
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and evaluation of isothiazolo[4,5-b]pyridines as cyclin G-associated kinase (GAK) inhibitors†
Yulia
Ivanova
 a,
Sander
Spittaels
a,
Ling-Jie
Gao
b,
Dominique
Schols
c,
Luc
Van Meervelt
d,
Mathy
Froeyen
b,
Wim
Dehaen
a and
Steven
De Jonghe
*c
a,
Sander
Spittaels
a,
Ling-Jie
Gao
b,
Dominique
Schols
c,
Luc
Van Meervelt
d,
Mathy
Froeyen
b,
Wim
Dehaen
a and
Steven
De Jonghe
*c
aKU Leuven, Department of Chemistry, Sustainable Chemistry for Metals and Molecules, Celestijnenlaan 200F, B-3001 Leuven, Belgium
bKU Leuven, Department of Pharmaceutical and Pharmacological Sciences, Rega Institute for Medical Research, Laboratory of Medicinal Chemistry, Herestraat 49, box 1030, 3000 Leuven, Belgium
cKU Leuven, Department of Microbiology, Immunology and Transplantation, Rega Institute for Medical Research, Laboratory of Virology and Chemotherapy, Herestraat 49, box 1043, 3000 Leuven, Belgium. E-mail: steven.dejonghe@kuleuven.be
dKU Leuven, Department of Chemistry, Biomolecular Architecture, Celestijnenlaan 200F, B-3001 Leuven, Belgium
First published on 22nd August 2024
Abstract
Isothiazolo[4,3-b]pyridines have been extensively explored as inhibitors of cyclin G-associated kinase (GAK). In order to expand the structure–activity relationship study and to discover other chemotypes that act as GAK inhibitors, the closely related isothiazolo[4,5-b]pyridine scaffold was explored. An easy and efficient synthetic procedure to access 3,5- and 3,6-dihalogenated isothiazolo[4,5-b]pyridines as key building blocks was developed. Regioselective functionalization with various substituents was performed. None of the newly synthesized isothiazolo[4,5-b]pyridines were active as GAK inhibitors. Molecular modeling was applied to rationalise their inactivity as GAK binders.
Introduction
Cyclin G-associated kinase (GAK), also known as auxilin II, is a serine-threonine protein kinase that belongs to the Numb-associated kinase (NAK) family.1 GAK plays a crucial role in clathrin-mediated endocytosis (CME) via phosphorylation of Thr144 and Thr156 of adaptor protein complexes 1 and 2 (AP1 and AP2).2 This phosphorylation is necessary for the recruitment of clathrin and AP-2 to the plasma membrane and is essential for the formation of coated vesicles during CME.3–6 In a later stage of CME, GAK is involved in the uncoating of these vesicles, allowing the recycling of clathrin back to the cell surface and finalising the cycle.7The GAK-regulated clathrin-mediated intracellular trafficking of cellular cargo proteins is a host process that is frequently hijacked by multiple viruses. It has been shown that GAK is an essential factor in the early and late stages of the lifecycle of various, unrelated viruses, such as the hepatitis C virus (HCV),8 the dengue virus (DENV),9 the Ebola virus (EBOV),9 and more recently the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).10 A preclinical proof-of-concept was achieved using erlotinib which is a potent, albeit non-selective GAK inhibitor, in DENV and EBOV murine models leading to reduced viremia, morbidity, and mortality.9 Overall, these data validate GAK inhibition as a promising strategy for the development of broad-spectrum antiviral agents. Besides the interest in GAK as a host target for antiviral therapy, GAK is also involved in various other human pathologies, ranging from neurodegenerative disorders (e.g. Parkinson's disease)11 and various cancers (such as osteosarcoma12 and prostate cancer13).
In the past years, our group extensively explored isothiazolo[4,3-b]pyridine chemistry as a central scaffold for the discovery of potent and selective GAK inhibitors as broad-spectrum antiviral agents. Previous structure–activity relationship (SAR) studies were mainly focused on modifications of the substituents at positions 3, 5 and 6 of the isothiazolo[4,3-b]pyridine core (Fig. 1).14–20 The first isothiazolo[4,3-b]pyridine-based GAK inhibitor (compound 1) possessed a substituted phenyl moiety at position 6 and a morpholine at position 3 and displayed high GAK affinity (IC50 = 51 nM). Later on, it was shown that the insertion of a hydrogen bond acceptor moiety on the substituent at position 3 (e.g. compound 2) led to an improved potency (IC50 = 14 nM). Switching the aryl substituent on the pyridine core from position 6 to position 5 resulted in compound 3 with an equally high GAK affinity (IC50 = 24 nM). Moreover, these isothiazolo[4,3-b]pyridine based GAK inhibitors exhibited in vitro antiviral activity against HCV, DENV, and EBOV, in the low micromolar range, while lacking cytotoxicity.
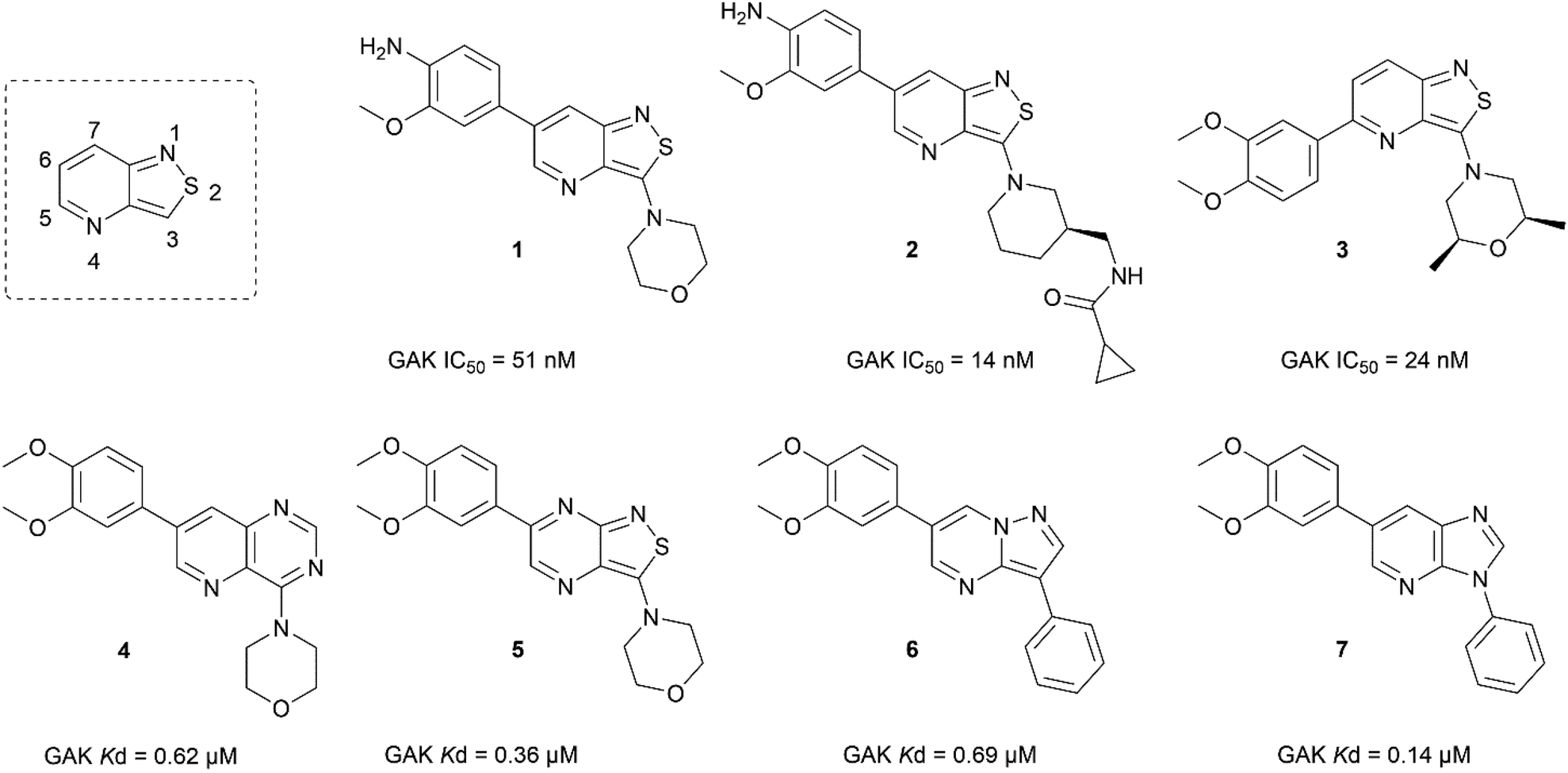 | ||
| Fig. 1 Previously reported GAK inhibitors. | ||
Besides modification of the substitution pattern, a scaffold hopping strategy has also been applied, while keeping the substitution pattern unchanged.17 A series of 13 novel scaffolds (including 6–5 and 6–6 bicyclic scaffolds) were prepared, from which 4 core structures displayed GAK affinity with dissociation constants (Kd values) below 1 μM (Fig. 1), including the quinazoline analogue 4, the isothiazolo[3,4-b]pyrazine derivative 5, the pyrazolo[1,5-a]pyrimidine 6 and imidazo[4,5-b]pyridine 7.
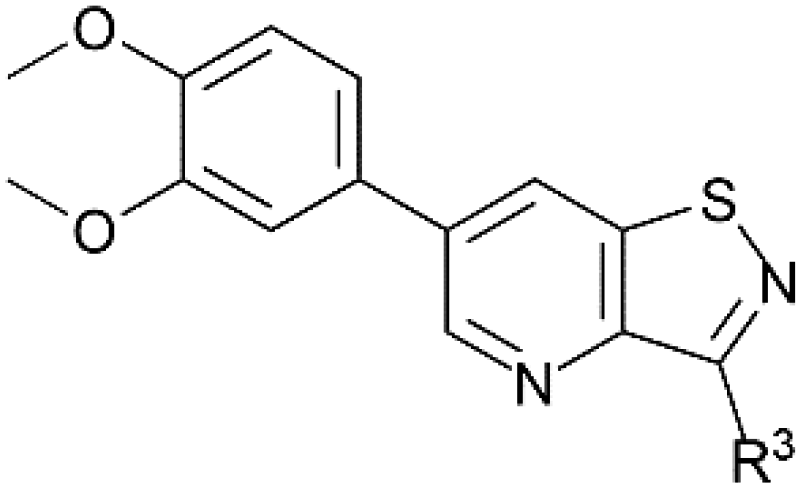
Despite this scaffold hopping approach demonstrating the potential of altering the 5-membered cycle, derivatives of the closely related isothiazolo[4,5-b]pyridine core have not been studied as potential GAK inhibitors. This structure results from a simple swap of the nitrogen and sulfur yielding a scaffold which is of similar size and shape to the parent isothiazolo[4,3-b]pyridine lead molecule (Fig. 2). In addition, a SciFinder™ search revealed that isothiazolo[4,5-b]pyridines are not very well studied in organic and medicinal chemistry, with only 889 compounds known based on this skeleton (accessed on May 2024). Hence, although the isothiazolo[4,5-b]pyridine scaffold is not completely novel, it is definitively less explored compared to other privileged scaffolds in medicinal chemistry, such as purines (661K compounds known, SciFinder™, accessed on May 2024) or pyrrolo[2,3-b]pyridines (358K compounds known, SciFinder™, accessed on May 2024). Therefore, in this study, a synthetic approach towards isothiazolo[4,5-b]pyridines is developed. A variety of groups was introduced onto the scaffold, including the substitution pattern of the known isothiazolo[4,3-b]pyridine based GAK inhibitors, since this allows for a side-by-side comparison of both series and for an evaluation of the importance of the central scaffold for GAK affinity.
