Total synthesis and structure–antifouling activity relationship of scabrolide F†
Received
30th April 2024
, Accepted 23rd May 2024
First published on 23rd May 2024
Abstract
An efficient synthetic strategy for scabrolide F (7), a norcembranolide diterpene that was isolated from the Taiwanese soft coral Sinularia scabra, has only recently been reported by our group. Herein, we report details of the first total synthesis of 7. The tetrahydrofuran domain of 7 was stereoselectively constructed via the 5-endo-tet cyclization of a hydroxy vinyl epoxide. The reaction of alkyl iodide 30 with dithiane 38, followed by the introduction of an alkene moiety, afforded allylation precursor 41. The coupling of alkyl iodide 42 and allylic stannane 43 was examined as a model experiment of allylation. Because the desired allylated product 44 was not obtained, an alternative synthetic route toward 7 was investigated instead. In the second synthetic approach, fragment–coupling between alkyl iodide 56 and aldehyde 58, macrolactonization, and transannular ring-closing metathesis were used as the key steps to achieve the first total synthesis of 7. We hope that this synthetic strategy provides access to the total synthesis of other macrocyclic norcembranolides. We also evaluated the antifouling activity and toxicity of 7 and its synthetic intermediates toward the cypris larvae of the barnacle Amphibalanus amphitrite. This study is the first to report the antifouling activity of norcembranolides as well as the biological activity of 7.
Introduction
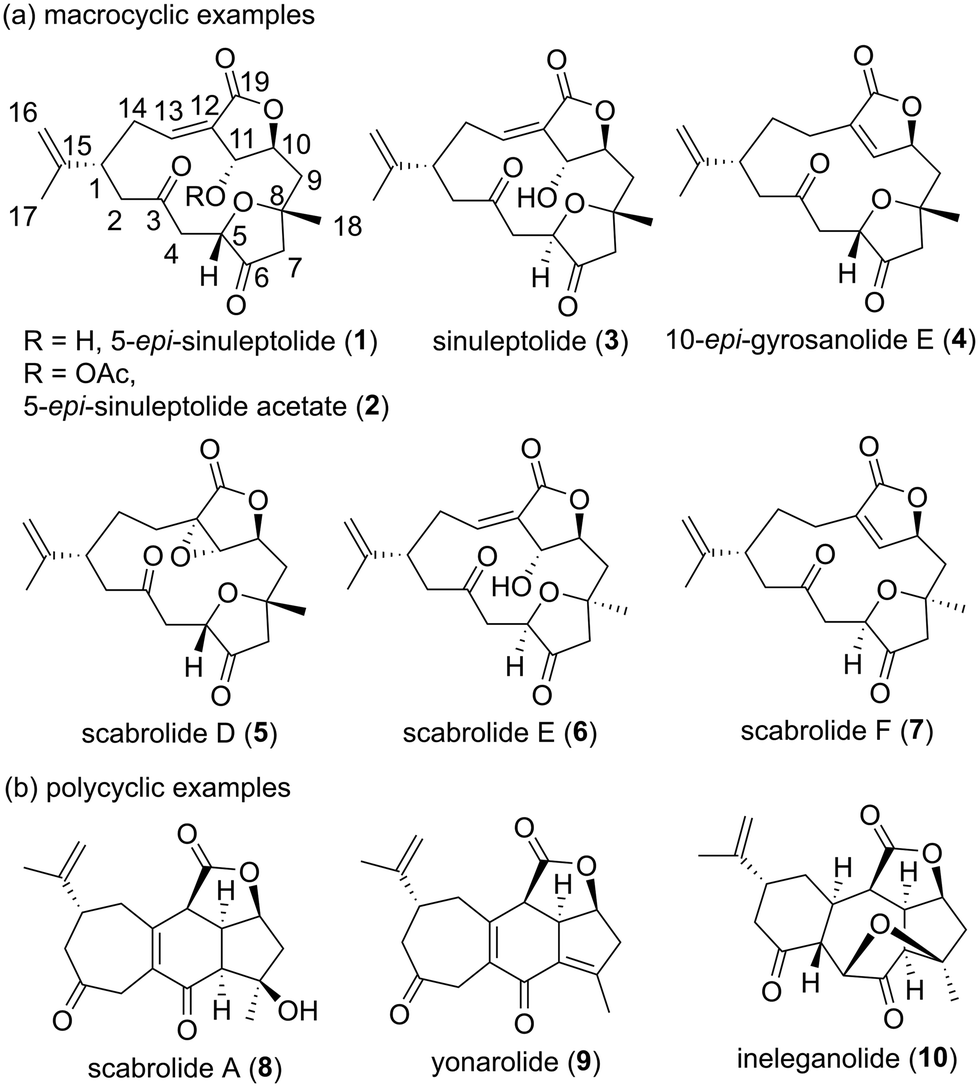
Marine organisms produce many secondary metabolites with diverse chemical structures and biological activities.1 These marine natural products are regarded as potential lead compounds in drug discovery and development.2 5-epi-Sinuleptolide (1, Fig. 1) was isolated from Sinularia leptoclados as the first example of a norcembranolide diterpene in 1978.3 Since then, a number of reports on macrocyclic and polycyclic norcembranolide diterpenes isolated from the soft corals of the genus Sinularia have been published.4 Norcembranoids exhibit various biological activities,4 and cembranoids are suggested to act as chemical defense substances against predators.5 The originally proposed relative configuration of 1, which was not named in the isolation paper, was determined using spectroscopic methods and X-ray crystallography.3 Fenical and Clardy's research group reported the isolation and structural determination of 1 and its C11 stereoisomer in 1985, which revised the C11 stereochemistry of natural product 1.6 The natural product sinuleptolide (3), which is the C5 stereoisomer of 1, was isolated and structurally elucidated by Umeyama et al. in 1993.7 Therefore, the name 5-epi-sinuleptolide is used for natural product 1. 5-epi-Sinuleptolide acetate (2) was obtained by both the derivatization of natural product 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8 and isolation from a natural source.9
8 and isolation from a natural source.9
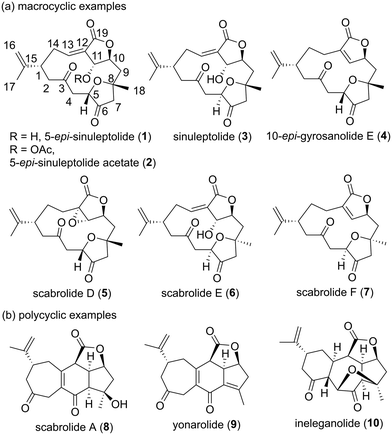 |
| | Fig. 1 Structures of selected norcembranolides 1–10. | |
Sheu et al. elucidated the absolute configurations of 1 and 3 by applying a modified Mosher method in 2005.10 The relative stereostructures of 10-epi-gyrosanolide E (4)6,11 and scabrolides D (5),12 E (6),13 and F (7)13 were clarified using spectroscopic analysis, X-ray crystallography, and comparison of their NMR data with those of 1 and 3. The relative configurations of scabrolide A (8)12,14 and yonarolide (9),14,15 which are representative polycyclic norcembranolides, were assigned based on a detailed 2D NMR analysis to be a [5,6,7] fused carbocyclic system linked to a γ-lactone moiety. Ineleganolide (10) has a highly fused and oxygenated structure including a [5,7,6] carbon skeleton, which was determined using spectroscopic and X-ray diffraction analyses.14,16 Among these norcembranolide diterpenoids, 5-epi-sinuleptolide (1) has been reported to show selective cytotoxicity against pancreatic cancer cells17 as well as decreased extracellular matrix expression during biofilm formation.18 5-epi-Sinuleptolide acetate (2) is moderately cytotoxic toward human tumor cell lines,9 and induces not only apoptosis in HL60 human leukemia cells by inhibiting Hsp90 but also mitochondrial stress.19 Sinuleptolide (3) exhibits antiproliferative activity against oral cancer Ca9–22 cells by inducing apoptosis, oxidative stress, and DNA damage.20
Compounds 1 and 3 have been reported to be dose-dependent inhibitors of TNF-α production;21 they also inhibit the growth of KB and Hepa59T/VGH cancer cells with 50% effective dose (ED50) values of 2.3–2.6 μg mL−1.12,22 Thao et al. reported the ability of scabrolide A (8) to inhibit IL-12 and IL-6 production with 50% inhibitory concentrations of 23.5 and 69.9 μM, respectively.23 The same group found 1 to exhibit peroxy radical-scavenging activity and electron-donating capacity to reduce Cu(II) to Cu(I).24 Yonarolide (9), a dehydration product of 8,4a,b displays inhibitory activity on NF-κB transcriptional activation in HepG2 cells.25 Ineleganolide (10) has been reported to inhibit the growth of P388 cells with an ED50 value of 3.82 μg mL−1.16 Polycyclic norcembranolides 8–10 have been proposed to be biosynthetically derived from macrocyclic norcembranolide 1 through transannular Michael addition and dehydration.4a,b,26 Therefore, the development of a synthetic route toward macrocyclic norcembranolides would lead to a consistent supply of polycyclic norcembranolides by total synthesis.
Norcembranolides have attracted much attention from the natural product synthesis community owing to their structural complexity, wide range of biological activities, and biosynthetic relationships between macrocyclic and polycyclic norcembranoids. Two reports on the total synthesis of macrocyclic norcembranolide diterpenoids have been published. Theodorakis et al. completed the racemic total synthesis of 4 and 5 in 2011, which revised the C11,C12-epoxide stereochemistry of natural product 5.27,28 In the same year, Lee et al. achieved the enantioselective total synthesis of (+)-4 (an enantiomer of 4), which revealed the absolute configuration of natural (−)-10-epi-gyrosanolide E to be 4.29 Synthetic efforts toward polycyclic norcembranolides have culminated in recent reports of the enantioselective total synthesis of (−)-scabrolide A (8) by the groups of Stoltz,30a,b Fürstner,31 Sarlah,32 and Zhang;33 (−)-yonarolide (9) by the groups of Stoltz30b and Sarlah;32 and (+)-ineleganolide (10) by the groups of Wood34 and Stoltz.30c In our work on the chemical synthesis and biological evaluation of cembranolides35 and norcembranolides, we reported the first total synthesis of (−)-scabrolide F (7) as a communication in 2022.36 However, we did not discuss our complete synthetic approaches to obtain this compound or investigate its biological properties. Herein, we disclose the full details of our first and second synthetic approaches toward 7, and describe the antifouling activity and toxicity of 7 and its synthetic intermediates toward the cypris larvae of the barnacle Amphibalanus amphitrite. Our successful synthetic strategy toward 7, which includes fragment–coupling, macrolactonization, and transannular ring-closing metathesis (RCM) as key steps, can be useful in the total synthesis of other macrocyclic norcembranolides. To the best of our knowledge, this study is the first to report the biological activity of 7 and the antifouling activity of norcembranolides.
