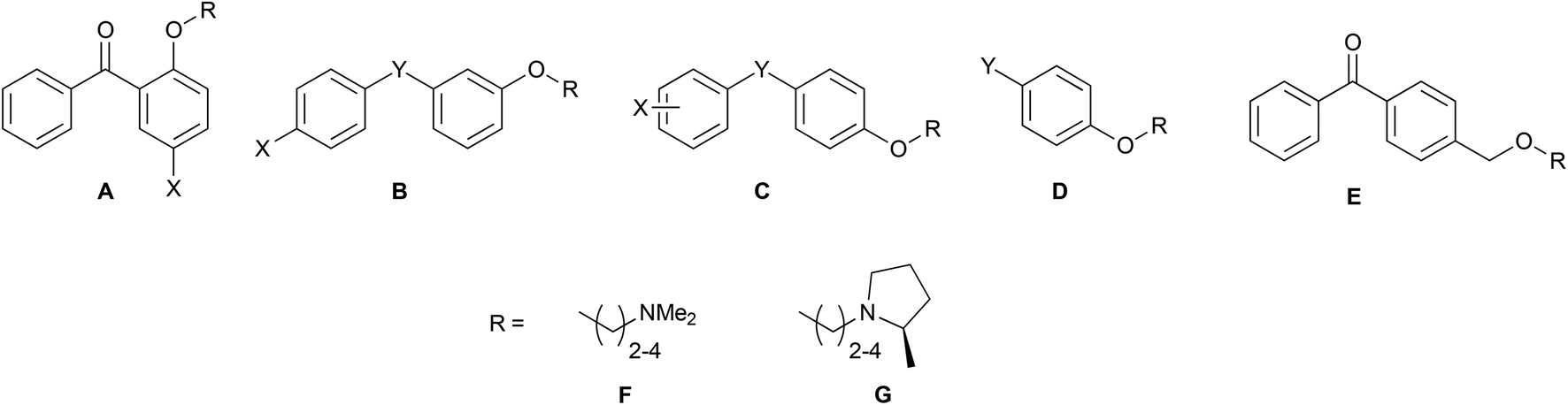
Combining the diaryl ethene from tamoxifen and the diaryl carbinol from clemastine led to a benzophenone being chosen as the parent scaffold that is both commercially available and chemically accessible, making modifications more approachable. Whilst scaffolds A, B, C, F and H were found commercially, the other scaffolds were synthesized in one-step through a Friedel–Crafts acylation protocol (Scheme 1A). Modifications introduced in this way included varying the position of the aminoalkoxy chain on ring B, as well as the addition of substituents into ring A to explore the essentiality of the clemastine chlorine substituent for activity as well as tolerance to bulkier substituents (Fig. 2). | ||
| Scheme 1 Synthesis of tamoxifen/clemastine hybrids. (A) Synthesis of para-hydroxybenzophenones D and E and meta-hydroxybenzophenones H, I and J. (B) Attachment of headgroups to hydroxybenzophenones. a – Na2CO3, KI, MeCN, 80 °C, 24 h; b – Cs2CO3, MeCN, RT, 24 h. | ||
 | ||
| Fig. 2 Benzophenone scaffolds designed for use in the synthesis of clemastine/tamoxifen hybrids. | ||
Each scaffold unit was then combined with two series of headgroups via a series of simple SN2 reactions summarized in Scheme 1B. These were the dimethylaminoalkyl motifs present in tamoxifen (Fig. 2L), and the (2R)-methyl-N-(ω-alkyl)pyrrolidines (Fig. 2M) that we have previously shown to have similar activity to clemastine but with easier chemical accessibility.30
Finally, two more focused set of compounds were prepared in which the benzophenone was replaced by a simple benzenoid scaffold (Scheme 2) and systems in which the ketone component was replaced by an oxygen (diphenyl ether) or a carbon (diphenylmethane) (Scheme 3).
