
Dual stimuli triggerable degradation of graft copolymers†
Samantha Marie
Clouthier
 a,
Jiajia
Li
ab,
Joji
Tanaka
*a and
Wei
You
*a
a,
Jiajia
Li
ab,
Joji
Tanaka
*a and
Wei
You
*a
aDepartment of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-3290, USA. E-mail: joji@email.unc.edu; wyou@unc.edu
bState and Local Joint Engineering Laboratory for Novel Functional Polymeric Materials, Jiangsu Key Laboratory of Advanced Functional Polymer Design and Application, Department of Polymer Science and Engineering, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou, China
First published on 29th November 2023
Abstract
Here we report one-pot stimuli-responsive tandem degradation of a graft copolymer with alternating backbone functionalities. A well-defined polymeric species is formed by selectively cleaving one of the structural repeat units in the main chain, and can be further ‘cut’ into half by cleaving the second structural unit. The high selectivity of these two orthogonal steps ensures that the order of the stimuli employed does not affect the outcome.
Over the last century, the use of synthetic polymers has increased in a variety of applications, from commodity plastics to drug delivery, owing to the tuneable properties of these polymers with synthetic design. One of the increasing issues is the non-degradable feature that is common to most synthetic polymers, leading to plastic pollution and health-related concerns. Owing to this reason, research in designing degradable polymers has gained much attraction.1 Moreover, employing an on-demand degradable functionality that is selective towards a specific stimulus, such as light,2 pH,3 chemicals,4 and temperature,5 has led to a new generation of environmentally responsive polymers.6,7
In the literature, degradable polymers synthesized by controlled radical polymerisation (CRP) techniques, such as nitroxide-mediated polymerisation (NMP),8 atom-transfer radical polymerisation (ATRP),9 and reversible addition-fragmentation chain transfer (RAFT),10 have only achieved limited incorporation of degradable units throughout the polymer backbone,11,12 typically via creative design of initiators and/or specific backbones. For example, disulfide-containing difunctional initiators have been implemented to synthesize block copolymers via ATRP that can subsequently be degraded by cleavage of the disulfide bond, resulting in polymers having half the molecular weight of the original species.13 In another example, Luo et al. prepared multiblock polymers with enzyme degradable units between the blocks, by clicking together a diazide peptide and a telechelic alkyne functional RAFT polymer.14
