
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploiting controlled transesterification as a “top down” approach to tailor poly(ε-caprolactone)-poly(lactic acid) copolymer structures with bis-Zn catalysts†
Thomas J.
Neal
 a,
Edward D.
Neal
b,
James
Cumby
a and
Jennifer A.
Garden
*a
a,
Edward D.
Neal
b,
James
Cumby
a and
Jennifer A.
Garden
*a
aEaStCHEM School of Chemistry, University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh, EH9 3FJ, Scotland, UK. E-mail: j.garden@ed.ac.uk
bSchool of Chemistry, University of Bristol, Bristol, BS8 1TS, UK
First published on 3rd April 2024
Abstract
Poly(ε-caprolactone)-poly(lactic acid) (PCL-PLA) copolymers have wide-ranging applications, where the polymer properties are controlled by the chain structure (i.e. block or random). Yet the synthesis of well-defined higher order multiblock PCL-PLA copolymers remains challenging, as competitive transesterification processes can occur that disrupt the polymer structure. Herein, we demonstrate that controlled transesterification can be harnessed as a “top down” method of tailoring PCL-PLA copolymer structures, instigated by the addition of ε-caprolactone to a living PCL-PLA-Zn copolymer chain. The extent of transesterification can be enhanced by increasing the ε-CL stoichiometry and decreasing the chain length of the diblock PCL-PLA precursors. While transesterification decreases the average length of the PCL and PLA blocks, the polymers retained their “blocky” nature as evidenced by DSC analysis. Novel computer simulations on simplified oligomeric systems show that transesterification occurs in both the PCL and PLA blocks of the original copolymer. This methodology also successfully transesterified an isolated PCL-block-PLA copolymer, suggesting that this zinc-catalysed approach may be a versatile post-polymerisation method for diversifying copolymer structures.
Introduction
Plastics are essential to everyday life and are used in a number of beneficial applications including packaging to reduce food waste,1 insulation to reduce energy consumption,2 and medical devices to improve quality of life.3 Yet despite these benefits, plastic pollution has caused significant environmental damage. Chemically recyclable and biodegradable polymers, such as poly(lactic acid) (PLA) and poly(ε-caprolactone) (PCL), are attractive alternatives to conventional commodity plastics.4–10 However, their applications are currently limited by their material properties.11 The synthesis of copolymers is a useful method of expanding and enhancing the range of properties available, by harnessing the beneficial properties of two (or more) different monomers. For example, combining lypophilic and hydrophilic monomers generates amphiphilic copolymers with applications spanning drug delivery, polymer coatings, and the compatibilisation of polymer blends,12–16 whilst copolymers combining biodegradable and non-degradable polymers offer a potential method of mitigating the release of micro-plastics into the environment.17–20 PCL/PLA copolymers are of particular interest as their complementary properties enable the formation of biodegradable thermoplastic elastomers that can be used in applications such as tissue engineering.21–23In addition to the choice of monomers, the copolymer properties are dictated by the composition and structure. The monomer distribution can vary from completely random to fully segmented block copolymers;24–31 the latter can give nanoscale phase-separation between the individual blocks, which can lead to favourable thermal and mechanical properties.32,33 Yet while the synthesis of PCL and PLA homopolymers via the ring opening polymerisation (ROP) of ε-caprolactone (ε-CL) or lactide (LA) is straightforward to achieve, the synthesis of PCL-block-PLA multiblock copolymers is challenging because transesterification can disrupt the block copolymer structure.34–36 Transesterification is particularly prevalent when extending a PLA* chain (* denotes a living polymer chain) with ε-CL, and very few catalysts have been successful in generating well-controlled di- or tri-block copolymers from PLA*.37,38 We previously reported that catalyst LZn2Et/BnOH (Fig. 1) delivered excellent activity and control in the synthesis of PCL-block-PLA copolymers (i.e. when ε-CL was polymerised first and LA second), yet gave random copolymer structures when ε-CL was added to a living PLA* chain.39
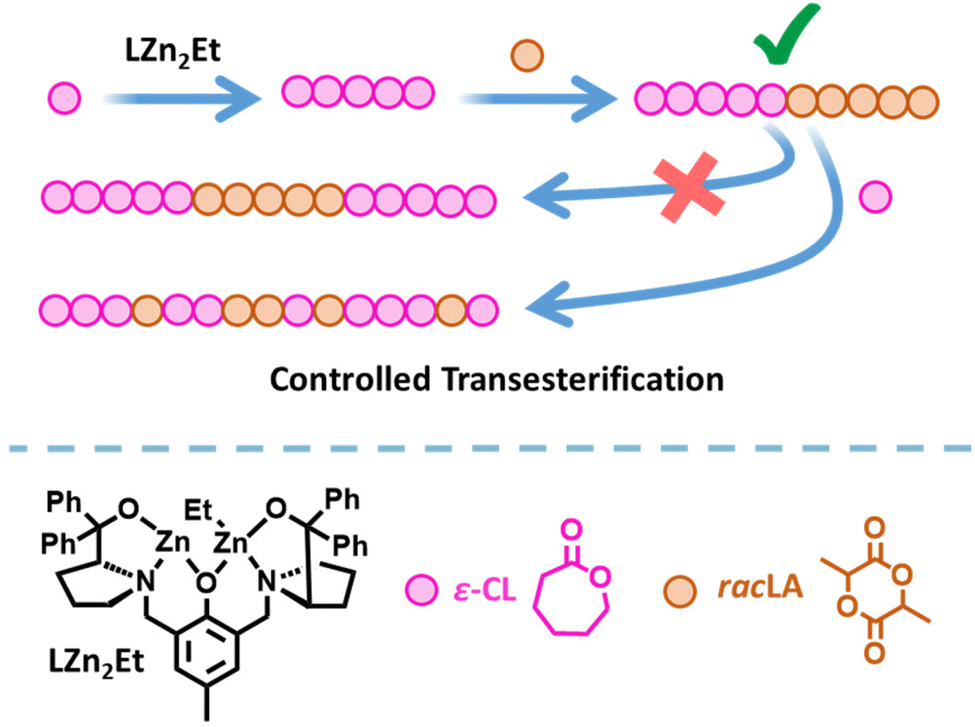 | ||
| Fig. 1 Synthesis of PCL-PLA diblock copolymers via ROP using LZn2Et/BnOH,39 and the strategy investigated herein to modify the copolymer structure using controlled transesterification. | ||
Other methods to prepare multiblock copolymers have included the use of diethylene glycol as a bifunctional alcohol initiator to prepare PLA-PCL-PLA triblock copolymers via formation of a PCL homopolymer with two active chain ends; this approach was limited to the formation of triblock copolymers.40 To achieve higher-order PCL-PLA multiblocks, relatively short hydroxy-end capped block copolymers have been reacted with a diisocyanate, to form urethane linkages between the copolymer chains.41 However, this method is often poorly controlled and leads to broad dispersities.
Could transesterification be a friend instead of a foe? The Platel group recently showed that transesterification can deliver tuneable copolymer structures from mixed LA/CL monomer feeds i.e. via a “bottom up” approach.42 Herein, we report an alternative “top down” method that uses a controlled degree of transesterification to tailor the structure of PCL-block-PLA copolymers to generate higher-order multi-block structures. Transesterification is instigated and controlled by the addition of ε-CL to a PCL-block-PLA* chain in the presence of a bis-zinc catalyst. Through novel modelling simulations underpinned by 13C NMR studies, we investigate the microstructures of the resultant PCL-PLA copolymers and show how this methodology can be utilised to modify isolated copolymers.