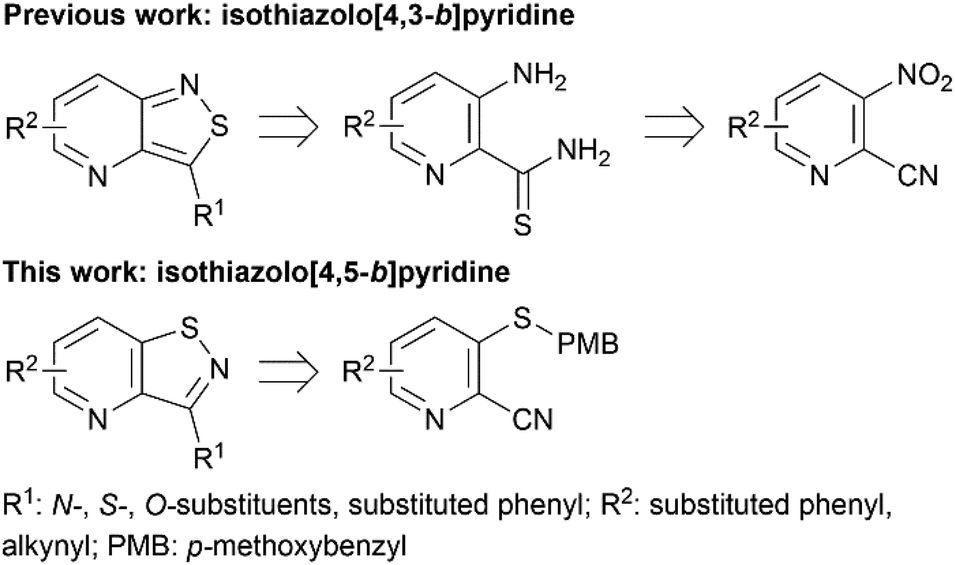 | ||
| Fig. 2 Medicinal chemistry strategy. | ||
Results and discussion
Chemistry
Potent GAK inhibitors based on a isothiazolo[4,3-b]pyridine scaffold bear an aryl residue (e.g. 3,4-dimethoxyphenyl) on the pyridine ring, whereas position 3 of the isothiazolo[4,3-b]pyridine scaffold tolerates more structural variety for GAK affinity. In contrast to isothiazolo[4,3-b]pyridines, which can be accessed from a 3-aminopyridine-2-carbothioamide analogue (derived from a suitable 3-nitropicolinonitrile derivative) by treatment with an oxidising agent to construct the isothiazole moiety, isothiazolo[4,5-b]pyridines require an appropriate 3-mercaptopicolinonitrile analogue that will then undergo oxidative ring closure to form the isothiazole ring (Fig. 2). Compound 12 was, thus, considered a key intermediate since it easily allows for structural modifications at position 3. Therefore, the 3,4-dimethoxyphenyl functionality was introduced as the first step (Scheme 1). A Suzuki coupling between the commercially available 3,5-dichloropicolinonitrile 8 and 3,4-dimethoxyphenylboronic acid, catalysed by Pd(PPh3)4, using K2CO3 as the base in a THF/H2O solvent mixture proceeds preferentially at position 5, resulting in compound 9, along with some minor amounts of the disubstituted side-product. The correct regiochemistry of compound 9 was elucidated via a 2D NMR NOESY experiment (see ESI†). The presence of clear NOE correlations between the proton at position 6 of the pyridine ring and both protons at the ortho position of the dimethoxyphenyl moiety confirmed that the 3,4-dimethoxyphenyl group was attached to position 5 of the pyridine scaffold.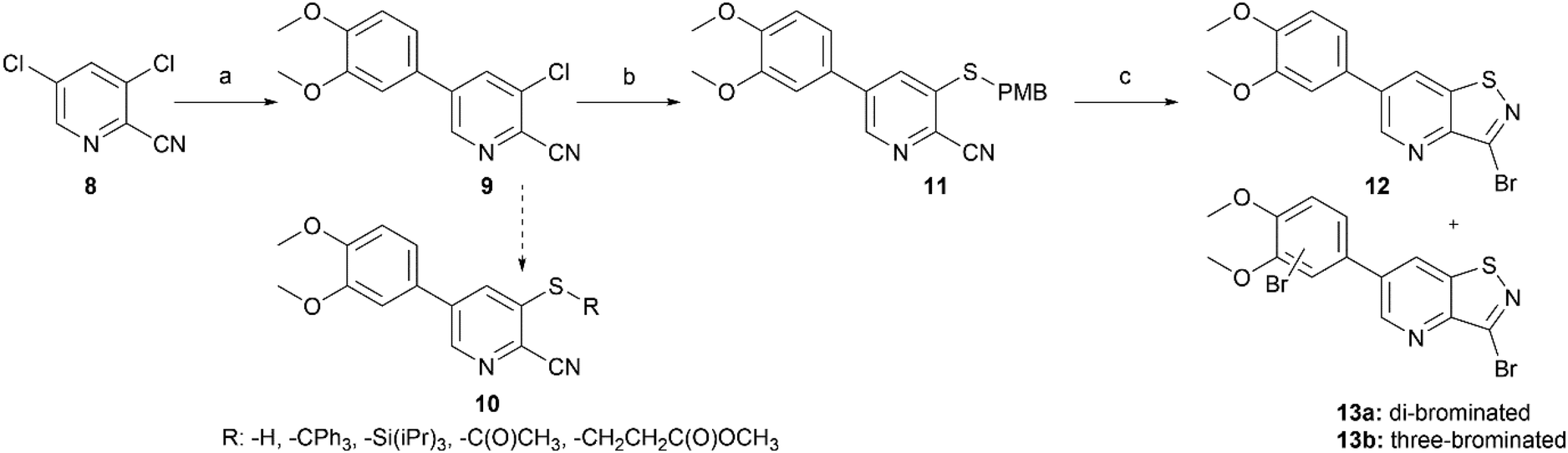 | ||
| Scheme 1 Initial synthetic approach towards isothiazolo[4,5-b]pyridines. Reagents and conditions: (a) 3,4-dimethoxyphenylboronic acid, Pd(PPh3)4, K2CO3, THF/H2O, 100 °C, 72%; (b) 4-methoxybenzylthiol, NaH, DMF, rt, 81%; (c) Br2, EtOAc, rt or reflux. | ||
For the introduction of sulfur onto the pyridine scaffold, various reagents were explored to obtain the compound with the general structure 10. Applying sodium sulfide or protected thiol reagents (such as triphenylmethanethiol, triisopropylsilanethiol and potassium thioacetate) only afforded messy reaction mixtures containing mainly side products. Although the reaction of pyridine 9 with sodium methanethiolate afforded the desired derivative, the subsequent cleavage of the methyl group under acidic conditions (HCl or HBr) was troublesome. Treatment of 9 with methyl 3-mercaptopropionate and sodium methoxide yielded the thioether, however, the desired compound was not isolated pure due to a low-yielding reaction. Gratifyingly, the reaction of 9 with p-methoxybenzylthiol at room temperature yielded (p-methoxybenzyl)thio-picolinonitrile 11 in good yield. The assembly of fused isothiazoles and isothiazolones by oxidative S–N bond formation is a known process.21 The synthesis of fused isothiazoles from the free thiol and neighbouring nitrile in the bromine atmosphere has been reported in several patents, however in rather unsatisfactory yields below 50%.22–24 In addition, these procedures start from an isolated free thiol, that is generated via acidic cleavage of a S-protected precursor, adding one more step to the synthetic sequence.25 Since p-methoxybenzyl (PMB) deprotection can be effected under oxidative conditions,26 we reasoned that both reactions (i.e. PMB cleavage and isothiazole formation) could be combined to deliver the isothiazolo[4,5-b]pyridine 12 by treatment of picolinonitrile 11 with bromine in a single step. The benefit of this procedure is the formation of the free thiol in situ and its immediate further conversion, avoiding dimer formation. However, bromination of the electron-rich 3,4-dimethoxyphenyl moiety hampered the isolation of the desired product 12. Mass spectral analysis of the crude reaction mixtures indicated that besides the desired compound 12, di- and three-brominated side products (13a and 13b, respectively) were formed when performing the reaction at reflux temperature, whereas the formation of a mono-brominated compound was observed at room temperature. To minimise bromination of the 3,4-dimethoxyphenyl moiety, various reaction circumstances (such as reducing the number of equivalents of bromine, decreasing the reaction temperature, or the slow and dropwise addition of bromine) were attempted. Unfortunately, none of these conditions was able to avoid overbromination, preventing the isolation of compound 12 as a pure substance.
In order to avoid bromination of the dimethoxyphenyl residue, an alternative strategy was developed (Scheme 2). Commercially available 5-bromo-3-fluoropicolinonitrile 14 was chosen as the starting material to ensure selectivity during the nucleophilic aromatic substitution. Treatment of 14 with p-methoxybenzylthiol using K2CO3 as base afforded compound 15 in good yield. The following oxidative cyclisation with bromine furnished the 3,6-dibromo-isothiazolo[4,5-b]pyridine 16 in a single step in high yield, offering convenient access to the previously not reported key intermediate. The structure of 16 was confirmed by single-crystal X-ray crystallography (CCDC 2357539† and Scheme 2B).
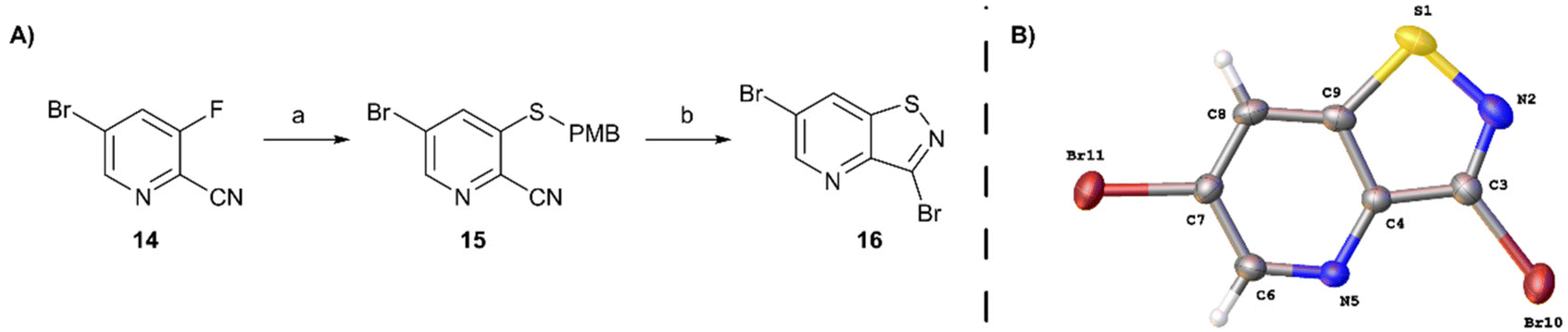 | ||
| Scheme 2 Two-step procedure towards 3,6-dibromo-isothiazolo[4,5-b]pyridine. Reagents and conditions: (A) (a) p-methoxybenzylthiol, K2CO3, DMA, 0 °C, 85%; (b) Br2, EtOAc, 80 °C, 88%. (B) The molecular structure of 16, showing the atom labeling and displacement ellipsoids drawn at the 30% probability level. | ||
Various groups were regioselectively introduced at position 3 of the isothiazolo[4,5-b]pyridine scaffold (Scheme 3). A nucleophilic aromatic substitution of 3,6-dibromoisothiazolo[4,5-b]pyridine 16 with different N-nucleophiles yielded compounds 17a–d and 17ea. The 3-amino congener 17a was obtained in a lower 62% yield due to its insoluble nature, hampering its purification. Reactions resulting in secondary (17b), and tertiary (17c–d and 17ea) amines proceeded smoothly with yields of 80% and 92–97%, respectively. Acidic cleavage of the Boc-protecting group of 17ea resulted in the intermediate 17eb, which was then followed by coupling with cyclopropanecarbonyl chloride, affording compound 17ec in good yield. The scope was extended to oxygen- and sulfur-containing nucleophiles to obtain corresponding compounds 17f and 17g. To access the 3-isopropoxy analogue, sodium hydride was used as a base, while the reactions with S- and N-nucleophiles proceeded without any additional base. Interestingly, position 3 was also susceptible to substitution with sodium azide, resulting in the formation of the 3-azido intermediate, which was directly used in a Cu(I)-catalyzed azide–alkyne cycloaddition affording the 1,4-disubstituted triazole analogue 17h in an overall yield of 28%. Using 3-ethynylpyridine and lithium bis(trimethylsilyl)amide (LiHMDS) as strong base allowed to isolate compound 17i. Position 3 was also susceptible to carbon–carbon bond formation. A Suzuki reaction allowed to install a 3,4-dimethoxyphenyl substituent simultaneously at positions 3 and 6, yielding compound 18j. To furnish the fully decorated scaffold, Pd-catalysed reactions were performed on intermediates 17a–i. A Suzuki procedure, utilizing tetrakis(triphenylphosphine)palladium, 3,4-dimethoxyphenylboronic acid and potassium carbonate, was used to prepare the final compounds 18a–h in fair to excellent yields (51–87%). Reaction with (4-fluorophenyl)boronic acid proceeded equally well, producing compound 18k in 85%. When a Suzuki coupling was applied to 3-(pyridin-3-ylethynyl)isothiazolo[4,5-b]pyridine 17i, this only resulted in the degradation of the triple bond, most likely due to Pd complexation, and no desired compound was isolated. The bromine at position 6 was also susceptible to the Sonogashira coupling, affording compound 18l in 71% yield. The correct regiochemistry was elucidated via a 2D NMR NOESY experiment, taking compound 18c as a model substrate (see ESI†).