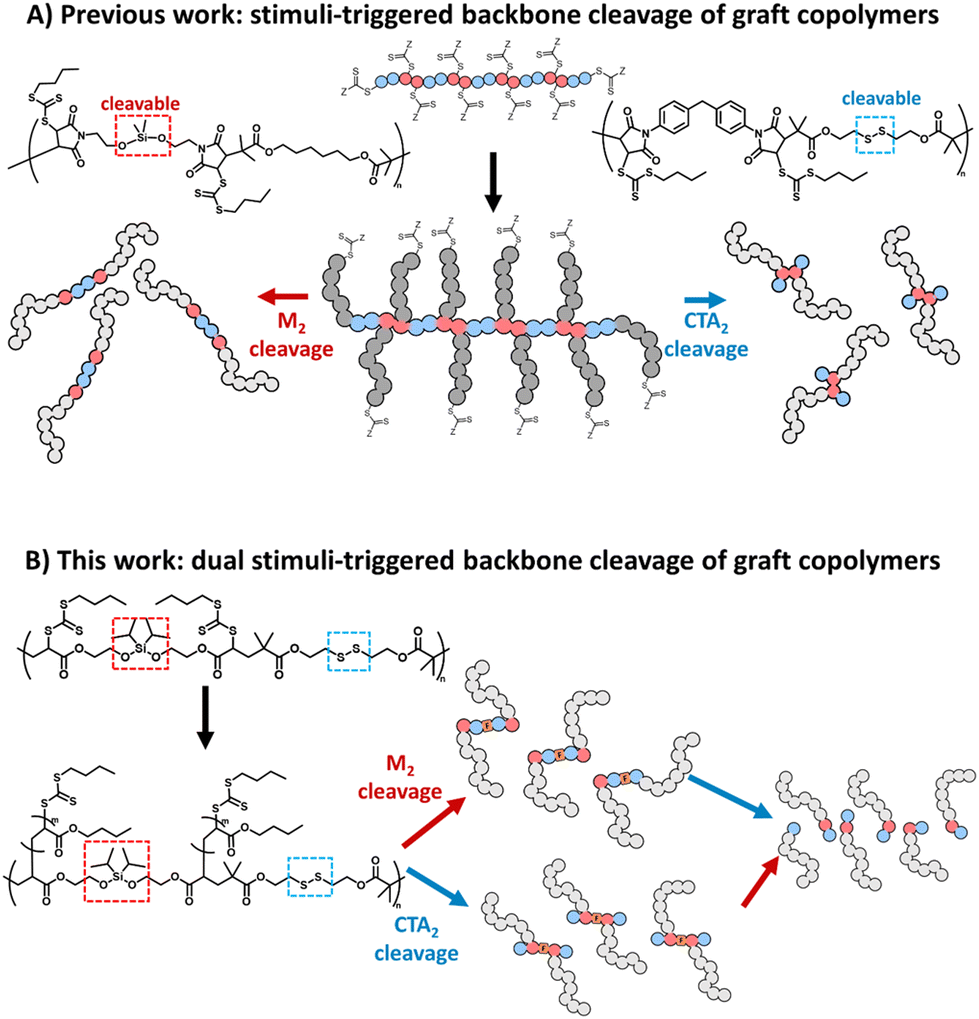
More recently, we reported RAFT step-growth polymerisation15 that combines the user-friendly nature and high functional group tolerance of RAFT polymerisation with the versatility in the polymer backbone functionality of step-growth, thus providing access to a highly functional polymer backbone in a facile manner.15–20 RAFT step-growth polymerisation is typically carried out using stoichiometrically equivalent bifunctional monomers and chain transfer agent (CTA) reagents that can undergo an efficient single unit monomer insertion (SUMI) process. Additionally, the in situ generated pendant RAFT agents along the backbone can be directly used to graft polymeric side chains via RAFT chain-growth polymerisation, permitting the synthesis of graft copolymers with a unique step-growth backbone functionality.15–18 A key advantage of A2 + B2 RAFT step-growth polymers is the integration of degradable linkers into the backbone using either the monomer or CTA units.15,17,18 As a result, degradation of the graft-copolymer can be triggered throughout the polymer backbone, revealing a unimolecular species with a narrow molecular weight distribution (Scheme 1A).15,17,18
 | ||
| Scheme 1 Dual stimuli triggerable graft copolymer in this work. | ||
Materials that can respond to more than one stimulus are becoming increasingly desirable for certain applications such as drug delivery where drug release kinetics can be optimized towards target sites;21–23 yet there are relatively few reported examples of polymers where two or more orthogonally degradable functionalities are incorporated into the polymer backbone. In one such example, Chang et al. demonstrated dual stimuli-responsive sequential degradation of a terpolymer containing both disulfide and phosphoester moieties prepared via ring opening metathesis polymerisation (ROMP).24
Since A2 + B2 step-growth polymerisation can allow an alternating sequence of functionalities to be embedded along the polymer backbone, we hypothesized that A2 + B2 RAFT step-growth could employ two orthogonally degradable functionalities, individually embedded in the bifunctional monomer and CTA reagents, to achieve orthogonal degradation. Furthermore, by grafting from this backbone we could prepare a dual stimuli-responsive graft copolymer that can selectively degrade with the desired linker, allowing for tuneability and tandem degradation with a high level of precision (Scheme 1B).
