
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of architectural design on the thermoresponsive properties of pyrrolidone-based terpolymers†
Lezhi
Wang
,
Haffsah
Iqbal
and
Theoni K.
Georgiou
 *
*
Department of Materials, Imperial College London, Exhibition Road, SW7 2AZ, London, UK. E-mail: t.georgiou@imperial.ac.uk
First published on 7th June 2024
Abstract
This study investigated a new series of amphiphilic, thermoresponsive terpolymers based on pyrrolidone. The terpolymers feature similar compositions and molar masses but differ in their architectures, i.e., the position of comonomers along the polymer chain. All polymers were synthesised via reversible addition–fragmentation chain transfer (RAFT) polymerisation. The study focused on the polymers’ thermoresponsive behaviour in aqueous solutions. Specifically, the cloud point temperatures (Tcp) and self-assembly conformations, as well as the thermally induced sol–gel transitions, were investigated. The terpolymers exhibit a solvent isotropic effect and display different Tcps in deuterium oxide (D2O) and deionised water (H2O), as determined through turbidimetry measurements. The phase transitions were further analysed using temperature-variation 1H NMR spectroscopy. Dynamic light scattering and transmission electron microscopy revealed that the triblock structure could self-assemble into micelles, whereas the statistical polymer could not. The micelle size varied depending on the pH. Visual testing and rheological studies showed how the polymer architecture influences thermoresponsive behaviour, with the BAC architecture exhibiting the widest gelation window. This research illustrates the importance of structure–property relationships and highlights the critical role of polymer architecture in their self-assembly and thermoresponsive properties.
1. Introduction
Polymers capable of undergoing a reversible phase transition in structure or solubility in response to changes in external stimuli, such as temperature,1–3 pH,4,5 light,6–8 and electrical or magnetic fields,9,10 are termed ‘smart polymers’. These polymers have attracted significant attention in fundamental research due to their diverse applications. Among the various stimuli-responsive polymers, thermo- and pH-responsive polymers are of particular interest, especially for their potential roles in the biomedical and pharmaceutical sectors, including the controlled delivery of proteins and drugs.11–13A prime example of such a polymer is poly[(2-dimethyl amino)ethyl methacrylate] (PDMAEMA), which exhibits responsiveness to both pH and temperature.14–16 At a pH of 8.5, this polymer has a lower critical solution temperature (LCST) of around 45 °C.17 In aqueous settings, its pKa lies between 7.3–7.5.18 PDMAEMA features a pendant side group that has a tertiary amine structure. By altering the pH of its environment, PDMAEMA can undergo protonation, becoming more hydrophilic and cationic at low pH levels, while deprotonation at elevated pH levels increases its hydrophobicity.19 The pH sensitivity of PDMAEMA-based hydrogels affects their volume phase transition behaviour near the LCST, a phenomenon attributed to the formation and disruption of hydrogen bonds between the tertiary amine group and water molecules under varying pH conditions.20,21 It has been observed that protonated PDMAEMA hydrogels display increased hydrophilicity and significant swelling in acidic environments, while in basic conditions, they dehydrate and contract.22–24 Thus, the dual responsiveness of PDMAEMA hydrogels to temperature and pH renders them ideal for diverse applications, such as drug delivery,25 contact lenses,26 and biosensors.27
Another promising category of polymers, which exhibits dual responsiveness in the biomedical domain, is the pyrrolidone-based polymers. Their notable biocompatibility, water solubility, and coordination capacity stem from the pyrrolidone group, which also imparts pH sensitivity to the polymer due to its amine group that can be protonated under acidic conditions.28,29 A renowned pyrrolidone-based polymer, poly(N-vinyl pyrrolidone) (PNVP), is utilised in medical devices and pharmaceutical applications due to its biocompatibility.30,31 However, synthesising PNVP poses challenges as NVP, its monomer, is less reactive, especially for the conventional reversible addition–fragmentation chain transfer (RAFT) polymerisation.32 Research indicates that synthesising PNVP polymers requires a careful choice of RAFT agents to ensure optimal reaction control.33,34
