
Lewis acid-catalyzed one-pot thioalkenylation of donor–acceptor cyclopropanes using in situ generated dithiocarbamates and propiolates†
Sanjeevni
Harikumar
a,
Labeeb Thazhe Kollorth
Kandy
b,
Avishek
Guin
 *a and
Akkattu T.
Biju
*a
*a and
Akkattu T.
Biju
*a
aDepartment of Organic Chemistry, Indian Institute of Science, Bangalore 560012, India. E-mail: avishekguin@iisc.ac.in; atbiju@iisc.ac.in; Web: https://orgchem.iisc.ac.in/atbiju/
bDepartment of Chemistry, University of Kannur-670327, Kerala, India
First published on 2nd February 2024
Abstract
Lewis acid-catalyzed one-pot 1,3-thioalkenylation of donor–acceptor (D–A) cyclopropanes has been demonstrated employing in situ generated dithiocarbamates (from amines and CS2) as nucleophilic triggers and alkyl propiolates as electrophiles. This method addresses the limitations of previously known carbothiolation approach, eliminating the need for extra filtration prior to the subsequent trapping with electrophiles. The anticipated thioalkenylated products were obtained in good to excellent yields with a moderate to good E/Z ratio. Three new bonds (C–N, C–S, and C–C) are formed during this 1,3-bisfunctionalization reaction. Notably, employing enantiomerically pure D–A cyclopropanes resulted in enantiopure 1,3-thioalkenylated products, underscoring the stereospecific nature of the developed reaction.
Introduction
Donor–acceptor cyclopropanes have been recognized as important three-carbon building blocks in organic synthesis.1 The merger of ring strain and bond polarization through the adjacent placement of an electron-donating and electron-withdrawing group facilitates a broad spectrum of chemical reactions, which are otherwise difficult to be envisioned.2 Familiar instances encompass cycloadditions, rearrangements, and ring-opening reactions, providing a quick and efficient means to attain intricate molecular scaffolds.3 These transformations have already been utilized in several total syntheses, incorporating D–A cyclopropane-derived motifs at intermediate stages.4 However, there have been limited investigations into the methods of 1,3-bisfunctionalization of D–A cyclopropanes.5 The primary hurdle involves the controlled delivery of a nucleophile and an electrophile in a manner that directs their preference for reacting with the donor–acceptor cyclopropanes, rather than spontaneously with each other. Achieving ring-opening 1,3-bisfunctionalization reactions can be accomplished using two distinct methods. The first method involves an insertion reaction, which is relatively straightforward due to its low risk of side reactions.6 The second option is a multicomponent coupling, which poses a comparatively greater challenge in execution (Scheme 1, eqn (1)).7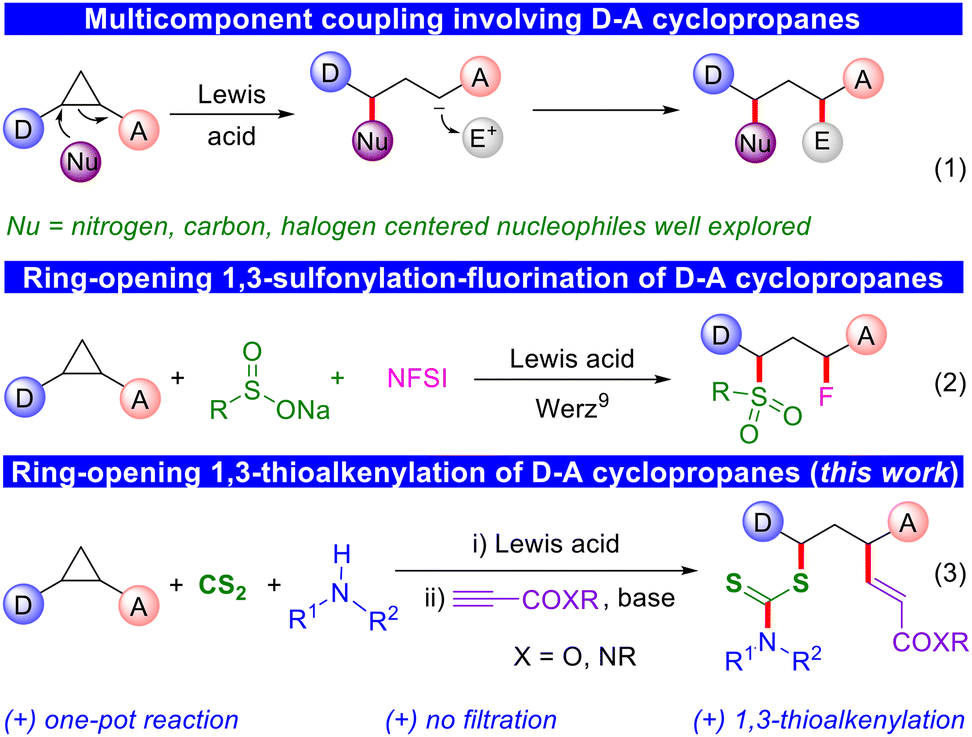 | ||
| Scheme 1 1,3-Bisfunctionalization of D–A cyclopropanes. | ||