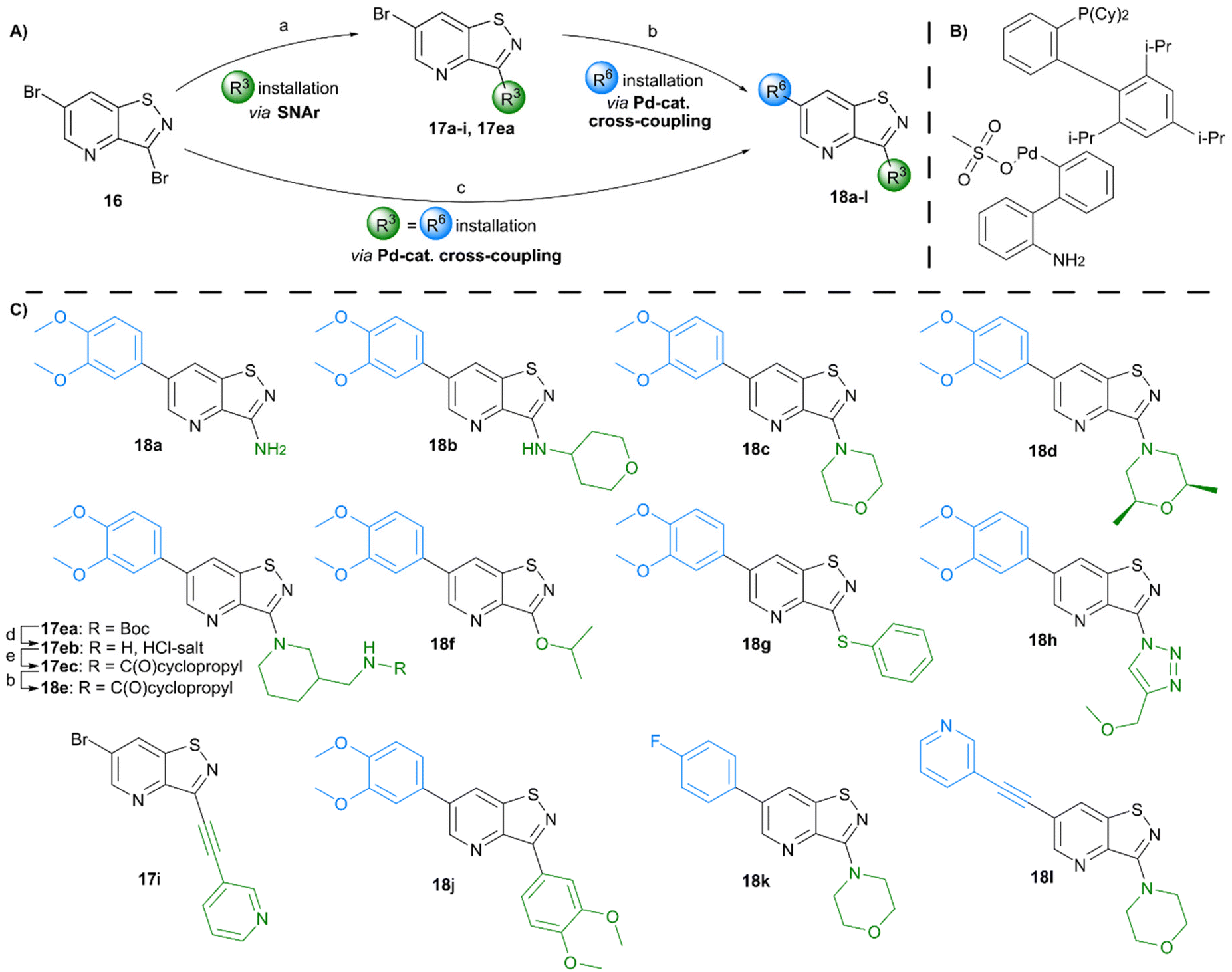 | ||
| Scheme 3 Synthesis of 3,6-disubstituted isothiazolo[4,5-b]pyridines. Reagents and conditions: (A) (a) for 17a–ea: N-nucleophile, EtOH or DMF, rt to 78 °C, 62–97%; for 17f: i-PrOH, NaH, DMF, 50 °C, 18%; for 17g: thiophenol, DMF, 50 °C, 53%; for 17h: (i) NaN3, DMF, 0 °C, (ii) methyl propargyl ether, CuSO4·5H2O, sodium ascorbate, DMSO/H2O, rt, 28%; for 17i: 3-ethynylpyridine, LiHMDS, THF, 0 °C, 80%; (b) for Suzuki coupling: 3,4-dimethoxyphenylboronic acid or 4-fluorophenylboronic acid, Pd(PPh3)4 or XPhos Pd G3, K2CO3 or Cs2CO3, dioxane/H2O, 80 °C or 100 °C, 51–87%; for Sonogashira coupling: 3-ethynylpyridine, Pd(PPh3)2Cl2, Et3N, DMF, 100 °C, 71%; (c) 3,4-dimethoxyphenylboronic acid, XPhos Pd G3, Cs2CO3, dioxane/H2O, 80 °C, 48%; (d) HCl/dioxane 4 M, 0 °C to rt, quant.; (e) cyclopropanecarbonyl chloride, pyridine, DCM, 0 °C to rt, 82%. (B) Structure of XPhos Pd G3 catalyst. (C) Reaction scope. | ||
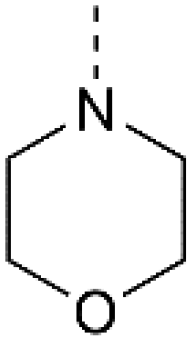
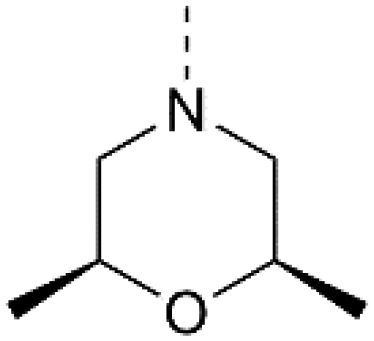
For the synthesis of 3,5-disubstituted isothiazolo[4,3-b]pyridines, an alternative synthetic scheme was established (Scheme 4). Commercially available 6-chloro-3-nitropicolinonitrile 19 was reacted with p-methoxybenzylthiol at −45 °C. This lower temperature allows for the selective substitution of the nitro group at position 3, leaving the chlorine at position 6 intact. The subsequent oxidative cyclisation was performed at 0 °C to avoid halogen exchange, yielding the 3-bromo-5-chloro-isothiazolo[4,5-b]pyridine 21 in good yield. The reaction of key intermediate 21 with morpholine and cis-2,6-dimethylmorpholine gave intermediates 22a and 22b, in yields of 57 and 71%, respectively. The lower yields, in comparison with the synthesis of 17c–d can be rationalised by the reduced selectivity, since position 5 of key intermediate 21 is activated by the presence of the neighbouring nitrogen. The final step consisted of a Suzuki coupling, which was performed using XPhos Pd G3 as a third-generation catalyst and cesium carbonate as the base to activate the less reactive chlorine functionality, furnishing the final compounds 23a–b in good yields.
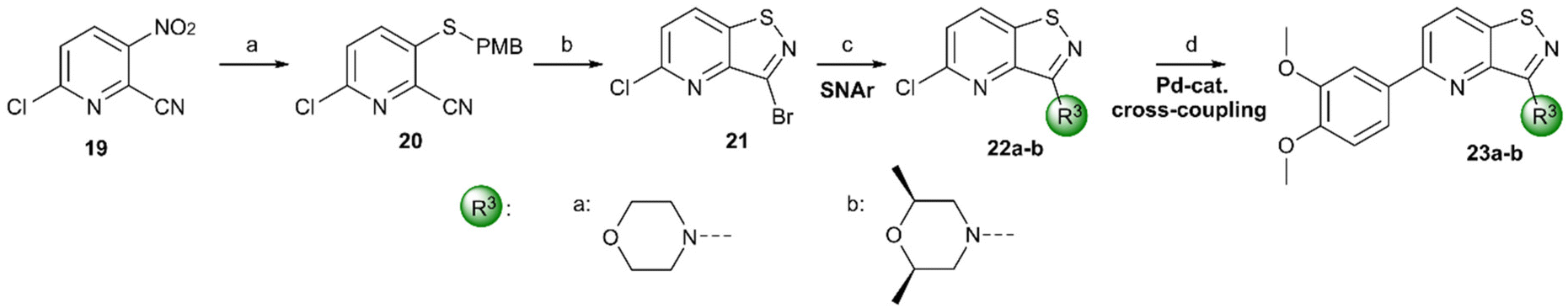 | ||
| Scheme 4 Synthesis of 3,5-disubstituted isothiazolo[4,5-b]pyridines. Reagents and conditions: (a) p-methoxybenzylthiol, K2CO3, DMF, −45 °C, 63%; (b) Br2, EtOAc, 0 °C, 75%; (c) (R3)2NH, EtOH, reflux, 57–71%; (d) 3,4-dimethoxyphenylboronic acid, XPhos Pd G3, Cs2CO3, dioxane/H2O, 80 °C, 89% – quant. PMB – p-methoxybenzyl. | ||
In order to shed a light on its mechanism, the oxidative cyclization reaction was run in the presence of 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO), a free radical trapping reagent (Scheme 5). Adding three equivalents of TEMPO to the reaction mixture allowed to isolate 3,6-dibromo-isothiazolo[4,5-b]pyridine 16 in 65% yield. This suggests that this reaction does not follow a radical pathway, but has rather an ionic nature. The lower yield in the control reaction with TEMPO is due to the purification technique (precipitate filtration and further recrystallization), which is dependent on the scale and solubilizing potential of the initial reaction mixture.
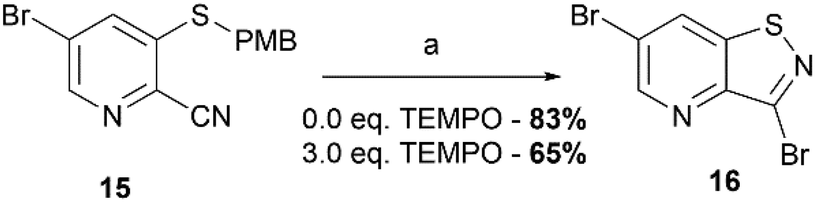 | ||
| Scheme 5 Free radical trapping experiment. Reagents and conditions: (a) Br2 (2.0 eq.), EtOAc, 80 °C, 4 h. | ||
The proposed ionic mechanism is depicted in Scheme 6. Intermediate A is formed by reaction of compound 15 with bromine. The subsequent elimination of cation E, similarly to the previously described mechanism of PMB deprotection,26 yields species B, containing a S–Br bond. Activation of the nitrile group via protonation by a catalytic amount of hydrogen bromide present in bromine, leads to intermediate C, which undergoes bromide attack to form the imine species D. An intramolecular cyclization, with concomitant elimination of HBr, furnishes the final product 16. The previously formed cation E is attacked by bromide forming PMB bromide F. Its electron-rich character makes intermediate F susceptible for overbromination affording compounds G, explaining the requirement of more than one equivalent of bromine. Moreover, upon the formation of compounds G, hydrogen bromide is released which acts as a catalyst in the synthesis of compound 16.
