DOI:
10.1039/D3PY01011B
(Paper)
Polym. Chem., 2024,
15, 40-45
Ligand pre-inserted α-diimine palladium catalysts and mechanism studies in olefin polymerization†
Received
6th September 2023
, Accepted 29th November 2023
First published on 30th November 2023
Abstract
Since the pioneering works of Brookhart and co-workers, extensive research efforts have been directed towards the development of high-performance late-transition-metal-based olefin polymerization catalysts. In this contribution, we designed α-diimine ligands bearing an allyl group and the ligand pre-inserted palladium catalysts Pd1-ACN and Pd2-ACN were generated. However, they demonstrated different behaviors due to their different steric hindrance, producing Pd1 and Pd2 intermediates, respectively. The palladium catalysts were utilized in ethylene polymerization and copolymerization. Specifically, Pd2-ACN with its larger steric hindrance exhibited higher activity, affording polyethylene with high molecular weight. Enhanced catalytic performance was also observed in ethylene copolymerization with acrylate-type monomers, demonstrating good polar group tolerance. To further understand this system, DFT calculations and mechanistic studies of the coordination–insertion process were conducted, indicating the formation of the Pd–H catalytically active species during the polymerization process. The pre-inserted α-diimine catalysts in this work demonstrated a unique form in olefin polymerization and provided some new information for catalyst design.
Introduction
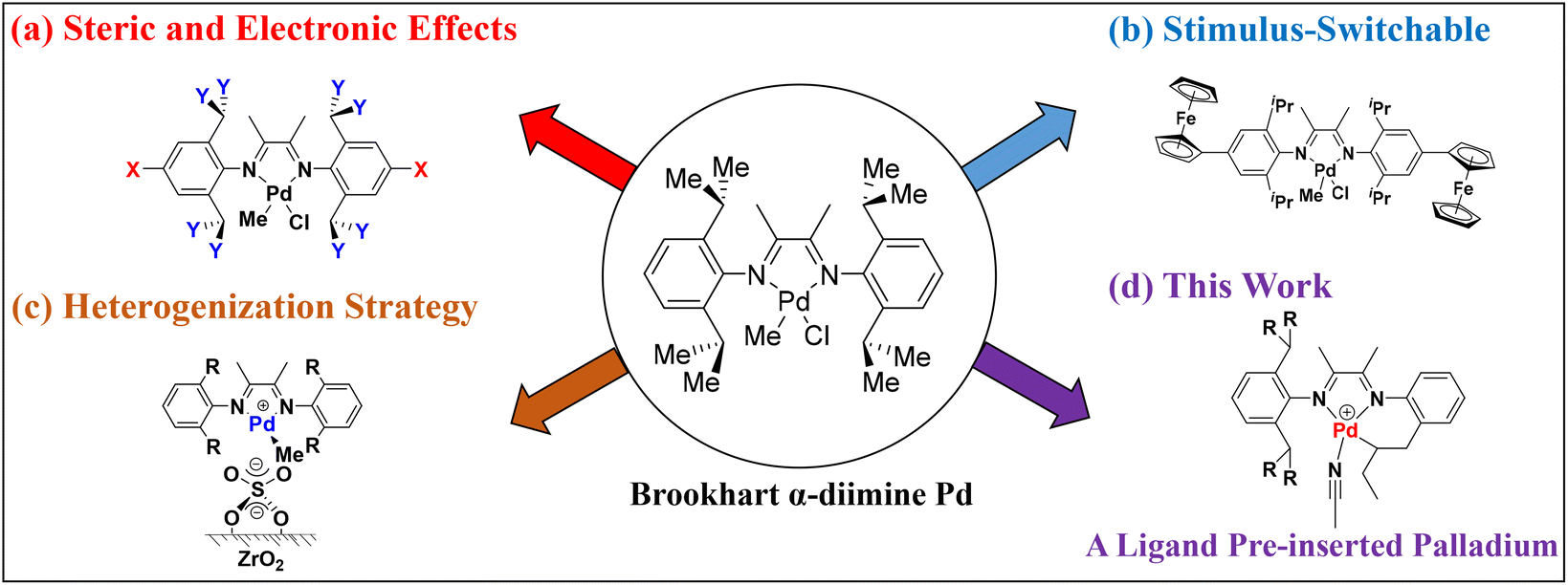
Polyolefins are some of the major synthetic polymers produced and consumed worldwide.1 In recent years, the design and development of high-performance olefin polymerization catalysts have attracted great attention from academia and industry.2–19 Inspired by the pioneering works of Brookhart and co-workers,20,21 late transition metal α-diimine palladium catalysts provide a method to synthesize branched polyolefins, benefitting from their unique “chain walking” behavior.22–24 These catalysts also promote the tolerance of polar groups, enabling the copolymerization of ethylene and polar monomers.25–31 The catalytic performance is usually influenced by the nature of α-diimine ligand structures and leads to polymers with different branching characteristics and microstructures. Thus, extensive research efforts are involved in designing various α-diimine palladium catalysts for olefin polymerization. For instance, electronic or steric tuning via different ligand substituents is the most widely practiced strategy (Fig. 1a).32–48 In addition, some alternative design strategies have emerged for the control of olefin polymerization and copolymerization processes, including a facile stimulus-switchable strategy (Fig. 1b)49–54 and a heterogenization strategy (Fig. 1c),55,56 affording a series of α-diimine palladium catalysts with unique catalytic properties.