To address this challenge, researchers have turned to N-(2-methacryloyloxy)ethyl pyrrolidone (NMEP) as a superior alternative to NVP due to its versatility and ease of synthesis. Unlike NVP, NMEP's polymerisable group is a methacrylate. This characteristic facilitates more straightforward re-initiation with acrylamides, acrylates, and other methacrylates using controlled polymerisation techniques, such as RAFT and atom transfer radical polymerisation (ATRP).35 Notably, Davis and his team successfully synthesised a series of pyrrolidone-based homopolymers with different side chain lengths using free radical polymerisations, showcasing their thermoresponsive attributes.36 Cai's group further refined PNMEP samples using visible light-activated RAFT polymerisation.37,38 They accurately determined the cloud points (Tcps) for PNMEP, which ranged from 71.5 °C to 52.8 °C as the molar mass (MM) increased from 20.6 kg mol−1 to 105.4 kg mol−1. This observed LCST range was subsequently verified by Armes's group.39 They employed RAFT solution polymerisation method and identified an LCST of 55 °C with a MM of around 100 kg mol−1. Additionally, Gibson et al., utilising a carboxylic acid-functional RAFT agent in ethanol, again through the RAFT solution polymerisation technique, produced PNMEP polymers with a pKa that was measured between 5.07 and 5.44.40
Researchers have exploited the stimuli-responsive capabilities of NMEP-based polymeric systems to harness a wide array of their applications in the biomedical field. Teunissen et al. developed thermoresponsive diblock copolymer brushes based on PNMEP using surface-initiated atom transfer radical polymerisation, showcasing their suitability for biomedical applications due to their exceptional antifouling characteristics and temperature-responsive behaviour.41,42 In a separate study, Magalhães et al. synthesised flexible and biocompatible hydrogels by combining NMEP with hyaluronic acid (HA), underscoring the potential of PNMEP-HA hydrogels in drug delivery systems tailored to the body's internal pH variations.43 Jia et al. employed an amphiphilic diblock copolymer composed of PNMEP to anchor haemoglobin, achieving impressive protein loading and maintaining bioactivity. This points to potential uses in biosensors and bioreactors.44 Additionally, Cheng et al. unveiled a novel pyrrolidone-based amphiphilic diblock copolymer consisting of PNMEP and poly(methyl methacrylate) (PMMA). Upon self-assembly in isopropanol, this polymer formed thermoresponsive organogels with 3D micellar networks, hinting at potential applications in transdermal drug delivery and nanomaterials.45
Herein, a series of terpolymers featuring novel combinations of comonomers was synthesised via RAFT polymerisation, harnessing the dual-responsive properties of DMAEMA and NMEP. Previous research explored DMAEMA-MMA copolymers in both diblock and statistical architectures, revealing that only the diblock copolymer, with a MM of 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 g mol−1, was able to form a thermoresponsive gel.46 Consequently, this study introduced an extra block and investigated the gelation properties of terpolymers, focusing on those comprising DMAEMA (A block), MMA (B block), and NMEP (C block), arranged in various architectural arrangements—ABC, BAC, BCA, and statistical—with similar MMs and compositions. Both PDMAEMA and PNMEP exhibited thermoresponsive behaviour and weak cationic polyelectrolyte characteristics, while PMMA was chosen as the non-ionic hydrophobic component due to its extensive use in biomedical devices. The thermal transitions and self-assembly behaviour of these terpolymers were explored. Special emphasis was placed on how the position of the blocks, i.e. architecture, affects the formation of thermogels.
900 g mol−1, was able to form a thermoresponsive gel.46 Consequently, this study introduced an extra block and investigated the gelation properties of terpolymers, focusing on those comprising DMAEMA (A block), MMA (B block), and NMEP (C block), arranged in various architectural arrangements—ABC, BAC, BCA, and statistical—with similar MMs and compositions. Both PDMAEMA and PNMEP exhibited thermoresponsive behaviour and weak cationic polyelectrolyte characteristics, while PMMA was chosen as the non-ionic hydrophobic component due to its extensive use in biomedical devices. The thermal transitions and self-assembly behaviour of these terpolymers were explored. Special emphasis was placed on how the position of the blocks, i.e. architecture, affects the formation of thermogels.
