
Design of substituted tetrahydrofuran derivatives for HIV-1 protease inhibitors: synthesis, biological evaluation, and X-ray structural studies†‡
Arun K.
Ghosh
 *ab,
Daniel
Lee
a,
Ashish
Sharma
a,
Megan E.
Johnson
a,
Ajay K.
Ghosh
a,
Yuan-Fang
Wang
c,
Johnson
Agniswamy
c,
Masayuki
Amano
d,
Shin-ichiro
Hattori
e,
Irene T.
Weber
c and
Hiroaki
Mitsuya
def
*ab,
Daniel
Lee
a,
Ashish
Sharma
a,
Megan E.
Johnson
a,
Ajay K.
Ghosh
a,
Yuan-Fang
Wang
c,
Johnson
Agniswamy
c,
Masayuki
Amano
d,
Shin-ichiro
Hattori
e,
Irene T.
Weber
c and
Hiroaki
Mitsuya
def
aDepartment of Chemistry, Purdue University, 560 Oval Drive, West Lafayette, IN 47907, USA. E-mail: akghosh@purdue.edu; Tel: +1 765 4945323
bDepartment of Medicinal Chemistry and Molecular Pharmacology, Purdue University, 560 Oval Drive, West Lafayette, IN 47907, USA
cDepartment of Biology, Georgia State University, Atlanta, Georgia 30303, USA
dDepartments of Infectious Diseases and Hematology, Kumamoto University Graduate School of Biomedical Sciences, Kumamoto 860-8556, Japan
eCenter for Clinical Sciences, National Center for Global Health and Medicine, Tokyo 162-8655, Japan
fExperimental Retrovirology Section, HIV and AIDS Malignancy Branch, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA
First published on 8th July 2024
Abstract
Substituted tetrahydrofuran derivatives were designed and synthesized to serve as the P2 ligand for a series of potent HIV-1 protease inhibitors. Both enantiomers of the tetrahydrofuran derivatives were synthesized stereoselectivity in optically active forms using lipase-PS catalyzed enzymatic resolution as the key step. These tetrahydrofuran derivatives are designed to promote hydrogen bonding and van der Waals interactions with the backbone atoms in the S2 subsite of the HIV-1 protease active site. Several inhibitors displayed very potent HIV-1 protease inhibitory activity. A high-resolution X-ray crystal structure of an inhibitor-bound HIV-1 protease provided important insight into the ligand binding site interactions in the active site.
Introduction
The development of HIV-1 protease inhibitor drugs and their introduction into combination therapy with reverse transcriptase inhibitors marked the beginning of an important breakthrough for the treatment of patients with HIV-1 infection and AIDS.1,2 The advent of this active antiretroviral therapy (ART) helped transform HIV-1 infection from an inevitably fatal disease into a manageable chronic ailment.3,4 Currently there is no treatment to eradicate HIV from an infected patient. However, ART treatment regimens significantly improve quality of life, enhance HIV management, and reduce mortality and morbidity in patients with HIV-1 infection and AIDS.5,6 Despite these advances, there are major drawbacks associated with the current treatment, particularly peptide-like features, poor ADME properties, drug toxicity and drug side effects.7,8 Another major alarming problem is the emergence of drug resistant HIV-1 strains, which may limit long-term treatment options.9,10 Therefore, the development of a new generation of protease inhibitors (PIs) with improved drug properties as well as broad-spectrum activity against multidrug resistant HIV-1 variants is critically important for future HIV/AIDS management.11,12Over the years, our research efforts have focused on the design and development of nonpeptide protease inhibitors incorporating stereochemically defined novel heterocycles to interact with the backbone residues in the active site of HIV-1 protease. We reported a variety of highly potent inhibitors that showed intriguing structural features and displayed broad-spectrum antiviral activity against multidrug resistant HIV-1 variants.13–15 One of these PIs is the FDA approved drug, darunavir (1, Fig. 1), which became a first-line therapy for the treatment of HIV/AIDS patients.16,17 One of the intriguing features of darunavir is the incorporation of a structure-based design nonpeptide ligand, 3(R),3a(S),6a(R)-bis-tetrahydrofuranylurethane (bis-THF).18,19 The P2 bis-THF ligand is specifically designed to promote maximum interactions, particularly hydrogen bonding interactions with the backbone atoms in the S2 subsite of the HIV-1 protease active site. This “backbone binding concept” emerged from the observation that the backbone conformation of the active site of mutant proteases shows only minimal distortion.20,21 Therefore, design strategies enhancing inhibitor protein backbone interactions may likely lead to inhibitors sustaining inhibitor affinity and potency.22,23 A number of X-ray crystallographic studies of darunavir-bound HIV-1 protease revealed that both oxygen atoms of the bis-THF ligand form very strong hydrogen bonding interactions with the backbone amide NHs of Asp29 and Asp30 in the S2 subsite.24,25 Furthermore, the bicyclic bis-THF scaffold nicely fills in the hydrophobic pocket, thereby enhancing van der Waals interactions with the enzyme active site residues. Darunavir's P2′ amino benzenesulfonamide ligand also forms hydrogen bonds with the Asp30 backbone NH as well as with the side chain carboxylic acid. These extensive interactions from the S2 to S2′ subsites of HIV-1 protease are likely to be responsible for darunavir's potency and effectiveness against multidrug-resistant HIV-1 variants.26,27
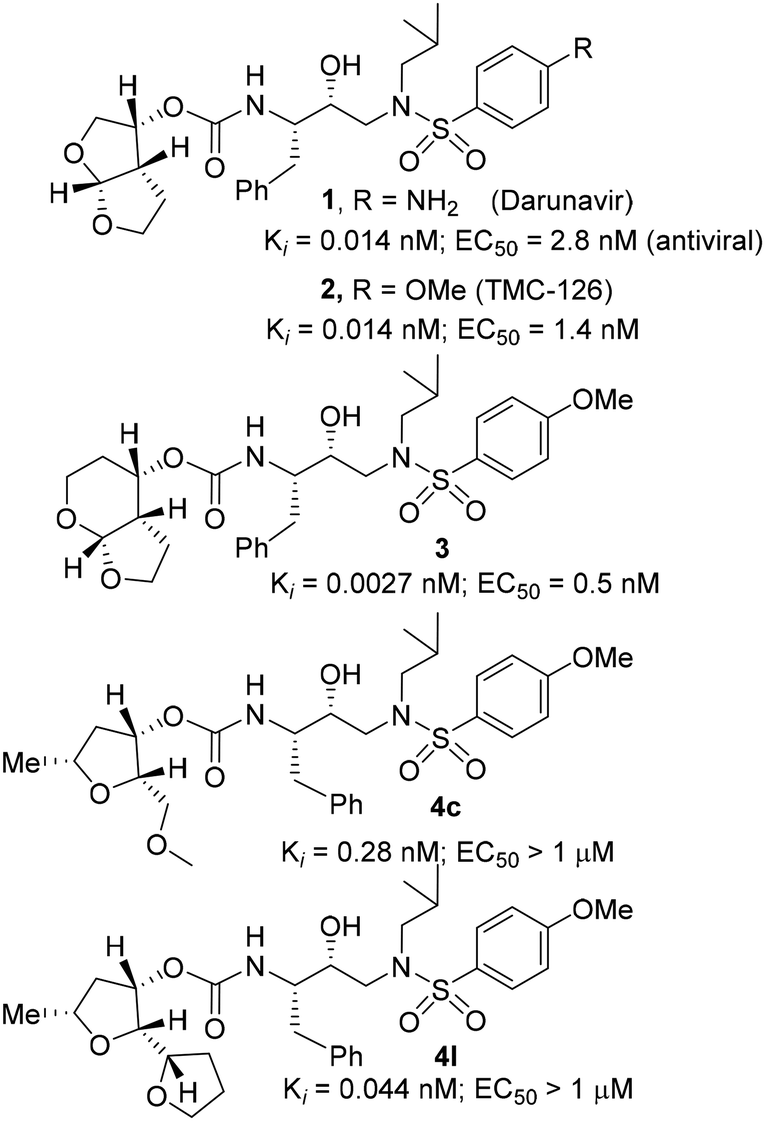 | ||
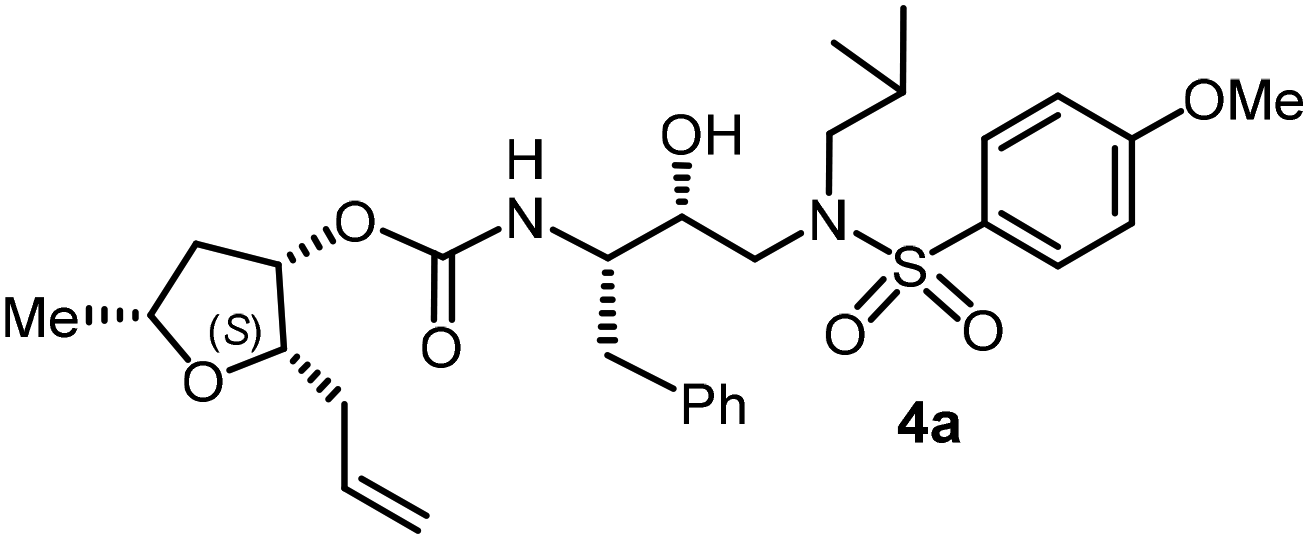
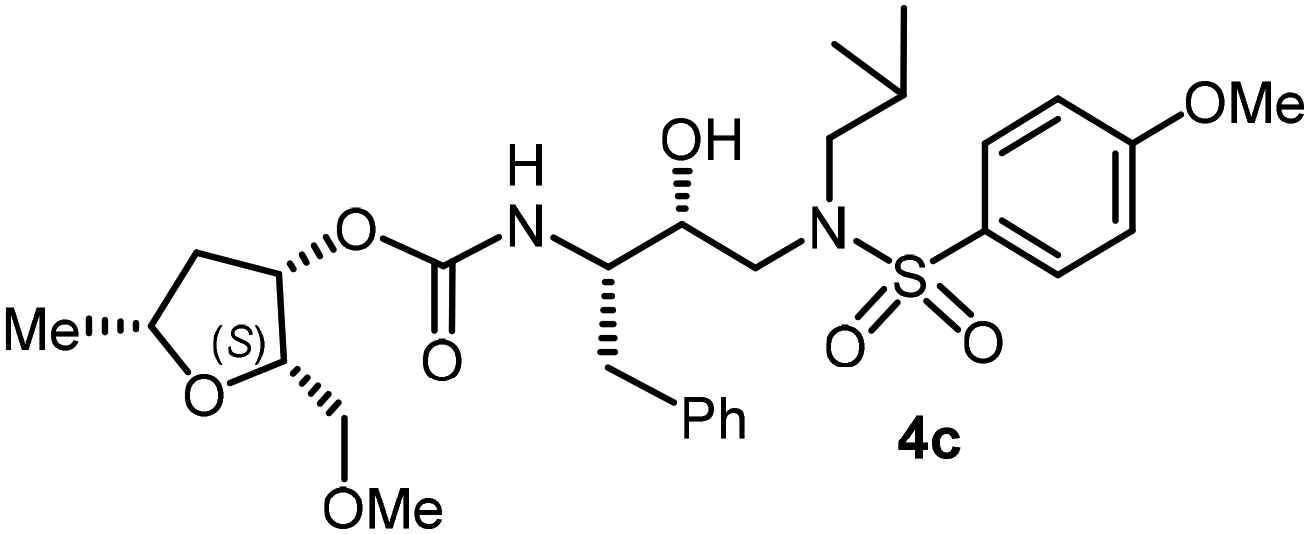
| Fig. 1 Structures of PIs 1–3, 4c, and 4l. | ||
Based upon the X-ray structures of darunavir and its P2′ methoxy derivative 2, we further designed a bicyclic acetal bearing a tetrahydropyranyl–tetrahydrofuran ring system in inhibitor 3.28 We speculated that a larger tetrahydropyranyl (THP) ring may improve hydrogen bonding with the active site aspartate backbone NHs due to increase in the dihedral angle of the bicyclic acetal oxygen atoms. Also, a larger THP-ring may enhance van der Waals interactions. Indeed, inhibitor 3 exhibited high enzyme affinity and antiviral activity and maintained excellent antiviral activity against a variety of multi-drug resistant clinical HIV-1 strains.29 Our PI design promoting backbone hydrogen bonding interactions also led to the development of diverse classes of PIs that exhibited marked antiviral activity and displayed robust activity against highly multidrug-resistant HIV-1 strains.13,20,21 Based upon the X-ray structure of darunavir bound HIV-1 protease, we have now investigated the potential of various substituted tetrahydrofuran derivatives with acyclic and cyclic functionalities to function as effective P2 ligands for the S2 subsite of the HIV-1 protease substrate binding site, exemplified in structures 4c and 4l. Herein, we report the design and syntheses of a variety of nonpeptide HIV-1 protease inhibitors incorporating functionalized tetrahydrofuran derivatives as the P2 ligands in combination with (R)-(hydroxyethylamine)sulfonamide isosteres. Functionalized tetrahydrofuran derivatives were synthesized stereoselectively in optically active form using lipase-PS catalyzed enzymatic resolution as the key step. All inhibitors were evaluated in HIV-1 protease inhibitory and antiviral assays. The X-ray crystal structure of an inhibitor-bound HIV-1 protease was determined to obtain molecular insights into the inhibitor protease interactions.
Results and discussion
Based on the X-ray crystal structure of darunavir and wild-type HIV-1 protease, we planned to explore new PIs containing acyclic tetrahydrofuran derivatives that can accommodate key ligand binding site interactions of the P2 bis-THF ligand of daruanvir.24,25 Furthermore, we envisioned that such substituted tetrahydrofuran derivatives can be optimized to improve the polar interactions with both Asp29 and Asp30 active site backbone atoms. Also, we speculated that installing appropriate alkyl substituents at the C2 and C5 positions would enhance hydrophobic contact with the S2 subsite residues. Accordingly, we designed several trisubstituted tetrahydrofuran derivatives with varying stereochemistry at the C2, C3, and C5 positions. For detailed assessment of the ligand-binding site interactions, the syntheses of these substituted tetrahydrofuran derivatives were carried out in an enantioselective manner. As shown in Scheme 1, commercially available α-methylene-γ-valerolactone 5 was subjected to ozonolysis to provide the corresponding keto lactone that was hydrogenated with 10% Pd–C in ethyl acetate under a hydrogen-filled balloon to afford the racemic α-hydroxy lactone 6 in 62% yield in two steps.30,31 Enzymatic resolution of racemic alcohol with amano-lipase PS-30 in a mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) of vinyl acetate and THF at 23 °C for 12 h resulted in the optically active alcohol 6 and acetate derivative 7 in excellent yield. Saponification of acetate 7 with aqueous NaOH at 23 °C for 12 h furnished the optically active alcohol ent-6 in 61% yield. Both enantiomeric alcohols were obtained with over 90% ee. Stereochemical identity of the enantiomers was previously determined by X-ray crystallographic analysis.31 Both enantiomeric alcohols were prepared in a multigram scale for the synthesis of various enantiomeric ligands for our studies.
:1) of vinyl acetate and THF at 23 °C for 12 h resulted in the optically active alcohol 6 and acetate derivative 7 in excellent yield. Saponification of acetate 7 with aqueous NaOH at 23 °C for 12 h furnished the optically active alcohol ent-6 in 61% yield. Both enantiomeric alcohols were obtained with over 90% ee. Stereochemical identity of the enantiomers was previously determined by X-ray crystallographic analysis.31 Both enantiomeric alcohols were prepared in a multigram scale for the synthesis of various enantiomeric ligands for our studies.
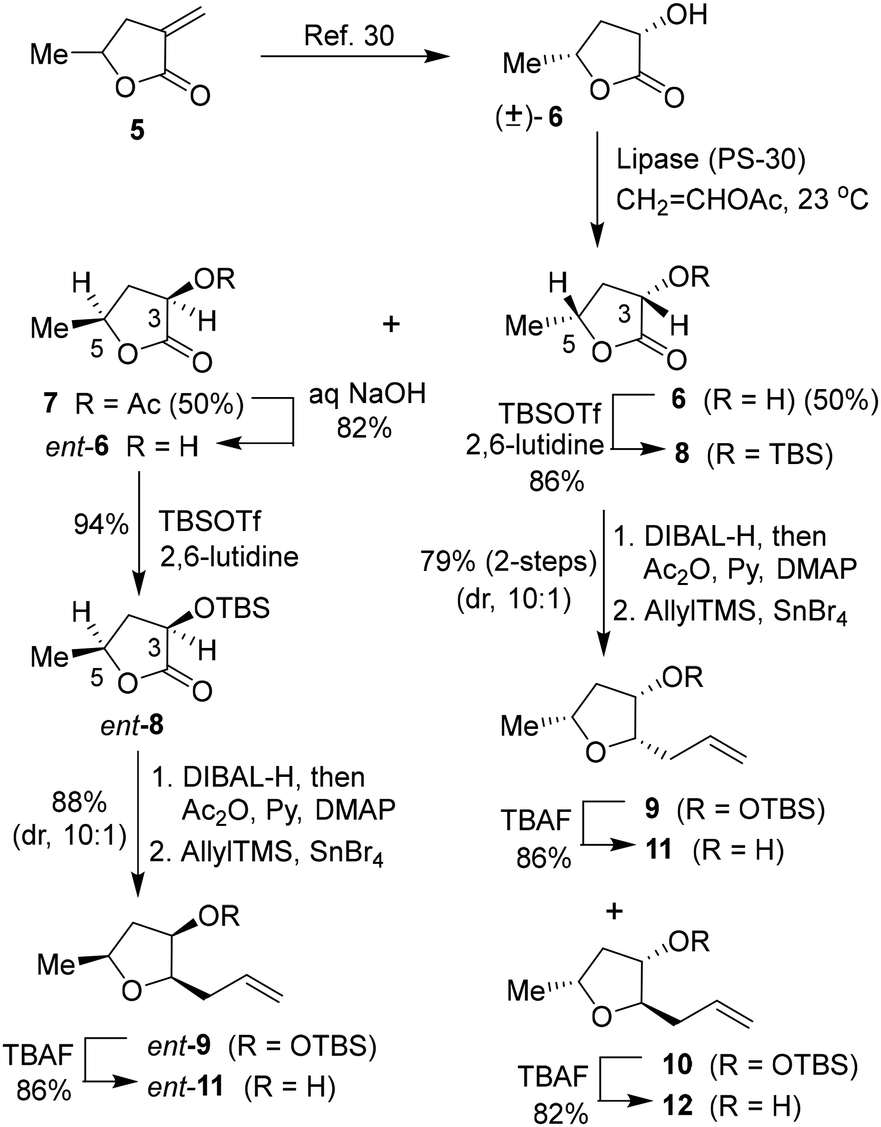 | ||
| Scheme 1 Synthesis of optically active (2S,3S,5R)-2-allyl-5-methyltetrahydrofuran-3-ol and its enantiomer. | ||
The optically active (3S,5R)-3-hydroxy-5-methyl 2-furanone 6 was reacted with TBSOTf and 2,6-lutidine in CH2Cl2 at 0 °C for 2 h to provide TBS-ether 8 in 96% yield. DIBAL-H reduction of lactone at −78 °C for 2 h gave the corresponding lactol that was immediately treated with acetic anhydride and triethylamine in the presence of a catalytic amount of DMAP at −78 °C for 2 h to furnish the corresponding acetate in 90% yield in two steps. Exposure of the resulting acetate to allyl trimethyl silane in the presence of SnBr4 in CH2Cl2 at −78 °C to 23 °C for 2 h resulted in the allyl derivative 9 as the major product (dr, 10:1 by 1 H-NMR analysis) along with a small amount of the minor diastereomer 10 in 88% yield.31,32 The diastereomers were separated by using silica gel chromatography. Treatment of the resulting allyl derivative 9 with TBAF in THF at 0 °C to 23 °C for 3 h furnished the trisubstituted ligand alcohol 11 in 86% isolated yield. Similarly, the removal of the TBS group from diastereomer 10 provided alcohol 12 in 82% yield. The enantiomeric ligand alcohol ent-11 was prepared from the optically active alcohol ent-6 using the same sequence of reactions as for the synthesis of ligand 11. The minor isomer ent-12 also formed in the reaction.
