
Chemodivergent phosphonylation of diazocarboxylates: light-on vs. light-off reactions†
Jalaj Kumar
Pathak
ab,
Ruchir
Kant
c and
Namrata
Rastogi
 *ab
*ab
aMedicinal & Process Chemistry Division, CSIR-Central Drug Research Institute, Lucknow-226031, India. E-mail: namrata.rastogi@cdri.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India
cBiochemistry & Structural Biology Division, CSIR-Central Drug Research Institute, Lucknow-226031, India
First published on 13th June 2024
Abstract
By tapping into the divergent reactivity of diazocarboxylates under thermal and photocatalytic conditions, we could develop chemodivergent phosphonylation protocols for α-diazocarboxylates with trialkyl phosphites. While the thermal reaction led to N–P bond formation affording phosphonylated hydrazones, the visible light-mediated reaction furnished phosphonylated aryl carboxylates through C–P bond formation. Both reactions are notable for their operational simplicity and mild conditions affording products in good yields without the requirement of a metal, base or photocatalyst.
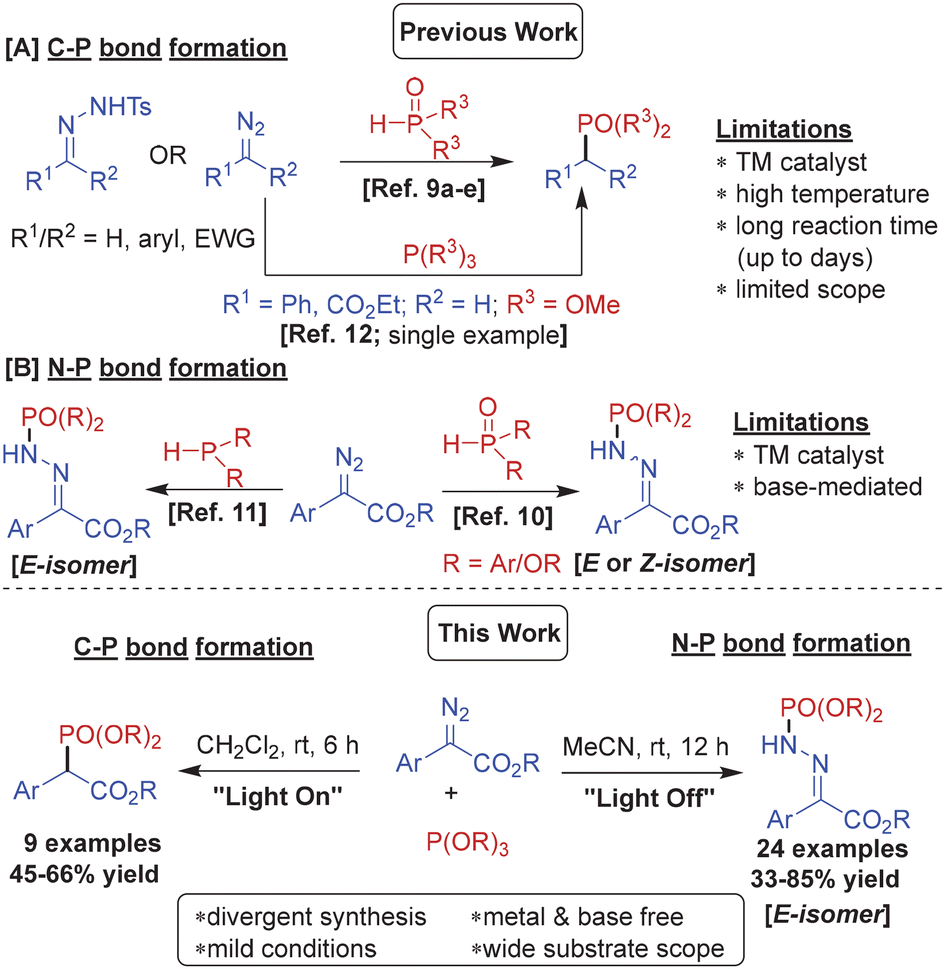
Introduction
Phosphorus containing organic scaffolds are found extensively among natural as well as synthetic medicinal compounds,1 agrochemicals2 and functional materials.3 Organophosphorus compounds are also valuable as catalysts, ligands, protecting groups and building blocks in organic chemistry.4 Therefore, numerous synthetic methods to incorporate phosphorus into organic scaffolds have been invented.5 Yet development of more efficient protocols to access various organophosphorus molecules is highly desired.The diazo compounds are undoubtedly amongst the most versatile organic substrates owing to their tendency to serve as dipoles, nucleophiles, electrophiles, and precursors to carbenes, ketenes and radical intermediates.6 This unique reactivity profile of the diazo group offers great opportunity to develop chemodivergent reactions by modulating the reaction conditions.7 For instance, aryl diazoacetates form free carbenes upon visible light irradiation8 whereas they serve as C-nucleophiles and N-electrophiles under thermal conditions. We envisioned that a nucleophile would undergo both an insertion reaction with the electrophilic