
Photocatalytic C2-trifluoroethylation and perfluoroalkylation of 3-substituted indoles using fluoroalkyl halides†
Shivani
Arora
a,
Tavinder
Singh
a and
Anand
Singh
 *abc
*abc
aDepartment of Chemistry, Indian Institute of Technology Kanpur, UP-208016, India. E-mail: anands@iitk.ac.in
bDepartment of Sustainable Energy Engineering, Kotak School of Sustainability, Indian Institute of Technology Kanpur, UP-208016, India
cChandrakanta Kesavan Center for Energy Policy and Climate Solutions, Indian Institute of Technology Kanpur, UP-208016, India
First published on 1st May 2024
Abstract
A photocatalytic reactivity platform for the C2-trifluoroethylation and perfluoroalkylation of 3-substituted indoles has been developed. A range of fluoroalkyl halides have been employed as radical precursors under mild, transition-metal-free conditions to access new (per)fluorinated chemical space featuring the indole substructure. This general protocol is also applicable to indole-containing peptides.
The chemical space populated by indole-derivatives is of special significance because this substructure is a constituent of various drug molecules such as acemetacin, panobinostat and oxypertine,1 and other compounds of biological importance (Fig. 1). The success of fluorinated molecules as drugs has led to the development and evaluation of fluorinated indole motifs as potential medicinal agents.2 There are several 2-trifluoroethylindole derivatives that display diverse bioactivities such as anticancer, nervous system and reproductive control agent qualities as depicted in Fig. 1.3 In this context, synthetic methods have been developed towards fluorinated indoles, especially those with such substitution at the 2-position.
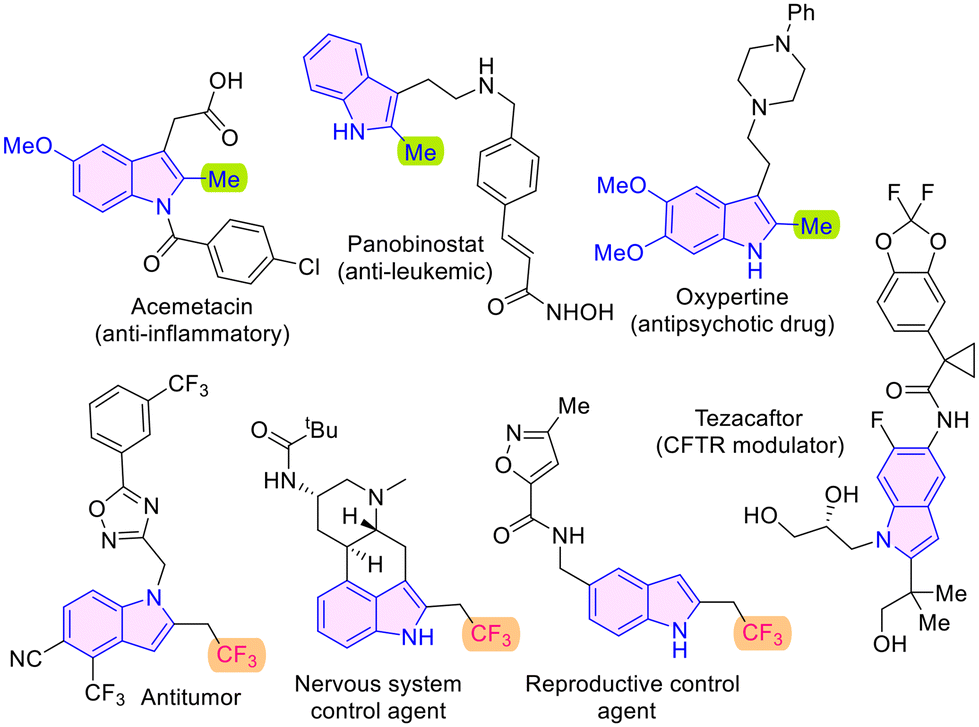 | ||
| Fig. 1 Bioactive molecules featuring the 2-alkylindole subunit. | ||
In 2018, the Liu group reported an interesting palladium catalyzed regioselective C-2 trifluoroethylation of indoles.4 While a tremendous feature of this method is its ability to provide C-2 selectivity without the use of an intramolecular directing group, no examples with other fluorinated motifs have been reported. The Weng group also developed a protocol for the synthesis of C-2 trifluoroethyl-substituted benzofused heterocycles using a copper catalyst.5 In this case, precursors which would lead to a 2,3-disubstituted product did not exhibit reactivity. Chen et al. developed a halogen-bonding mediated perfluoroalkylation of arenes and alkenes.6 We envisioned a mechanistically distinct strategy wherein photochemically generated trifluoroethyl and polyfluorinated radicals could be added to indoles at the C-2 position. Visible-light photoredox catalysis7 has been employed extensively for a variety of trifluoromethylation reactions8 and for the alkylation of electron rich heterocycles.9 In a majority of cases, activated radical precursors are usually required for radical generation.