2. Experimental
2.1 Materials
1-(2-Hydroxyethyl)-2-pyrrolidone (MM = 129.16 g mol−1, 98%), methacryloyl chloride (MM = 104.54 g mol−1, 97%), DMAEMA (MM = 157.21 g mol−1, 98%), MMA (MM = 100.12 g mol−1, 99%), 2-cyano-2-propyl benzodithioate (CPDB, >97%, employed as the chain transfer agent (CTA) for RAFT polymerisation), 2,2′-azobis(2-methylpropionitrile) (AIBN, 98%, free radical initiator), 1,4-dioxane (99.8%, anhydrous), and N,N-dimethylformamide (DMF, HPLC grade ≥99.9% and anhydrous ≥99.8%) were procured from Sigma-Aldrich, UK. Sodium carbonate (Na2CO3, ≥98%, purified), sodium chloride (NaCl, ≥99%, purified), magnesium sulphate (MgSO4, ≥98.0%, anhydrous) and n-hexane (≥97%, HPLC grade) were obtained from VMR Chemicals. Triethylamine (99%, anhydrous) was purchased from Fluorochem. Lithium Bromide (LiBr, 99%, purified), chloroform (99%, anhydrous), diethyl ether (≥99%, HPLC grade), and polytetrafluoroethylene (PTFE) hydrophilic syringe filters (0.45 μm pore size, 25 mm diameter) were sourced from Thermo Fisher Scientific. The deionised water was produced using the ELGA Purelab Option Water Purification System. The SEC calibration standards, PMMA samples with molecular masses (MM) of 0.54, 1.09, 1.81, 7.20, 13.90, 30.78, 72.80, 146.50, 260.90, 538.50, 1020.00, and 2210.00 kg mol−1, were purchased from Agilent Technologies.In addition to the above, other chemicals essential for the monomer and solvent purification, as well as polymer characterisation, were also acquired from Sigma-Aldrich, UK. These include calcium hydride (CaH2, ≥97%, drying agent), basic aluminum oxide (Al2O3·KOH), 2,2-diphenyl-1-picrylhydrazyl hydrate (DPPH), deuterated chloroform (CDCl3, 99.8%), phosphate-buffered saline (PBS, tablets, pH = 7.4 at 25 °C), sodium hydroxide pellets (NaOH, 97%) and hydrochloric acid solution (volumetric, 1 M).
2.2 Synthesis of N-(2-(methacryloyloxy)ethyl) pyrrolidone (NMEP) monomer
N-(2-(Methacryloyloxy)ethyl) pyrrolidone (NMEP) was in-house synthesised via the reaction between 1-(2-hydroxyethyl)-2-pyrrolidone and methacryloyl chloride. This procedure has been documented in previous studies.36,38,47–49 Specifically, 1-(2-hydroxyethyl)-2-pyrrolidone (114.3 g, 0.89 mol), anhydrous triethylamine (179.1 g, 1.77 mol), and 400 mL anhydrous chloroform were added into a 2L dried round-bottom flask. The reaction flask was then immersed in a thermostatic ice bath at 0 °C. A solution of methacryloyl chloride (120.3 g, 1.15 mol) diluted in 100 mL of anhydrous chloroform was added dropwise to the flask over a period of 2 hours. The mixture was stirred, allowed to return to room temperature, and reacted overnight.Upon completion of the reaction, the white ammonium salt was removed through filtration. The remaining solution was concentrated using rotary evaporation and extracted with a 5% Na2CO3 solution, followed by a saturated NaCl solution. The mixture was then dried under anhydrous MgSO4 and filtered. It was then passed through basic alumina and distilled under reduced pressure to yield the purified target monomer. The successful synthesis of the monomer was verified using 1H NMR (δ, in CDCl3). δ 6.01 (m, 1H, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CH3)–), δ. 5.49 (m, 1H, CH2C(CH3)–), δ 4.18 (m, 2H, –OCH2–), δ 3.50 (m, 2H, –CH2N(CO)CH2CH2CH2), δ 3.39 (m, 2H, –N(CO)CH2CH2CH2), δ 2.33 (m, 2H, –N(CO)CH2CH2CH2), δ 1.90 (m, 2H, –N(CO)CH2CH2CH2) and δ 1.80 (d, 3H, CH3C(CH2)–), as shown in Fig. S1.† Prior to use, the NMEP monomers were distilled at 140 °C under reduced pressure.
C(CH3)–), δ. 5.49 (m, 1H, CH2C(CH3)–), δ 4.18 (m, 2H, –OCH2–), δ 3.50 (m, 2H, –CH2N(CO)CH2CH2CH2), δ 3.39 (m, 2H, –N(CO)CH2CH2CH2), δ 2.33 (m, 2H, –N(CO)CH2CH2CH2), δ 1.90 (m, 2H, –N(CO)CH2CH2CH2) and δ 1.80 (d, 3H, CH3C(CH2)–), as shown in Fig. S1.† Prior to use, the NMEP monomers were distilled at 140 °C under reduced pressure.