To demonstrate this concept, we chose to employ silyl ether and disulfides as orthogonally degradable functionalities (Scheme 1B). Following closely our previous report, the disulfide functionality was incorporated into the bifunctional CTA (CTA2SS) and silyl ether was incorporated into the bifunctional monomer (M2). In contrast to our previous report, we selected acrylate as the monomer unit, as it can be readily synthesized compared to bis-maleimides. Additionally, a more sterically substituted diisopropyl silyl ether was selected to prevent inadvertent hydrolysis that was observed in our previous report with less sterically substituted dimethyl silyl ether.25
We first investigated the synthesis of the backbone using thermally-initiated RAFT step-growth between the diisopropyl silyl ether tethered diacrylate monomer (M2) and the disulfide tethered RAFT agent (CTA2SS) (Fig. 1). We employed stoichiometrically balanced reaction conditions for diacrylates in dioxane and used AIBN as the initiator at 70 °C as previously reported ([CTA2SS]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[M2]0:[AIBN]0 = 1.0 M:1.0 M:0.05 M).171H-NMR was used to determine monomer conversion and SEC analysis was used to determine the molecular weights relative to polystyrene standards in THF.17 The polymerisation proceeded nicely reaching a modest molecular weight and conversion after 4.0 hours (p = 93%, Mw = 12.2k) (Table S1, Fig. S2 and S3,† and Fig. 1B). Furthermore, the polymerisation followed the expected step growth molecular weight evolution (Mn, Mw, and Mz) with conversion as predicted using Flory's equations (Fig. 1A).26
:[M2]0:[AIBN]0 = 1.0 M:1.0 M:0.05 M).171H-NMR was used to determine monomer conversion and SEC analysis was used to determine the molecular weights relative to polystyrene standards in THF.17 The polymerisation proceeded nicely reaching a modest molecular weight and conversion after 4.0 hours (p = 93%, Mw = 12.2k) (Table S1, Fig. S2 and S3,† and Fig. 1B). Furthermore, the polymerisation followed the expected step growth molecular weight evolution (Mn, Mw, and Mz) with conversion as predicted using Flory's equations (Fig. 1A).26
 | ||
| Fig. 1 A2 + B2 RAFT step-growth polymerisation. (A) Evolution of the molecular weight averages (Mw, Mn, and Mz) from SEC analysis using polystyrene calibration, plotted with monomer conversion (p) determined by 1H-NMR spectroscopy. These are plotted with the theoretical line for step-growth molecular weight evolution that assumes no cyclization.26 (B) THF-SEC (normalized dRI) analysis of RAFT step-growth polymerisation. The molecular weight is relative to polystyrene calibration. | ||
We next employed our backbone with the embedded silyl ether and disulfide moieties (P(CTA2SS-alt-M2)) to graft poly(butyl acrylate) (PBA) via RAFT polymerisation with a monomer to CTA functionality ratio of 40 ([BA]0/[CTA]0 = 40) in dioxane ([BA]0 = 3.0 M). Using AIBN as the thermal initiator ([CTA]0/[AIBN]0 = 20) at 65 °C, the reaction proceeded to 84% monomer conversion after 4.0 hours, yielding a theoretical Mn value of about 4.8k per PBA sidechain (Fig. S4 and S5†). A visible shift in the SEC trace of the graft copolymer towards a lower retention time from the precursor backbone was observed, consistent with the formation of the graft copolymer (Fig. 2B and Fig. S6†). Moreover, the absolute Mn value obtained from the light scattering detector was consistent with the theoretical Mn value calculated from the number of structural repeat units approximated from the absolute Mn value of the precursor backbone, and the theoretical Mn value per PBA sidechain (4.8k). Additionally, the Mark–Houwink plot obtained from the triple detection SEC (dRI, LS, VS) analysis (Fig. S6†) of both the isolated backbone and the graft copolymer reveals α values of 0.634 and 0.479, respectively (Fig. 2A). An α value of 0.634 is consistent with the molecular weight distribution for linear polymers (where typical α values range from 0.5 to 0.8), and a significant decrease in the alpha value for the graft copolymer (α = 0.479) confirms the shift to a branched conformation (Fig. 2A).27