Within the realm of 1,3-bisfunctionalization of donor–acceptor (D–A) cyclopropanes, the exploration has primarily focused on nitrogen, carbon, and halogen centred nucleophiles. For instance, Studer and co-workers showcased the 1,3-aminobromination of D–A cyclopropanes,7a while Werz and co-workers employed a related strategy to demonstrate the 1,3-aminothiolation of D–A cyclopropanes.7b Furthermore, Saha and co-workers revealed a 1,3-bisarylation of D–A cyclopropanes using a multicomponent coupling approach.7c However, the utilization of sulfur-based nucleophilic triggers in the 1,3-bisfunctionalization of D–A cyclopropanes has received limited consideration. Along these lines, our group reported a two-step carbothiolation of D–A cyclopropanes, necessitating a filtration step before the subsequent alkylation.8 More recently, the Werz group also demonstrated 1,3-sulfonylation–fluorination using sulfinate salts as nucleophiles (eqn (2)).9 Considering the scarce existing literature on sulfur-based nucleophilic triggers to open D–A cyclopropanes, we conceived a one-pot thioalkenylation method for D–A cyclopropanes (eqn (3)). This method involves the in situ generation of dithiocarbamates (from amines and CS2) as nucleophilic triggers and the use of alkyl propiolates as electrophiles. A significant advantage of this approach is that it overcomes the limitation of the previous two-pot carbothiolation process, which required additional filtration. A similar one-pot strategy for 1,3-carbocarbonation was already reported by Werz and co-workers due to the compatibility issue of nucleophiles and electrophiles.10 Notably, organosulfur motifs hold considerable importance in pharmaceuticals, materials, and agrochemicals.11
Results and discussion
With the envisioned thioalkenylation method for D–A cyclopropanes in mind, the present studies were initiated by the treatment of cyclopropane 1a with piperidine 2a and CS2 in the presence of Yb(OTf)3 in THF at 25 °C for 16 h, and this was followed by the treatment of methyl propiolate 3a and Cs2CO3. Interestingly, under these one-pot reaction conditions, the expected 1,3-thioalkenylated product 4a was isolated in 77% yield with a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 E/Z ratio (Table 1, entry 1). Changing the stoichiometry of CS2 led to a reduction in the yield of 4a to 63% with a 4:1 E/Z ratio (entry 2). Similarly, decreasing the quantities of 2a and 3a also resulted in diminished product yields, while maintaining a similar E/Z ratio (entries 3 and 4). Variations in solvents further resulted in diminished product yields (entries 5 and 6). Further optimizations by reducing the catalyst loading or use of other Lewis acids proved ineffective (entries 7–9). The use of organic bases, such as DBU and DABCO, did not yield any product, and only a 24% yield of 4a with a 4:1 E/Z ratio was obtained in the presence of K2CO3 (entries 10–12).
:1 E/Z ratio (Table 1, entry 1). Changing the stoichiometry of CS2 led to a reduction in the yield of 4a to 63% with a 4:1 E/Z ratio (entry 2). Similarly, decreasing the quantities of 2a and 3a also resulted in diminished product yields, while maintaining a similar E/Z ratio (entries 3 and 4). Variations in solvents further resulted in diminished product yields (entries 5 and 6). Further optimizations by reducing the catalyst loading or use of other Lewis acids proved ineffective (entries 7–9). The use of organic bases, such as DBU and DABCO, did not yield any product, and only a 24% yield of 4a with a 4:1 E/Z ratio was obtained in the presence of K2CO3 (entries 10–12).