Results and discussion
Synthesis of PCL-PLA copolymers
In the presence of BnOH, LZn2Et was previously established as a highly active catalyst for the preparation of well-controlled PCL50-block-PLA50 copolymers through the sequential addition of ε-CL then LA (where 50 represents the targeted degree of polymerisation, DP, of each block).39 Here, the reaction conditions were optimised to prepare extended diblock structures (PCLn-block-PLAn, n = 100, 200 or 400), by increasing the monomer concentration and temperature (ε-CL loading of 22% w/w in toluene, 70 °C). The polymerisation of ε-CL was rapid and well-controlled (≥95% in <5 min, Đ = 1.14, Fig. S2a†), and LA was subsequently added to extend PCL into a diblock copolymer (Scheme 1b). 1H NMR analysis showed high conversion of rac-LA to PLA in under 1 h (≥80%), and SEC analysis confirmed extension of the PCL block (refer to Fig. S2† for details). Notably, this also showcases the tolerance of the LZn2Et/BnOH catalyst system towards higher monomer loadings of n = 400. | ||
| Scheme 1 The one-pot synthesis of PCL-PLA copolymers via ROP using LZn2Et/BnOH where step (a) forms the PCL homopolymer, step (b) forms the PCL-PLA diblock copolymer, and step (c) uses transesterification to tailor the PCL-PLA copolymer structures. | ||
The resultant copolymer composition, structure, and the extent of transesterification within the targeted PCL100-block-PLA100, PCL200-block-PLA200 and PCL400-block-PLA400 copolymers was investigated using 1H NMR spectroscopy. For PCL100-block-PLA100, integration of the PCL and PLA resonances determined the PCL![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PLA composition to be 116:84 (Fig. S2b†), which was comparable to the targeted value. The 1H NMR spectrum also shows distinct resonances for the PCL units that are directly adjacent to PLA units compared to the other PCL units (Fig. S2b†). The relative integrations of these signals can be used to approximate the number average sequence length of each block (lCL and lLA) and the randomness character of the copolymer (R) (see ESI† for details).43 An R value close to 0 suggests a well-defined block copolymer structure whereas an R value close to 1 suggests a random distribution of monomer units. For PCL100-block-PLA100, lCL and lLA were calculated to be 37 and 50, respectively, with an R value of 0.05. These values are close to the theoretical values for PCL100-block-PLA100 (where R, lCL and lLA are calculated to be 0.02, 100 and 100, respectively) suggesting that minimal structure-disrupting transesterification occurs. Intriguingly, the relative integrations remain constant after 24 h at 70 °C (Fig. S3†), suggesting that no additional transesterification occurs without an external source, and that the structure of the living PCL100-block-PLA100* copolymer is stable under these conditions.
:PLA composition to be 116:84 (Fig. S2b†), which was comparable to the targeted value. The 1H NMR spectrum also shows distinct resonances for the PCL units that are directly adjacent to PLA units compared to the other PCL units (Fig. S2b†). The relative integrations of these signals can be used to approximate the number average sequence length of each block (lCL and lLA) and the randomness character of the copolymer (R) (see ESI† for details).43 An R value close to 0 suggests a well-defined block copolymer structure whereas an R value close to 1 suggests a random distribution of monomer units. For PCL100-block-PLA100, lCL and lLA were calculated to be 37 and 50, respectively, with an R value of 0.05. These values are close to the theoretical values for PCL100-block-PLA100 (where R, lCL and lLA are calculated to be 0.02, 100 and 100, respectively) suggesting that minimal structure-disrupting transesterification occurs. Intriguingly, the relative integrations remain constant after 24 h at 70 °C (Fig. S3†), suggesting that no additional transesterification occurs without an external source, and that the structure of the living PCL100-block-PLA100* copolymer is stable under these conditions.
Factors influencing the extent of transesterification
