
Shining light for organophotocatalysed site-selective sulfonylation of anilides†‡
Swati
Singh
 ,
Gopal
Chakrabortty
,
Kajal
Tiwari
,
Neha
Dagar
and
Sudipta
Raha Roy
*
,
Gopal
Chakrabortty
,
Kajal
Tiwari
,
Neha
Dagar
and
Sudipta
Raha Roy
*
Department of Chemistry, Indian Institute of Technology Delhi, Hauz Khas, New Delhi, 110016, India. E-mail: srr@chemistry.iitd.ac.in; Tel: (+91) 11-2659-7954
First published on 22nd August 2024
Abstract
The site-selective sulfonylation of C(sp2)–H bonds of anilide and quinoline amide derivatives has been developed using organophotocatalysis. This mild and sustainable protocol, which operates at room temperature, precludes the requirement for any metal-based catalyst or photocatalyst and oxidant, which are the challenges associated with existing methodologies. Furthermore, the generation of aryl sulfonyl radicals from commercially available aryl sulfonyl chlorides has been achieved through the use of Rose Bengal as an organophotocatalyst, an approach that was previously unexplored. The detailed mechanistic investigation unveiled the underlying mechanism for site-selective sulfonylation at both the proximal and distal positions, thereby establishing a straightforward approach for building valuable aryl sulfone scaffolds.
Introduction
Inclusion of sulfonyl moieties into organic scaffolds is a persistent challenge to the synthetic community owing to their ubiquitous presence in bioactive molecules.1 The sulfonylation of aromatic compounds, which has traditionally been accomplished through established Friedel–Crafts-type reactions, remains a subject of ongoing interest.2 As a result, considerable endeavors have been devoted to enhancing the sulfonylation protocol through the utilization of diverse sulfonyl precursors (Scheme 1a).3 Nevertheless, the generation of sulfonyl radicals continues to require the application of oxidants and high temperatures.4 In the era of the prevailing inclination towards sustainable and environmentally conscious methodologies, photo-redox catalysis presents itself as a potentially viable solution to address certain challenges by offering substantial benefits in terms of controllability, cost-effectiveness, and environmental compatibility.5 In fact, sulfonyl radicals could be produced through direct photoreduction or homolytic sulfur–halogen bond cleavage using readily available, inexpensive precursors such as sulfonyl chlorides.6 Despite the extensive investigation of synthetic methodologies that utilize metallophotoredox catalysis to directly activate sulfonyl chlorides, there is still a need to address the challenges associated with the identification of suitable organophotocatalytic systems that can efficiently and sustainably dissociate sulfur–halogen bonds through homolytic processes.6f Furthermore, the existence of synthetic challenges becomes evident when considering the site-selective sulfonylation of aniline derivatives.7 The task is challenging due to the necessity of navigating sulfonyl radicals towards the aromatic C–H bonds of aniline and, at the same time, impeding the sulfonylation of competing N–H bonds while preserving proximal and distal site-selectivity.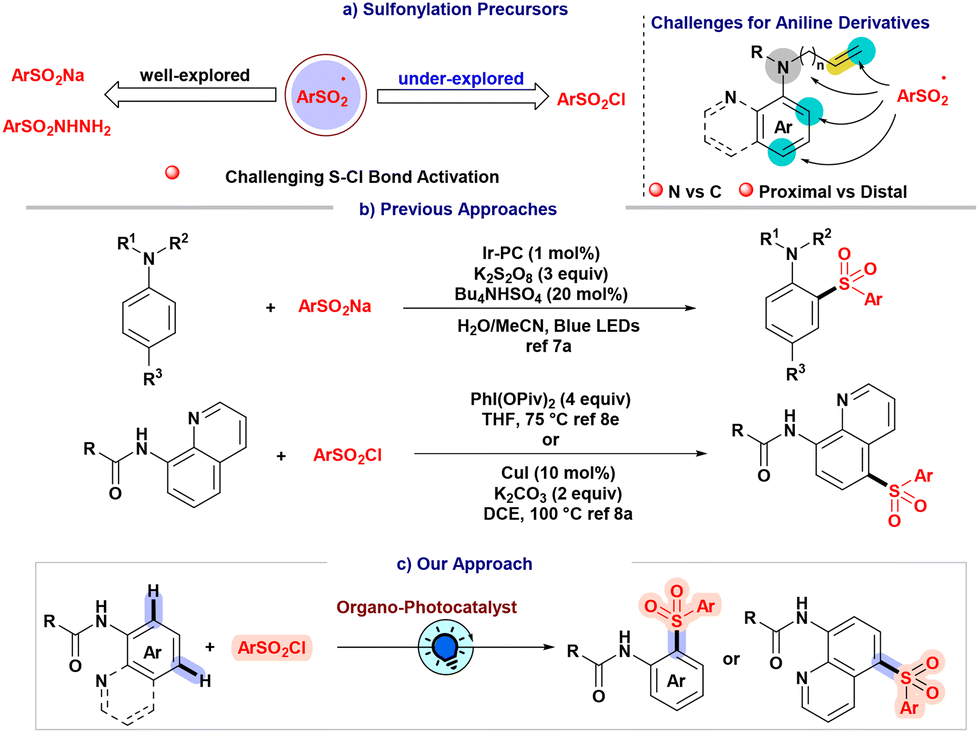 | ||
| Scheme 1 Photoinduced sulfonylation of anilides and the reported approaches. | ||
We postulated that the installation of the acyl group on the aniline compound could diminish the reactivity of aniline, restraining it from directly participating in nucleophilic substitution with sulfonyl chloride, which results in a change from N–H to C–H bond selectivity of the sulfonyl radical and, therefore, increases the possibility of directly attaching the sulfonyl radical to the aromatic ring. Although our strategy to achieve selectivity is encouraging, site-selectivity remains a subject of contention.
