
Photoinduced metal-free trifluoro/perfluoroalkylation of heteroarenes†
Shashank
Singh
 and
Ravi P.
Singh
*
and
Ravi P.
Singh
*
Department of Chemistry, Indian Institute of Technology, Delhi Hauz Khas, New Delhi 110016, India. E-mail: ravips@chemistry.iitd.ac.in
First published on 29th April 2024
Abstract
A practical and straightforward protocol to access trifluoromethylated/perfluoroalkylated heteroarenes via radical-type nucleophilic substitution rather than typical radical-type electrophilic substitution is described here. The substrate scope was observed to be broad and diverse—covering arenes, heteroarenes (containing N, O, S), bioactive cores, and allylic cores. Mechanistic studies confirmed a radical-mediated reaction pathway.
Over several decades, strategies for synthesizing fluorinated scaffolds have seen an incredible transformation and even today constitute a subject of immense interest.1 This unparalleled interest is attributed to the major influence of the fluorine group on the physiochemical and biological activities of organic molecules.2 Organofluoro compounds are widely used in the fields of materials science3a–c and agriculture3d (Scheme 1A). The ability to add trifluoromethyl and/or perfluoroalkyl groups to arenes and heteroarenes is thus important—but has become a specific bottleneck in efforts at effecting easy translation of the above strategies into commercially viable methods. Derivatizing arenes and heteroarenes with trifluoromethyl and/or perfluoroalkyl groups is also important industrially as such processes are necessary for generating the precursors used to manufacture commercial medications such as mapracorat,4a fluoxetine,4b and penthiopyrad.4c
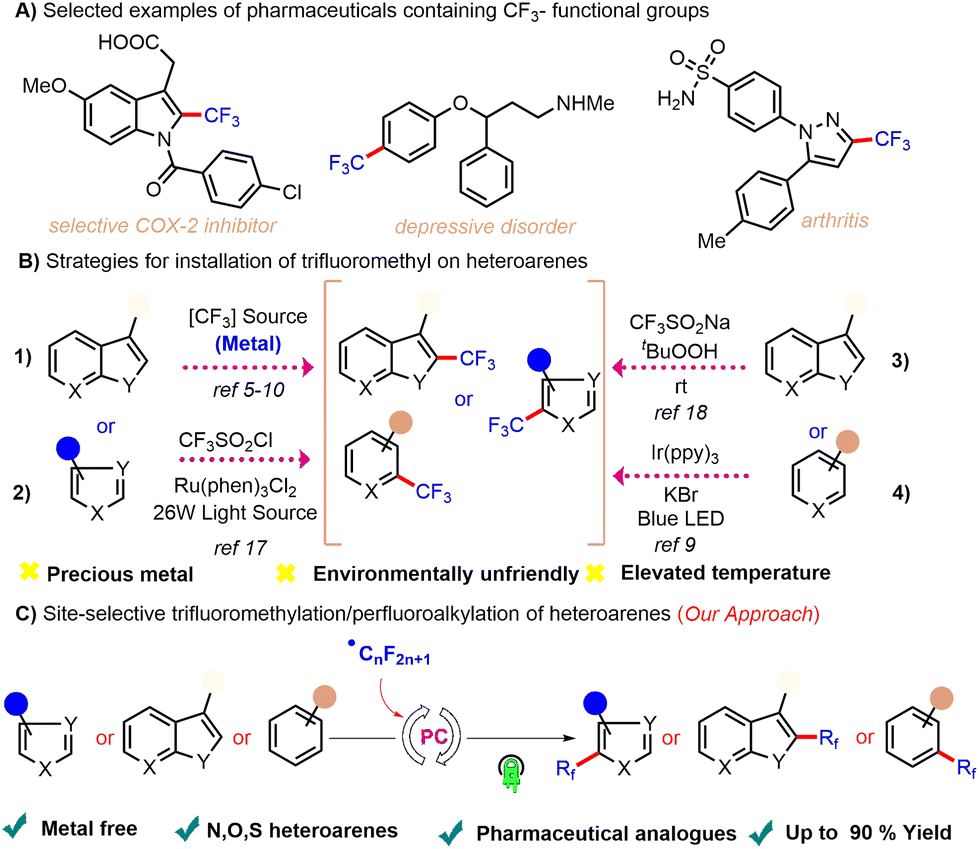 | ||
| Scheme 1 (A) Biologically relevant trifluoromethylated scaffolds. (B) Previous strategies for trifluoromethylation. (C) Our work: trifluoromethylation/perfluoroalkylation of heteroarenes. | ||
To address the above-described bottleneck, various attempts at designing methods for the practical synthesis of trifluoromethylated/perfluoroalkylated motifs have been reported. Most methods involving heteroarene trifluoromethylation documented in the literature involve the use of metals such as Pd,5 Ag,6 Cu,7 Ru,8 Ir,9 and Ni.10 These approaches, though elegant, suffer from various limitations including the need to carry out pre-functionalizations such as with halides or boronic acids the need to use directing groups, and by-product formation. In addition, some of the traditional strategies also exploit oxidative trifluoromethylation using corrosive gaseous, namely CF3I11 and CF3SO2Cl,12 electrophiles such as Umemoto's reagent,13 Togni reagent,14 and Shibata's reagent,15 nucleophiles such as Rupert-Prakash reagent (CF3SiMe3),16etc. In addition to these methods being impractical due to their use of expensive metals and designer trifluoromethylating reagents, they also display limited utilization due to functional group intolerance for various classes of heterocycles.