diazocarbene and nucleophilic addition reaction with the terminal nitrogen of the diazo group, under suitable conditions. We opted to explore the phosphorus nucleophiles for these reactions considering the significance of organophosphorus scaffolds, as mentioned above, and also because phosphorus can adopt various oxidation states offering options for further transformation of the products. Notably, several reactions of diazo compounds with P(O)H compounds to access organophosphorus scaffolds featuring C–P or N–P linkages have been reported in the recent literature. For instance, phosphine oxides undergo transition metal-mediated P–H insertion or reductive catalytic coupling with diazo substrates (or diazo intermediates in situ generated from tosylhydrazones) forming C(sp3)–P bonds (Scheme 1A).9 On the other hand, Zhu and co-workers reported a base-mediated nucleophilic addition of phosphonates with α-diazoesters resulting in E-phosphinamides with N–P bonds (Scheme 1B).10a However, during the preparation of this manuscript Nan and Yi published photocatalytic phosphonylation of H-phosphine oxides with α-diazoesters to afford Z-phosphinic hydrazones.10b Similar N–P bond formation was reported by Pullarkat and Leung through palladium-catalyzed asymmetric diarylphosphine addition to α-diazoesters leading to the enantioenriched phosphinic hydrazones (Scheme 1B).11
 | ||
| Scheme 1 Carbon vs. nitrogen phosphonylation of diazo compounds. | ||
To the best of our knowledge phosphites have never been used as substrates with diazo compounds except for an iron-catalyzed reaction of trimethyl phosphite with phenyldiazomethane and ethyl diazoacetate forming dimethyl benzylphosphonate and ethyl 2-(dimethoxyphosphoryl)acetate, respectively, through a phosphorus ylide intermediate12 (Scheme 1A). We hereby report diazocarboxylate phosphonylation with phosphites via nucleophilic substitution or carbene insertion generating divergent organophosphorus scaffolds featuring N–P or C–P bonds, respectively, by simple “switch-off” or “switch-on” of blue light (Scheme 1; this work).
We first started investigating the nucleophilic substitution reaction with methyl 2-diazo-2-phenylacetate 1a and trimethyl phosphite 2a as model substrates (Table 1).