Earlier, Wu and co-workers developed a sulfonylation strategy of quinoline amide with sulfonyl chloride utilizing a copper catalyst at elevated temperature (Scheme 1b).8a The same group further disclosed a sulfonylation reaction with directing group-assisted naphthyl amide derivatives utilizing two metal catalyst systems along with a stoichiometric amount of an oxidant using sulfinate salts.8d Another group utilized 4 equivalents of hypervalent iodine at elevated temperatures for the sulfonylation of anilides and quinoline amides (Scheme 1b).8e In the sulfonylated aniline protocols, the Willis group designed an elegant strategy for the site-selective sulfonylation of aniline using a transition metal-based photocatalyst and an inorganic oxidant.7a Therefore, precedent reports on sulfonylation protocols of anilide moieties have described utilizing precious transition metal systems, stoichiometric amounts of radical initiators, and high temperatures that lead toward identifying alternate sulfonyl radical precursors and chemical strategies (Scheme 1b).8 In order to circumvent the inherent challenges associated with the transition metal catalyst systems herein, we implemented a milder organophotocatalytic strategy for site-selective sulfonylation of anilides using aryl sulfonyl chlorides as sulfonyl radical precursors (Scheme 1c).
Results and discussion
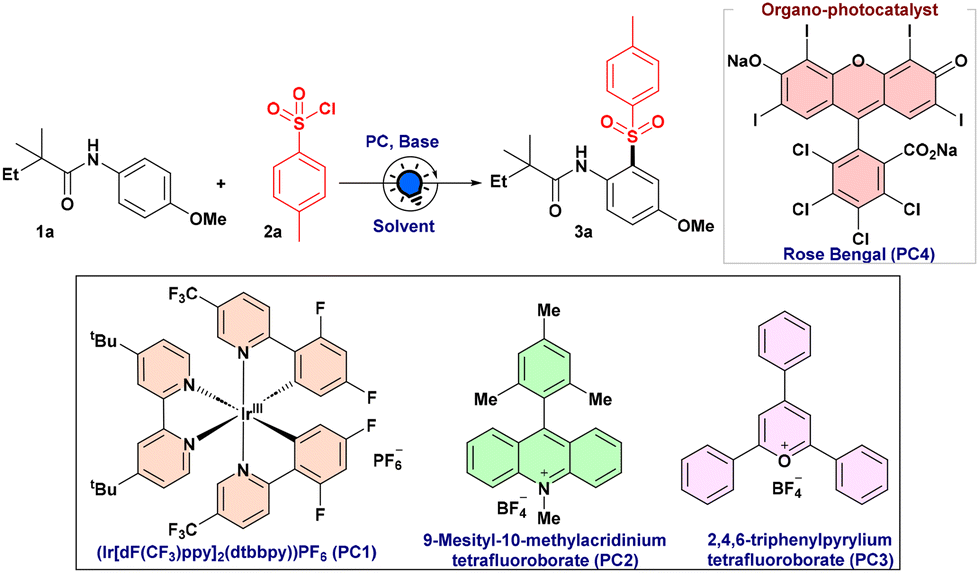
In pursuit of establishing a sulfonylation protocol, a model substrate of N-(4-methoxyphenyl)-2,2-dimethylbutanamide (1a) was used in the presence of 4-methylbenzene sulfonyl chloride (2a) in DCE under 440 nm photoirradiation. At first, we studied a series of commercially available photocatalysts. Encouragingly, with [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (PC1), sulfonylated product 3a was obtained in 55% yield as a single regioisomer (Table 1, entry 1). While performing the reaction with other organocatalysts, MesAcr+BF4− (PC2) and 2,4,6-triphenylpyrylium tetrafluoroborate (PC3), the efficiency of the reaction decreased significantly (Table 1, entries 2 and 3). To our delight, Rose Bengal (PC4) was found to be an effective photocatalyst, delivering the desired product 3a in 86% yield (Table 1, entry 4). Furthermore, we found that KOAc and DIPEA both provided inferior results (Table 1, entry 5). Subsequently, Cs2CO3 turned out to be an appropriate base for this sulfonylation reaction, producing a 92% yield of the desired product 3a (Table 1, entry 6). The fact that acetate is less basic than the carbonate base might be a contributing factor. In contrast, DIPEA can undergo oxidation and engage in electron transfer with Rose Bengal. It was observed that the yield of 3a was reduced significantly when CH3CN, tBuOH and EtOAc were used as solvents instead of DCE (Table 1, entries 7 and 8). As anticipated, the absence of a photocatalyst, a base, or aerobic conditions had an adverse effect on our expected product formation (Table 1, entries 9–11).|
|
|||
|---|---|---|---|
| Entry | Catalyst | Base | Yielda,b (3a %) |
| a Reaction conditions: unless otherwise specified, 1a (0.2 mmol), 2a (0.3 mmol), PC (2 mol%), and Cs2CO3 (0.24 mmol) in 2 mL DCE irradiated with a Kessil blue LED (440 nm) for 24 h under an argon atmosphere. b Yields determined by gas chromatography using benzophenone as the internal standard. c CH3CN is used as a solvent. d t BuOH is used as a solvent. e EtOAc is used as a solvent. f Reaction was performed under aerobic conditions. | |||