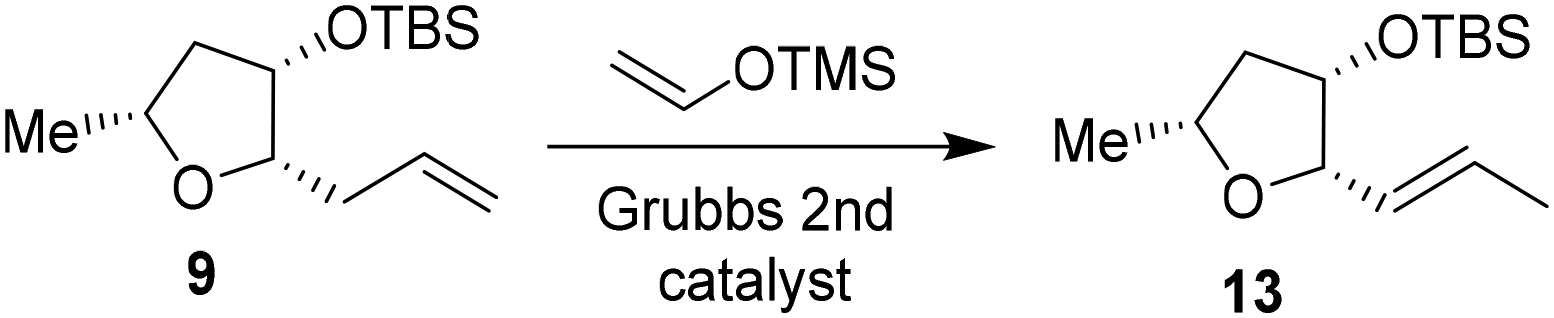
We then planned to investigate the substituted tetrahydrofuran derivative containing a C2 methoxymethyl side chain. The synthesis of (2S,3S,5R)-2(methoxymethyl)-5-methyl-3-tetrahydrofuranol is shown in Scheme 2. The synthesis of the requisite aldehyde requires the introduction of a C2 allyl chain followed by olefin isomerization and cleavage of the resulting olefin. We investigated the olefin isomerization of the C2 substituted tetrahydrofuran derivative under a variety of conditions and the results are shown in Table 1. We first examined terminal alkene isomerization of 9 using 20 mol% Grubbs 2nd generation catalyst in MeOH at 60 °C for 4 h, as described by Hanessian and co-workers.33 This condition provided the disubstituted olefin 13 in 67% yield (entry 1). We then examined olefin isomerization using 5 mol% Grubbs 2nd generation catalyst in the presence of 10 equivalents of trimethyl(vinyloxy)silane in CH2Cl2 at 60 °C in a sealed tube for 24 h, as reported by Nishida and co-workers.34 This condition provided the isomerized olefin product 13 in 61% yield (entry 2). The reaction of 9 with 10 mol% Grubbs 2nd generation catalyst and 10 equivalents of trimethyl(vinyloxy)silane in CH2Cl2 at 110 °C in a sealed tube for 24 h furnished the isomerization product 13 in 95% yield (entry 3). We also investigated 15 mol% catalyst at 60 °C for 72 h, which provided a lower yield of 13. Interestingly, the isomerization in toluene resulted in a significant reduction in product yields (entries 5 and 6). For the synthesis of the ligand alcohol 14, olefin 13 was subjected to ozonolysis in a mixture (1:1) of CH2Cl2 and MeOH at −78 °C for 2 h followed by reduction with NaBH4 at −78 °C for an additional 2 h. These conditions resulted in alcohol 14 in 92% yield in two steps. The treatment of alcohol 14 with NaH in THF at 0 °C followed by the reaction with MeI at 0 °C to 23 °C afforded the corresponding methyl ether derivative in 63% yield. The reaction of the resulting methyl ether with TBAF at 0 °C to 23 °C for 3 h furnished the ligand alcohol 15 in near-quantitative yield. The enantiomeric ligand alcohol ent-15 was synthesized from ent-9 following the same sequence of reaction as for methyl ether 15.
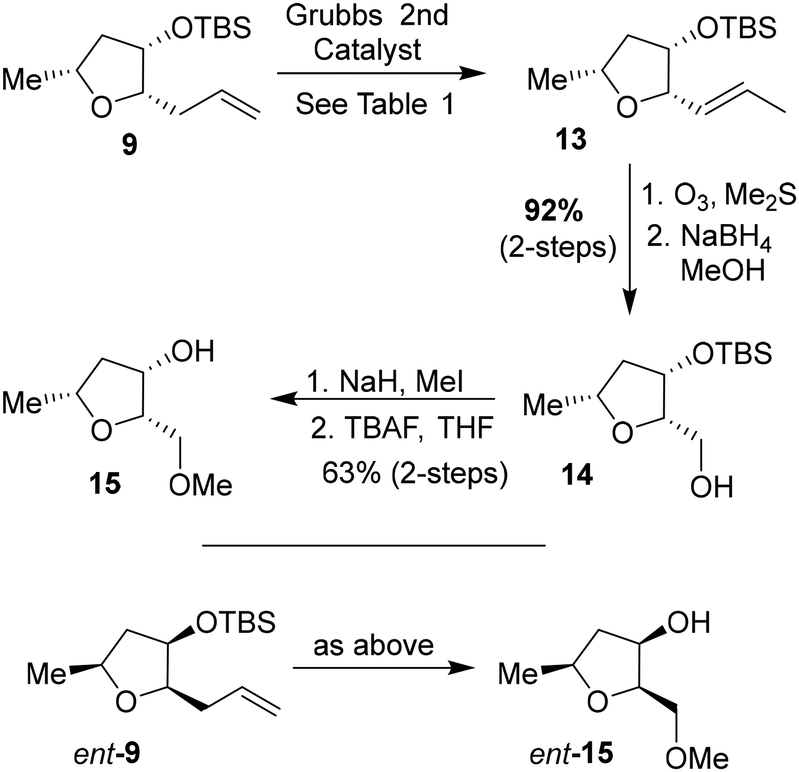 | ||
| Scheme 2 Synthesis of (2S,3S,5R)-2-(methoxymethyl)-5-methyltetrahydrofuranol 15 and ent-15. | ||
|
|
|||
|---|---|---|---|
| Entry | Grubbs 2nd (mol%) | Reaction conditionsc | Yieldb (%) |
| a Reaction were carried out at the 0.1 mmol scale and in 0.012 M solution. b Isolated yield at chromatography. c Trimethyl(vinyloxy)silane (10 equiv.) was used for all experiments except those shown in entry 1. | |||
| 1 | 20 | MeOH, 60 °C, 4 h | 67 |
| 2 | 5 | CH2Cl2, 60 °C, 24 h | 61 |
| 3 | 10 | CH2Cl2, 110 °C, 24 h | 95 |
| 4 | 15 | CH2Cl2, 60 °C, 72 h | 54 |
| 5 | 5 | Toluene, 110 °C, 24 h | 60 |
| 6 | 10 | Toluene, 110 °C, 24 h | 47 |
To investigate the effect of the stereochemistry at the C2 methoxymethyl side chain, we prepared the corresponding diastereomeric tetrahydrofuran derivatives in a stereoselective manner, as shown in Scheme 3. TBS protected lactone 8 was treated with allyl magnesium bromide in ether at −78 °C for 3 h to provide the corresponding hemiketal intermediate in 63% yield. Reduction of the resulting hemiketal using triethylsilane in the presence of BF3·OEt2 in CH2Cl2 at −78 °C for 3 h furnished the allyl derivative 10 as a major product (dr, 10:1 by 1H-NMR) in 58% yield. Olefin isomerization using Grubbs second generation catalyst as described above resulted in olefin 16 in 61% yield.33,34 The ozonolysis of 16 followed by reduction with NaBH4 afforded alcohol 17 in 85% yield over 2 steps. Alcohol 17 was converted to its methyl ether with NaH and MeI. Removal of the TBS ether using TBAF in THF furnished (2R,3S,5R)-2(methoxymethyl)-5-methyl tetrahydrofuran-3-ol 18 in 84% yield over 2 steps. The corresponding enantiomeric ligand alcohol ent-18 was prepared following the same sequence of reactions as compound 18.
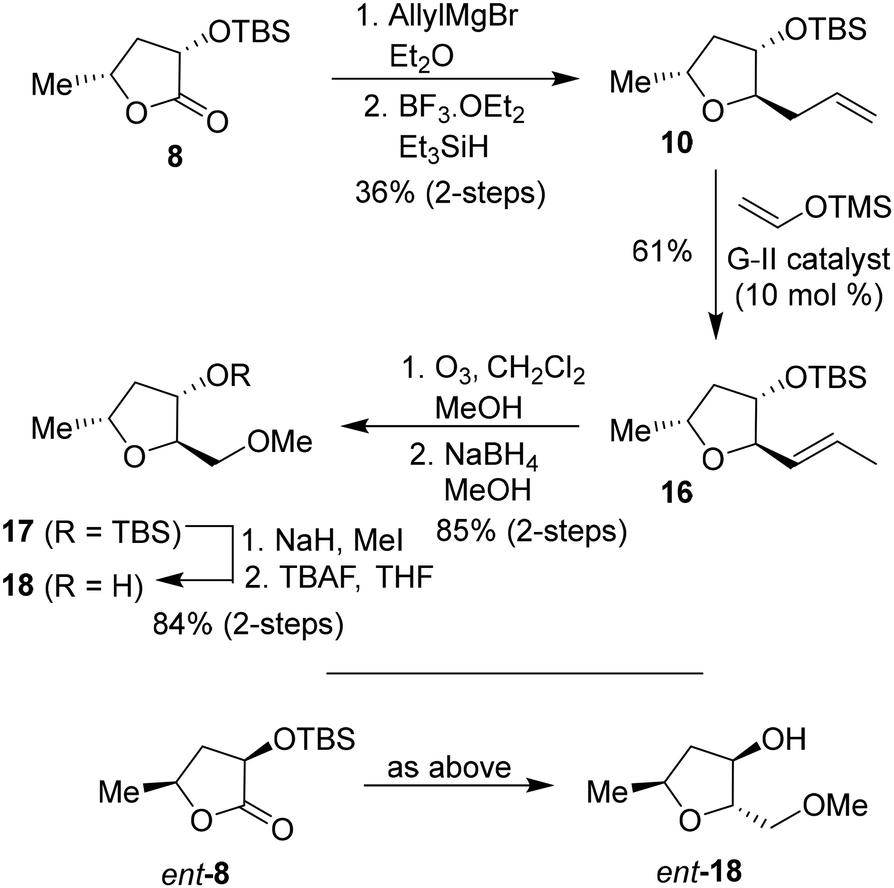 | ||
| Scheme 3 Synthesis of (2R,3S,5R)-2-(methoxymethyl)-5-methyltetrahydrofuranol 18 and ent-18. | ||
Based upon our previous design of the bicyclic fused bis tetrahydrofuran (bis-THF) ligand, we planned to examine the feasibility of substituted 2-tetrahydrofuranyl–tetrahydrofuran derivatives as the P2 ligands. The synthesis of diastereomeric ligands utilizing isomerized olefin 13 is shown in Scheme 4. The ozonolytic cleavage of olefin 13 with Ph3P at −78 °C for 1.5 h provided aldehyde 19 in 76% yield. Aldehyde 19 was subjected to allylation under a variety of conditions. The results are shown in Table 2. The reaction of 19 with allyl trimethylsilane in the presence of 3 equivalents of BF3·OEt2 at −78 °C for 4 h provided a mixture (82:18) of diastereomers 20 and 21 in 78% combined yield (entry 1). The diastereomeric ratios were determined by 1H-NMR (500 MHz) analysis. The use of allyl tributyl stannane under the above conditions afforded a mixture (83:17) of alcohols 20 and 21 in 79% yield (entry 2). The use of MgBr2·OEt2 as the Lewis acid resulted in a marginal diastereoselective (45:55 ratio) and reduction in yield (entry 3). Treatment of 19 with allylMgBr in ether at −78 °C for 3 h provided diastereomers 20 and 21 as a mixture (45:55) in 59% yield (entry 4). The use of ZnCl2 as the bidentate Lewis acid and allyl tributyl stannane at −78 °C to 23 °C for 18 h resulted in the reversal of diastereoselectivity and diastereomers 20 and 21 were obtained as a mixture (25:75) in 75% yield. These diastereomers were separated by silica gel chromatography and the major diastereomer 20 (from entry 2) was treated with 9-BBN in THF at 0 °C to 23 °C for 6 h.
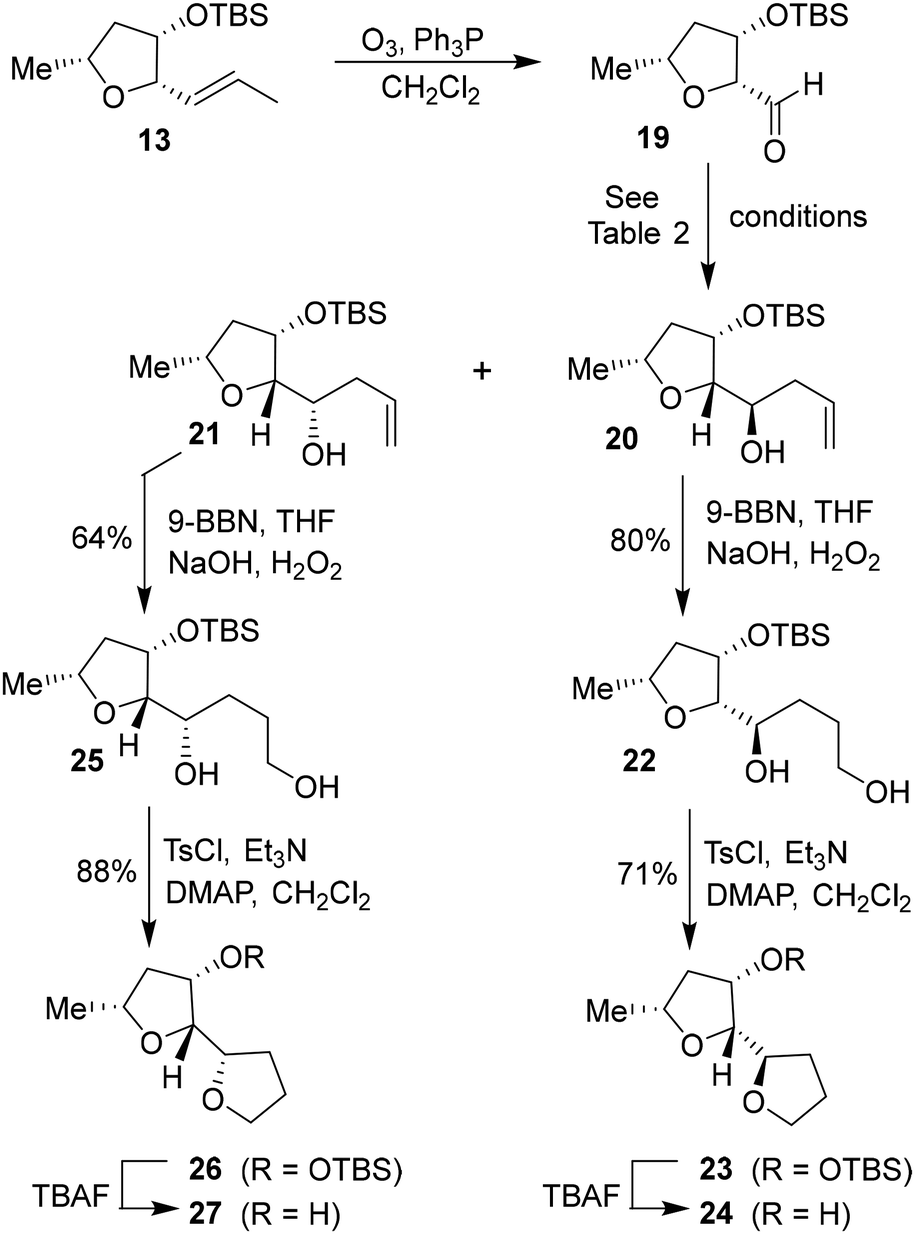 | ||
| Scheme 4 Synthesis of (2R,2′R,3S,5R)-5-methyloctahydro-[2,2′-bifuran]-3-ol 24 and its diastereomer 27. | ||
a
|
|
||||
|---|---|---|---|---|
| Entry | M | Lewis acid and conditions | Yieldb (%) | Ratio (20:21)c |
| a Reactions were carried at the 0.1 mmol scale. b Isolated yield at chromatography. c Ratios were determined by 1H-NMR analysis. | ||||
| 1 | SiMe3 | BF3·OEt2, CH2Cl2, −78 °C, 4 h | 78 | 4.2:1 |
| 2 | SnBu3 | BF3·OEt2, CH2Cl2, −78 °C, 18 h | 79 | 4.3:1 |
| 3 | SnBu3 | MgBr2·OEt2, CH2Cl2, 23 °C, 4 h | 75 | 1:1.3 |
| 4 | MgBr | ZnCl2, Et2O, −78 °C, 3 h | 59 | 1:1.2 |
| 5 | SnBu3 | ZnCl2, CH2Cl2, −78 °C, 18 h | 75 | 1:3 |
| 6 | SiMe3 | ZnCl2, CH2Cl2, −78 °C, 18 h | 66 | 1:2.5 |
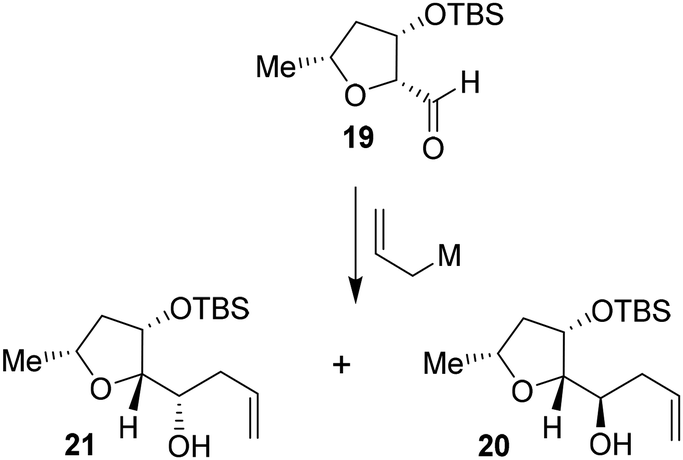
Oxidation of the resulting organoborane with 3 N NaOH and 30% H2O2 at 23 °C for 3 h furnished diol 22 in 80% yield. Treatment of diol 22 with tosyl chloride and Et3N in the presence of a catalytic amount of DMAP in CH2Cl2 at 0 °C to 23 °C for 48 h furnished the bicyclic tetrahydrofuran derivative 23 in 71% yield. Deprotection of the TBS group using TBAF in THF at 0 °C to 23 °C for 3 h afforded the ligand alcohol 24 in near-quantitative yields. The diastereomeric alcohol 21 (from entry 5) was then converted to the diastereomeric tetrahydrofuranyl–tetrahydrofuran derivative 27 using the same sequence of reactions.