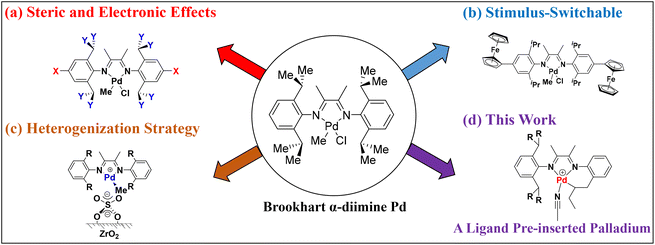 |
| | Fig. 1 Different strategies to design unique α-diimine palladium catalysts. | |
Therefore, the development of a catalyst will always be a frontier scientific problem in this field, influencing the olefin polymerization process through catalyst structural design. As part of our continuous efforts in this area, we wish to explore novel α-diimine palladium catalysts to modulate olefin (co)polymerization. In this contribution, we designed α-diimine ligands bearing a reactive allyl group to prepare unique ligand pre-inserted palladium catalysts (Fig. 1d). The objective was to investigate if the pre-inserted palladium catalyst could initiate olefin polymerization and explore the exact catalytically active species during the polymerization process. This work will demonstrate a unique form of olefin polymerization and provide some new information for catalyst design.
Results and discussion
Ligand and catalyst synthesis
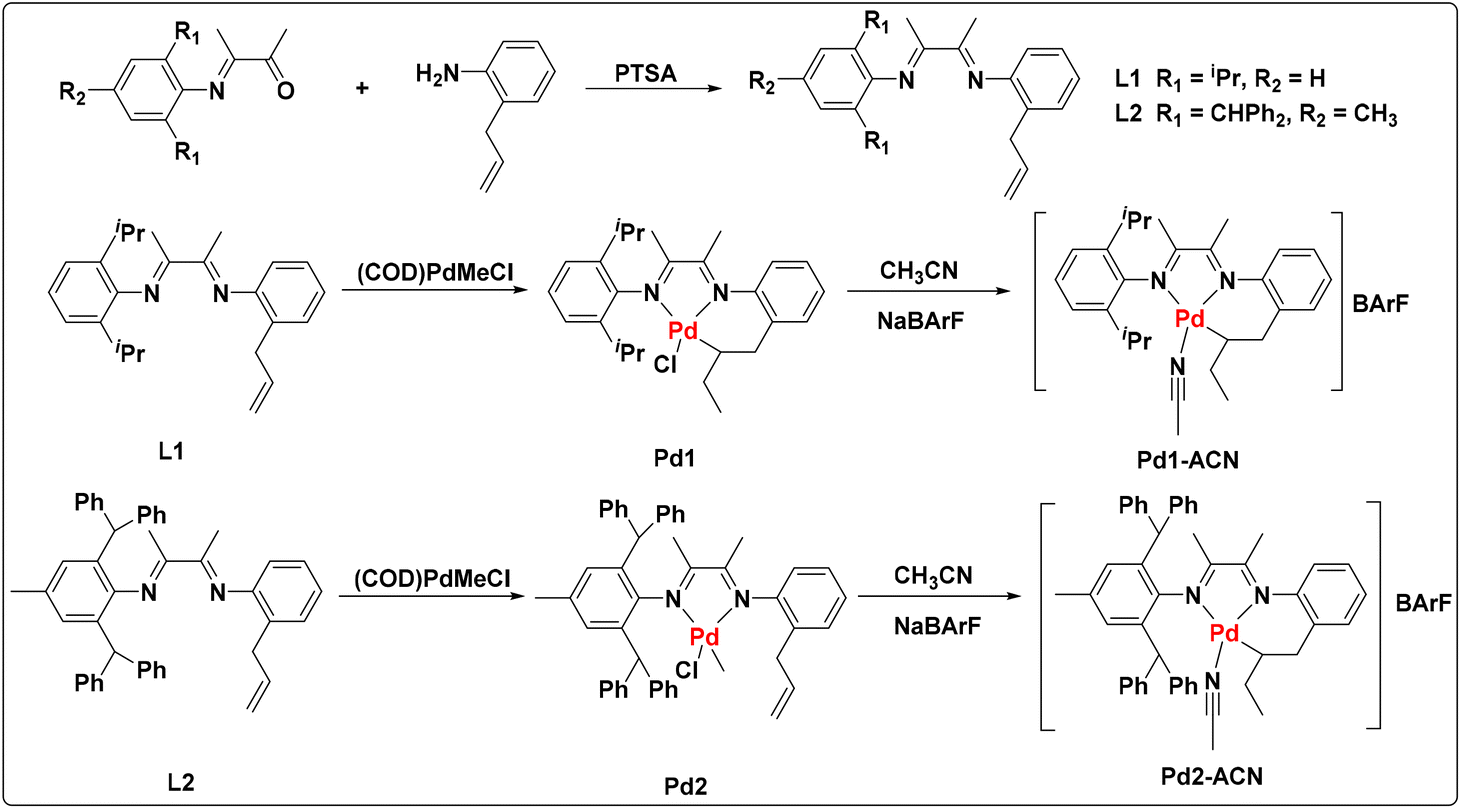
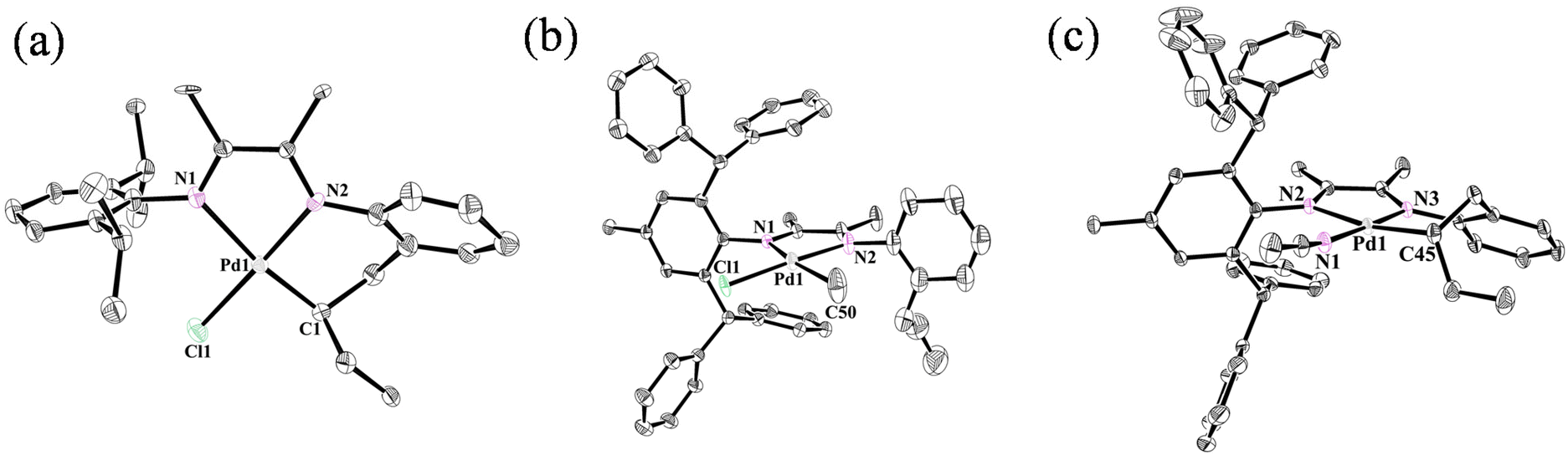
Ligands L1 and L2 were easily prepared starting from α-iminoketone and 2-allylaniline through condensation reactions in high yield (Fig. 2) catalyzed by p-toluenesulfonic acid (PTSA). The reaction of (COD)PdMeCl (COD = 1,5-cyclooctadiene) with ligands afforded the palladium complexes Pd1 and Pd2 bearing different steric hindrance. In order to improve the straightforwardness of subsequent polymerization studies, the single-component cationic palladium species (Pd1-ACN and Pd2-ACN) were generated by reacting Pd–Cl with sodium tetrakis(3,5-bis(trifluoromethyl)phenyl)borate (NaBArF) in the presence of MeCN. Single crystals of Pd1 (Fig. 3a), Pd2 (Fig. 3b) and Pd2-ACN (Fig. 3c) were grown from mixtures of dichloromethane/hexane and their molecular structures are shown in Fig. 3. These indicate that the Pd centers adopt a distorted square-planar geometry.