| 1 | PC1 | K2CO3 | 55 |
| 2 | PC2 | K2CO3 | Trace |
| 3 | PC3 | K2CO3 | n.r. |
| 4 | PC4 | K2CO3 | 86 |
| 5 | PC4 | KOAc/DIPEA | Trace |
| 6 | PC4 | Cs 2 CO 3 | 92 |
| 7c,d | PC4 | Cs2CO3 | Trace |
| 8e | PC4 | Cs2CO3 | 40 |
| 9 | PC4 | — | n.r. |
| 10 | — | Cs2CO3 | n.r. |
| 11f | PC4 | Cs2CO3 | n.r. |
Keeping the optimized conditions in mind, we set out to explore the scope of sulfonyl chlorides for the sulfonylation reaction of anilides. Electron-donating group (such as –Me, –OMe, and –Et) substituted aryl sulfonyl chlorides performed well under our developed photochemical conditions and generated sulfonylated products (3a–c) (Scheme 2).
Also, the electron-withdrawing halo groups at the para- and meta-positions were well tolerated, giving good yields of the expected products (3d–f). Moreover, polyaromatic ring containing sulfonyl chlorides provided the desired product 3g, with a good yield. When we carried out the reaction of a thiophene-containing sulfonyl chloride, to our delight, we got the sulfonylated product 3h with an excellent yield. Next, we varied the alkyl group of anilide derivatives and performed the reaction with different aryl sulfonyl chlorides. Isopropyl, tert-butyl, and 2,2-dimethyl butyl group containing anilides were competent substrates for delivering the corresponding sulfonylated products (3i–k). In addition, X-ray crystallography and NMR analysis pinpointed the sulfonyl group on the aromatic ring and validated the formation of 3i (CCDC 2333703‡) through proximal sulfonylation. Additionally, when we checked the viability of isopropyl sulfonyl chloride as an alkyl sulfonyl source, we were unable to produce our desired product 3l. However, our optimized conditions allowed us to use cyclohexyl-containing anilide to access aromatic sulfone 3m in a moderate yield. When we performed the reaction with 2-phenylacetanilide and acetanilide derivatives, we got our expected products (3n and 3o) with good yields. Moreover, anilide derivatives containing unsaturated bonds were found to be amenable under this sulfonylation protocol, giving excellent yields of site-selective sulfonylated products (3p and 3q) exclusively, and we did not observe any intramolecular radical cyclized product or hydrosulfonylation of the olefinic bond.6c,9 We showed that this strategy could be expanded by conducting the reaction with various substitutions on the aromatic ring of anilide derivatives, and notably, proximal sulfonylated products were formed exclusively (3r–x). To our delight, we were able to synthesize the anticipated sulfonylated products (3r–t) after substituting with methyl and tert-butyl groups without affecting the reaction yield, even when the ortho position of the anilide was substituted with a methyl group. Next, we explored the scope of substitution on the anilide derivatives for the photoinduced sulfonylation reaction to demonstrate functional group tolerance. It was noteworthy that both isopropoxy and aryloxy substituted anilides were tolerated under these conditions and gave the corresponding proximal sulfonylated products (3u and 3v) in good yields (Scheme 2). Furthermore, thioether reacted well in our developed protocol, providing 63% yield of sulfone derivative 3w.