Results and discussion
Retrosynthetic analysis
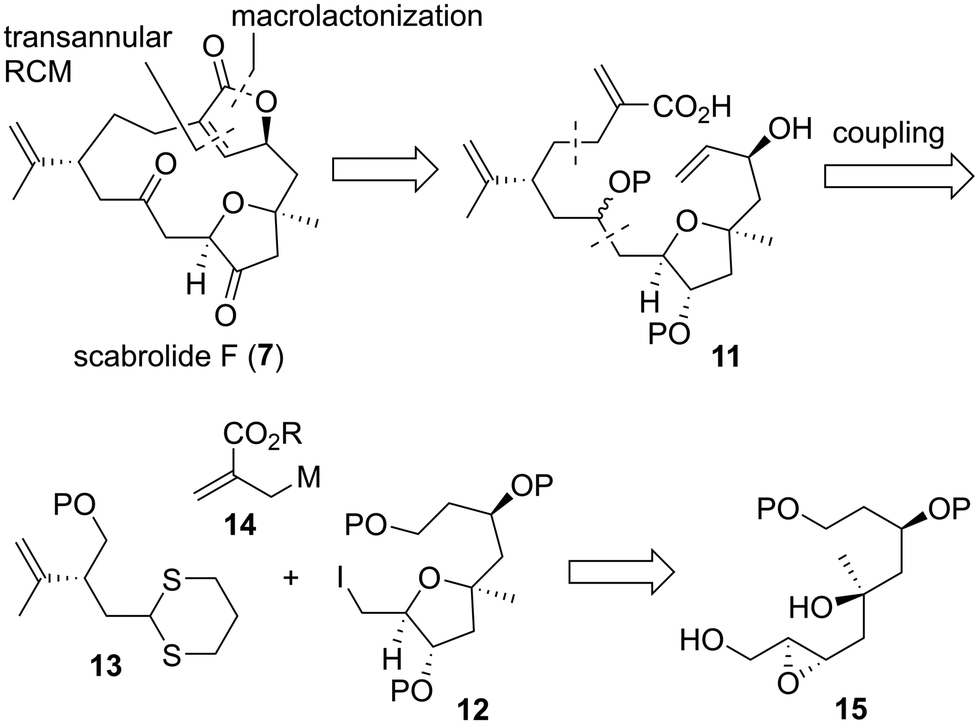
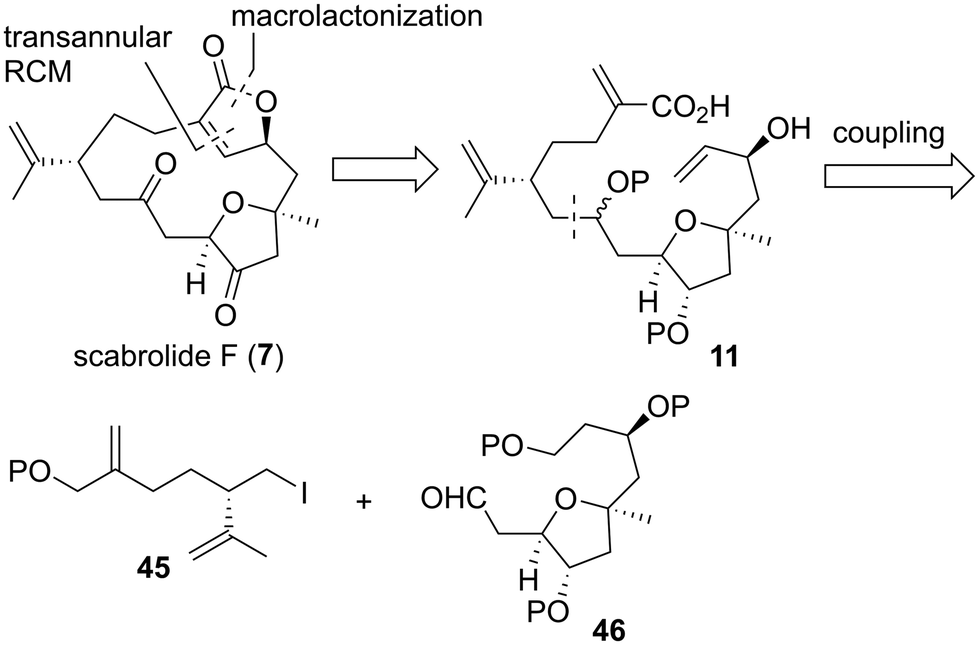
Macrocyclic norcembranolides have a 14-membered carbon framework and butenolide or butenolide-related structures, as shown in Fig. 1. Initially, we analyzed the retrosynthesis of 7 (Scheme 1), which would be applicable to other macrocyclic norcembranolides. Hydroxycarboxylic acid 11 was designed as the key synthetic intermediate to reach 7. We investigated two possible transformations from 11 to 7, namely, macrolactonization37/transannular RCM38 and RCM/lactonization sequences. We selected the former because this sequence could control the alkene geometry of the RCM product.39 We planned to synthesize 11 by connecting alkyl iodide 12, dithiane 13, and allylic metal reagent 14. 2,5-cis-Disubstituted-3-oxygenated tetrahydrofuran 1240 could be obtained via the 5-endo-tet cyclization of epoxy diol 15.
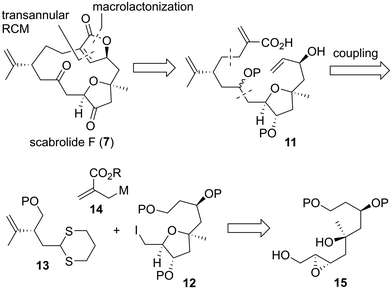 |
| | Scheme 1 First generation retrosynthetic analysis of scabrolide F (7). | |
Synthesis of the tetrahydrofuran fragment
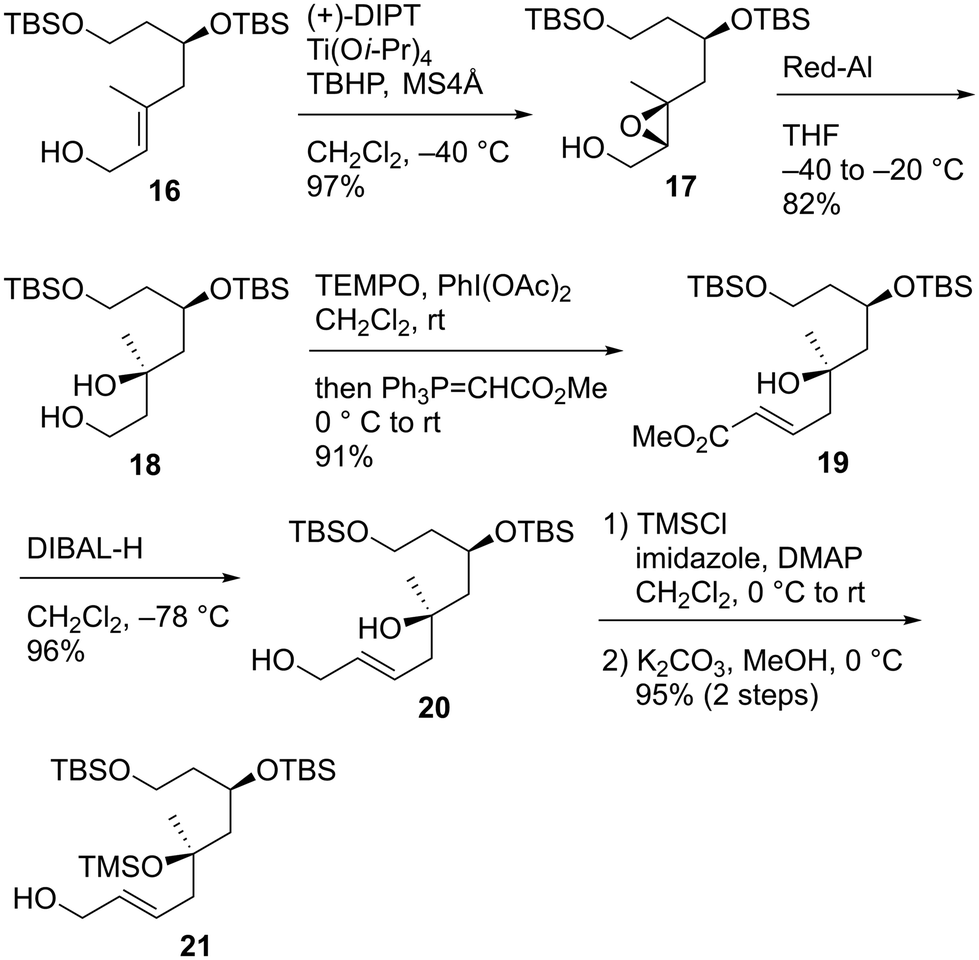
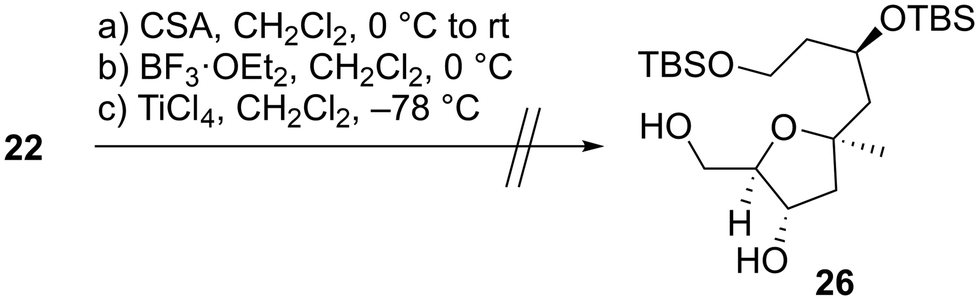
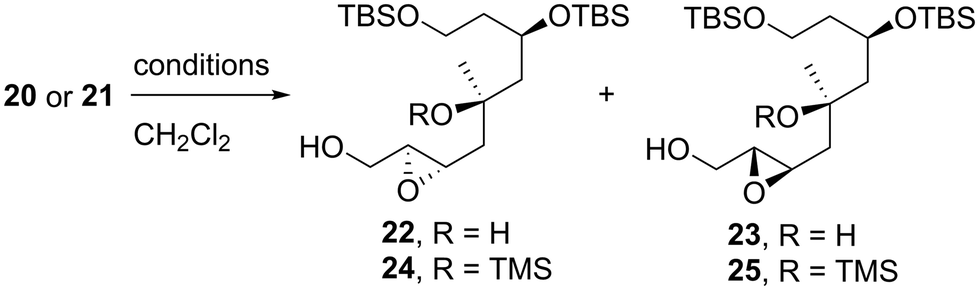
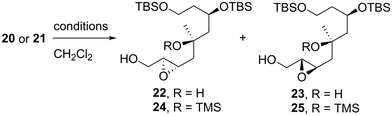
We first examined the stereoselective synthesis of the tetrahydrofuran fragment. The treatment of the known allylic alcohol 16, which was prepared from L-aspartic acid in an optically pure form,41 with (+)-diisopropyl tartrate (DIPT)/Ti(Oi-Pr)4/tert-butyl hydroperoxide (TBHP)/molecular sieves (MS) 4Å (ref. 42) gave epoxy alcohol 17 in 97% yield as a single diastereomer (Scheme 2).43 The obtained epoxide 17 was subjected to regioselective reductive ring-opening with sodium bis(2-methoxyethoxy)aluminum hydride (Red-Al)44 to afford 1,3-diol 18. The oxidation of 18 with 2,2,6,6-tetramethyl-1-piperidinyloxyl (TEMPO)/PhI(OAc)245 and Wittig olefination of the resulting aldehyde with Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO2Me were carried out in a one-pot operation to provide α,β-unsaturated ester 19. The reduction of 19 with diisobutylaluminum hydride (DIBAL-H), protection of the resulting diol 20 as the bis-trimethylsilyl (TMS) ether, and selective removal of the primary TMS group afforded allylic alcohol 21. We then optimized the reaction conditions in the diastereoselective epoxidation of allylic alcohols 20 and 21 (Table 1). When we applied Sharpless epoxidation conditions42 with (+)-diethyl tartrate (DET) to 20, the desired epoxy diol 22 was produced as a single diastereomer;43 however, the chemical yield of 22 was 22%, and the starting material 20 was recovered in 73% yield (entry 1). Changing the starting material from tertiary alcohol 20 to tertiary TMS ether 21 under the same conditions quantitatively provided the desired epoxy alcohol 24 as the sole product (entry 2). When we treated diol 20 with meta-chloroperoxybenzoic acid (mCPBA) under substrate-directed conditions, we fortunately observed the quantitative formation of 22 as a single diastereomer (entry 3).46 The mCPBA-mediated epoxidation of tertiary TMS ether 21 afforded a diastereomeric mixture of 24 and 25 in a 1:1.2 ratio and 86% combined yield (entry 4). A comparison of the results obtained in entries 3 and 4 revealed that the tertiary alcohol moiety was essential for high diastereoselectivity during mCPBA epoxidation. With cyclization precursor 22 in hand, we attempted the 5-endo-tet cyclization of 22 (Scheme 3). The treatment of 22 with camphorsulfonic acid (CSA)47c as a Brønsted acid gave a complex mixture without the desired product 26. The use of BF3·OEt2 (OEt = diethyl ether)47a,c and TiCl447b as Lewis acids did not produce tetrahydrofuran 26. Given these unsuccessful results, we changed the cyclization precursor from epoxy diol 22 to the corresponding hydroxy vinyl epoxide48 to promote 5-endo-tet cyclization.