|
|
|||
|---|---|---|---|
| Entry | Variation of the initial conditionsa | Yield of 4ab (%) |
E:Zc |
| a Initial conditions: 1a (0.2 mmol), CS2 (0.4 mmol), 2a (0.4 mmol), Yb(OTf)3 (20 mol%), THF (1.5 mL) for 16 h, and then a base (2.5 equiv.) and 3a (2.0 equiv.) for 12 h. b The yield was determined on the basis of 1H NMR of the crude reaction mixture using CH2Br2 as the internal standard. The isolated yield of 4a is given in parentheses. c The E/Z ratio was determined from 1H NMR of the crude reaction mixture. Nd indicates not determined. | |||
| 1 | None | 78 (77) |
5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :1 :1
|
| 2 | 1.5 equiv. of CS2 instead of 2.0 equiv. | 63 | 4:1 |
| 3 | 1.5 equiv. of 2a instead of 2.0 equiv. | 68 | 4:1 |
| 4 | 1.5 equiv. of 3a instead of 2.0 equiv. | 65 | 4:1 |
| 5 | DCE instead of THF | 70 | 5:1 |
| 6 | CH2Cl2 instead of THF | 16 | 4:1 |
| 7 | 10 mol% of Yb(OTf)3 instead of 20 mol% | 30 | 4:1 |
| 8 | Sc(OTf)3 instead of Yb(OTf)3 | 18 | 5:1 |
| 9 | Sn(OTf)2 instead of Yb(OTf)3 | 10 | 4:1 |
| 10 | DBU instead of Cs2CO3 | <5 | Nd |
| 11 | DABCO instead of Cs2CO3 | <5 | Nd |
| 12 | K2CO3 instead of Cs2CO3 | 24 | 4:1 |
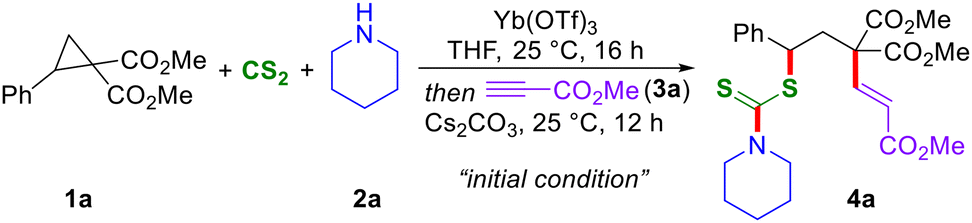
Consequently, entry 1 was selected as the optimized reaction condition and was subsequently employed for further analysis of substrate scope.12
With the optimized reaction conditions in hand, first, we evaluated the scope of D–A cyclopropanes (Scheme 2). A diverse range of structurally and electronically different D–A cyclopropanes bearing electron-rich, -neutral, and -poor functional groups at the 4-position of the arene ring on the donor end underwent a smooth reaction, and the corresponding products were formed in good to excellent yields with a good E/Z ratio (4a–4e). Furthermore, D–A cyclopropanes having substitution at the 3- and 2-positions on the benzene ring reacted effectively under the current optimized conditions, leading to the formation of the anticipated products in good yields (4f–4h). The presence of a di-substituted aryl ring and naphthyl-containing D–A cyclopropanes resulted in the formation of the desired 1,3-thioalkenylated products in moderate yields and with a moderate E/Z ratio (4i and 4j). Additionally, the reaction can be extended to incorporate heteroatoms and diverse ester moieties on D–A cyclopropanes, thereby expanding the scope of the reaction (4k and 4l).
