
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the mechanism of carboxylate elimination from carbohydrate monoester-derived radicals†
Julia A.
Turner
a,
Hendrik
Zipse
 *b and
Mark S.
Taylor
*a
*b and
Mark S.
Taylor
*a
aDepartment of Chemistry, University of Toronto, 80 St. George St, Toronto, ON M5S 3H6, Canada. E-mail: mtaylor@chem.utoronto.ca
bDepartment Chemie, Ludwig-Maximilians-Universität München, Butenandtstrasse 5-13, Haus F, 81377 München, Germany. E-mail: zipse@cup.uni-muenchen.de
First published on 10th April 2024
Abstract
A computational study of the mechanism of hydrogen atom transfer-induced carboxylate elimination from monoacylated 1,2-diol groups in pyranosides is presented. A comprehensive analysis of the 1,2-migration, elimination and fragmentation pathways reveals that concerted elimination via a 7-membered, hydrogen-bonded transition state is favored. Relative rates of elimination inferred from an intramolecular competition experiment are consistent with the trends obtained from the calculations.
Introduction
Fragmentations and rearrangements of β-acyloxy and β-phosphatoxy radicals have been implicated in the chemistry of nucleic acids and lipids, and form the basis of useful preparative methods in organic chemistry.1,2 The impact of such reactions in the domain of carbohydrate synthesis, initiated by Giese's development of a method for the synthesis of 2-deoxyglycosides,3 has been significant; new developments continue to emerge, including the use of transition metal (photo)catalysis to promote acyloxy migrations to the anomeric position.4 The processes have attracted sustained interest from the mechanistic perspective, as they traverse pathways at the boundary of radical and polar reactivity; studies along this line include experimental determinations of rate constants, solvent effects and isotope labelling5,6 along with computational modelling of proposed reaction pathways.7,8Recently, our group took advantage of the reactivity of β-acyloxy radicals along with photocatalytic hydrogen atom transfer (HAT) to develop a method for the redox isomerization9 of pyranosides to ketodeoxysugars (Scheme 1a).10 The yield was dependent on the identity of the leaving group, and in the case of the optimal pyranoside monoester substrates, deoxygenation took place exclusively at the site of O-acylation. This second observation, which implied an acceleration of C–O bond cleavage due to the presence of the ester substituent, was in keeping with prior work from Lenz and Giese, who found that 2-O-acylation facilitated heterolytic β-C–O bond cleavage from a C-3 adenosyl radical.11 Considering the utility of ketodeoxysugars as synthetic building blocks, we sought to gain insight into the mechanism of HAT-induced elimination of carboxylate and sulfonate from pyranosides, focusing on the effects of the leaving group on the rate of C–O bond cleavage. Herein, we describe a computational study that points towards a concerted elimination via a hydrogen-bonded transition state as the lowest-energy pathway for C–O bond cleavage. Calculated effects of leaving group basicity on the rate of elimination are consistent with experimentally determined product ratios for redox isomerizations of unsymmetrical 1,3-bis-O-functionalized glycerol model systems.
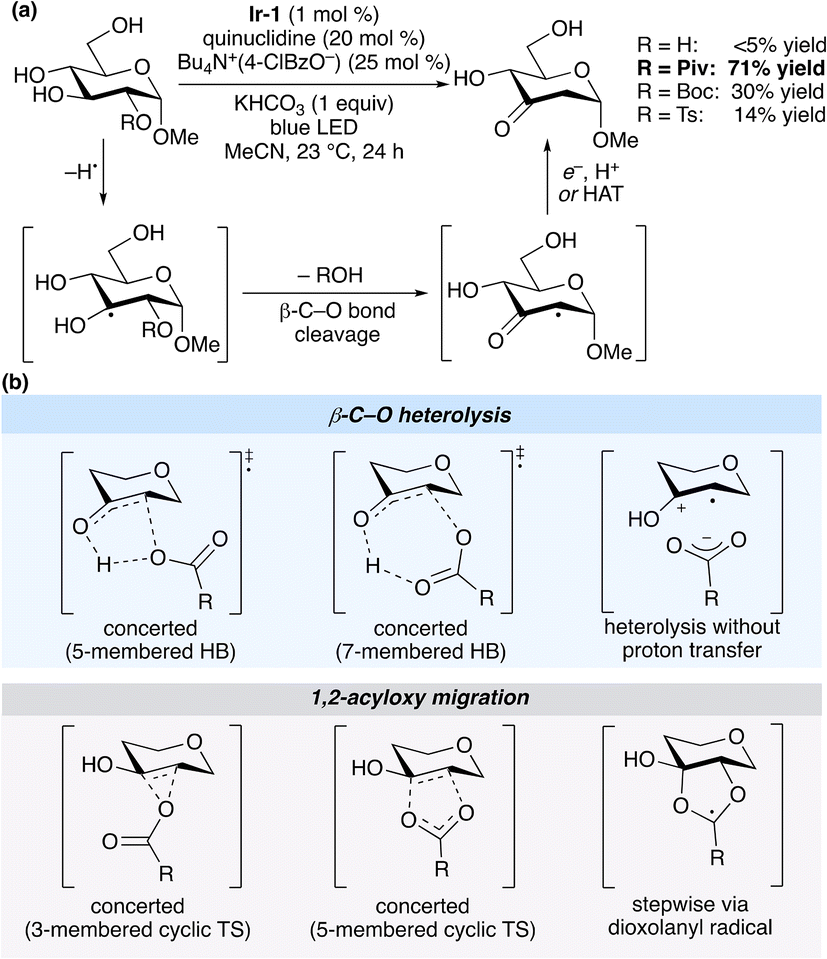 | ||