For the synthesis of inhibitors with substituted tetrahydrofurans, various ligand alcohols were converted to the corresponding mixed activated carbonate derivatives, as shown in Scheme 5. Reactions of optically active ligand alcohols (11, 12, 15, ent-15, 18, ent-18, 24 and 27) were reacted with 4-nitrophenyl chloroformate and pyridine in CH2Cl2 at 0 °C to 23 °C for 12 h, furnishing mixed carbonates 28a–h in good yields.26 Syntheses of inhibitors with (R)-(hydroxyethylamine)sulfonamide isosteres containing 4-methoxysulfonamide and 4-aminosulfonamide as the P2′-ligands are shown in Scheme 6. Treatment of known amine derivatives 29 and 30 with mixed activated carbonates 28a–e in the presence of diisopropylethylamine (DIPEA) in CH3CN at 23 °C for 12–36 h provided inhibitors 4a–l.26 The full structures of these inhibitors are shown in Table 3.
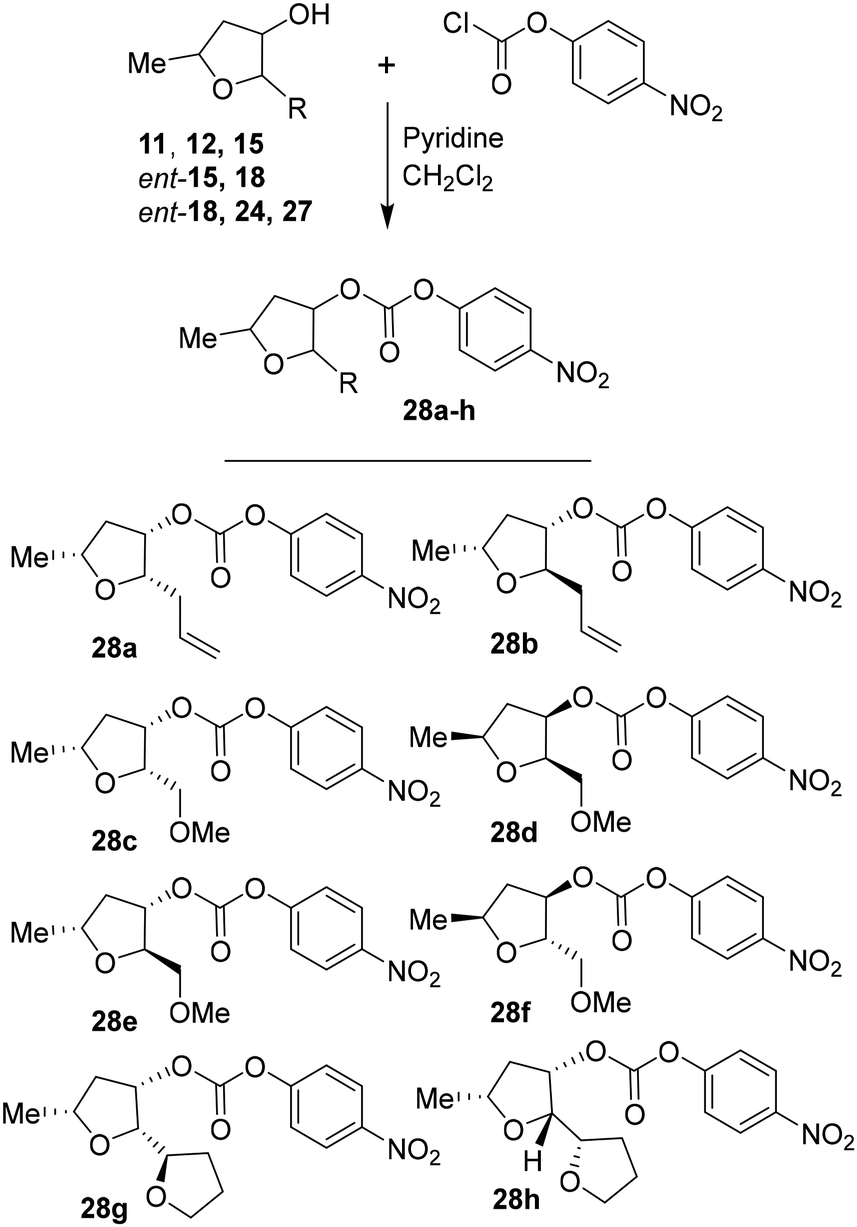 | ||
| Scheme 5 Synthesis of activated carbonates 28a–h. | ||
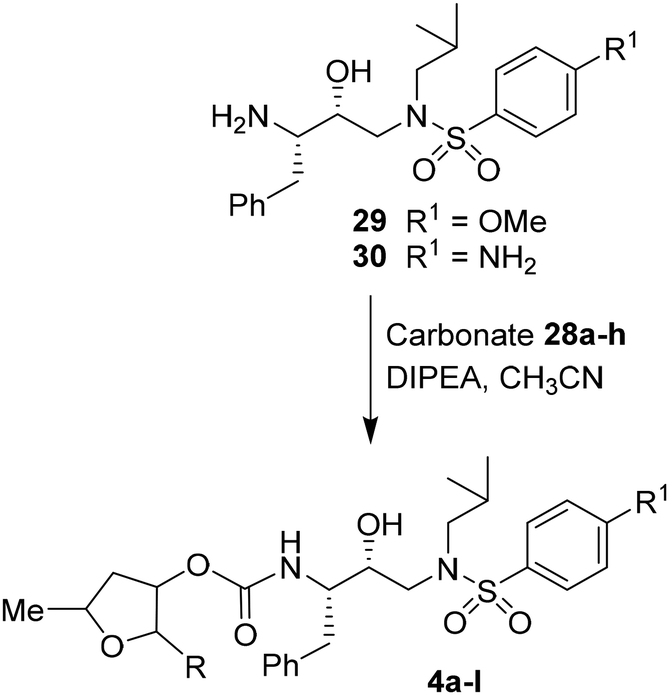 | ||
| Scheme 6 Synthesis of protease inhibitors 4a–l. | ||
| Entry | Inhibitor |
K
ia (nM) |
EC50b (μM) |
|---|---|---|---|
| a K i values represent at least 5 data points. Standard error in all cases was less than 7%. Darunavir exhibited Ki = 16 pM. b Values are means of at least three experiments in MT-2 cells. Darunavir exhibited EC50 = 1.6 nM. | |||
| 1 |
|
0.4 | >1 |
| 2 |
|
26 | >1 |
| 3 |
|
0.28 | >1 |
| 4 |
|
88 | >1 |
| 5 |
|
1.2 | >1 |
| 6 |
|
35.3 | >1 |
| 7 |
|
0.78 | >1 |
| 8 |
|
0.049 | >1 |
| 9 |
|
9.4 | >1 |
| 10 |
|
2.6 | >1 |
| 11 |
|
0.31 | >1 |
| 12 |
|
0.044 | >1 |
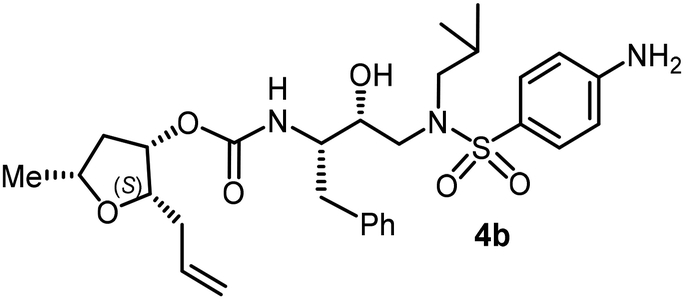
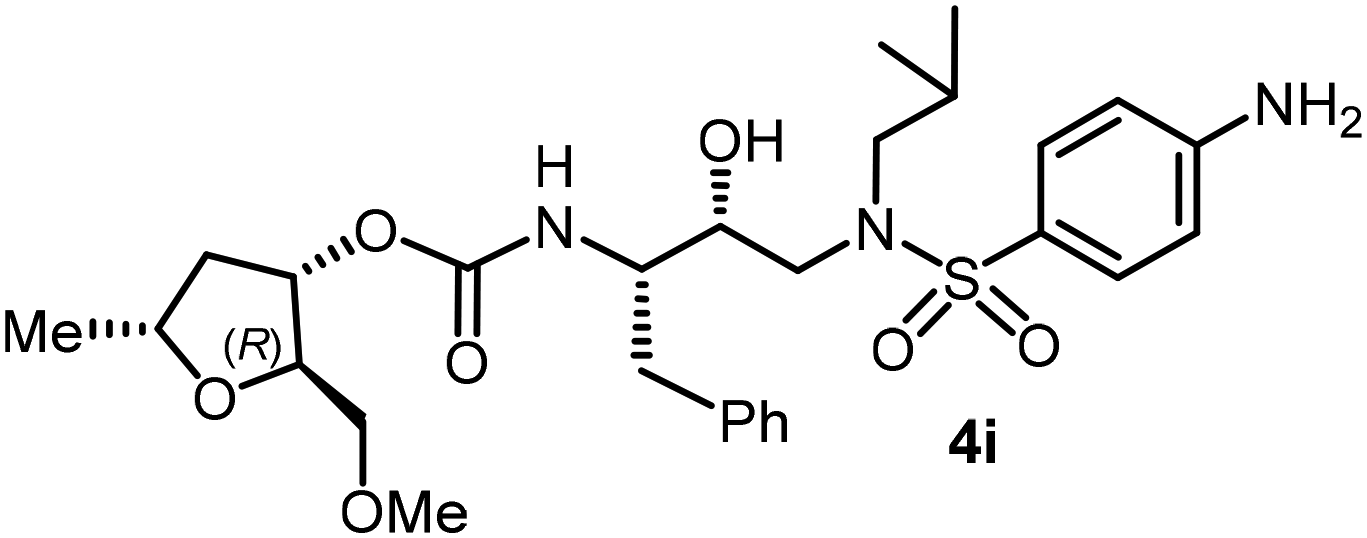
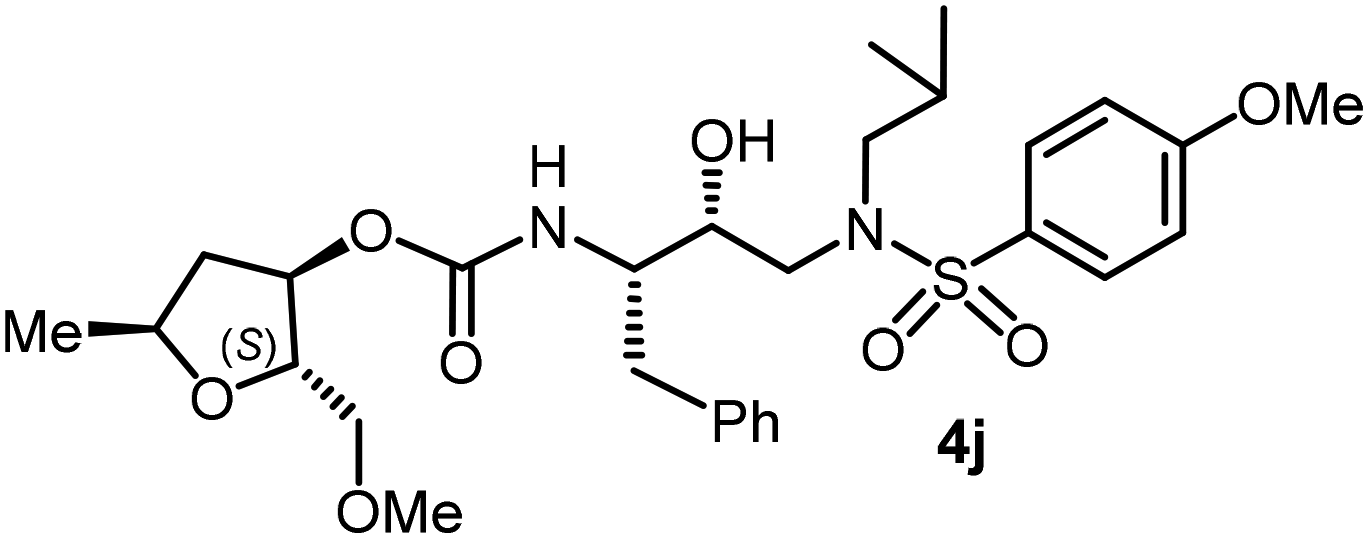
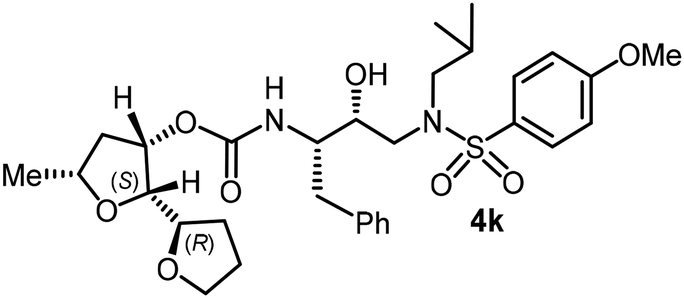
The selected PIs containing tri-substituted tetrahydrofuran derivatives as the P2-ligands were designed and synthesized to assess their interactions in the S2-subsite with respect to the bis-THF ligand of darunavir.16 The structure and activity of these new PIs are shown in Table 3. The synthetic PIs were first evaluated using the spectrofluorometric assay described by Toth and Marshall.35 As shown, inhibitor 4a with C-2(S) allyl and C-5(R) methyl substituents showed a HIV-1 protease inhibitory Ki value of 0.4 nM. We then determined the antiviral activity of these PIs in MT-2 human-T-lymphoid cells exposed to HIVLAI.30 In this cell-based antiviral assay, inhibitor 4a did not exhibit any appreciable antiviral activity.36,37 Compound 4b with 4-aminosulfonamide as the P2′-ligand displayed significant loss of HIV-1 protease inhibitory activity (entry 2). Compound 4c with a C2(S) methoxymethyl substituent was designed to make hydrogen bonding interactions in the S2-subsite. However, compound 4c exhibited marginal improvement of protease inhibitory activity over compound 4a without a polar substituent at the C2 position (entry 3). It turns out that the choice of P2′-ligand is important for activity as the 4-methoxysulfonamide P2′ ligand showed enhanced potency over the 4-aminosulfonamide P2′ ligand as compound 4d exhibited over 300-fold reduction of protease inhibitory activity over compound 4c. Interestingly, none of these derivatives showed any appreciable antiviral activity (entries 1–4). We then examined the effect of the enantiomeric P2 ligand in compounds 4e and 4f. However, compound 4e showed a nearly 4-fold reduction of enzyme activity over 4c which contains an enantiomeric P2-ligand. In compound 4g, we altered the C2 lipophilic allyl side chain stereochemistry. However, compound 4g displayed HIV-1 protease inhibitory activity that was comparable to that of compound 4a containing the C2(S)-allyl substituent (entries 1 and 7). Compound 4h with a C2(R)-methoxymethyl substituent exhibited very potent enzyme inhibitory activity (Ki 49 pM) compared to compound 4c with a C2(S)-methoxymethyl substituent. The corresponding compound 4i with a P2′-aminosulfonamide ligand showed significant reduction of enzyme Ki value (entries 8 and 9). Compound 4j with an enantiomeric P2 ligand is significantly less potent than compound 4h, indicating that the C2(R)-methoxymethyl stereochemistry is more suitable for polar and van der Waals interactions in the S2 subsite. We then examined the effect of a C2-tetrahydrofuranyl substituent in compounds 4k and 4l, differing the stereochemistry at the C2′-position. Interestingly, compound 4k with a 2(S),2′(R) tetrahydrofuranyl–tetrahydrofuran ligand exhibited HIV-1 protease inhibitory activity (Ki 0.31 nM) that was comparable to that of compound 4c with a C2 methoxymethyl side chain (entries 3 and 11). Compound 4l, on the other hand, with a diastereomeric tetrahydrofuranyl–tetrahydrofuran ligand (2S,2S′ configuration) displayed very potent HIV-1 protease inhibitory activity with a Ki value of 44 pM comparable to that of darunavir (entry 12). However, both compounds 4k and 4l did not exhibit any appreciable antiviral activity. The reason for the lack of antiviral activity is not clear. The observed high EC50 values may be due to fact that these compounds do not penetrate the cell. We and others observed such differences between HIV-1 inhibitory activity and cell-based antiviral activity, previously.38,39 The compounds in Table 3 have also shown relatively higher clogP (lipophilicity) values (4l, clogP 5) than darunavir (clogP 2.9). Compound 4l showed marked HIV-1 protease affinity, possibly due to strong hydrogen bonding interactions with the backbone residues as well as van der Waals interactions in the S2 subsite of HIV-1 protease.