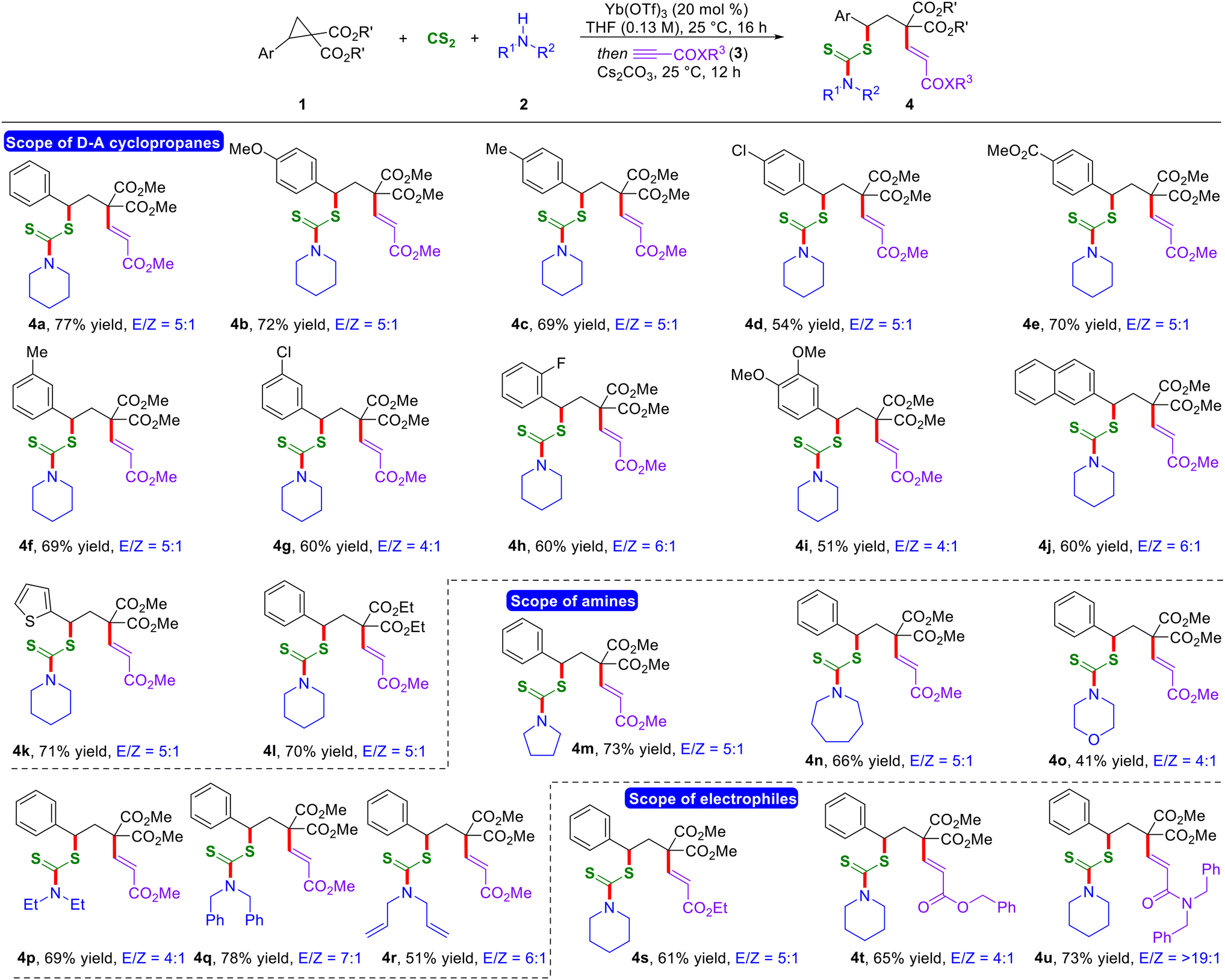 | ||
| Scheme 2 Substrate scope of the reaction. aReaction conditions: 1 (0.2 mmol, 1.0 equiv.), CS2 (0.4 mmol), 2 (0.4 mmol), Yb(OTf)3 (20 mol%), THF (1.5 mL), 25 °C for 16 h, and then Cs2CO3 (2.5 equiv.), alkyl propiolate 3 (2.0 equiv.), 12 h. Yields of the isolated products are given. The E/Z ratio was determined using 1H NMR of the crude reaction mixture. | ||
Next, we examined the variation on secondary amines. A variety of cyclic secondary aliphatic amines with different ring sizes displayed successful reactivity under the optimized one-pot reaction conditions, resulting in the formation of the corresponding 1,3-thioalkenylated products in good yields and with a moderate E/Z ratio (4m–4o). Moreover, various acyclic secondary aliphatic amines delivered the expected 1,3-bisfunctionalized products in good yields and with a moderate to good E/Z ratio (4p–4r). Notably, alkyl propiolates having different ester substitutions also worked well under the present reaction conditions, thus expanding the scope of the reaction further (4s and 4t). Interestingly, the utilization of N,N-dibenzyl propiolamide as the electrophilic fourth component resulted in the formation of the anticipated thioalkenylated product 4u in 73% yield with a >19:1 E/Z ratio.13
To gain insight into the mechanism of the present four-component reaction, few mechanistic experiments were performed. Initially, all the components were added together to check whether the desired 1,3-thioalkenylated product is formed or