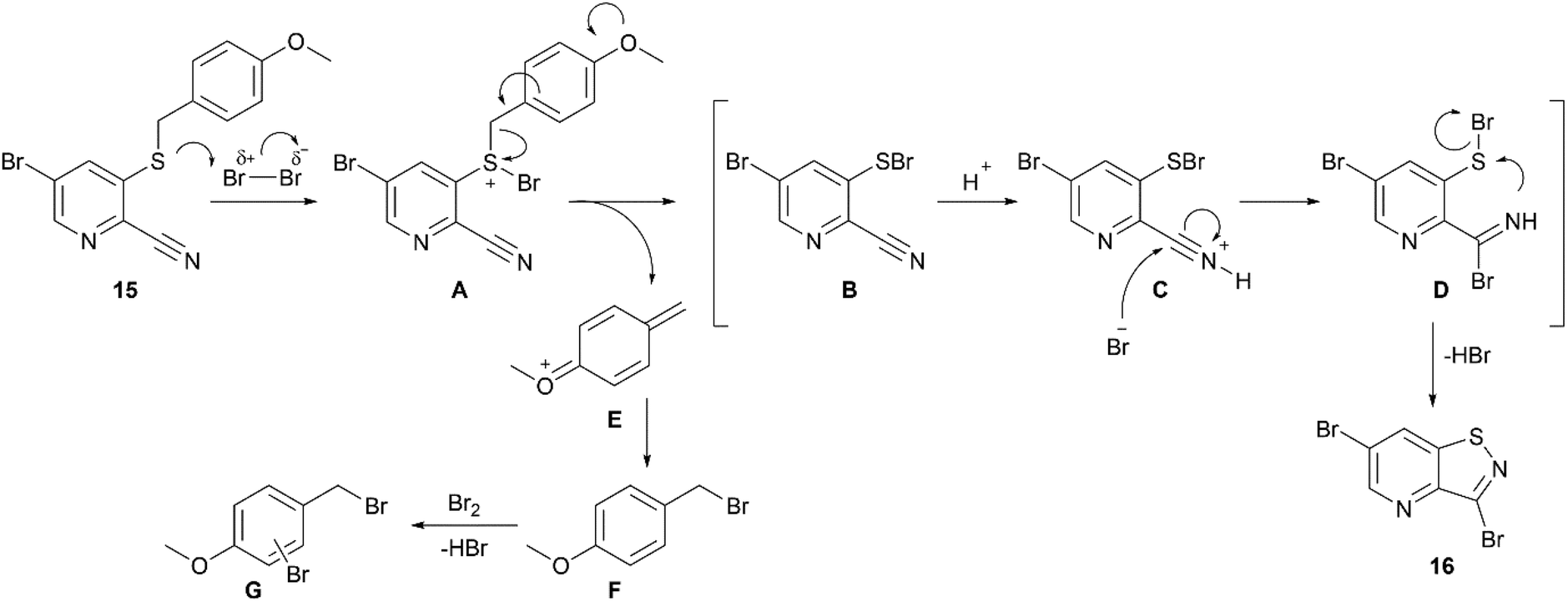 | ||
| Scheme 6 Proposed mechanism of cyclization. | ||
GAK binding affinity evaluation
To assess GAK binding affinity of a selection of the isothiazolo[4,5-b]pyridines (18c–f and 23a–b) the commercially available LanthaScreen™ Europium kinase binding assay was employed (Table 1).27 In this competitive assay, the ATP binding site is exposed to a Alexa Fluor 647-labeled conjugate or tracer and the potential GAK ligands. A high time-resolved fluorescence resonance energy transfer (TR-FRET) signal resulting from the antibody and tracer binding, indicates the occupation of the ATP site. When a compound is endowed with high GAK binding affinity, it displaces the tracer leading to the reduction in the TR-FRET signal. The purity of the analysed samples was above 95% (ESI, Table S1†).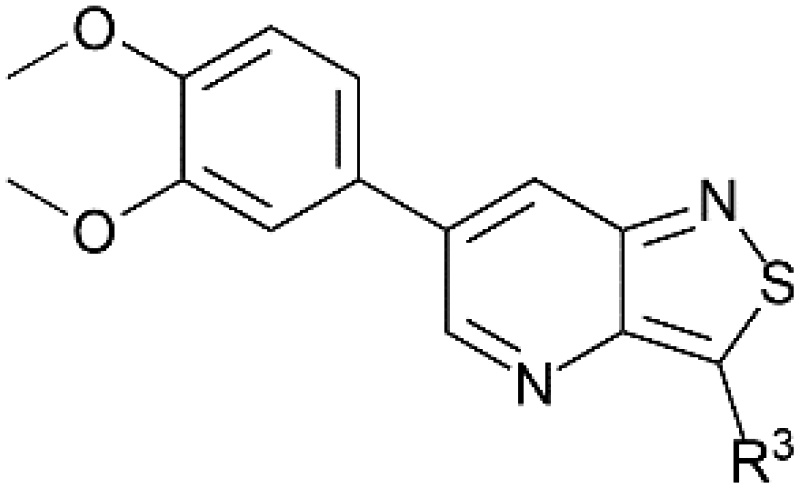
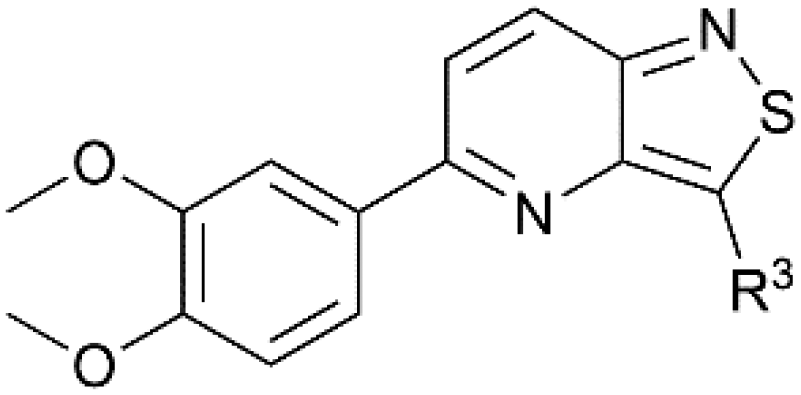
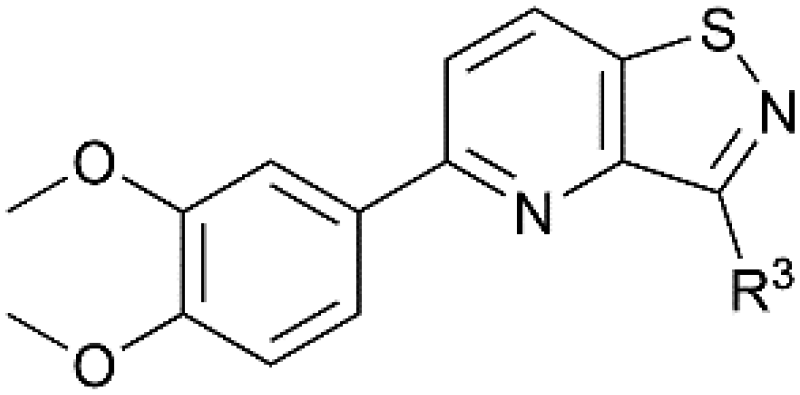
| Isothiazolo[4,3-b]pyridinesa | Isothiazolo[4,5-b]pyridinesa | |
|---|---|---|
| a IC50 = half-maximal inhibitory concentration. Values are the mean of two independent experiments. b At higher concentrations, these compounds exhibited intrinsic fluorescence against the acceptor wavelength, interfering with the assay readout. c Data from ref. 19. d Data from ref. 20. | ||
| R3 |
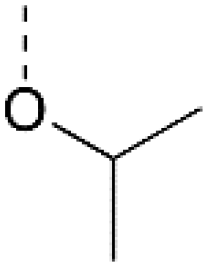
|
|
|
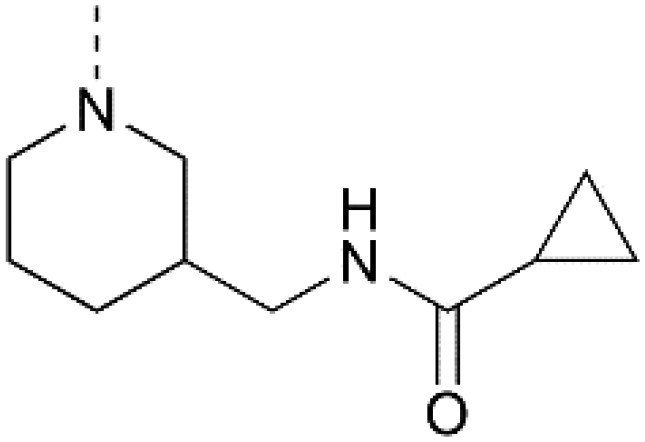
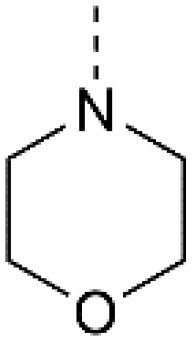
28 | 23a |
IC50 = 0.124 μM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c c |
IC50 > 3.33 μMb |
|
|
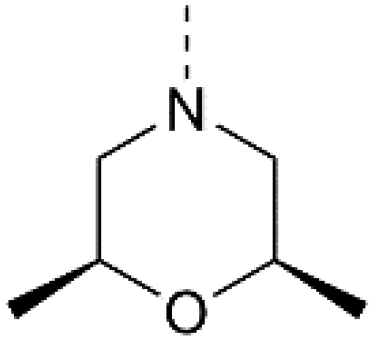
3 | 23b |
| IC50 = 0.024 μMc |
IC50 > 3.33 μMb |
|
The goal of this research was to evaluate the importance of the central isothiazolo[4,3-b]pyridine scaffold for GAK affinity, by the synthesis of a series of isothiazolo[4,5-b]pyridines. It is well-known from previous research that an electron-rich aryl group (such as a dimethoxyphenyl, trimethoxyphenyl or amino-methoxyphenyl) at position 5 or 6 of the central core is optimal for GAK affinity. On the other hand, prior SAR studies revealed that position 3 of the isothiazolo[4,3-b]pyridine scaffold tolerates more structural variety, such as various N- and O-nucleophiles. Hence, within the newly synthesized isothiazolo[4,5-b]pyridines, 6-(3,4-dimethoxyphenyl) (compounds 18c–f), as well as 5-(3,4-dimethoxyphenyl) derivatives (compounds 23a–b) were selected for GAK affinity evaluation due to their high structural similarity to the previously reported potent inhibitors.
In order to study the influence of swapping the sulfur and nitrogen atoms within the isothiazole moiety, a side-by-side comparison of the GAK binding affinity of both scaffolds was performed, keeping the substitution pattern intact. Therefore, the previously reported isothiazolo[4,3-b]pyridines 3 and 24–28 (Fig. 3) were included for comparison purposes. Isothiazolo[4,3-b]pyridines 24 and 27 were previously shown to be endowed with potent GAK affinity in the KINOMEscan™ platform of DiscoverX. To confirm their GAK binding in the Lanthascreen™ assay (which was used to evaluate GAK affinity of the newly synthesized isothiazolo[4,5-b]pyridines) compounds 24 and 27 were resynthesized according to known procedures.14,15 Biochemical evaluation in the Lanthascreen™ binding assay revealed that these compounds behaved as potent GAK ligands (Table 1), as expected based on the DiscoverX data. A good correlation between both assay formats was observed, which is in agreement with previous literature findings.28
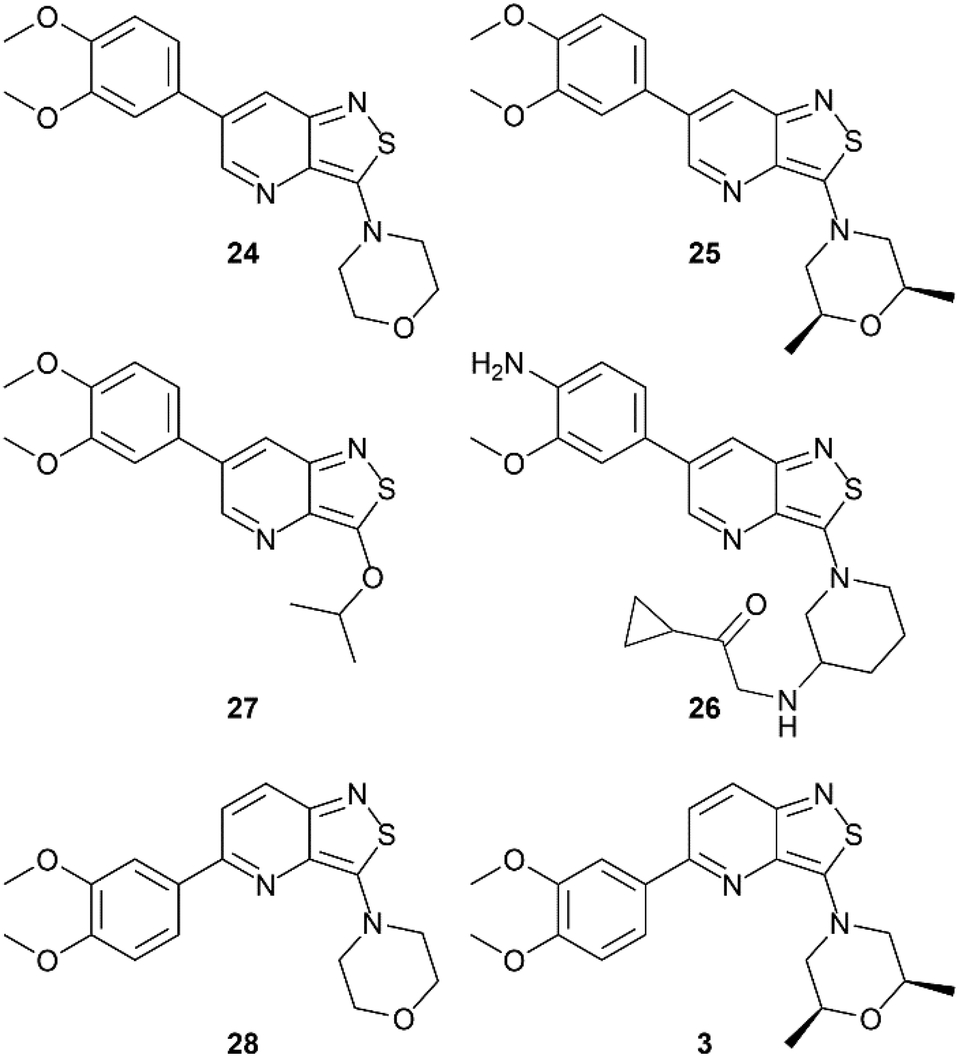 | ||
| Fig. 3 Known isothiazolo[4,3-b]pyridines included in current SAR study. | ||
As can be clearly derived from the data in Table 1, all isothiazolo[4,5-b]pyridines completely lack GAK affinity, whereas the parent isothiazolo[4,3-b]pyridines do show GAK affinity. Hence, swapping the sulfur and nitrogen atoms within the isothiazole moiety had a detrimental effect on GAK inhibition.