Recently, visible light photoredox catalysis has emerged as a powerful tool for achieving easy activation of C–C bonds. Photoredox-mediated radical trifluoromethylation protocols complement the traditional approaches. In pioneering work reported by Nagib and Macmillan, they showcased photolytic trifluoromethylation of heteroarenes using trifluorosulfonyl chloride (CF3SO2Cl).17 Baran et al. presented a direct C–H trifluoromethylation of a heterocycle using CF3SO2Na as a CF3 radical precursor.18 Li et al. disclosed trifluoromethylation of heteroarenes using CF3SO2Na and UV light irradiation.19 Bottecchia et al. developed a trifluoromethylation protocol of heteroarenes using CF3I and 5,5′-bis(((2-trifluoromethyl)phenyl)ethynyl)-2,2′bithiophene as a photocatalyst.20 Díaz Díaz et al. presented trifluoromethylation of heteroarenes using CF3SO2Cl and Ir(ppy)3 as a photocatalyst.21 Xiao disclosed trifluoromethylation of heteroarenes via trifluoromethylsulfonyl-pyridinium salts (TFSP) in the presence of Ir(ppy)3 photocatalyst.9 However, note that similar to the traditional procedures involving metal catalysis, these strategies are also impractical as they too use either expensive/toxic metal catalysts or are restricted to just a few select fluoromethylation products due to narrow substrate scopes (Scheme 1B). Therefore, there remains a dire need to develop a straightforward, environmentally benign, scalable and economical method that enables the synthesis of various classes of trifluoromethylated/perfluoroalkylated heterocycles.
To develop a protocol addressing the trifluoromethylation gap, the indole-ring-containing motif was chosen as the heterocycle. Of the important classes of pharmaceutically relevant scaffolds, alkaloids such as tryptamines,22 which contain each an indole ring as a core nucleus, holds an important place in synthetic organic chemistry. The tryptamine skeleton serves as the basis for many synthetic, semi-synthetic, and naturally occurring pharmacological medications. Our interest23 and previous work on heteroarene24 motifs led us to hypothesize that the relatively low energy required for the activation of heteroaromatic C–H in the tryptamine skeleton can be easily provided by a photoredox reaction, allowing us to investigate a mild strategy for achieving trifluoromethylation and perfluoroalkylation (Scheme 1C).