|
|
|||
|---|---|---|---|
| Entry |
1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2a :2a |
Solvent | Yield of 3ac (%) |
| a Unless otherwise mentioned, the reactions were carried out on a 0.2 mmol scale of 1a with specified amounts of 2a in 2 mL solvent at rt under N2. b Time taken for reaction completion: 10–12 h. c Isolated yields. d Complex mixture. e Reaction in 1 mL solvent. | |||
| 1d |
1:1 |
DMSO | 0 |
| 2d |
1:1 |
DMF | 0 |
| 3 |
1:1 |
PhMe | 15 |
| 4 |
1:1 |
MeOH | 40 |
| 5 |
1:1 |
CH2Cl2 | 42 |
| 6 |
1:1 |
CHCl3 | 48 |
| 7 |
1:1 |
(Me)2CO | 46 |
| 8 |
1:1 |
MeCN | 50 |
| 9 |
2:1 |
MeCN | 45 |
| 10 |
1:2 |
MeCN | 66 |
| 11 |
1:3 |
MeCN | 78 |
| 12 |
1:4 |
MeCN | 72 |
| 13e |
1:3 |
MeCN | 75 |
The initial experiments with 1a and 2a in a 1:1 ratio employing DMSO or DMF as the solvent at room temperature failed to furnish any identifiable product (entries 1 and 2). Upon changing the solvent to toluene, the expected phosphinamide product 3a was isolated in 15% yield (entry 3). Further solvent screening revealed moderate and almost similar yields in methanol (40%) and dichloromethane (42%) (entries 4 and 5). However, the yield improved to 50% in acetonitrile with comparable yields in chloroform (48%) and acetone (46%) (entries 6–8). Further, the reaction with a 2:1 substrate ratio of 1a and 2a furnished 3a in 45% yield (entry 9) but reversing the ratio to 1:2 led to the isolation of 3a in 66% yield (entry 10). Further optimization of the reaction conditions established that a 1:3 ratio of 1a and 2a in acetonitrile were the best conditions affording 3a in 78% isolated yield (entries 8–12). Apparently, the yields were largely independent of the reaction concentration since 3a was isolated in 75% yield in a more concentrated mixture of 1a and 2a taken in a 1:3 ratio (entry 13). The product was isolated as exclusively the E-isomer, as identified by 1H NMR and single crystal X-ray analysis of 3k.13