As previous studies from our group showed that the addition of 50 eq. of ε-CL to a PLA50* homopolymer gave a random copolymer structure,39 we decided to investigate the influence of adding a lower quantity of ε-CL to a PCL200-block-PLA200* diblock copolymer, to investigate whether a lower degree of transesterification could give a copolymer with a more “blocky” structure. Therefore, 10, 20, 40, and 80 equivalents of ε-CL were added to a living PCL200-block-PLA200* diblock copolymer (prepared in situ using the bis-zinc catalyst) as a neat liquid, and the resultant transesterification was assessed by 1H NMR spectroscopy. The high concentration of ε-CL drives the ring-opening of the third monomer, a feature that has been previously reported for other systems.44,45 The l and R values showed a clear increase in transesterification upon increasing the ε-CL stoichiometry (Fig. 2 and Table 1). Upon addition of 10 equivalents of ε-CL to the PCL200-block-PLA200* chain, the R value remained close to 0 (0.08), indicating a relatively blocky structure. However, it is worth noting that the R value has increased compared to the diblock copolymer precursor (R = 0.02), indicating that some transesterification has occurred. Upon increasing the ε-CL equivalents to 20, 40 and 80 eq., the R value further increased to 0.18, 0.26, and 0.30, respectively (Fig. 2b), showing that the degree of transesterification and thus the blocky/random nature of the copolymer is dictated by the quantity of ε-CL added. For example, the average sequence length for each copolymer unit was relatively high when 10 eq. of ε-CL was added (lLA = 33; lCL = 21), but this was significantly reduced when 80 eq. of ε-CL was added (lLA = 7; lCL = 6). Notably, the addition of 20 eq. of ε-CL to PCL200-block-PLA200 gave very similar R and l values independent of whether all 20 eq. were added at the same time (sample 2, Fig. S5†), or as two portions of 10 eq. (sample 5, refer to ESI† for details). In all cases, SEC analysis shows that addition of ε-CL increases the dispersity, providing additional evidence for transesterification (Table S1†).46 In some instances, the molar mass decreased after ε-CL was added, suggesting that some intramolecular transesterification/degradation has occurred in these samples.46 Moreover, the DOSY NMR spectra of sample 4, where the highest reduction of molar mass is observed, shows two observable diffusion coefficients that each contain both PCL and PLA units (Fig. S6†), which may be due to intramolecular transesterification resulting in the formation of lower molar mass cyclic species (refer to ESI† for details).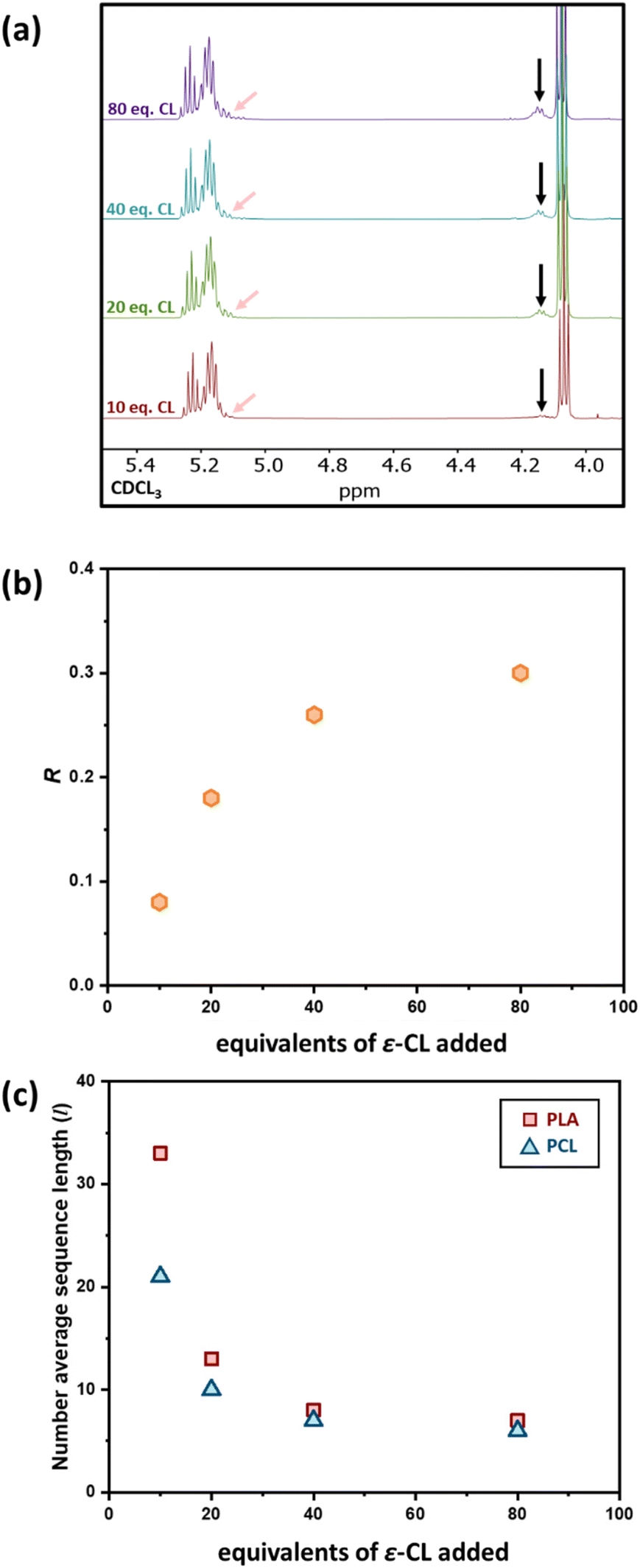 | ||
| Fig. 2 (a) 1H NMR spectra of PCL200-block-PLA200 with various eq. of ε-CL added (indicated on the spectra), where the black arrow indicates PCL protons adjacent to a PLA unit and the pink arrow indicates PLA protons adjacent to a PCL unit. Plots of (b) the randomness value R and (c) the number average sequence length (l) calculated using 1H NMR spectroscopy against the equivalents of ε-CL added. | ||
| Sample | Diblock copolymer | Transesterification | Post-transesterification copolymer structure | |||
|---|---|---|---|---|---|---|
| Targeted PCL DPa | Targeted PLA DPb | Eq. of ε-CL added | l LA | l CL | R | |
| a Conversion of ε-CL was ≥93% in all cases (see Table S1† for additional details). b Conversion of rac-LA was ≥80% in all cases (see Table S1† for additional details). c Calculated using 1H NMR spectroscopy. d 10 equivalents of ε-CL added initially and then another 10 equivalents were added (refer to Fig. S5†). | ||||||
| Control | 200 | 200 | 0 | 100 | 101 | 0.02 |
| 1 | 200 | 200 | 10 | 33 | 21 | 0.08 |
| 2 | 200 | 200 | 20 | 13 | 10 | 0.18 |
| 3 | 200 | 200 | 40 | 8 | 7 | 0.26 |
| 4 | 200 | 200 | 80 | 7 | 6 | 0.30 |
| 5 | 200 | 200 | 20 | 11 | 9 | 0.20 |
| 6 | 100 | 100 | 20 | 5 | 5 | 0.42 |
| 7 | 400 | 400 | 20 | 33 | 27 | 0.07 |
The effect of the initial block length also influences the blockiness of the copolymer structure upon transesterification. This was investigated by adding 20 eq. of ε-CL to PCL100-block-PLA100* (sample 6) and PCL400-block-PLA400* (sample 7). Specifically, higher number-average sequence lengths were achieved when a longer diblock copolymer was used (Table 1 and Fig. 3a). Overall, these results show that the R and l values, i.e. the level of “blockiness” of the resulting copolymer, can be fine-tuned by varying the number of equivalents of ε-CL added to a living PCL-block-PLA-Zn chain as well as the block length of the diblock copolymer precursor. This raises the question: could Zn-mediated transesterification provide a simple method of modifying the copolymer structure from blocky to tailored random copolymers? To answer this, it is important to understand the discrepancy between the number-average sequence length and the true sequence lengths along a polymer backbone.
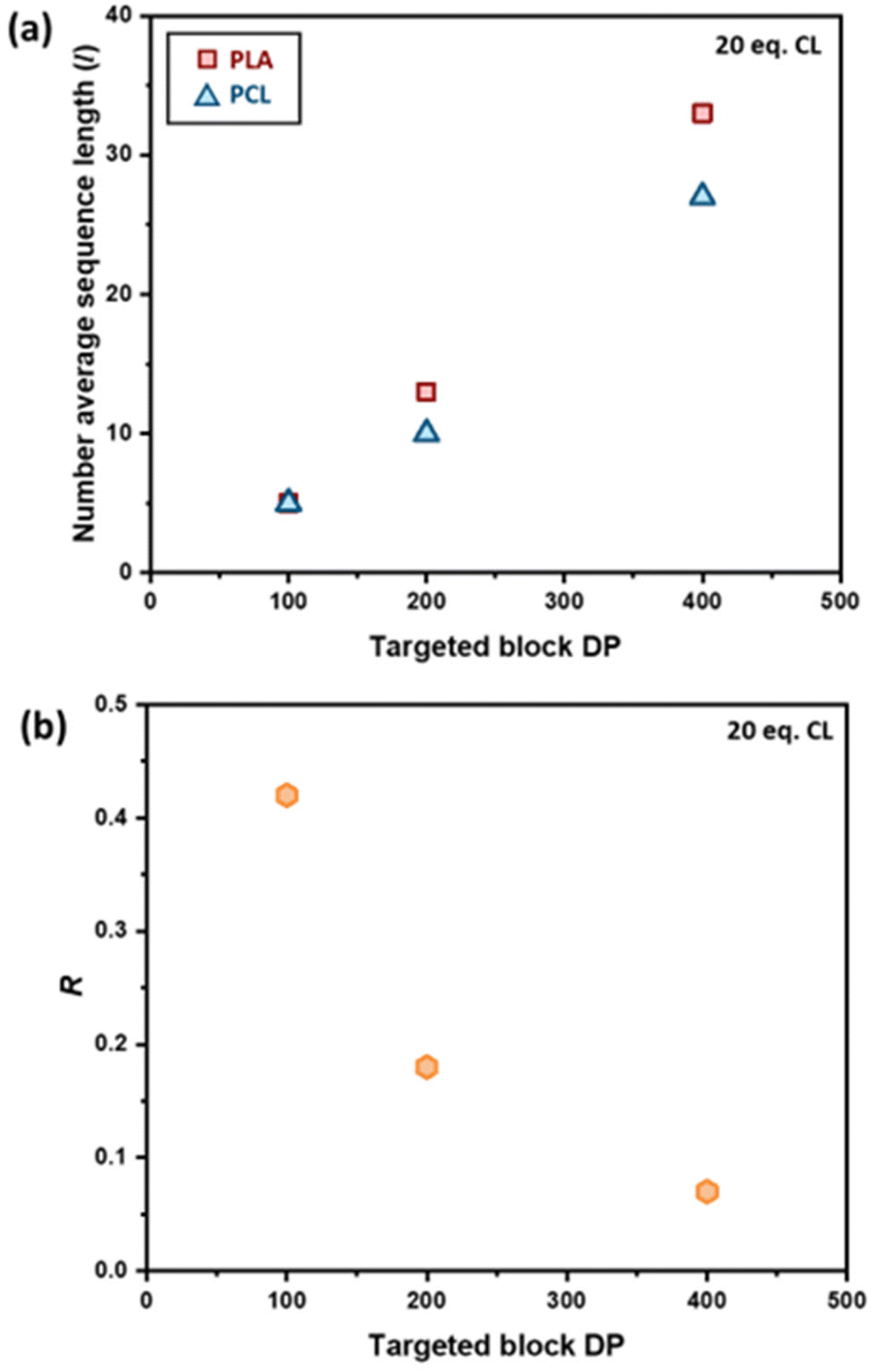 | ||
| Fig. 3 Plots of (a) the number average sequence length (l) and (b) the randomness value R calculated using 1H NMR spectroscopy against the targeted block DP for adding 20 equivalents of ε-CL to PCL100-block-PLA100*, PCL200-block-PLA200* and PCL400-block-PLA400*. | ||
Further understanding the influence of transesterification on the copolymer structure