To obtain molecular insight into the HIV-1 protease–inhibitor interactions, we determined the X-ray crystal structure of HIV-1 protease complexed with compound 4l. An active site interaction of 4l-bound HIV-1 protease structure is shown in Fig. 2. The X-ray structure of wild-type HIV-1 protease co-crystallized with inhibitor 4l (GRL-072-17A) was determined and refined to 1.32 Å resolution. Two conformations of the inhibitor with a C2-symmetry were visible in the active site of the protease dimer at relative occupancy of 0.55/0.45. The protease dimer structure is similar to that of the PR/DRV complex with a RMSD of 0.17 Å for 198 equivalent Cα atoms.25 Most differences in Cα positions are less than 0.4 Å. The largest disparities of about 0.6 Å for Pro81′ and the adjacent Gly49 and Gly49′ are likely due to the large P2 group of (2S,2′S,5R)-5-methyloctahydro-2,2′-bifuran instead of the bis-tetrahydrofuran of DRV.25
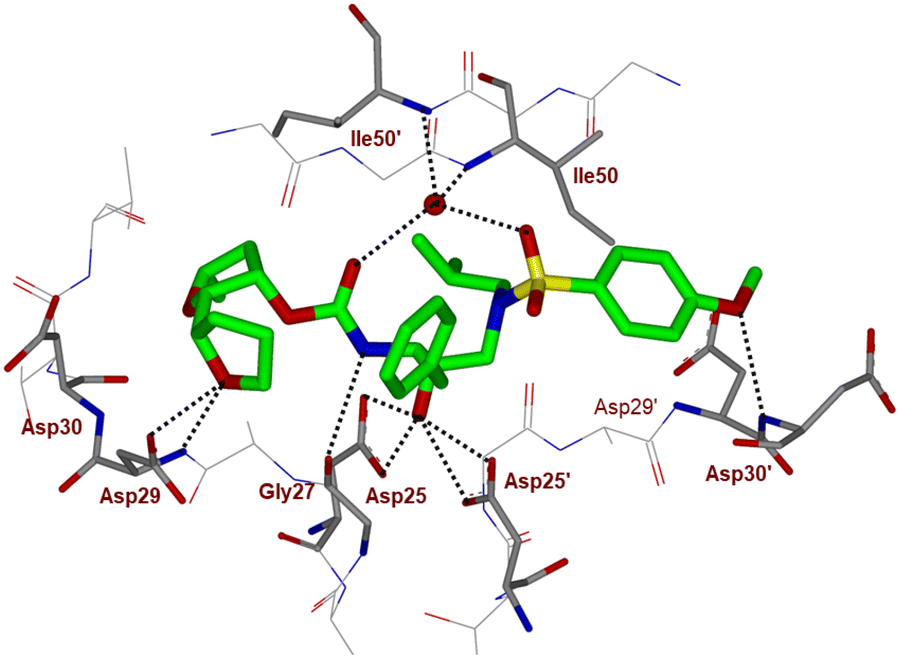 | ||
| Fig. 2 X-ray structure of inhibitor 4l (green color)-bound to the active site of wild-type HIV-1 protease (PDB code: 9B2H). All strong hydrogen bonding interactions are shown as dotted lines. | ||
The inhibitor retains the majority of hydrogen bonds observed between DRV and the main chain atoms of protease with the exception of the altered P2 group. An overlay of X-ray structures of darunavir-bound HIV-1 protease and compound 4l-bound HIV-1 protease is shown in Fig. 3. The number of van der Waals interactions for the P2 tetrahydrofuranyl–tetrahydrofuran ligand of inhibitor 4l with the protease are reduced compared to those of protease and darunavir interactions. The new P2 group has an added methyl group and a single bond to separate the two tetrahydrofurans in the opposite direction to create a tetrahydrofuranyl–tetrahydrofuran derivative. The changes in the P2 group increase its flexibility in the binding cavity of the HIV protease. The P1–P2′ scaffold which links the P2 group with the rest of the inhibitor shifts toward the flap to create space for the additional methyl group. The atom bearing the methyl group sinks into a hydrophobic bowl-shaped depression and forms new van der Waals interactions with the hydrophobic side chains of Ala28, Val32 and Ile84, while maintaining a weak C–H⋯O interaction with the carboxyl oxygen of Asp30. The flexible linkage allows the outer tetrahydrofuran ring to reduce the unfavorable interaction with the carboxyl oxygen of Gly48 to 3.5 Å compared with 3.0 Å in the darunavir-bound HIV-1 protease structure.25 The oxygen atom of the outer tetrahydrofuran in the P2 group preserves hydrogen bonds with the amide nitrogen atom and side chain oxygen of Asp29 at distances of 2.9 Å and 3.3 Å, respectively. However, the oxygen atom of the inner tetrahydrofuran has lost both hydrogen bonds to the amide nitrogen atoms of Asp29 and Asp30 (interatomic distances of 3.8 Å) unlike the bis-THF ligand of darunavir. Overall, the alteration in the P2 group decreases the inhibitor interactions with HIV protease compared to those seen in the bis-THF ligand with the HIV-1 protease active site.24,25
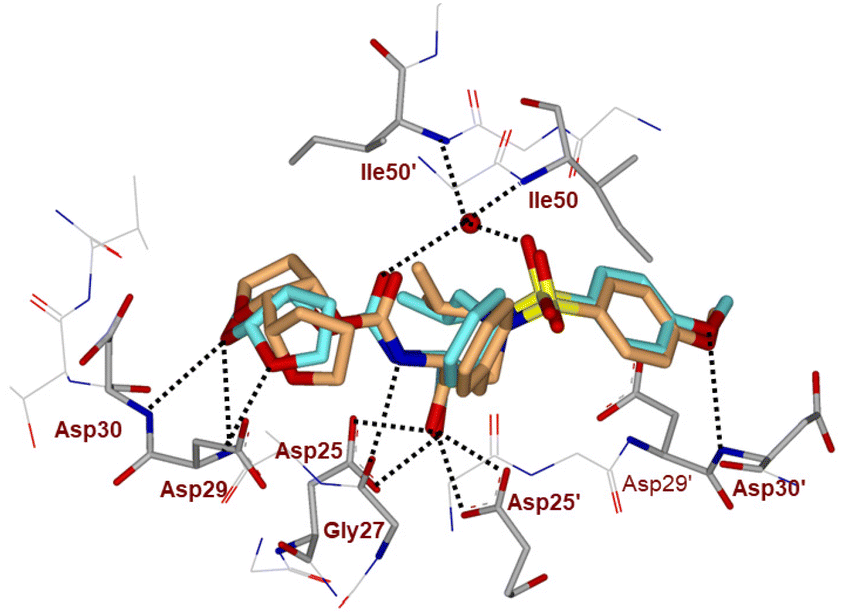 | ||
| Fig. 3 Overlay of the X-ray structure of inhibitor 4l (orange color)-HIV-1 protease (PDB code: 9B2H) and X-ray structure of TMC-126 (turquoise color)-bound HIV-1 protease (PDB code: 3I7E). | ||
Conclusion
In summary, we investigated the use of a series of substituted tetrahydrofuran derivatives as P2 ligands for HIV-1 protease inhibition. These ligands were designed based upon the X-ray crystal structure of darunavir-bound HIV-1 protease. In particular, the substituted tetrahydrofuran derivatives containing polar and lipophilic substituents are designed to form hydrogen bonds and fill in the hydrophobic pocket similar to the bis-THF ligand of darunavir in the S2 subsite of HIV-1 protease. The syntheses of the substituted tetrahydrofuran were carried out in a stereoselective manner in an optically active form. In general, inhibitors containing these ligands exhibited very potent HIV-1 protease inhibitory activity and a number of compounds displayed protease inhibitory activity comparable to darunavir. The influence of ligand stereochemistry is clearly evident. Compound 4h with a 2(R)-methoxymethyl side chain exhibited over 5-fold improvement of enzyme affinity compared to compound 4c with a 2(S)-methoxymethyl side chain. Also, inhibitors 4k and 4l with tetrahydrofuranyl–tetrahydrofuran moieties with C2(R) and C2(S) configuration, respectively, displayed preference for the C2(S) configuration as compound 4l exhibited a HIV-1 protease inhibitory Ki of 44 pM, comparable to that of darunavir (Ki 14 pM). Several inhibitors containing these stereochemically defined ligands displayed very potent HIV-1 protease inhibitory activity; however they did not exhibit any appreciable antiviral activity. Inhibitors containing the P′2-methoxysulfonamide ligand were significantly more potent than inhibitors with the P′2-aminosulfonamide ligand. We determined the high resolution X-ray structure of the inhibitor 4l-bound HIV-1 protease. The structure revealed that C2 substituted THF oxygen forms a strong hydrogen bond with the backbone Asp29 amide NH in the S2 subsite. However, the oxygen atom of the inner THF ring does not form any hydrogen bond with the Asp29 and Asp30 backbone NH, like the bis-THF ligand of darunavir. This loss of the backbone hydrogen bonding interaction as well higher lipophilicity may be responsible for the lack of antiviral activity of these PIs. Further design and optimization of inhibitor properties are in progress in our laboratory.Experimental section
All reactions were carried out under an inert atmosphere, either N2 or Ar, using magnetic stirring and oven-dried glassware. All solvents were anhydrous and distilled prior to use. Dichloromethane and triethylamine were distilled from calcium hydride. Tetrahydrofuran, diethyl ether, and benzene were distilled from sodium/benzophenone. All other solvents were of HPLC grade or better. Flash column chromatography was performed using EM Science 60–200 mesh silica gel. Thin-layer chromatography was performed using 60 F-254 E. Merck silica gel plates. 1H- and 13C-NMR were recorded using Bruker AV-400, Avance DRX-500, Varian Mercury-Vx-300, and Gemini-2300 spectrometers and Me4Si as an internal standard. Optical rotations were recorded on a PerkinElmer 341 polarimeter. A Thermo Finnigan LCQ Classic mass was used for MS analyses. The purity of the test compounds was determined by HRMS and HPLC analysis. All test compounds showed ≥95% purity.3-Hydroxy-5-methyldihydrofuran-2(3H)-one ((±)-6)
To a stirred solution of α-methylene-γ-butyrolactone 5 (6.12 g, 54.6 mmol) in CH2Cl2 (20 mL) at −78 °C was bubbled a stream of ozone until blue color persisted. The ozone stream was then stopped and purged with a stream of oxygen to remove the excess ozone. After adding dimethyl sulfide (12 mL, 163.7 mmol), the reaction mixture was warmed to 23 °C and stirred for 4 h. The reaction mixture was concentrated under reduced pressure and the crude product (4.9 g, 79% yield) was used for the next step without any further purification.To a stirred solution of the unsaturated lactone intermediate (3.5 g, 31 mmol) in EtOAc (20 mL) was added Pd–C (150 mg, 10 wt%). The resulting solution was stirred at 23 °C under 1 atm H2 gas over 24 h. Upon completion of the reaction, the mixture was filtered through a plug of Celite, and the solvents were removed under reduced pressure. The crude product was purified by silica gel column chromatography (50% EtOAc in hexanes) to give racemic lactone 6 (2.87 g, 79%). 1H NMR (400 MHz, CDCl3) δ: 4.57 (dd, J = 11.3, 8.4 Hz, 1H), 4.54–4.45 (m, 1H), 4.03 (brs, 1H), 2.70 (ddd, J = 12.6, 8.4, 5.1 Hz, 1H), 1.90–1.80 (m, 1H), 1.43 (d, J = 6.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ, 178.2, 73.9, 69.1, 38.8, 20.9.
(3S,5R)-3-Hydroxy-5-methyldihydrofuran-2(3H)-one ((+)-6)
To a solution of racemic lactone 6 (960 mg, 8.23 mmol) in THF (15 mL) were added vinyl acetate (13.3 mL, 144.9 mmol) and lipase PS-30 (0.9 g) at 23 °C under an argon atmosphere. The reaction mixture was stirred for 2 h (50:50 by 1H NMR). After this period, the reaction mixture was filtered through a plug of Celite and solvents were removed under reduced pressure. The crude product was purified by silica gel column chromatography (20% to 50% EtOAc in hexanes) to give alcohol (+)-6 (485 mg, 50% yield) as a colorless oil and acetate 7 (650 mg, 50% yield). Alcohol (+)-6; 1H NMR (400 MHz, CDCl3) δ: 4.58 (dd, J = 11.2, 8.4 Hz, 1H), 4.52–4.44 (m, 1H), 4.18 (brs, 1H), 2.69 (ddd, J = 12.9, 8.3, 5.1 Hz, 1H), 1.89–1.79 (m, 1H), 1.42 (d, J = 6.3 Hz, 3H); 13C (100 MHz, DMSO-d6) δ: 178.3, 73.9, 69.0, 38.8, 20.9; LRMS-ESI (m/z): 139.2 (M + Na)+; [α]20D +2.8 (c 1.0, CHCl3). Acetate 7; 1H NMR (400 MHz, CDCl3) δ: 5.45 (dd, J = 10.9, 8.6 Hz, 1H), 4.59–4.47 (m, 1H), 2.76 (ddd, J = 12.7, 8.5, 5.3 Hz, 1H), 2.10 (s, 3H), 1.83 (dt, J = 12.5, 10.6 Hz, 1H), 1.43 (d, J = 6.3 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ, 172.5, 169.8, 73.6, 69.0, 36.7, 21.0, 20.6; LRMS-ESI (m/z): 181.2 (M + Na)+; [α]20D −18.45 (c 1.0, CHCl3).
(3R,5S)-3-Hydroxy-5-methyldihydrofuran-2(3H)-one (ent-6)
To a stirred solution of acetate 7 (320 mg, 2.0 mmol) in MeOH (5 mL) was added aqueous NaOH (10% solution, 5 mL) and the mixture was stirred at 23 °C for 12 h. After this period, the reaction mixture was acidified with 1 N HCl solution and solvents were concentrated under reduced pressure. The aqueous layer was extracted multiple times with EtOAc. The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography (60% EtOAc in hexane) to give alcohol ent-6 (144 mg, 61%) as a colorless oil. LRMS-ESI (m/z): 139.2 (M + Na)+; [α]20D −2.3 (c 1.0, CH3OH); for NMR data, please see lactone (+)-6.(3S,5R)-3-((tert-Butyldimethylsilyl)oxy)-5-methyldihydrofuran-2(3H)-one (8)
To a stirred solution of the optically active alcohol (+)-6 (485 mg, 4.2 mmol) in CH2Cl2 (10 mL) were added 2,6-lutidine (1.45 ml, 12.5 mmol) and TBSOTf (1.45 mL, 6.27 mmol) at 0 °C under an argon atmosphere. The reaction mixture was warmed to 23 °C and stirred for 1 h. The reaction was quenched by the addition of saturated aqueous NaHCO3 and extracted with CH2Cl2. The extracts were washed with saturated aqueous NaCl, dried (Na2SO4), and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (10% EtOAc in hexanes) and TBS lactone 8 (923 mg, 96%) was obtained as a white amorphous solid; 1H NMR (400 MHz, CDCl3) δ: 4.46–4.34 (m, 2H), 2.56 (ddd, J = 12.6, 8.1, 5.3 Hz, 1H), 1.77 (dt, J = 12.4, 10.3 Hz, 1H), 1.37 (d, J = 6.3 Hz, 3H), 0.85 (d, J = 3.8 Hz, 9H), 0.10 (d, J = 17.0 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ, 175.8, 72.6, 69.8, 40.0, 25.7, 21.1, 18.2, −4.7, −5.3; LRMS-ESI (m/z): 253.2 (M + Na)+; HRMS (ESI), calcd for C11H23O3Si: m/z 231.1416 [M + H]+, found 231.1408; [α]20D −16.0 (c 1.0, CHCl3).(((2S,3S,5R)-2-Allyl-5-methyltetrahydrofuran-3-yl)oxy)(tert-butyl)dimethylsilane (9)
To a stirred solution of lactone 8 (415 mg, 1.80 mmol) in CH2Cl2 (16 mL) at −78 °C was added DIBAL-H (1 M in hexanes, 2.70 ml, 2.70 mmol) under an argon atmosphere and stirred at the same temperature for 2 h. The reaction was quenched by the addition of MeOH (3 mL) and warmed to 23 °C. Then, a saturated aqueous solution of sodium potassium tartrate was added and stirred vigorously at 23 °C for 2 h until it formed a white suspension. The white suspension was filtered through a plug of Celite and the filtrate was concentrated under reduced pressure. To crude lactol (417 mg) were added DMAP (44 mg, 0.36 mmol), Et3N (1 mL, 7.19 mmol) and Ac2O (0.42 mL, 4.50 mmol) at 0 °C under an argon atmosphere and stirred for 2 h. Upon completion of the reaction, the solvents were concentrated under reduced pressure and the crude residue was purified by silica gel column chromatography (15% EtOAc in hexane) to give the acetate intermediate (445 mg, 90% yield in two steps).To a solution of the acetate intermediate (344 mg, 1.26 mmol) in CH2Cl2 (12.6 mL, 0.1 M) was added allyl trimethylsilane (0.8 mL, 5 mmol) at 23 °C under an argon atmosphere and then cooled to −78 °C. After the addition of SnBr4 (660 mg, 1.50 mmol), the mixture was warmed to 23 °C over 2 h. Upon completion, the reaction was quenched by the addition of saturated aqueous Na2HPO4 and extracted with CH2Cl2. The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. 1H NMR analysis of the unpurified crude product showed a pair of diastereomers in a 10:1 ratio. The crude product was purified by silica gel column chromatography (70% CH2Cl2 in hexanes) to give the major olefin 9 (256 mg, 80%) as a colorless oil and the minor olefin 10 (25 mg, 8%) as a colorless oil. Olefin 9: 1H NMR (400 MHz, CDCl3) δ: 5.85 (ddt, J = 17.1, 10.2, 6.9 Hz, 1H), 5.09 (d, J = 17.2 Hz, 1H), 5.02 (d, J = 10.2 Hz, 1H), 4.22 (dt, J = 6.4, 3.7 Hz, 1H), 3.96–3.89 (m, 1H), 3.63 (q, J = 6.2 Hz, 1H), 2.42–2.32 (m, 2H), 2.26 (dt, J = 13.4, 6.9 Hz, 1H), 1.47 (ddd, J = 13.0, 6.6, 2.9 Hz, 1H), 1.29 (d, J = 6.2 Hz, 3H), 0.89 (s, 9H), 0.04 (d, J = 4.3 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ, 135.9, 116.4, 83.2, 73.7, 73.5, 43.5, 34.4, 25.9, 22.2, 18.2, −4.4, −4.9; LRMS-ESI (m/z): 279.3 (M + Na)+; HRMS (APCI), calcd for C14H29O2Si: m/z 257.1936 [M + H]+ found m/z 257.1935; [α]20D +16.8 (c 1.0, CHCl3).
(2S,3S,5R)-2-Allyl-5-methyltetrahydrofuran-3-ol (11)
To a stirred solution of olefin 9 (21 mg, 0.08 mmol) in THF (2 mL) was added TBAF solution (1M in THF, 0.20 mL, 0.20 mmol) at 0 °C under an argon atmosphere. The reaction mixture was warmed to 23 °C and stirred for 3 h. After this period, the reaction was quenched with water and the aqueous layer was extracted with EtOAc. The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography (20% EtOAc in hexane) to give alcohol 11 (10 mg, 86%) as a white amorphous solid. 1H NMR (400 MHz, CDCl3) δ: 5.94–5.82 (m, 1H), 5.20–5.05 (m, 2H), 4.27–4.17 (m, 1H), 3.98–3.88 (m, 1H), 3.66–3.55 (m, 1H), 2.50–2.35 (m, 3H), 1.70 (brs, 1H), 1.54–1.44 (m, 1H), 1.33 (d, J = 6.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ, 134.7, 117.0, 82.3, 73.5, 73.1, 43.1, 33.4, 21.9; LRMS-ESI (m/z): 165.1 (M + Na)+; HRMS (APCI), calcd for C8H15O2: m/z 143.1072 [M + H]+, found m/z 143.1069; [α]20D +8.6 (c 0.33, CHCl3).(3R,5S)-3-((tert-Butyldimethylsilyl)oxy)-5-methyldihydrofuran-2(3H)-one (ent-8)
Lactone ent-8 (268 mg, 94% yield) was obtained as a white amorphous solid from alcohol ent-6 (144 mg, 1.24 mmol) by following the procedure described above for its enantiomer 8. For NMR data, please see lactone 8; LRMS-ESI (m/z): 253.2 (M + Na)+; HRMS (ESI), calcd for C11H23O3Si: m/z 231.1416 [M + H]+, found 231.1414; [α]20D +15.5 (c 1.0, CHCl3).(((2R,3R,5S)-2-Allyl-5-methyltetrahydrofuran-3-yl)oxy)(tert-butyl)dimethylsilane (ent-9)
By following the procedure outlined for the preparation 9, olefin ent-9 was obtained as a colorless oil (210 mg, 0.77 mmol) with dr 10:1 after the diastereoselective allylation reaction. LRMS-ESI (m/z): 257 (M + H)+; HRMS (APCI), calcd for C14H29O2Si: m/z 257.1936 [M + H]+ found m/z 257.1934; [α]20D −14.0 (c 1.0, CHCl3). For NMR data, please see the enantiomeric compound 9.