| Scheme 1 (a) Redox isomerizations of glucopyranosides illustrating the effect of the substituent at the 2-position. (b) Conceivable pathways for β-C–O bond cleavage from a 2-O-acylated radical intermediate. Ir-1 denotes [Ir(dF(CF3)py)2(dtbpy)]PF6. | ||
Results and discussion
Conceivable pathways for β-C–O bond cleavage in a 2-O-acylated, pyranoside-derived radical are summarized in Scheme 1b. Concerted proton transfer and β-C–O bond heterolysis can take place through five- or seven-membered cyclic transition states; alternatively, heterolysis without proton transfer leads to a radical cation–carboxylate ion pair. 1,2-Acyloxy migration (Surzur–Tanner-type rearrangement) could take place in a concerted fashion via three- and five-membered cyclic transition states, or through a stepwise mechanism involving a dioxolanyl radical intermediate. A subsequent elimination of carboxylic acid from the resulting monoacylated hemiketal-derived radical yields the stabilized α-carbonyl radical.A preliminary computational investigation of the redox isomerization of an α-glucopyranoside monoester identified intermediates and transition states for the stepwise, 1,2-acyloxy migration pathway.10 Earlier work by Giese and co-workers, supported by calculations at the PMP2/6-31G(d)//UHF/3-21G(d) level of theory, pointed towards the seven-membered cyclic transition state for concerted proton transfer/β-C–O bond heterolysis from 1,3-diacylglycerol-derived radicals.12 The authors noted that the β-elimination was accelerated by the presence of a free OH group at the α-position, and was suppressed for such substrates in protic solvent, consistent with the proposed stabilization of the transition state by intramolecular hydrogen bonding. A similar transition state has been proposed for the elimination of toluenesulfonic acid from a C-3′ radical of 2′-tosyl-homouridine.13 We aimed to use computational modeling to evaluate each of the pathways depicted in Scheme 1b for a pyranoside model system, focusing on the effects of the leaving group on the rate of ketodeoxysugar formation.
Scheme 2 depicts the calculated Gibbs free energies of intermediates and transition states for the transformation of monoacylated tetrahydropyran-3,4-diol-derived radical Ac-I to tetrahydropyranone-derived radical V and AcOH (Ac-VI). Elimination and fragmentation pathways are depicted in blue, while those involving 1,2-acyloxy migration are shown in black. Transition state structures are depicted in Fig. 1, annotated with the C–O bond distances and the sum of charges for atoms in the carboxylate group calculated from natural population analysis (NPA) (q(CO2CH3), in red). Gas phase geometry optimizations were conducted at the (U)M06-2X/def2-TZVP level of theory, followed by single-point energy calculations using the SMD model for acetonitrile.14 These calculations are consistent (R2 = 0.99) with energies calculated by solvent phase re-optimization of intermediates and transition states at the SMD(AcCN)/(U)M06-2X/def2-TZVP level of theory (Fig. S02†). Further benchmarking of (U)M06-2X/def2-TZVP//SMD(acetonitrile) results against DLPNO-CCSD(T)/CBS or G3B3 levels of theory indicate a high level of agreement (R2 of 0.99) providing justification for application of the former level of theory in the present study (see ESI† for details).