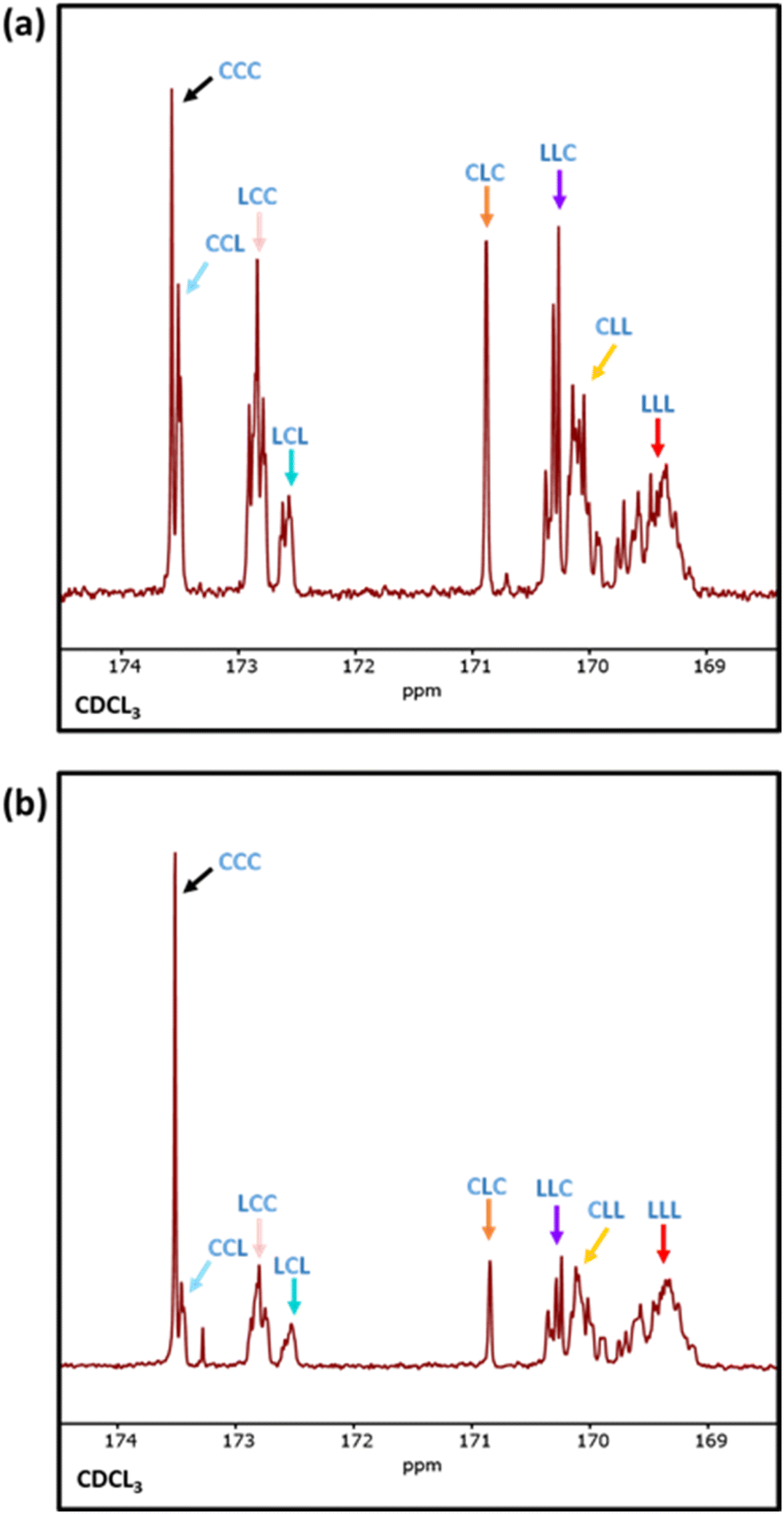
Differential scanning calorimetry (DSC) analysis indicated that copolymer samples 1–4 (Table 1) have a largely segmented structure post-transesterification, as all four polymers displayed thermal behaviours that are associated with block copolymers. Each thermogram showed two distinct glass transition temperatures (Tg) for each component of the copolymer, with no significant reduction in the crystallinity (Fig. S7a†). In contrast, DSC analysis of a fully statistical PCL200-PLA200 copolymer showed a single broad Tg and only a small melting peak (Tm), respectively indicating a lack of block structure and crystallinity (Fig. S8† and Table S2†). These results suggest that transesterified copolymers 1–4 have a somewhat “blocky” structure despite the relatively short average sequence lengths (Table 1). No significant trend was observed between the PCL Tg or Tm and the extent of transesterification (Table S2†). This was largely expected as the thermal transitions of PCL have been reported to remain fairly constant to lower molar masses in block copolymers.47 However, the PLA Tg decreased upon reduction of the number average sequence length (Fig. S7b†). Furthermore, the observed PLA Tg (33–45 °C) is significantly lower than the literature value reported for PLA homopolymers (ca. 60 °C),48 indicating that the presence of PCL/short nature of the PLA blocks has a significant effect on the thermal transitions of the PLA blocks. These results indicate that there is enough ‘blocky’ structure for the individual copolymer components to phase separate and provide two distinct Tg values.We therefore probed the polymer structures by quantitative 13C NMR analysis, as the 13C carbonyl resonances are very sensitive to the sequence of monomer units along the PCL-PLA chain (Fig. 4).49,50 With copolymers 1–4, the most intense signals observed were the CCC and LLL triads, where C and L indicate ring-opened caprolactone and lactic acid units (i.e. half a lactide monomer), respectively. The intensity of these resonances suggest that there are some large blocks of PCL and PLA present even after transesterification, which agrees with the observation of two distinct Tg values in the DSC thermograms. However, transesterification is evident, as baseline resonances due to the LCC, CCL, LCL, CLC, LLC and CLL triads were observed with greater intensities than expected for the triblock copolymer. Importantly, the presence of the CLC signal at 170.8 ppm confirms that transesterification has occurred, as this triad requires the disruption of LL units formed through the ring opening polymerisation of a lactide monomer. The lLA and lCL values were also calculated using the triad intensities from the quantitative 13C NMR spectra.35 For example, sample 3 (Table 1) was calculated to have lLA and lCL values of 12 and 10, respectively, which gave relatively good agreement with the values calculated from the 1H NMR spectra (vide supra). The 13C NMR spectra provided further support for increased transesterification upon increasing the quantity of ε-CL added to PCL200-block-PLA200 (Table 1 and Fig. 4). For sample 1 (10 eq. ε-CL), no signals for the CLC or LCL triads were observed, although small baseline signals for the LCC, CCL, LLC and CLL triads were present. Conversely, all the triad signals are observed for sample 4 (80 eq. ε-CL), with an increase in the CLC signal emphasizing a greater degree of transesterification.
 | ||
| Fig. 4 (a) Expanded 13C NMR spectra of PCL200-block-PLA200 with 10–80 eq. of ε-CL added and the respective peak assignments indicated on the spectra. (b) Shows the same 13C NMR spectra with a magnified baseline. | ||
Overall, the observation of the CLC and LCL resonances shows that the structure is not completely “blocky”, and there are some individual caprolactone and lactic acid units along the polymer chain. It is important to note that, while useful, the 1H and 13C NMR spectra give information that is an average of the entire system. This means that transesterification may only occur in a fraction of the copolymers with others remaining unaffected. Therefore, polymer sample 3 was separated into different molar mass fractions using SEC, and the R value of each fraction was subsequently examined by 1H NMR spectroscopy (Fig. S9†). To confirm that the copolymer sample had been successfully fractionated, SEC analysis was performed and showed that each fraction had a different peak molar mass (Mp, Fig. S9†). Importantly, the 1H NMR spectra showed the R values to be relatively constant across the molar mass fractions, suggesting that the transesterified copolymer structure is fairly consistent across the copolymer sample.