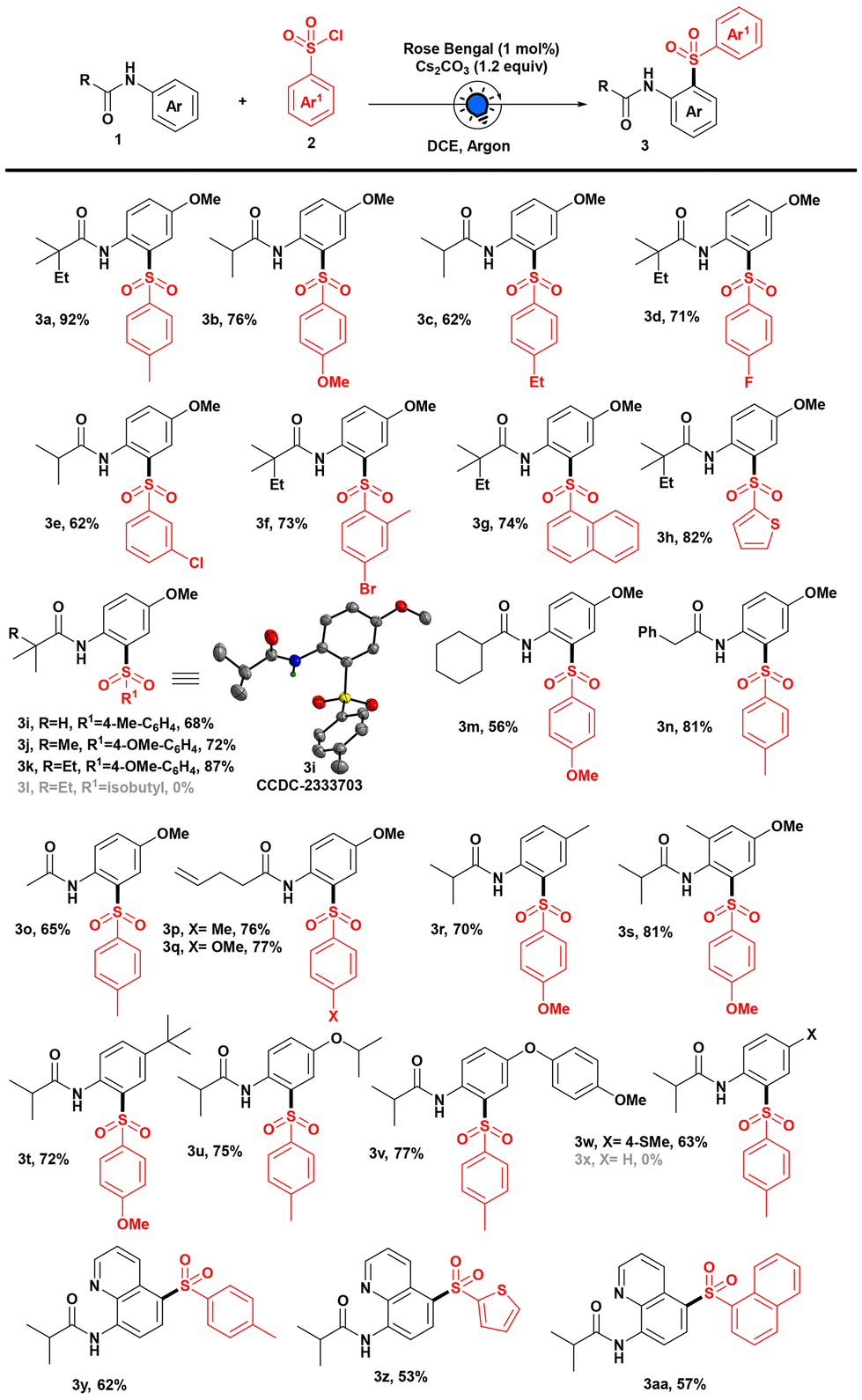 | ||
| Scheme 2 Substrate scope for the site-selective sulfonylation. Reaction conditions: unless otherwise specified, 1 (0.2 mmol), 2 (0.3 mmol), Rose Bengal (2 mol%, 2 mg), and Cs2CO3 (0.24 mmol) in 2 mL DCE irradiated with a blue LED (440 nm) for 24 h under an argon atmosphere. | ||
Subsequently, in an effort to gain a more comprehensive understanding of the site-selective outcome of the reaction, we attempted to execute it using N-phenylisobutyramide; unfortunately, we did not observe the formation of the desired product 3x, and the starting material was fully recovered (for other unsuccessful substrates, see the ESI‡). As a result, we sought to deploy our model reaction for the site-selective installation of sulfone on medicinally important quinoline scaffolds. In fact, to check the viability of quinoline amide, when we performed the reaction with various aryl sulfonyl chlorides, we got the corresponding distal C-5 aryl sulfones (3y–aa) exclusively in good yields. Therefore, to understand the proximal and distal sulfonylation of anilide derivatives, we performed computational studies (see the ESI‡). The formation of electrophilic ArSO2˙ through the cleavage of the SVI–Cl bond enables subsequent reactions with anilides.10 Consequently, to gain insight into the electron density of the anilide derivative for the addition of the electrophilic sulfonyl radical, we computed the charge density of the optimized structure N-(4-methoxyphenyl)isobutyramide (1i) and N-(quinolin-8-yl)isobutyramide (1y) using the B3LYP method with the 6-31+G(d,p) basis set considering SMD solvation in DCE. It was observed that the electron density is notably higher at the C2 position in the case of 1i, leading to proximal sulfonylation. In contrast, the electron density was found to be significantly higher at the C5 position of quinoline amide (1y), resulting in distal sulfonylation of the delivered C5 sulfonylated product (3y–3aa).