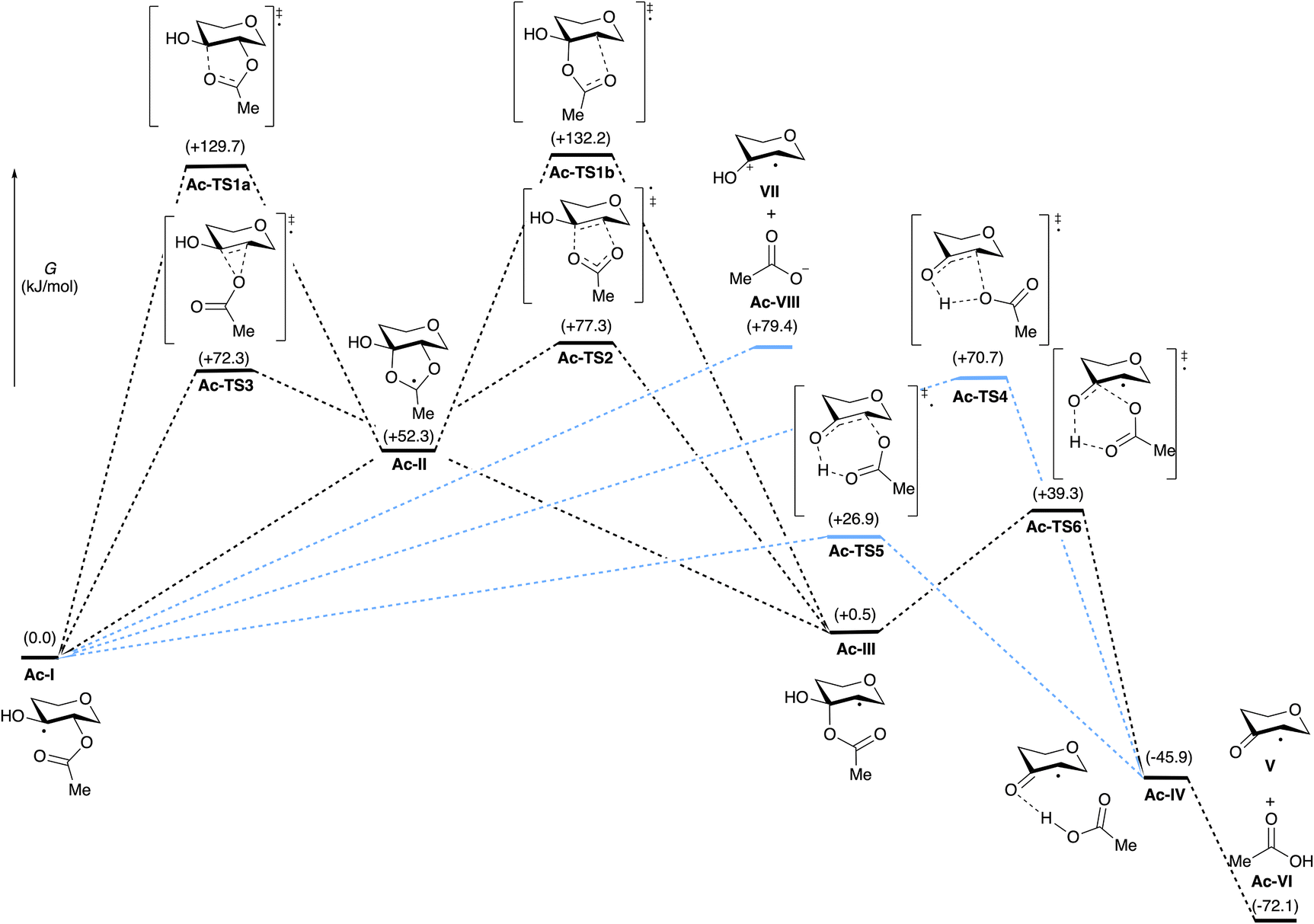 | ||
| Scheme 2 Calculated Gibbs free energies (kJ mol−1) of proposed intermediates and transition states for the rearrangement (black) and heterolysis (blue) pathways leading to the formation of V and Ac-VI from Ac-I ((U)M06-2X/def2-TZVP//SMD(acetonitrile) level of theory). | ||
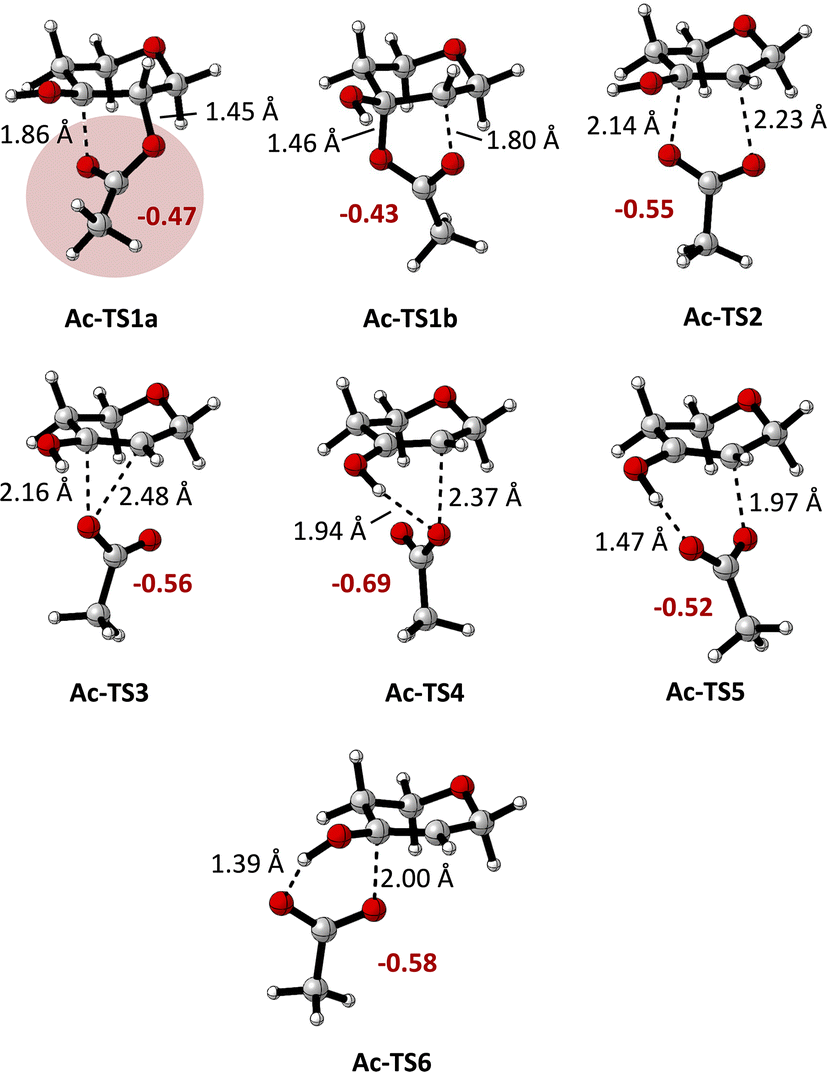 | ||
| Fig. 1 Calculated transition state structures for the rearrangement of Ac-I to Ac-IV ((U)M06-2X/def2-TZVP level of theory), as defined in Scheme 2. Carboxylate group charges q(CO2CH3) are shown in red (NPA/(U)M06-2X/def2-TZVP results). | ||
Acyloxy group migration (Ac-I → Ac-III) is without notable driving force (ΔG298 = +0.5 kJ mol−1). The most facile pathway for the rearrangement involves 3-membered ring transition state Ac-TS3 with a barrier ΔG‡298 of +72.3 kJ mol−1. Transition state Ac-TS1a leading to ring-closure/ring-opening is significantly higher in free energy (ΔG‡298 = +129.7 kJ mol−1). Direct elimination of AcOH through seven-membered transition state Ac-TS5 is the preferred pathway (ΔG‡298 = +26.9 kJ mol−1). The tetrahydropyran