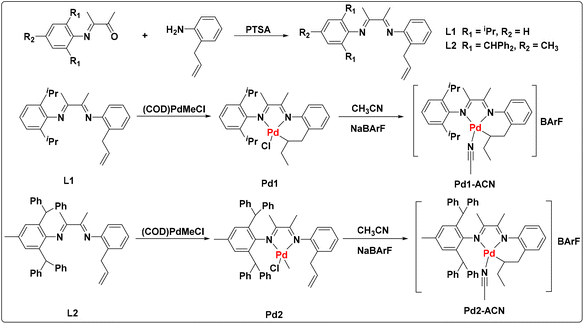 |
| | Fig. 2 Synthesis of ligands and the corresponding palladium complexes. | |
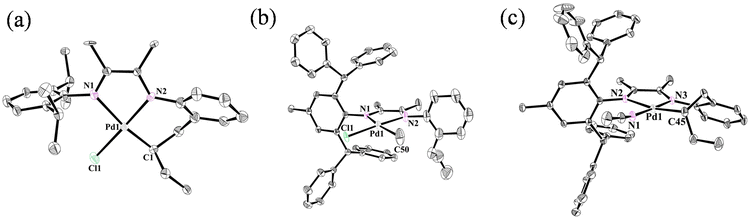 |
| | Fig. 3 Molecular structures of Pd1, Pd2 and Pd2-ACN (CCDC numbers, 2266075, 2266073, 2266074). Hydrogen atoms and the anion are omitted for clarity. The ellipsoids are set at 30% probability. Selected bond lengths (Å) and angles (°): Pd1 (a) Pd1–N1 2.190 (18), Pd1–N2 2.038 (19), Pd1–Cl1 1.94 (2), Pd1–C1 2.299 (6), N1–Pd1–Cl1 94.5 (8), N1–Pd1–N2 78.2 (6), N2–Pd1–C1 100.1 (5), Cl1–Pd1–C1 87.1 (7); Pd2 (b) Pd1–N1 2.126 (3), Pd1–N2 2.053 (4), Pd1–Cl1 2.167 (4), Pd1–C50 2.085 (13), N1–Pd1–Cl1 98.56 (11), N1–Pd1–N2 77.39 (13), N2–Pd1–C50 85.9 (5), Cl1–Pd1–C50 97.5 (5); and Pd2-ACN (c) Pd1–N1 2.005 (3), Pd1–N2 2.153 (2), Pd1–N3 2.036 (3), Pd1–C45 2.041 (3), N1–Pd1–N2 100.98 (10), N3–Pd1–N2 77.27 (10), N3–Pd1–C45 94.60 (12), C45–Pd1–N1 87.02 (12). | |
These palladium complexes were well characterized by 1H, H–H COSY, and 13C NMR spectra, elemental analysis and MALDI-TOF-MS (see the ESI†). As is shown, a pre-inserted Pd1 complex was observed even without any additions of cocatalyst NaBArF. The characteristic peaks of proton signals at 3.5, 3.1, 1.9, 1.3 and 0.6 ppm were attributed to the inserted allyl group in the 1H NMR spectrum and the assignments were further determined by the H–H COSY spectrum. The corresponding cationic species Pd1-ACN was accessed quantitatively through the abstraction of the –Cl atom with NaBArF cocatalyst. In contrast, Pd2 bearing the benzhydryl group showed the coordination mode without ligand pre-insertion. When abstracting the –Cl atom, the pre-inserted complex Pd2-ACN was also generated in high yield. As depicted in Fig. 2, all the cyclometallated palladium complexes (Pd1, Pd1-ACN and Pd2-ACN) were generated through the ligand 2, 1-insertion process, enabling the formation of the stable six-membered ring.