not. As expected, the four-component coupling product 4a was not formed. Instead, the in situ generated dithiocarbamate was directly added to the methyl propiolate to furnish product 5a in 46% yield (Scheme 3, eqn (4)). This observation underscored the significance of the one-pot protocol, wherein the expected thioalkenylated products were consistently obtained as the major products in all cases. Moreover, to elucidate the mode of addition of dithiocarbamates to D–A cyclopropanes, an experiment was performed using enantiomerically pure D–A cyclopropanes. When the bisfunctionalization reaction was conducted in the presence of (S)-1a, the 1,3-thioalkenylated product (R)-4a was isolated in 76% yield with 97% enantiomeric excess (eqn (5)). This study provides insights into the SN2-type addition of the in situ generated dithiocarbamates to D–A cyclopropanes.14,15
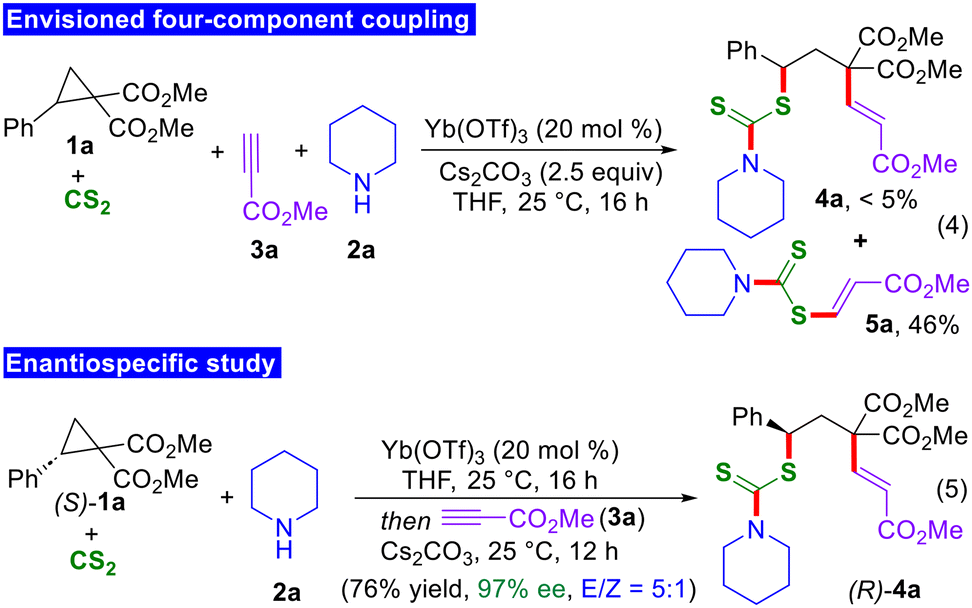 | ||
| Scheme 3 Mechanistic experiments. | ||
In order to illustrate the practical applicability of the current 1,3-bisfunctionalization reaction, the reaction was conducted on a 2.0 mmol scale. Gratifyingly, during the scale-up experiment, the anticipated product 4a was generated in a 79% yield and with a 5:1 E/Z ratio, without compromising reactivity and selectivity (Scheme 4). This underscores the practical and scalable nature of the present methodology. In addition, selective reduction of the α,β-unsaturated ester moiety of 4a was accomplished by treatment with LiAlH4 to furnish the allylic alcohol derivative 6 in 75% yield.