ring adopts a boatlike conformation to facilitate alignment of the SOMO with 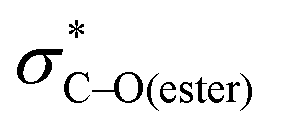 in TS5. Both the five-membered ring transition state Ac-TS4 (ΔG‡298 = +70.7 kJ mol−1) and the ion pair arising from β-C–O bond heterolysis (radical cation VII and acetate Ac-VIII, ΔG298 = +79.4 k mol−1) are higher in energy than Ac-TS5. Given the low barrier for concerted radical mediated elimination viaAc-TS5, the chemistry of radical Ac-I will be dominated by AcOH elimination and rapid formation of radical V unless an extraordinarily fast bimolecular process traps radical Ac-I (e.g., by HAT).
in TS5. Both the five-membered ring transition state Ac-TS4 (ΔG‡298 = +70.7 kJ mol−1) and the ion pair arising from β-C–O bond heterolysis (radical cation VII and acetate Ac-VIII, ΔG298 = +79.4 k mol−1) are higher in energy than Ac-TS5. Given the low barrier for concerted radical mediated elimination viaAc-TS5, the chemistry of radical Ac-I will be dominated by AcOH elimination and rapid formation of radical V unless an extraordinarily fast bimolecular process traps radical Ac-I (e.g., by HAT).