In order to probe the trade-off between transesterification and ε-CL propagation, we investigated the impact of adding 20 eq. of ε-CL to PLA* homopolymer with a DP of 200 (Fig. S10†). 13C NMR signals for the CCL, LCL, CLC, LLC, and CLL triads confirmed that the ε-CL units have been inserted into the copolymer chain. However, the CCC triad was absent. This suggests that no significant propagation of ε-CL occurs, although the observation of the CCL signal indicates the potential for sequential insertion of two ε-CL monomers. This indicates that transesterification occurs more rapidly than ε-CL propagation, which is further supported by the observation of the LCL triad. On the basis of these results, we tentatively propose that monomer insertion is likely to proceed via insertion of a single ring-opened ε-CL monomer to the PLA* chain (i.e. PLA-CL*), which subsequently initiates transesterification of an exogenous PLA* chain resulting in a shorter PLA* homopolymer and a longer PLA* copolymer with a single ring-opened ε-CL unit inserted into the chain (Fig. S11†). Due to the living nature of these chains, which are capped by Zn, this procedure is repeated until maximum conversion of monomeric ε-CL is reached. If the point along the polymer chain at which transesterification occurs is randomly selected, then the distribution of ε-CL units along the chain will also be random.
We were curious to understand whether transesterification of PCL-block-PLA* disrupted solely the PLA* block, or also the initial PCL block. Therefore, we compared the 1H and 13C NMR spectra produced from the reaction of PLA20* oligomers with 20 eq. of ε-CL, to that of PCL10-PLA20* oligomers with 10 eq. of ε-CL (Fig. 5 and S12†). Oligomers were used as simpler model systems to facilitate analysis of the polymer structure, with a relatively large ε-CL:polymer ratio used to increase the extent of transesterification.
 | ||
| Fig. 5 Expanded 13C NMR spectra of (a) PLA20* with 20 eq. of ε-CL (sample 8) and (b) PCL10-block-PLA20 with 10 eq. of ε-CL (sample 9), showing the respective peak assignments. | ||
Firstly, 20 eq. of monomeric ε-CL was added to the PLA20* homopolymer. This increased the Mn value from 6.2 kg mol−1 to 11.8 kg mol−1, confirming incorporation of ring-opened ε-CL into the PLA20* chains (sample 8, Table 2 and Fig. S13a†). Significant transesterification occurred, as evidenced by a substantial signal at 4.13 ppm in the 1H NMR spectrum (Fig. S12a†), and CLC triads in the 13C NMR spectrum (Fig. 5a). The resulting copolymer was determined to have a random structure; both lLA and lCL were calculated to be 1 and 2, respectively (by 1H NMR spectroscopy), and the R value was determined to be 1.33. Notably, this value is greater than 1, which indicates a high concentration of alternating sequences (i.e. CLC and LCL).51 This value is also much greater than the R values of copolymers 1–7, despite using a homopolymer precursor rather than a diblock copolymer, corroborating the influence of block length and ε-CL stoichiometry upon the extent of transesterification. As PLA20* has a low chain length, and the stoichiometry of ε-CL:LA is 1:1, ring-opening and insertion of all ε-CL leads to a highly random copolymer structure. A notable difference between the addition of 20 eq. of ε-CL to PLA20* or PLA200* (vide supra) is that the CCC triad is observed in the former, but not the latter. This suggests that the ε-CL:PLAn ratio influences the relative rates of ε-CL propagation vs. transesterification, with an increased quantity of ε-CL promoting propagation.
| Sample | (Co)polymer | Transesterification | Post-transesterification copolymer structure | ||||
|---|---|---|---|---|---|---|---|
| Targeted PCL DP | Targeted PLA DP | Eq. of ε-CL added | l LA | l L | l CL | R | |
| a Calculated using 1H NMR spectroscopy and rounded to the nearest integer. b Average sequence length of lactic acid unit, L, which is half an LA unit, rounded to the nearest integer. c Calculated using 1H NMR spectroscopy. | |||||||
| 8 | — | 20 | 20 | 1 | 2 | 2 | 1.33 |
| 9 | 10 | 20 | 10 | 2 | 3 | 2 | 1.05 |
Secondly, 10 eq. of ε-CL was added to a PCL10-block-PLA20 oligomer (sample 9, Table 2) and SEC analysis confirmed uptake of ε-CL into the polymer structure (Mn increased from 6.6 kg mol−1 to 9.0 kg mol−1, Fig. S13b†). 1H and 13C NMR analysis confirmed the formation of a random copolymer (lLA = lCL = 2 and R = 1.05, with observation of all eight triads, Fig. 5b and S12b†). Importantly, the relative intensity of both the CCC and LLL resonances was reduced after transesterification with ε-CL. The percentage of ring-opened ε-CL in CCC sequences reduced to 37% (see Fig. S14† for calculation) from 91% (theoretical value of 90%, i.e. 9 PCL units in CCC sequences and 1 in a CCL sequence) after transesterification, and the percentage of LA present in LLL sequences reduced to 52% (see Figure) from 96% (theoretical value of 95%, i.e. 19 PLA units in LLL sequences and 1 in a CLL sequence). If ε-CL was exclusively inserted into the PLA block of the PCL10-PLA20 precursor then the minimum percentage of ε-CL in CCC sequences would be 45% (refer to Fig. S13 in the ESI† for details). Therefore, a reduction to 37% suggests that the PCL block is also disrupted during the transesterification.
DSC analysis of samples 8 and 9 confirmed the highly random post-transesterification structure suggested by the 13C NMR analysis, as only one Tg was observed at −22 °C and −25 °C respectively (Fig. S15†), with no evidence of PCL crystallinity. Additionally, a PCL20-block-PLA20 diblock copolymer was synthesised and used as a reference, which displayed two distinct Tg values (−57 °C and 25 °C) and PCL crystallinity.