We commenced our initial investigation with NHCbz-tryptamine 1a and bench-stable Langlois reagent 2a under photoredox conditions. After extensive tests, the optimal conditions were established to be 1a (1.0 equiv.), 2a (3.0 equiv.), eosin Y (2.5 mol%), and (NH4)2S2O8 (2.0 equiv.) in MeCN (0.1 M), under photoirradiation (12 W blue LED irradiation (525 nm)) and stirring at rt for 12 h. The reaction conditions achieved the desired coupling, which afforded 3a in 72% isolated yield (Table 1, entry 1).
|
|
||
|---|---|---|
| Entry | Deviation from “condition A” | Yieldb (%) |
| a Reactions were carried out with 1a (0.2 mmol), CF3SO2Na 2a (0.6 mmol), (NH4)2S2O8 (3 equiv.), eosin Y (2.5 mol%) in 2 mL MeCN under 12 W green LED irradiation at room temperature for 12 h. b Isolated yield. MeCN = acetonitrile, DCM = dichloromethane, DIPEA = diisopropyl ethyl amine, ND: no reaction. | ||
| 1 | None | 72 |
| 2 | Absence of light(dark) | N.D. |
| 3 | Np photcatalyst | N.D. |
| 4 | No oxidant | N.D. |
| 5 | No inert atmosphere | N.D. |
| 6 | TPT instead of Eosin Y | 11 |
| 7 | [Mes-Arc-Me+]BF4− instead of Eosin Y | 60 |
| 8 | RoseBengal instead of Eosin Y | 25 |
| 9 | [Ru(bpy)3] instead of Eosin Y | 68 |
| 10 | DCM instead of MeCN | 60 |
| 11 | DCE instead of MeCN | 35 |
| 12 | Dioxance instead of MeCN | 65 |
| 13 | With DIPEA as additive | 69 |

Control experiments demonstrated that the exclusion of any component in the reaction, specifically light, photocatalyst or oxidant, failed to give the desired product (Table 1, entries 2–5). A screening of various photocatalysts revealed eosin Y as optimal and high yielding; others were less effective (Table 1, entries 6–9). A survey of various solvents indicated MeCN to be the optimal solvent, and replacing it led to a significant decrease in reactivity (Table 1, entries 10–12). Note that inclusion of additives did not result in any significant increase in the yield of the desired product (Table 1, entry 13). After successfully optimizing the reaction conditions for NHCbz -tryptamine, we then investigated the tolerance of our protocol for trifluoromethylation of functional indole cores and other classes of heteroarenes (Table 2). Boc-protected tryptamine derivative performed well, affording C-2 product in excellent yield (Table 2, 3b).
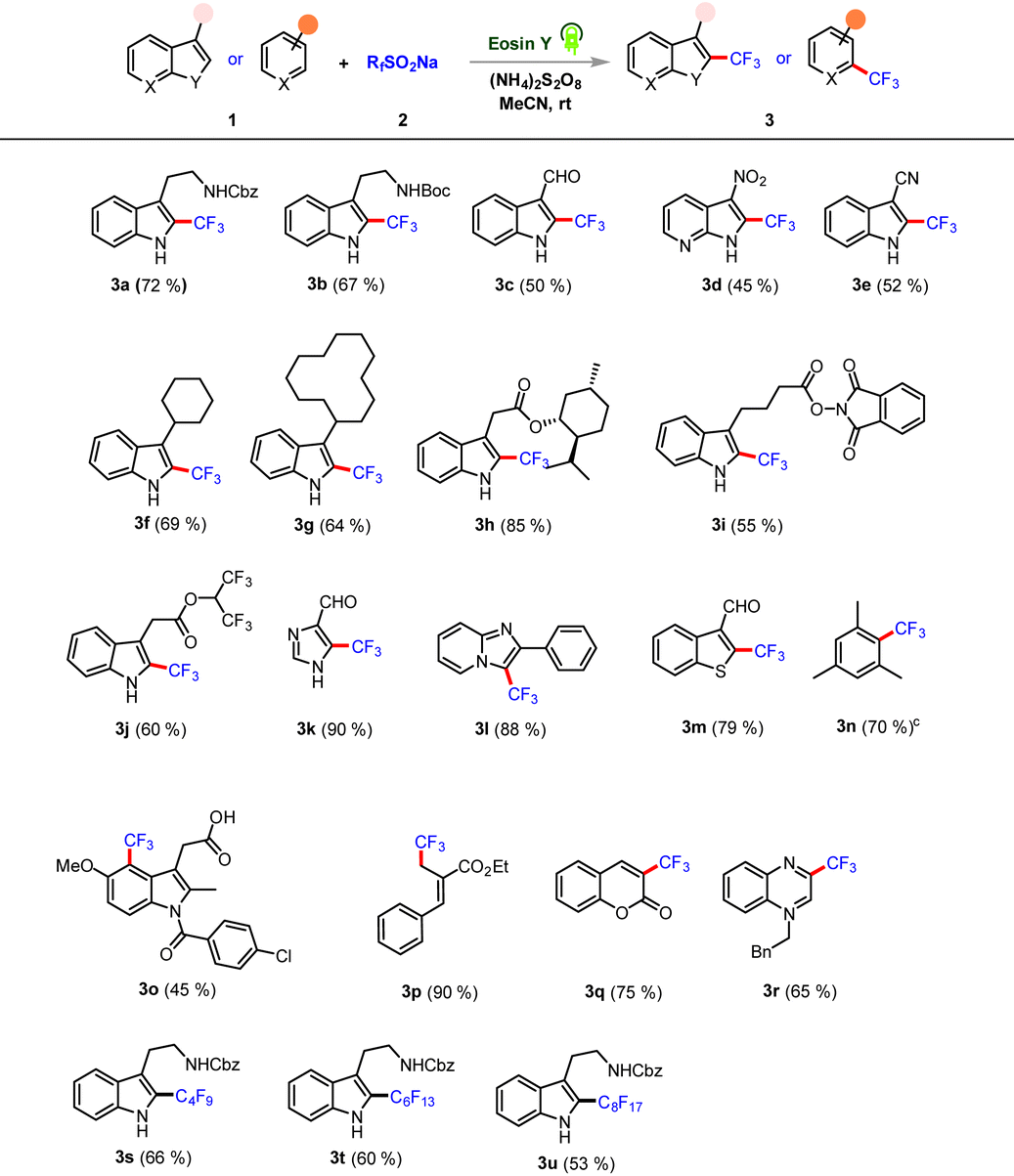