CHCO2Me were carried out in a one-pot operation to provide α,β-unsaturated ester 19. The reduction of 19 with diisobutylaluminum hydride (DIBAL-H), protection of the resulting diol 20 as the bis-trimethylsilyl (TMS) ether, and selective removal of the primary TMS group afforded allylic alcohol 21. We then optimized the reaction conditions in the diastereoselective epoxidation of allylic alcohols 20 and 21 (Table 1). When we applied Sharpless epoxidation conditions42 with (+)-diethyl tartrate (DET) to 20, the desired epoxy diol 22 was produced as a single diastereomer;43 however, the chemical yield of 22 was 22%, and the starting material 20 was recovered in 73% yield (entry 1). Changing the starting material from tertiary alcohol 20 to tertiary TMS ether 21 under the same conditions quantitatively provided the desired epoxy alcohol 24 as the sole product (entry 2). When we treated diol 20 with meta-chloroperoxybenzoic acid (mCPBA) under substrate-directed conditions, we fortunately observed the quantitative formation of 22 as a single diastereomer (entry 3).46 The mCPBA-mediated epoxidation of tertiary TMS ether 21 afforded a diastereomeric mixture of 24 and 25 in a 1:1.2 ratio and 86% combined yield (entry 4). A comparison of the results obtained in entries 3 and 4 revealed that the tertiary alcohol moiety was essential for high diastereoselectivity during mCPBA epoxidation. With cyclization precursor 22 in hand, we attempted the 5-endo-tet cyclization of 22 (Scheme 3). The treatment of 22 with camphorsulfonic acid (CSA)47c as a Brønsted acid gave a complex mixture without the desired product 26. The use of BF3·OEt2 (OEt = diethyl ether)47a,c and TiCl447b as Lewis acids did not produce tetrahydrofuran 26. Given these unsuccessful results, we changed the cyclization precursor from epoxy diol 22 to the corresponding hydroxy vinyl epoxide48 to promote 5-endo-tet cyclization.
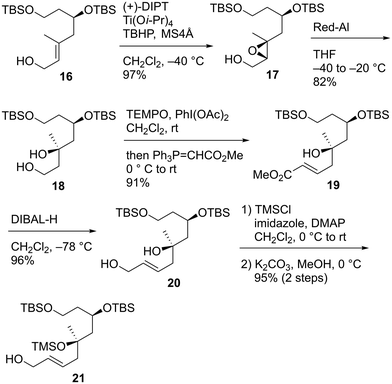 |
| | Scheme 2 Synthesis of epoxidation precursors 20 and 21. | |
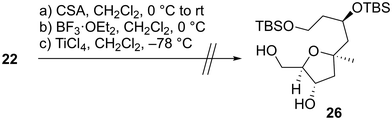 |
| | Scheme 3 Unsuccessful attempt for the 5-endo-tet cyclization of epoxy diol 22. | |
Table 1 Diastereoselective epoxidation of allylic alcohols 20 and 21
|
|
| Entry |
Allylic alcohol |
Conditions |
Yielda |
Diastereomeric ratiob |
|
Isolated yield.
Based on 1H NMR analysis.
73% Recovery of starting material 20.
|
| 1c |
20
|
(+)-DET, Ti(Oi-Pr)4, TBHP, MS4Å, −40 to −20 °C |
22% |
22:23 > 20:1 |
| 2 |
21
|
(+)-DET, Ti(Oi-Pr)4, TBHP, MS4Å, −40 to −20 °C |
quant |
24:25 > 20:1 |
| 3 |
20
|
mCPBA, −40 to 0 °C |
quant |
22:23 > 20:1 |
| 4 |
21
|
mCPBA, −40 to 0 °C |
86% |
24:25 = 1:1.2 |
The synthetic route toward tetrahydrofuran 30 is described in Scheme 4. The silylation of diol 22 with trimethylsilyl chloride (TMSCl), followed by the reaction of the resulting bis-TMS ether with K2CO3 in methanol (MeOH), gave alcohol 24 in 95% yield in two steps; this product was also stereoselectively obtained by the Sharpless epoxidation of 21, as shown in Table 1. Epoxy alcohol 24 was subjected to TEMPO oxidation45 and Wittig methylenation to quantitatively afford vinyl epoxide 27 in two steps. The TMS protecting group of tris-silyl ether 27 was selectively removed using citric acid monohydrate in MeOH to give the tertiary alcohol. The 5-endo-tet-cyclization of the obtained hydroxy vinyl epoxide48 proceeded smoothly with CSA at temperatures ranging from −40 to −10 °C to produce tetrahydrofuran 28 in 98% yield. The resulting hydroxy group of 28 was protected with methoxymethyl chloride (MOMCl)/N,N-diisopropylethylamine (i-Pr2NEt)/tetra-n-butylammonium iodide (TBAI) to yield 29, which was subjected to ozonolysis/reduction and the subsequent iodination of the obtained alcohol with I2/triphenylphosphine (Ph3P)/imidazole to afford alkyl iodide 30.