(2R,3R,5S)-2-Allyl-5-methyltetrahydrofuran-3-ol (ent-11)
By following the procedure outlined for the preparation of its enantiomer 11, alcohol ent-11 was obtained as an amorphous solid. LRMS-ESI (m/z): 165.1; HRMS (APCI), calcd for C8H15O2: m/z 143.1072 [M + H]+, found m/z 143.1077. Using similar deprotection to TBS ether 10 provided alcohol 12.tert-Butyldimethyl(((2S,3S,5R)-5-methyl-2-(prop-1-en-1-yl)tetrahydrofuran-3-yl)oxy)silane 13
To a stirred mixture of olefin 9 (144 mg, 0.56 mmol) and vinyloxytrimethylsilane (0.84 mL, 5.60 mmol) in CH2Cl2 (45 mL) was added the Grubb's second generation catalyst (48 mg, 0.056 mmol) at 23 °C under an argon atmosphere. The reaction mixture was refluxed at 110 °C for 24 h in a sealed tube. The mixture was cooled to 23 °C and concentrated under reduced pressure to remove solvents. The crude residue was purified by silica gel column chromatography (2% diethyl ether in hexanes) to give olefin 13 (136 mg, 95%) as a colorless oil; 1H NMR (400 MHz, CDCl3) δ: 5.73–5.62 (m, 2H), 4.24–4.23 (m, 1H), 4.04–3.97 (m, 2H), 2.31–2.28 (m, 1H), 1.71 (d, J = 5.2 Hz, 3H), 1.60–1.50 (m, 1H), 1.33 (d, J = 5.2 Hz, 3H), 0.88 (s, 9H), 0.02 (s, 6H); 13C NMR (100 MHz, CDCl3) δ, 129.4, 128.4, 84.5, 74.9, 73.6, 43.4, 25.7. 22.0, 18.1, 17.8, −4.4, −4.7; LRMS-ESI (m/z): 279.3 (M + Na)+; HRMS (APCI), calcd for C14H29O2Si: m/z 257.1936 [M + H]+ found m/z 257.1936; [α]20D +28.0 (c 1.0, CHCl3).((2S,3S,5R)-3-((tert-Butyldimethylsilyl)oxy)-5-methyltetrahydrofuran-2-yl)methanol 14
A solution of olefin 13 (36 mg, 0.14 mmol) in a 1:1 mixture of CH2Cl2 and MeOH (3.6 mL) was cooled down to −78 °C. Ozone was passed into the solution until the color of the solution turned pale blue. The reaction mixture was then purged with oxygen for 5 min and NaBH4 (16 mg, 0.43 mmol) was added. After stirring for 2 h at −78 °C, the reaction mixture was diluted with EtOAc and quenched with saturated aqueous NH4Cl. The aqueous layer was extracted with EtOAc. The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography (10% EtOAc in hexanes to 30% EtOAc in hexanes) to give alcohol 14 (32 mg, 92% yield in two steps) as a white amorphous solid; 1H NMR (400 MHz, CDCl3) δ: 4.54–4.46 (m, 1H), 3.98–3.92 (m, 1H), 3.83–3.77 (m, 3H), 2.32–2.27 (m, 1H), 1.36–1.27 (m, 1H), 1.24 (d, J = 6.2 Hz, 3H), 0.90 (s, 9H), 0.04 (s, 6H); 13C NMR (100 MHz, CDCl3) δ, 81.2, 74.5, 73.5, 62.7, 43.4, 25.6, 21.3, 17.8, −4.7, −5.3; LRMS-ESI (m/z): 269.2 (M + Na)+; HRMS (APCI), calcd for C12H27O3Si: 247.1729 m/z [M + H]+, found m/z 247.1725; [α]20D +13.9 (c 1.0, CHCl3).
(2S,3S,5R)-2-(Methoxymethyl)-5-methyltetrahydrofuran-3-ol 15
To a stirred solution of alcohol 14 (32 mg, 0.13 mmol) in THF (2.6 mL) was added NaH (60% dispersion in mineral oil, 31 mg, 0.77 mmol) at 0 °C under an argon atmosphere. After stirring for 10 min at 0 °C, MeI (88 μL, 1.42 mmol) was added and the reaction mixture was slowly warmed to 23 °C over 3 h. Upon completion, the reaction was quenched with saturated aqueous NH4Cl and the aqueous layer was extracted with diethyl ether. The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by silica gel column chromatography gave the corresponding methyl ether 14a (21 mg, 0.08 mmol) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 4.35–4.29 (m, 1H), 3.98 (q, J = 6.8 Hz, 1H), 3.81 (q, J = 6.4 Hz, 1H), 3.58–3.52 (m, 2H), 3.38 (s, 3H), 2.32–2.25 (m, 1H), 1.54–1.44 (m, 1H), 1.32 (d, J = 6.4 Hz, 3H), 0.88 (s, 9H), 0.04 (s, 6H); 13C NMR (100 MHz, CDCl3) δ, 81.8, 74.1, 73.1, 72.1, 59.1, 43.4, 25.6, 21.8, 17.9, −4.8, −5.3; LRMS-ESI (m/z): 283.2 (M + Na)+; [α]20D +18.7 (c 1.0, CHCl3).To a stirred solution of the above-mentioned methyl ether (16 mg, 0.06 mmol) in THF (2 mL) was added TBAF solution (1M in THF, 0.15 mL, 0.15 mmol) at 0 °C under an argon atmosphere. The reaction mixture was warmed to room temperature and stirred for 3 h. The reaction was quenched with water and the aqueous layer was extracted with CH2Cl2. The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude residue was purified by flash silica gel column chromatography (40% EtOAc in hexanes) to give alcohol 15 (8 mg, quantitative) as a white amorphous solid. 1H NMR (400 MHz, CDCl3) δ: 4.44–4.39 (m, 1H), 3.99–3.89 (m, 1H), 3.85–3.60 (m, 3H), 3.41 (s, 3H), 2.75 (brs, 1H), 2.44–2.32 (m, 1H), 1.56–1.45 (m, 1H), 1.33 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ, 80.2, 73.9, 73.8, 71.6, 59.4, 43.2, 21.4; LRMS-ESI (m/z): 169.1 (M + Na)+; HRMS (APCI), calcd for C7H15O3: 147.1021 m/z [M + H]+, found m/z 147.1015; [α]20D +3.8 (c 0.33, CHCl3).
(2R,3R,5S)-2-(Methoxymethyl)-5-methyltetrahydrofuran-3-ol (ent-15)
Alcohol ent-15 (9 mg, 93% yield) was prepared as a white amorphous solid from compound ent-9 by following the procedure described above for its enantiomer 9. LRMS-ESI (m/z): 169.1 (M + Na)+; HRMS (APCI), calcd for C7H15O3: 147.1021 m/z [M + H]+, found m/z 147.1017; [α]20D −4.2 (c 1.0, CHCl3). For NMR data, please see compound 15.(((2R,3S,5R)-2-Allyl-5-methyltetrahydrofuran-3-yl)oxy)(tert-butyl)dimethylsilane (10)
To a stirred solution of lactone 8 (317 mg, 1.38 mmol) in CH2Cl2 (14 mL) was added allyl magnesium bromide solution (1.0 M in Et2O, 1.65 mL, 1.65 mmol) at −78 °C under an argon atmosphere. After stirring for 3 h at the same temperature, the reaction was quenched by the addition of saturated aqueous NH4Cl and the aqueous layer was extracted with EtOAc. The combined organic layers were washed with brine, dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude product was passed through a flash silica gel column to afford the hemiketal (282 mg, 61%).To the hemiketal (282 mg, 1.04 mmol) in CH2Cl2 (8.5 mL) were consecutively added Et3SiH (1.03 mL, 6.47 mmol) and BF3 OEt2 (0.38 mL, 3.11 mmol) at −78 °C under an argon atmosphere. After stirring for 2 h at the same temperature, the reaction was quenched by the addition of saturated aqueous NaHCO3 and the aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with brine, dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (5% EtOAc in hexanes) to afford 10 (154 mg, 58%) as a yellow oil; 1H NMR (400 MHz, CDCl3) δ: 5.84 (ddt, J = 17.0, 9.8, 6.8 Hz, 1H), 5.14–5.01 (m, 2H), 4.20–4.11 (m, 1H), 4.04–3.97 (m, 1H), 3.85–3.79 (m, 1H), 2.35–2.27 (m, 1H), 2.28–2.16 (m, 2H), 1.57–1.50 (m, 1H), 1.28 (d, J = 6.2 Hz, 3H), 0.88 (s, 9H), 0.05 (s, 6H); 13C NMR (101 MHz, CDCl3) δ: 134.8, 116.8, 83.5, 76.1, 73.6, 42.4, 37.6, 25.7, 22.1, 17.8, −4.6, −4.9; LRMS-ESI (m/z): 279.3 (M + Na)+; HRMS (ESI), calcd for C14H29O2Si: m/z 257.1936 [M + H]+, found m/z 257.1930; [α]20D +30.8 (c 1.0, CHCl3).
tert-Butyldimethyl(((2R,3S,5R)-5-methyl-2-(prop-1-en-1-yl)tetrahydrofuran-3-yl)oxy)silane 16
By following the procedure outlined for the preparation of olefin 13, olefin 16 (40 mg, 34%; 61% brsm) was obtained as a colorless oil from compound 10 (115 mg, 0.45 mmol) after terminal olefin migration; 51 mg of the starting material 10 was recovered. 1H NMR (400 MHz, CDCl3) δ: 5.78–5.65 (m, 1H), 5.44–5.31 (m, 1H), 4.23–4.15 (m, 1H), 4.12–3.95 (m, 2H), 2.30–2.21 (m, 1H), 1.69 (d, J = 4.1 Hz, 3H), 1.68–1.55 (m, 1H), 1.28 (d, J = 4.1 Hz, 3H), 0.87 (s, 9H), 0.03 (s, 6H); 13C NMR (100 MHz, CDCl3) δ, 130.2, 128.6, 85.2, 76.6, 73.4, 42.4, 25.6, 22.2, 18.0, 17.6, −4.7, −5.3. LRMS-ESI (m/z): 279.3 (M + Na)+; HRMS (ESI), calcd for C14H29O2Si: m/z 257.1936 [M + H]+, found m/z 257.1938.((2R,3S,5R)-3-((tert-Butyldimethylsilyl)oxy)-5-methyltetrahydrofuran-2-yl)methanol (17)
By following the procedure outlined for the preparation of alcohol 14, alcohol 17 (16 mg, 85% yield in two steps) was obtained as a yellowish oil from olefin 16 (19 mg, 0.08 mmol) after ozonolysis and NaBH4 reduction. 1H NMR (400 MHz, CDCl3) δ: 4.28–4.14 (m, 2H), 3.86–3.79 (m, 1H), 3.80–3.50 (m, 2H), 2.31–2.20 (m, 1H), 2.01 (brs, 1H), 1.65–1.58 (m, 1H), 1.30 (d, J = 5.2 Hz, 3H), 0.88 (s, 9H), 0.06 (s, 6H); 13C NMR (100 MHz, CDCl3) δ, 84.6, 74.4, 72.8, 62.2, 42.9, 26.6, 21.8, 17.8, −4.7, −5.3. LRMS-ESI (m/z): 269.2 (M + Na)+; HRMS (APCI), calcd for C12H27O3Si: 247.1729 m/z [M + H]+, found m/z 247.1724; [α]20D +33.0 (c 1.0, CHCl3).(2R,3S,5R)-2-(Methoxymethyl)-5-methyltetrahydrofuran-3-ol (18)
By following the procedure outlined for the preparation of methyl ether 15, the corresponding methyl ether derivative (14 mg, 90% yield) was obtained as a yellow oil from alcohol 17 (15 mg, 0.06 mmol) after methylation. 1H NMR (400 MHz, CDCl3) δ: 4.44–4.15 (m, 2H), 3.90–3.85 (m, 1H), 3.51–3.47 (m, 1H), 3.40–3.38 (m, 1H), 3.38 (s, 3H), 2.28–2.20 (m, 1H), 1.60–1.52 (m, 1H), 1.29 (d, J = 6.2 Hz, 3H), 0.88 (s, 9H), 0.01 (s, 6H); 13C NMR (100 MHz, CDCl3) δ: 83.3, 74.1, 73.3, 72.8, 59.2, 42.7, 25.6, 21.8, 17.9, −4.6, −5.3; LRMS-ESI (m/z): 283.2 (M + Na)+; [α]20D +37.0 (c 0.67, CHCl3).By following the procedure outlined for the preparation of alcohol 15, alcohol 18 (7 mg, 93% yield) was obtained as a colorless oil (14 mg, 0.05 mmol) after TBAF deprotection. 1H NMR (400 MHz, CDCl3) δ: 4.30–4.14 (m, 2H), 3.96–3.88 (m, 1H), 3.54–3.47 (m, 1H), 3.46–3.40 (m, 1H), 3.38 (s, 3H), 2.43–2.35 (m, 1H), 2.12 (brs, 1H), 1.63–1.57 (m, 1H), 1.30 (d, J = 6.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 82.6, 74.4, 74.2, 73.4, 59.3, 42.2, 21.8; LRMS-ESI (m/z): 169.1 (M + Na)+; HRMS (APCI), calcd for C7H15O3: 147.1021 m/z [M + H]+, found m/z 147.1014; [α]20D +17.3 (c 0.33, CHCl3).
(((2S,3R,5S)-2-Allyl-5-methyltetrahydrofuran-3-yl)oxy)(tert-butyl)dimethylsilane (ent-10)
The allyl derivative ent-10 (43 mg, 28% in two steps) was obtained as a single isomer from lactone ent-8 (144 mg, 0.63 mmol) in two steps as a colorless oil by following the procedure described above for the allyl derivative 10. For NMR data, please see compound 10. LRMS-ESI (m/z): 279.3 (M + Na)+; HRMS (APCI), calcd for C14H29O2Si: m/z 257.1936 [M + H]+ found m/z 257.1937; [α]20D −35.2 (c 1.0, CHCl3).tert-Butyldimethyl(((2S,3R,5S)-5-methyl-2-(prop-1-en-1-yl)tetrahydrofuran-3-yl)oxy)silane (ent-16)
To a stirred mixture of olefin ent-10 (43 mg, 0.17 mmol) and vinyloxytrimethylsilane (0.25 mL, 1.68 mmol) in CH2Cl2 (8.2 mL) was added the second-generation Grubb's catalyst (7 mg, 0.008 mmol) at 23 °C under an argon atmosphere. The reaction mixture was heated to 110 °C and vigorously stirred for 24 h. After this period, the mixture was cooled to 23 °C and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography (2% diethyl ether in hexanes) to give olefin ent-16 (20 mg, 47%) as a colorless oil. For NMR data, please see compound 16. LRMS-ESI (m/z): 279.3 (M + Na)+; HRMS (ESI), calcd for C14H29O2Si: m/z 257.1936 [M + H]+, found m/z 257.1933.((2S,3R,5S)-3-((tert-Butyldimethylsilyl)oxy)-5-methyltetrahydrofuran-2-yl)methanol (ent-17)
Alcohol ent-17 (15 mg, 78%) was obtained as a yellow oil from olefin ent-16 (20 mg, 0.08 mmol) by following the procedure described above for compound 17. For NMR data, please see compound 17. LRMS-ESI (m/z): 269.2 (M + Na)+; HRMS (APCI), calcd for C12H27O3Si: 247.1729 m/z [M + H]+, found m/z 247.1729; [α]20D −32.2 (c 0.50, CHCl3).(2S,3R,5S)-2-(Methoxymethyl)-5-methyltetrahydrofuran-3-ol (ent-18)
Alcohol ent-17 (14.5 mg, 0.06 mmol) was converted to methyl ether (9.5 mg, 63%) and obtained as a yellow oil by following the procedure described above. Compound ent-18 (4.6 mg, 96%) was obtained after the removal of TBS ether (8 mg, 0.03 mmol) by following the procedure described above for compound 18. For NMR data, please see compound 18. LRMS-ESI (m/z): 169.1 (M + Na)+; HRMS (APCI), calcd for C7H15O3: 147.1021 m/z [M + H]+, found m/z 147.1015; [α]20D −16.7 (c 0.13, CHCl3).(R)-1-((2S,3S,5R)-3-((tert-Butyldimethylsilyl)oxy)-5-methyltetrahydrofuran-2-yl)but-3-en-1-ol (20)
Ozone gas was bubbled into a solution of isomerized olefin 13 (78 mg, 0.30 mmol) in CH2Cl2 (5 mL) at −78 °C for 10 min. After the solution turned pale blue, oxygen was bubbled into the solution for 10 min and triphenylphosphine (96 mg, 0.36 mmol) was added. The reaction mixture was stirred for 15 min at −78 °C and warmed to rt for 1 h. After this period, the solvent was concentrated under reduced pressure and the crude residue was passed through a flash silica gel column to afford aldehyde 19 (56 mg, 76% yield) as a colorless oil. This sensitive aldehyde was used immediately after the quick purification step with 50% EtOAc in hexanes.To a stirred solution of aldehyde 19 (56 mg, 0.23 mmol) in distilled CH2Cl2 (5 mL) were consecutively added allyl tributyl stannane (0.29 mL, 0.91 mmol) and BF3 OEt2 (85 μL, 0.69 mmol) at −78 °C under an argon atmosphere. The reaction mixture was slowly warmed to room temperature over 6 h and stirred at room temperature for 12 h. After this period, the reaction was quenched with saturated aqueous NH4Cl and the aqueous layer was extracted with CH2Cl2. The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography (5% EtOAc in hexanes to 10% EtOAc in hexanes) to give olefin 20 (52 mg, 79%) as a colorless oil with a dr of 4.2:1; 1H NMR (400 MHz, CDCl3) δ: 5.98–5.86 (m, 1H), 5.21–5.05 (m, 2H), 4.56 (dt, J = 6.6, 5.5 Hz, 1H), 4.02–3.84 (m, 2H), 3.56 (dd, J = 7.5, 5.4 Hz, 1H), 3.00 (brs, 1H), 2.57–2.48 (m, 1H), 2.35–2.23 (m, 2H), 1.54 (ddd, J = 12.6, 8.1, 5.5 Hz, 1H), 1.29 (d, J = 6.1 Hz, 3H), 0.91 (s, 9H), 0.11 (d, J = 2.3 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ, 135.2, 117.1, 82.6, 74.3, 73.4, 70.2, 42.7, 38.3, 25.7, 21.7, 17.8, −4.4, −5.3; LRMS-ESI (m/z): 309.2 (M + Na)+; HRMS (APCI), calcd for C15H31O3Si: m/z 287.2042 [M + H]+ found m/z 287.2043; [α]20D +24.8 (c 1.0, CHCl3).
(S)-1-((2S,3S,5R)-3-((tert-Butyldimethylsilyl)oxy)-5-methyltetrahydrofuran-2-yl)but-3-en-1-ol (21)
Aldehyde 19 (43 mg, 65% yield) was obtained from olefin 13 (57 mg, 0.22 mmol) as described above. To a stirred solution of aldehyde 19 (43 mg, 0.18 mmol) in distilled CH2Cl2 (5 mL) were consecutively added ZnCl2 (72 mg, 0.53 mmol) and allyl tributyl stannane (0.27 mL, 0.88 mmol) at −78 °C under an argon atmosphere. The reaction mixture was slowly warmed to room temperature over 6 h and stirred at room temperature for 12 h. After this period, the reaction was quenched with saturated aqueous NaHCO3 and the aqueous layer was extracted with CH2Cl2. The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography (5% EtOAc in hexanes to 10% EtOAc in hexanes) to afford olefin 21 (38 mg, 75%) as a major product with a dr of 3:1; 1H NMR (400 MHz, CDCl3) δ: 5.90 (ddt, J = 17.2, 10.2, 7.0 Hz, 1H), 5.18–5.04 (m, 2H), 4.42 (dt, J = 6.5, 4.7 Hz, 1H), 4.01–3.88 (m, 2H), 3.57 (t, J = 4.6 Hz, 1H), 3.14 (brs, 1H), 2.33 (dt, J = 19.5, 6.4 Hz, 3H), 1.54 (td, J = 7.8, 3.8 Hz, 1H), 1.34 (d, J = 6.1 Hz, 3H), 0.90 (s, 9H), 0.09 (s, 6H); 13C NMR (100 MHz, CDCl3) δ, 135.1, 116.9, 83.2, 74.6, 73.4, 70.1, 43.7, 37.7, 25.6, 21.4, 17.8, −4.5, −5.3; LRMS-ESI (m/z): 309.2 (M + Na)+; HRMS (APCI), calcd for C15H31O3Si: m/z 287.2042 [M + H]+ found m/z 287.2045; [α]20D +16.7 (c 0.67, CHCl3).