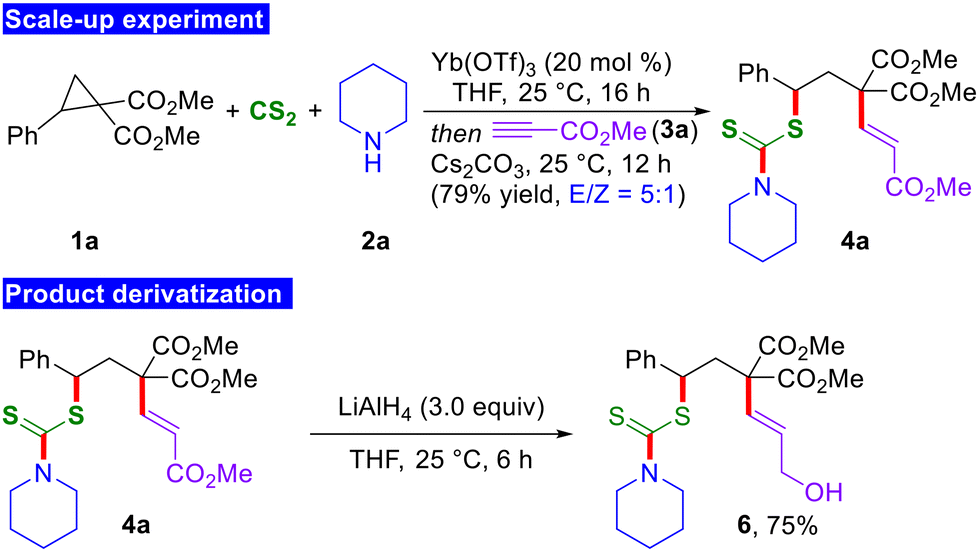 | ||
| Scheme 4 Scale-up experiment and product derivatization. | ||
Mechanistically, the reaction proceeds via the nucleophilic addition of amine 2 to CS2, resulting in the formation of a dithiocarbamate intermediate A (Scheme 5). This intermediate A is then added to the Lewis acid-activated D–A cyclopropane B, forming the ring-opened intermediate C by the SN2-type addition. Following this, intermediate C undergoes a proton-transfer step to form the monofunctionalized intermediate D. Subsequently, deprotonation of intermediate D with Cs2CO3 followed by alkenylation ultimately resulted in the formation of the thioalkenylated product 4. The observed diastereoselectivity can be rationalized based on the thermodynamic stability of the diastereomer, wherein the most stable diastereomer forms predominantly.
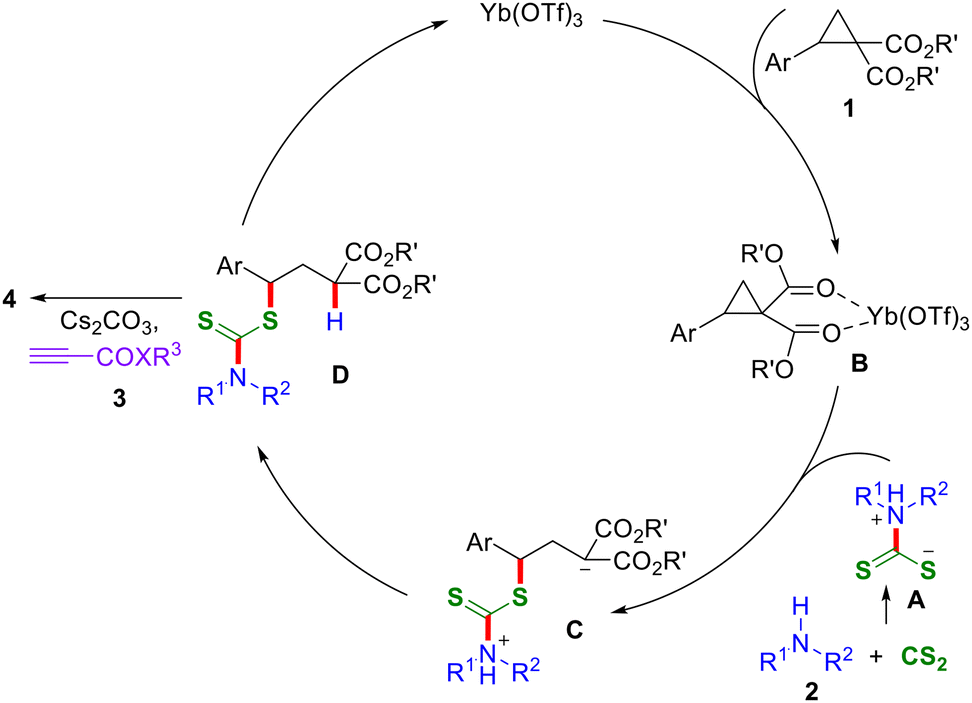 | ||
| Scheme 5 Tentative mechanism of the reaction. | ||
Conclusions
In conclusion, we have established a versatile and mild one-pot procedure for the 1,3-thioalkenylation of D–A cyclopropanes. The current reaction conditions exhibit compatibility with diverse functional groups, consistently yielding the anticipated products in moderate to good yields and with a good E/Z ratio in all instances. Notably, enantiopure D–A cyclopropanes produce the corresponding enantiopure products, showcasing the SN2 type addition of dithiocarbamates to D–A cyclopropanes. Related multicomponent coupling reactions involving D–A cyclopropanes are being explored in the ongoing research in our laboratory.Data availability
Details on the experimental procedures and the characterization data of all 1,3-thioalkenylation products.Author contributions
A. G. and A. T. B. conceived the idea and designed the experiments. A. G. performed the optimization studies. S. H. and L. T. K. K. performed the substrate scope analysis and mechanistic studies. A. G. and A. T. B. wrote the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Science and Engineering Research Board (SERB), Government of India (File Number: SCP/2022/000837) is greatly acknowledged. A. G. thanks MHRD (for PMRF). We thank Shiksha Deswal (IISc-OC) for helpful discussions.References