To understand this photoinduced process better, when we carried out the model reaction with a radical scavenger BHT, formation of 3a was suppressed completely (Scheme 3), and the sulfonyl adduct of BHT 4 was detected by HRMS, confirming the radical intermediacy of the reaction. Furthermore, the sulfonyl radical was trapped with a diphenyl ethylene moiety, which was confirmed by HRMS. We also synthesized N-sulfonylated anilide 6 separately and subjected it to our reaction, but the desired product 3a was not formed, which ruled out the in situ photo-Fries rearrangement for the site-selective sulfonylation on the aryl ring of the anilide.11
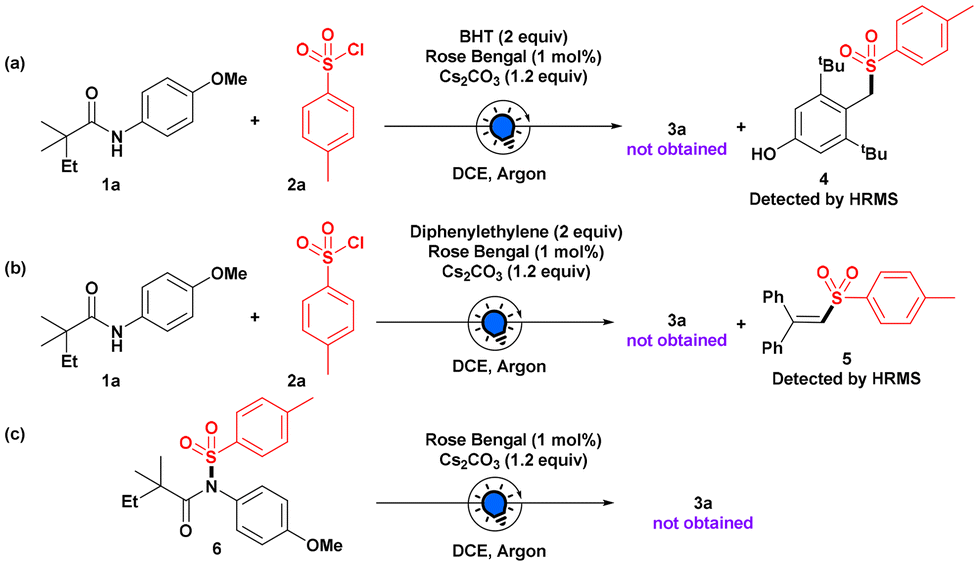 | ||
| Scheme 3 Control Experiments. | ||
In order to ascertain the exact involvement of the photocatalyst at the excited state during this photoinduced sulfonylation process, we conducted a fluorescence lifetime decay study with a 485 nm laser source (Fig. 1). The amplitude-weighted average fluorescence lifetime (τav) of PC4 in EtOAc was recorded at 2.53 ns. When we studied the lifetime decay of PC4 after the sequential addition of 2a and 1a separately, in fact, with both of these quenchers, we observed a gradual decrease in the τav value (Fig. 1A and B). However, the associated Stern–Volmer plot indicated the degree of dynamic quenching, with 2a greater than 1a (Fig. 1C). Therefore, it might be anticipated that aryl sulfonyl chloride was involved in the dynamic quenching with the excited photocatalyst. Furthermore, the significance of continuous visible-light irradiation was confirmed by light on/off studies, and the graph depicted in Fig. 1D shows the necessity of light irradiation for the sulfonylation reaction.