(R)-1-((2S,3S,5R)-3-((tert-Butyldimethylsilyl)oxy)-5-methyltetrahydrofuran-2-yl)butane-1,4-diol (22)
To a stirred solution of olefin 20 (45 mg, 0.16 mmol) in distilled THF (1.6 mL) was added dropwise a solution of 9-BBN (0.5 M in THF, 0.80 mL, 0.40 mmol) at 0 °C under an argon atmosphere. The mixture was warmed to room temperature and stirred for 6 h. Upon completion of the reaction, aqueous 3 N NaOH solution (0.5 mL) and 30% H2O2 (1 mL) were added and the resulting mixture was stirred for 3 h at room temperature. Water (2 mL) was added and the aqueous layer was extracted with Et2O (3 × 20 mL). The combined organic layers were washed with brine, dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography (60% EtOAc in hexanes to 80% EtOAc in hexanes) to provide diol 22 (38 mg, 80%) as a colorless oil; 1H NMR (400 MHz, CDCl3) δ: 4.57 (q, J = 6.1 Hz, 1H), 3.99–3.89 (m, 1H), 3.84 (ddd, J = 10.2, 8.1, 2.6 Hz, 1H), 3.66 (dtd, J = 15.8, 11.0, 5.4 Hz, 2H), 3.55 (dd, J = 7.7, 5.7 Hz, 1H), 2.27 (dt, J = 12.8, 6.4 Hz, 1H), 1.90 (dtd, J = 13.9, 6.7, 2.6 Hz, 1H), 1.76 (dq, J = 9.1, 6.9, 6.4 Hz, 2H), 1.53 (ddd, J = 12.9, 8.5, 6.0 Hz, 2H), 1.27 (d, J = 6.1 Hz, 3H), 0.90 (s, 9H), 0.11 (s, 6H); 13C NMR (100 MHz, CDCl3) δ, 82.5, 74.6, 73.3, 71.2, 63.0, 42.5, 30.8, 29.3, 25.6, 21.6, 17.8, −4.4, −5.3; LRMS-ESI (m/z): 327.2 (M + Na)+; HRMS (ESI), calcd for C15H33O4Si: m/z 305.2148 [M + H]+, found m/z 305.2146; [α]20D +18.7 (c 1.0, CHCl3).tert-Butyldimethyl(((2S,2′R,3S,5R)-5-methyloctahydro-[2,2′-bifuran]-3-yl)oxy)silane (23)
To a stirred solution of diol 22 (35 mg, 0.11 mmol) in CH2Cl2 (1.8 mL) were added triethylamine (0.10 mL, 0.71 mmol) and DMAP (2.6 mg, 0.02 mmol) at 0 °C under an argon atmosphere. After stirring for 10 min, 4-toluenesulfonyl chloride (27.2 mg, 0.14 mmol) was added and the resulting mixture was stirred for 24 h at room temperature. The reaction was quenched with saturated aqueous NH4Cl and the aqueous layer was extracted with CH2Cl2. The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography (20% EtOAc in hexanes) to give bicyclic ether 23 (23 mg, 71%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 4.35 (ddd, J = 6.6, 4.3, 2.7 Hz, 1H), 4.06–3.97 (m, 2H), 3.85 (td, J = 7.8, 7.3, 3.7 Hz, 1H), 3.71–3.60 (m, 2H), 2.26 (ddd, J = 13.4, 7.7, 6.0 Hz, 1H), 2.05–1.96 (m, 1H), 1.94–1.82 (m, 3H), 1.49 (ddd, J = 13.1, 6.1, 2.8 Hz, 1H), 1.30 (d, J = 6.2 Hz, 3H), 0.89 (s, 9H), 0.06 (s, 6H); 13C NMR (100 MHz, CDCl3) δ, 80.1, 77.0, 73.9, 73.0, 67.7, 43.2, 28.1, 25.9, 25.7, 22.0, 18.0, −5.0, −5.2; LRMS-ESI (m/z): 309.2 (M + Na)+; HRMS (APCI), calcd for C15H31O3Si: m/z 287.2042 [M + H]+ found m/z 287.2044; [α]20D +4.3 (c 1.0, CHCl3).(2R,2′R,3S,5R)-5-Methyloctahydro-[2,2′-bifuran]-3-ol (24)
To a stirred solution of bicyclic ether 23 (18 mg, 0.06 mmol) was added TBAF solution (1M in THF, 0.10 mL, 0.10 mmol) at 0 °C under an argon atmosphere. The mixture was stirred for 3 h at room temperature. The reaction was quenched with saturated aqueous NH4Cl and the aqueous layer was extracted with CH2Cl2. The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography (80% EtOAc in hexanes) to give alcohol 24 (10 mg, 99%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 4.46 (dt, J = 7.4, 3.8 Hz, 1H), 4.11 (q, J = 7.1 Hz, 1H), 4.02–3.92 (m, 1H), 3.89 (dt, J = 8.2, 6.4 Hz, 1H), 3.75 (dt, J = 8.2, 6.9 Hz, 1H), 3.49 (dd, J = 7.8, 4.4 Hz, 1H), 2.93 (s, 1H), 2.36 (dt, J = 13.8, 7.0 Hz, 1H), 2.19–2.10 (m, 1H), 1.98–1.88 (m, 2H), 1.87–1.75 (m, 1H), 1.60–1.51 (m, 1H), 1.33 (d, J = 6.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ, 84.5, 77.8, 74.6, 73.3, 68.2, 42.1, 29.7, 25.4, 21.6. LRMS-ESI (m/z): 195.1 (M + Na)+; HRMS (APCI), calcd for C9H17O3: m/z 173.1177 [M + H]+ found m/z 173.1172; [α]20D +10.0 (c 0.34, CHCl3).(S)-1-((2S,3S,5R)-3-((tert-Butyldimethylsilyl)oxy)-5-methyltetrahydrofuran-2-yl)butane-1,4-diol (25)
To a stirred solution of olefin 21 (33 mg, 0.12 mmol) in distilled THF (1.6 mL) was added dropwise a solution of 9-BBN (0.5 M in THF, 0.80 mL, 0.40 mmol) at 0 °C under an argon atmosphere. The mixture was warmed to room temperature and stirred for 6 h. Upon completion of the reaction, aqueous 3 N NaOH solution (0.5 mL) and 30% H2O2 (1 mL) were added and the resulting mixture was stirred for 3 h at room temperature. Water (2 mL) was added and the aqueous layer was extracted with Et2O (3 × 20 mL). The combined organic layers were washed with brine, dried (MgSO4), filtered, and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography (60% EtOAc in hexanes to 80% EtOAc in hexanes) to provide diol 25 (22 mg, 64%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 4.47–4.38 (m, 1H), 3.94 (dt, J = 7.6, 6.3 Hz, 1H), 3.87 (dt, J = 8.0, 4.0 Hz, 1H), 3.66 (dt, J = 7.5, 5.6 Hz, 2H), 3.60–3.51 (m, 1H), 2.30 (dt, J = 13.1, 6.7 Hz, 1H), 1.79–1.70 (m, 2H), 1.67 (ddd, J = 11.9, 5.7, 2.8 Hz, 2H), 1.52 (ddd, J = 13.0, 7.7, 4.3 Hz, 1H), 1.32 (d, J = 6.2 Hz, 3H), 0.89 (s, 9H), 0.08 (s, 6H); 13C NMR (100 MHz, CDCl3) δ, 84.1, 74.6, 73.4, 70.8, 62.8, 43.7, 30.6, 29.8, 25.6, 21.4, 17.8, −4.5, −5.3; LRMS-ESI (m/z): 327.2 (M + Na)+; HRMS (ESI), calcd for C15H33O4Si: m/z 305.2148 [M + H]+, found m/z 305.2149; [α]20D +20.4 (c 1.0, CHCl3).tert-Butyldimethyl(((2S,2′S,3S,5R)-5-methyloctahydro-[2,2′-bifuran]-3-yl)oxy)silane (26)
To a stirred solution of diol 25 (22 mg, 0.07 mmol) in CH2Cl2 (1.8 mL) were added triethylamine (0.10 mL, 0.71 mmol) and DMAP (2.6 mg, 0.02 mmol) at 0 °C under an argon atmosphere. After stirring for 10 min, 4-toluenesulfonyl chloride (27.2 mg, 0.14 mmol) was added and the resulting mixture was stirred for 24 h at room temperature. The reaction was quenched with saturated aqueous NH4Cl and the aqueous layer was extracted with CH2Cl2. The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography (20% EtOAc in hexanes) to give compound 26 (18 mg, 88%). 1H NMR (400 MHz, CDCl3) δ: 4.29 (ddd, J = 6.5, 4.1, 2.7 Hz, 1H), 4.18–4.08 (m, 1H), 4.06–3.98 (m, 1H), 3.93–3.83 (m, 1H), 3.80–3.72 (m, 1H), 3.48 (dd, J = 8.4, 4.2 Hz, 1H), 2.33–2.24 (m, 1H), 2.10–1.98 (m, 1H), 1.90–1.83 (m, 2H), 1.55–1.44 (m, 2H), 1.34 (d, J = 6.1 Hz, 3H), 0.88 (s, 9H), 0.05 (d, J = 4.7 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ, 86.2, 78.1, 74.4, 73.4, 67.9, 43.5, 28.0, 25.8, 25.6, 22.0, 17.8, −4.2, −5.3; LRMS-ESI (m/z): 309.2 (M + Na)+; HRMS (APCI), calcd for C15H31O3Si: m/z 287.2042 [M + H]+ found m/z 287.2041; [α]20D +30.9 (c 0.50, CHCl3).(2R,2′S,3S,5R)-5-Methyloctahydro-[2,2′-bifuran]-3-ol (27)
To a stirred solution of compound 26 (17 mg, 0.06 mmol) was added TBAF solution (1M in THF, 0.10 mL, 0.10 mmol) at 0 °C under an argon atmosphere. The mixture was stirred for 3 h at room temperature. The reaction was quenched with saturated aqueous NH4Cl and the aqueous layer was extracted with CH2Cl2. The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography (80% EtOAc in hexanes) to give alcohol 27 (11 mg, 99%). 1H NMR (400 MHz, CDCl3) δ: 4.36 (dt, J = 6.5, 4.5 Hz, 1H), 4.16 (dt, J = 7.0, 3.5 Hz, 1H), 4.02–3.88 (m, 2H), 3.86–3.78 (m, 1H), 3.65 (dd, J = 5.0, 3.9 Hz, 1H), 2.40–2.29 (m, 1H), 2.10–1.80 (m, 5H), 1.58–1.52 (m, 1H), 1.32 (d, J = 6.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ, 83.2, 78.1, 74.2, 73.4, 68.9, 43.7, 28.0, 25.6, 21.7. LRMS-ESI (m/z): 195.1 (M + Na)+; HRMS (APCI), calcd for C9H17O3: m/z 173.1177 [M + H]+ found m/z 173.1171; [α]20D +24.6 (c 0.33, CHCl3).General procedure for the preparation of activated carbonate
To a solution of alcohol in CH2Cl2 (10 mL per mmol of alcohol) was added pyridine (5 equiv.) at 23 °C under an argon atmosphere, and the reaction mixture was cooled to 0 °C followed by the addition of 4-nitrophenyl chloroformate (2.2 equiv.). The reaction mixture was warmed to 23 °C and stirred for 12 h. Upon completion of the reaction, the solvents were removed under reduced pressure and the crude product was purified by silica gel column chromatography.Activated carbonates (28a)
Following the general procedure A, activated carbonate 28a (5 mg, 24% yield) was prepared as a white amorphous solid from alcohol 11 (10 mg, 0.07 mmol). 1H NMR (400 MHz, CDCl3) δ: 8.28 (d, J = 9.2 Hz, 2H), 7.38 (d, J = 9.2 Hz, 2H), 5.91–5.80 (m, 1H), 5.26 (ddd, J = 6.6, 3.9, 2.3 Hz, 1H), 5.22–5.07 (m, 2H), 4.02 (ddt, J = 13.5, 7.3, 6.2 Hz, 1H), 3.83 (ddd, J = 7.3, 6.5, 3.9 Hz, 1H), 2.64–2.55 (m, 1H), 2.55–2.46 (m, 2H), 1.77–1.69 (m, 1H), 1.37 (d, J = 6.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ, 155.4, 152.1, 145.3, 133.9, 125.2, 121.6, 117.4, 80.9, 80.7, 73.7, 40.5, 33.4, 21.3; LRMS-ESI (m/z): 330.1 (M + Na)+. HRMS (APCI), calcd for C15H18NO6: m/z 308.1134 [M + H]+, found m/z 308.1129.Activated carbonates (28b)
Following the general procedure, activated alcohol 28b (10 mg, 30% yield) was prepared as a yellow oil from alcohol 12 (16 mg, 0.11 mmol). 1H NMR (500 MHz, CDCl3) δ: 8.28 (d, J = 9.2 Hz, 2H), 7.39 (d, J = 9.2 Hz, 2H), 5.89–5.78 (m, 1H), 5.20–5.11 (m, 2H), 5.03 (ddd, J = 7.1, 3.4, 2.7 Hz, 1H), 4.36–4.28 (m, 1H), 4.26 (td, J = 6.7, 2.7 Hz, 1H), 2.60–2.51 (m, 1H), 2.34 (tt, J = 6.8, 1.3 Hz, 2H), 1.82 (ddd, J = 13.9, 5.8, 3.5 Hz, 1H), 1.34 (d, J = 6.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ, 155.4, 152.1, 145.4, 133.4, 125.3, 121.8, 118.0, 83.3, 82.1, 73.9, 53.4, 38.6, 37.3, 21.7; LRMS-ESI (m/z): 330.1 (M + Na)+; [α]20D +15.1 (c 0.64, CHCl3). HRMS (APCI), calcd for C15H18NO6: m/z 308.1134 [M + H]+, found m/z 308.1136.Activated carbonates (28c and 28d)
Following the general procedure, activated carbonate 28c (17 mg, 91% yield) was prepared as a white amorphous solid from alcohol 15 (8 mg, 0.06 mmol). 1H NMR (400 MHz, CDCl3) δ: 8.28 (d, J = 9.0 Hz, 2H), 7.38 (d, J = 9.0 Hz, 2H), 5.34 (p, J = 3.8 Hz, 1H), 4.10–3.95 (m, 2H), 3.67 (d, J = 5.7 Hz, 2H), 3.41 (s, 3H), 2.66–2.53 (m, 1H), 1.76–1.70 (m, 1H), 1.37 (d, J = 6.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ, 155.4, 152.0, 145.3, 125.2, 121.6, 80.2, 79.7, 74.1, 70.6, 59.3, 40.3, 21.1; LRMS-ESI (m/z): 312 (M + H)+; HRMS (APCI), calcd for C14H18NO7: m/z 312.1083 [M + H]+, found m/z 312.1082; [α]20D +5.8 (c 0.33, CHCl3). Following the general procedure, activated carbonate 28d (16 mg, 70% yield) was prepared as a white amorphous solid from alcohol ent-15 (9 mg, 0.07 mmol). For NMR data, please see compound 28c; LRMS-ESI (m/z): 312 (M + H)+; HRMS (APCI), calcd for C14H18NO7: m/z 312.1083 [M + H]+, found m/z 312.1091; [α]20D −6.2 (c 0.33, CHCl3).Activated carbonates (28e and 28f)
Following the general procedure, activated carbonate 28e (13 mg, 78% yield) was prepared as a white amorphous solid from alcohol 18 (7 mg, 0.05 mmol). 1H NMR (400 MHz, CDCl3) δ: 8.28 (d, J = 9.2 Hz, 2H), 7.38 (d, J = 9.2 Hz, 2H), 5.22 (ddd, J = 7.2, 4.1, 3.1 Hz, 1H), 4.40–4.32 (m, 1H), 4.29 (td, J = 4.5, 3.1 Hz, 1H), 3.57–3.48 (m, 2H), 3.39 (s, 3H), 2.57 (dt, J = 13.9, 7.1 Hz, 1H), 1.82 (ddd, J = 13.6, 6.2, 4.0 Hz, 1H), 1.34 (d, J = 6.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ, 155.4, 152.2, 145.5, 125.3, 121.7, 81.9, 81.7, 75.2, 73.1, 59.5, 39.2, 21.5; LRMS-ESI (m/z): 312 (M + H)+; HRMS (APCI), calcd for C14H18NO7: m/z 312.1083 [M + H]+, found m/z 312.1082 [α]20D +18.9 (c 0.13, CHCl3).By following the general procedure, activated alcohol 28f (4 mg, 40% yield) was prepared as a white amorphous solid from alcohol ent-18 (4.6 mg, 0.03 mmol). For NMR data, please see compound 28e. LRMS-ESI (m/z): 312 (M + H)+; HRMS (APCI), calcd for C14H18NO7: m/z 312.1084 [M + H]+, found m/z 312.1081; [α]20D −19.1 (c 0.13, CHCl3).