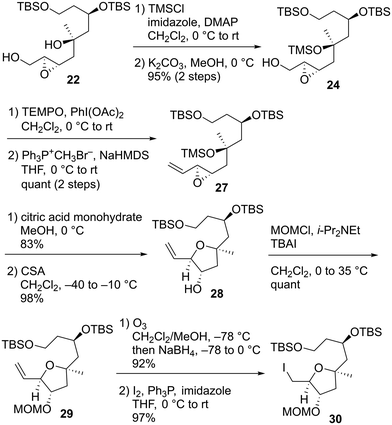 |
| | Scheme 4 Synthesis of tetrahydrofuran 30. | |
First synthetic approach
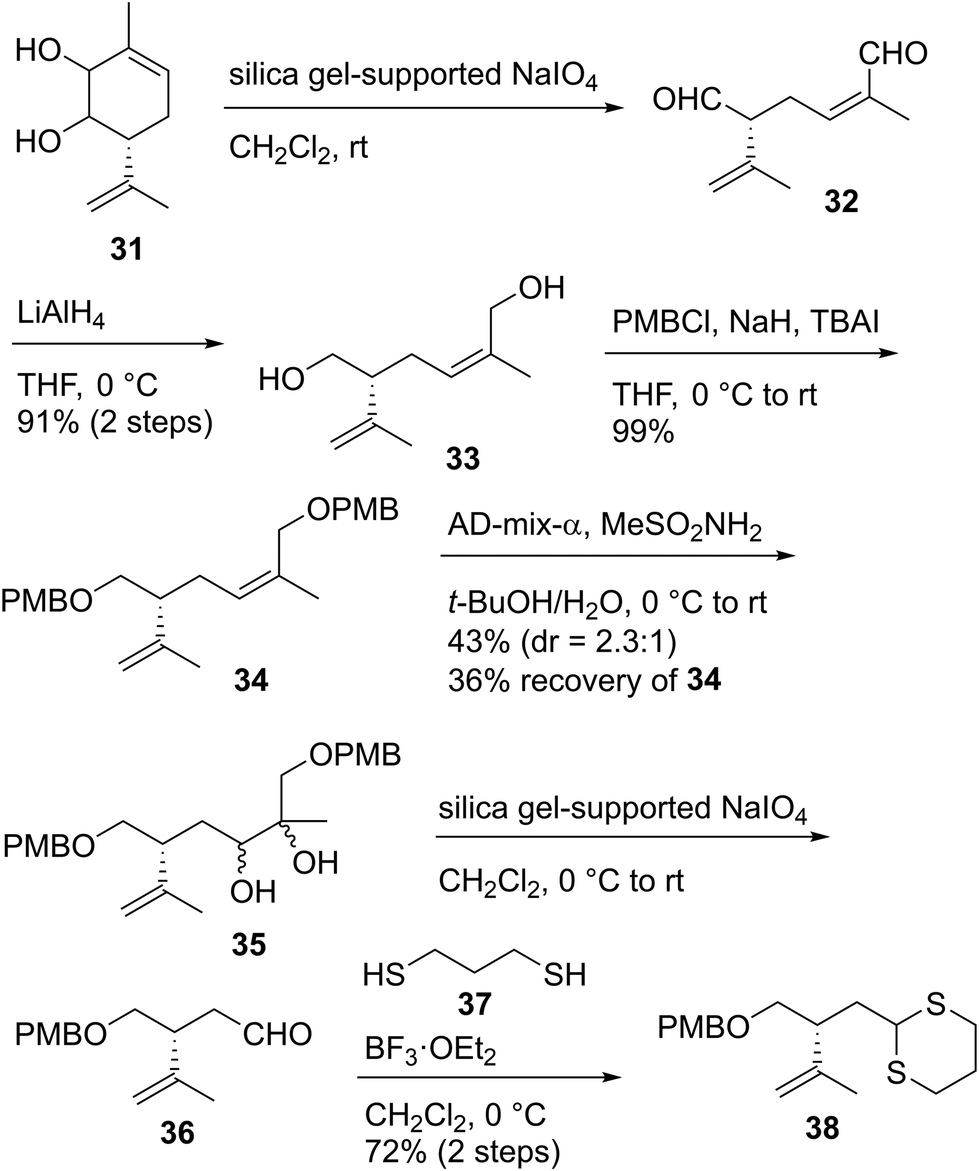
Having stereoselectively synthesized tetrahydrofuran fragment 30, we investigated the enantioselective synthesis of dithiane 38, which is the coupling partner of 30 (Scheme 5). The oxidative cleavage of 1,2-diol 31, which was prepared from (S)-carvone in three steps,49 was conducted with silica gel-supported NaIO450 to give the known dialdehyde 32.49 The unstable compound 32 was immediately reduced with LiAlH4 to afford diol 33 in 91% yield in two steps. The protection of 33 with p-methoxybenzyl chloride (PMBCl)/NaH/TBAI provided bis-PMB ether 34 in 99% yield. The regioselective dihydroxylation of the trisubstituted alkene moiety of 34 possessing the disubstituted terminal alkene group was performed using AD-mix-α51 to produce diol 35 as a 2.3:1 diastereomeric mixture in 43% yield, along with recovered 34 in 36% yield. When we extended the time or increased the temperature in this reaction, the formation of a byproduct in which the disubstituted alkene part was also dehydroxylated was observed. The use of AD-mix-β or OsO4/N-methylmorpholine N-oxide (NMO) did not improve the chemical yield. Diol 35 was subjected to scission oxidation using silica gel-supported NaIO450 to yield aldehyde 36, which was subsequently reacted with 1,3-propanedithiol (37)/BF3·OEt2 to furnish dithiane 38.
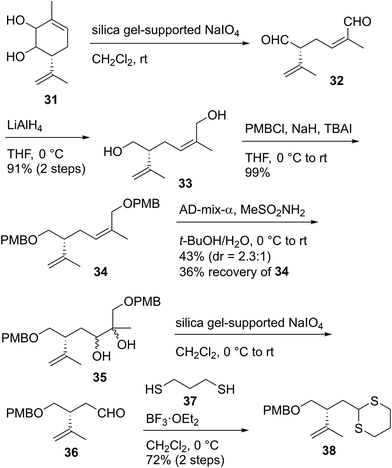 |
| | Scheme 5 Synthesis of dithiane 38. | |
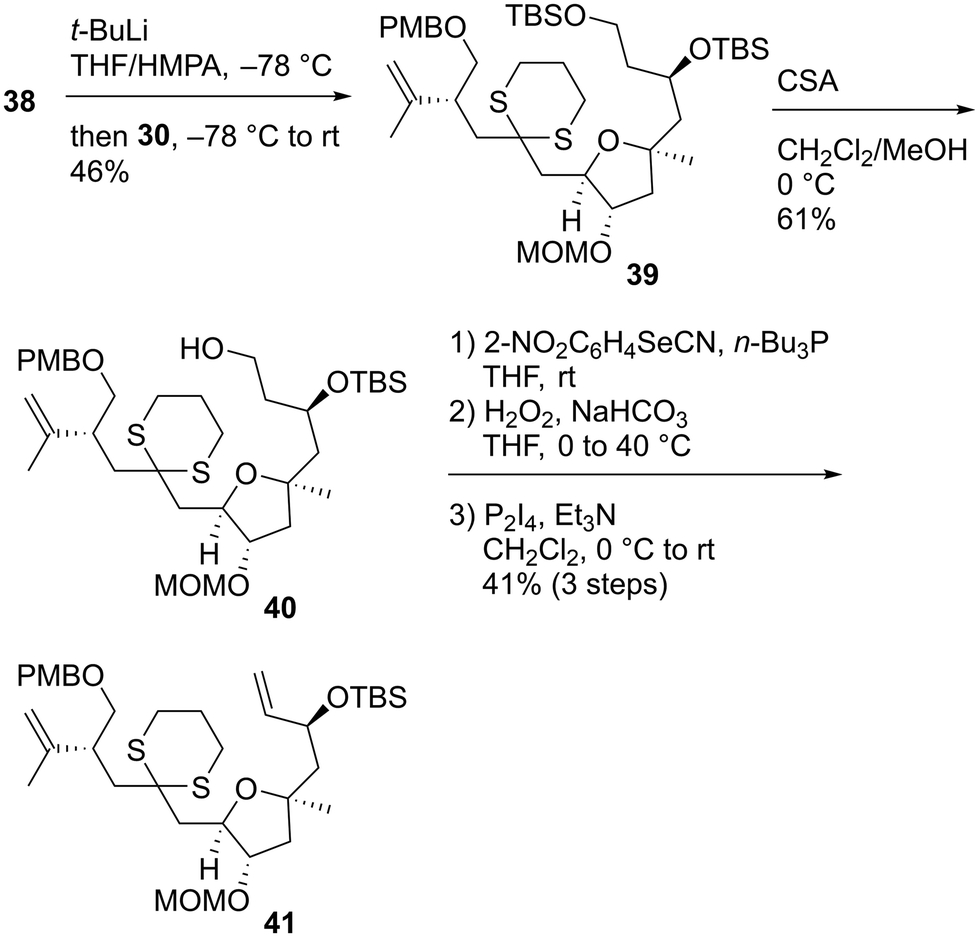
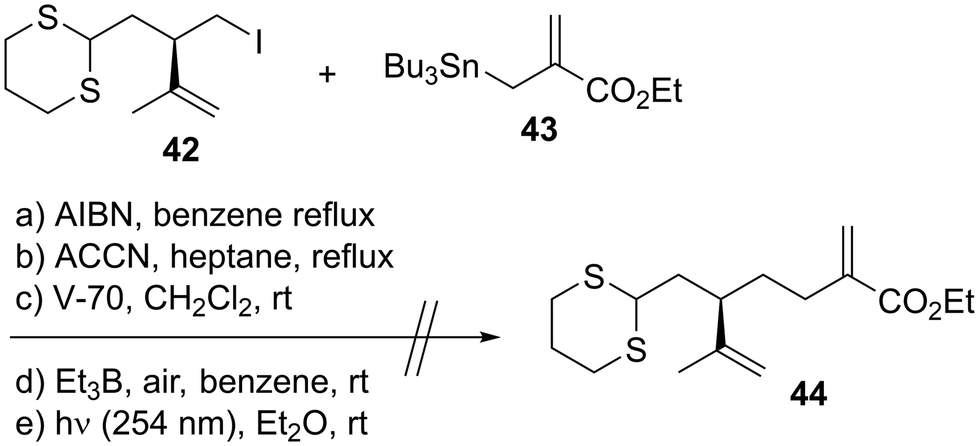
With both coupling precursors 30 and 38 in hand, we proceeded to connect these two compounds (Scheme 6). The deprotonation of dithiane 38 with t-BuLi in tetrahydrofuran (THF)/hexamethylphosphoric acid triamide (HMPA) was followed by a reaction with alkyl iodide 30 to produce the desired coupling product 39 in 46% yield.52 The primary tert-butyldimethylsilyl (TBS) moiety of 39 was selectively removed using CSA in CH2Cl2/MeOH to afford alcohol 40. The reaction of 40 with 2-NO2C6H4SeCN/n-Bu3P53 and subsequent oxidation with H2O2 gave the corresponding monosulfoxide to which the terminal alkene moiety was successfully introduced. The resulting dithiane monoxide was deoxygenated using P2I4/Et3N,54 and dithiane 41 was obtained in 41% yield in three steps. Our next task was to transform 41 to the macrolactonization precursor, which corresponds to compound 11 in Scheme 1. We conducted a model study of allylation using alkyl iodide 4255 and allylic stannane 43,56 as described in Scheme 7. Although we applied several radical reaction conditions, we did not obtain the desired product 44. The use of 2,2′-azobisisobutyronitrile (AIBN)56 as a radical initiator in benzene under reflux gave a complex mixture. When we used 1,1′-azobis(cyclohexanecarbonitrile) (ACCN)57d in heptane under reflux, the starting material 42 was consumed, but several byproducts with unidentified structures were formed. When we conducted the reaction using 2,2′-azobis(2,4-dimethyl-4-methoxyvaleronitrile) (V-70)57c at room temperature, compound 42 was recovered in 52% yield. The use of Et3B/air57a at room temperature recovered 42 in 55% yield. The photoirradiation reaction at 254 nm (ref. 57b) yielded a complex mixture. These unsuccessful outcomes motivated us to change our synthetic plan toward 7.