 | ||
| Scheme 2 Synthesis of analogues 35 and 36. (a) K2CO3, TEBA–Cl, EtOAc; (b) K2CO3, MeCN. | ||
 | ||
| Scheme 3 Synthesis of analogues: (A) 19 and 20; (B) 34. | ||
2.2. Antileishmanial screening of clemastine/tamoxifen hybrids
A screening workflow was established with the aim of selecting the hybrids with the most promising therapeutic properties (Fig. 3). Using the well-established resazurin cell viability assay,31 all compounds were initially screened against L. major promastigotes, an Old World CL-causing species. Compounds exhibiting an EC50 < 10 μM were selected for further screening against L. amazonensis as a representative New World CL-causing species. In this step, compounds were also assayed against HepG2 cells to assess their cytotoxicity to mammalian cells (Table 1). The most active compounds that displayed an EC50 ≤ 2 μM against both Leishmania species as well as a selectivity index (SI) above 10 were singled out for further characterisation (Fig. 4). This included assays against promastigotes of L. braziliensis and L. infantum, causative agents of CL and VL in South America, respectively, as well as anti-intracellular amastigote assays against L. amazonensis and L. infantum. Additionally, these compounds were assayed against bone-marrow derived macrophages (BMDM), a host cell considered as model of infection for drug susceptibilities studies in Leishmania.32 | ||
| Fig. 3 Screening workflow for the selection of the most active antileishmanial compounds. | ||
 | ||
| Fig. 4 Relative activity of compounds against L. major and L. amazonensis promastigotes. Dots represent compounds with an EC50 ≤ 10 μM for both species. Red dot is clemastine fumarate; green dot is tamoxifen; grey dots are compounds with SI < 10 for at least one species. Dotted line is the 2 μM cut-off for selection of compounds with highest activity, where compounds within the bottom left quadrant were selected. Data are the average EC50 values (in μM) of three independent experiments. | ||
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compounds | Scaffold | X | Y | Carbon chain | R | EC50L. majora | EC50L. amazonensisa | CC50 HepG2a | SI (L. major)b | SI (L. amazonensis)b |
| a Values represent the average and standard error of the mean in micromolar (μM) of three independent experiments in triplicate. b The SI (selectivity index) was determined as the ratio between the CC50 of HepG2 cells and EC50 of promastigotes of the indicated Leishmania species. c G-head groups is (2S)-methylpyrrolidine stereoisomer. d G-head groups is 2,2-dimethylpyrrolidine. ND, data not determined. | ||||||||||
| 1 | A | H | — | 2 | F | >100 | ND | ND | ND | ND |
| 2 | A | H | — | 3 | F | 32.51 ± 5.77 | ND | ND | ND | ND |
| 3 | A | Cl | — | 2 | F | 68.91 ± 15.92 | ND | ND | ND | ND |
| 4 | A | Cl | — | 3 | F | 11.76 ± 1.1 | ND | ND | ND | ND |
| 5 | A | H | — | 2 | G | 45.93 ± 15.68 | ND | ND | ND | ND |
| 6 | A | H | — | 3 | G | 30.11 ± 9.04 | ND | ND | ND | ND |
| 7 | A | H | — | 4 | G | 11.14 ± 0.23 | ND | ND | ND | ND |
| 8 | A | Cl | — | 2 | G | 35.78 ± 5.5 | ND | ND | ND | ND |
| 9 | A | Cl | — | 3 | G | 18.53 ± 3.24 | ND | ND | ND | ND |
| 10 | A | Cl | — | 4 | G | 1.62 ± 1.48 | 3.07 ± 0.92 | >100 | >61 | >32 |
| 11 | B | H | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
2 | G | 4.58 ± 1.65 | >100 | >100 | >22 | — |
| 12 | B | H | CO |
3 | G | 1.4 ± 0.0025 | 0.74 ± 0.13 | >100 | >71 | >135 |
| 13 | B | H | CO |
4 | G | 0.93 ± 0.16 | 3.51 ± 1.44 | >100 | >107 | >48 |
| 14 | B | Cl | CO |
2 | G | 5.14 ± 2.71 | 24.8 ± 10.51 | >100 | >19 | >4 |
| 15 | B | Cl | CO |
3 | G | 0.27 ± 0.026 | 0.37 ± 0.14 | >100 | >370 | >270 |
| 16 | B | Cl | CO |