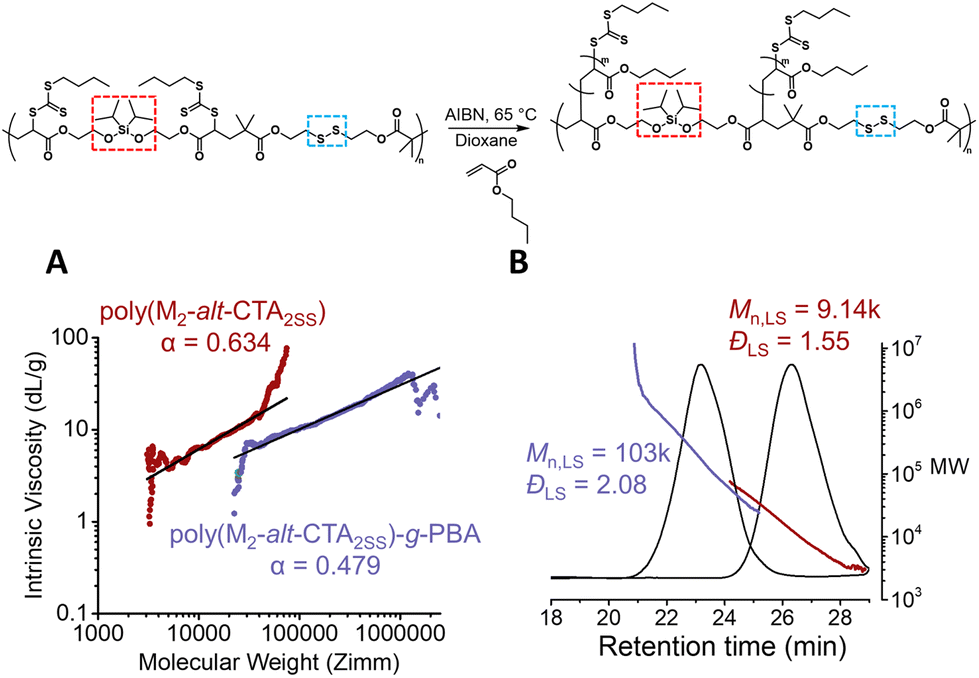 | ||
| Fig. 2 Graft polymerisation of the RAFT step-growth backbone via RAFT. (A) The Mark–Houwink plot for the degradable RAFT step-growth backbone and the graft copolymer; the slope is determined from linear regression across the same range of data points used in molecular weight analysis (red line in Fig. S6†). (B) LS-SEC of P(M2-alt-CTA2SS) and P(M2-alt-CTA2SS)-g-PBA. | ||
We next screened various conditions to target selective cleavage of the silyl ether and disulfide groups embedded in the main chain of the graft copolymer backbone, and used conventional SEC analysis to characterize the degraded polymers. Indeed, selectively cleaving only one of the structural units along an alternating step-growth backbone will result in a uniform species that consists of two polymeric side chains. We anticipated that the efficiency of each cleavage may strongly depend on the solvent; therefore, we investigated two different solvents for each reaction, butanol as a protic solvent and THF as an aprotic solvent.
We first examined the selective cleavage of the silyl ether groups using a fluoride source.28 Experimentally, we employed 20 equivalents of tetra-n-butylammonium fluoride (TBAF) and acetic acid (AcOH) relative to the silyl ether units.15 We observed successful selective cleavage of the graft copolymer backbone – resulting from selective cleavage of the silyl ether – after 2.0 hours in THF (Mn = 14k, Đ = 1.09). In contrast, partial cleavage of the graft copolymer backbone was observed after 3.0 hours in butanol (Mn = 36k, Đ = 1.44, Fig. S7†). We attributed this solvent-dependent behaviour to the reaction mechanism which likely proceeds through the SN2 pathway and favours aprotic solvents.29
We next screened the appropriate conditions to selectively degrade the disulfide bond using redox chemistry. Here, we used 20 equivalents of tributyl phosphine (PBu3) with respect to the disulfide bond.17 Although mechanistically the reduction of the disulfide with phosphines has been reported to proceed through the SN2 pathway, a proton source is inherently required for the reduction.30 Typically the presence of water is necessary to drive the reaction, although previously we found that the reduction could proceed to completion in alcoholic solvents without the addition of water.15 Here we found the selective cleavage of the disulfide in butanol to reach completion in under 1.5 hours (Mn = 14k, Đ = 1.13). It is important to emphasize that the silyl ether did not undergo metathesis with this solvent (butanol) and remained intact in the polymer backbone during this process. Interestingly, reduction of the disulfide was also observed in THF, albeit to a lesser extent (Mn = 16k, Đ = 1.39 after 2.0 hours, Fig. S8†) despite the lack of a proton source. We believe that this is likely due to the residual methanol present from the purification process as methanol was used to precipitate the PBA graft copolymer. Nevertheless, employing a protic solvent was necessary for complete cleavage of the disulfide bonds.
Using these conditions we identified above, we investigated one-pot, two-step tandem degradation of the graft copolymer by first employing 20 equivalents of TBAF/AcOH in THF, obtaining a unimodal molecular weight distribution with a narrow dispersity corresponding to two PBA side chains tethered by a disulfide bond (Mn = 14.9k, Đ = 1.11, Fig. 3A). As a protic solvent was necessary for the second degradation step, THF was removed under reduced pressure and replaced with butanol prior to the addition of PBu3 (20 eq.). Pleasingly, this resulted in a noticeable shift in the SEC chromatogram corresponding to one PBA side chain (Mn = 7.6k, Đ = 1.07, Fig. 3A).