Recently, C-2 alkylation of indoles was reported by the Melchiorre group wherein tryptophan containing peptides were alkylated using pyridinium salts as activated radical precursors,9b employing an EDA complex strategy. Important lessons from this study are that C2 functionalization is challenging as evidenced by moderate yields, and that achieving the C2 selectivity for the indole motif is a key aspect when using peptide substrates. Indeed, the contrasting selectivity for the β-position of tryptophan derivatives observed by Shi et al. under photoredox conditions further attests to the difficulty of finding a unified solution to this problem.10 Our reaction design was predicated on the applicability of an entire range of fluoroalkyl electrophiles which would provide access to trifluoroethylated and perfluorinated indoles, including those that are not accessible by known methods. The salient features and advantages include the ability to employ unfunctionalized precursors, a transition-metal-free protocol, works under mild conditions, and applicability to peptidic substrates. The solution to achieve C2-selectivity over β-selectivity (which occurs via SET oxidation of indole) was envisioned to invoke a reductive quenching mechanism involving a sacrificial electron donor which exhibits a lower reduction potential than the indole motif. In such a catalytic cycle, the SET activation of relatively unactivated alkyl halides would also be possible, thereby obviating the need for prefunctionalized, activated precursors (Scheme 1).
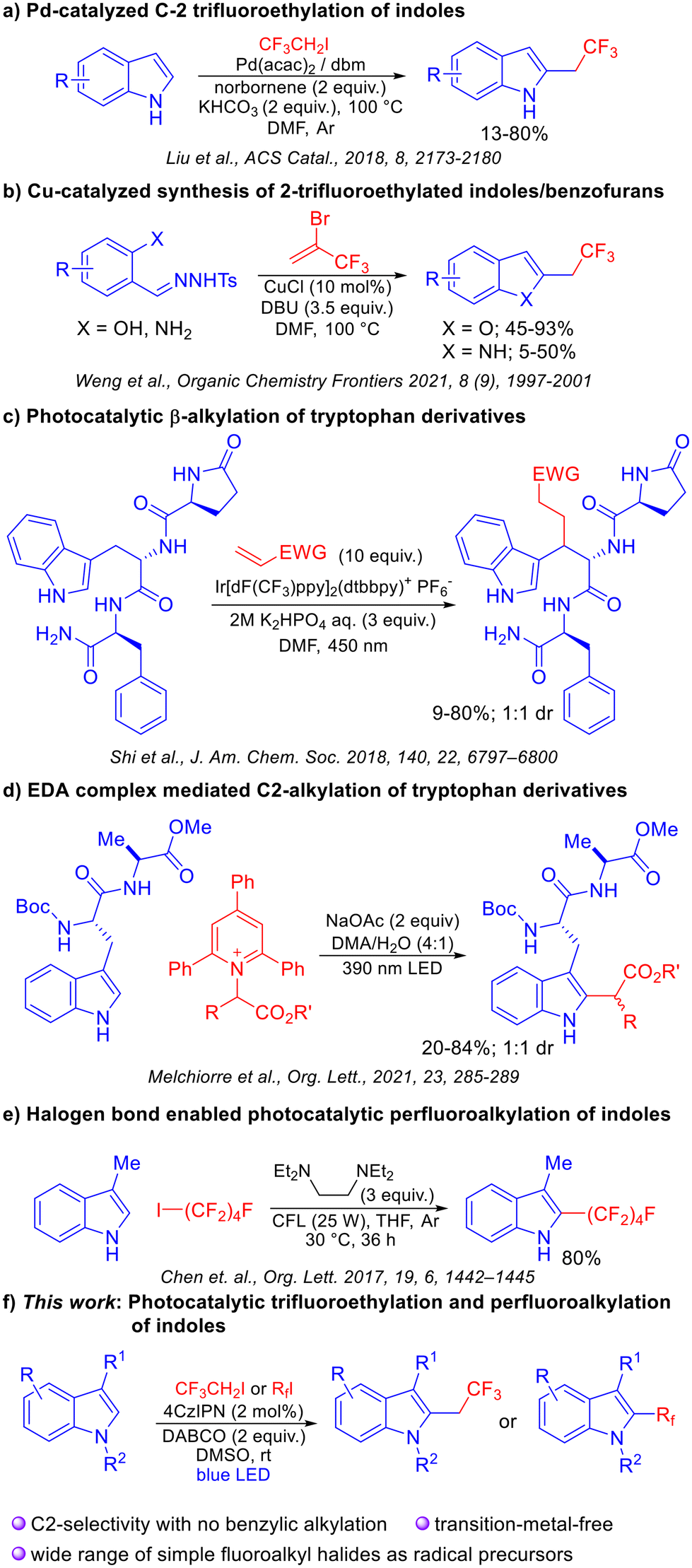 | ||
| Scheme 1 Strategies for indole alkylation and trifluoroethylation. | ||