Furthermore, the broad scope of this protocol was validated by the observation of heteroarenes bearing sensitive functional groups (such as aldehyde and nitro groups) providing C-2 products in moderate yields (Table 2, 3c–3d). The electron-withdrawing cyano group was tolerated, affording the desired C-2 adduct in good yield (Table 2, 3e). We found that the reaction proceeded with high substrate generality as aliphatic cyclic chain substituents afforded the respective desired C-2 adducts in good yields (Table 2, 3f–3g). Additionally, trifluoromethylation of indole cores bearing bioactive scaffolds and redox-active component substrates were also felicitous and gave corresponding C-2 adducts in good to excellent yields (Table 2, 3h–3i). Similarly, a hydrolysis-prone hexafluoro-isopropanol-derived ester component was well tolerated in this C–H trifluoromethylation, giving product with a good yield (Table 2, 3j). Interestingly, an unsymmetrical heteroarene bearing multiple sites of reaction also provided site-selective C-5-coupled products in excellent yield (Table 2, 3k). Subsequently, imidazo[1,2-α]pyridine and benzothiophene 3-carbaldehyde scaffolds were tested; they performed well, and the corresponding adducts were formed in good to excellent yields (Table 2, 3l–3m). We next turned our attention towards arene functionalization, due to it offering the possibility of a valuable pharmaceutically active core. Delightfully our protocol demonstrated complementary reactivity for trifluoromethylation of an arene in a site-selective fashion in moderate to good yields (Table 2, 3n). The nonsteroidal drug indomethacin was directly functionalized and yielded a C-4-trifluoromethylated adduct in moderate yield (Table 2, 3o). A highly reactive bromo-derived MBH ester scaffold also reacted efficiently under the optimized protocol, yielding the corresponding trifluoromethylated adduct in excellent yield (Table 2, 3p). The generality of the methodology was further established by extending the reaction to coumarin and N-benzyl quinoxalinone. These reactions afforded C-3-trifluoromethylated adducts in good yields (Table 2, 3q–3r). We next evaluated whether the transformation could be performed with other perfluoroalkyl sulfinate salts. Gratifyingly, our protocol was found to tolerate a wide range of perfluoroalkyl substituents and resulted in C-2-perfluoroalkylated tryptamine adducts in good yields (Table 2, 3s–3u).