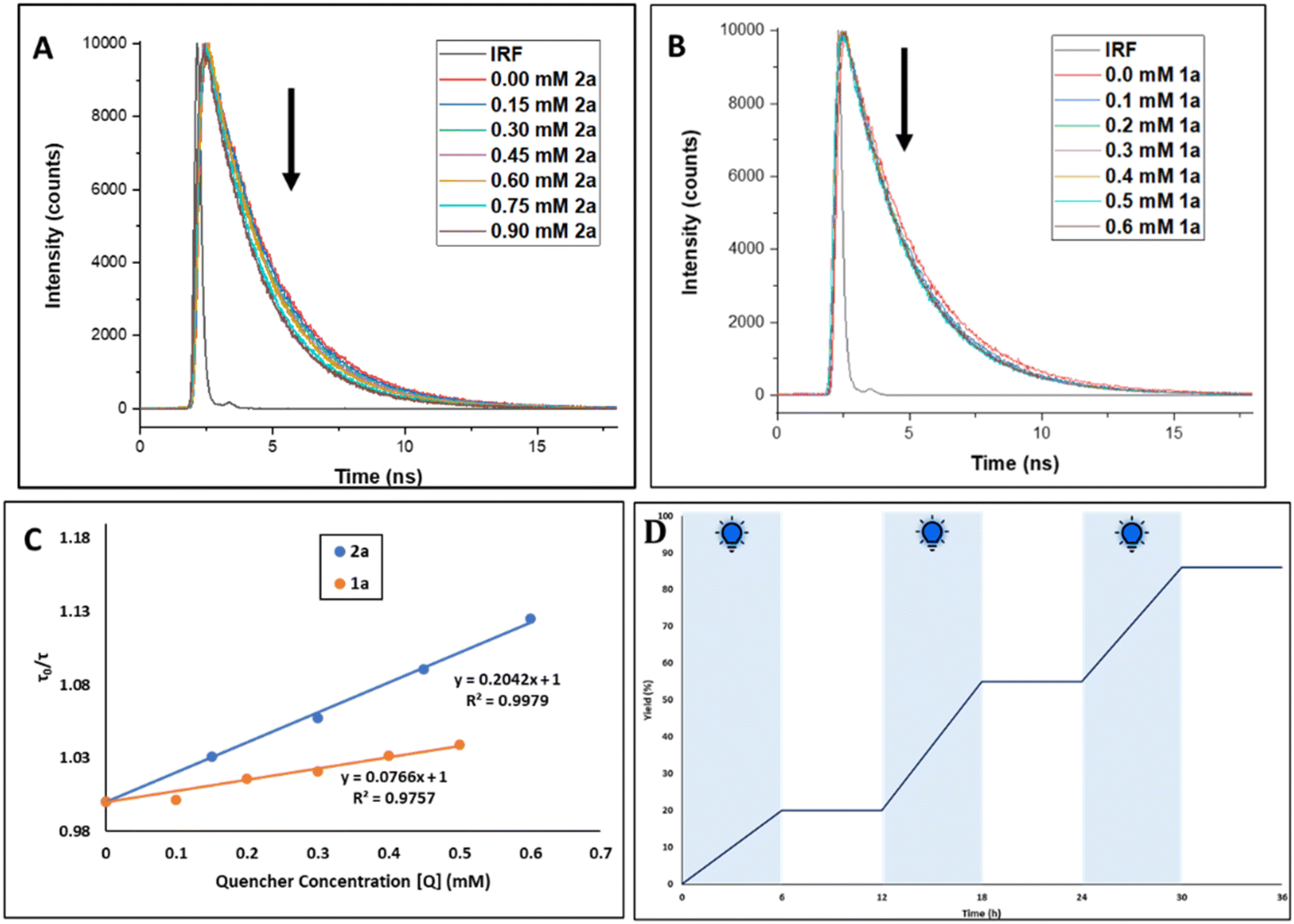 | ||
| Fig. 1 (A) Fluorescence lifetime quenching studies for 3.75 mM PC4 in ethyl acetate with increasing concentration of 2a as the quencher (addition from 0 to 0.9 mM). (B) Fluorescence lifetime quenching studies for 3.75 mM PC4 in ethyl acetate with increasing concentration of 1a as the quencher (addition from 0 to 0.6 mM). (C) Combined Stern–Volmer plot of a solution of 3.75 mM PC4 in ethyl acetate with increasing concentration of 1a (orange line) and 2a (blue line) as the quencher. (D) Switch on/off experiment. | ||
Based on the aforementioned observations and literature precedents, a plausible reaction path for the sulfonylation strategy is depicted in Scheme 4. Initially, RB when exposed to light produced the excited state RB*, which may be involved in the quenching with 1i and 2a, respectively, as evidenced by fluorescence lifetime quenching studies. However, after comparing the redox potential of anilide 1i with RB* (for details, see the ESI‡), it was observed that reductive quenching of RB is not feasible. Furthermore, the oxidative quenching pathway is the most promising reaction pathway since our technique only works with aryl sulfonyl chloride, as the S–Cl bond dissociation energy of alkyl sulfonyl chloride is somewhat higher than that of aryl sulfonyl chloride12 and perhaps because of this, we were unable to produce our desired product 3l utilizing isopropyl sulfonyl chloride as an alkyl sulfonyl source. Thus, after photoexcitation, RB* (ERB·+/RB* = −1.34 V vs. SCE) is involved in oxidative quenching with 2a (Ered = −1.25 V vs. SCE) to produce sulfonyl radical I, with RB* converting into RB˙+. Direct addition of radical I to the nucleophilic center of anilide 1i produces radical intermediate II. Then intermediate II was involved in the SET process with RB˙+ to regenerate RB and get transformed into the carbocation intermediate III, followed by deprotonation with the help of chloride ion to produce the required sulfonylated product 3i.