Molecular modeling
To rationalise the lack of GAK affinity of the various isothiazolo[4,5-b]pyridines, a close inspection of the experimentally determined binding mode of isothiazolo[4,3-b]pyridine 1 to GAK (PDB entry 4Y8D15) was performed (Fig. 4, top panel). Besides van der Waals interactions of the morpholinyl and 4-amino-3-methoxyphenyl moieties, four interactions of the isothiazolo[4,5-b]pyridine scaffold are observed.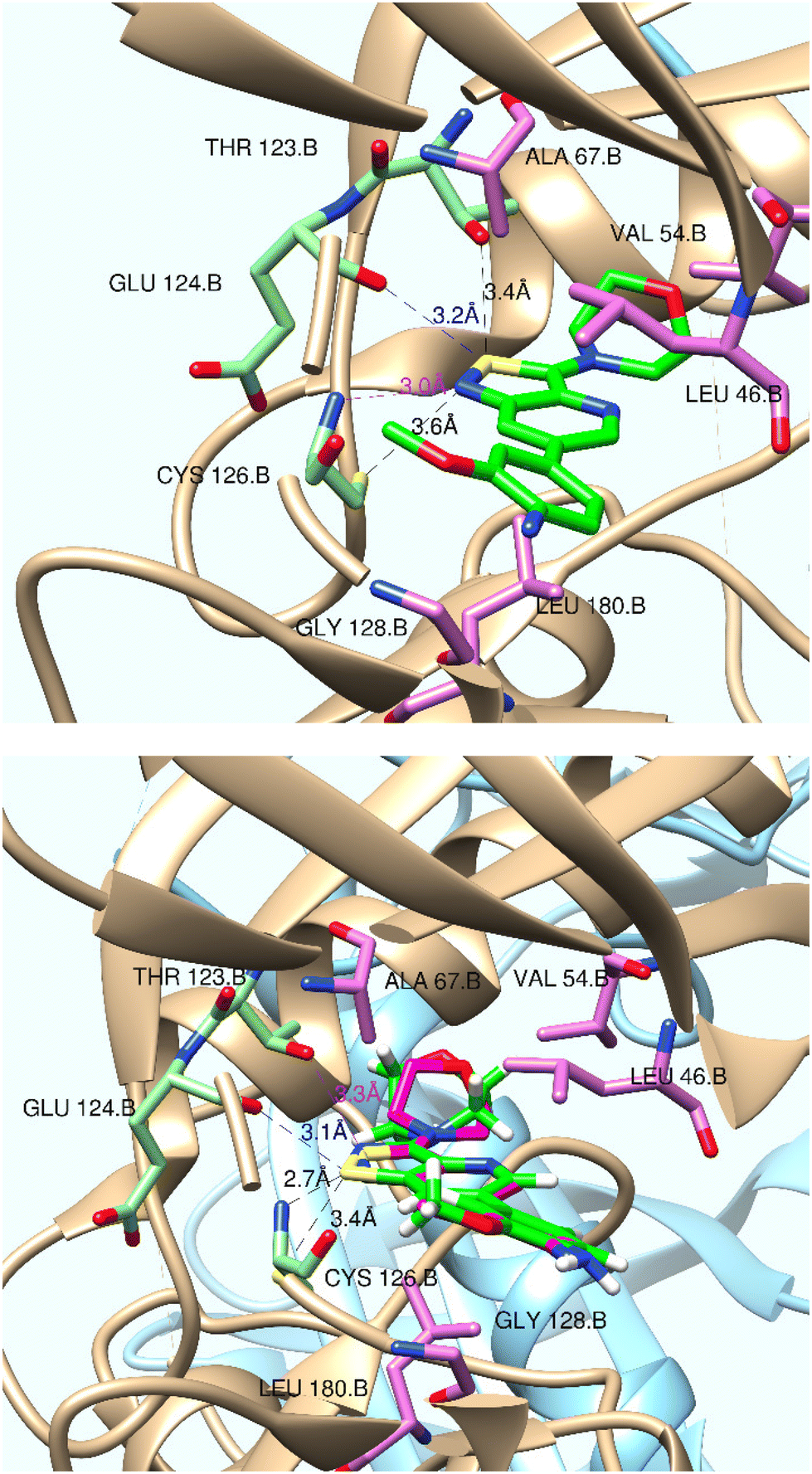 | ||
| Fig. 4 Top: interactions between isothiazolo[4,3-b]pyridine 1 (green) and GAK in PDB 4Y8D (resolution 2.1 Å). Bottom: isothiazolo[4,5-b]pyridine 29 (green), superimposed on compound 1 (magenta). Figures were created using UCSF Chimera.31 | ||
(i) A classical hydrogen bond between the nitrogen of the isothiazole moiety and the backbone NH of Cys126 (N/N distance 3.0 Å); this bond is well-known as a stabilizing interaction.
(ii) A non-conventional hydrogen bond between cys126.SG acting as a hydrogen bond donor and the isothiazole nitrogen (SG/N distance 3.6 Å); this type of hydrogen bond is an important interaction.29
(iii) A non-conventional hydrogen bond between the sulfur of the isothiazole moiety and the hydroxyl group of Thr123 (S/O distance 3.4 Å); since sulfur is a rather poor H-bond acceptor, this H-bond is rather weak.
(iv) A chalcogen bond (ChB) interaction between the sulfur of the isothiazole and the backbone oxygen of Glu124 (S/O distance 3.2 Å, C–S⋯O angle is 153.2°).30 The ability of sulfur to act as a ChB donor is favoured because of the electron-withdrawing capacity of the nitrogen, covalently attached to the sulfur.
Overall, the combination of these interactions contributes to the excellent GAK binding affinity of compound 1.
The corresponding isothiazolo[4,5-b]pyridine (compound 29) was then superimposed (by adjusting the flexible dihedral angles) on the isothiazolo[4,3-b]pyridine 1 (which displays potent GAK affinity). A different interaction pattern of compound 29 with GAK was observed, when compared to GAK inhibitor 1 (Fig. 4, bottom).
(i) The sulfur atom of the isothiazole moiety is engaged in a hydrogen bonding with the backbone nitrogen of Cys126 (S/N distance 2.7 Å). This interaction is rather weak as sulfur functions as hydrogen bond acceptor.
(ii) The thiol group of Cys126 (as H-bond donor) makes a weak hydrogen bond with the isothiazole sulfur as H-bond acceptor (S/S distance 3.3 Å).
(iii) The isothiazole sulfur has a ChB interaction with Glu124.O (distance S/O 3.1 Å; angle O–S–C 150.94°).
(iv) The nitrogen of the isothiazole can hydrogen bond to Thr123.OG/HOG. However, the N/O distance of 3.4 Å makes this classical hydrogen bond rather weak.
It is clear that the superimposition model with the isothiazolo[4,5-b]pyridine 29 still shows important interactions with GAK. Only the classical hydrogen bond might be weaker based on the distance criterion. However, a comparison of the interactions based on this superimposition model does not offer a satisfying explanation for the complete lack of GAK affinity of isothiazolo[4,5-b]pyridines.
The dipole moment is a widely used parameter to explain bioactivities of molecules. In order to investigate if differences in the polarity of a molecule are related to different GAK inhibition, the dipole moment of both scaffolds was calculated (Table 2). The dipole moment of the novel isothiazolo[4,5-b]pyridine 29 is drastically increased, when compared to the original isothiazolo[4,3-b]pyridine analogue 1 (Table 2).
| Cmpd | Structure | Dipole moment (Debye) |
|---|---|---|
| 1 |
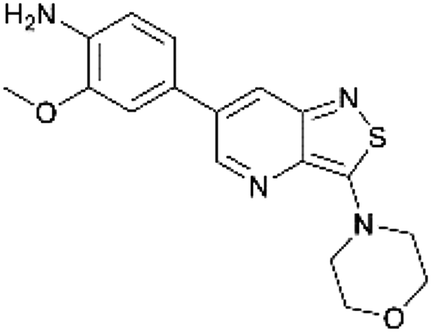
|
0.785 |
| 29 |
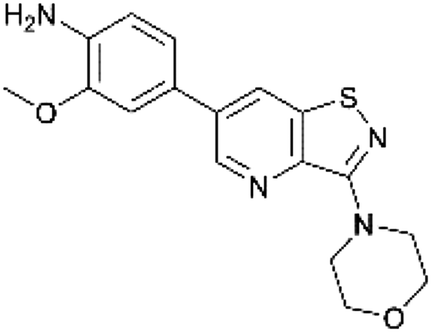
|
4.647 |
To further investigate how dissimilar the isothiazolo[4,3-b]pyridine 1 and the isothiazolo[4,5-b]pyridine 29 are in terms of electrostatics, the molecular electrostatic potential surfaces around both scaffolds (having a similar conformation as compound 1 in 4Y8D) was calculated. The blue (positive) potential around the sulfur of the isothiazolo[4,3-b]pyridine 1 (Fig. 5, top) matches the red negative potential in the GAK binding pocket (Fig. 6). This electrostatic complementarity between GAK and isothiazolo[4,3-b]pyridine 1 is beneficial for GAK affinity.
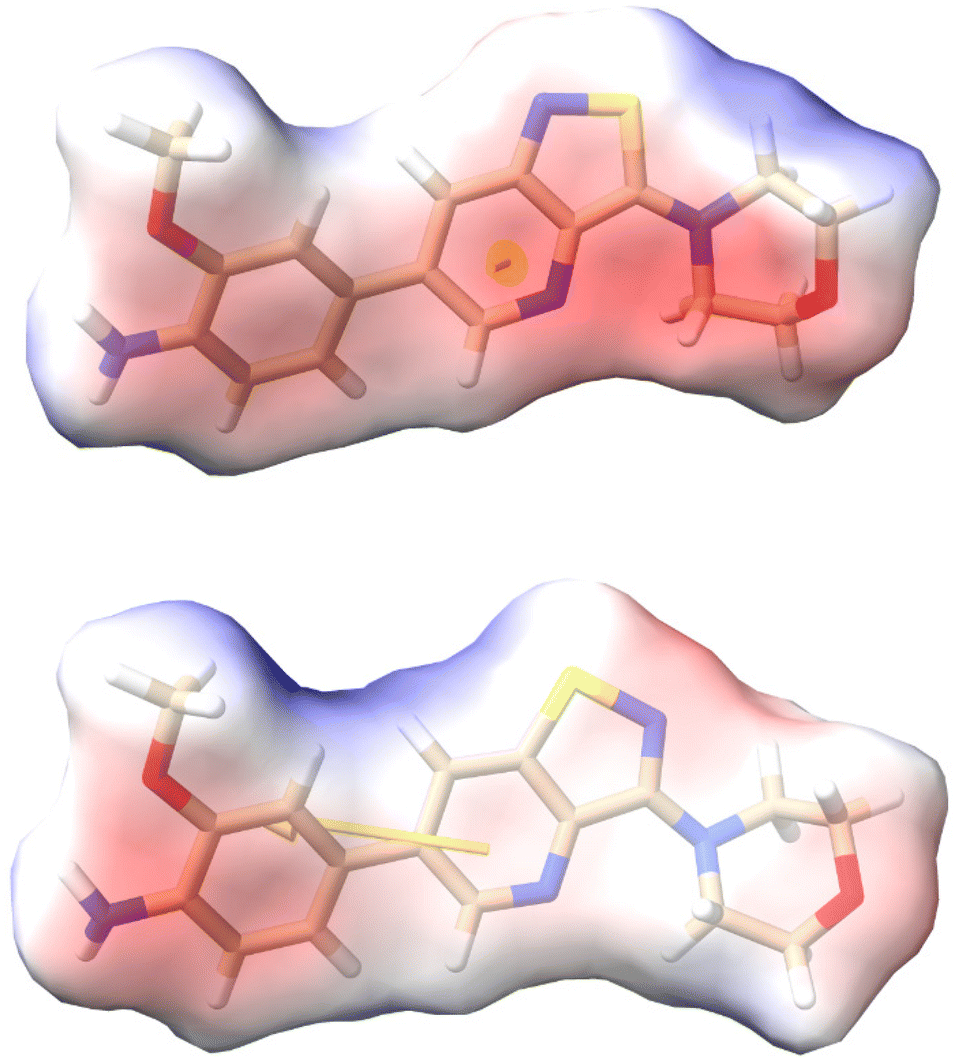 | ||
| Fig. 5 Electrostatic potential calculated by APBS around the 2 scaffolds (having a conformation similar to compound 1 in PDB 4Y8D). Top: isothiazolo[4,3-b]pyridine 1. Bottom: isothiazolo[4,5-b]pyridine 29. The yellow arrow is the dipole vector starting at the centre of mass origin. The dipole points from − to + charges. Colors: red = negative potential; blue = positive potential; white = neutral. Figure created in ChimeraX.33 | ||
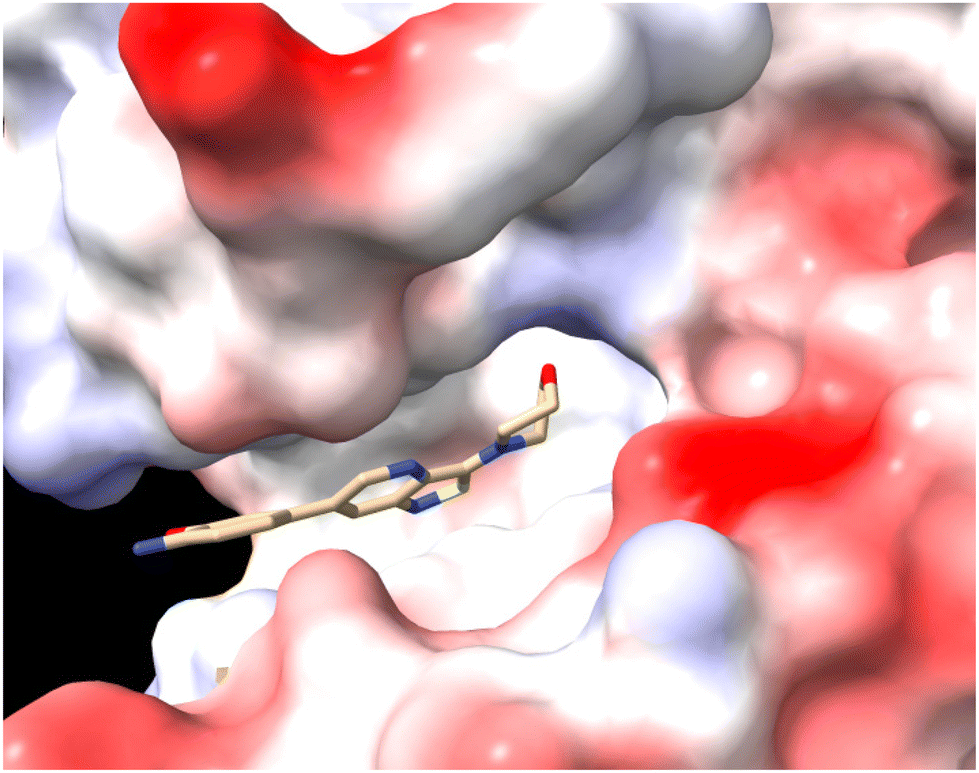 | ||
| Fig. 6 The GAK ATP binding site in PDB 4Y8D, as coloured by Coulomb potential (red = negative signal, white = neutral, blue = positive potential). | ||
In contrast, for the isothiazolo[4,5-b]pyridine 29 (Fig. 5, bottom), a more negative potential (red) is observed at the nitrogen of the isothiazole moiety, which is incompatible with the red negative potential at this position in the ATP binding site of GAK (Fig. 6), which explains the inactivity of isothiazolo[4,5-b]pyridines as GAK binders.