Variation of the acyl group substituent, and replacement of acyl with sulfonyl,15 allow for evaluation of steric and electronic effects on the free energy barriers for the transformation (Table 1). Substitution of the acetyl group for pivaloyl (Piv-I) has minimal effects on the calculated energies for the rearrangement and elimination pathways, indicating that steric factors do not have a major influence. In contrast, pronouned electronic effects are evident from the calculations; systematically decreasing the Brønsted basicity of the leaving group (Bz-I → ClAc-I → Cl3Ac-I → F3Ac-I → Ts-I) results in a decrease in the barriers for all pathways, with concerted elimination viaTS5 remaining the dominant one in each case. This finding is consistent with previous work indicating that rearrangement and migration reactions are accelerated by electron-withdrawing groups on the acyloxyl and phosphatoxy substituent,1a,b,16 and with the calculated negative charge buildup at the acetate group in the transition states (Fig. 1). Stepwise migration viaTS1a and TS1b is the least sensitive to electronic effects on rate, consistent with the lower degree of charge buildup at the carboxyl oxygens. For the best leaving groups, (F3Ac-I and Ts-I), fragmentation to the radical cation VII and carboxylate/sulfonate VIII is exergonic. A linear relationship between the free energy of activation viaTS5 and the free energy of heterolytic dissociation ΔG(VII+VII) is observed (Fig. 2), consistent with charge separation in the transition state for elimination (TS5). The calculated pKa of the leaving group in acetonitrile also relates linearly to the free energy of activation viaTS5 (see the ESI†). Overall, the results indicate that increasing leaving group ability of the β-substituent accelerates elimination from radicals I, with barriers of less than 10 kJ mol−1 being calculated for expulsion of Cl3AcOH, F3AcOH and TsOH.
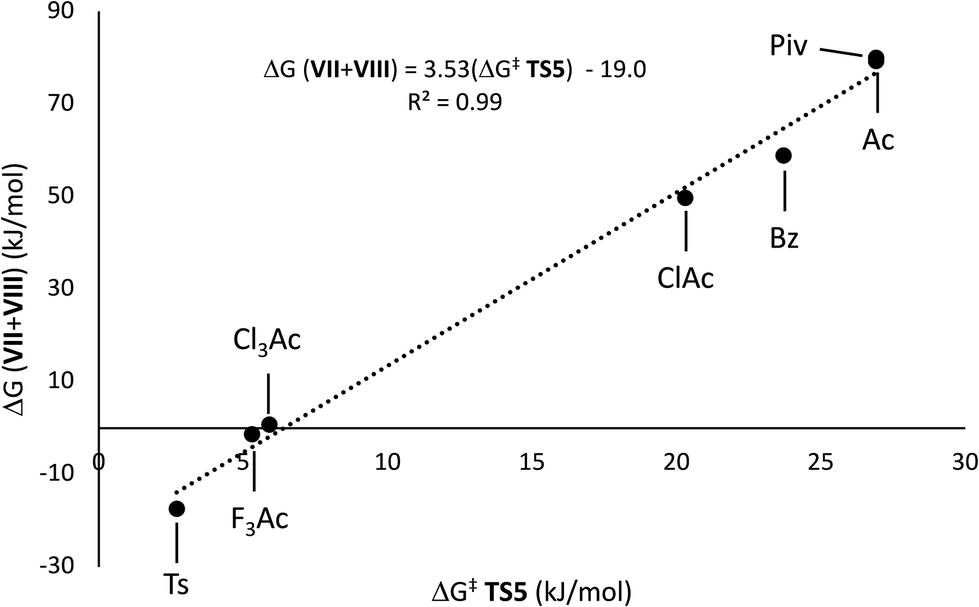 | ||
| Fig. 2 Free energy of activation (ΔG‡, kJ mol−1) for TS5vs. reaction energy for heterolytic dissociation (ΔG(VII + VIII), kJ mol−1) for carboxylate and sulfonate groups ((U)M06-2X/def2-TZVP//SMD(acetonitrile) level of theory). | ||
| Species | Substituent | ||||||
|---|---|---|---|---|---|---|---|
| Ac | Piv | Bz | ClAc | Cl3Ac | F3Ac | Ts | |
| I | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| TS1a | +129.7 | +127.8 | +90.9 | +105.6 | +82.7 | +87.4 | — |
| II | +52.3 | +46.5 | +8.0 | +20.2 | +7.2 | +12.9 | — |
| TS1b | +132.2 | +131.1 | +102.3 | +114.7 | +100.8 | +104.7 | — |
| III | +0.5 | –1.1 | –0.9 | –1.9 | –7.2 | –5.8 | +4.5 |
| TS2 | +77.3 | +76.5 | +68.9 | +59.6 | +25.1 | +24.6 | +16.5 |