Activated carbonates (28g)
By following the general procedure, activated carbonate 28g (17 mg, 82%) was prepared from alcohol 24 (10 mg, 0.06 mmol). 1H NMR (400 MHz, CDCl3) δ: 8.27 (d, J = 9.2 Hz, 2H), 7.39 (d, J = 9.2 Hz, 2H), 5.40 (ddd, J = 7.0, 4.3, 2.9 Hz, 1H), 4.18–4.10 (m, 1H), 4.07 (td, J = 7.3, 6.1 Hz, 1H), 3.91–3.84 (m, 1H), 3.76 (dt, J = 8.3, 6.8 Hz, 1H), 3.68 (dd, J = 7.9, 4.3 Hz, 1H), 2.58 (dt, J = 14.2, 7.1 Hz, 1H), 2.19–2.05 (m, 1H), 2.01–1.84 (m, 3H), 1.75 (ddd, J = 14.2, 7.1, 2.9 Hz, 1H), 1.35 (d, J = 6.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ, 125.2, 121.7, 83.2, 80.0, 76.2, 74.2, 68.3 40.2, 29.2, 25.6, 21.3; LRMS-ESI (m/z): 360.2 (M + Na)+; HRMS (APCI), calcd for C16H20NO7: m/z 338.1239 [M + H]+ found m/z 338.1249; [α]20D +21.9 (c 0.30, CHCl3).Activated carbonates (28h)
By following the general procedure, activated carbonate 28h (8 mg, 44%) was prepared from alcohol 27 (9 mg, 0.05 mmol). 1H NMR (400 MHz, CDCl3) δ: 8.28 (d, J = 9.1 Hz, 2H), 7.38 (d, J = 9.1 Hz, 2H), 5.29 (p, J = 3.3 Hz, 1H), 4.15 (q, J = 7.4 Hz, 1H), 4.12–4.04 (m, 1H), 3.97–3.88 (m, 1H), 3.88–3.78 (m, 1H), 3.72 (dd, J = 7.2, 4.3 Hz, 1H), 2.61 (dt, J = 14.2, 7.1 Hz, 1H), 2.09–2.01 (m, 1H), 1.95–1.91 (m, 1H), 1.76 (ddd, J = 14.2, 7.4, 2.9 Hz, 1H), 1.65 (dq, J = 11.8, 8.1 Hz, 2H), 1.39 (d, J = 6.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ, 155.3, 152.1, 145.4, 125.2, 121.6, 83.8, 80.6, 74.2, 68.4, 60.3, 40.6, 28.1, 25.7, 21.1. LRMS-ESI (m/z): 360.1 (M + Na)+; HRMS (APCI), calcd for C16H20NO7: m/z 338.1239 [M + H]+ found m/z 338.1240; [α]20D +9.6 (c 0.27, CHCl3).General procedure for inhibitor synthesis using amine 29 or 30
To a stirred solution of activated carbonate and amine 29 or 30 in acetonitrile (2 mL) was added DIPEA (8 equiv.) at 0 °C under an argon atmosphere. The reaction mixture was stirred at room temperature until completion of the reaction. Upon completion, the solvents were removed under reduced pressure and the crude residue was purified by silica gel column chromatography.(2S,3S,5R)-2-Allyl-5-methyltetrahydrofuran-3-yl((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (4a)
Following the general procedure, inhibitor 4a (7 mg, 97% yield) was prepared as a white amorphous solid from amine 29 and activated carbonate 28a (5 mg, 0.02 mmol). 1H NMR (500 MHz, CDCl3) δ: 7.72 (d, J = 8.9 Hz, 2H), 7.30–7.19 (m, 5H), 6.98 (d, J = 8.9 Hz, 2H), 5.70 (ddt, J = 18.8, 9.1, 6.9 Hz, 1H), 5.08–4.97 (m, 3H), 4.85 (d, J = 8.2 Hz, 1H), 3.88 (s, 3H), 3.86–3.81 (m, 3H), 3.62 (ddd, J = 7.8, 5.9, 4.0 Hz, 1H), 3.15 (dd, J = 15.1, 7.7 Hz, 1H), 3.07–2.93 (m, 3H), 2.90–2.75 (m, 2H), 2.41 (dt, J = 14.3, 7.2 Hz, 1H), 2.19 (dt, J = 14.5, 7.4 Hz, 1H), 2.14–2.06 (m, 1H), 1.89–1.77 (m, 1H), 1.45 (ddd, J = 14.0, 7.3, 2.4 Hz, 1H), 1.28 (d, J = 6.2 Hz, 3H), 0.92 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ, 163.0, 155.8, 137.4, 134.4, 129.4, 129.3, 128.5, 126.5, 116.9, 114.3, 81.3, 75.6, 73.5, 72.7, 58.7, 55.6, 54.9, 53.7, 53.4, 40.7, 35.3, 33.2, 27.2, 21.3, 20.1, 19.8; LRMS-ESI (m/z): 575.0 (M + H)+; HRMS-ESI (m/z): C30H42N2O7S; calc'd for [M + Na]+: 597.2605, found 597.2611.(2S,3S,5R)-2-Allyl-5-methyltetrahydrofuran-3-yl((2S,3R)-4-((4-amino-N-isobutylphenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (4b)
Following the general procedure, inhibitor 4b (7 mg, 62% yield) was prepared as a yellow amorphous solid from amine 30 and activated carbonate 28a (7 mg, 0.02 mmol). 1H NMR (400 MHz, CDCl3) δ: 7.55 (d, J = 8.2 Hz, 2H), 7.34–7.14 (m, 5H), 6.69 (d, J = 8.2 Hz, 2H), 5.72–5.62 (m, 1H), 5.07–5.03 (m, 1H), 5.02–4.96 (m, 2H), 4.85 (d, J = 7.8 Hz, 1H), 3.92–3.85 (m, 1H), 3.90–3.84 (m, 2H), 3.62–3.60 (m, 1H), 3.18–2.72 (m, 6H), 2.44–2.30 (m, 1H), 2.25–2.05 (m, 2H), 1.88–1.78 (m, 1H), 1.49–1.41 (m, 1H), 1.27 (d, J = 6.2 Hz, 3H), 0.92 (d, J = 6.6 Hz, 3H), 0.86 (d, J = 6.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ, 155.9, 137.6, 134.5, 129.5, 129.4, 128.5, 126.6, 117.0, 114.2, 81.42, 75.7, 73.6, 72.8, 58.8, 55.0, 53.8, 40.8, 35.4, 33.3, 27.3, 21.4, 20.2, 19.9; LRMS-ESI (m/z): 560.0 (M + H)+; HRMS-ESI (m/z): C29H41N3O6S; calc'd for [M + Na]+: 582.2608, found 582.2615.(2S,3S,5R)-2-(Methoxymethyl)-5-methyltetrahydrofuran-3-yl((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (4c)
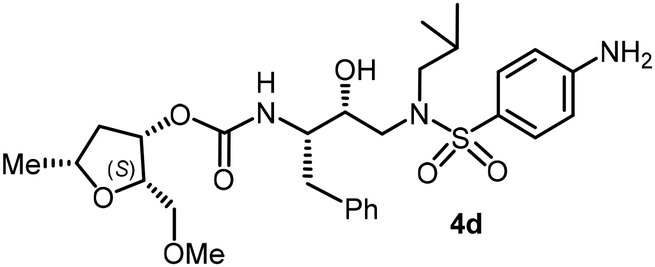
Following the general procedure, inhibitor 4c (8 mg, 58% yield) was prepared as a white amorphous solid from amine 29 and activated carbonate 28c (7 mg, 0.02 mmol). 1H NMR (400 MHz, CDCl3) δ: 7.71 (d, J = 8.9 Hz, 2H), 7.34–7.17 (m, 5H), 6.98 (d, J = 8.9 Hz, 2H), 5.18–5.11 (m, 1H), 4.84 (d, J = 8.3 Hz, 1H), 3.93 (q, J = 6.6 Hz, 1H), 3.87 (s, 3H), 3.86–3.77 (m, 4H), 3.38–3.31 (m, 2H), 3.30 (s, 3H), 3.14 (dd, J = 15.2, 8.3 Hz, 1H), 3.07–2.91 (m, 3H), 2.86 (dd, J = 14.1, 8.4 Hz, 1H), 2.79 (dd, J = 13.4, 6.7 Hz, 1H), 2.47–2.36 (m, 1H), 1.87–1.76 (m, 1H), 1.46 (dd, J = 14.0, 7.7 Hz, 1H), 1.29 (d, J = 6.1 Hz, 3H), 0.91 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ, 163.0, 148.4, 137.5, 129.7, 129.4, 129.3, 128.5, 126.5, 114.26, 80.4, 75.5, 74.0, 72.6, 70.8, 59.2, 58.7, 55.5, 54.9, 55.6, 40.6, 35.2, 27.2, 21.2, 20.1, 19.8; LRMS-ESI (m/z): 578.0 (M + H)+; HRMS-ESI (m/z): C29H42N2O8S; calc'd for [M + H]+: 579.2735, found 579.2740.(2S,3S,5R)-2-(Methoxymethyl)-5-methyltetrahydrofuran-3-yl((2S,3R)-4-((4-amino-N-isobutylphenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (4d)
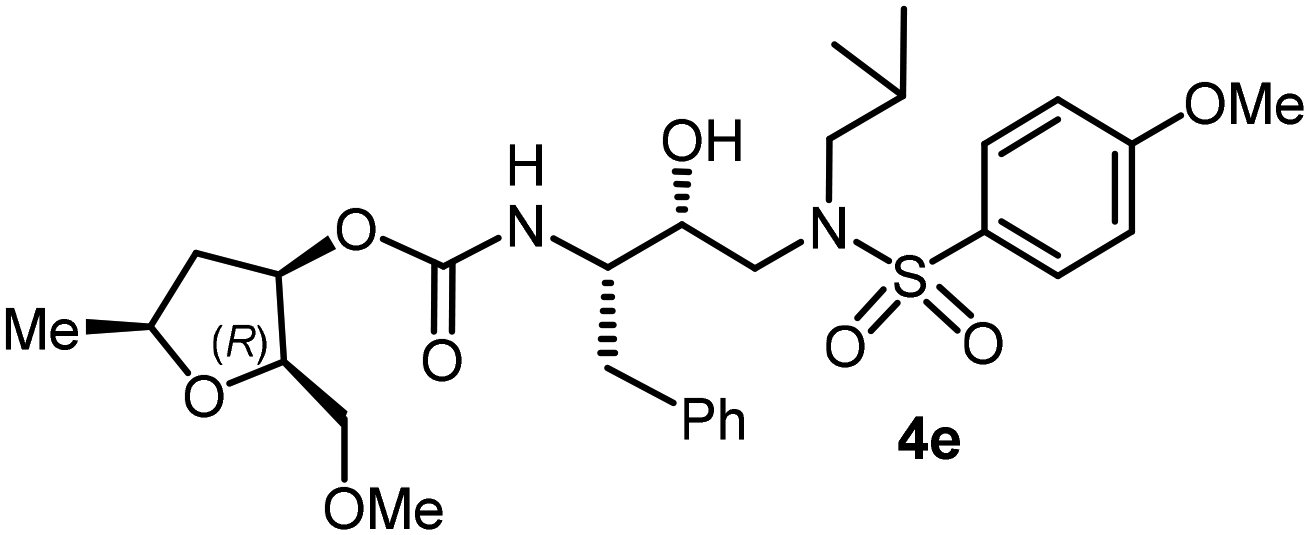
Following the general procedure, inhibitor 4d (7 mg, 69% yield) was prepared as a yellow amorphous solid from amine 30 and activated carbonate 28c (6 mg, 0.02 mmol). 1H NMR (400 MHz, CDCl3) δ: 7.55 (d, J = 8.7 Hz, 2H), 7.32–7.20 (m, 5H), 6.68 (d, J = 8.7 Hz, 2H), 5.18–5.12 (m, 1H), 4.85 (d, J = 8.4 Hz, 1H), 3.98–3.89 (m, 1H), 3.86–3.81 (m, 3H), 3.40–3.32 (m, 2H), 3.31 (s, 3H), 3.14 (dd, J = 15.1, 8.3 Hz, 1H), 3.08–2.81 (m, 5H), 2.76 (dd, J = 13.4, 6.6 Hz, 1H), 2.41 (dt, J = 14.1, 7.2 Hz, 1H), 1.88–1.74 (m, 2H), 1.52–1.42 (m, 2H), 1.30 (d, J = 6.1 Hz, 3H), 0.92 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ, 155.7, 150.6, 137.6, 129.4, 129.3, 128.4, 128.3, 126.5, 114.0, 80.4, 77.1, 75.5, 74.0, 72.7, 70.9, 59.2, 58.8, 54.9, 53.7, 40.6, 35.3, 27.2, 21.2, 20.1, 19.8; LRMS-ESI (m/z): 564.0 (M + H)+; HRMS-ESI (m/z): C28H41N3O7S; calc'd for [M + H]+: 564.2738, found 564.2733.(2R,3R,5S)-2-(Methoxymethyl)-5-methyltetrahydrofuran-3-yl((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (4e)
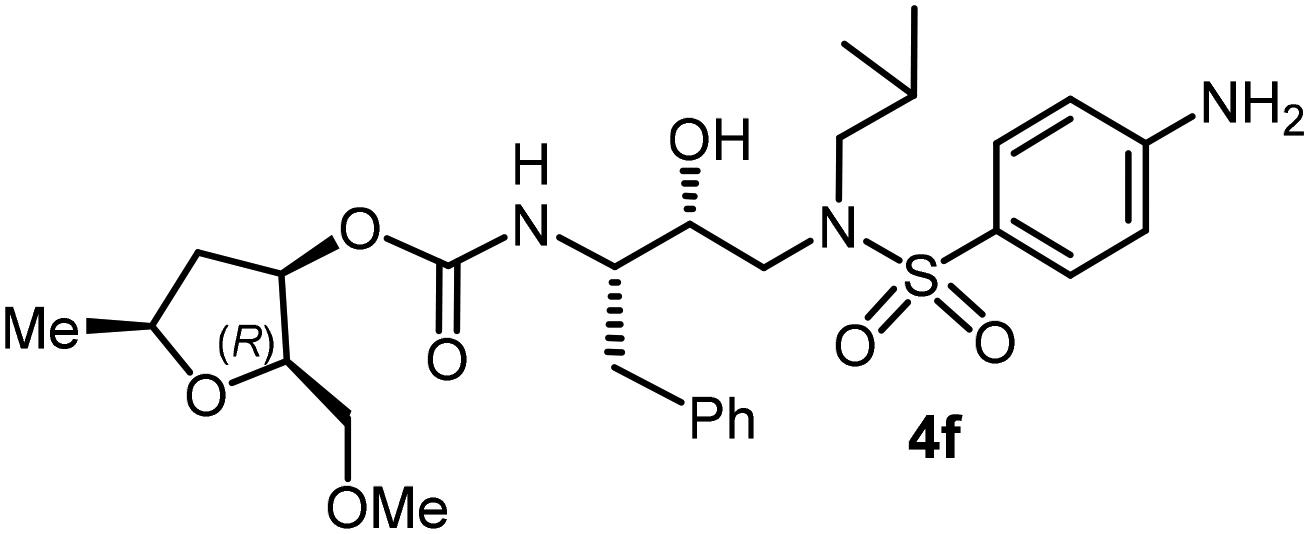
Following the general procedure, inhibitor 4e (7 mg, 71% yield) was prepared as a white amorphous solid from amine 29 and activated carbonate 28d (5 mg, 0.02 mmol). 1H NMR (400 MHz, CDCl3) δ: 7.71 (d, J = 8.8 Hz, 2H), 7.33–7.17 (m, 5H), 6.99 (d, J = 8.8 Hz, 2H), 5.11–5.05 (m, 1H), 4.84 (d, J = 8.6 Hz, 1H), 3.94 (h, J = 6.4 Hz, 1H), 3.88 (s, 3H), 3.86–3.77 (m, 4H), 3.52–3.49 (m, 2H), 3.34 (s, 3H), 3.14 (dd, J = 15.1, 8.4 Hz, 1H), 3.05–2.94 (m, 3H), 2.89–2.76 (m, 3H), 2.38–2.30 (m, 1H), 1.90–1.77 (m, 1H), 1.60 (brs, 1H), 1.33–1.28 (m, 2H), 0.93 (d, J = 6.6 Hz, 3H), 0.88 (d, J = 6.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ, 163.0, 155.7, 137.5, 129.7, 129.4, 128.4, 126.5, 114.3, 80.1, 75.8, 74.0, 72.5, 71.1, 59.2, 58.7, 55.5, 54.9, 53.6, 40.6, 35.3, 27.2, 21.3, 20.1, 19.8; LRMS-ESI (m/z): 578.0 (M + Na)+; HRMS-ESI (m/z): C29H42N2O8S; calc'd for [M + Na]+: 601.2554, found 601.2557.(2R,3R,5S)-2-(Methoxymethyl)-5-methyltetrahydrofuran-3-yl((2S,3R)-4-((4-amino-N-isobutylphenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (4f)
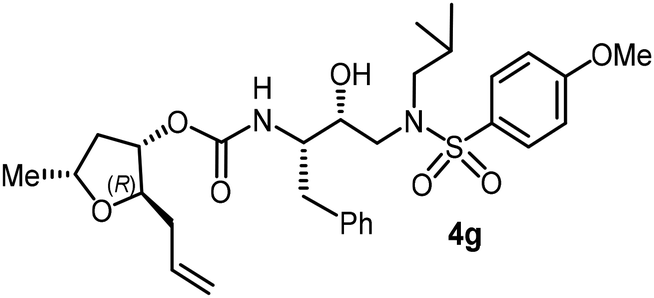
Following the general procedure, inhibitor 4f (16 mg, 54% yield) was prepared from amine 30 and activated carbonate 28d (16 mg, 0.05 mmol). 1H NMR (500 MHz, CDCl3) δ: 7.54 (d, J = 8.7 Hz, 2H), 7.33–7.16 (m, 5H), 6.67 (d, J = 8.7 Hz, 2H), 5.06 (ddd, J = 6.8, 4.3, 2.5 Hz, 1H), 4.87 (d, J = 8.6 Hz, 1H), 4.18 (brs, 1H), 3.94 (ddt, J = 13.6, 7.4, 6.2 Hz, 1H), 3.89–3.81 (m, 4H), 3.50 (d, J = 4.7 Hz, 2H), 3.33 (s, 3H), 3.13 (dd, J = 15.1, 8.2 Hz, 1H), 3.05–2.91 (m, 3H), 2.83 (dd, J = 14.0, 8.6 Hz, 1H), 2.76 (dd, J = 13.3, 6.6 Hz, 1H), 2.33 (dt, J = 14.2, 7.1 Hz, 1H), 1.88–1.75 (m, 1H), 1.27 (d, J = 15.5 Hz, 3H), 0.92 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ, 155.6, 150.7, 137.6, 129.4, 128.4, 126.5, 126.4, 126.0, 114.0, 80.1, 75.7, 74.1, 72.6, 71.1, 59.2, 58.8, 54.9, 53.7, 40.6, 35.3, 27.2, 21.3, 20.1, 19.8; LRMS-ESI (m/z): 564.0 (M + Na)+; HRMS-ESI (m/z): C28H41N3O7S; calc'd for [M + Na]+: 586.2557, found 586.2562.(2R,3S,5R)-2-Allyl-5-methyltetrahydrofuran-3-yl((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (4g)
Following the general procedure, inhibitor 4g (8.5 mg, 68% yield) was prepared as a white amorphous solid from amine 29 and activated carbonate 28b (6.5 mg, 0.02 mmol). 1H NMR (800 MHz, CDCl3) δ: 7.71 (d, J = 8.8 Hz, 2H), 7.33–7.20 (m, 5H), 6.98 (d, J = 8.5 Hz, 2H), 5.81–5.70 (m, 1H), 5.12–5.03 (m, 2H), 4.83 (d, J = 9.0 Hz, 1H), 4.81–4.78 (m, 1H), 4.22–4.15 (m, 1H), 3.88 (s, 3H), 3.87–3.77 (m, 3H), 3.12 (dd, J = 15.0, 8.5 Hz, 1H), 3.04–2.98 (m, 2H), 2.96 (dd, J = 13.2, 8.4 Hz, 1H), 2.89 (dd, J = 14.0, 8.8 Hz, 1H), 2.79 (dd, J = 13.5, 6.7 Hz, 1H), 2.40–2.33 (m, 1H), 2.25–2.16 (m, 2H), 1.86–1.78 (m, 1H), 1.65–1.53 (m, 2H), 1.24 (d, J = 6.4 Hz, 3H), 0.91 (d, J = 6.2 Hz, 3H), 0.87 (d, J = 6.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ, 163.1, 156.0, 137.6, 134.0, 129.8, 129.5, 129.5, 128.5, 126.6, 117.4, 114.4, 82.3, 78.9, 73.7, 72.6, 58.8, 55.6, 55.1, 53.7, 39.0, 37.3, 35.4, 27.3, 21.7, 20.1, 19.9; LRMS-ESI (m/z): 575.0 (M + H)+; HRMS-ESI (m/z): C30H42N2O7S; calc'd for [M + Na]+: 597.2605, found 597.2613.(2R,3S,5R)-2-(Methoxymethyl)-5-methyltetrahydrofuran-3-yl((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (4h)
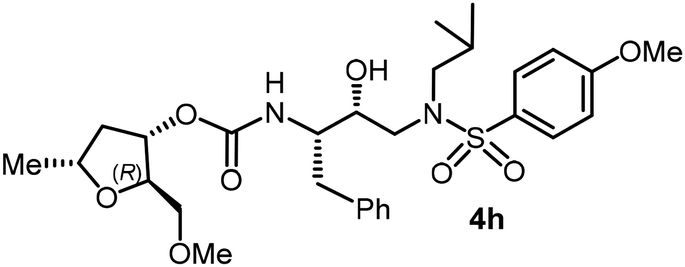
Following the general procedure, inhibitor 4h (10 mg, 76% yield) was prepared as a white amorphous solid from amine 29 and activated carbonate 28e (7 mg, 0.02 mmol). 1H NMR (400 MHz, CDCl3) δ: 7.70 (d, J = 8.9 Hz, 2H), 7.31–7.20 (m, 5H), 6.97 (d, J = 8.8 Hz, 2H), 4.96–4.91 (m, 1H), 4.88 (d, J = 8.4 Hz, 1H), 4.28–4.19 (m, 1H), 3.87 (s, 3H), 3.85–3.79 (m, 1H), 3.43–3.35 (m, 2H), 3.33 (s, 3H), 3.16–3.07 (m, 1H), 3.03–2.85 (m, 5H), 2.79 (dd, J = 13.3, 6.6 Hz, 1H), 2.41–2.29 (m, 1H), 1.87–1.75 (m, 1H), 1.63–1.51 (m, 2H), 1.29–1.26 (m, 3H), 0.90 (d, J = 6.6 Hz, 3H), 0.86 (d, J = 6.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ, 163.0, 156.0, 137.5, 129.7, 129.4, 128.4, 126.5, 114.3, 108.6, 82.1, 76.8, 74.6, 73.1, 72.5, 59.2, 58.7, 55.5, 55.1, 53.6, 39.3, 35.3, 27.2, 21.4, 20.1, 19.8. LRMS-ESI (m/z): 579.0 (M + H)+; HRMS-ESI (m/z): C29H42N2O8S; calc'd for [M + Na]+: 601.2554, found 601.2558.(2R,3S,5R)-2-(Methoxymethyl)-5-methyltetrahydrofuran-3-yl((2S,3R)-4-((4-amino-N-isobutylphenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (4i)
Following the general procedure, inhibitor 4i (6.5 mg, 60% yield) was prepared as a yellow amorphous solid from amine 30 and activated carbonate 28e (6 mg, 0.02 mmol). 1H NMR (500 MHz, CDCl3) δ: 7.54 (d, J = 8.7 Hz, 2H), 7.33–7.16 (m, 5H), 6.68 (d, J = 8.5 Hz, 2H), 4.95–4.90 (m, 1H), 4.88 (d, J = 8.8 Hz, 1H), 4.29–4.18 (m, 2H), 3.94–3.87 (m, 2H), 3.87–3.75 (m, 2H), 3.41–3.36 (m, 2H), 3.34 (s, 3H), 3.11 (dd, J = 15.1, 8.3 Hz, 1H), 3.03–2.84 (m, 5H), 2.76 (dd, J = 13.3, 6.7 Hz, 1H), 2.42–2.31 (m, 1H), 1.88–1.74 (m, 1H), 1.60–1.49 (m, 1H), 1.24 (d, J = 4.1 Hz, 3H), 0.91 (d, J = 6.6 Hz, 3H), 0.86 (d, J = 6.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ, 155.9, 150.6, 137.5, 129.4, 129.4, 128.4, 126.5, 114.0, 82.1, 74.6, 73.1, 72.5, 59.2, 58.8, 55.0, 53.7, 39.3, 35.3, 27.2, 21.4, 20.1, 19.8; LRMS-ESI (m/z): 564.0 (M + H)+; HRMS-ESI (m/z): C28H41N3O7S; calc'd for [M + Na]+: 586.2557, found 586.2564.(2S,3R,5S)-2-(Methoxymethyl)-5-methyltetrahydrofuran-3-yl((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (4j)
Following the general procedure, inhibitor 4j (4.3 mg, 66% yield) was prepared as a white amorphous solid from amine 29 and activated carbonate 28f (3.5 mg, 0.01 mmol). 1H NMR (400 MHz, CDCl3) δ: 7.70 (d, J = 8.7 Hz, 2H), 7.34–7.17 (m, 5H), 6.98 (d, J = 8.7 Hz, 2H), 4.93–4.88 (m, 1H), 4.85–4.80 (m, 1H), 4.26–4.20 (m, 1H), 4.07–4.04 (m, 1H), 3.88 (s, 3H), 3.87–3.82 (m, 2H), 3.41 (d, J = 4.3 Hz, 2H), 3.34 (s, 3H), 3.20–3.10 (m, 1H), 3.05–2.89 (m, 5H), 2.84–2.75 (m, 1H), 2.38–2.26 (m, 1H), 1.68–1.61 (m, 1H), 1.51–1.44 (m, 1H), 1.22 (d, J = 6.2 Hz, 3H), 0.91 (d, J = 6.4 Hz, 3H), 0.87 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ, 163.1, 156.0, 137.6, 129.8, 129.5, 129.3, 128.5, 126.6, 114.4, 82.0, 77.2, 74.8, 73.2, 72.6, 59.3, 58.8, 55.6, 55.0, 53.8, 39.4, 35.6, 27.3, 21.6, 20.1, 19.9; LRMS-ESI (m/z): 579.0 (M + H)+; HRMS-ESI (m/z): C29H42N2O8S; calc'd for [M + Na]+: 601.2554, found 601.2560.(2S,2′S,3S,5R)-5-Methyloctahydro-[2,2′-bifuran]-3-yl((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (4k)