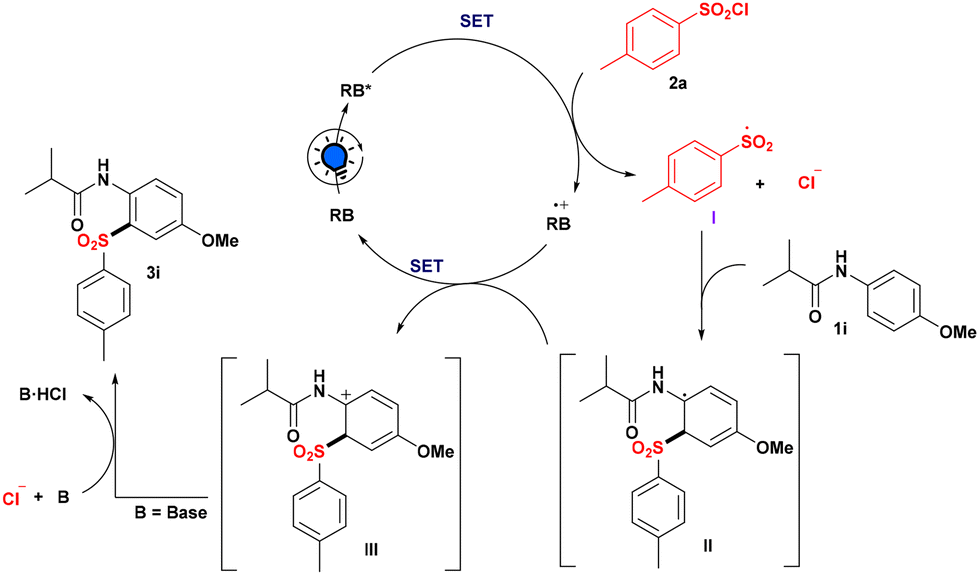 | ||
| Scheme 4 Plausible Reaction Mechanism. | ||
Conclusions
In conclusion, we developed a site-selective photochemical approach for aryl sulfonylation of direct C(sp2)–H bonds of anilide derivatives utilizing easily accessible aryl sulfonyl chlorides as a sulfonylating agent. We generated sulfonyl radicals using a less expensive organophotocatalyst. The extensive scope evaluation illustrated the synthetic utility with anilides, sulfonyl chlorides, and biologically relevant heterocycle-containing anilides. Detailed mechanistic experiments and computational studies were conducted to elucidate the mechanism and exclusive site-selectivity.Data availability
The data supporting this article have been included as part of the ESI. All experimental data, detailed procedures, NMR spectra and characterization data for all compounds can be found in the ESI.‡Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
S. R. R. thanks CSIR (02(0416)/21/EMR-II) and SERB (CRG/2022/002306) for financial support. S. S. and N. D. thank CSIR, New Delhi, for the research fellowship. G. C. and K. T. thank UGC, New Delhi, for the research fellowship. The authors acknowledge CRF, IIT Delhi for instrument facilities.References
- (a) W. Tadrent, A. Alabdul Magid, A. Kabouche, D. Harakat, L. Voutquenne-Nazabadioko and Z. Kabouche, Nat. Prod. Res., 2017, 31, 1437–1445 CrossRef CAS PubMed; (b) C. Zhao, K. P. Rakesh, L. Ravidar, W.-Y. Fang and H.-L. Qin, Eur. J. Med. Chem., 2019, 162, 679–734 CrossRef CAS PubMed; (c) U. Lücking, Org. Chem. Front., 2019, 6, 1319–1324 RSC; (d) M. Mustafa and J.-Y. Winum, Expert Opin. Drug Discovery, 2022, 17, 501–512 CrossRef CAS PubMed.
- (a) G. A. Olah, S. Kobayashi and J. Nishimura, J. Am. Chem. Soc., 1973, 95, 564–569 CrossRef CAS; (b) K. Bahrami, M. M. Khodei and F. Shahbazi, Tetrahedron Lett., 2008, 49, 3931–3934 CrossRef CAS; (c) A. Falconnet, J.-D. Arndt, A. S. K. Hashmi and T. Schaub, Eur. J. Org. Chem., 2022, 2022, e202200477 CrossRef CAS.
- (a) S. Liang, K. Hofman, M. Friedrich and G. Manolikakes, Eur. J. Org. Chem., 2020, 2020, 4664–4676 CrossRef CAS; (b) F.-L. Yang and S.-K. Tian, Tetrahedron Lett., 2017, 58, 487–504 CrossRef CAS; (c) G. Qiu, K. Zhou and J. Wu, Chem. Commun., 2018, 54, 12561–12569 RSC; (d) D.-Q. Dong, Q.-Q. Han, S.-H. Yang, J.-C. Song, N. Li, Z.-L. Wang and X.-M. Xu, ChemistrySelect, 2020, 5, 13103–13134 CrossRef CAS.
- D. Joseph, M. A. Idris, J. Chen and S. Lee, ACS Catal., 2021, 11, 4169–4204 CrossRef CAS.
- (a) R. C. McAtee, E. J. McClain and C. R. J. Stephenson, Trends Chem., 2019, 1, 111–125 CrossRef CAS PubMed; (b) L. Guillemard and J. Wencel-Delord, Beilstein J. Org. Chem., 2020, 16, 1754–1804 CrossRef CAS PubMed; (c) S. Singh, V. J. Roy, N. Dagar, P. P. Sen and S. R. Roy, Adv. Synth. Catal., 2021, 363, 937–979 CrossRef CAS.