| TS3 | +72.3 | +71.7 | +68.5 | +56.8 | +24.6 | +24.4 | +21.2 |
| TS4 | +70.7 | +71.1 | +63.2 | +54.7 | +23.6 | +23.2 | — |
| TS5 | +26.9 | +26.9 | +23.7 | +20.3 | +5.9 | +5.3 | +2.7 |
| TS6 | +39.3 | +41.0 | +34.8 | +32.6 | +13.2 | +12.6 | +12.5 |
| IV | –45.9 | –44.8 | –51.0 | –50.8 | –62.0 | –62.7 | –56.1 |
| V + VI | –72.1 | –73.1 | –75.2 | –74.3 | –75.9 | –75.3 | –64.5 |
| VII + VIII | +79.4 | +80.0 | +59.0 | +49.8 | +0.8 | –1.3 | –14.4 |
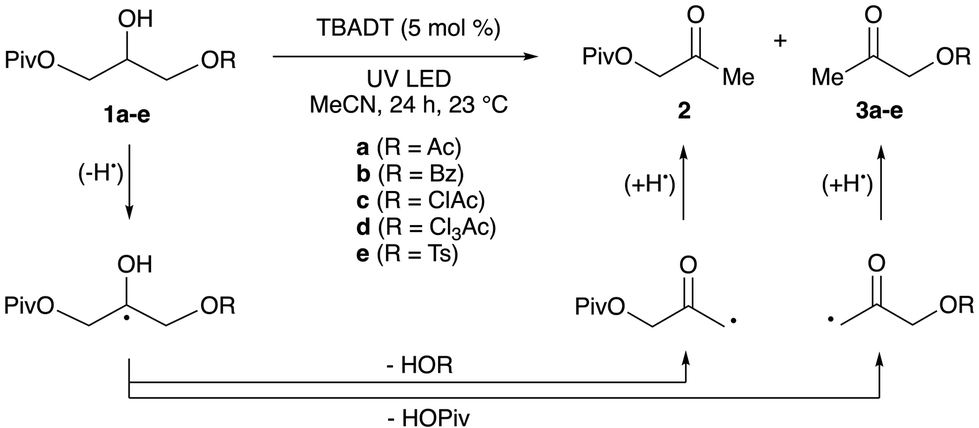
To obtain experimental data on relative rates of elimination from β-substituted α-hydroxyalkyl radicals, we investigated photocatalytic redox isomerizations of unsymmetrical, disubstituted glycerol derivatives 1a–1e (Table 2). This intramolecular competition experiment allows for ratios of elimination rates to be determined from product distributions. Giese and co-workers used a related experiment to determine that a 2-glyceryl radical bearing diethylphosphatoxy and acetoxy groups undergoes selective elimination of (EtO)2PO2H.12 Irradiation of acetonitrile solutions of 1a–1e with a 370 nm light-emitting diode (LED) in the presence of catalytic tetra-n-butylammonium decatungstate (TBADT) resulted in hydroxyacetone derivatives 2 and 3a–3e. (Photocatalytic HAT with quinuclidine (see Scheme 1a) was inefficient for reactions of 1a–1e.) A 1.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of products 2 and 3a was obtained from the reaction of diester 1a, consistent with the calculations that showed nearly equivalent free energies of activation (Ac-TS5 and Piv-TS5, ΔΔG‡298 = 0.0 kcal mol−1). (A perfect correlation between calculated free energies of activation and log(krel) was not expected, because the tetrahydropyran model system was used for the calculations.) BzOH, ClAcOH and Cl3AcOH underwent elimination in preference to PivOH, with the magnitude of the effect increasing in the order 1b < 1c < 1d as anticipated based on the computational results. Likewise, TsOH underwent elimination significantly faster than PivOH (2:3e > 20:1). Conversions were low (∼5%) for the reactions of benzoate 1b and tosylate 1e, perhaps due to inhibition of the photocatalyst by the reaction byproducts; nonetheless, the relative concentrations of products 2 and 3 could be assessed reliably by 1H NMR spectroscopy. For substrate 1d, competitive migration of the Cl3Ac group to the 2-position was observed. The results indicate that considerable variation of the rate of HAT-induced elimination from diol derivatives can be achieved by changing the acyl substituent. The ClAc substituent appears to offer a useful balance of reactivity towards elimination and stability towards intramolecular acyl transfer.