Conclusion
To probe the importance of the isothiazolo[4,3-b]pyridine scaffold for GAK binding, the closely related isothiazolo[4,5-b]pyridines (with a substitution pattern that is known to be beneficial for GAK affinity) were prepared. Surprisingly, the isothiazolo[4,5-b]pyridines were completely devoid of GAK affinity. Molecular modeling allowed to rationalise the lack of GAK binding due to a number of weaker hydrogen interactions, as well as of a less favourable electrostatic complementarity.Despite the negative GAK data, the chemistry presented in this work is still of high value. A straightforward and high-yielding two-step synthesis of the dihalo-isothiazolo[4,5-b]pyridine building blocks 16 and 21 was established. These key intermediates can be used in various synthetic transformations (such as nucleophilic aromatic substitutions and palladium-catalyzed cross-coupling reactions), allowing regioselective functionalisation of the isothiazolo[4,5-b]pyridine scaffold. It allows the preparation of isothiazolo[4,5-b]pyridine based compound libraries with broad structural diversity that can be used in various drug discovery screening programs.
Experimental section
Chemistry
000 (FWHM) using leucine enkephalin as lock mass.
MP 164–167 °C 1H NMR (400 MHz, CDCl3) δ (ppm): 8.79 (d, J = 2.0, 1H), 7.97 (d, J = 2.0, 1H), 7.19 (dd, J = 8.3, 2.1, 1H), 7.06 (d, J = 2.1, 1H), 7.01 (d, J = 8.3, 1H), 3.98 (s, 3H), 3.96 (s, 3H). 13C NMR (101 MHz, CDCl3) δ (ppm): 151.0, 150.1, 147.0, 140.9, 136.0, 134.6, 130.6, 127.2, 120.6, 115.1, 112.0, 110.2, 56.3, 56.2. HRMS (ESI+): m/z calculated for C14H11ClN2O2 [M + H]+: 275.05817; found: 275.0585.
MP 136–137 °C 1H NMR (400 MHz, CDCl3) δ (ppm): 8.66 (d, J = 2.0, 1H), 7.67 (d, J = 2.0, 1H), 7.26–7.23 (m, 2H), 7.05 (dd, J = 8.3, 2.1, 1H), 6.96 (d, J = 8.3, 1H), 6.88 (d, J = 2.1, 1H), 6.85–6.82 (m, J = 8.6, 2H), 4.23 (s, 2H), 3.94 (s, 3H), 3.93 (s, 3H), 3.77 (s, 3H). 13C NMR 159.4, 150.6, 149.9, 146.7, 139.3, 138.6, 136.1, 132.7, 130.3, 128.2, 127.6, 120.3, 116.2, 114.3, 111.9, 110.1, 56.3, 56.2, 55.4, 38.3. HRMS (ESI+): m/z calculated for C22H20N2O3S [M + H]+: 393.12673; found: 393.1264.
MP 92–93 °C 1H NMR (600 MHz, CDCl3) δ (ppm): 8.50 (d, J = 2.0 Hz, 1H), 7.77 (d, J = 2.0 Hz, 1H), 7.25–7.22 (m, 2H), 6.87–6.84 (m, 2H), 7.37–7.33 (m, 2H), 4.20 (s, 2H), 3.80 (s, 3H). 13C NMR (101 MHz, CDCl3) δ (ppm): 159.5, 148.9, 140.9, 139.4, 131.9, 130.2, 126.2, 124.4, 115.2, 114.3, 55.3, 37.8. HRMS (ESI+): m/z calculated for C14H11BrN2OS [M + H]+: 334.9849; found: 334.9845.
MP 185–186 °C 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.06 (d, J = 2.0 Hz, 1H), 8.97 (d, J = 2.0 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ (ppm): 150.8, 146.6, 146.1, 137.9, 132.7, 120.6. HRMS (ESI+): m/z calculated for C6H2Br2N2S [M + H]+: 292.8379; found: 292.8379.
Nucleophilic substitution
MP 109–112 °C 1H NMR (400 MHz, CDCl3) δ (ppm): 8.42 (d, J = 2.1 Hz, 1H), 8.28 (d, J = 2.1 Hz, 1H), 2.87 (br s, 2H). 13C NMR (101 MHz, CDCl3) δ (ppm): 149.2, 148.0, 133.7, 125.5, 124.5, 114.7. HRMS (ESI+): m/z calculated for C6H4BrN3S [M + H]+: 229.93825; found: 229.9390.
MP 125–127 °C 1H NMR (600 MHz, CDCl3) δ (ppm): 8.43 (d, J = 2.0 Hz, 1H), 8.16 (d, J = 2.0 Hz, 1H), 4.01–3.98 (m, 2H), 3.38 (dt, J = 2.0, 11.8 Hz, 2H), 3.02–2.97 (m, 1H), 2.94 (d, J = 5.1 Hz, 1H), 1.99–1.96 (m, 2H), 1.57–1.50 (m, 2H). 13C NMR (151 MHz, CDCl3) δ (ppm): 148.5, 148.3, 134.1, 125.4, 124.9, 114.7, 66.6, 57.4, 33.8. HRMS (ESI+): m/z calculated for C11H12BrN3OS [M + H]+: 313.99577; found: 313.9964.
MP 154–156 °C 1H NMR (400 MHz, CDCl3) δ (ppm): 8.63 (d, J = 2.0 Hz, 1H), 8.28 (d, J = 2.0 Hz, 1H), 4.02–4.00 (m, 4H), 3.91–3.89 (m, 4H). 13C NMR (101 MHz, CDCl3) δ (ppm): 160.3, 147.9, 147.3, 142.0, 130.6, 119.6, 66.9, 47.9. HRMS (ESI+): m/z calculated for C10H10BrN3OS [M + H]+: 299.9801; found: 299.9804.
MP 94–96 °C 1H NMR (400 MHz, CDCl3) δ (ppm): 8.63 (d, J = 2.0 Hz, 1H), 8.25 (d, J = 2.0 Hz, 1H), 4.81–4.78 (m, 2H), 3.88–3.83 (m, 2H), 2.75 (dd, J = 10.5, 12.9 Hz, 2H), 1.27 (d, J = 6.4 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ (ppm): 159.9, 147.8, 147.3, 142.0, 130.6, 119.5, 71.7, 52.9, 19.1. HRMS (ESI+): m/z calculated for C12H14BrN3OS [M + H]+: 328.0114; found: 328.0110.
MP 110–112 °C 1H NMR (600 MHz, CDCl3) δ (ppm): 8.65 (d, J = 1.8 Hz, 1H), 8.26 (d, J = 1.8 Hz, 1H), 5.73 (br s, 1H), 4.08–4.03 (m, 2H), 3.73–3.70 (m, 1H), 3.58–3.55 (m, 1H), 3.30–3.26 (m, 1H), 3.19–3.15 (m, 1H), 1.95 (br s, 1H), 1.86–1.78 (m, 2H), 1.70–1.66 (m, 1H), 1.48 (s, 9H), 1.46–1.43 (m, 1H). 13C NMR (151 MHz, CDCl3) δ (ppm): 160.9, 156.4, 147.8, 147.2, 141.9, 130.8, 119.4, 79.1, 51.3, 48.5, 42.4, 35.7, 28.6, 28.1, 23.4. HRMS (ESI+): m/z calculated for C17H23BrN4O2S [M + H]+: 427.0798; found: 427.0794.
MP 209–210 °C 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.89 (d, J = 2.2 Hz, 1H), 8.77 (d, J = 2.2 Hz, 1H), 8.17 (br s, 3H), 4.70–4.67 (m, 1H), 4.47–4.43 (m, 1H), 3.20–3.13 (m, 1H), 3.02 (dd, J = 9.9, 13.0 Hz, 1H), 2.86–2.70 (m, 2H), 2.80–1.99 (m, 1H), 1.94–1.86 (m, 1H), 1.79–1.73 (m, 1H), 1.61–1.50 (m, 1H), 1.36–1.27 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ (ppm): 159.6, 147.5, 147.2, 141.3, 132.0, 118.9, 50.4, 47.5, 41.5, 33.5, 27.6, 23.6. HRMS (ESI+): m/z calculated for C12H15BrN4S [M + H]+: 327.0274; found: 327.0278.
MP 162–163 °C 1H NMR (600 MHz, CDCl3) δ (ppm): 8.62 (d, J = 2.0 Hz, 1H), 8.27 (d, J = 2.0 Hz, 1H), 6.51 (br s, 1H), 4.15–4.11 (m, 2H), 3.67–3.60 (m, 2H), 3.52–3.48 (m, 1H), 3.25–3.20 (m, 1H), 2.00–1.94 (m, 1H), 1.88–1.78 (m, 2H), 1.71–1.65 (m, 1H), 1.50–1.45 (m, 1H), 1.43–1.39 (m, 1H), 1.00–0.98 (m, 2H), 0.78–0.75 (m, 2H). 13C NMR (151 MHz, CDCl3) δ (ppm): 173.6, 160.8, 147.9, 147.1, 142.0, 130.9, 119.4, 51.2, 48.5, 41.6, 35.7, 28.3, 23.5, 15.1, 7.2. HRMS (ESI+): m/z calculated for C16H19BrN4OS [M + H]+: 395.0536; found: 395.0531.
MP 69–70 °C 1H NMR (600 MHz, CDCl3) δ (ppm): 8.75 (d, J = 1.9 Hz, 1H), 8.30 (d, J = 1.9 Hz, 1H), 5.45 (sept, J = 6.8 Hz, 1H), 1.53 (d, J = 6.8 Hz, 6H). 13C NMR (151 MHz, CDCl3) δ (ppm): 161.9, 149.4, 146.4, 139.5, 130.8, 120.6, 73.2, 22.1. HRMS (ESI+): m/z calculated for C9H9BrN2OS [M + H]+: 272.9692; found: 272.9691.
MP 118–121 °C 1H NMR (600 MHz, CDCl3) δ (ppm): 8.80 (d, J = 1.8 Hz, 1H), 8.40 (d, J = 1.8 Hz, 1H), 7.74–7.71 (m, 2H), 7.48–7.46 (m, 3H). 13C NMR (101 MHz, CDCl3) δ (ppm): 160.9, 149.6, 147.1, 146.5, 135.1, 130.6, 129.7, 129.6, 128.1, 120.6. HRMS (ESI+): m/z calculated for C12H7BrN2S2 [M + H]+: 322.93073; found: 322.9312.
MP 192–193 °C 1H NMR (600 MHz, DMSO-d6) δ (ppm): 9.17 (d, J = 1.8 Hz, 1H), 9.11 (s, 1H), 9.07 (d, J = 1.8 Hz, 1H), 4.64 (s, 2H), 3.36 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 150.9, 148.6, 144.2, 140.3, 132.9, 124.2, 120.3, 64.5, 57.5. HRMS (ESI+): m/z calculated for C10H8BrN5OS [M + H]+: 325.97061; found: 325.9709.
MP 139–141 °C 1H NMR (400 MHz, CDCl3) δ (ppm): 8.80 (dd, J = 0.9, 2.2 Hz, 1H), 8.65 (dd, J = 1.7, 4.9 Hz, 1H), 8.60 (d, J = 2.0 Hz, 1H), 8.25 (d, J = 2.0 Hz, 1H), 7.87–7.84 (ddd, J = 1.7, 2.2, 7.9 Hz, 1H), 7.37–7.33 (ddd, J = 0.9, 4.9, 7.9 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ (ppm): 152.9, 150.2, 150.0, 139.3, 138.3, 137.2, 128.5, 125.7, 123.4, 118.8, 114.2, 98.4, 74.0. HRMS (ESI+): m/z calculated for C13H6BrN3S [M + H]+: 315.95390; found: 315.9532.