Computational study
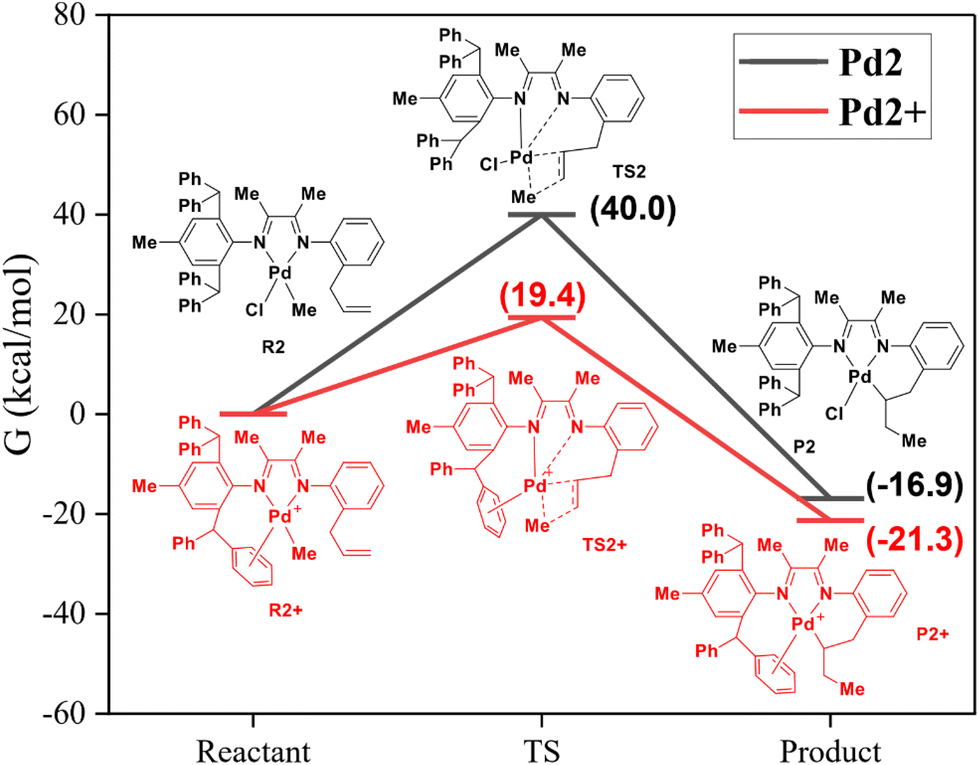
In order to understand the different reaction modes, density functional theory (DFT) calculations were carried out with the above palladium complexes with varying steric hindrance. Recent work by Harth et al. has proposed a pathway by confirming that the dissociation of the Pd–N bond occurred when activating 1-butene using the neutral palladium complex.57 It explains the phenomenon of intramolecular pre-insertion for the Pd1 complex in this work. In contrast, the neutral Pd2 complex cannot mediate the intramolecular coordination–insertion reaction. Therefore, DFT studies of the Pd2 complex were performed. As shown in Fig. 4, the corresponding energy barrier of the intramolecular reaction is as high as 40.0 kcal mol−1 (Fig. 4, black). This may be due to the significantly increased steric hindrance caused by the benzhydryl substituents in Pd2. Under the activation of NaBArF, cationic Pd2+ species can mediate the intramolecular reaction (energy barrier = 19.4 kcal mol−1), generating an intramolecular cyclized product (Fig. 4, red). These DFT calculation results support the N-dissociation proposal, thus releasing a coordination site for the insertion of the allyl group.
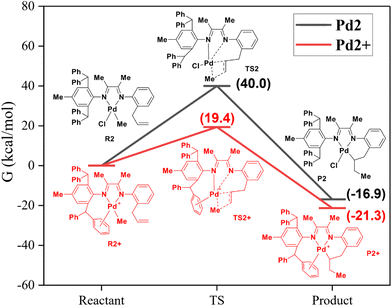 |
| | Fig. 4 DFT calculations of the coordination–insertion reactions with palladium complexes. | |
Ethylene polymerization
The Pd1-ACN and Pd2-ACN complexes were used in ethylene polymerization processes at different temperatures (Table 1). At 5 °C, the catalyst Pd1-ACN generated traces of polyethylene within 1 hour (Table 1, entry 1). Prolonging the polymerization time to 8 hours, Pd1-ACN was used in ethylene polymerization, and the produced polyethylene was highly branched (branching density, 94/1000 C) with a molecular weight of up to 61![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 (Table 1, entry 2). Increasing the polymerization temperature, the generated polyethylene showed decreased molecular weight and higher branching density (Table 1, entries 3–5). This phenomenon was ascribed to the increased chain transfer rate with rising temperature.
700 (Table 1, entry 2). Increasing the polymerization temperature, the generated polyethylene showed decreased molecular weight and higher branching density (Table 1, entries 3–5). This phenomenon was ascribed to the increased chain transfer rate with rising temperature.