2.3 Synthesis of a PMMA or PDMAEMA macro-CTA
PDMAEMA and PMMA served as macro-CTAs for subsequent chain extensions. Both DMAEMA and MMA are commercially available monomers. Their purification involved: (i) passing through basic alumina to remove inhibitors and impurities; (ii) adding DPPH tprevent unintended polymerisation; (iii) introducing CaH2 to eliminate moisture; and (iv) performing vacuum distillation on the day of polymerisation to ensure the purity of the monomers.The detailed synthetic procedure for the macro-CTA is outlined below, using PMMA as an example. MMA monomer (12 g, 0.120 mol) was added to a 100 mL round-bottom Schlenk flask, along with the following components: CPDB (CTA, 1.06 g, 4.79 mmol; target DP = 25), AIBN (78.73 mg, 0.48 mmol; [CPDB]:[AIBN] = 10:1), and DMF (20.0 g). The Schlenk flask underwent freeze–pump–thaw cycles for 45 minutes and was then placed in a preheated oil bath at 70 °C for 12 hours. One portion of the PMMA sample was extracted for SEC and 1H NMR analysis. Using 1H NMR spectroscopy, the degree of polymerisation (DP) of the resulting macro-CTA was calculated, as shown in Fig. S2and S3.† Two peaks were selected for calculating DP: specifically, PMMA exhibited a signal at δ = 3.6 ppm attributed to the methyl protons at the end of its side chain, and a signal at δ = 7.8–8.0 ppm due to the aromatic protons on the end group of the RAFT agent.
2.4 Synthesis of PNMEP-based terpolymers and their diblock precursors
Following the successful synthesis of PMMA and PDMAEMA macro-CTAs, four amphiphilic terpolymers with different block orders (i.e. ABC, BAC, BCA and statistical) were prepared by sequential RAFT polymerisations on PMMA or PDMAEMA precursors, as depicted in Fig. 1.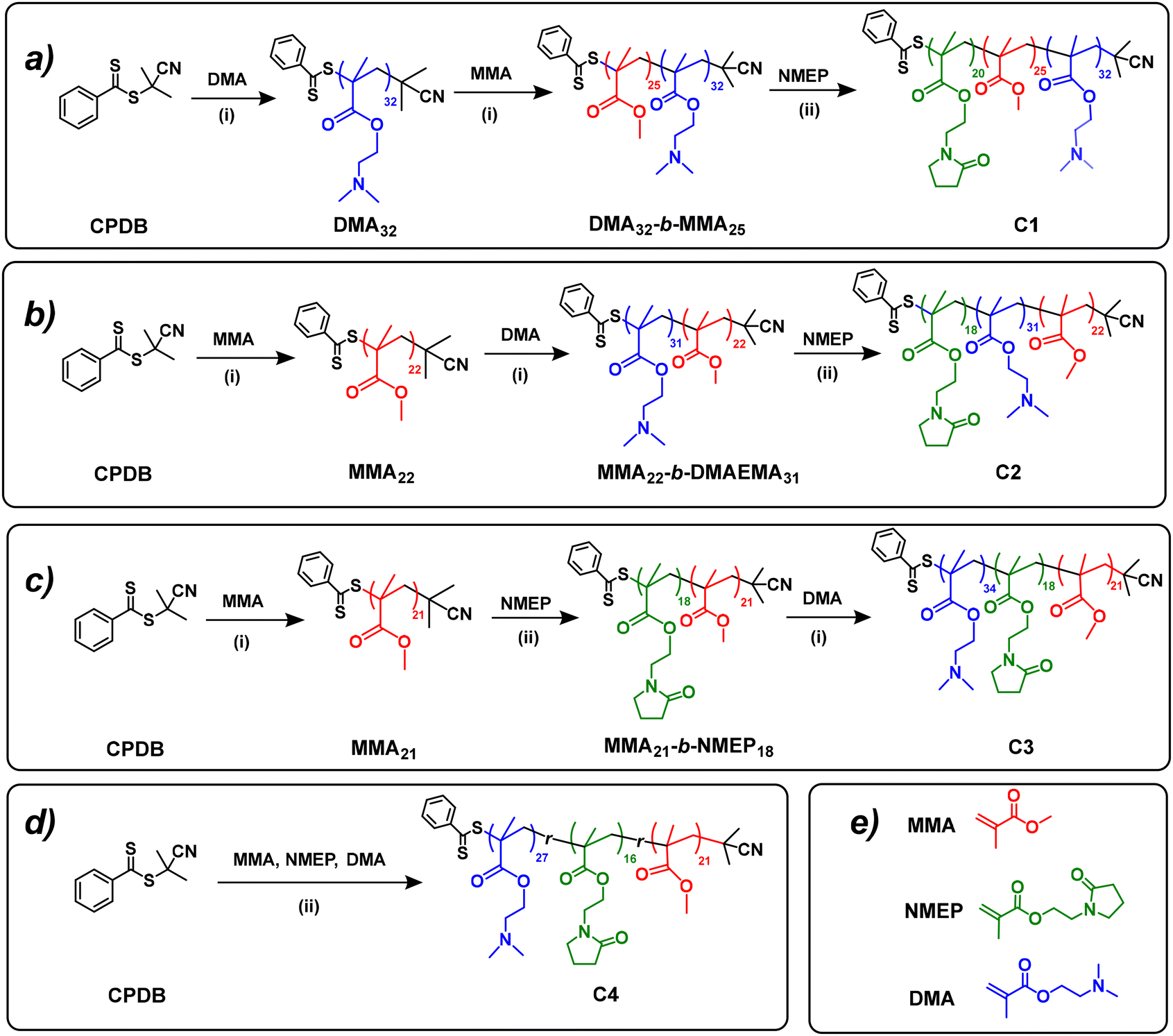 | ||
| Fig. 1 Synthetic route of all amphiphilic terpolymers (C1–C4) in this study via RAFT polymerisation. Roman numerals (i) and (ii) indicate the reaction conditions: (i) reaction temperature of 70 °C with AIBN as the initiator and anhydrous DMF as the solvent, (ii) reaction temperature of 70 °C with AIBN as the initiator and 1,4-dioxane as the solvent. | ||