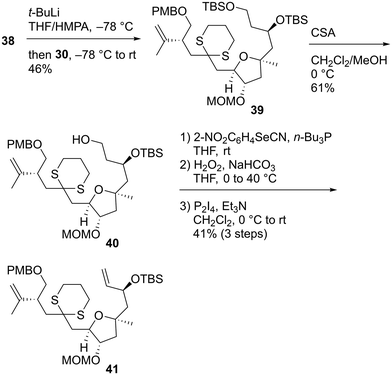 |
| | Scheme 6 Synthesis of dithiane 41. | |
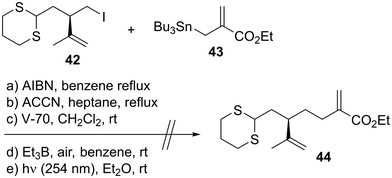 |
| | Scheme 7 Unsuccessful model study of allylation. | |
Second synthetic approach
As in the case of the first retrosynthetic analysis, hydroxycarboxylic acid 11 was set as the key synthetic intermediate toward 7 in the second synthetic plan (Scheme 8). We planned to synthesize 11 in a convergent manner by fragment–coupling between alkyl iodide 45 and aldehyde 46.
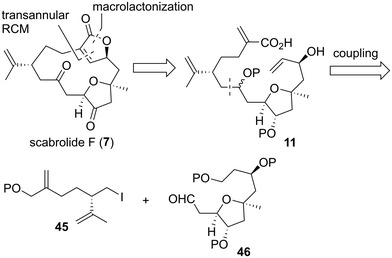 |
| | Scheme 8 Second generation retrosynthetic analysis of scabrolide F (7). | |
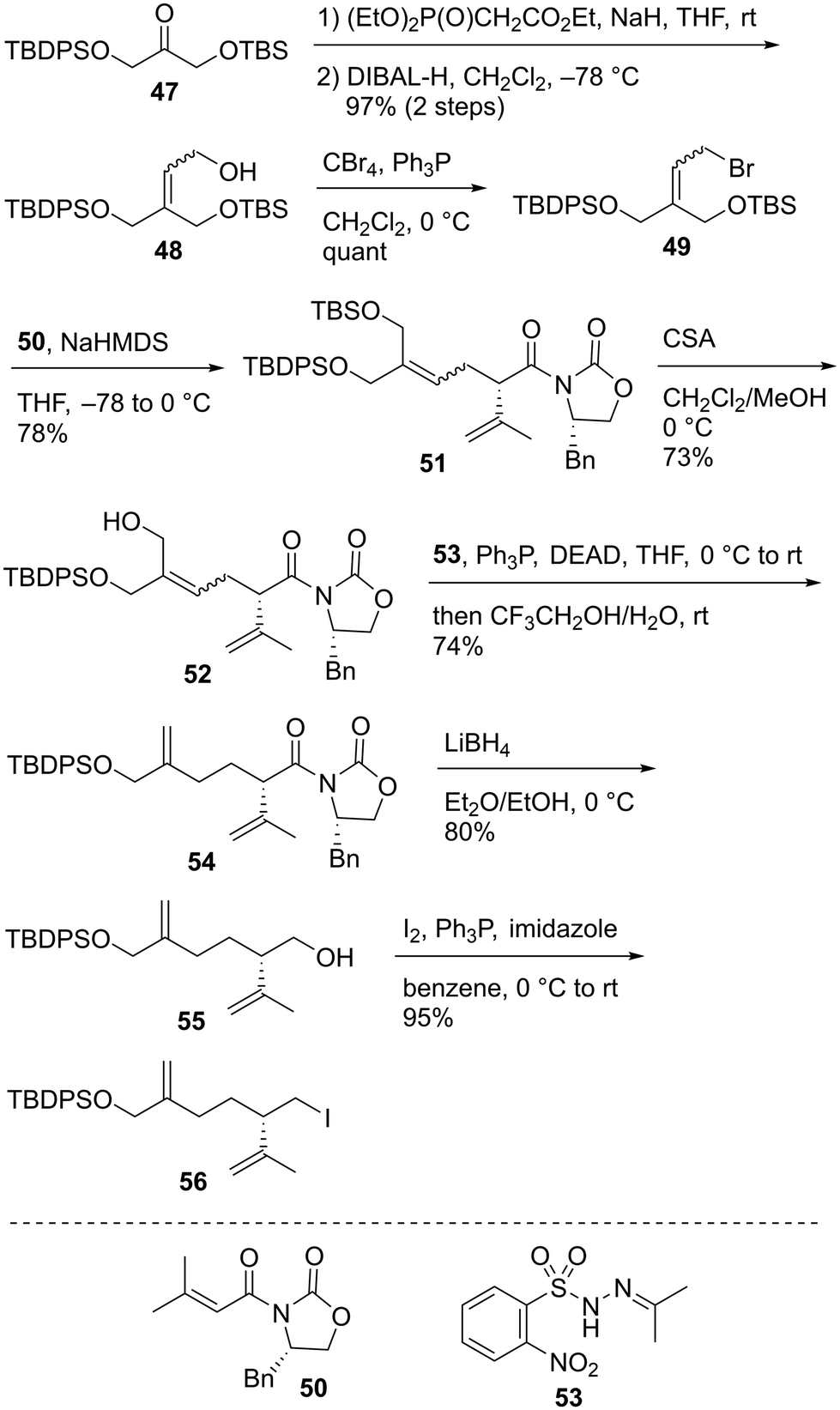
The enantioselective synthesis of alkyl iodide 56 commenced from the known ketone 47 (Scheme 9).58 The treatment of 47 with (EtO)2P(O)CH2CO2Et/NaH and subsequent reduction of the resulting α,β-unsaturated ester using DIBAL-H afforded allylic alcohol 48 in 97% yield in two steps as a 1.5:1 mixture of geometric isomers. Compound 48 was reacted with CBr4/Ph3P to provide allylic bromide 49. The obtained compound 49 was subjected to Evans asymmetric alkylation59 using chiral imide 5060 to produce the allylated product 51 in 78% yield. The TBS protective group of bis-silyl ether 51 was selectively removed using CSA to give allylic alcohol 52. The reductive transposition of allylic alcohol 52 was realized using the conditions reported by Movassaghi et al.61 The reaction of 52 with N-isopropylidene-N′-2-nitrobenzenesulfonyl hydrazine (53)/Ph3P/diethyl azodicarboxylate (DEAD) afforded the corresponding Mitsunobu adduct, which was subjected to hydrolysis using CF3CH2OH/H2O with the subsequent sigmatropic release of N2 to give diene 54 in 74% yield. Imide 54 was reduced to alcohol 55 using LiBH4 in 80% yield, and the reaction of 55 with I2/Ph3P/imidazole furnished alkyl iodide 56 in 95% yield.