After optimizing the reaction conditions, we set out to determine the scope of the thermal phosphonylation reaction. For this purpose, several α-diazo esters 1a–1q and trialkylphosphites 2a–2d were subjected to the optimized reaction conditions (Table 2). Initially, trimethyl phosphite 2a was used as the reaction partner with various electron-rich and electron-deficient aryl diazocarboxylates to afford the corresponding phosphinamides 3a–3l in high yields ranging from 33–85%. Notably, commonly encountered functional groups such as –OMe, –OCF3, –Ph, –CF3, –NO2 and halogens (Cl, Br, F) on the aryl ring of the diazo substrate were tolerated well in the reaction. The reaction of trimethyl phosphite 2a also proceeded smoothly with methyl 2-diazo-2-(naphthalen-1-yl)acetate and heteroaryl substrates such as ethyl 2-(benzo[d][1,3]dioxol-5-yl)-2-diazoacetate and ethyl 2-diazo-2-(thiophen-3-yl)acetate to furnish the corresponding products 3m, 3n and 3o in high yields. Moreover, a phenyldiazoester with an allyl acetate group 1p and a propargyl acetate group 1q also reacted well with 2a to yield 3p and 3q in 63 and 60% yields, respectively. Further, the N-phosphonylation of aryldiazocarboxylate 1a could also be carried out efficiently with other trialkyl phosphites such as triethyl phosphite 2b, triisopropyl phosphite 2c and triallyl phosphite 2d to afford 3r–3t in 65–72% yields.
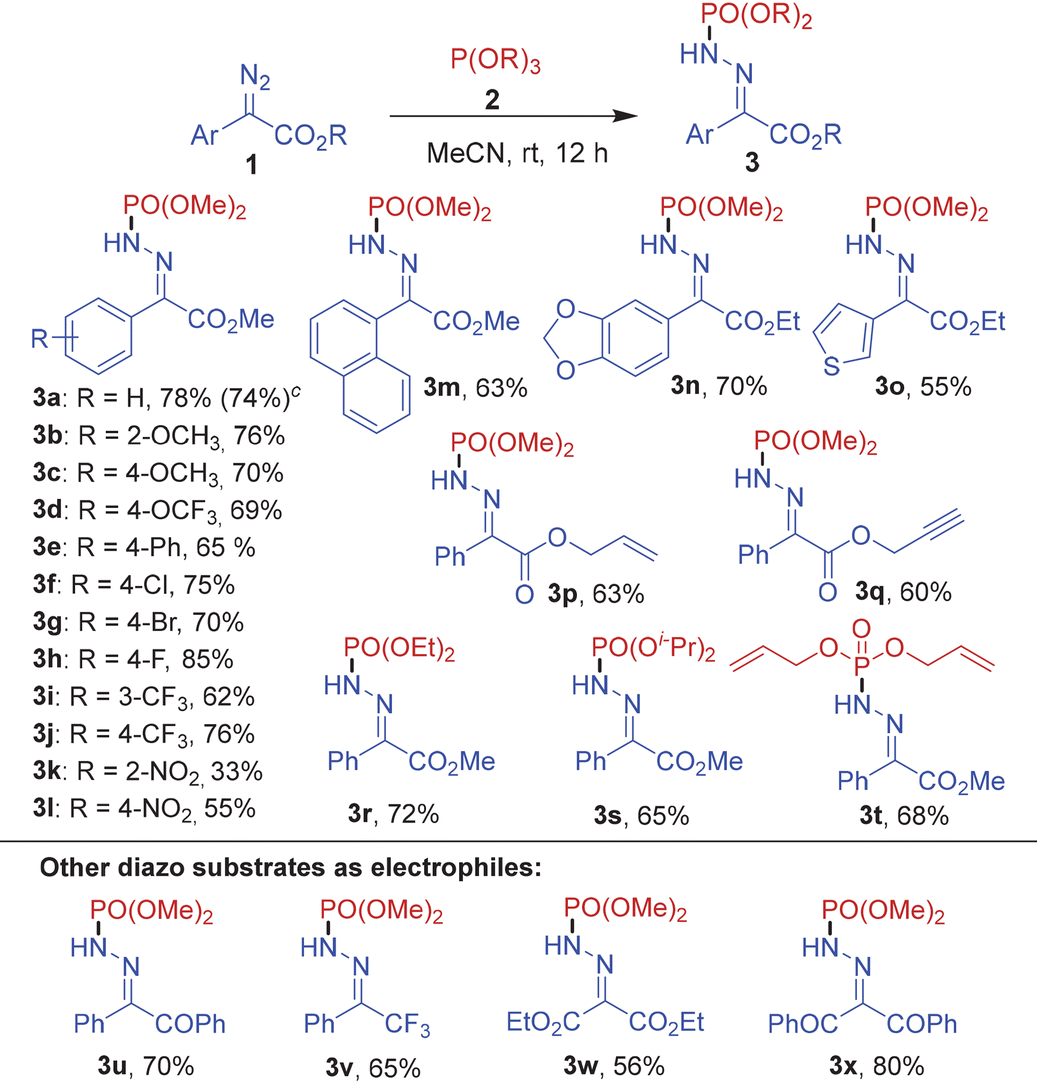
Eventually, the N-phosphonylation reaction could be successfully extended to other diazo substrates bearing at least one acceptor group including 2-diazo-1,2-diphenylethan-1-one (1r), (1-diazo-2,2,2-trifluoroethyl)benzene (1s), diethyl 2-diazomalonate (1t) and 2-diazo-1,3-diphenylpropane-1,3-dione (1u) furnishing the corresponding products 3u–3x in excellent yields.
The E-configuration was assigned to the products on the basis of analogy with 3k, the structure of which was confirmed by single X-ray crystallography (Fig. 1).13
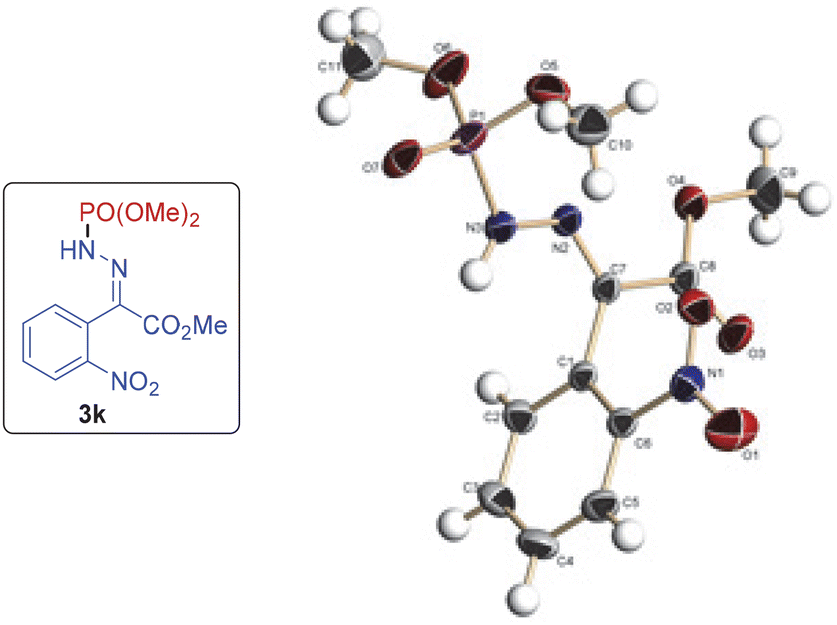 | ||
| Fig. 1 ORTEP diagram drawn with 30% ellipsoid probability for non-H atoms of the crystal structure of compound 3k determined at 294 K. | ||
Next, we set out to establish the suitable conditions for the proposed photochemical phosphonylation reaction between 1a and 2a (Table 3).