To gain mechanistic insights into this coupling reaction, radical scavengers 2,2,6,6-tetramethyl-1-peperidyloxy (TEMPO) and BHT were added; they suppressed the reactivity and the corresponding radical adduct was detected using GC-MS (Scheme 2). Based on putting together these pieces of experimental evidence, the reaction was concluded to proceed via a radical mechanism. Note that in situ EPR studies of the corresponding reaction mixture also indicated the presence of a radical intermediate in the reaction system (Fig. 1A). To eliminate the possibility of an interaction between eosin Y and the substrates during the catalytic cycle and to observe the ground/excited state of the reactants, experiments involving UV studies were performed. These studies revealed a lack of formation of electron donor–acceptor (EDA) complex (see the ESI†), indicating no possibility of an EDA complex in the reaction medium (Fig. 1B). Based on the established mechanism of action for such photoactive dyes, it was speculated that eosin Y, on being irradiated with green light, would undergo excitation with it further undergoing single-electron oxidation with (NH4)2S2O8 to produce oxidized eosin Y˙+ and corresponding sulfate dianion (SO42−) and sulfate radical anion (SO4˙−), which would then oxidize the Langlois reagent to generate the reactive CF3 radical. To further validate our hypothesis, fluorescence quenching experiments were carried out with 1a, 2a and (NH4)2S2O8. A fluorescence quenching effect was witnessed in the presence of (NH4)2S2O8 (Fig. 1C). The corresponding Stern–Volmer plot indicated that the excited state of eosin Y was quenched by (NH4)2S2O8 and an increase in quenching was noticed with an increasing oxidant concentration (see the ESI†).
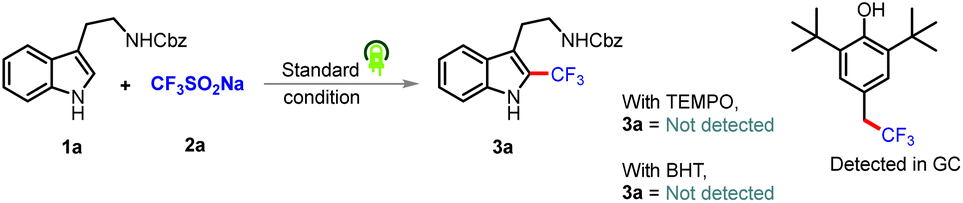 | ||
| Scheme 2 Mechanistic studies. | ||
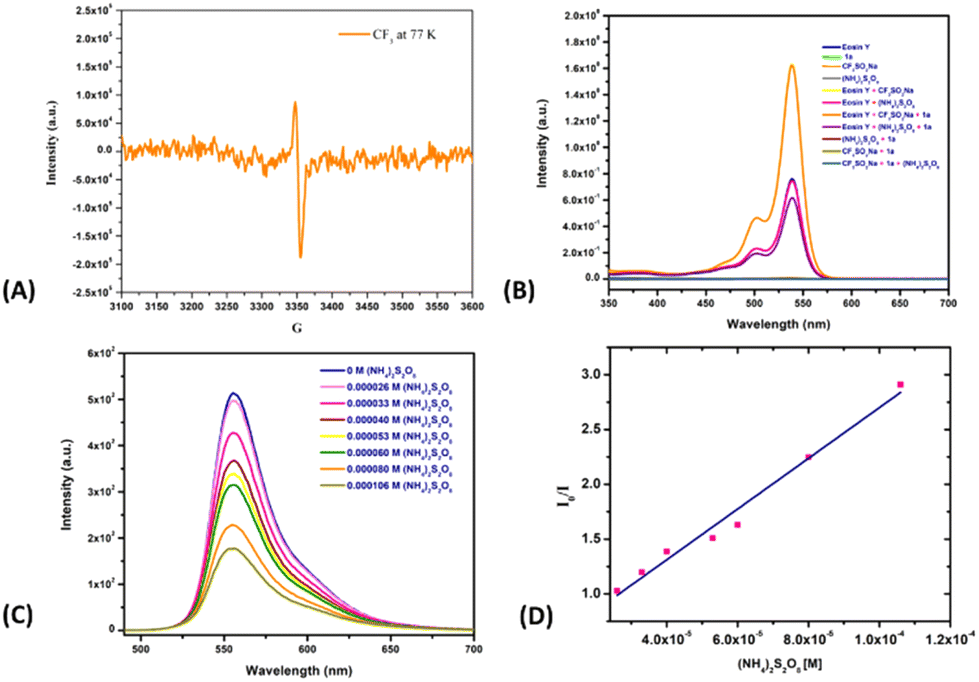 | ||
| Fig. 1 (A) EPR experiment. (B) UV-visible studies. (C) Fluorescence quenching with (NH4)2S2O8. (D) Stern–Volmer plot for (NH4)2S2O8. | ||