Suzuki coupling
MP 125–127 °C 1H NMR (600 MHz, CDCl3) δ (ppm): 8.83 (d, J = 2.0 Hz, 1H), 8.23 (d, J = 2.0 Hz, 1H), 7.22 (dd, J = 1.8, 8.0 Hz, 1H), 7.14 (d, J = 1.8 Hz, 1H), 7.02 (d, J = 8.0 Hz, 1H), 5.40 (br s, 2H), 3.98 (s, 3H), 3.96 (s, 3H). 13C NMR (151 MHz, CDCl3) δ (ppm): 158.7, 149.9, 149.8, 146.7, 144.9, 140.8, 135.6, 130.1, 125.9, 120.5, 111.9, 110.8, 56.3, 56.2. HRMS (ESI+): m/z calculated for C14H13N3O2S [M + H]+: 288.08011; found: 288.0789.
MP 167–168 °C 1H NMR (600 MHz, CDCl3) δ (ppm): 8.78 (d, J = 1.9 Hz, 1H), 8.21 (d, J = 1.9 Hz, 1H), 7.21 (dd, J = 1.9, 8.2 Hz, 1H), 7.13 (d, J = 1.9 Hz, 1H), 7.01 (d, J = 8.2 Hz, 1H), 5.76 (d, J = 7.9 Hz, 2H), 4.23–4.16 (m, 1H), 4.07–4.04 (m, 2H), 3.98 (s, 3H), 3.96 (s, 3H), 3.60 (dt, J = 1.9, 11.5 Hz, 2H), 2.22–2.19 (m, 2H), 1.72–1.65 (m, 2H). 13C NMR (101 MHz, CDCl3) δ (ppm): 157.6, 149.9, 149.8, 146.2, 144.6, 140.9, 135.9, 130.2, 126.0, 120.4, 111.9, 110.8, 67.0, 56.24, 56.20, 48.7, 33.6. HRMS (ESI+): m/z calculated for C19H21N3O3S [M + H]+: 372.13762; found: 372.1377.
MP 132–134 °C 1H NMR (400 MHz, CDCl3) δ (ppm): 8.82 (d, J = 2.2 Hz, 1H), 8.20 (d, J = 2.2 Hz, 1H), 7.21 (dd, J = 2.0, 8.2 Hz, 1H), 7.13 (d, J = 2.0, 1H), 7.00 (d, J = 8.2, 1H), 4.06–4.04 (m, 4H), 3.96 (s, 3H), 3.94 (s, 3H), 3.92–3.90 (m, 4H). 13C NMR (101 MHz, CDCl3) δ (ppm): 160.7, 149.9, 149.7, 146.7, 145.6, 142.3, 134.4, 129.9, 125.6, 120.3, 111.9, 110.6, 66.9, 56.2, 56.1, 47.9. HRMS (ESI+): m/z calculated for C18H19N3O3S [M + H]+: 358.1219; found: 358.1212.
MP 151–152 °C 1H NMR (600 MHz, CDCl3) δ (ppm): 8.84 (d, J = 1.9 Hz, 1H), 8.20 (d, J = 1.9 Hz, 1H), 7.22 (dd, J = 2.1, 8.1 Hz, 1H), 7.14 (d, J = 2.1 Hz, 1H), 7.01 (d, J = 8.1 Hz, 1H), 4.88 (d, J = 11.4 Hz, 2H), 3.97 (s, 3H), 3.95 (s, 3H), 3.93–3.87 (m, 2H), 2.79 (dd, J = 2.2, 11.4 Hz, 2H), 1.29 (d, J = 6.3 Hz, 6H). 13C NMR (151 MHz, CDCl3) δ (ppm): 160.4, 149.9, 149.8, 146.7, 145.6, 142.4, 134.4, 129.9, 125.6, 120.3, 111.9, 110.7, 71.8, 56.2, 53.0, 19.1. HRMS (ESI+): m/z calculated for C20H23N3O3S [M + H]+: 386.1533; found: 386.1527.
MP 166–167 °C 1H NMR (600 MHz, CDCl3) δ (ppm): 8.82 (d, J = 2.0 Hz, 1H), 8.22 (d, J = 2.0 Hz, 1H), 7.21 (dd, J = 2.2, 8.2 Hz, 1H), 7.13 (d, J = 2.2 Hz, 1H), 7.02 (d, J = 8.2 Hz, 1H), 6.82 (br s, 1H), 4.10–4.05 (m, 2H), 3.97 (s, 3H), 3.96 (s, 3H), 3.89–3.86 (m, 1H), 3.76–3.73 (m, 1H), 3.60–3.55 (m, 1H), 3.29–3.25 (m, 1H), 2.02–2.01 (m, 1H), 1.89–1.81 (m, 2H), 1.74–1.69 (m, 1H), 1.56–1.51 (m, 1H), 1.47–1.43 (m, 1H), 1.02–0.99 (m, 2H), 0.77–0.74 (m, 2H). 13C NMR (151 MHz, CDCl3) δ (ppm): 173.6, 161.3, 149.9, 149.8, 146.8, 145.5, 142.3, 134.6, 129.9, 126.0, 120.4, 111.9, 110.8, 56.24, 56.21, 51.3, 48.6, 41.3, 35.6, 28.3, 23.4, 15.2, 7.2. HRMS (ESI+): m/z calculated for C24H28N4O3S [M + H]+: 453.1955; found: 453.1947.
MP 154–155 °C 1H NMR (400 MHz, CDCl3) δ (ppm): 8.92 (d, J = 2.0 Hz, 1H), 8.22 (d, J = 2.0 Hz, 1H), 7.22 (dd, J = 2.2, 8.3 Hz, 1H), 7.13 (d, J = 2.2 Hz, 1H), 7.01 (d, J = 8.3 Hz, 1H), 5.49 (sept, J = 6.3 Hz, 1H), 3.97 (s, 3H), 3.95 (s, 3H), 1.54 (d, J = 6.3 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ (ppm): 162.2, 149.9, 149.8, 147.7, 145.5, 139.8, 135.8, 129.9, 125.8, 120.6, 111.9, 110.8, 72.8, 56.3, 56.2, 22.2. HRMS (ESI+): m/z calculated for C17H18N2O3S [M + H]+: 331.1111; found: 331.1112.
MP 161–163 °C 1H NMR (600 MHz, CDCl3) δ (ppm): 9.00 (d, J = 2.0 Hz, 1H), 8.32 (d, J = 2.0 Hz, 1H), 7.77–7.76 (m, 2H), 7.49–7.47 (m, 3H), 7.24 (dd, J = 1.9, 8.3 Hz, 1H), 7.15 (d, J = 1.9 Hz, 1H), 7.03 (d, J = 8.3 Hz, 1H), 3.99 (s, 3H), 3.96 (s, 3H). 13C NMR (151 MHz, CDCl3) δ (ppm): 160.5, 150.1, 149.9, 147.9, 147.5, 145.5, 135.7, 135.0, 129.8, 129.53, 129.52, 128.6, 125.5, 120.6, 112.0, 110.8, 56.3, 56.2. HRMS (ESI+): m/z calculated for C20H16N2O2S2 [M + H]+: 381.07259; found: 381.0721.
MP 195–197 °C 1H NMR (600 MHz, CDCl3) δ (ppm): 9.24 (s, 1H), 9.13 (d, J = 2.0 Hz, 1H), 8.44 (d, J = 2.0 Hz, 1H), 7.29 (dd, J = 2.3, 8.4 Hz, 1H), 7.19 (d, J = 2.3 Hz, 1H), 7.06 (d, J = 8.4 Hz, 1H), 4.78 (s, 2H), 4.00 (s, 3H), 3.98 (s, 3H), 3.50 (s, 3H). 13C NMR (101 MHz, CDCl3) δ (ppm): 150.5, 150.0, 149.4, 148.8, 148.1, 145.3, 140.5, 136.2, 129.0, 125.5, 122.9, 120.8, 112.1, 110.8, 66.0, 58.6, 56.3, 56.2. HRMS (ESI+): m/z calculated for C18H17N5O3S [M + H]+: 384.11247; found: 384.1125.
MP 123–124 °C 1H NMR (600 MHz, CDCl3) δ (ppm): 9.08 (d, J = 1.8 Hz, 1H), 8.41 (dd, J = 1.9, 8.4 Hz, 1H), 8.39 (d, J = 1.8 Hz, 1H), 8.16 (d, J = 1.9 Hz, 1H), 7.28 (dd, J = 2.3, 8.4 Hz, 1H), 7.20 (d, J = 2.3 Hz, 1H), 7.06–7.03 (m, 2H), 4.04 (s, 3H), 4.00 (s, 3H), 3.98 (s, 3H), 3.97 (s, 3H). 13C NMR (151 MHz, CDCl3) δ (ppm): 161.7, 150.7, 149.9, 149.8, 149.1, 148.3, 147.9, 147.2, 134.2, 129.9, 127.4, 125.2, 121.9, 120.5, 111.9, 111.5, 110.9, 110.8, 56.24, 56.21, 56.16, 56.1. HRMS (ESI+): m/z calculated for C22H20N2O4S [M + H]+: 409.1216; found: 409.1217.
MP 139–141 °C 1H NMR (600 MHz, CDCl3) δ (ppm): 8.81 (d, J = 2.0 Hz, 1H), 8.21 (d, J = 2.0 Hz, 1H), 7.63–7.61 (m, 2H), 7.23–7.20 (m, 2H), 4.07–4.05 (m, 4H), 3.94–3.92 (m, 4H). 13C NMR (151 MHz, CDCl3) δ (ppm): 163.4 (JC–F = 249.3 Hz), 160.7, 146.7, 145.6, 142.7, 133.7, 133.4 (JC–F = 3.3 Hz), 129.5 (JC–F = 8.3 Hz), 126.1, 116.5 (JC–F = 22.0 Hz), 67.0, 47.9. HRMS (ESI+): m/z calculated for C16H14FN3OS [M + H]+: 316.09143; found: 316.0915.
MP 180–181 °C 1H NMR (600 MHz, CDCl3) δ (ppm): 8.83 (d, J = 1.2 Hz, 1H), 8.72 (d, J = 1.8 Hz, 1H), 8.62 (dd, J = 1.2, 4.9 Hz, 1H), 8.26 (d, J = 1.8 Hz, 1H), 7.88–7.86 (m, 1H), 7.36–7.33 (ddd, J = 0.6, 4.9, 7.8 Hz, 1H), 4.05–4.03 (m, 4H), 3.93–3.91 (m, 4H). 13C NMR (101 MHz, CDCl3) δ (ppm): 160.7, 152.6, 149.6, 148.4, 145.8, 142.3, 138.8, 130.9, 123.3, 119.6, 117.5, 90.6, 89.2, 67.0, 47.9. HRMS (ESI+): m/z calculated for C17H14N4OS [M + H]+: 323.09610; found: 323.0960.
MP 118–119 °C 1H NMR (600 MHz, CDCl3) δ (ppm): 7.58 (d, J = 8.7 Hz, 1H), 7.35 (d, J = 8.6 Hz, 1H), 7.21–7.19 (m, 2H), 6.84–6.82 (m, 2H), 4.18 (s, 2H), 3.79 (s, 3H). 13C NMR (151 MHz, CDCl3) δ (ppm): 159.5, 149.9, 141.1, 138.2, 134.1, 130.2, 127.8, 126.9, 114.8, 114.4, 55.5, 38.4. HRMS (ESI+): m/z calculated for C14H11ClN2OS [M + H]+: 291.0353; found: 291.0356.
MP 123–124 °C 1H NMR (600 MHz, CDCl3) δ (ppm): 8.26 (d, J = 8.5 Hz, 1H), 7.54 (d, J = 8.6 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ (ppm): 151.7, 148.4, 144.2, 137.9, 130.8, 124.3. HRMS (ESI+): m/z calculated for C6H2BrClN2S [M + H]+: 248.8884; found: 248.8917.
MP 144–145 °C 1H NMR (400 MHz, CDCl3) δ (ppm): 8.07 (d, J = 8.6 Hz, 1H), 7.36 (d, J = 8.6 Hz, 1H), 4.01–3.98 (m, 4H), 3.92–3.89 (m, 4H). 13C NMR (101 MHz, CDCl3) δ (ppm): 159.9, 147.6, 145.3, 142.9, 130.9, 1225, 66.9, 47.8. HRMS (ESI+): m/z calculated for C10H10ClN3OS [M + H]+: 256.0306; found: 256.0312.
MP 128–129 °C 1H NMR (600 MHz, CDCl3) δ (ppm): 8.06 (d, J = 8.6 Hz, 1H), 7.35 (d, J = 8.6 Hz, 1H), 4.77 (d, J = 10.8 Hz, 2H), 3.89–3.84 (m, 2H), 2.77 (dd, J = 2.1, 10.5 Hz, 2H), 8.06 (d, J = 6.2 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ (ppm): 159.6, 147.5, 145.3, 142.8, 130.9, 122.5, 71.7, 52.9, 19.1. HRMS (ESI+): m/z calculated for C12H14ClN3OS [M + H]+: 284.0619; found: 284.0624.