4 | G | 0.30 ± 0.065 | 0.85 ± 0.46 | 25.64 ± 0.94 | 85 | 30 |
| 17 | B | Methyl | CO |
3 | G | 0.63 ± 0.17 | 1.44 ± 0.44 | >100 | >158 | >69 |
| 18 | B | Isopropyl | CO |
3 | G | 5.35 ± 1.04 | 8.49 ± 2.64 | 16.45 ± 3.99 | 3 | 2 |
| 19 | B | H | CH2 | 3 | G | 0.85 ± 0.23 | 3.07 ± 0.19 | >100 | >117 | >32 |
| 20 | B | H | O | 3 | G | 1.33 ± 0.3 | 1.77 ± 0.30 | 22.71 ± 3.50 | 17.07 | 12.83 |
| 21 | B | H | CO |
3 | Gc | ND | 3.03 ± 0.43 | ND | ND | ND |
| 22 | B | H | CO |
3 | Gd | ND | 0.62 ± 0.063 | ND | ND | ND |
| 23 | C | H | CO |
2 | F | 11.61 ± 2.68 | ND | ND | ND | ND |
| 24 | C | H | CO |
3 | F | 7.07 ± 1.4 | 4.69 ± 1.38 | ND | ND | ND |
| 25 | C | H | CO |
4 | F | 2.91 ± 0.26 | 1.58 ± 0.25 | >100 | >34 | >63 |
| 26 | C | H | CO |
2 | G | 4.51 ± 0.35 | 1.2 ± 0.35 | >100 | >22 | >83 |
| 27 | C | H | CO |
3 | G | 4.25 ± 0.12 | 3.69 ± 1.51 | >100 | >23 | >27 |
| 28 | C | H | CO |
4 | G | 5.37 ± 0.42 | 2.56 ± 0.84 | >100 | >18 | >39 |
| 29 | C | p-Cl | CO |
2 | G | 2.02 ± 0.005 | 0.8 ± 0.27 | >100 | >49 | >125 |
| 30 | C | p-Cl | CO |
3 | G | 2.37 ± 0.18 | 1.56 ± 0.59 | 31.07 ± 11.04 | 13 | 19 |
| 31 | C | p-Cl | CO |
4 | G | 2.2 ± 0.56 | 1.42 ± 0.52 | 16.05 ± 2.13 | 7 | 11 |
| 32 | C | m-Cl | CO |
3 | G | 2.96 ± 0.4 | 1.14 ± 0.39 | 25.11 ± 2.03 | 8 | 22 |
| 33 | C | o-Cl | CO |
3 | G | 0.63 ± 0.001 | 6.77 ± 0.93 | >100 | >158 | >15 |
| 34 | C | H | CH2 | 3 | G | 0.62 ± 0.3 | 7.25 ± 0.61 | 27.06 ± 6.67 | 44 | 3.73 |
| 35 | D | MeCO |
— | 3 | G | >100 | >100 | >100 | ND | ND |
| 36 | D | H | — | 3 | G | >100 | >100 | >100 | ND | ND |
| 37 | E | — | — | 2 | F | 8.01 ± 2.39 | 10.27 ± 2.9 | ND | ND | ND |
| 38 | E | — | — | 3 | F | >10 | >10 | ND | ND | ND |
| Clemastine | — | — | — | — | — | 0.035 ± 0.012 | 0.038 ± 0.003 | 25.57 ± 1.76 | 730 | 673 |
| Tamoxifen | — | — | — | — | — | 4.03 ± 0.33 | 2.39 ± 0.17 | 31.32 ± 1.91 | 8 | 13 |
| Miltefosine | — | — | — | — | — | 6.57 ± 0.89 | 20.71 ± 3.54 | ND | ND | ND |
The screenings against promastigotes of L. major and L. amazonensis allowed for insights regarding the structure–activity relationship (SAR) of these compounds. Firstly, there was a notable difference in activity dependent on the position of the aminoalkoxy side chain on ring B in relation to the central ketone. Compounds with a meta-arrangement (Table 1, entries 11–16) were the most active with 15 and 16, for example, having an EC50 ≤ 1 μM against both species tested as well as a promising selectivity index. In contrast, the set of ortho-substituted analogues contained the least active compounds (Table 1, entries 1–10), and the para-substituted group showed intermediate activity (Table 1, entries 23–33). The presence of a chlorine substituent in either ring A (ortho-substituted analogues) or ring B (meta- and para-substituted analogues) resulted in enhanced activity. Although the para- and meta-chloro substituents were equally effective, the first is a more chemically accessible position and was chosen for further studies. Replacing the chlorine atom by a methyl group is tolerated, however a bulkier substituent such as the isopropyl group caused loss in activity and increased cytotoxicity. The mono-aromatic analogues tested were inactive against both promastigote species, rendering the diaryl system essential for activity (Table 1, entries 35 and 36). Benzophenones with a benzylic aminoalkoxy side chain (Table 1, entries 37 and 38) did not display satisfactory activity. Replacing the carbonyl group with an oxygen or methylene linker was tolerated, however these analogues had lower overall activity and/or selectivity against L. amazonensis promastigotes (Table 1, entries 19, 20 and 34) when compared to analogue 12.