Table 1 Ethylene polymerization catalyzed by the palladium catalystsa
| Entry |
Cat. |
T (°C) |
t (h) |
Yieldb (g) |
Act.b (105) |
M
nc |
PDIc |
B
|
|
Conditions: 10 μmol of Pd catalyst in 2 mL of CH2Cl2, 18 mL of toluene and ethylene at 8 atm.
The yields and activities are the average of at least two runs. Activity is in units of 105 g mol−1 h−1.
Determined by GPC in THF at 40 °C with polystyrene standards.
Branching numbers are given per 1000 carbon atoms. Branches per 1000 C = (CH3/3)/[(CH + CH2 + CH3)/2] × 1000 determined by 1H NMR spectroscopy in CDCl3.
|
| 1 |
Pd1-ACN
|
5 |
1 |
Trace |
— |
— |
— |
— |
| 2 |
Pd1-ACN
|
5 |
8 |
0.42 |
0.05 |
61700 |
3.0 |
94 |
| 3 |
Pd1-ACN
|
30 |
1 |
0.18 |
0.18 |
6900 |
3.1 |
99 |
| 4 |
Pd1-ACN
|
50 |
1 |
0.39 |
0.39 |
6500 |
3.4 |
101 |
| 5 |
Pd1-ACN
|
80 |
1 |
0.17 |
0.17 |
4800 |
1.8 |
105 |
| 6 |
Pd2-ACN
|
5 |
8 |
0.77 |
0.10 |
68100 |
2.0 |
88 |
| 7 |
Pd2-ACN
|
30 |
1 |
0.42 |
0.42 |
24800 |
2.4 |
91 |
| 8 |
Pd2-ACN
|
50 |
1 |
2.05 |
2.05 |
17700 |
2.7 |
92 |
| 9 |
Pd2-ACN
|
80 |
1 |
2.03 |
2.03 |
7900 |
2.6 |
96 |
For the catalyst Pd2-ACN, bearing larger steric hindrance, the produced polyethylene presented higher molecular weight and lower branching density compared to polyethylene from Pd1-ACN under the same conditions (Table 1, entries 6–9). This may originate from the chain transfer process that was suppressed by the enhanced steric hindrance in Pd2-ACN. Higher activity was also observed in ethylene polymerization for Pd2-ACN compared to that for Pd1-ACN at 30 °C and 50 °C (Table 1, entries 3 vs. 7 and entries 4 vs. 8). The activity difference between these two catalysts was more dramatic at 80 °C. The catalyst Pd2-ACN showed 12 times higher activity with a value of 2 × 105 g mol−1 h−1 compared to Pd1-ACN, demonstrating the good thermal stability of this catalyst (Table 1, entries 5 and 9). The generated polyethylenes were characterized by NMR and GPC analyses (see the ESI†).
Ethylene-polar monomer copolymerization
For ethylene copolymerization, acrylate-type monomers such as methyl acrylate (MA) and tert-butyl acrylate (t-BA) were investigated. In E-MA copolymerization, the catalyst Pd1-ACN produced E-MA copolymers with comparable activity and molecular weight at 30 °C and 50 °C (Table 2, entries 1 and 2). In contrast to Pd1-ACN, Pd2-ACN generated E-MA copolymers with higher catalytic activity and the molecular weight can be up to 14500 g mol−1 (Table 2, entry 4) and 12500 g mol−1 (Table 2, entry 5) with MA incorporation ratios of 0.7 mol% and 1.0 mol%, respectively. When the temperature was increased to 80 °C, a higher MA incorporation ratio of 1.9 mol% was achieved for Pd2-ACN (Table 2, entry 6). However, decreased catalytic activity and molecular weight for the produced E-MA copolymer were observed, although it still behaved better than its Pd1-ACN counterpart (Table 2, entry 3). These results can be explained by the enhanced steric hindrance around the palladium species, which could mediate slow chain transfer processes, enabling copolymers with higher activity, higher molecular weight and lower polar monomer incorporation (Table 2, entries 1, 2 vs. entries 4, 5).
Table 2 Ethylene–polar monomer copolymerization catalyzed by the palladium catalystsa
| Entry |
Cat. |
Monomer |
T (°C) |
Yieldb (g) |
Act.b (104) |
X
Mc (%) |
M
nd |
PDId |
|
Polymerization conditions: 20 μmol of Pd catalyst in 2 mL of CH2Cl2, total volume of toluene and polar monomer (2.5 M L−1) = 18 mL, ethylene at 8 atm and 3 h.
The yields and activities are the average of at least two runs. Activity is in units of 104 g mol−1 h−1.
Determined by 1H NMR spectroscopy in CDCl3.
Determined by GPC in THF at 40 °C with polystyrene standards.
|
| 1 |
Pd1-ACN
|
MA |
30 |
0.27 |
0.45 |
1.6 |
2700 |
1.69 |
| 2 |
Pd1-ACN
|
MA |
50 |
0.30 |
0.50 |
2.0 |
2300 |
1.47 |
| 3 |
Pd1-ACN
|
MA |
80 |
Trace |
— |
— |
— |
— |
| 4 |
Pd2-ACN
|
MA |
30 |
0.85 |
1.42 |
0.7 |
14500 |
1.53 |
| 5 |
Pd2-ACN
|
MA |
50 |
1.46 |
2.43 |
1.0 |
12500 |
1.75 |
| 6 |
Pd2-ACN
|
MA |
80 |
0.39 |
0.65 |
1.9 |
4900 |
1.60 |
| 7 |
Pd2-ACN
|
t-BA |
30 |
0.91 |
1.52 |
0.6 |
13600 |
1.18 |
| 8 |
Pd2-ACN
|
t-BA |
50 |
1.60 |
2.67 |
0.8 |
11800 |
1.22 |
| 9 |
Pd2-ACN
|
t-BA |
80 |
0.52 |
0.87 |
1.5 |
4200 |
1.48 |
Excluding methyl acrylate, the polar monomer tert-butyl acrylate with its bulkier group was also studied using Pd2-ACN in ethylene copolymerization. The synthesized E-t-BA copolymers showed similar molecular weight to the E-MA copolymers at different polymerization temperatures (Table 2, entries 7–9). A slight decrease in the polar monomer incorporation ratio was observed for the copolymerization, which resulted from the larger steric hindrance of tert-butyl acrylate. These copolymers were well characterized by NMR and GPC analyses (see the ESI†).