Simulation of copolymer structure
The ratio of triads present in the 13C NMR spectra is a rich source of structural information, and computer simulations offer an opportunity to convert this into insight into the polymer structure. Therefore, we simulated potential structures of oligomeric copolymers 8 and 9 using a reverse Monte Carlo procedure (Fig. 6 and S16–S18, see ESI† for more details).52 For 5 × 104 iterations, approximately 6500 sequences with a minimum score of 14 were found for sample 8 whereas approximately 1500 lowest scoring sequences (11) were found for sample 9 (Fig. S16†). For comparison, a random sequence gives a score of 28. For sample 8 the modal mean average sequence lengths for ε-CL and lactic acid (L, which is half an LA unit as LA contains two ester groups that can undergo transesterification, and is denoted as lL = 2lLA) were found to be 1.8 (52% of lowest score sequences) and 2.9 (57% of lowest score sequences), respectively. Furthermore, the standard deviation of the sequence length was 0.93 for ε-CL sequences and 1.79 for L. Sample 9 showed somewhat similar model mean average sequence lengths of 2.1 for ε-CL (53% of lowest score sequences) and 4.0 for L (69% of lowest score sequences), with standard deviations of 1.66 and 2.50, respectively. Examples of the structures simulated for samples 8 and 9 are shown in Fig. S17.† In all cases, largely random copolymer structures were generated, which is concordant with the conclusions manually drawn from the experimental data.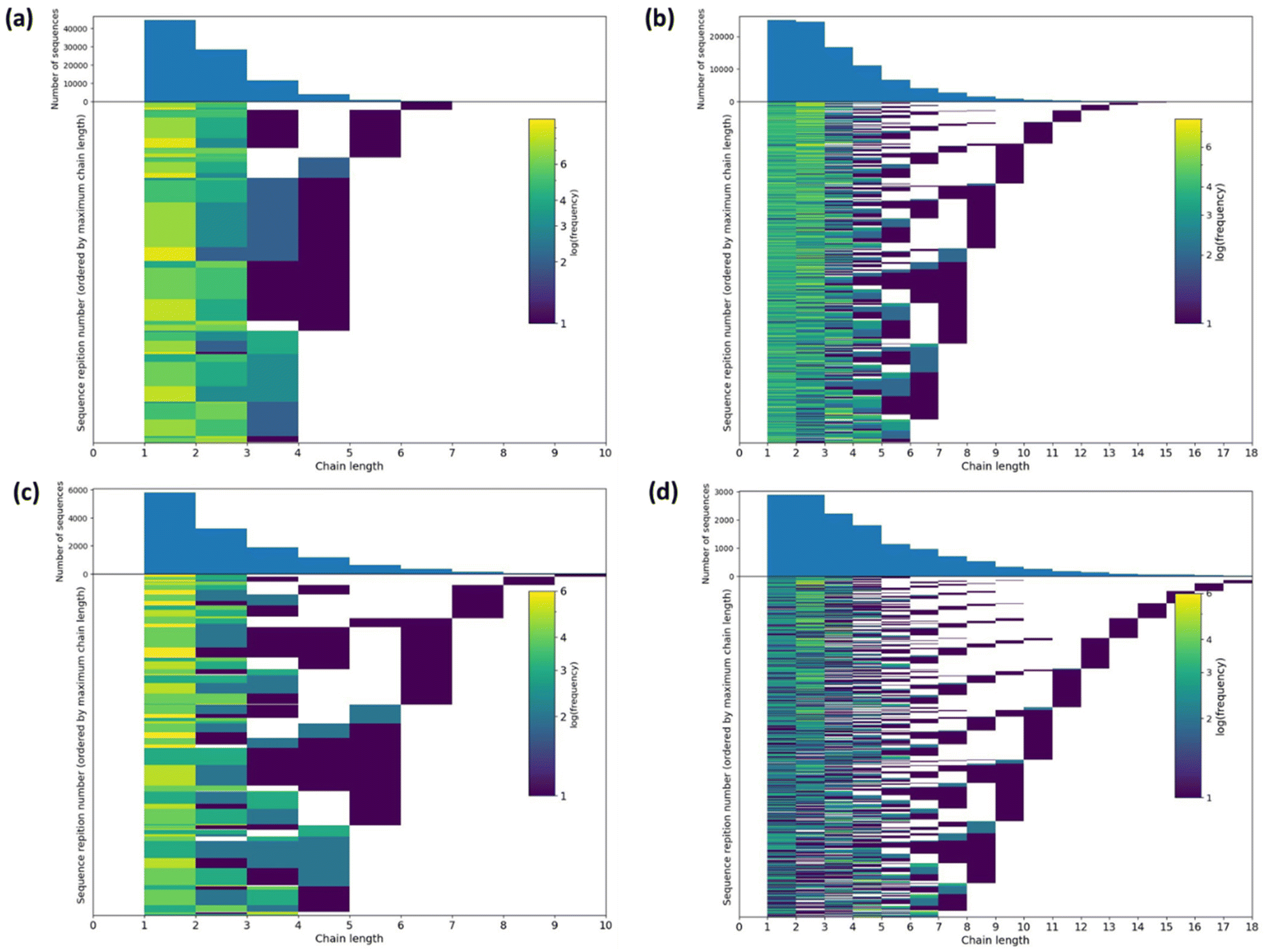 | ||
| Fig. 6 2D histograms of sequence number against sequence length, with the 1D histogram above, for PLA20-PCL20 (sample 8), where (a) is ε-CL and (b) is L, and for PCL10-PLA20-PCL10 (sample 9), where (c) is ε-CL and (d) is L. | ||
Individual sequence lengths for each monomer in the lowest scoring sequences were extracted for samples 8 and 9. To show the frequency with which these sequence lengths occur in the lowest scoring sequences, 2D histograms of sequence number against sequence length were prepared (Fig. 6). These histograms illustrate that there are a high number of low sequence lengths (1–4) present in the majority of the lowest scoring patterns, and that the frequency of sequence lengths present decreases rapidly for higher sequence lengths (>4). The histogram obtained from sample 9 shows a slightly longer tail, which may arise from the lower number of equivalents of ε-CL added compared to sample 8, giving less transesterification. These histograms further demonstrate the randomness of the oligomeric structures produced via transesterification.
Examination of δ-valerolactone
In the trade-off between propagation and transesterification, the monomer propagation rate could play a key role in determining the copolymer structure. δ-Valerolactone (δ-VL) has a lower ring-strain than both rac-LA and ε-CL,53 and so it was hypothesised that adding δ-VL as the third monomer would tip the balance towards transesterification instead of propagation. Therefore, 20 equivalents of δ-VL was added to PLA20* and PCL20* homopolymers (samples 10 and 11, respectively, Table 3).| Sample | Initial polymer | Transesterification | |
|---|---|---|---|
| Targeted PCL DP | Targeted PLA DP | Eq. of δ-VL added | |
| 10 | — | 20 | 20 |
| 11 | 20 | — | 20 |
1H NMR analysis of copolymer 10 showed a significant resonance at 4.14 ppm, indicative of δ-VL units adjacent to LA units and providing clear evidence for δ-VL initiating transesterification (Fig. 7a).54 The 1H NMR spectra of sample 11 does not provide useful information on transesterification due to overlapping resonances for PCL and PVL. However, the 13C NMR spectrum clearly distinguishes between the different α-carbon environments (i.e. the carbon adjacent to the carbonyl), and four resonances are observed between 63.5–64.5 ppm relating to the VC, CC, VV, and CV diads (V and C represent ring-opened δ-VL and ε-CL units, respectively).55 The presence of these diads confirms that δ-VL can be used to transesterify a PCL homopolymer. As the ring-strain of the cyclic esters decreases in the order LA > ε-CL > δ-VL,53 this may explain why δ-VL can transesterify both PCL* and PLA* homopolymers, and why ε-CL transesterifies PLA* homopolymers, yet LA does not transesterify PCL* homopolymers.