:1 ratio of products 2 and 3a was obtained from the reaction of diester 1a, consistent with the calculations that showed nearly equivalent free energies of activation (Ac-TS5 and Piv-TS5, ΔΔG‡298 = 0.0 kcal mol−1). (A perfect correlation between calculated free energies of activation and log(krel) was not expected, because the tetrahydropyran model system was used for the calculations.) BzOH, ClAcOH and Cl3AcOH underwent elimination in preference to PivOH, with the magnitude of the effect increasing in the order 1b < 1c < 1d as anticipated based on the computational results. Likewise, TsOH underwent elimination significantly faster than PivOH (2:3e > 20:1). Conversions were low (∼5%) for the reactions of benzoate 1b and tosylate 1e, perhaps due to inhibition of the photocatalyst by the reaction byproducts; nonetheless, the relative concentrations of products 2 and 3 could be assessed reliably by 1H NMR spectroscopy. For substrate 1d, competitive migration of the Cl3Ac group to the 2-position was observed. The results indicate that considerable variation of the rate of HAT-induced elimination from diol derivatives can be achieved by changing the acyl substituent. The ClAc substituent appears to offer a useful balance of reactivity towards elimination and stability towards intramolecular acyl transfer.
|
|
||
|---|---|---|
| Entry | Substrate | Product ratio (2:3a–e)b |
| a Substrate (0.1 mmol), tetra-n-butylammonium decatungstate (TBADT, 5 mol%), acetonitrile ([substrate]0 = 0.125 M), 32 W UV LED, rt, 24 h. b Ratios determined by 1H NMR spectroscopic analysis of unpurified reaction mixtures. | ||
| 1 | 1a | 1.1:1 |
| 2 | 1b | 1.7:1 |
| 3 | 1c | 5.4:1 |
| 4 | 1d | >20:1 |
| 5 | 1e | >20:1 |
Conclusions
In summary, a computational investigation of the mechanism of expulsion of carboxylic acids from α-hydroxy-β-acyloxy radicals in a pyranoside model system has been conducted. Concerted elimination through a seven-membered, intramolecular hydrogen bonded transition state is the lowest-energy pathway, consistent with earlier findings from Giese's group. Due to charge separation in the transition state, the introduction of electron-withdrawing carboxyl substituents, or the replacement of carboxylate for sulfonate, has an accelerating effect. This trend is apparent from the results of intramolecular competition experiments using unsymmetrical glycerol derivatives. To apply these findings to redox isomerizations of carbohydrate derivatives, the effect of the acyl substituent on the rate and selectivity of HAT, as well as its ease of installation and stability towards migration must be taken into account; efforts along this line are underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NSERC, the Canada Foundation for Innovation and the Province of Ontario. The authors thank the Leibniz-Rechenzentrum (LRZ) for generous allocation of computational resources.References
- (a) A. L. J. Beckwith, D. Crich, P. J. Duggan and Q. Yao, Chem. Rev., 1997, 97, 3273–3312 CrossRef CAS PubMed; (b) D. Crich, F. Brebion and D.-H. Suk, Top. Curr. Chem., 2006, 263, 1–38 CAS; (c) B. Matsuo, A. Granados, J. Majhi, M. Sharique, G. Levitre and G. A. Molander, ACS Org. Inorg. Au, 2022, 2, 435–454 CrossRef CAS PubMed.
- (a) B. Giese, X. Beyrich-Graf, P. Erdmann, M. Petretta and U. Schwitter, Chem. Biol., 1995, 2, 367–375 CrossRef CAS PubMed; (b) J. Stubbe and W. A. van der Donk, Chem. Biol., 1995, 2, 793–801 CrossRef CAS PubMed; (c) J. C. Walton, Chem. Soc. Rev., 2021, 50, 7496–7512 RSC.
- B. Giese, K. S. Gröninger, T. Witzel, H.-G. Korth and R. Sustmann, Angew. Chem., Int. Ed. Engl., 1987, 26, 233–234 CrossRef.
- (a) G. Zhao, W. Yao, M. N. Mauro and M.-Y. Ngai, J. Am. Chem. Soc., 2021, 143, 1728–1734 CrossRef CAS PubMed; (b) G. Zhao, W. Yao, I. Kevlishvili, J. N. Mauro, P. Liu and M.-Y. Ngai, J. Am. Chem. Soc., 2021, 143, 8590–8596 CrossRef CAS PubMed.