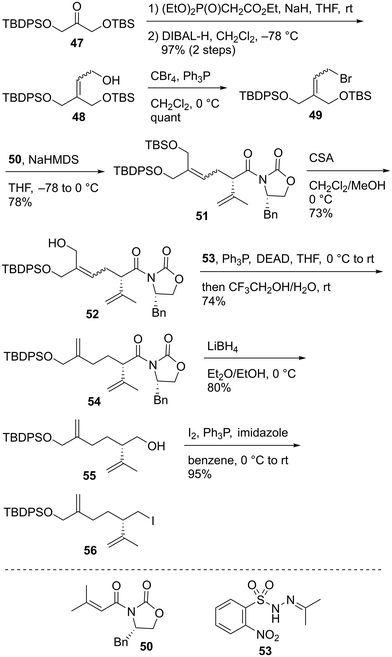 |
| | Scheme 9 Synthesis of alkyl iodide 56. | |
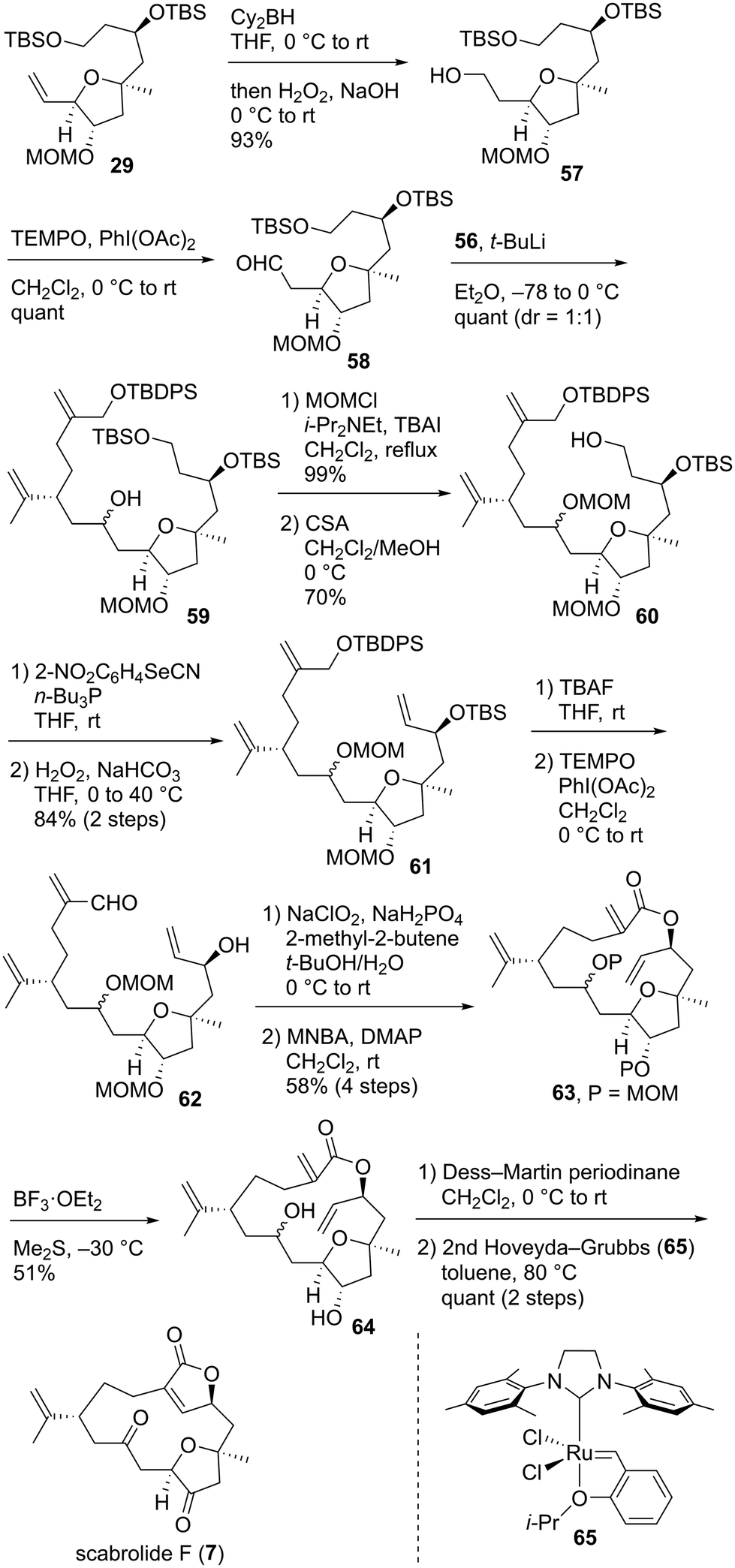
Having successfully prepared coupling precursor 56, we focused on the connection of the fragments and their subsequent transformation toward the total synthesis of 7. The hydroboration of alkene 29 and oxidative work-up afforded alcohol 57, which was oxidized with TEMPO/PhI(OAc)245 to give coupling precursor 58 (Scheme 10). Convergent fragment–coupling was achieved by the lithium–iodine exchange of alkyl iodide 56 with t-BuLi and reaction of the resulting anion with aldehyde 58, quantitatively affording the desired alcohol 59 as a 1:1 diastereomeric mixture. Alcohol 59 was protected as the methoxymethyl (MOM) ether, and the primary TBS group was selectively removed to yield alcohol 60. Dienol 60 was converted to triene 61 by the formation of the corresponding selenide53 and subsequent oxidation in 84% yield in two steps. Bis-silyl ether 61 was treated with tetra-n-butylammonium fluoride (TBAF) to give the corresponding diol, which was subjected to TEMPO oxidation45 to provide hydroxyaldehyde 62. The Pinnick oxidation of 62 with NaClO2/NaH2PO4/2-methyl-2-butene62 afforded the hydroxycarboxylic acid, which was immediately treated with 2-methyl-6-nitrobenzoic anhydride (MNBA)/4-dimethylaminopyridine (DMAP)63 to furnish macrolactone 63 in 58% yield in four steps. Bis-MOM ether 63 was deprotected using BF3·OEt2/Me2S35a,c to give diol 64 in 51% yield.64 Prolonging the reaction time reduced the chemical yield of 64 owing to the formation of byproducts. Therefore, this reaction was stopped after 1 h and repeated three times. 64 was oxidized to the corresponding diketone using Dess–Martin periodinane.65 Finally, the transannular RCM38 of the triene proceeded smoothly in the presence of a second-generation Hoveyda–Grubbs catalyst (65)66 in toluene at 80 °C, leading to the quantitative formation of scabrolide F (7) in two steps. After the completion of the total synthesis of 7, we analyzed its 2D NMR spectra. The 1H and 13C NMR data of the synthesized 7 were nearly identical to those reported for the natural product.13,67 The measured specific rotation of the synthetic product 7 was [α]25D −5.1 (c 0.075, CHCl3), which was consistent with that of the natural product, [α]27D −6.3 (c 0.48, CHCl3).13 Therefore, the absolute configuration of natural scabrolide F was elucidated to be that shown in 7.
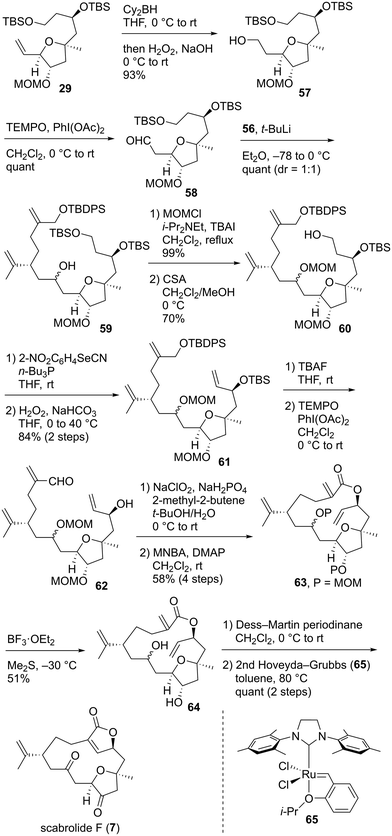 |
| | Scheme 10 Total synthesis of scabrolide F (7). | |
Antifouling activity and toxicity
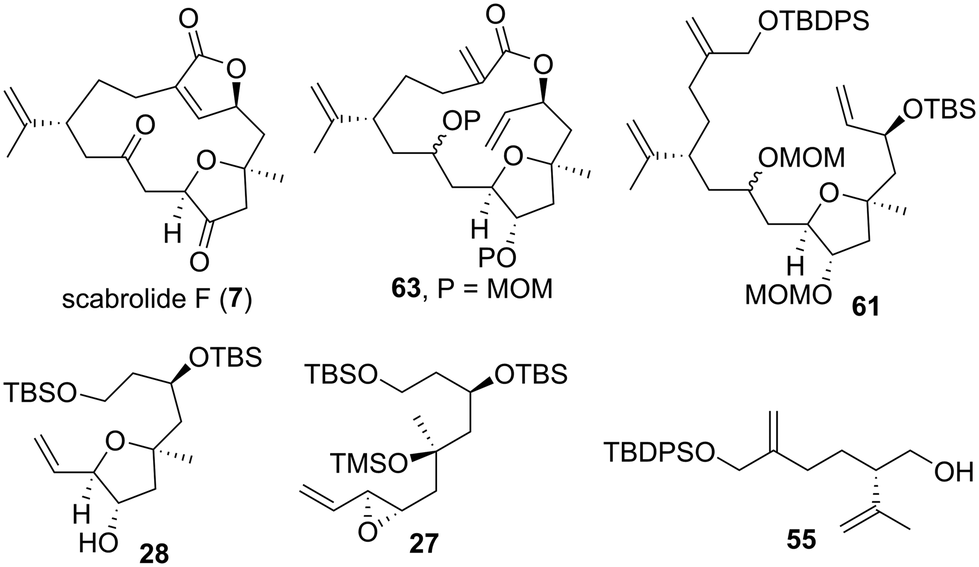
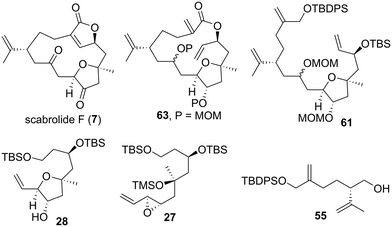
Having completed the total synthesis of 7 and elucidation of the absolute configuration of natural scabrolide F, we examined the biological activity of the synthetic product 7. We previously evaluated the antifouling activity of sarcophytonolides H and J, which are cembranolide diterpenes isolated from the soft coral S. latum, and found these compounds to be antifouling-active.35b,c Thus, we envisioned that 7, a norcembranolide isolated from the same genus, would also exhibit the antifouling activity demonstrated by sarcophytonolides H and J. We first evaluated the antifouling activity and toxicity of CuSO4 as a positive control against the cypris larvae of the barnacle A. amphitrite after a 96 h incubation period. In this study, antifouling activity and toxicity were represented by 50% effective concentration (EC50) and 50% lethal concentration (LC50) values, respectively. The EC50 (0.29 μg mL−1) and LC50 (2.10 μg mL−1) values of CuSO4 were nearly equivalent to those reported in the literature (Table 2).68 As expected, the synthetic product 7 inhibited the settlement of cypris larvae with an EC50 value of 9.46 μg mL−1. Encouraged by this result, we assessed the antifouling activity of several synthetic intermediates of 7. Transannular RCM precursor 63 exhibited approximately twice as high antifouling activity (EC50 = 4.51 μg mL−1) as 7, potentially proposing that the butenolide structure is not essential for exerting antifouling activity. Macrocyclization precursor 61 demonstrated a settlement-inhibitory effect, with an EC50 value of 5.74 μg mL−1. Tetrahydrofuran 28 and epoxide 27 also exhibited antifouling activity regardless of the differences in their molecular skeleton (28: EC50 = 5.89 μg mL−1, 27: EC50 = 5.60 μg mL−1). Interestingly, acyclic compound 55 showed higher antifouling activity (EC50 = 2.65 μg mL−1) than the five other compounds. The relatively low solubility of 7 in water may explain why its antifouling effect is weaker than that of the five other compounds. Furthermore, we evaluated the toxicity of 7, 63, 61, 28, 27, and 55, and found these compounds to be nontoxic at a concentration of 50 μg mL−1 (LC50 > 50 μg mL−1). Among the six compounds evaluated, 55, which showed the highest antifouling activity without toxicity, is a candidate for the development of environmentally friendly antifouling agents.