The length of the side chain appeared to be important with the shorter ethyl linker giving lower activity against at least one of the two initial species tested. The only exception was compound 29, which displayed activity below 2 μM against both species tested. Dimethylamine-containing headgroups were also less active when compared to the pyrrolidine-containing compounds. For the latter, the R-stereoisomer (Table 1, entry 12) was approximately four-fold more effective than the S-enantiomer (Table 1, entry 21) against L. amazonensis. Consistent with this, the gem dimethyl analogue (Table 1, entry 22) was examined and found to be equipotent with the R isomer, but as this precursor amine is less readily available this was not explored further.
Finally, the seven most active compounds (12, 15–17, 20, 29 and 30) were then further characterised for activity against promastigotes of L. braziliensis and L. infantum (Table 2) and intracellular amastigotes of L. amazonensis and L. infantum, following cytotoxicity evaluation in BMDM (Table 3). These analyses indicated that these chimeric compounds show high efficacy and selectivity against all four species tested. Additionally, good levels of activity were observed in the intramacrophage amastigote assay, with four compounds (16, 17, 20 and 29) displaying an EC50 ≤ 2 μM and a SI > 10. Overall, when compared with both clemastine and tamoxifen, these hybrids exhibit intermediatory activity against L. major and L. amazonensis and in the case of analogues 15 and 16 comparable activity to clemastine against L. braziliensis and L. infantum. As with clemastine, the hybrids have a significantly lower sp2 atom count and a higher degree of flexibility than tamoxifen, which may contribute to this higher activity. However, whilst more active, the synthesis of clemastine is non-trivial, challenging further structural variations. The short and simple synthetic route to the hybrids renders these structures as highly promising templates for further lead optimisation.
| Compounds | EC50aL. braziliensis |
EC50aL. infantum |
SIb (L. braziliensis) | SIb (L. infantum) |
|---|---|---|---|---|
| a Values represent the average and standard error of the mean in micromolar (μM) of three independent experiments in triplicate. b The SI (selectivity index) was determined as the ratio between the CC50 of HepG2 cells and EC50 of promastigotes of the indicated Leishmania species. | ||||
| 12 | 0.53 ± 0.14 | 4.42 ± 0.27 | >189 | >23 |
| 15 | 0.38 ± 0.054 | 1.32 ± 0.12 | >263 | >76 |
| 16 | 0.21 ± 0.044 | 1.00 ± 0.14 | 124 | 26 |
| 17 | 0.65 ± 0.12 | 2.93 ± 0.11 | >154 | >34 |
| 20 | 0.26 ± 0.10 | 2.84 ± 0.16 | 88 | 8 |
| 29 | 0.41 ± 0.091 | 2.32 ± 0.091 | >243 | >43 |
| 30 | 0.084 ± 0.015 | 1.25 ± 0.11 | 369 | 25 |
| Clemastine | 0.14 ± 0.002 | 0.71 ± 0.043 | 186 | 36 |
| Tamoxifen | 9.55 ± 2.045 | 4.97 ± 0.49 | 3 | 6 |
| Compounds | CC50a BMDM |
EC50aL. amazonensis |
EC50aL. infantum |
SIb (L. amazonensis) | SIb (L. infantum) |
|---|---|---|---|---|---|
| a Values represent the average and standard error of the mean in micromolar (μM) of three independent experiments in triplicate. b The SI (selectivity index) was determined by the ratio between the CC50 of BMDM and EC50 of promastigotes of the indicated Leishmania species. ND, data not determined. | |||||