|
|
||||
|---|---|---|---|---|
| Entry |
1a:2a |
Solvent | Yield of 4ac (%) | Yield of 3ac (%) |
| a Unless otherwise mentioned, the reactions were carried out on a 0.2 mmol scale of 1a with specified amounts of 2a in 2 mL solvent at rt irradiated with 455 nm blue LEDs under N2. b Time taken for reaction completion: 5–6 h. c Isolated yields. d Complex mixture. | ||||
| 1d |
1:1 |
DMSO | 0 | 0 |
| 2d |
1:1 |
DMF | 0 | 0 |
| 3 |
1:1 |
PhMe | 15 | 20 |
| 4 |
1:1 |
MeOH | 28 | 13 |
| 5 |
1:1 |
CH2Cl2 | 35 | 10 |
| 6 |
1:1 |
CHCl3 | 26 | 16 |
| 7 |
1:1 |
(Me)2CO | 30 | 15 |
| 8 |
1:1 |
MeCN | 30 | 20 |
| 9 |
1:2 |
CH2Cl2 | 38 | 30 |
| 10 |
2:1 |
CH2Cl2 | 55 | <5 |
| 11 |
3:1 |
CH2Cl2 | 52 | <5 |
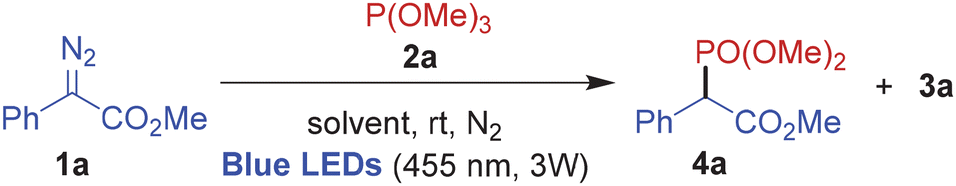
To begin with, equivalent amounts of 1a and 2a were reacted in various solvents under blue light irradiation. The reactions in both dimethyl sulfoxide and dimethyl formamide were too complex to be conclusive (entries 1 and 2). In toluene, although the reaction furnished the anticipated C–P phosphonylation product 4a in 15% yield, the phosphinamide 3a was also isolated in 20% yield (entry 3). In other solvents including methanol, dichloromethane, chloroform, acetone and acetonitrile 4a was obtained in moderate yields (26–35%) along with 3a (10–20%) (entries 4–8). The reaction when carried out in dichloromethane with a 1:2 ratio of 1a and 2a provided 4a in 38% yield, but 3a was also isolated in 30% yield (entry 9). Fortunately, formation of 3a was completely suppressed and 4a could be isolated in 55% yield upon changing the ratio of 1a and 2a to 2:1 (entry 10). Further, a slight reduction in the yield of 4a was noticed upon changing the substrate ratio to 1:3 (entry 11), therefore the conditions in entry 10 were finally selected as the optimal reaction conditions.