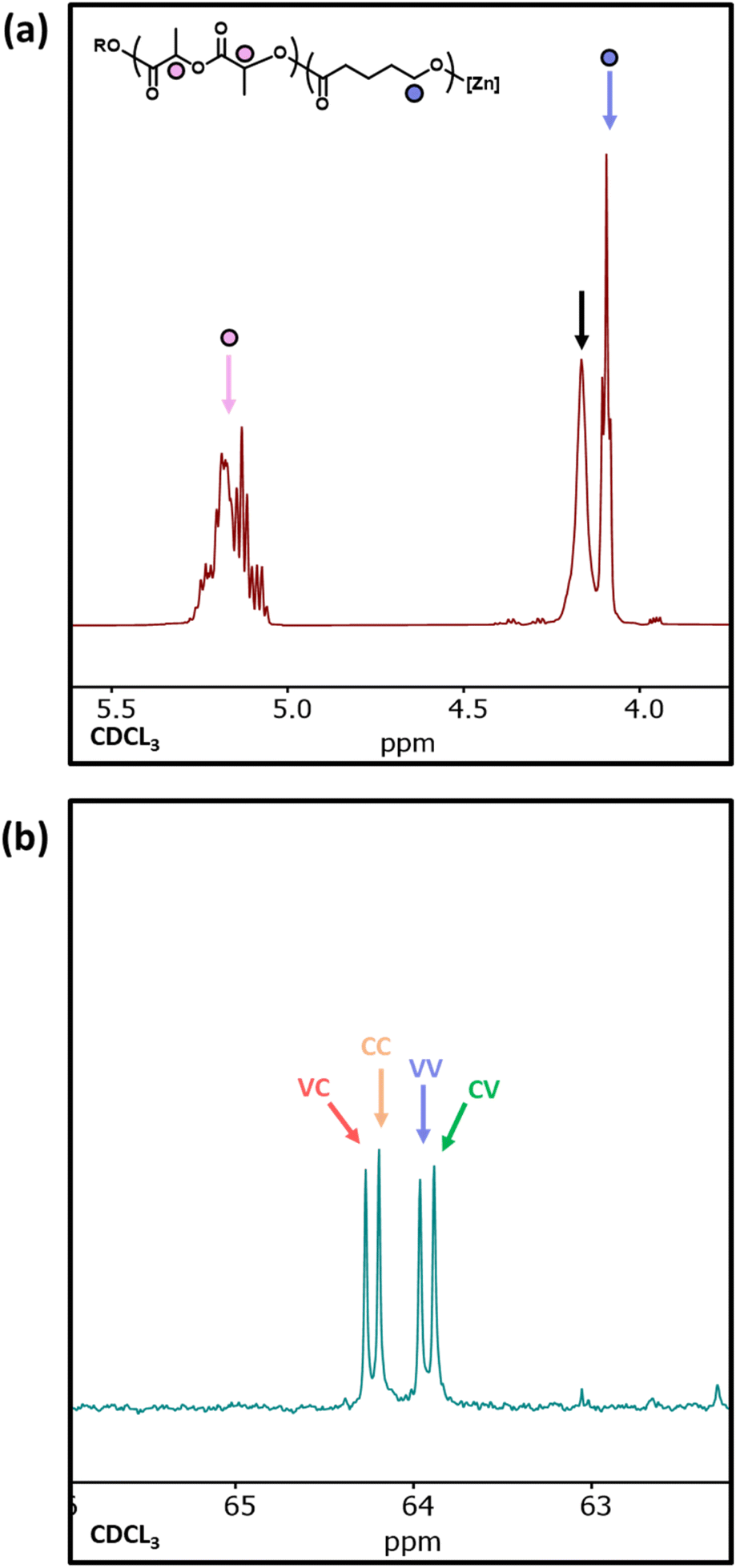 | ||
| Fig. 7 (a) 1H NMR spectra of PLA20-PVL20 (sample 10) where, the black arrow indicates PVL protons adjacent to a PLA unit. (b) Expanded 13C NMR spectra of PCL20-PVL20 (sample 11) with the respective peak assignments indicated on the spectra where V and C represent ring-opened δ-VL and ε-CL units, respectively. | ||
Transesterification of an isolated diblock copolymer
Having demonstrated that transesterification can be used to modulate (co)polymer structures in the presence of a bis-Zn catalyst, we wondered whether this approach could be used as a post-polymerisation modification method for isolated PCL-block-PLA copolymers. Therefore, a PCL200-block-PLA200 copolymer was prepared and isolated through precipitation from methanol. This α-benzoxy, ω-hydroxy-end capped copolymer was subsequently reacted with LZn2Et to generate an active, PCL200-block-PLA200*-Zn copolymer. Subsequent addition of 40 eq. of ε-CL instigated transesterification, as confirmed by 1H and 13C NMR spectroscopy (Fig. S19†). Notably, the copolymer structure obtained through this post-polymerisation modification route deviated slightly from the comparable one-pot synthesis route (sample 3, Table 1). Specifically, lLA, lCL and R were calculated to be 15, 13 and 0.15, respectively (cf 8, 7 and 0.26 for sample 3). This deviation may arise through inefficiencies in forming the activated copolymer, i.e., not all copolymer chains are activated by LZn2Et. Furthermore, no CLC or LCL triads were observed in the 13C NMR spectra, which shows that the extent of transesterification is lower when using an isolated diblock copolymer precursor. Yet transesterification clearly occurs, as there is a marked decrease in the lLA, lCL and R values compared to the PCL200-block-PLA200 precursor. Therefore, this novel post-polymerisation modification via Zn-mediated transesterification has the potential to be a useful methodology to modify polymer structures.Conclusions
Overall, these results show that transesterification can be used as a one-pot method of modulating PCL-block-PLA copolymer structures in the presence of a bis-zinc catalyst. Transesterification was instigated by the addition of ε-CL, and the randomness of the copolymer structure was increased by increasing the ε-CL:PCL-block-PLA* copolymer ratio and decreasing the PCL-block-PLA* block length. In contrast, using a low quantity of ε-CL and a high PCL-PLA block length delivers copolymers with a relatively “blocky” structure after transesterification, as confirmed by DSC and 13C NMR analysis. The trade-off between transesterification and propagation is key, and low ε-CL concentration or the use of δ-VL as a monomer with low ring-strain both tipped the balance towards transesterification instead of propagation.
NMR studies combined with novel reverse Monte Carlo simulations of oligomeric copolymer structures showed that transesterification occurs in both the PCL and PLA blocks of the PCL-block-PLA* oligomer precursors to generate highly random structures with low average sequence lengths. The use of computer simulations is a potentially powerful yet currently underused tool for determining and predicting polymer structures, and provides an interesting avenue for future exploration.
The synthesis of targeted copolymer architectures often relies on an understanding of the monomer reactivity ratios and careful catalyst selection i.e. a “bottom up” approach.41 Here, we show that the post-synthesis addition of ε-CL to a living PCL-block-PLA-Zn chain can disrupt block copolymer structures, providing a complementary “top down” route to generate a diverse range of tailored polymer structures. This approach was successfully used in the post-polymerisation modification of an isolated PCL-block-PLA copolymer. Overall, these results indicate that Zn-mediated transesterification can be used as a method of decreasing the average block length, while maintaining a significantly “blocky” structure.
Author contributions
T. J. N. and J. A. G. designed the project. T. J. N. performed the experiments. E. D. N. wrote the reverse Monte Carlo simulation with consultation from J. C. The manuscript was written by all authors. J. A. G. and J. C. edited the manuscript. J. A. G. acquired the funding and directed the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the UKRI Future Leaders Fellowship (Grant MR/T042710/1), the EPSRC Aerosol Science Centre for Doctoral Training (EP/S023593/1), and the British Royal Society (J. A. G. RSG\R1\180101) for funding. Dr Weronika Gruszka, and Katharine Welch are gratefully acknowledged for preliminary investigations on the synthesis of PCL-PLA copolymers using the LZn2Et/BnOH catalyst system. We also thank the University of Edinburgh NMR facility team (Dr Juraj Bella and Dr Richard York). Finally, we would like to thank Monica Chandwani and Hannah Logan for DSC instrument training and UK Ministry of Defence for funding the equipment.References
- R. Coles and M. Kirwan, Food and Beverage Packaging Technology, Wiley-Blackwell, Chichester, 2nd edn, 2011 Search PubMed.
- H. T. Mohan, K. Jayanarayanan and K. M. Mini, Constr. Build. Mater., 2021, 271, 121520 CrossRef.
- L. W. McKeen, Plastics Used in Medical Devices, Elsevier Inc., 2014 Search PubMed.
- Y. Kumar, P. Shukla, P. Singh, P. P. Prabhakaran, V. K. Tanwar and Y. Kumar, J. Food Prod. Dev. Packag., 2014, 1, 1–6 Search PubMed.
- T. Mekonnen, P. Mussone, H. Khalil and D. Bressler, J. Mater. Chem. A, 2013, 1, 13379–13398 RSC.
- C. R. Álvarez-Chávez, S. Edwards, R. Moure-Eraso and K. Geiser, J. Cleaner Prod., 2012, 23, 47–56 CrossRef.