| 12 | >100 | 3.95 ± 0.38 | 1.30 ± 0.35 | 25 | 77 |
| 15 | 36.38 ± 6.25 | 5.35 ± 1.055 | 0.69 ± 0.18 | 7 | 53 |
| 16 | 32.48 ± 5.01 | 2.11 ± 0.43 | 2.43 ± 0.58 | 15 | 13 |
| 17 | 48.73 ± 9.77 | 2.44 ± 0.59 | 1.19 ± 0.29 | 20 | 41 |
| 20 | 49.46 ± 0.36 | 0.80 ± 0.096 | 1.076 ± 0.14 | 62 | 46 |
| 29 | >100 | 2.27 ± 0.26 | 1.72 ± 0.44 | 44 | 58 |
| 30 | 43.13 ± 2.30 | 3.89 ± 0.70 | 0.86 ± 0.17 | 11 | 50 |
| Clemastine | 22.35 ± 3.78 | 0.46 ± 0.10 | ND | 49 | ND |
| Tamoxifen | 52.44 ± 2.34 | 4.90 ± 0.69 | 2.4 ± 0.3 | 10 | 22 |
3. Conclusions
The standard cut off values suggested by the Drugs for Neglected Diseases initiative (DNDi) for lead discovery programs for antileishmanials is an EC50 ≤ 2 μM against both intracellular amastigotes and promastigotes as well as a selectivity index (SI) above 10.33 Importantly for the development of new antileishmanial drugs, the results obtained demonstrate that this benchmark has been met across a range of parasite species of both Old and New World origin, and suggest that these easy-to-synthesise chimeric structures therefore represent versatile starting points for a lead optimisation programme. The success of this simple phenotypic driven fragment linking strategy suggest that this may be a productive method for the development of other promising compounds. As with many anti-leishmanials, including all the existing approved drugs, although both clemastine fumarate and tamoxifen target IPCS they also exhibit polypharmacology with other modes of action.19,34 It is likely that these hybrids retain these characteristics, and these different activities will be impacted in different ways by the structural changes made which may account for the variable levels of activity observed. It is possible to speculate that the more consistent level of activity exhibited by 15 and 16 across a range of Leishmania species may indicate a more focused mode of action. A better understanding of the target(s) and mechanisms of action of both these compounds and the parent drug and is needed to clarify this and direct further studies. The easier access to these hybrids facilitates such studies and work towards target identification is ongoing and will be described in due course.4. Experimental
4.1. Chemical synthesis
4.2. Leishmania culture
Leishmania amazonensis (MHOM/BR/1975/JOSEFA), Leishmania major (MHOM/IL/80/Friedlin), Leishmania braziliensis (MHOM/BR/1994/H3227) and Leishmania infantum (MHOM/BR/1972/LD) were cultivated at 25 °C in Schneider's insect medium at pH 7, supplemented with 15% heat-inactivated fetal bovine serum (FBS) and penicillin/streptomycin 100 μg ml−1. Transgenic lines of L. amazonensis and L. infantum expressing luciferase were previously generated, maintained in media containing 32 μg mL−1 hygromycin B35,36 and used for anti-amastigote dose–response assays (item 4.6).4.3. HepG2 cell culture
HepG2 cells were cultivated at 37 °C in a 5% CO2 atmosphere in DMEM supplemented with 10% FBS, containing 100 μg mL−1 penicillin/streptomycin. Cells were split every 3 days by removing the media and adding trypsin, which were then incubated for 10 min at 37 °C in a 5% CO2 atmosphere. Trypsin was deactivated by adding 20 mL fresh media, which was homogenised and added to a new culture flask in a 1:4 proportion.