Our optimization studies began with N-methyl-3-methyl indole (1a) and trifluoroethyl iodide (2a) as the carbon radical precursor and the results are outlined in Table 1. The optimized conditions include the use of DABCO as the base, 4CzIPN as the photocatalyst, and DMSO solvent under blue LED irradiation, affording 72% yield of product 3a (Table 1, entry 1). A range of solvents such as acetonitrile, DCE, THF and acetone were also evaluated but DMSO provided the best results (Table 1, entries 2 and 3). Among bases, K2CO3 and iPr2NEt provided significantly lower yields of the product (Table 1, entries 4 and 5).
|
|
||
|---|---|---|
| Entry | Change from standard conditions | 3a (%) |
| a Reaction conditions: 1a (0.34 mmol), 2a (0.69 mmol), DABCO (2 equiv.), 4CzIPN (2 mol%), DMSO (2 mL), 18 W blue LEDs under N2. | ||
| 1 | None | 72 |
| 2 | MeCN, DCE, THF instead of DMSO | 52, 00, 00 |
| 3 | Acetone instead of DMSO | 25 |
| 4 | K2CO3 instead of DABCO | 42 |
| 5 | iPr2NEt instead of DABCO | 19 |
| 6 | Ru(bpy)3Cl2·6H2O instead of 4CzIPN | 64 |
| 7 | Rose Bengal instead of 4CzIPN | 00 |
| 8 | Without a photocatalyst | 00 |
| 9 | Without light | 00 |
| 10 | Without DABCO | 00 |
Changing the photocatalyst to Ru(bpy)3Cl2·6H2O resulted in a diminished yield of the product and no product formation was observed when Rose Bengal was employed (Table 1, entries 6 and 7). Various control experiments were performed which revealed that light, photocatalyst, and base were crucial for a successful transformation (Table 1, entries 8–10). The structure of 3a was also confirmed by X-ray crystallography.