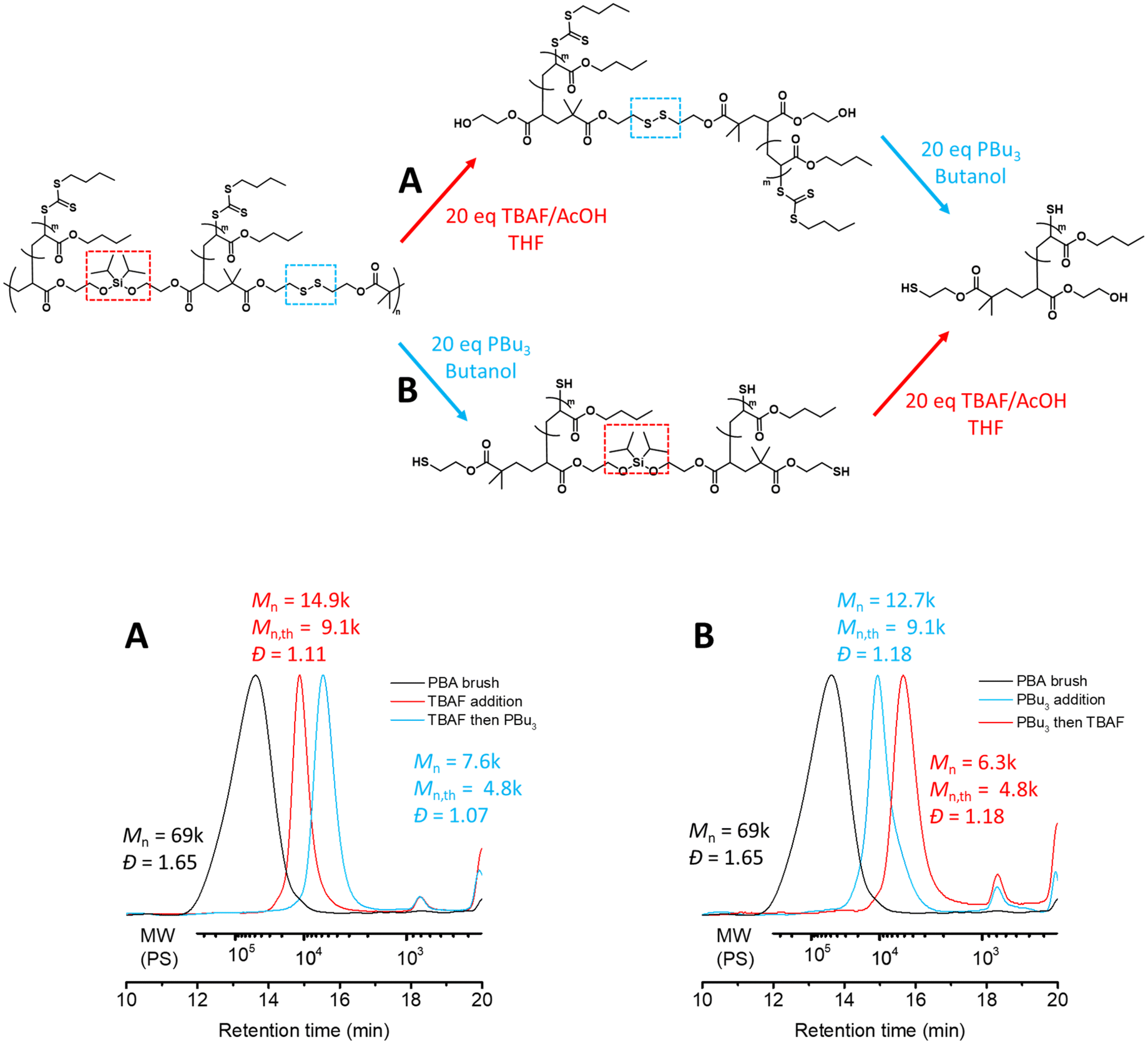 | ||
| Fig. 3 Orthogonal degradation of the graft copolymer backbone. (A) Dual stimuli degradation of the silyl ether group (red), then the disulfide bond (blue). (B) THF-SEC (normalized dRI) chromatograms of P(CTA2SS-alt-M2)-g-PBA (black curve, Mn = 69k, Đ = 1.65), dual stimuli degradation of the disulfide bond (blue), then the silyl ether group (red). | ||
To further demonstrate the versatility of our dual stimuli triggerable degradation of the graft copolymer backbone, we carried out the tandem degradation in the reverse order to the above, specifically, cleaving the disulfide bonds with PBu3 (20 eq.) in butanol (Mn = 12.7k, Đ = 1.18, Fig. 3B) followed by cleaving the silyl ether units with TBAF/AcOH (20 eq.) in THF (Mn = 6.3k, Đ = 1.18, Fig. 3B). It is worth noting that in practice removing butanol is more challenging than removing THF due to its higher boiling point; nonetheless it can be removed by blowing compressed gas on the surface of the solution. Here we used argon to prevent inadvertent oxidation of the liberated thiols during the solvent removal. In summary, we successfully demonstrated that tandem degradation of the alternating cleavable functionalities embedded in the main chain of the graft copolymer backbone can be performed sequentially independent of the order.