Mechanism investigation
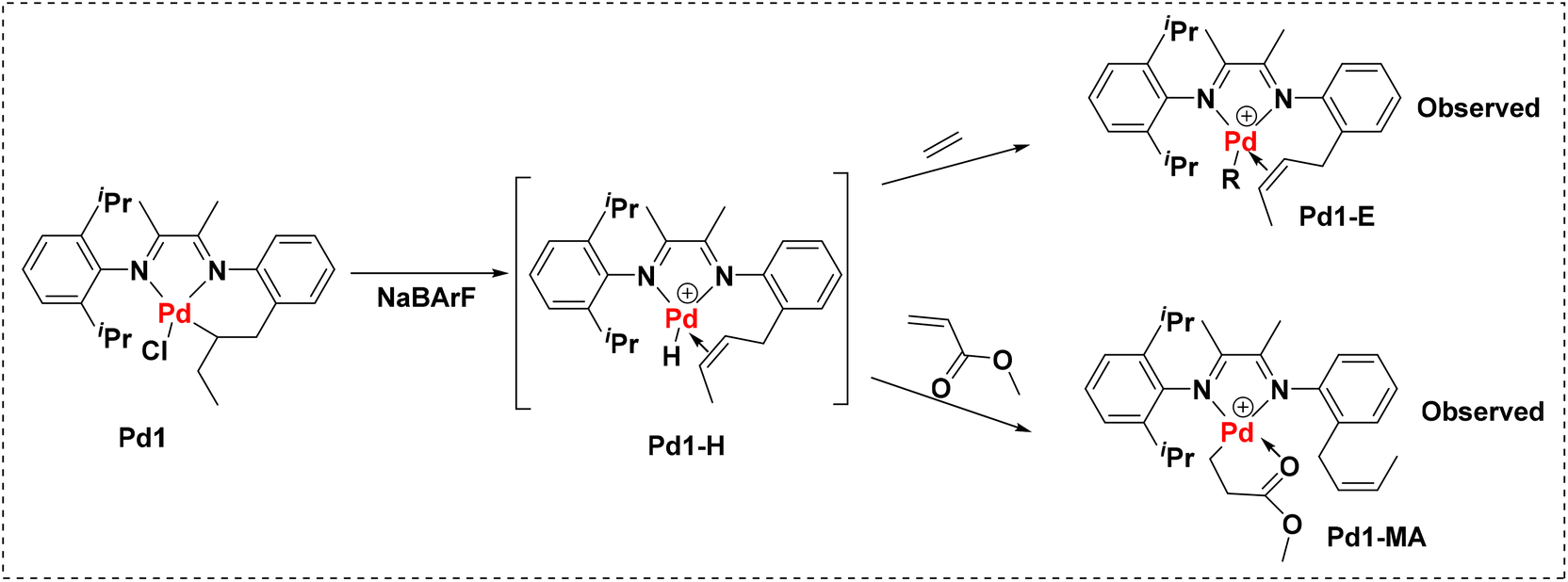
To further understand this system, mechanistic studies of the coordination–insertion process were conducted. Due to the competing coordination of CH3CN in Pd1-ACN, the base-free precatalyst Pd1 was used to explore the catalytically active species during the polymerization process (Fig. 5). For ethylene polymerization, the reaction of Pd1 and stoichiometric ethylene in CDCl3 at room temperature was conducted with the activation of the cocatalyst NaBArF. After 30 min, the ethylene was almost consumed with the formation of the alkyl long chain as indicated by the 1H NMR analysis (Fig. S1†). The 2-butenyl-substituted palladium intermediate (Pd1-E) was determined as the major product analysed by observing the H–H COSY spectrum (Fig. S2†). The generated Pd1-E indicates the occurrence of a β-H elimination intermediate (Pd1-H, unobserved) before ethylene chain growth starting from Pd1 under the activation of NaBArF. When the same experiment was carried out with 10 eq. of MA using Pd1, the product Pd1-MA could be identified from the 1H NMR and H–H COSY spectra (Fig. S3 and S4†), proving the MA insertion into the Pd–H bond and the release of the 2-butenyl group.
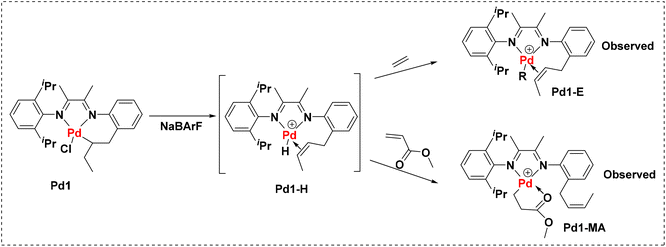 |
| | Fig. 5 Supposed coordination–insertion mechanism of the Pd1 complex towards ethylene and methyl acrylate. | |
Excluding the major catalytically active Pd–H species, the original Pd–C bond may initiate with minor possibility the polyethylene chain growth and this could explain the generation of slightly bimodal polyethylene as presented in Table 1 under real polymerization conditions. As shown, the original secondary Pd–C bond presented higher steric hindrance than the Pd–H bond. Therefore, it was difficult for the monomer to be inserted into the Pd–C bond straightforwardly, making it susceptible to inducing β-H elimination steps especially in E-MA copolymerization. Thus, a narrow polymer distribution index (PDI) was achieved for the produced E-MA copolymers as presented in Table 2 compared to the polyethylene detailed in Table 1.