Furthermore, quenching constants were determined by carrying out Stern–Volmer analyses (Fig. 1D).
Based on the information in the literature and the observations made in the present study (see ESI†), a plausible mechanism was derived for this reaction. According to this proposed mechanism, photoirradiation of eosin Y* with green light followed by single-electron oxidation of catalyst resulted in the formation of sulfate dianion (SO42−), which in turn oxidized CF3SO2Na to generate a trifluoromethyl radical. This radical was intercepted by heteroarenes 1 to form 1′, which subsequently underwent single-electron oxidation (SET) and protonation to ultimately afford the desired compounds 3 (Scheme 3).
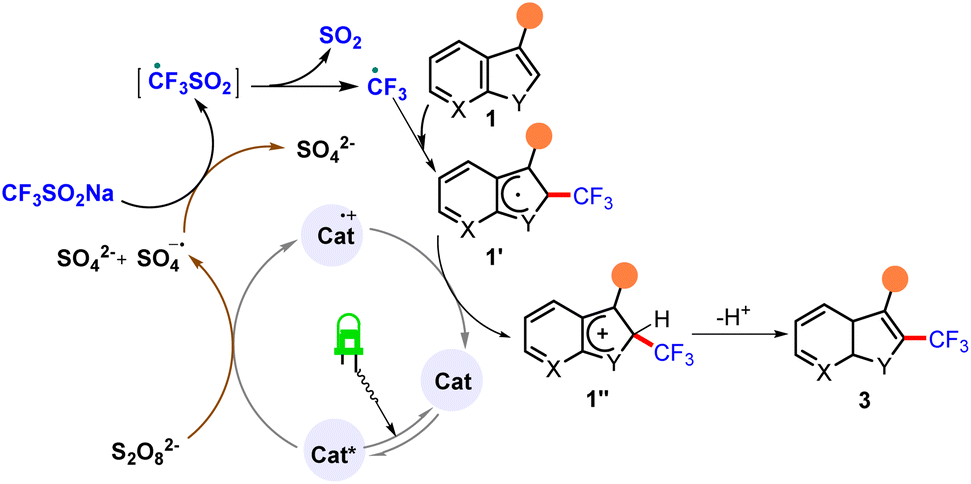 | ||
| Scheme 3 Plausible mechanism. | ||
Conclusions
In summary, we have developed a transition-metal-free, visible-light-mediated trifluoromethylation/perfluoroalkylation of heteroarenes. This simple protocol employs bench-stable, easy-to-handle and inexpensive sodium triflinate (Langlois reagent) as a CF3 source and eosin Y as a photoredox catalyst. This strategy allows direct installation of both trifluoromethyl and perfluoroalkyl groups into various classes of heterocycles, thus providing an opportunity for a one-step direct functionalization of heteroarenes, and exhibits excellent functional group tolerance, and is thus suited for late-stage functionalization of natural products and pharmaceuticals.Conflicts of interest
“There are no conflicts to declare”.Acknowledgements
Generous financial support from (SERB/SCP/2022/000599) is greatly acknowledged. S. S. is grateful to MHRD for the fellowship.References
- (a) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed; (b) M. G. Campbell and T. Ritter, Chem. Rev., 2015, 115, 612–633 CrossRef CAS PubMed; (c) P. A. Champagne, J. Desroches, J.-D. Hamel, M. Vandamme and J.-F. Paquin, Chem. Rev., 2015, 115, 9073–9174 CrossRef CAS PubMed; (d) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed.