The detailed synthetic route for MMA22-b-DMAEMA31-b-NMEP18 (C2) serves as an illustrative example as follows: PMMA22, used as the macro-CTA (2.27 g, 0.908 mmol), was combined in a 50 mL Schlenk flask with the DMAEMA monomer (5.0 g, 31.8 mmol), AIBN (22.4 mg, 0.136 mmol; [macro-CTA]:[AIBN] = 6.7:1), and DMF(6.75 g). After undergoing freeze–pump–thaw cycles, the reaction mixture was sealed, with the deoxygenated solution later heated in a preheated oil bath at 70 °C for 24 hours. The resulting diblock was then characterised using 1H NMR and SEC to determine its structure and MM. The identified structure, MMA22-b-DMAEMA31, was precipitated in diethyl ether and served as the macro-CTA for the subsequent step.
The diblock macro-CTA (5.01 g, 0.66 mmol), NMEP monomer (2.6 g, 13.18 mmol), and AIBN (16.2 mg, 0.10 mmol; [macro-CTA]:[AIBN] ratio = 6.6:1) were dissolved in 1,4-dioxane (6.9 g) within a 50 mL Schlenk flask, and the mixture was deoxygenated using freeze–thaw cycles. Following a 24-hour reaction in a 70 °C-oil bath, the final product was obtained through precipitation in n-hexane. The 1H NMR spectra of the final polymers and the precursors are presented in Fig. S4.† The statistical terpolymer (C4) was synthesised similarly to the triblock ones, with all comonomers, the CTA, and the free radical initiator added together at the beginning, yielding the final product in a single reaction step under identical conditions. The detailed reaction ratios for the RAFT polymerisations of all the terpolymers are listed in Table S1.†
2.5 Polymer characterisations
The theoretical MM for polymer synthesis could be calculated by employing the equation:
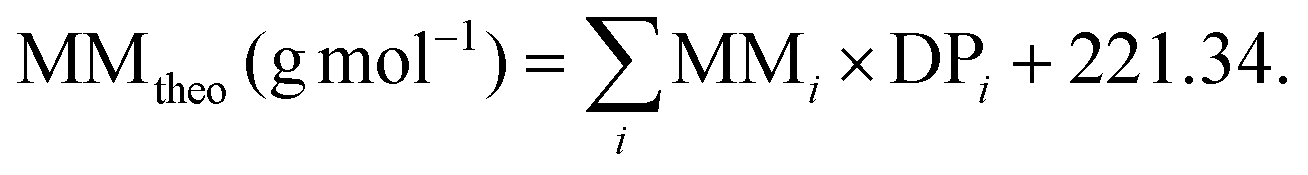
In this equation, the targeted DP of each block is multiplied by the MM of each comonomer and then added to the MM of the chain transfer agent CPDB.