Suzuki coupling
MP 188–189 °C 1H NMR (600 MHz, CDCl3) δ (ppm): 8.15 (d, J = 8.7 Hz, 1H), 7.80 (d, J = 8.7 Hz, 1H), 7.69 (d, J = 2.0 Hz, 1H), 7.60 (dd, J = 2.0, 8.5 Hz, 1H), 6.99 (d, J = 8.5 Hz, 1H), 4.15–4.13 (m, 4H), 3.99 (s, 3H), 3.96 (s, 3H), 3.96–3.94 (m, 4H). 13C NMR (151 MHz, CDCl3) δ (ppm): 160.8, 153.9, 150.6, 149.5, 144.2, 143.7, 131.7, 129.3, 119.8, 118.4, 111.4, 109.9, 67.0, 56.2, 55.9, 48.1. HRMS (ESI+): m/z calculated for C18H19N3O3S [M + H]+: 358.1219; found: 358.1213.
MP 162–163 °C 1H NMR (600 MHz, CDCl3) δ (ppm): 8.15 (d, J = 8.7 Hz, 1H), 7.81 (d, J = 8.7 Hz, 1H), 7.73 (d, J = 2.1 Hz, 1H), 7.59 (dd, J = 2.0, 8.3 Hz, 1H), 6.99 (d, J = 8.3 Hz, 1H), 5.04 (d, J = 12.2 Hz, 1H), 4.00 (s, 3H), 3.97 (s, 3H), 3.96–3.91 (m, 2H), 2.84 (dd, J = 2.2, 12.7 Hz, 2H), 1.30 (d, J = 6.3 Hz, 6H). 13C NMR (151 MHz, CDCl3) δ (ppm): 160.4, 153.8, 150.5, 149.5, 144.3, 143.6, 131.7, 129.3, 119.8, 118.3, 111.3, 109.8, 71.8, 56.2, 55.9, 53.0, 19.2. HRMS (ESI+): m/z calculated for C20H23N3O3S [M + H]+: 386.1533; found: 386.1526.
X-ray cocrystal structure of GAK with compound 1. PDB entry 4Y8D was used for all modelling calculations.15
Superposition of compound 29. The conformational energy of compound 29 was optimized in vacuum conditions using Open MOPAC at the PM6 level.32 Compound 29 was then fitted onto compound 1 in the ATP binding site of GAK by adjusting the flexible dihedral angles.
Calculation of dipole moments. Open MOPAC was used to calculate the dipole moment of isothiazolo[4,3-b]pyridine 1 and isothiazolo[4,5-b]pyridine 29 at the PM6 while maintaining the binding conformations level.32
Calculation of electrostatic potentials. The atoms were assigned charges and sizes from the amber gaff2 force field using pdb2pqr. Then the electrostatic potential around the molecules was calculated by APBS using default parameters.38 The potentials were then visualized in chimerax.31,33
Author contributions
W. D. and S. D. J. supervised the project. W. D., D. S., S. D. J., and Y. I. conceptualised the idea. S. S. performed the investigation of the initially proposed synthetic method depicted in Scheme 1 in course of his Master's thesis. L. J. G. performed the synthesis of some reference isothiazolo[4,3-b]pyridines. Y. I. performed the rest of the experimental work. Y. I. wrote the first draft of the manuscript. Y. I. constructed the ESI. L. V. M. performed the crystal structure determination. M. F. performed molecular modeling and wrote the related part of manuscript and ESI. All authors discussed and agreed to the final version of the manuscript.Data availability
Crystallographic data for compound 16 have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 2357539. Other data supporting this article have been included as part of the ESI.†Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
This research was supported by the Research Foundation Flanders (FWO) through the strategic basic research grant (1S04023N) to Y. I. and infrastructure grants (I002720N and I001920N). This project was supported by award number W81XWH2110456 from the Department of Defense (DoD, United States Army Medical Research Acquisition Activity, USAMRAA). Mass spectrometry was made possible by the support of the Hercules Foundation of the Flemish Government (grant 20100225-7). L. V. M. thanks the Hercules Foundation for supporting the purchase of the diffractometer through project AKUL/09/0035. The authors acknowledge Bart Van Huffel and Gert Steurs for technical assistance with the NMR spectrometry and Jef Rozenski for the HRMS measurements. S. S. acknowledges Yenthel Verhaegen and Demian Kalebić for guidance during the Master's thesis.References
- C. Huang, C. Ji and J. Wang, Mol. Biol. Rep., 2023, 50, 4645–4652 CrossRef CAS PubMed.
- C. X. Zhang, Å. E. Y. Engqvist-Goldstein, S. Carreno, D. J. Owen, E. Smythe and D. G. Drubin, Traffic, 2005, 6, 1103–1113 CrossRef CAS PubMed.
- A. Young, Semin. Cell Dev. Biol., 2007, 18, 448–458 CrossRef CAS PubMed.
- H. T. McMahon and E. Boucrot, Nat. Rev. Mol. Cell Biol., 2011, 12, 517–533 CrossRef CAS PubMed.
- M. A. Edeling, C. Smith and D. Owen, Nat. Rev. Mol. Cell Biol., 2006, 7, 32–44 CrossRef CAS PubMed.
- D. Fessart and F. Delom, Int. J. Cell Biol., 2011, 2011, 14 Search PubMed.
- D. W. Lee, X. Zhao, F. Zhang, E. Eisenberg and L. E. Greene, J. Cell Sci., 2005, 118, 4311–4321 CrossRef CAS PubMed.
- G. Neveu, A. Ziv-Av, R. Barouch-Bentov, E. Berkerman, J. Mulholland and S. Einav, J. Virol., 2015, 89, 4387–4404 CrossRef CAS PubMed.
- E. Bekerman, G. Neveu, A. Shulla, J. Brannan, S. Y. Pu, S. Wang, F. Xiao, R. Barouch-Bentov, R. R. Bakken, R. Mateo, J. Govero, C. M. Nagamine, M. S. Diamond, S. De Jonghe, P. Herdewijn, J. M. Dye, G. Randall and S. Einav, J. Clin. Invest., 2017, 127, 1338–1352 CrossRef PubMed.
- M. Karim, S. Saul, L. Ghita, M. K. Sahoo, C. Ye, N. Bhalla, C. W. Lo, J. Jin, J. G. Park, B. Martinez-Gualda, M. P. East, G. L. Johnson, B. A. Pinsky, L. Martinez-Sobrido, C. R. M. Asquith, A. Narayanan, S. De Jonghe and S. Einav, Antiviral Res., 2022, 204, 105367 CrossRef CAS PubMed.
- A. Beilina, I. N. Rudenko, A. Kaganovich, L. Civiero, H. Chau, S. K. Kalia, L. V. Kalia, E. Lobbestael, R. Chia, K. Ndukwe, J. Ding, M. A. Nalls, M. Olszewski, D. N. Hauser, R. Kumaran, A. M. Lozano, V. Baekelandt, L. E. Greene, J. M. Taymans, E. Greggio and M. R. Cookson, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 2626–2631 CrossRef CAS PubMed.
- M. Susa, E. Choy, X. Liu, J. Schwab, F. J. Hornicek, H. Mankin and Z. Duan, Mol. Cancer Ther., 2010, 9, 3342–3350 CrossRef CAS PubMed.
- M. R. Ray, L. A. Wafa, H. Cheng, R. Snoek, L. Fazli, M. Gleave and P. S. Rennie, Int. J. Cancer, 2006, 118, 1108–1119 CrossRef CAS PubMed.
- J. Li, S. Kovackova, S. Pu, J. Rozenski, S. De Jonghe, S. Einav and P. Herdewijn, MedChemComm, 2015, 6, 1666–1672 RSC.
- S. Kovackova, L. Chang, E. Bekerman, G. Neveu, R. Barouch-Bentov, A. Chaikuad, C. Heroven, M. Šála, S. De Jonghe, S. Knapp, S. Einav and P. Herdewijn, J. Med. Chem., 2015, 58, 3393–3410 CrossRef CAS PubMed.
- S. Y. Pu, R. Wouters, S. Schor, J. Rozenski, R. Barouch-Bentov, L. I. Prugar, C. M. O'Brien, J. M. Brannan, J. M. Dye, P. Herdewijn, S. De Jonghe and S. Einav, J. Med. Chem., 2018, 61, 6178–6192 CrossRef CAS PubMed.
- R. Wouters, J. Tian, P. Herdewijn and S. De Jonghe, ChemMedChem, 2019, 14, 237–254 CrossRef CAS PubMed.
- R. Wouters, S. Y. Pu, M. Froeyen, E. Lescrinier, S. Einav, P. Herdewijn and S. De Jonghe, Eur. J. Med. Chem., 2019, 163, 256–265 CrossRef CAS PubMed.
- B. Martinez-Gualda, S. Y. Pu, M. Froeyen, P. Herdewijn, S. Einav and S. De Jonghe, Bioorg. Med. Chem., 2020, 28, 115188 CrossRef CAS PubMed.
- B. Martinez-Gualda, S. Saul, M. Froeyen, D. Schols, P. Herdewijn, S. Einav and S. De Jonghe, Eur. J. Med. Chem., 2021, 213, 113158 CrossRef CAS PubMed.
- Y. Ivanova, M. Smoljo, S. De Jonghe and W. Dehaen, ARKIVOC, 2024, 202312146 Search PubMed.
- F. Jakob, J. Alen, S. Kruger, D. Friebe, S. Hennen and Gruenenthal GMBH, Substituted pyrrolidine amides IV, World Intellectual Property Organization, 2020254551, 2020 Search PubMed.
- R. T. Skerlj, E. M. J. Bourque, W. J. Greenlee, P. T. Lansbury and Lysosomal Therapeutics INC, Substituted imidazo[1,2-b]pyridazines, substituted imidazo[1,5-b]pyridazines, related compounds, and their use in the treatment of medical disorders, World Intellectual Property Organization, WO2017192930, 2017 Search PubMed.
- S. Wang, E. Kumpulainen, J. Pystynen, A. Pohjakallio, A. Haikarainen and Orion Corporation, 2-(1-Heteroarylpiperazin-4-yl)methyl-1,4-benzodioxane derivatives as alpha2c antagonists, World Intellectual Property Organization, WO2016193551, 2016 Search PubMed.
- G. Zhou, Y. Wei, W. Li, H. Yuan, K. C. Liu and Acerand Therapeutics (Shanghai) CO, Acerand Therapeutics (Hong Kong) limited, Pyrazole-containing derivative, pharmaceutically acceptable salt thereof, preparation method therefor and application thereof World Intellectual Property Organization, WO2023116865, 2023.
- N. Rival, A. Albornoz Grados, L. Schiavo, F. Colobert and G. Hanquet, Tetrahedron Lett., 2015, 56, 6823–6826 CrossRef CAS.
- C. S. Lebakken, S. M. Riddle, U. Singh, W. J. Frazee, H. C. Eliason, Y. Gao, L. J. Reichling, B. D. Marks and K. W. Vogel, SLAS Discovery, 2009, 14, 924–935 CrossRef CAS PubMed.
- S. Verdonck, S. Y. Pu, F. J. Sorrell, J. M. Elkins, M. Froeyen, L. J. Gao, L. I. Prugar, D. E. Dorosky, J. M. Brannan, R. Barouch-Bentov, S. Knapp, J. M. Dye, P. Herdewijn, S. Einav and S. De Jonghe, J. Med. Chem., 2019, 62, 5810–5831 CrossRef CAS PubMed.
- P. Zhou, F. Tian, F. Lv and Z. Shang, Proteins: Struct., Funct., Bioinf., 2009, 76, 151–163 CrossRef CAS PubMed.
- A. Pizzi, A. Daolio, R. Beccaria, N. Demitri, F. Viani and G. Resnati, Chem. – Eur. J., 2023, 29, e202300571 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- J. J. P. Stewart, J. Mol. Model, 2007, 13, 1173–1213 CrossRef CAS PubMed.
- E. C. Meng, T. D. Goddard, E. F. Pettersen, G. S. Couch, Z. J. Pearson, J. H. Morris and T. E. Ferrin, Protein Sci., 2023, 32, e4792 CrossRef CAS PubMed.
- CrysAlis PRO, Agilent Technologies UK Ltd, Yarnton, Oxfordshire, England, 2012 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- E. Jurrus, D. Engel, K. Star, K. Monson, J. Brandi, L. E. Felberg, D. H. Brookes, L. Wilson, J. Chen, K. Liles, M. Chun, P. Li, D. W. Gohara, T. Dolinsky, R. Konecny, D. R. Koes, J. E. Nielsen, T. Head-Gordon, W. Geng, R. Krasny, G. W. Wei, M. J. Holst, J. A. McCammon and N. A. Baker, Protein Sci., 2018, 27, 112–128 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2357539. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob00908h |
| This journal is © The Royal Society of Chemistry 2024 |