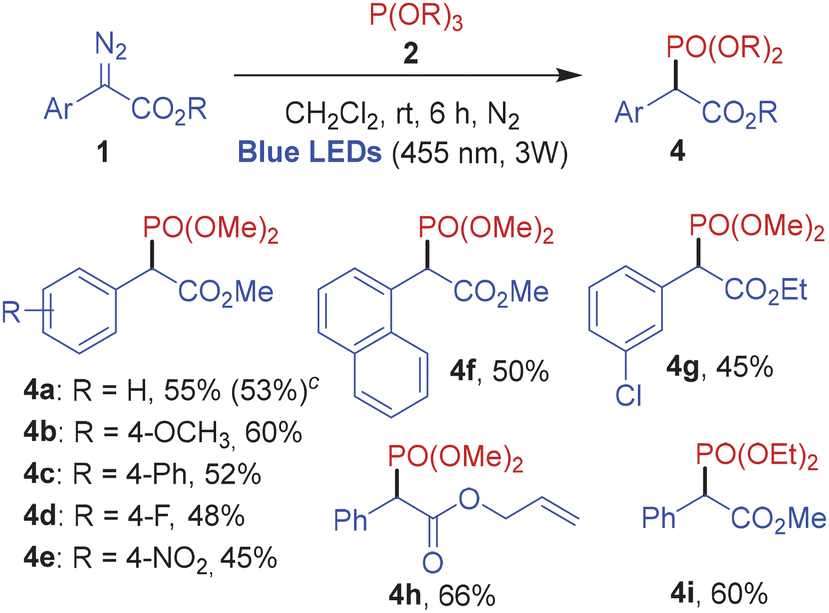
Subsequently in order to investigate the scope of the photochemical phosphonylation reaction, several aryl α-diazoeters were employed in the blue light-mediated reaction with trimethylphosphite 2a and triethylphosphite 2b (Table 4). Evidently, phenyl diazo methylesters bearing substituents with various electronic properties performed well under the photoredox conditions to furnish the corresponding C-phosphonylated products 4a–4f in good yields. The diazo substrates with an ethyl ester group and an allyl ester group also reacted efficiently with methylphosphite to afford 4g and 4h in 45% and 66% yield, respectively. The reaction of methyl 2-diazo-2-phenylacetate 1a with ethylphosphite 2b under the optimized photoredox conditions furnished 4i in 60% yield.
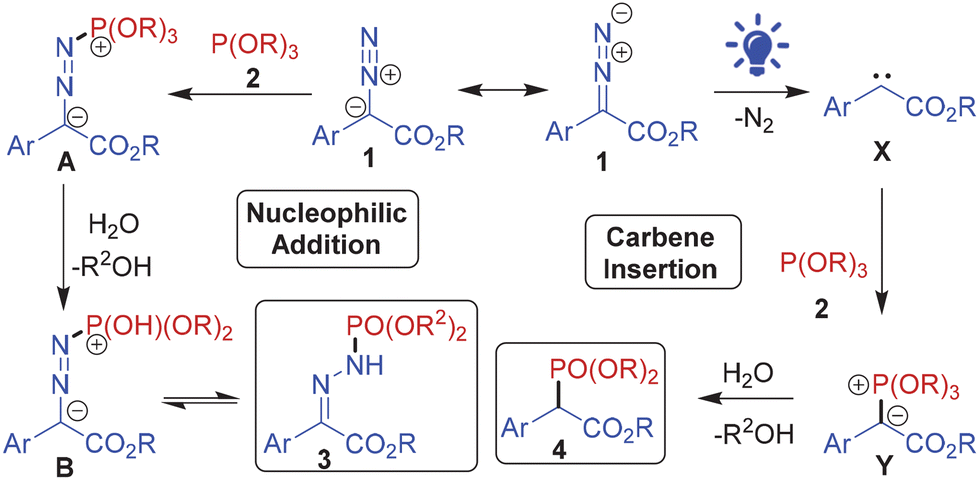
Mechanistically, the two phosphonylations follow divergent routes, as depicted in Scheme 2. The N-phosphonylation proceeds through nucleophilic addition of the phosphite to the terminal nitrogen of the diazo group. The resulting zwitterionic intermediate A upon dealkylation during aqueous workup forms intermediate B which tautomerizes to the N-phosphonylated product 3. On the other hand, the aryl diazoester upon visible light-mediated photolysis generates free carbene species X which undergoes insertion with the phosphite to form ylide intermediate Y. Subsequent decomposition of the intermediate Y upon workup leads to the C-phosphonylated product 4. The carbene intermediate in the reaction was confirmed by the formation of dimethyl 2,3-diphenylmaleate, the carbene dimerization product, in the visible light-mediated reaction between 1a and 2a (confirmed by ESMS of crude 4a; Fig. S108 in the ESI†).