- (a) H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151 CrossRef CAS PubMed; (b) Y. Xia, X. Liu and X. Feng, Angew. Chem., Int. Ed., 2021, 60, 9192 CrossRef CAS PubMed; (c) V. Pirenne, B. Muriel and J. Waser, Chem. Rev., 2021, 121, 227 CrossRef CAS PubMed; (d) A. U. Augustin and D. B. Werz, Acc. Chem. Res., 2021, 54, 1528 CrossRef CAS PubMed; (e) T. F. Schneider, J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504 CrossRef CAS PubMed; (f) D. B. Werz and A. T. Biju, Angew. Chem., Int. Ed., 2020, 59, 3385 CrossRef CAS PubMed; (g) P. Singh, R. K. Varshnaya, R. Dey and P. Banerjee, Adv. Synth. Catal., 2020, 362, 1447 CrossRef CAS.
- (a) M. S. Gordon, J. Am. Chem. Soc., 1980, 102, 7419 CrossRef CAS; (b) R. D. Bach and O. Dmitrenko, J. Am. Chem. Soc., 2004, 126, 4444 CrossRef CAS PubMed.
- (a) P. D. Pohlhaus, S. D. Sanders, A. T. Parsons, W. Li and J. S. Johnson, J. Am. Chem. Soc., 2008, 130, 8642 CrossRef CAS PubMed; (b) A. Ortega, R. Manzano, U. Uria, L. Carrillo, E. Reyes, T. Tejero, P. Merino and J. L. Vicario, Angew. Chem., Int. Ed., 2018, 57, 8225 CrossRef CAS PubMed; (c) M. Petzold, P. G. Jones and D. B. Werz, Angew. Chem., Int. Ed., 2019, 58, 6225 CrossRef CAS; (d) F. de Nanteuil and J. Waser, Angew. Chem., Int. Ed., 2011, 50, 12075 CrossRef CAS PubMed; (e) K. Verma, I. M. Taily and P. Banerjee, Org. Biomol. Chem., 2019, 17, 8149 RSC; (f) N. L. Ahlburg, P. G. Jones and D. B. Werz, Org. Lett., 2020, 22, 6404 CrossRef CAS PubMed; (g) M. Faltracco, K. N. A. van de Vrande, M. Dijkstra, J. M. Saya, T. A. Hamlin and E. Ruijter, Angew. Chem., Int. Ed., 2021, 60, 14410 CrossRef CAS PubMed; (h) A. E. Vartanova, A. Y. Plodukhin, N. K. Ratmanova, I. A. Andreev, M. N. Anisimov, N. B. Gudimchuk, V. B. Rybakov, I. I. Levina, O. A. Ivanova, I. V. Trushkov and I. V. Alabugin, J. Am. Chem. Soc., 2021, 143, 13952 CrossRef CAS PubMed; (i) G. A. Oliver, M. N. Loch, A. U. Augustin, P. Steinbach, M. Sharique, U. K. Tambar, P. G. Jones, C. Bannwarth and D. B. Werz, Angew. Chem., Int. Ed., 2021, 60, 25825 CrossRef CAS PubMed; (j) S. Kolb, M. Petzold, F. Brandt, P. G. Jones, C. R. Jacob and D. B. Werz, Angew. Chem., Int. Ed., 2021, 60, 15928 CrossRef CAS PubMed; (k) S. S. Hosseini, A. Abdi, A. Nikbakht, H. R. Bijanzadeh, F. Rominger, D. B. Werz and S. Balalaie, Synlett, 2022, 468 CAS; (l) V. Pirenne, E. G. L. Robert and J. Waser, Chem. Sci., 2021, 12, 8706 RSC; (m) S. Nicolai and J. Waser, Angew. Chem., Int. Ed., 2022, 61, e202209006 CrossRef CAS.