Table 2 Antifouling activity (EC50) and toxicity (LC50) of CuSO4, synthetic scabrolide F (7), and its synthetic intermediatesa
|
|
| Compound |
EC50 (μg mL−1) |
LC50 (μg mL−1) |
|
Against the cypris larvae of the barnacle Amphibalanus amphitrite.
|
| CuSO4 |
0.29 |
2.10 |
|
7
|
9.46 |
>50 |
|
63
|
4.51 |
>50 |
|
61
|
5.74 |
>50 |
|
28
|
5.89 |
>50 |
|
27
|
5.60 |
>50 |
|
55
|
2.65 |
>50 |
Conclusions
We performed the first total synthesis and evaluation of the antifouling activity of scabrolide F (7), a norcembranolide diterpene isolated from S. scabra. The 2,5-cis-disubstituted-3-oxygenated tetrahydrofuran moiety of 7 was constructed using the 5-endo-tet cyclization of a hydroxy vinyl epoxide. The coupling between alkyl iodide 30 and dithiane 38 and introduction of an alkene moiety afforded compound 41. As our model study on allylation using alkyl iodide 42 and allylic stannane 43 was unsuccessful, we investigated an alternative synthetic approach toward 7 instead. In the revised synthetic route, convergent fragment–coupling between alkyl iodide 56 and aldehyde 58, macrocyclization, and transannular RCM were successfully accomplished as key steps, leading to the first total synthesis of 7. We then assayed the antifouling activity and toxicity of 7 and its key synthetic intermediates 63, 61, 28, 27, and 55 against the cypris larvae of A. amphitrite; these six compounds were found to be antifouling-active (EC50 = 2.65–9.46 μg mL−1) and nontoxic (LC50 > 50 μg mL−1). Among these six active compounds, acyclic compound 55, which showed the most potent settlement-inhibitory activity without toxicity, was proposed as a candidate for the preparation of environmentally benign antifoulants. The synthetic route toward 7 proposed in this study can be useful in the total synthesis of other macrocyclic norcembranolides. Further synthetic and biological studies of other norcembranolides are underway in our laboratory.
Author contributions
H. T. conceived and directed the study. Y. S. and R. M. conducted the syntheses and collected the compound data. T. Y. collected and maintained the barnacles. H. T. evaluated the biological activities. All authors analyzed the collected data. H. T. prepared the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to Prof. Jyh-Horng Sheu (National Sun Yat-sen University) and Prof. Atallah F. Ahmed (King Saud University) for their valuable discussions on natural scabrolide F. We are also grateful to Dr Noriyuki Endo and Mr Yusuke Murata (Himeji EcoTech Co., Ltd) for their valuable discussions on the biological evaluations conducted in this study. We thank Mr Shunya Kanemitsu (Okayama University) and Ms. Shiori Kitade (University of Hyogo) for their experimental support. We are grateful to the Division of Instrumental Analysis at Okayama University for their assistance with the NMR and HRMS measurements. This work was supported by JSPS KAKENHI Grant Numbers JP21H01938 and JP23K21115 to H. T. and JP20K15576 to T. Y.
References
-
(a) D. Uemura, Chem. Rec., 2006, 6, 235–248 CrossRef CAS PubMed;
(b) A. R. Carroll, B. R. Copp, R. A. Davis, R. A. Keyzers and M. R. Prinsep, Nat. Prod. Rep., 2023, 40, 275–325 RSC.
-
(a) D. J. Newman and G. M. Cragg, Planta Med., 2016, 82, 775–789 CrossRef CAS PubMed;
(b) K.-H. Altmann, Chimia, 2017, 71, 646–652 CrossRef CAS PubMed;
(c) C. Alves, J. Silva, S. Pinteus, H. Gaspar, M. C. Alpoim, L. M. Botana and R. Pedrosa, Front. Pharmacol., 2018, 9, 777 CrossRef PubMed.
-
(a) B. F. Bowden, J. C. Coll, S. J. Mitchell, J. Mulder and G. J. Stokie, Aust. J. Chem., 1978, 31, 2049–2056 CrossRef CAS;
(b) K. E. Turner, B. F. Bowden, G. J. Stokie and C. J. Howard, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 1283–1284 CrossRef.
-
(a) Y. Li and G. Pattenden, Nat. Prod. Rep., 2011, 28, 429–440 RSC;
(b) R. A. Craig and B. M. Stoltz, Chem. Rev., 2017, 117, 7878–7909 CrossRef CAS PubMed;
(c) X. Yan, J. Liu, X. Leng and H. Ouyang, Mar. Drugs, 2021, 19, 335 CrossRef CAS PubMed.
-
(a) J. C. Coll, Chem. Rev., 1992, 92, 613–631 CrossRef CAS;
(b) J. C. Coll, I. R. Price, G. M. König and B. F. Bowden, Mar. Biol., 1987, 96, 129–135 CrossRef.
- A. Sato, W. Fenical, Z. Qi-tai and J. Clardy, Tetrahedron, 1985, 41, 4303–4308 CrossRef CAS.
- N. Shoji, A. Umeyama and S. A. Arihara, J. Nat. Prod., 1993, 56, 1651–1653 CrossRef CAS.
- Y. Li and G. Pattenden, Tetrahedron, 2011, 67, 10045–10052 CrossRef CAS.
- W.-H. Yen, L.-C. Hu, J.-H. Su, M.-C. Lu, W.-H. Twan, S.-Y. Yang, Y.-C. Kuo, C.-F. Weng, C.-H. Lee, Y.-H. Kuo and P.-J. Sung, Molecules, 2012, 17, 14058–14066 CrossRef CAS PubMed.
- Y.-J. Tseng, A. F. Ahmed, C.-F. Dai, M.-Y. Chiang and J.-H. Sheu, Org. Lett., 2005, 7, 3813–3816 CrossRef CAS PubMed.
- S.-Y. Cheng, C.-T. Chuang, Z.-H. Wen, S.-K. Wang, S.-F. Chiou, C.-H. Hsu, C.-F. Dai and C.-Y. Duh, Bioorg. Med. Chem., 2010, 18, 3379–3385 CrossRef CAS PubMed.
- J.-H. Sheu, A. F. Ahmed, R.-T. Shiue, C.-F. Dai and Y.-H. Kuo, J. Nat. Prod., 2002, 65, 1904–1908 CrossRef CAS PubMed.
- A. F. Ahmed, J.-H. Su, Y.-H. Kuo and J.-H. Sheu, J. Nat. Prod., 2004, 67, 2079–2082 CrossRef CAS PubMed.
- W.-X. Cui, M. Yang, H. Li, S.-W. Li, L.-G. Yao, G. Li, W. Tang, C.-H. Wang, L.-F. Liang and Y.-W. Guo, Bioorg. Chem., 2020, 94, 103350 CrossRef CAS PubMed.
- K. Iguchi, K. Kajiyama and Y. Yamada, Tetrahedron Lett., 1995, 36, 8807–8808 CrossRef CAS.
- C.-Y. Duh, S.-K. Wang, M.-C. Chia and M. Y. Chiang, Tetrahedron Lett., 1999, 40, 6033–6035 CrossRef CAS.
- W.-C. Tsai, W.-H. Wang, B.-C. Huang, C.-Y. Huang and J.-H. Sheu, Molecules, 2021, 26, 6932 CrossRef CAS PubMed.
- S.-P. Tseng, W.-C. Hung, C.-Y. Huang, Y.-S. Lin, M.-Y. Chan, P.-L. Lu, L. Lin and J.-H. Sheu, Mar. Drugs, 2016, 14, 143 CrossRef PubMed.
- K.-J. Huang, Y.-C. Chen, M. El-Shazly, Y.-C. Du, J.-H. Su, C.-W. Tsao, W.-H. Yen, W.-B. Chang, Y.-D. Su, Y.-T. Yeh and M.-C. Lu, Molecules, 2013, 18, 2924–2933 CrossRef CAS PubMed.
- Y.-T. Chang, C.-Y. Huang, K.-T. Li, R.-N. Li, C.-C. Liaw, S.-H. Wu, J.-R. Liu, J.-H. Sheu and H.-W. Chang, Arch. Oral Biol., 2016, 66, 147–154 CrossRef CAS PubMed.
- H. Takaki, R. Koganemaru, Y. Iwakawa, R. Higuchi and T. Miyamoto, Biol. Pharm. Bull., 2003, 26, 380–382 CrossRef CAS PubMed.
- A.-F. Ahmed, R.-T. Shiue, G.-H. Wang, C.-F. Dai, Y.-H. Kuo and J.-H. Sheu, Tetrahedron, 2003, 59, 7337–7344 CrossRef CAS.
- N. P. Thao, N. H. Nam, N. X. Cuong, T. H. Quang, P. T. Tung, L. D. Dat, D. Chae, S. Kim, Y.-S. Koh, P. V. Kiem, C. V. Minh and Y. H. Kim, Bioorg. Med. Chem. Lett., 2013, 23, 228–231 CrossRef PubMed.
- N. P. Thao, B. T. T. Luyen, S. H. Lee, H. D. Jang, P. V. Kiem, C. V. Minh and Y. H. Kim, Med. Chem. Res., 2015, 24, 3551–3560 CrossRef CAS.
- N. P. Thao, N. H. Nam, N. X. Cuong, B. T. T. Luyen, B. H. Tai, J. E. Kim, S. B. Song, P. V. Kiem, C. V. Minh and Y. H. Kim, Arch. Pharmacal Res., 2014, 37, 706–712 CrossRef CAS PubMed.
- The Pattenden research group succeeded in the biomimetic synthesis of ineleganolide (10) from 5-epi-sinuleptolide (1), which supported the biosynthetic proposal. See ref. 8.
- A. Saitman, P. Rulliere, S. D. E. Sullivan and E. A. Theodorakis, Org. Lett., 2011, 13, 5854–5857 CrossRef CAS PubMed.
- Norcembrenolide B is used as the compound name of 10-epi-gyrosanolide E in ref. 27.
- M. S. Kwon, S. H. Sim, Y. K. Chung and E. Lee, Tetrahedron, 2011, 67, 10179–10185 CrossRef CAS.
-
(a) N. J. Hafeman, S. A. Loskot, C. E. Reimann, B. P. Pritchett, S. C. Virgil and B. M. Stoltz, J. Am. Chem. Soc., 2020, 142, 8585–8590 CrossRef CAS PubMed;
(b) N. J. Hafeman, S. A. Loskot, C. E. Reimann, B. P. Pritchett, S. C. Virgil and B. M. Stoltz, Chem. Sci., 2023, 14, 4745–4758 RSC;
(c) B. M. Gross, S.-J. Han, S. C. Virgil and B. M. Stoltz, J. Am. Chem. Soc., 2023, 145, 7763–7767 CrossRef CAS PubMed.