With the optimized conditions in hand, we examined the scope and generality of various substituted indoles and fluorinated radical precursors as depicted in Scheme 2. N-Ethyl-3-methyl indole afforded the corresponding product 3b in good yield (Scheme 2). Indoles featuring a free NH group and a pendant ester moiety such as indole-3-ethylacetate, indole-3-ethylpropanoate, and indole-3-methylpropanoate afforded products in moderate to good yields (3c–3e, Scheme 2). An indole derivative with a Boc-protected primary amine also reacted smoothly, affording product 3f in 53% yield. The yield was diminished when an acetyl-containing side chain was appended on the indole precursor (3g, Scheme 2). Indole-3-acetonitrile afforded product 3h in a significantly lower yield likely due to unfavorable single electron transfer (SET) oxidation of the benzylic radical intermediate leading to sluggish formation of the carbocation intermediate. Next, we focused on perfluoroalkylation as depicted in Scheme 2 and discovered that the reaction proceeded smoothly regardless of the extent of fluorination. The reaction of N-alkylated indole precursors with heptafluoropropyl iodide (C3F7I), nonafluorobutyl iodide (C4H9I) and tridecatrifluorohexyl iodide (C6F13I) afforded products in moderate to good yields (3i–3l, Scheme 2). A free NH group on the indole is also tolerated well and the corresponding products were obtained in good yields (3m–n, Scheme 2). These products also demonstrate that indole precursors with ester and Boc-protected amine functionalities at the 3-position are compatible with the reaction conditions. In the case of C3 unsubstituted indole, an inseparable mixture of products was obtained (Scheme 2, 3o).