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed; (b) H.-J. Böhm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637–643 CrossRef PubMed; (c) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC; (d) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- (a) F. Babudri, G. M. Farinola, F. Naso and R. Ragni, Chem. Commun., 2007, 1003–1022 RSC; (b) R. Milad, J. Shi, A. Aguirre, A. Cardone, B. Milián-Medina, G. M. Farinola, M. Abderrabba and J. Gierschner, J. Mater. Chem. C, 2016, 4, 6900–6906 RSC; (c) M. Pagliaro and R. Ciriminna, J. Mater. Chem., 2005, 15, 4981–4991 RSC; (d) Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, iScience, 2020, 23, 101467 CrossRef CAS PubMed.
- (a) Y. Yanase, H. Katsuta, K. Tomiya, M. Enomoto and O. Sakamoto, J. Pest Sci., 2013, 38, 167–168 CrossRef; (b) C. J. Wenthur, M. R. Bennett and C. W. Lindsley, ACS Chem. Neurosci., 2014, 5, 14–23 CrossRef CAS; (c) M. Baiula and S. Spampinato, Inflammation Allergy: Drug Targets, 2014, 13, 289–298 CrossRef CAS PubMed.
- (a) X. Wang, L. Truesdale and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 3648–3649 CrossRef CAS PubMed; (b) M. Miura, C.-G. Feng, S. Ma and J.-Q. Yu, Org. Lett., 2013, 15, 5258–5261 CrossRef CAS PubMed; (c) K. Natte, R. V. Jagadeesh, L. He, J. Rabeah, J. Chen, C. Taeschler, S. Ellinger, F. Zaragoza, H. Neumann, A. Brückner and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 2782–2786 CrossRef CAS PubMed; (d) Q. Zhao, T. Besset, T. Poisson, J.-P. Bouillon and X. Pannecoucke, Eur. J. Org. Chem., 2016, 76–82 CrossRef CAS.
- (a) Y. Ye, S. H. Lee and M. S. Sanford, Org. Lett., 2011, 13, 5464–5467 CrossRef CAS PubMed; (b) G. Shi, C. Shao, S. Pan, J. Yu and Y. Zhang, Org. Lett., 2015, 17, 38–41 CrossRef CAS PubMed.
- (a) L. Chu and F.-L. Qing, J. Am. Chem. Soc., 2010, 132, 7262–7263 CrossRef CAS PubMed; (b) T. D. Senecal, A. T. Parsons and S. L. Buchwald, J. Org. Chem., 2011, 76, 1174–1176 CrossRef CAS PubMed; (c) L. Chu and F.-L. Qing, Org. Lett., 2012, 14, 2106–2109 CrossRef CAS PubMed; (d) Y. Ye and M. S. Sanford, J. Am. Chem. Soc., 2012, 134, 9034–9037 CrossRef CAS PubMed; (e) S. Arimori and N. Shibata, Org. Lett., 2015, 17, 1632–1635 CrossRef CAS PubMed; (f) J. Lei, X. Wu and Q. Zhu, Org. Lett., 2015, 17, 2322–2325 CrossRef CAS PubMed.