- Z. Meng and A. Fürstner, J. Am. Chem. Soc., 2022, 144, 1528–1533 CrossRef CAS PubMed.
- R. Serrano, Y. D. Boyko, L. W. Hernandez, A. Lotuzas and D. Sarlah, J.
Am. Chem. Soc., 2023, 145, 8805–8809 CrossRef CAS PubMed.
- Y.-P. Zhang, S. Du, Y. Ma, W. Zhan, W. Chen, X. Yang and H. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202315481 CrossRef CAS PubMed.
- J. P. Tuccinardi and J. L. Wood, J. Am. Chem. Soc., 2022, 144, 20539–20547 CrossRef CAS PubMed.
-
(a) H. Takamura, K. Iwamoto, E. Nakao and I. Kadota, Org. Lett., 2013, 15, 1108–1111 CrossRef CAS;
(b) H. Takamura, T. Kikuchi, N. Endo, Y. Fukuda and I. Kadota, Org. Lett., 2016, 18, 2110–2113 CrossRef CAS PubMed;
(c) H. Takamura, T. Kikuchi, K. Iwamoto, E. Nakao, N. Harada, T. Otsu, N. Endo, Y. Fukuda, O. Ohno, K. Suenaga, Y.-W. Guo and I. Kadota, J. Org. Chem., 2018, 83, 11028–11056 CrossRef CAS PubMed.
- H. Takamura, Y. Sugitani, R. Morishita and I. Kadota, Org. Lett., 2022, 24, 7845–7849 CrossRef CAS PubMed.
-
(a) A. Parenty, X. Moreau and J.-M. Campagne, Chem. Rev., 2006, 106, 911–939 CrossRef CAS PubMed;
(b) A. Parenty, X. Moreau, G. Niel and J.-M. Campagne, Chem. Rev., 2013, 113, PR1–PR40 CrossRef CAS PubMed;
(c) Y. Li, X. Yin and M. Dai, Nat. Prod. Rep., 2017, 34, 1185–1192 RSC.
- For reviews of metathesis, see:
(a) A. Deiters and S. F. Martin, Chem. Rev., 2004, 104, 2199–2238 CrossRef CAS;
(b) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4490–4527 CrossRef CAS;
(c) I. Cheng-Sánchez and F. Sarabia, Synthesis, 2018, 50, 3749–3786 CrossRef.
- For strategies of macrocyclization and postcyclization, see: A. Fürstner, Acc. Chem. Res., 2021, 54, 861–874 CrossRef PubMed.
- For reviews of the synthesis of tetrahydrofurans, see:
(a) J. P. Wolfe and M. B. Hay, Tetrahedron, 2007, 63, 261–290 CrossRef CAS PubMed;
(b) G. Jalce, X. Franck and B. Figadère, Tetrahedron: Asymmetry, 2009, 20, 2537–2581 CrossRef CAS.
- H. Takamura, Y. Kadonaga, Y. Yamano, C. Han, I. Kadota and D. Uemura, Tetrahedron, 2009, 65, 7449–7456 CrossRef CAS.
-
(a) T. Katsuki and K. B. Sharpless, J. Am. Chem. Soc., 1980, 102, 5974–5976 CrossRef CAS;
(b) R. M. Hanson and K. B. Sharpless, J. Org. Chem., 1986, 51, 1922–1925 CrossRef CAS.
- The resulting stereochemistry at the epoxide moiety was determined after constructing the tetrahydrofuran ring. See the ESI for details.†.
- J. M. Finan and Y. Kishi, Tetrahedron Lett., 1982, 23, 2719–2722 CrossRef CAS.
- A. De Mico, R. Margarita, L. Parlanti, A. Vescovi and G. Piancatelli, J. Org. Chem., 1997, 62, 6974–6977 CrossRef CAS.
- For reviews of substrate-directed reactions, see:
(a) A. H. Hoveyda, D. A. Evans and G. C. Fu, Chem. Rev., 1993, 93, 1307–1370 CrossRef CAS;
(b) S. G. Davies, A. M. Fletcher and J. E. Thomson, Org. Biomol. Chem., 2014, 12, 4544–4549 RSC.
-
(a) B. Kang, J. Mowat, T. Pinter and R. Britton, Org. Lett., 2009, 11, 1717–1720 CrossRef CAS PubMed;
(b) E. Pavlakos, T. Georgiou, M. Tofi, T. Montagnon and G. Vassilikogiannakis, Org. Lett., 2009, 11, 4556–4559 CrossRef CAS PubMed;
(c) B. Das and D. N. Kumar, Tetrahedron Lett., 2010, 51, 6011–6013 CrossRef CAS.
-
(a) T. Oka, K. Fujiwara and A. Murai, Tetrahedron, 1996, 52, 12091–12110 CrossRef CAS;
(b) R. S. Narayan, M. Sivakumar, E. Bouhlel and B. Borhan, Org. Lett., 2001, 3, 2489–2492 CrossRef CAS PubMed;
(c) Y. Chen, J. Jin, J. Wu and W.-M. Dai, Synlett, 2006, 1177–1180 CAS.
- H. J. Kim, L. Su, H. Jung and S. Koo, Org. Lett., 2011, 13, 2682–2685 CrossRef CAS PubMed.
- Y.-L. Zhong and T. K. M. Shing, J. Org. Chem., 1997, 62, 2622–2624 CrossRef CAS PubMed.
- H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483–2547 CrossRef CAS.
-
(a) J. B. Shotwell and W. R. Roush, Org. Lett., 2004, 6, 3865–3868 CrossRef CAS PubMed;
(b) S. Sakamoto, H. Sakazaki, K. Hagiwara, K. Kamada, K. Ishii, T. Noda, M. Inoue and M. Hirama, Angew. Chem., Int. Ed., 2004, 43, 6505–6510 CrossRef CAS PubMed.
- P. A. Grieco, S. Gilman and M. Nishizawa, J. Org. Chem., 1976, 41, 1485–1486 CrossRef CAS.
-
(a) J. N. Denis and A. Krief, Tetrahedron Lett., 1979, 20, 3995–3996 CrossRef;
(b) H. Suzuki, N. Sato and A. Osuka, Chem. Lett., 1980, 9, 143–144 CrossRef;
(c) J. V. Allen, A. P. Green, S. Hardy, N. M. Heron, A. T. L. Lee and E. J. Thomas, Tetrahedron Lett., 2008, 49, 6352–6355 CrossRef CAS.
- For the synthesis of alkyl iodide 42, see the ESI.†.
- J. E. Baldwin, R. M. Adlington, D. J. Birch, J. A. Crawford and J. B. Sweeney, J. Chem. Soc., Chem. Commun., 1986, 1339–1340 RSC.
-
(a) K. Miura, Y. Ichinose, K. Nozaki, K. Fugami, K. Oshima and K. Utimoto, Bull. Chem. Soc. Jpn., 1989, 62, 143–147 CrossRef CAS;
(b) J. E. Baldwin, R. Fieldhouse and A. T. Russell, Tetrahedron Lett., 1993, 34, 5491–5494 CrossRef CAS;
(c) Y. Kita, A. Sano, T. Yamaguchi, M. Oka, K. Gotanda and M. Matsugi, Tetrahedron Lett., 1997, 38, 3549–3552 CrossRef CAS;
(d) E. Bacqué, F. Pautrat and S. Z. Zard, Org. Lett., 2003, 5, 325–328 CrossRef PubMed.
- L. C. Garcia, L. G. Donadío, E. Mann, S. Kolusheva, N. Kedei, N. E. Lewin, C. S. Hill, J. S. Kelsey, J. Yang, T. E. Esch, M. Santos, M. L. Peach, J. A. Kelley, P. M. Blumberg, R. Jelinek, V. E. Marquez and M. J. Comin, Bioorg. Med. Chem., 2014, 22, 3123–3140 CrossRef CAS PubMed.
-
(a) D. A. Evans, M. D. Ennis and D. J. Mathre, J. Am. Chem. Soc., 1982, 104, 1737–1739 CrossRef CAS;
(b) M. M. Heravi, V. Zadsirjan and B. Farajpour, RSC Adv., 2016, 6, 30498–30551 RSC.
- H. P. Pepper, S. J. Tulip, Y. Nakano and J. H. George, J. Org. Chem., 2014, 79, 2564–2573 CrossRef CAS PubMed.
- M. Movassaghi and O. K. Ahmad, J. Org. Chem., 2007, 72, 1838–1841 CrossRef CAS PubMed.
-
(a) G. A. Kraus and B. Roth, J. Org. Chem., 1980, 45, 4825–4830 CrossRef CAS;
(b) B. S. Bal, W. E. Childers and H. W. Pinnick, Tetrahedron, 1981, 37, 2091–2096 CrossRef CAS.
- I. Shiina, M. Kubota, H. Oshiumi and M. Hashizume, J. Org. Chem., 2004, 69, 1822–1830 CrossRef CAS PubMed.
- We investigated the sequence of the transannular RCM of 63 and removal of two MOM groups. The transannular RCM of 63 using a second-generation Hoveyda–Grubbs catalyst (65) gave the desired butenolide in 71% yield. Although we applied several reaction conditions in the deprotection of the resulting bis-MOM ether, we did not obtain the desired diol in reproducible yields. Therefore, we conducted the deprotection of bis-MOM ether 63.
-
(a) D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155–4156 CrossRef CAS;
(b) D. B. Dess and J. C. Martin, J. Am. Chem. Soc., 1991, 113, 7277–7287 CrossRef CAS.
- S. B. Garber, J. S. Kingsbury, B. L. Gray and A. H. Hoveyda, J. Am. Chem. Soc., 2000, 122, 8168–8179 CrossRef CAS.
- See the ESI for details.†.
- Y. Kitano, T. Ito, T. Suzuki, Y. Nogata, K. Shinshima, E. Yoshimura, K. Chiba, M. Tada and I. Sakaguchi, J. Chem. Soc., Perkin Trans. 1, 2002, 2251–2255 RSC.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Yuki
Sugitani
a,
Ryohei
Morishita
a,
Takefumi
Yorisue
*a,
Yuki
Sugitani
a,
Ryohei
Morishita
a,
Takefumi
Yorisue