4.4. Anti-promastigote dose–response assay
Log-phase promastigotes were quantified and suspended in fresh medium to a density of 1 × 106 parasites per ml. In a 96-well plate, 1 × 105 parasites were applied per well and parasites were incubated at 25 °C in increasing concentrations of each compound for 44 hours. Then, 10 μL of 0.1 mg mL−1 resazurin solution (Fisher Scientific, Leicestershire, UK) were added per well and the plate was incubated for 4 hours at 25 °C. Fluorescence (excitation at 555 nm and emission at 585 nm) was detected using the PolarStar Omega luminometer (BMGLabTech). EC50 values were obtained by GraphPad Prism 6, after determination of sigmoidal regression curves. At least three independent experiments in triplicates were performed for each molecule. Tamoxifen and clemastine were used as control drugs.4.5. Cytotoxicity assays
HepG2 cells were detached when cell culture was approximately 80% confluent and quantified to be resuspended in fresh DMEM medium to a density of 5 × 104 cells per mL which 200 μL were added to each well (1 × 104 cells) in a 96-well plate incubated overnight at 37 °C in a 5% CO2 atmosphere. For BMDM, the cells were plated as described in the section 4.6. In both cases, the supernatant was removed and fresh media containing several concentrations of compounds was added, and plates incubated at 37 °C on a 5% CO2 atmosphere for 44 hours. Next, 10 μL of 0.1 mg mL−1 resazurin solution (Fisher Scientific, Leicestershire, UK) were added per well and incubated for 4 h before fluorescence intensity was determined, using a plate reader (excitation at 555 nm and emission at 585 nm). At least three independent experiments were performed for each molecule with all samples in triplicates. Every plate had tamoxifen or clemastine as the control. EC50 values were obtained by GraphPad Prism 6, after determination of sigmoidal regression curves.4.6. Anti-intramacrophage amastigote dose–response assay
Bone-marrow-derived macrophages (BMDM) were obtained from female BALB/c mice, which were cultivated in RPMI 1640 medium (Thermo Fisher Scientific) supplemented with 10% FBS, 0.1 M sodium pyruvate, and 100 μg mL−1 penicillin/streptomycin and incubated in a 5% CO2 atmosphere at 37 °C, as previously described.36 The macrophages were plated in 96-well plates with a density of 8 × 104 cells per well in 100 μL per well. After incubation for 24 hours, the macrophages were infected with stationary-phase promastigotes at a ratio of 20:1 (parasites/macrophage) for both L. amazonensis and L. infantum. After 4 hours of incubation at 34 °C (except for L. infantum that was incubated at 37 °C), 200 μL of fresh medium containing several concentrations of the compounds tested was added to the wells, and the plates were incubated for 72 hours at 34 °C or 37 °C for L. infantum. Next, 15 μL of luciferin (One-Glo Luciferase Assay System, Promega) were then added per well and bioluminescence was measured in a PolarStar Omega luminometer (BMGLabTech). Parasite survival in treated samples was determined based on the ratio of treated/untreated cells. EC50 values were obtained by GraphPad Prisma 6, after determination of sigmoidal regression curves. At least three independent experiments in triplicate were performed for each molecule with all samples.
For experiments using mice, protocols and procedures were approved by the Ethics Committee for Animal Experimentation of the Instituto de Biologia, Universidade Estadual de Campinas (UNICAMP) (protocol: 5719-1/2021). Animals were obtained from Centro Multidisciplinar para Investigação Biológica (CEMIB) of UNICAMP and kept in mini-isolators, receiving food and water at libitum.
4.7. Statistical analysis
All statistical analyses were performed by GraphPad Prisma 6 (CA, USA), using ANOVA one-way test and multiple comparisons Dunnett's test. P value <0.05 was considered as statistically significant.Author contributions
PGS and SRBU conceived and designed the project. PGS, PWD and ACC supervised the project. VSA and MBB designed and conducted the experiments and collected the data. VSA, PGS, PWD and ACC analysed the results. VSA and PGS drafted the manuscript. All authors read, reviewed, and approved the final draft.Conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.Acknowledgements
We thank Durham University GCRF CDT (studentship to VSA), The Royal Society (The Royal Society International Collaboration Awards for Research Professors 2016: IC160044), the UKRI Global Challenges Research Fund under grant agreement ‘A Global Network for Neglected Tropical Diseases’ (grant number MR/P027989/1), and the GCRF and Newton Fund Consolidation Account (GNCA) EP/X527713/1 for financial support. We also thank Camilo C. Janeri for technical assistance with animal-related experiments.References
- S. Burza, S. L. Croft and M. Boelaert, Lancet, 2018, 392, 951–970 CrossRef PubMed.