- (a) S. K. Pagire, A. Hossain and O. Reiser, Org. Lett., 2018, 20, 648–651 CrossRef CAS PubMed; (b) S. K. Pagire, N. Kumagai and M. Shibasaki, Org. Lett., 2020, 22, 7853–7858 CrossRef CAS PubMed; (c) S. M. Hell, C. F. Meyer, A. Misale, J. B. I. Sap, K. E. Christensen, M. C. Willis, A. A. Trabanco and V. Gouverneur, Angew. Chem., Int. Ed., 2020, 59, 11620–11626 CrossRef CAS PubMed; (d) S. Cao, D. Kim, W. Lee and S. Hong, Angew. Chem., Int. Ed., 2023, 62, e202312780 CrossRef CAS PubMed; (e) X. Wu, W. Zhang, G. Sun, X. Zou, X. Sang, Y. He and B. Gao, Nat. Commun., 2023, 14, 5168 CrossRef CAS PubMed; (f) C. He, M. Wang, B. Dong, Y. Su, X.-H. Duan and L. Liu, Green Synth. Catal., 2024, 5, 117–121 CrossRef CAS.
- (a) T. C. Johnson, B. L. Elbert, A. J. M. Farley, T. W. Gorman, C. Genicot, B. Lallemand, P. Pasau, J. Flasz, J. L. Castro, M. MacCoss, D. J. Dixon, R. S. Paton, C. J. Schofield, M. D. Smith and M. C. Willis, Chem. Sci., 2018, 9, 629–633 RSC; (b) J. Nikl, D. Ravelli, D. Schollmeyer and S. R. Waldvogel, ChemElectroChem, 2019, 6, 4450–4455 CrossRef CAS; (c) X.-Q. Li, Q.-Q. Liao, J. Lai and Y.-Y. Liao, Front. Chem., 2023, 11 DOI:10.3389/fchem.2023.1267223.
- (a) H. Qiao, S. Sun, F. Yang, Y. Zhu, W. Zhu, Y. Dong, Y. Wu, X. Kong, L. Jiang and Y. Wu, Org. Lett., 2015, 17, 6086–6089 CrossRef CAS PubMed; (b) H.-W. Liang, K. Jiang, W. Ding, Y. Yuan, L. Shuai, Y.-C. Chen and Y. Wei, Chem. Commun., 2015, 51, 16928–16931 RSC; (c) J.-M. Li, J. Weng, G. Lu and A. S. C. Chan, Tetrahedron Lett., 2016, 57, 2121–2124 CrossRef CAS; (d) P. Bai, S. Sun, Z. Li, H. Qiao, X. Su, F. Yang, Y. Wu and Y. Wu, J. Org. Chem., 2017, 82, 12119–12127 CrossRef CAS PubMed; (e) Y. Wang, Y. Wang, Q. Zhang and D. Li, Org. Chem. Front., 2017, 4, 514–518 RSC; (f) S. Liang, M. Bolte and G. Manolikakes, Chem. – Eur. J., 2017, 23, 96–100 CrossRef CAS PubMed; (g) Y. Sun, C. Feng, P. Wang, F. Yang and Y. Wu, Org. Chem. Front., 2021, 8, 5710–5715 RSC.
- (a) S. M. Hell, C. F. Meyer, A. Misale, J. B. I. Sap, K. E. Christensen, M. C. Willis, A. A. Trabanco and V. Gouverneur, Angew. Chem., Int. Ed., 2020, 59, 11620–11626 CrossRef CAS PubMed; (b) T. Zhong, J. T. Yi, Z. Da Chen, Q. C. Zhuang, Y. Z. Li, G. Lu and J. Weng, Chem. Sci., 2021, 12, 9359–9365 RSC.
- (a) F. De Vleeschouwer, V. Van Speybroeck, M. Waroquier, P. Geerlings and F. De Proft, Org. Lett., 2007, 9, 2721–2724 CrossRef CAS PubMed; (b) B. Lipp, L. M. Kammer, M. Kücükdisli, A. Luque, J. Kühlborn, S. Pusch, G. Matulevičiūtė, D. Schollmeyer, A. Šačkus and T. Opatz, Chem. – Eur. J., 2019, 25, 8965–8969 CrossRef CAS PubMed.
- (a) K. K. Park, J. J. Lee and J. Ryu, Tetrahedron, 2003, 59, 7651–7659 CrossRef CAS; (b) E. Torti, S. Protti, D. Merli, D. Dondi and M. Fagnoni, Chem. – Eur. J., 2016, 22, 16998–17005 CrossRef CAS PubMed.
- R. J. O'Reilly and M. Balanay, Chem. Data Collect., 2019, 19, 100180 CrossRef.
Footnotes |
| † Dedicated to Prof. Brindaban C. Ranu on the occasion of his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available. 1H and 13C NMR spectra of 3 and CCDC 2333703. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob01169d |
| This journal is © The Royal Society of Chemistry 2024 |