- (a) G. Koltzenburg, G. Behrens and D. Schulte-Frohlinde, J. Am. Chem. Soc., 1982, 104, 7311–7312 CrossRef CAS; (b) M. Newcomb, J. H. Horner, P. O. Whitted, D. Crich, X. Huang, Q. Yao and H. Zipse, J. Am. Chem. Soc., 1999, 121, 10685–10694 CrossRef CAS; (c) M. Newcomb, N. Miranda, X. Huang and D. Crich, J. Am. Chem. Soc., 2000, 122, 6128–6129 CrossRef CAS; (d) J. H. Horner, L. Bagnol and M. Newcomb, J. Am. Chem. Soc., 2004, 126, 14979–14987 CrossRef CAS PubMed.
- (a) L. R. C. Barclay, J. Lusztyk and K. U. Ingold, J. Am. Chem. Soc., 1984, 106, 1793–1796 CrossRef CAS; (b) A. L. J. Beckwith and P. J. Duggan, J. Chem. Soc., Chem. Commun., 1988, 1000–1002 RSC; (c) H.-G. Korth, R. Sustmann, K. S. Gröninger, M. Leisung and B. Giese, J. Org. Chem., 1988, 53, 4364–4369 CrossRef CAS; (d) A. L. J. Beckwith and P. J. Duggan, J. Chem. Soc., Perkin Trans. 2, 1992, 1777–1783 RSC; (e) D. Crich, Q. Yao and G. F. Filzen, J. Am. Chem. Soc., 1995, 117, 11455–11470 CrossRef CAS; (f) D. Crich and G. F. Filzen, J. Org. Chem., 1995, 60, 4834–4837 CrossRef CAS; (g) M. E. Jung and Y. Xu, Org. Lett., 1999, 1, 1517–1519 CrossRef CAS PubMed; (h) S.-Y. Choi, D. Crich, J. H. Horner, X. Huang, M. Newcomb and P. O. Whitted, Tetrahedron, 1999, 55, 3317–3326 CrossRef CAS; (i) D. Crich and D.-H. Suk, Can. J. Chem., 2004, 82, 75–79 CrossRef CAS; (j) C. H. Witt and K. A. Woerpel, J. Org. Chem., 2023, 88, 12802–12807 CrossRef CAS PubMed.
- (a) S. Saebo, A. L. J. Beckwith and L. Radom, J. Am. Chem. Soc., 1984, 106(18), 5119–5122 CrossRef CAS; (b) H. Zipse, J. Am. Chem. Soc., 1997, 119, 1087–1093 CrossRef CAS; (c) H. Zipse and M. Bootz, J. Chem. Soc., Perkin Trans. 2, 2001, 1566–1572 RSC.
- H. Zipse, Adv. Phys. Org. Chem., 2003, 38, 111–130 CrossRef CAS.
- (a) V. Dimakos, D. Gorelik, H. Y. Su, G. E. Garrett, G. Hughes, H. Shibayama and M. S. Taylor, Chem. Sci., 2020, 11, 1531–1537 RSC; (b) Y. Masuda, H. Tsuda and M. Murakami, Angew. Chem., Int. Ed., 2020, 59, 2755–2759 CrossRef CAS PubMed; (c) H. M. Carder, C. E. Suh and A. E. Wendlandt, J. Am. Chem. Soc., 2021, 143, 13798–13805 CrossRef CAS PubMed.
- J. A. Turner, N. Rosano, D. J. Gorelik and M. S. Taylor, ACS Catal., 2021, 11, 11171–11179 CrossRef CAS.
- R. Lenz and B. Giese, J. Am. Chem. Soc., 1997, 119, 2784–2794 CrossRef CAS.
- S. N. Müller, R. Batra, M. Senn, B. Giese, M. Kisel and O. Shadyro, J. Am. Chem. Soc., 1997, 119, 2795–2803 CrossRef.
- (a) M. J. Robins, Z. Guo and S. F. Wnuk, J. Am. Chem. Soc., 1997, 119, 3637–3638 CrossRef CAS; (b) M. J. Robins, Z. Guo, M. C. Samano and S. F. Wnuk, J. Am. Chem. Soc., 1998, 121, 1425–1433 CrossRef.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed; (b) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC; (c) M. J. Frisch, et al., Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- (a) E. Taxil, L. Bagnol, J. H. Horner and M. Newcomb, Org. Lett., 2003, 5, 827–830 CrossRef CAS PubMed; (b) S. F. Lancelot, F. L. Cozens and N. P. Schepp, Org. Biomol. Chem., 2003, 1, 1972–1979 RSC.
- B. Giese, X. Beyrich-Graf, J. Burger, C. Kesselheim, M. Senn and T. Schäfer, Angew. Chem., Int. Ed. Engl., 1993, 32, 1742–1744 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Full computational details and results of benchmarking calculations. See DOI: https://doi.org/10.1039/d4ob00241e |
| This journal is © The Royal Society of Chemistry 2024 |