 | ||
| Scheme 2 Plausible mechanism. | ||
Conclusions
In conclusion, we developed mild protocols for accessing phosphonylated hydrazones and phosphonylated aryl carboxylates through thermal and photochemical phosphonylation, respectively, of α-diazocarboxylates with trialkyl phosphites. The former reaction is an example of a nucleophilic substitution reaction whereas the latter reaction follows a carbene insertion pathway. Both reactions proceed under mild reaction conditions, show wide substrate scope and afford the corresponding products in good yields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
JP thanks DST, New Delhi for an INSPIRE Ph. D. fellowship. We thank the SAIF division of CSIR-CDRI for the analytical support. We also thank Dr T. S. Thakur of the Biochemistry and Structural Biology Division, CSIR-CDRI for help in X-ray crystal structure determination studies of 3k. CDRI Communication No: 10806.References
- (a) U. Pradere, E. C. Garnier-Amblard, S. J. Coats, F. Amblard and R. F. Schinazi, Chem. Rev., 2014, 114, 9154–9218 CrossRef CAS PubMed; (b) S. Demkowicz, J. Rachon, M. Daśko and W. Kozak, RSC Adv., 2016, 6, 7101–7112 RSC; (c) J. B. Rodriguez and C. Gallo-Rodriguez, ChemMedChem, 2019, 14, 190–216 CrossRef CAS PubMed.
- (a) R. G. Hall, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 258–265 CrossRef CAS; (b) C. Zhou, X. Luo, N. Chen, L. Zhang and J. Gao, J. Agric. Food Chem., 2020, 68, 3344–3353 CrossRef CAS.
- (a) T. Baumgartner, Acc. Chem. Res., 2014, 47, 1613–1622 CrossRef CAS PubMed; (b) Z. Wang and T. Baumgartner, Chem. Rec., 2015, 15, 199–217 CrossRef CAS PubMed; (c) M. A. Shameem and A. Orthaber, Chem. – Eur. J., 2016, 22, 10718–10735 CrossRef CAS PubMed; (d) D. Joly, P.-A. Bouit and M. Hissler, J. Mater. Chem. C, 2016, 4, 3686–3698 RSC.
- (a) D. Heift, Z. Benkő and H. Grützmacher, Chem. – Eur. J., 2014, 20, 11326–11330 CrossRef CAS PubMed; (b) C. Xie, A. J. Smaligo, X.-R. Song and O. Kwon, ACS Cent. Sci., 2021, 7, 536–558 CrossRef CAS PubMed; (c) T. Gensch, G. P. Gomes, P. Friederich, E. Peters, T. Gaudin, R. Pollice, K. Jorner, A. K. Nigam, M. Lindner-D'Addario, M. S. Sigman and A. Aspuru-Guzik, J. Am. Chem. Soc., 2022, 144, 1205–1217 CrossRef CAS PubMed.
- (a) Y. Gao, G. Tang and Y. Zhao, Phosphorus, Sulfur Silicon Relat. Elem., 2017, 192, 589–596 CrossRef CAS; (b) K. Neog and P. Gogoi, Org. Biomol. Chem., 2020, 18, 9549–9561 RSC; (c) M. Arisawa, Synthesis, 2020, 2795–2806 CrossRef CAS; (d) D. J. Jones, E. M. O'Leary and T. P. O'Sullivan, Adv. Synth. Catal., 2020, 362, 1825–1830 CrossRef CAS; (e) Y. Mei, Z. Yan and L. L. Liu, J. Am. Chem. Soc., 2022, 144(4), 1517–1522 CrossRef CAS; (f) Y. Liu, X. Chen and B. Yu, Chem. – Eur. J., 2023, 29, e202302142 CrossRef CAS PubMed; (g) X. Liu, L. Zhou, R. Yang, X.-R. Song and Q. Xiao, Adv. Synth. Catal., 2023, 365, 2280–2298 CrossRef CAS.