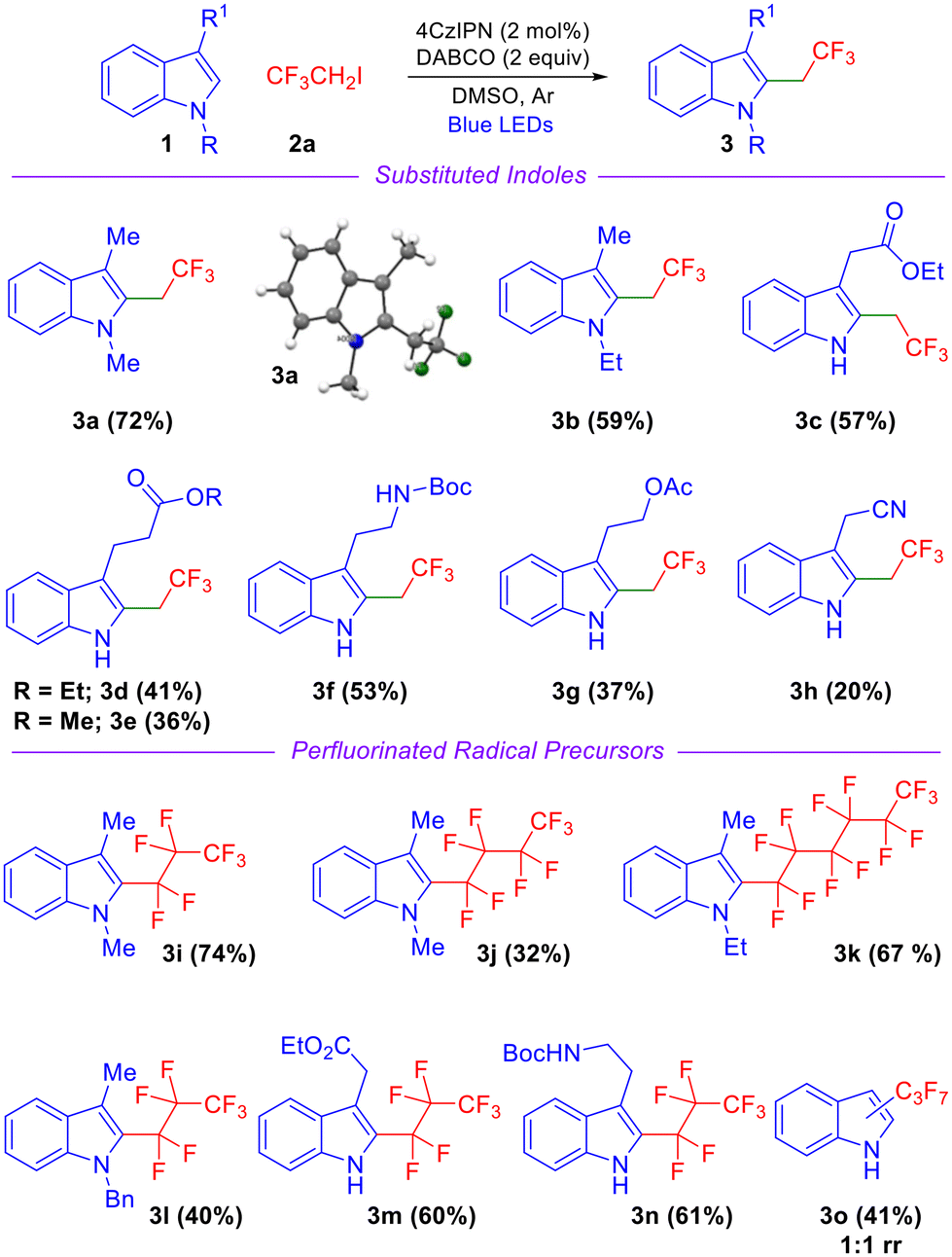 | ||
| Scheme 2 Reaction scope: evaluation of indoles and fluorinated electrophiles. | ||
We further extended this methodology to the functionalization of indoles featuring esters and amide functionalities derived from natural products and peptides, including those with defined stereocenters. A general observation was that yields were in the same (moderate) range as those reported by Melchiorre,9b Shi,10 and Weng,5 although no benzylic alkylation (or β-alkylation in the case of tryptophan derivatives) was observed in any case. Among these, the menthol ester afforded the corresponding product in good yield (6a, Scheme 3). Tryptophan and melatonin derivatives also resulted in the corresponding perfluoroalkylated products in 41% and 26% yields, respectively (6b–c). Dipeptides derived from tryptophan (Trp-Gly-OEt, Trp-Ala-OEt, Trp-Leu-OEt) also underwent fluoroethylation and afforded products 6d–f in moderate yields as depicted in Scheme 3.
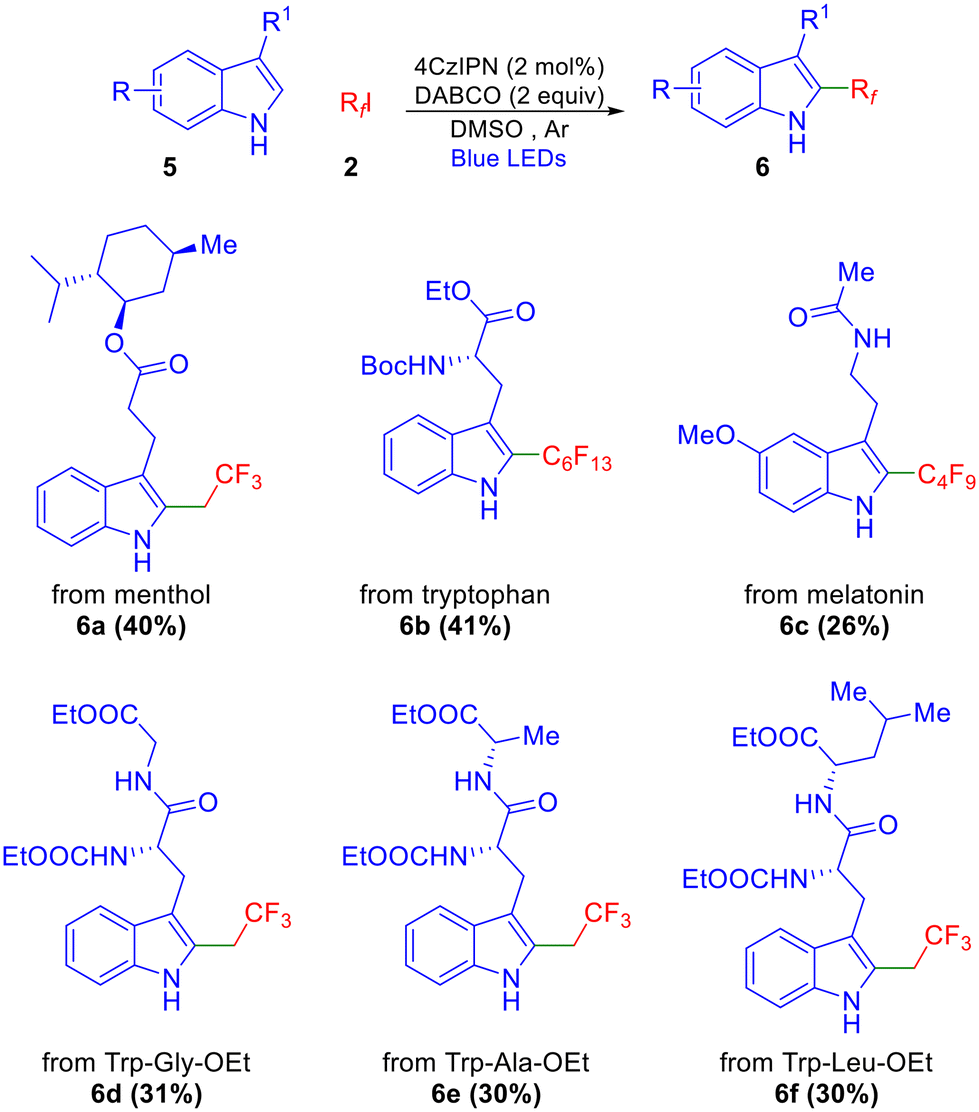 | ||
| Scheme 3 Trifluoethylation and perfluoroalkylation of indole-derived natural products. | ||
In order to gain insight into the reaction mechanism, we performed radical quenching experiments using TEMPO, BHT, and 1,1-diphenylethylene under otherwise identical conditions and the results are depicted in Scheme 4. No product formation was observed in the presence of TEMPO, suggesting the formation of radical intermediates as the reaction was completely suppressed.