- (a) C. L. Morales and B. L. Pagenkopf, Org. Lett., 2008, 10, 157 CrossRef CAS PubMed; (b) C. A. Carson and M. A. Kerr, Chem. Soc. Rev., 2009, 38, 3051 RSC; (c) A. F. G. Goldberg and B. M. Stoltz, Org. Lett., 2011, 13, 4474 CrossRef CAS PubMed; (d) A. F. G. Goldberg, R. A. Craig II, N. R. O'Connor and B. M. Stoltz, Tetrahedron Lett., 2015, 56, 2983 CrossRef CAS PubMed; (e) N. R. O'Connor, J. L. Wood and B. M. Stoltz, Isr. J. Chem., 2016, 56, 431 CrossRef; (f) Y. Kimura, Y. Sone, T. Saito, T. Mochizuki and Y. Nishii, Asian J. Org. Chem., 2017, 6, 977 CrossRef CAS.
- (a) S. Das, C. G. Daniliuc and A. Studer, Org. Lett., 2016, 18, 5576 CrossRef CAS PubMed; (b) D. Saha, I. M. Taily and P. Banerjee, Eur. J. Org. Chem., 2021, 5053 CrossRef CAS; (c) Z. Zuo and A. Studer, Org. Lett., 2022, 24, 949 CrossRef CAS PubMed; (d) C. Sparr and R. Gilmour, Angew. Chem., Int. Ed., 2011, 50, 8391 CrossRef CAS PubMed.
- (a) A. Guin, T. Rathod, R. N. Gaykar, T. Roy and A. T. Biju, Org. Lett., 2020, 22, 2276 CrossRef CAS; (b) J. Wallbaum, L. K. B. Garve, P. G. Jones and D. B. Werz, Org. Lett., 2017, 19, 98 CrossRef CAS PubMed.
- (a) S. Das, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2017, 56, 11554 CrossRef CAS; (b) A. U. Augustin, P. G. Jones and D. B. Werz, Chem. – Eur. J., 2019, 25, 11620 CrossRef CAS PubMed; (c) B. Mondal, D. Das and J. Saha, Org. Lett., 2020, 22, 5115 CrossRef CAS PubMed; (d) S. Das, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 4053 CrossRef CAS PubMed; (e) S. Deswal, A. Guin and A. T. Biju, Org. Lett., 2023, 25, 1643 CrossRef CAS PubMed.
- A. Guin, S. Deswal and A. T. Biju, J. Org. Chem., 2022, 87, 6504 CrossRef CAS PubMed.
- G. A. Oliver and D. B. Werz, Org. Lett., 2023, 25, 3568 CrossRef CAS PubMed.
- H. F. Von Köller, P. G. Jones and D. B. Werz, Chem. – Eur. J., 2023, 29, e202203986 CrossRef PubMed.
- (a) K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2018, 376, 5 CrossRef PubMed; (b) P. Devendar and G.-F. Yang, Top. Curr. Chem., 2017, 375, 8 CrossRef PubMed.
- See the ESI† for details.
- Disappointingly, the use of activated internal alkynes such as dimethyl acetylene dicarboxylate (DMAD) as the alkyne component did not afford the desired four-component product under the present conditions.
- For related SN2-like ring opening of D–A cyclopropanes, see: (a) A. Jacob, P. G. Jones and D. B. Werz, Org. Lett., 2020, 22, 8720 CrossRef CAS PubMed; (b) L. K. B. Garve, P. G. Jones and D. B. Werz, Angew. Chem., Int. Ed., 2017, 56, 9226 CrossRef CAS PubMed.
- For related enantiospecific ring opening of D–A cyclopropanes, see: P. D. Pohlhaus and J. S. Johnson, J. Am. Chem. Soc., 2005, 127, 16014 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob00053f |
| This journal is © The Royal Society of Chemistry 2024 |