Furthermore, to improve atom efficiency we examined the selective degradation conditions with reduced equivalents of the reagents. We found 10 equivalents of TBAF/AcOH in THF were sufficient to fully cleave the silyl ether units within 2 hours (Mn = 14.9k, Đ = 1.12), while further lowering the equivalents to 5 equivalents required a longer reaction time of 48 hours (Mn = 14.6k, Đ = 1.07, Fig. S9†). Additionally, we found 10 equivalents of PBu3 in butanol were also sufficient to fully cleave the disulfide units within 2.0 hours (Mn = 14.1k, Đ = 1.13, Fig. S10†).
Finally, combining these two new conditions, we examined one-pot tandem degradation with reduced equivalents. Pleasingly, 10 equivalents of TBAF/AcOH in THF followed by 10 equivalents of PBu3 in butanol revealed a unimolecular species with narrow dispersity corresponding to two PBA side chains (Mn = 14.9k, Đ = 1.12, Fig. S11†) and one PBA side chain (Mn = 7.6k, Đ = 1.07, Fig. S11†), respectively.
To summarize, an alternating functional backbone was easily prepared by A2 + B2 RAFT step-growth polymerisation using a functional bifunctional monomer and CTA reagents, and was directly grafted via RAFT controlled chain-growth polymerisation. Here, a graft copolymer with alternating cleavable functional groups of silyl ether and disulfide bonds embedded in the structural repeat units of the main-chain backbone was prepared. The orthogonal nature of these two stimulus-triggerable functionalities allowed us to accomplish dual-stimuli selective tandem degradation of the graft copolymer backbone into a well-defined polymeric species, consisting of two polymeric grafts after the first stimulus, which can be further precisely ‘cut’ into half after the second stimulus. This was carried out by employing TBAF/AcOH and PBu3 to selectively cleave the silyl ether and disulfide bond units respectively, which can be done independently of the order. By judiciously selecting different (orthogonal) stimulus-triggerable units, one can expand our methodology to a wide range of multiple stimuli triggerable degradation of graft copolymers.
Author contributions
The manuscript was written through contributions of all authors.Conflicts of interest
The authors declare the following competing financial interest(s): J.T. and W.Y. are named inventors on a patent application owned by UNC-Chapel Hill (PCT/US2022/042087) which laid the foundation for this work. Dr You is also a co-founder of Delgen Biosciences, a startup company that has licensed this UNC patent application.Acknowledgements
This work was financially supported by the National Science Foundation (NSF) under Award CHE-2108670. A Bruker AVANCE III Nanobay 400 MHz NMR spectrometer was supported by NSF under Grant No. CHE-0922858, and a Bruker NEO 600 MHz NMR spectrometer was supported by NSF under Grant No. CHE-1828183. The authors thank Dr Marc A. ter Horst from University of North Carolina's Department of Chemistry NMR Core Laboratory for the use of the NMR spectrometers.References
- B. Jothimani, B. Venkatachalapathy, N. S. Karthikeyan and C. Ravichandran, Materials Horizons: From Nature to Nanomaterials, in Green Biopolymers and their Nanocomposites, ed. D. Gnanasekaran, Springer, Singapore, 2019, pp. 403–422, DOI:10.1007/978-981-13-8063-1_17.