Conclusions
In summary, two α-diimine ligands bearing allyl groups were prepared and showed different reaction modes with (COD)PdMeCl. The generated palladium Pd1 was the allyl group pre-inserted complex, but Pd2 was not. Under the activation of NaBArF, the pre-inserted cationic palladium species Pd1-ACN and Pd2-ACN were accessed in high yield. These palladium catalysts were used in ethylene polymerization and copolymerization. Compared to Pd1-ACN, Pd2-ACN with its larger steric hindrance exhibited higher activity during ethylene polymerization, affording polyethylene with high molecular weight. Performance enhancement was also observed in ethylene copolymerization with acrylate-type monomers (MA and t-BA), demonstrating the great polar group tolerance of this catalyst. In addition, mechanistic studies were conducted with Pd1, indicating that it was susceptible to inducing β-H elimination to form the Pd-H catalytically active species during the polymerization process. The pre-inserted α-diimine catalysts in this work demonstrated a unique form in olefin polymerization and will provide some important information for catalyst design.
Author contributions
Min Chen conceived the idea, designed the experiments and evaluated the data. Chen Tan performed the DFT calculations and revised the original manuscript. Min Chen and Chao Li wrote the manuscript together. Min Chen, Chao Li and Chen Tan acquired financial funding support for this project.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC, 21971230, U19B6001, 22001004 and 22201003) and the Anhui Province Key Laboratory of Environment-Friendly Polymer Materials, Excellent Research and Innovation Team Project of Anhui Province (2022AH010001). We are grateful for the mentorship and great support from Professor Changle Chen (USTC).
References
- M. Stürzel, S. Mihan and R. Mülhaupt, Chem. Rev., 2016, 116, 1398–1433 CrossRef.
- C. Tan and C. L. Chen, Angew. Chem., 2019, 131, 7168–7276 Search PubMed.
- H. Mu, L. Pan and Y. Li, Chem. Rev., 2015, 115, 12091–12137 CrossRef CAS.
- H. L. Mu, G. L. Zhou, X. Q. Hu and Z. B. Jian, Coord. Chem. Rev., 2021, 435, 213802 CrossRef CAS.
- C. L. Chen, Nat. Rev. Chem., 2018, 2, 6–14 CrossRef CAS.
- T. V. Tran and L. H. Do, Eur. Polym. J., 2021, 142, 110100 CrossRef CAS.
- A. Nakamura, T. M. Anselment, J. Claverie, B. Goodall, R. F. Jordan, S. Mecking, B. Rieger, A. Sen, P. W. N. M. van Leeuwen and K. Nozaki, Acc. Chem. Res., 2013, 46, 1438–1449 CrossRef CAS.
- A. Nakamura, S. Ito and K. Nozaki, Chem. Rev., 2009, 109, 5215–5244 CrossRef CAS.
- H. Makio, H. Terao, A. Iwashita and T. Fujita, Chem. Rev., 2011, 111, 2363–2449 CrossRef CAS PubMed.
- F. Lin and S. Mecking, Angew. Chem., 2022, 134, e202203923 CrossRef.
- T. V. Tran, E. Lee, Y. H. Nguyen, H. D. Nguyen and L. H. Do, J. Am. Chem. Soc., 2022, 144, 17129–17139 CrossRef CAS.
- M. Baur, F. Lin, T. O. Morgen, L. Odenwald and S. Mecking, Science, 2021, 374, 604–607 CrossRef CAS PubMed.
- H. Zhang, C. Zou, H. Zhao, Z. G. Cai and C. L. Chen, Angew. Chem., 2021, 133, 17586–17591 CrossRef.
- S. Xiong, M. M. Shoshani, X. Zhang, H. A. Spinney, A. J. Nett, B. S. Henderson, T. F. Miller and T. Agapie, J. Am. Chem. Soc., 2021, 143, 6516–6527 CrossRef CAS PubMed.
- G. Ji, Z. Chen, X. Y. Wang, X. S. Ning, C. J. Xu, X. M. Zhang, W. J. Tao, J. F. Li, Y. S. Gao, Qi. Shen, X. L. Sun, H. Y. Wang, J. B. Zhao, Bo. Zhang, Y. L. Guo, Y. N. Zhao, J. J. Sun, Y. Luo and Y. Tang, Nat. Commun., 2021, 12, 6283 CrossRef CAS.
- Y. Jiang, Z. Zhang, S. Li and D. M. Cui, Angew. Chem., 2022, 134, e202112966 CrossRef.
- Y. Wu, T. H. Nan, X. L. Ji, B. Liu and D. M. Cui, Angew. Chem., Int. Ed., 2022, 61, e202205894 CrossRef CAS PubMed.
- C. Zou, W. Pang and C. L. Chen, Sci. China: Chem., 2018, 61, 1175–1178 CrossRef CAS.
- Y. Sun, Q. Wang, Y. Pan, W. M. Pang, C. Zou and M. Chen, Chin. J. Chem., 2022, 40, 2773–2779 CrossRef CAS.
- L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414–6415 CrossRef CAS.
- L. K. Johnson, S. Mecking and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 267–268 CrossRef CAS.
- F. Z. Wang and C. L. Chen, Polym. Chem., 2019, 10, 2354–2369 RSC.
- Y. Zhang, X. Hu, C. Wang and Z. B. Jian, ACS Catal., 2022, 12, 14304–14320 CrossRef CAS.