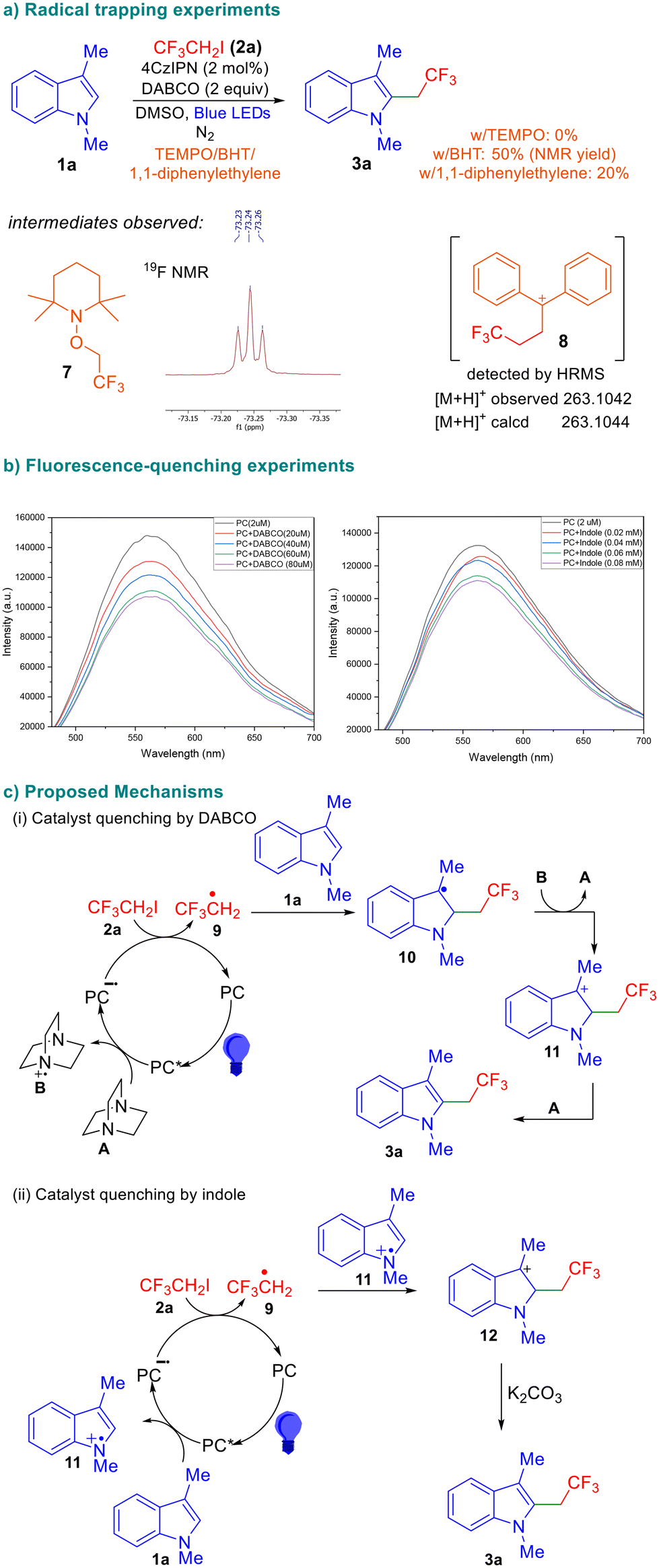 | ||
| Scheme 4 Mechanistic studies and proposed mechanisms. | ||
The trifluoroethyl radical was trapped by 1,1-diphenylethylene and we were able to observe the corresponding intermediate 8 using mass spectroscopy. We also performed fluorescence quenching experiments and observed that among all reaction components, the excited-state photocatalyst was quenched by DABCO, indicating that it is involved in the first single electron transfer. Based on the information available in the literature,11 control experiments described in Table 1, and the above-mentioned results, a plausible reaction mechanism is proposed in Scheme 4. The excited-state photocatalyst undergoes reductive quenching with DABCO (A) to form the DABCO radical cation B and the reduced photocatalyst radical anion (PC˙−). Subsequent SET from this species to the fluoroalkyl iodide electrophile results in the formation of the fluoroalkyl radical 9. Upon trapping of 9 by the indole, radical 10 is formed, which is subsequently oxidized by the DABCO radical cation generated during the first SET. The involvement of DABCO in these two steps is supported by the observation that it is indispensable for successful transformation (Table 1, entry 8). The formation of the benzylic cation 11 is followed by elimination to afford the desired product 3a. The reaction also leads to product formation when certain inorganic