- P. Zhao, J. Xia, M. Cao and H. Xu, ACS Macro Lett., 2020, 9, 163–168 CrossRef CAS PubMed.
- G. Kocak, C. Tuncer and V. Bütün, Polym. Chem., 2017, 8, 144–176 RSC.
- Y. L. Colson and M. W. Grinstaff, Adv. Mater., 2012, 24, 3878–3886 CrossRef CAS PubMed.
- A. Lv, Y. Cui, F.-S. Du and Z.-C. Li, Macromolecules, 2016, 49, 8449–8458 CrossRef CAS.
- M. Wei, Y. Gao, X. Li and M. J. Serpe, Polym. Chem., 2017, 8, 127–143 RSC.
- M. A. Stuart, W. T. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- M. Lages, N. Gil, P. Galanopoulo, J. Mougin, C. Lefay, Y. Guillaneuf, M. Lansalot, F. D'Agosto and J. Nicolas, Macromolecules, 2022, 55, 9790–9801 CrossRef CAS.
- M. Mizuntani, K. Satoh and M. Kamigaito, Macromolecules, 2011, 44, 2382–2386 CrossRef CAS.
- C. A. Bell, G. G. Hedir, R. K. O'Reilly and A. P. Dove, Polym. Chem., 2015, 6, 7447–7454 RSC.
- V. Delplace and J. Nicolas, Nat. Chem., 2015, 7, 771–784 CrossRef CAS PubMed.
- M. D. Rikkou and C. S. Patrickios, Prog. Polym. Sci., 2011, 36, 1079–1097 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2002, 35, 9009–9014 CrossRef CAS.
- K. Luo, J. Yang, P. Kopeckova and J. Kopecek, Macromolecules, 2011, 44, 2481–2488 CrossRef CAS PubMed.
- J. Tanaka, N. E. Archer, M. J. Grant and W. You, J. Am. Chem. Soc., 2021, 143, 15918–15923 CrossRef CAS PubMed.
- P. Boeck, N. Archer, J. Tanaka and W. You, Polym. Chem., 2022, 13, 2589–2594 RSC.
- N. E. Archer, P. T. Boeck, Y. Ajirniar, J. Tanaka and W. You, ACS Macro Lett., 2022, 11, 1079–1084 CrossRef CAS PubMed.
- S. M. Clouthier, J. Tanaka and W. You, Polym. Chem., 2022, 13, 6114–6119 RSC.
- P. T. Boeck, J. Tanaka, W. You, B. S. Sumerlin and A. S. Veige, Polym. Chem., 2023, 14, 2592–2598 RSC.
- J. Tanaka, J. Li, S. M. Clouthier and W. You, Chem. Commun., 2023, 59, 8168–8198 RSC.
- K. Miyata, Y. Kakizawa, N. Nishiyama, A. Harada, Y. Yamasaki, H. Koyama and K. Kataoka, J. Am. Chem. Soc., 2004, 126, 2355–2361 CrossRef CAS PubMed.
- A. M. Jazani and J. K. Oh, Polym. Chem., 2022, 13, 4557–4568 RSC.
- K. Maruya-Li, C. Shetty, A. M. Jazani, N. Arezi and J. K. Oh, ACS Omega, 2020, 5, 3734–3742 CrossRef CAS PubMed.
- C.-C. Chang and T. Emrick, Macromolecules, 2014, 47, 1344–1350 CrossRef CAS.
- P. Shieh, H. V. Nguyen and J. A. Johnson, Nat. Chem., 2019, 11, 1124–1132 CrossRef CAS PubMed.
- P. J. Flory, J. Am. Chem. Soc., 1936, 58, 1877–1885 CrossRef CAS.
- Y. Lu, L. An and Z.-G. Wang, Macromolecules, 2013, 46, 5731–5740 CrossRef CAS.
- T. D. Nelson and D. R. Crouch, Synthesis, 1996, 1996, 1031–1069 CrossRef.
- K. C. Westaway and Z.-G. Lai, Can. J. Chem., 1989, 67, 345–349 CrossRef CAS.
- O. Dmitrenko, C. Thorpe and R. D. Bach, J. Org. Chem., 2007, 72, 8298–8307 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py01105d |
| This journal is © The Royal Society of Chemistry 2024 |