- L. Guo, S. Y. Dai, X. Sui and C. L. Chen, ACS Catal., 2016, 6, 428–441 CrossRef CAS.
- Z. Chen and M. Brookhart, Acc. Chem. Res., 2018, 51, 1831–1839 CrossRef CAS PubMed.
- S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169–1204 CrossRef CAS PubMed.
- Y. Zhang and Z. B. Jian, Macromolecules, 2020, 53, 8858–8866 CrossRef CAS.
- Z. Chen, W. Liu, O. Daugulis and M. Brookhart, J. Am. Chem. Soc., 2016, 138, 16120–16129 CrossRef CAS PubMed.
- S. Y. Chen, R. C. Pan, Y. Liu and X. B. Lu, Organometallics, 2021, 40, 3703–3711 CrossRef CAS.
- S. Zhong, Y. Tan, L. Zhong, J. Gao, H. Liao, L. Jiang and Q. Wu, Macromolecules, 2017, 50, 5661–5669 CrossRef CAS.
- Z. Xiao, L. Zhong, C. Du, W. Du, H. Zheng, C. S. Cheung and H. Gao, Macromolecules, 2021, 54, 687–695 CrossRef CAS.
- S. Zhong, Y. Tan, L. Zhong, J. Gao, H. Liao, L. Jiang, H. Gao and Q. Wu, Macromolecules, 2017, 50, 5661–5669 CrossRef CAS.
- K. E. Allen, J. Campos, O. Daugulis and M. Brookhart, ACS Catal., 2015, 5, 456–464 CrossRef CAS.
- S. Y. Dai and C. L. Chen, Angew. Chem., Int. Ed., 2020, 59, 14884–14890 CrossRef CAS.
- D. N. Vaccarello, K. S. O'Connor, P. Iacono, J. M. Rose, A. E. Cherian and G. W. Coates, J. Am. Chem. Soc., 2018, 140, 6208–6211 CrossRef CAS.
- Y. Zhang, C. Wang, S. Mecking and Z. B. Jian, Angew. Chem., 2020, 132, 14402–14408 CrossRef.
- Y. Liu and E. Harth, Angew. Chem., 2021, 133, 24309–24317 CrossRef.
- Y. Ge, S. Li, W. Fan and S. Y. Dai, Inorg. Chem., 2021, 60, 5673–5681 CrossRef CAS.
- M. Li, X. Wang, Y. Luo and C. L. Chen, Angew. Chem., 2017, 129, 11762–11767 CrossRef.
- S. Y. Dai and C. L. Chen, Angew. Chem., 2016, 128, 13475–13479 CrossRef.
- S. Y. Dai, X. L. Sui and C. L. Chen, Angew. Chem., 2015, 127, 10086–10091 CrossRef.
- Z. Xiao, H. Zheng, C. Du, L. Zhong, H. Liao, J. Gao and Q. Wu, Macromolecules, 2018, 51, 9110–9121 CrossRef CAS.
- W. Lu, H. Wang, W. Fan and S. Y. Dai, Inorg. Chem., 2022, 61, 6799–6806 CrossRef CAS PubMed.
- F. Zhai, J. B. Solomon and R. F. Jordan, Organometallics, 2017, 36, 1873–1879 CrossRef CAS.
- F. Zhai and R. F. Jordan, Organometallics, 2014, 33, 7176–7192 CrossRef CAS.
- S. Y. Dai, S. Zhou, W. Zhang and C. L. Chen, Macromolecules, 2016, 49, 8855–8862 CrossRef CAS.
- M. H. Ji, G. F. Si, Y. Pan, C. Tan and M. Chen, J. Catal., 2022, 415, 51–57 CrossRef CAS.
- G. Liao, Z. Xiao, X. Chen, C. Du, L. Zhong, C. S. Cheung and H. Gao, Macromolecules, 2019, 53, 256–266 CrossRef.
- M. Zhao and C. L. Chen, ACS Catal., 2017, 7, 7490–7494 CrossRef CAS.
- Y. Miyamura, K. Kinbara, Y. Yamamoto, V. K. Praveen, K. Kato, M. Takata and T. Aida, J. Am. Chem. Soc., 2010, 132, 3292–3294 CrossRef CAS PubMed.
- G. Wang, D. Peng, Y. Sun and C. L. Chen, CCS Chem., 2020, 3, 2025–2034 CrossRef.
- D. Peng and C. L. Chen, Angew. Chem., Int. Ed., 2021, 60, 22195–22200 CrossRef CAS.
- H. Dau, A. Keyes, H. E. Basbug Alhan, E. Ordonez, E. Tsogtgerel, A. P. Gies, E. Auyeung, Z. Zhou, A. Maity, A. Das, D. C. Powers, D. B. Beezer and E. Harth, J. Am. Chem. Soc., 2020, 142, 21469–21483 CrossRef CAS PubMed.
- A. Keyes, H. Dau, K. Matyjaszewski and E. Harth, Angew. Chem., 2022, 134, e202112742 CrossRef.
- J. Gao, R. W. Dorn, G. P. Laurent, F. A. Perras, A. J. Rossini and M. P. Conley, Angew. Chem., Int. Ed., 2022, 61, e202117279 CrossRef CAS PubMed.
- D. B. Culver, H. Tafazolian and M. P. Conley, Organometallics, 2018, 37, 1001–1006 CrossRef CAS.
- D. Nguyen, S. Wang, L. C. Grabow and E. Harth, J. Am. Chem. Soc., 2023, 145, 9755–9770 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2266073–2266075. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3py01011b |
| ‡ These authors conducted the experiments and contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 * and
Min
Chen
* and
Min
Chen