- S. Mohan, P. Revill, S. Malvolti, M. Malhame, M. Sculpher and P. M. Kaye, PLoS Neglected Trop. Dis., 2022, 16, 1–17 Search PubMed.
- A. Poulaki, E. T. Piperaki and M. Voulgarelis, Microorganisms, 2021, 9, 759 CrossRef CAS PubMed.
- T. F. Silva, F. Tomiotto-Pellissier, A. K. S. Pasquali, F. Pinto-Ferreira, W. R. Pavanelli, I. Conchon-Costa, I. T. Navarro and E. T. Caldart, Acta Trop., 2021, 221, 106018 CrossRef CAS PubMed.
- I. Abadías-Granado, A. Diago, P. A. Cerro, A. M. Palma-Ruiz and Y. Gilaberte, Actas Dermo-Sifiliogr., 2021, 112, 601–618 CrossRef PubMed.
- WHO. World Health Organization, Status of endemicity of cutaneous leishmanisis, 2022 Search PubMed.
- WHO. World Health Organization, Status of endemicity of visceral leishmaniasis, 2022 Search PubMed.
- F. Baileyid, K. Mondragon-Shem, L. R. Haines, A. Olabi, A. Alorfi, J. A. Ruiz-Postigo, J. Alvar, P. Hotez, E. R. Adams, I. D. Vélez, W. Al-Salem, J. Eaton, Á. Acosta-Serrano and D. H. Molyneux, PLoS Neglected Trop. Dis., 2019, 13, 1–22 Search PubMed.
- M. Pires, B. Wright, P. M. Kaye, V. da Conceição and R. C. Churchill, PLoS One, 2019, 14, 1–21 Search PubMed.
- M. Yanik, M. S. Gurel, Z. Simsek and M. Kati, Clin. Exp. Dermatol., 2004, 29, 464–467 CrossRef CAS PubMed.
- S. R. B. Uliana, C. T. Trinconi and A. C. Coelho, Parasitology, 2017, 1–17 Search PubMed.
- S. Salari, M. Bamorovat, I. Sharifi and P. G. N. Almani, J. Clin. Lab. Anal., 2022, 36, 1–16 CrossRef PubMed.
- M. O. Harhay, P. L. Olliaro, M. Vaillant, F. Chappuis, M. A. Lima, K. Ritmeijer, C. H. Costa, D. L. Costa, S. Rijal, S. Sundar and M. Balasegaram, Am. J. Trop. Med. Hyg., 2011, 84, 543–550 CrossRef PubMed.
- D. C. Swinney and J. Anthony, Nat. Rev. Drug Discovery, 2011, 10, 507–519 CrossRef CAS PubMed.
- S. Pushpakom, F. Iorio, P. A. Eyers, K. J. Escott, S. Hopper, A. Wells, A. Doig, T. Guilliams, J. Latimer, C. McNamee, A. Norris, P. Sanseau, D. Cavalla and M. Pirmohamed, Nat. Rev. Drug Discovery, 2018, 18, 41–58 CrossRef PubMed.
- S. S. Braga, Eur. J. Med. Chem., 2019, 183 Search PubMed.
- D. C. Miguel, J. K. U. Yokoyama-Yasunaka, W. K. Andreoli, R. A. Mortara and S. R. B. Uliana, J. Antimicrob. Chemother., 2007, 60, 526–534 CrossRef CAS PubMed.
- C. T. Trinconi, J. Q. Reimão, V. I. Bonano, C. R. Espada, D. C. Miguel, J. K. U. Yokoyama-Yasunaka and S. R. B. Uliana, Parasitology, 2018, 145, 490–496 CrossRef PubMed.
- J. G. M. Mina, R. L. Charlton, E. Alpizar-Sosa, D. O. Escrivani, C. Brown, A. Alqaisi, M. P. G. Borsodi, C. P. Figueiredo, E. V. De Lima, E. A. Dickie, W. Wei, R. Coutinho-Silva, A. Merritt, T. K. Smith, M. P. Barrett, B. Rossi-Bergmann, P. W. Denny and P. G. Steel, ACS Infect. Dis., 2021, 7, 47–63 CrossRef CAS PubMed.