bases are used, albeit in a low yield (Table 1). This indicates that there is a possibility of an alternate mechanism wherein the excited-state catalyst is quenched by the indole to result in the radical cation 11 which intercepts the fluorinated radical 9 leading to cation 12. Subsequent deprotonation of 12 affords the desired product.
Conclusions
In summary, we have developed a photocatalytic route towards 2-trifluoroethylated and 2-perfluoroalkylated indole derivatives. The transformation can employ a wide range of simple, unfunctionalized fluoroalkyl halide precursors, works under mild conditions, and utilizes a reductive quenching mechanism to achieve C2-selectivity. Application of this protocol to tryptophan containing peptides was also demonstrated.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the SERB, New Delhi (CRG/2022/0082850).References
- (a) M. T. Samara, H. Cao, B. Helfer, J. M. Davis and S. Leucht, Eur. Neuropsychopharmacol., 2014, 24, 1046–1055 CrossRef CAS PubMed; (b) D. E. Biancur, J. A. Paulo, B. Małachowska, M. Quiles Del Rey, C. M. Sousa, X. Wang, A. S. Sohn, G. C. Chu, S. P. Gygi and J. W. Harper, Nat. Commun., 2017, 8, 15965 CrossRef CAS PubMed; (c) J. Nicholson, S. J. Jevons, B. Groselj, S. Ellermann, R. Konietzny, M. Kerr, B. M. Kessler and A. E. Kiltie, Cancer Res., 2017, 77, 3027–3039 CrossRef CAS PubMed; (d) S. Gahr, C. Mayr, T. Kiesslich, R. Illig, D. Neureiter, B. Alinger, M. Ganslmayer, T. Wissniowski, P. D. Fazio and R. Montalbano, Int. J. Oncol., 2015, 47, 963–970 CrossRef CAS PubMed.