Following the general procedure, inhibitor 4k (16 mg, 83% yield) was prepared from amine 29 and activated carbonate 28g (11 mg, 0.03 mmol) as a white amorphous solid. 1H NMR (400 MHz, CDCl3) δ: 7.71 (d, J = 8.7 Hz, 2H), 7.36–7.17 (m, 5H), 6.98 (d, J = 8.9 Hz, 2H), 5.25–5.20 (m, 1H), 4.82 (d, J = 7.9 Hz, 1H), 3.98 (q, J = 6.6 Hz, 1H), 3.90 (s, 1H), 3.88 (s, 3H), 3.86–3.80 (m, 3H), 3.67–3.63 (m, 2H), 3.16–2.91 (m, 6H), 2.81 (dd, J = 13.4, 6.8 Hz, 1H), 2.43 (dt, J = 14.2, 7.1 Hz, 1H), 1.89–1.78 (m, J = 6.5 Hz, 5H), 1.58–1.40 (m, 1H), 1.27 (d, J = 6.1 Hz, 3H), 0.91 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ, 162.9, 155.9, 137.5, 129.8, 129.4, 129.4, 128.4, 126.4, 114.2, 83.3, 76.3, 75.3, 73.9, 72.4, 68.0, 58.6, 55.5, 54.9, 53.5, 40.6, 35.0, 28.1, 27.1, 25.9, 21.4, 20.1, 19.8; LRMS-ESI (m/z): 605.03 (M + H)+; HRMS-ESI (m/z): C31H45N2O8S; calc'd for [M + H]+: 605.2896, found 605.2906.(2S,2′R,3S,5R)-5-Methyloctahydro-[2,2′-bifuran]-3-yl((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (4l)
Following the general procedure, inhibitor 4l (4.8 mg, 62% yield) was prepared from amine 29 and activated carbonate 28h (4 mg, 0.01 mmol). 1H NMR (800 MHz, CDCl3) δ: 7.72 (d, J = 8.8 Hz, 2H), 7.28 (t, J = 7.6 Hz, 2H), 7.21 (d, J = 7.6 Hz, 3H), 6.99 (d, J = 8.4 Hz, 2H), 5.14–5.05 (m, 1H), 4.84 (d, J = 8.4 Hz, 1H), 3.99–3.95 (m, 1H), 3.88 (s, 3H), 3.86–3.80 (m, 2H), 3.79–3.71 (m, 2H), 3.51 (dd, J = 7.8, 4.4 Hz, 1H), 3.35 (s, 1H), 3.16 (dd, J = 15.2, 8.1 Hz, 1H), 3.08 (dd, J = 14.3, 4.0 Hz, 1H), 3.03–2.93 (m, 2H), 2.82–2.78 (m, 2H), 2.42–2.34 (m, 1H), 1.85–1.78 (m, 2H), 1.77–1.72 (m, 1H), 1.51–1.46 (m, 1H), 1.35–1.31(m, 1H), 1.30 (d, J = 6.1 Hz, 3H), 0.93 (d, J = 6.6 Hz, 3H), 0.88 (d, J = 2.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ, 163.1, 155.6, 137.6, 129.8, 129.5, 129.4, 128.5, 126.5, 114.4, 84.3, 77.5, 75.6, 74.3, 73.0, 68.2, 58.8, 55.6, 55.0, 53.7, 41.0, 35.6, 27.2, 25.9, 21.4, 20.2, 19.9. LRMS-ESI (m/z): 605.0 (M + H)+; HRMS-ESI (m/z): C31H45N2O8S; calc'd for [M + H]+: 605.2896, found 605.2899.Methods: determination of X-ray structures of the HIV-1 protease–inhibitor complex
HIV-1 protease was expressed and purified as described.25,40 The protease–inhibitor complex (PR/GRL-072-17A) was crystallized by hanging drop vapor diffusion method with a solution of 1.7 M NaCl and 0.1 M sodium acetate at pH 5.5 in the well. X-ray diffraction data were collected on a single crystal cooled to 90 K at SER-CAT (22-ID beamline), Advanced Photon Source, Argonne National Lab (Chicago, USA) with X-ray wavelength of 1.0 Å. X-ray data were processed by HKL-200041 to give an Rmerge of 8.9%. The crystal structure was solved by PHASER42 in CCP4i Suit43–45 using one of the previously reported isomorphous structures46 as the initial model, and refined by SHELX-201447,48 and Refmac549 using X-ray data at 1.32 Å resolution. PRODRG-250 and JLigand51 were used to construct the inhibitor and geometric restraints for refinement. COOT52,53 was used to modify the PR–inhibitor structure. Alternative conformations were modeled, and anisotropic atomic displacement parameters (B factors) were applied for all protein atoms including solvent molecules. The final refined solvent structure comprised two Na+ ions, four Cl− ions, two glycerol molecules, one formic acid, and 221 water molecules. The crystallographic statistics are listed in Table 1. The coordinates and structural factors of the protease complexes with GRL-072-17A have been deposited in the Protein Data Bank54 with accession codes 9B2H.Author contributions
Arun K. Ghosh: writing – review & editing, writing – original draft, supervision, resources, project administration, methodology, investigation, funding acquisition, formal analysis, and conceptualization. Daniel Lee: methodology and investigation. Ashish Sharma: methodology and investigation. Megan E. Johnson: methodology and data curation. Ajay K. Ghosh: methodology and data curation. Yuan-Fang Wang: methodology and investigation. Johnson Agniswamy: methodology and investigation. Masayuki Amano: methodology and investigation. Shin-ichiro Hattori: methodology and investigation. Irene T. Weber: writing, editing, supervision, methodology, and data curation. Hiroaki Mitsuya: supervision, methodology, data curation, and funding acquisition.Data availability
The authors confirm that the data supporting these studies are available within the article and its ESI.‡Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Institutes of Health (Grant AI150466, AKG and Grant AI150461, ITW). X-ray data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) beamline 22BM at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the US Department of Energy, Basic Energy Sciences, Office of Science, under Contract No. W-31-109-Eng-38. This work was also supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health (HM), and in part by grants for the promotion of AIDS research from the Ministry of Health; grants from Welfare and Labor of Japan (HM); grants for the Research Program on HIV/AIDS from the Japan Agency for Medical Research and Development (AMED) under grant numbers JP15fk0410001 and JP16fk041001 (HM); a grant from the National Center for Global Health and Medicine (NCGM) Research Institute (HM); and a Grant-in-Aid for Scientific Research and a Grant-in-Aid for Challenging Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Monbu Kagakusho) (HM). The authors would like to thank the Purdue University Center for Cancer Research, for the shared NMR and mass spectrometry facilities.References
- R. C. Ogden and C. W. Flexner, Protease Inhibitors in AIDS Theraphy, New York, 2011, pp. 1–300 Search PubMed.
- J. A. Esté and T. C. Cihlar, Antiviral Res., 2010, 85, 25–33 CrossRef.
- C. W. Diffenbach and A. S. Fauci, Ann. Intern. Med., 2011, 154, 766–771 CrossRef PubMed.
- M. S. Cohen, Y. Q. Chen and M. McCauley, N. Engl. J. Med., 2011, 365, 493–505 CrossRef CAS PubMed.
- J. Maenza and C. Flexner, Am. Fam. Physician, 1998, 57, 2789–2798 CAS.
- M. Vitoria, A. Rangaraj, N. Ford and M. Doherty, Curr. Opin. HIV AIDS, 2019, 14, 143–149 CrossRef PubMed.
- G. Barbaro, Lancet, 2004, 363, 900–901 CrossRef PubMed.
- A. Chawla, C. Wang, C. Patton, M. Murray, Y. Punekar, A. de Ruiter and C. Steinhart, Infect. Dis. Ther., 2018, 183–195 CrossRef.
- C. W. Dieffenbach and A. S. Fauci, Ann. Intern. Med., 2011, 154, 766–771 CrossRef PubMed.
- K. A. Bosh, A. S. Johnson, A. L. Hernandez, J. Prejean, J. Taylor, R. Wingard, L. A. Valleroy and H. I. Hall, Morb. Mortal. Wkly. Rep., 2020, 69, 1717–1724 CrossRef PubMed.
- L. Waters and M. Nelson, Int. J. Clin. Pract., 2007, 61, 983–990 CrossRef CAS.
- C. E. Reust, Am. Fam. Physician, 2011, 83, 1443–1451 Search PubMed.
- A. K. Ghosh, I. T. Weber and H. Mitsuya, Chem. Commun., 2022, 58, 11762–11782 RSC.
- A. K. Ghosh, H. L. Osswald and G. Prato, J. Med. Chem., 2016, 59, 5172–5208 CrossRef CAS.
- A. K. Ghosh and B. D. Chapsal, Aspartic Acid Proteases as Therapeutic Targets, Wiley-VCH, Weinheim, Germany, 2010, pp. 169–204 Search PubMed.
- A. K. Ghosh, Z. L. Dawson and H. Mitsuya, Bioorg. Med. Chem., 2007, 15, 7576–7580 CrossRef CAS.
- A. Curran and E. R. Pascuet, Enfermedades Infecciosas y Microbiología Clínica, 2008, vol. 10, pp. 14–22 Search PubMed.
- A. K. Ghosh, P. R. Sridhar, N. Kumaragurubaran, Y. Koh, I. T. Weber and H. Mitsuya, ChemMedChem, 2006, 1, 939–950 CrossRef CAS.
- A. K. Ghosh, B. Chapsal and H. Mitsuya, Aspartic Acid Proteases as Therapeutic Targets, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010, pp. 205–243 Search PubMed.
- A. K. Ghosh, D. D. Anderson, I. T. Weber and H. Mitsuya, Angew. Chem., Int. Ed., 2012, 51, 1778–1802 CrossRef CAS.
- A. K. Ghosh, B. Chapsal, I. T. Weber and H. Mitsuya, Acc. Chem. Res., 2008, 41, 78–86 CrossRef CAS.
- A. K. Ghosh and B. D. Chapsal, in From Introduction to Biological and Small Molecule Drug Research and Development, ed. R. Jefferis, 2013, pp. 355–384 Search PubMed.
- J. Mallolas, AIDS Rev., 2017, 19, 105–112 Search PubMed.
- A. Y. Kovalevsky, F. Liu, S. Leshchenko, A. K. Ghosh, J. M. Louis, R. W. Harrison and I. T. Weber, J. Mol. Biol., 2006, 363, 161–173 CrossRef CAS PubMed.
- Y. Tie, P. I. Boross, Y.-F. Wang, L. Gaddis, A. K. Hussain, S. Leshchenko, A. K. Ghosh, J. M. Louis, R. W. Harrison and I. T. Weber, J. Mol. Biol., 2004, 338, 341–352 CrossRef CAS PubMed.
- Y. Koh, H. Nakata, K. Maeda, H. Ogata, G. Bilcer, T. Devasamudram, J. F. Kincaid, P. Boross, Y.-F. Wang, Y. Tie, P. Volarath, L. Gaddis, R. W. Harrison, I. T. Weber, A. K. Ghosh and H. Mitsuya, Antimicrob. Agents Chemother., 2003, 47, 3123–3129 CrossRef CAS.
- S. De Meyer, H. Azijn, D. Surleraux, D. Jochmans, A. Tahri, R. Pauwels, P. Wigerinck and M.-P. de Béthune, Antimicrob. Agents Chemother., 2005, 49, 2314–2321 CrossRef CAS.
- A. K. Ghosh, B. D. Chapsal, A. Baldridge, M. P. Steffey, D. E. Walters, Y. Koh, M. Amano and H. Mitsuya, J. Med. Chem., 2011, 54, 622–634 CrossRef CAS.
- M. Amano, P. M. Salcedo-Gómez, R. S. Yedidi, R. Zhao, H. Hayashi, K. Hasegawa, T. Nakamura, C. D. Martyr, A. K. Ghosh and H. Mitsuya, Antimicrob. Agents Chemother., 2019, 63, e00466–e00419 CrossRef CAS PubMed.
- A. K. Ghosh and P. R. Nyalapatla, Tetrahedron, 2017, 73, 1820–1830 CrossRef CAS PubMed.
- A. K. Ghosh and Y. Chen, Tetrahedron Lett., 1995, 36, 505–508 CrossRef CAS PubMed.
- A. K. Ghosh and P. R. Nyalapatla, Org. Lett., 2016, 18, 2296–2299 CrossRef CAS PubMed.
- S. Hanessian, S. Giroux and A. Larsson, Org. Lett., 2006, 8, 5481–5484 CrossRef CAS PubMed.
- M. Arisawa, Y. Terada, K. Takahashi, M. Nakagawa and A. Nishida, J. Org. Chem., 2006, 71, 4255–4261 CrossRef CAS PubMed.
- M. V. Toth and G. R. Marshall, Int. J. Pept. Protein Res., 1990, 36, 544–550 CrossRef CAS.
- Y. Koh, M. Amano, T. Towata, M. Danish, S. Leshchenko-Yashchuk, D. Das, M. Nakayama, Y. Tojo, A. K. Ghosh and H. Mitsuya, J. Virol., 2010, 84, 11961–11969 CrossRef CAS PubMed.
- K. Yoshimura, R. Kato, M. F. Kavlick, A. Nguyen, V. Maroun, K. Maeda, K. A. Hussain, A. K. Ghosh, S. V. Gulnik, J. W. Erickson and H. Mitsuya, J. Virol., 2002, 76, 1349–1358 CrossRef CAS PubMed.
- A. K. Ghosh, Z. Xia, S. Kovela, W. L. Robinson, M. E. Johnson, D. W. Kneller, Y. F. Wang, M. Aoki, Y. Takamatsu, I. T. Weber and H. Mitsuya, Potent HIV-1 Protease Inhibitors Containing Carboxylic and Boronic Acids: Effect on Enzyme Inhibition and Antiviral Activity and Protein-Ligand X-ray Structural Studies, ChemMedChem, 2019, 14, 1863–1872 CrossRef CAS.
- N. M. Midde, B. J. Patters, P. S. S. Rao, T. J. Cory and S. Kumar, Expert Opin. Invest. Drugs, 2016, 25, 1189–1200 CrossRef CAS.
- J. Agniswamy, D. W. Kneller, R. Brothers, Y.-F. Wang, R. W. Harrison and I. T. Weber, ACS Omega, 2019, 4, 8707–8719 CrossRef CAS PubMed.
- Z. Otwinowski, W. Minor, C. W. Carter Jr. and R. M. Sweet, Processing of X-ray Diffraction Data Collected in Oscillation Mode, Academic Press, New York, 1997, pp. 307–326 Search PubMed.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS PubMed.
- J. Agirre, M. Atanasova, H. Bagdonas and C. B. Ballard, et al. , Acta Crystallogr., 2023, 79, 449–461 CAS.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- Collaborative Computational Project, Number 4, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1994, 50, 760–763 CrossRef.
- C.-H. Shen, Y.-F. Wang, A. Y. Kovalevsky, R. W. Harrison and I. T. Weber, FEBS J., 2010, 277, 3699–3714 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
- G. M. Sheldrick and T. R. Schneider, Methods Enzymol., 1997, 277, 319–343 CAS.
- G. N. Murshudov, A. A. Vagin and E. J. Dodson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53, 240–255 CrossRef CAS PubMed.
- A. W. Schuettelkopf and D. M. F. van Aalten, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 1355–1363 CrossRef.
- A. A. Lebedev, P. Young, M. N. Isupov, O. V. Moroz, A. A. Vagin and G. N. Murshudov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 431–440 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS.
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
Footnotes |
| † This manuscript is dedicated to Professor Sukh Dev, an exemplary teacher, a scholar, and a pioneer, on the occasion of his 100th birthday. |
| ‡ Electronic supplementary information (ESI) available: Crystallographic data collection and refinement statistics. See DOI: https://doi.org/10.1039/d4ob00506f |
| This journal is © The Royal Society of Chemistry 2024 |