- For selected reviews on diazo compounds, see: (a) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981–10080 CrossRef CAS PubMed; (b) N. R. Candeias, R. Paterna and P. M. P. Gois, Chem. Rev., 2016, 116, 2937–2981 CrossRef CAS; (c) Y. Xiang, C. Wang, Q. Ding and Y. Peng, Adv. Synth. Catal., 2019, 361, 919–944 CrossRef CAS; (d) Z. Yang, M. L. Stivanin, I. D. Jurberg and R. M. Koenigs, Chem. Soc. Rev., 2020, 49, 6833–6847 RSC; (e) P. K. Mykhailiuk, Chem. Rev., 2020, 120, 12718–12755 CrossRef CAS; (f) J. Durka, J. Turkowska and D. Gryko, ACS Sustainable Chem. Eng., 2021, 9, 8895–8918 CrossRef CAS; (g) S. Dong, X. Liu and X. Feng, Acc. Chem. Res., 2022, 55, 415–428 CrossRef CAS PubMed; (h) A. Dasgupta, E. Richards and R. L. Melen, ACS Catal., 2022, 12, 442–452 CrossRef CAS PubMed; (i) C. Empel, C. Pei and R. M. Koenigs, Chem. Commun., 2022, 58, 2788–2798 RSC.
- (a) M. Wang, L. Kong, Q. Wu and X. Li, Org. Lett., 2018, 20(15), 4597–4600 CrossRef CAS PubMed; (b) G. G. Faura, T. Nguyen and S. France, J. Org. Chem., 2021, 86, 10088–10104 CrossRef PubMed; (c) Z. Qi and S. Wang, Org. Lett., 2021, 23(21), 8549–8553 CrossRef CAS PubMed; (d) C. Empel, C. Pei, F. He, S. Jana and R. M. Koenigs, Chem. – Eur. J., 2022, 28, e202104397 CrossRef CAS PubMed; (e) G. D. Titov, G. I. Antonychev, M. S. Novikov, A. F. Khlebnikov, E. V. Rogacheva, L. A. Kraeva and N. V. Rostovskii, Org. Lett., 2023, 25(15), 2707–2712 CrossRef CAS PubMed.
- I. D. Jurberg and H. M. L. Davies, Chem. Sci., 2018, 9, 5112–5118 RSC.
- (a) A. M. Polozov, N. A. Polezhaeva, A. H. Mustaphin, A. V. Khotinen and B. A. Arbuzov, Synthesis, 1990, 515–517 CrossRef CAS; (b) W. Miao, Y. Gao, X. Li, Y. Gao, G. Tang and Y. Zhao, Adv. Synth. Catal., 2012, 354, 2659–2664 CrossRef CAS; (c) L. Wu, X. Zhang, Q.-Q. Chen and A.-K. Zhou, Org. Biomol. Chem., 2012, 10, 7859–7862 RSC; (d) H. E. Bartrum, D. C. Blakemore, C. J. Moody and C. J. Hayes, Tetrahedron, 2013, 69, 2276–2282 CrossRef CAS; (e) L. Wang, Y. Wu, Y. Liu, H. Yang, X. Liu, J. Wang, X. Li and J. Jiang, Org. Lett., 2017, 19, 782–785 CrossRef CAS PubMed.
- (a) H. Jiang, H. Jin, A. Abdukader, A. Lin, Y. Cheng and C. Zhu, Org. Biomol. Chem., 2013, 11, 3612–3615 RSC; (b) Y. Feng, H. Ding, R. Liu, W. Wei, G. Nan and D. Yi, Asian J. Org. Chem., 2024, 13, e202400015 CrossRef CAS.
- L. B. Baĺazs, Y. Huang, J. B. Khalikuzzaman, Y. Li, S. A. Pullarkat and P.-K. Leung, J. Org. Chem., 2020, 85, 14763–14771 CrossRef PubMed.
- V. K. Aggarwal, J. R. Fulton, C. G. Sheldon and J. de Vicente, J. Am. Chem. Soc., 2003, 125, 6034–6035 CrossRef CAS PubMed.
- The crystal structure of compound 3k has been deposited at the Cambridge Crystallographic Data Centre and the reference no. CCDC 2342436 was allotted.†.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C, 31P and 19F NMR spectra of all new compounds. CCDC 2342436. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob00573b |
| This journal is © The Royal Society of Chemistry 2024 |