- N. Iqbal, S. Choi, E. Kim and E. J. Cho, J. Org. Chem., 2012, 77, 11383–11387 CrossRef CAS PubMed.
- F. Xiao, J.-H. Lin, F. Hao, X. Zheng, Y. Guo and J.-C. Xiao, Org. Chem. Front., 2022, 9, 1982–1985 RSC.
- (a) Y. Wu, H.-R. Zhang, R.-X. Jin, Q. Lan and X.-S. Wang, Adv. Synth. Catal., 2016, 358, 3528–3533 CrossRef CAS; (b) S. Deolka, R. Govindarajan, E. Khaskin, R. R. Fayzullin, M. C. Roy and J. R. Khusnutdinova, Angew. Chem., Int. Ed., 2021, 60, 24620–24629 CrossRef CAS PubMed.
- T. Kino, Y. Nagase, Y. Ohtsuka, K. Yamamoto, D. Uraguchi, K. Tokuhisa and T. Yamakawa, J. Fluor. Chem., 2010, 131, 98–105 CrossRef CAS.
- H. Chachignon, H. Guyon and D. Cahard, Beilstein J. Org. Chem., 2017, 13, 2800–2818 CrossRef CAS PubMed.
- (a) C. Zhang, Org. Biomol. Chem., 2014, 12, 6580–6589 RSC; (b) S. Tsujibayashi, Y. Kataoka, S. Hirano and H. Matsubara, Org. Biomol. Chem., 2018, 16, 4517–4526 RSC.
- (a) M. S. Wiehn, E. V. Vinogradova and A. Togni, J. Fluor. Chem., 2010, 131, 951–957 CrossRef CAS; (b) E. Mejía and A. Togni, ACS Catal., 2012, 2, 521–527 CrossRef.
- N. Shibata, A. Matsnev and D. Cahard, Beilstein J. Org. Chem., 2010, 6, 65 Search PubMed.
- (a) P. Ramaiah, R. Krishnamurti and G. K. S. Prakash, in Organic Syntheses, 2003, pp. 232–232 Search PubMed; (b) G. K. S. Prakash, P. V. Jog, P. T. D. Batamack and G. A. Olah, Science, 2012, 338, 1324–1327 CrossRef CAS PubMed.
- D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224–228 CrossRef CAS PubMed.
- Y. Ji, T. Brueckl, R. D. Baxter, Y. Fujiwara, I. B. Seiple, S. Su, D. G. Blackmond and P. S. Baran, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14411–14415 CrossRef CAS PubMed.
- L. Li, X. Mu, W. Liu, Y. Wang, Z. Mi and C.-J. Li, J. Am. Chem. Soc., 2016, 138, 5809–5812 CrossRef CAS PubMed.
- C. Bottecchia, R. Martín, I. Abdiaj, E. Crovini, J. Alcazar, J. Orduna, M. J. Blesa, J. R. Carrillo, P. Prieto and T. Noël, Adv. Synth. Catal., 2019, 361, 945–950 CrossRef CAS.
- B. Maiti, A. Abramov, R. Pérez-Ruiz and D. Díaz Díaz, Acc. Chem. Res., 2019, 52, 1865–1876 CrossRef CAS PubMed.
- B. E. Blough, A. Landavazo, A. M. Decker, J. S. Partilla, M. H. Baumann and R. B. Rothman, Psychopharmacology, 2014, 231, 4135–4144 CrossRef CAS PubMed.
- S. Singh, K. S. Naskar, A. Kundu and R. P. Singh, Synthesis, 2023, 3685–3692 CrossRef CAS.
- (a) K. N. Tripathi, S. Singh, N. Akhtar, K. Manna and R. P. Singh, Chem. Commun., 2022, 58, 9674–9677 RSC; (b) S. Singh and R. P. Singh, Org. Biomol. Chem., 2022, 20, 7891–7895 RSC; (c) S. Singh, K. N. Tripathi and R. P. Singh, Org. Biomol. Chem., 2022, 20, 5716–5720 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob00511b |
| This journal is © The Royal Society of Chemistry 2024 |