- D. E. Yerien, S. Bonesi and A. Postigo, Org. Biomol. Chem., 2016, 14, 8398–8427 RSC.
- Y. Wan, Y. Li, C. Yan, M. Yan and Z. Tang, Eur. J. Med. Chem., 2019, 183, 111691 CrossRef CAS PubMed.
- H. Zhang, H.-Y. Wang, Y. Luo, C. Chen, Y. Cao, P. Chen, Y.-L. Guo, Y. Lan and G. Liu, ACS Catal., 2018, 8, 2173–2180 CrossRef CAS.
- W. Wu, B. Luo, Y. You and Z. Weng, Org. Chem. Front., 2021, 8, 1997–2001 RSC.
- Y. Wang, J. Wang, G.-X. Li, G. He and G. Chen, Org. Lett., 2017, 19, 1442–1445 CrossRef CAS PubMed.
- (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (c) J. D. Bell and J. A. Murphy, Chem. Soc. Rev., 2021, 50, 9540–9685 RSC; (d) A. A. Festa, L. G. Voskressensky and E. V. Van der Eycken, Chem. Soc. Rev., 2019, 48, 4401–4423 RSC.
- B. Ding, Y. Weng, Y. Liu, C. Song, L. Yin, J. Yuan, Y. Ren, A. Lei and C.-W. Chiang, Eur. J. Org. Chem., 2019, 7596–7605 CrossRef CAS.
- (a) Ł. W. Ciszewski, J. Durka and D. Gryko, Org. Lett., 2019, 21, 7028–7032 CrossRef PubMed; (b) B. Laroche, X. Tang, G. Archer, R. Di Sanza and P. Melchiorre, Org. Lett., 2021, 23, 285–289 CrossRef CAS PubMed; (c) J. W. Tucker, J. M. R. Narayanam, S. W. Krabbe and C. R. J. Stephenson, Org. Lett., 2010, 12, 368–371 CrossRef CAS PubMed; (d) L. Furst, B. S. Matsuura, J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, Org. Lett., 2010, 12, 3104–3107 CrossRef CAS PubMed; (e) M. J. James, F. Strieth-Kalthoff, F. Sandfort, F. J. R. Klauck, F. Wagener and F. Glorius, Chem. – Eur. J., 2019, 25, 8240–8244 CrossRef CAS PubMed; (f) H. Baguia, J. Beaudelot, C. Moucheron and G. Evano, Chem. Commun., 2022, 58, 9080–9083 RSC; (g) N. J. W. Straathof, H. P. L. Gemoets, X. Wang, J. C. Schouten, V. Hessel and T. Noël, ChemSusChem, 2014, 7, 1612–1617 CrossRef CAS PubMed; (h) N. J. Straathof, D. J. van Osch, A. Schouten, X. Wang, J. C. Schouten, V. Hessel and T. Noël, J. Flow Chem., 2014, 4, 12–17 CrossRef CAS.
- Y. Yu, L.-K. Zhang, A. V. Buevich, G. Li, H. Tang, P. Vachal, S. L. Colletti and Z.-C. Shi, J. Am. Chem. Soc., 2018, 140, 6797–6800 CrossRef CAS PubMed.
- L. Li, X. Chen, C. Pei, J. Li, D. Zou, Y. Wu and Y. Wu, Tetrahedron Lett., 2021, 83, 153407 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2322591. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob00392f |
| This journal is © The Royal Society of Chemistry 2024 |