- A. Munshi and P. Singh, Breast, 2008, 17, 121–124 CrossRef PubMed.
- M. N. Singh, H. F. Stringfellow, E. Paraskevaidis, P. L. Martin-Hirsch and F. L. Martin, Cancer Treat. Rev., 2007, 33, 91–100 CrossRef CAS PubMed.
- D. C. Miguel, J. K. U. Yokoyama-Yasunaka and S. R. B. Uliana, PLoS Neglected Trop. Dis., 2008, 2, e249 CrossRef PubMed.
- D. C. Miguel, R. C. Zauli-Nascimento, J. K. U. Yokoyama-Yasunaka, S. Katz, C. L. Barbiéri and S. R. B. Uliana, J. Antimicrob. Chemother., 2009, 63, 365–368 CrossRef CAS PubMed.
- C. T. Trinconi, J. Q. Reimao, J. K. U. Yokoyama-Yasunaka, D. C. Miguel and S. R. B. Uliana, Antimicrob. Agents Chemother., 2014, 58, 2608–2613 CrossRef PubMed.
- C. T. Trinconi, J. Q. Reimão, A. C. Coelho and S. R. B. Uliana, J. Antimicrob. Chemother., 2016, 71, 1314–1322 CrossRef PubMed.
- H. B. Kaiser, J. Allergy Clin. Immunol., 1990, 86, 1000–1003 CrossRef CAS PubMed.
- C. T. Trinconi, D. C. Miguel, A. M. Silber, C. Brown, J. G. M. Mina, P. W. Denny, N. Heise and S. R. B. Uliana, Int. J. Parasitol.: Drugs Drug Resist., 2018, 8, 475–487 Search PubMed.
- K. M. Kasiotis and S. A. Haroutounian, Curr. Org. Chem., 2012, 16, 335–352 CrossRef CAS.
- A. M. Fournier, R. A. Brown, W. Farnaby, H. Miyatake-Ondozabal and J. Clayden, Org. Lett., 2010, 12, 2222–2225 CrossRef CAS PubMed.
- P. Steel, P. Denny, R. Charlton and B. Rossi-Bergmann, WO application No. WO2023047107A1, 2022 Search PubMed.
- A. Kulshrestha, V. Bhandari, R. Mukhopadhyay, V. Ramesh, S. Sundar, L. Maes, J. C. Dujardin, S. Roy and P. Salotra, Parasitol. Res., 2013, 112, 825–828 CrossRef PubMed.
- R. C. Zauli-Nascimento, D. C. Miguel, J. K. U. Yokoyama-Yasunaka, L. I. A. Pereira, M. A. Pelli De Oliveira, F. Ribeiro-Dias, M. L. Dorta and S. R. B. Uliana, Trop. Med. Int. Health, 2010, 15, 68–76 CAS.
- K. Katsuno, J. N. Burrows, K. Duncan, R. H. Van Huijsduijnen, T. Kaneko, K. Kita, C. E. Mowbray, D. Schmatz, P. Warner and B. T. Slingsby, Nat. Rev. Drug Discovery, 2015, 14, 751–758 CrossRef CAS PubMed.
- C. T. Trinconi, D. C. Miguel, A. M. Silber, C. Brown, J. G. M. Mina, P. W. Denny, N. Heise and S. R. B. Uliana, Int. J. Parasitol.: Drugs Drug Resist., 2018, 8, 475–487 Search PubMed.
- J. Q. Reimão, C. T. Trinconi, J. K. Yokoyama-Yasunaka, D. C. Miguel, S. P. Kalil and S. R. B. Uliana, J. Microbiol. Methods, 2013, 93, 95–101 CrossRef PubMed.
- J. Q. Reimão, J. C. Oliveira, C. T. Trinconi, P. C. Cotrim, A. C. Coelho and S. R. B. Uliana, PLoS Neglected Trop. Dis., 2015, 9, e0003556 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob02091f |
| This journal is © The Royal Society of Chemistry 2024 |