- R. L. Reddy, V. S. Reddy and G. A. Gupta, Int. J. Emerging Technol. Adv. Eng., 2013, 3, 82–89 Search PubMed.
- M. M. Reddy, S. Vivekanandhan, M. Misra, S. K. Bhatia and A. K. Mohanty, Prog. Polym. Sci., 2013, 38, 1653–1689 CrossRef CAS.
- G. Q. Chen and M. K. Patel, Chem. Rev., 2012, 112, 2082–2099 CrossRef CAS PubMed.
- K. Sudesh and T. Iwata, Clean: Soil, Air, Water, 2008, 36, 433–442 CAS.
- S. Farah, D. G. Anderson and R. Langer, Adv. Drug Delivery Rev., 2016, 107, 367–392 CrossRef CAS PubMed.
- A. Rösler, G. W. M. Vandermeulen and H. A. Klok, Adv. Drug Delivery Rev., 2012, 64, 270–279 CrossRef.
- Y. Morishima, S. Nomura, T. Ikeda, M. Seki and M. Kamachi, Macromolecules, 1995, 28, 2874–2881 CrossRef CAS.
- J. Xu, J. M. Eagan, S. S. Kim, S. Pan, B. Lee, K. Klimovica, K. Jin, T. W. Lin, M. J. Howard, C. J. Ellison, A. M. Lapointe, G. W. Coates and F. S. Bates, Macromolecules, 2018, 51, 8585–8596 CrossRef CAS.
- J. M. Eagan, J. Xu, R. Di Girolamo, C. M. Thurber, C. W. Macosko, A. M. La Pointe, F. S. Bates and G. W. Coates, Science, 2017, 355, 814–816 CrossRef CAS PubMed.
- S. M. King, R. K. Heenan, S. C. Purkiss, R. J. Barlow, P. R. Gellert and C. Washington, Langmuir, 2003, 8428–8435 Search PubMed.
- W. A. Phillip, B. O′neill, M. Rodwogin, M. A. Hillmyer and E. L. Cussler, ACS Appl. Mater. Interfaces, 2010, 2, 847–853 CrossRef CAS PubMed.
- Y. You, C. Hong, W. Wang, W. Lu and C. Pan, Macromolecules, 2004, 37, 9761–9767 CrossRef CAS.
- J. K. Oh, Soft Matter, 2011, 7, 5096–5108 RSC.
- R. Z. Xiao, Z. W. Zeng, G. L. Zhou, J. J. Wang, F. Z. Li and A. M. Wang, Int. J. Nanomed., 2010, 5, 1057–1065 CAS.
- D. Cohn and A. Hotovely Salomon, Biomaterials, 2005, 26, 2297–2305 CrossRef CAS PubMed.
- M. H. Huang, S. Li, D. W. Hutmacher, J. Coudane and M. Vert, J. Appl. Polym. Sci., 2006, 102, 1681–1687 CrossRef CAS.
- M. H. Huang, S. Li and M. Vert, Polymer, 2004, 45, 8675–8681 CrossRef CAS.
- P. A. Lovell and R. J. Young, Introduction to Polymers, CRC Press, Boca Raton, 2011 Search PubMed.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32–38 CrossRef CAS.
- M. Lazzari, G. Liu and S. Lecommandoux, Block Copolymers in Nanoscience, John Wiley & Sons, Weinheim, 2007 Search PubMed.
- P. Alexandridis and B. Lindman, Amphiphilic Block Copolymers: Self-Assembly and Applications, Elsevier, Amsterdam, 2000 Search PubMed.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- A. Rösler, G. W. Vandermeulen and H. A. Klok, Adv. Drug Delivery Rev., 2001, 53, 95–108 CrossRef PubMed.
- N. A. Lynd, A. J. Meuler and M. A. Hillmyer, Prog. Polym. Sci., 2008, 33, 875–893 CrossRef CAS.
- A. J. Ryan, S. M. Mai, J. P. A. Fairclough, I. W. Hamley and C. Booth, Phys. Chem. Chem. Phys., 2001, 3, 2961–2971 RSC.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969 RSC.
- M. Naddeo, A. Sorrentino and D. Pappalardo, Polymers, 2021, 13, 1–19 CrossRef PubMed.
- V. T. Lipik, L. K. Widjaja, S. S. Liow, M. J. M. Abadie and S. S. Venkatraman, Polym. Degrad. Stab., 2010, 95, 2596–2602 CrossRef CAS.
- D. Dakshinamoorthy and F. Peruch, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2161–2171 CrossRef CAS.
- V. T. Lipik and M. J. M. Abadie, Int. J. Biomater., 2012, 2012, 1–11 CrossRef PubMed.
- M. Florczak, J. Libiszowski, J. Mosnacek, A. Duda and S. Penczek, Macromol. Rapid Commun., 2007, 28, 1385–1391 CrossRef CAS.
- D. Pappalardo, L. Annunziata and C. Pellecchia, Macromolecules, 2009, 42, 6056–6062 CrossRef CAS.
- W. Gruszka, L. C. Walker, M. P. Shaver and J. A. Garden, Macromolecules, 2020, 53, 4294–4302 CrossRef CAS.
- Z. Jing, X. Huang, X. Liu, M. Liao, Z. Zhang and Y. Li, RSC Adv., 2022, 12, 13180–13191 RSC.
- T. Yu, J. Ren, S. Gu and M. Yang, Polym. Adv. Technol., 2010, 21, 183–188 CrossRef CAS.
- B. Beament, D. Britton, T. Malcomson, G. R. Akien, N. R. Halcovitch, M. P. Coogan and R. H. Platel, Inorg. Chem., 2024, 63, 280–293 CrossRef CAS PubMed.
- J. Fernández, A. Etxeberria and J. R. Sarasua, J. Mech. Behav. Biomed. Mater., 2012, 9, 100–112 CrossRef PubMed.
- S. Petrovic, Electrochem. Crash Course Eng, 2021, pp. 27–30 Search PubMed.
- S. Penczek, J. Pretula and S. Slomkowski, Chem. Teach. Int., 2021, 3, 33–57 CrossRef.
- P. H. Dubois, I. Barakat, R. Jérême and P. H. Teyssié, Macromolecules, 1993, 26, 4407–4412 CrossRef CAS.
- M. Brogly, S. Bistac, C. Delaite and C. Alzina, Polym. Int., 2020, 69, 1105–1112 CrossRef CAS.
- K. Stefaniak and A. Masek, Materials, 2021, 14, 5254 CrossRef CAS PubMed.
- K. Zółtowska, U. Piotrowska, E. Oledzka, M. Sobczak and D. J. McPhee, Molecules, 2015, 20, 21909–21923 CrossRef PubMed.
- X. X. Zheng and Z. X. Wang, RSC Adv., 2017, 7, 27177–27188 RSC.
- P. Dobrzynski, S. Li, J. Kasperczyk, M. Bero, F. Gasc and M. Vert, Biomacromolecules, 2005, 6, 483–488 CrossRef CAS PubMed.
- E. Neal and T. Neal, Zenodo., 2023 DOI:10.5281/zenodo.10410624.
- A. Duda and S. Penczek, Polymers from renewable resources: biopolyesters and biocatalysis, ACS symposium series, Washington DC, 2000 Search PubMed.
- A. Sangroniz, L. Sangroniz, S. Hamzehlou, N. Aranburu, H. Sardon, J. R. Sarasua, M. Iriarte, J. R. Leiza and A. Etxeberria, Polymers, 2022, 14, 1–14 Search PubMed.
- R. M. Abdur, B. Mousavi, H. M. Shahadat, N. Akther, Z. Luo, S. Zhuiykov and F. Verpoort, New J. Chem., 2021, 45, 11313–11316 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00169a |
| This journal is © The Royal Society of Chemistry 2024 |