O)OCH3), (ii) for PNMEP, the distinctive peak is observed with proton resonance signals at around δ = 2.00–2.13 ppm (–N(CO)CH2CH2CH2), (iii) for PDMAEMA, the distinctive peak is seen at around δ = 2.53–2.70 ppm ((CO)OCH2CH2).
The temperature-dependent 1H NMR analysis was also performed on the same machine for polymers dissolved in D2O at a concentration of 10 mg mL−1. Measurements were conducted at five different temperatures for each sample, including 25 °C, 30 °C, 35 °C, 40 °C, and 45 °C. The samples were allowed to settle at each temperature for 5 minutes prior to each measurement.
The hydrodynamic diameters of triblock terpolymers, derived from the DLS, were subsequently compared to theoretically modelled values for a spherical micelle with fully extended chains. These values were based on the projected length of one methacrylate unit (0.254 nm) and the corresponding experimental DP. Specifically, the following equations were used: (i) for the ABC architecture, where the hydrophobic MMA is positioned as the middle block, the theoretical dh is calculated as dh (nm) = (DPMMA + 2 × DPDMAEMA) × 0.254 nm; (ii) for the BAC and BCA architectures, where the hydrophobic MMA forms a distinct block at the end of the polymer chain, the theoretical dh is calculated as dh (nm) = [DPMMA + 2 × (DPDMAEMA + DPNMEP)] × 0.254 nm; (iii) for the statistical terpolymer, the theoretical dh is calculated as 〈dg2〉1/2 = 2 × [2 × 2.20 × (DPNMEP + DPDMAEMA + DPMMA)/3]1/2 × 0.154 nm, assuming the formation of a random polymer coil.50 The experimental DPs were calculated from their composition and MM values, which were determined previously via SEC and 1H NMR analyses.
3. Results and discussion
3.1 Synthesis and characterisation of the terpolymers: molar mass, composition and pKa
This research synthesised a series of PNMEP-based terpolymers through RAFT polymerisation, a method well-established in existing studies. Notably, prior works have utilised RAFT solution, dispersion, and emulsion polymerisation techniques,40 which can be activated by either visible light37,38 or temperature.39,40 In this investigation, four unique PNMEP-based terpolymer structures – ABC, BAC, BCA, and statistical – were developed. The RAFT polymerisation process, employing CPDB as the CTA and AIBN as the initiator, was conducted under conditions of 70 °C for durations of either 12 or 24 hours, as detailed in Table S1.†The terpolymers in this study were synthesised from three distinct comonomers: A (DMAEMA), B (MMA), and C (NMEP). Comonomers A and C (DMAEMA and NMEP) are noted for their thermoresponsive properties, while B (MMA) imparts hydrophobic characteristics to the polymer. The synthesis targeted a consistent MM of 12171 g mol−1, with compositions across all architectures aiming for a specific ratio of 46% w/w DMAEMA, 21% w/w MMA, and 33% w/w NMEP. This ratio was selected based on preliminary findings that indicated the terpolymer's thermoresponsive behaviour is compromised, rendering it insoluble in deionised water when the MMA content exceeds 25% w/w. Specifically, a triblock copolymer of MMA-b-DMAEMA-b-NMEP was fabricated with a targeted 25% w/w MMA, which was found to be insoluble in water. Table 1 presents detailed structural information of these polymers, demonstrating compositions that closely align with the targeted values, within an acceptable deviation range. Further details, including SEC traces and 1H NMR spectra with peak assignments and DP calculations, are provided in the ESI,† section 5–6.
| No. | Experimental polymer structurea,b | MM(Target) (g mol−1) |
M
n (SEC)c (g mol−1) |
Đ | Compositiond (% w/w) | pKae |
|---|---|---|---|---|---|---|
| a Abbreviations: NMEP, DMA, and MMA correspond to N-(2-(methacryloyloxy)ethyl) pyrrolidone, 2-(dimethylamino)ethyl methacrylate, and methyl methacrylate, respectively. Note that DMA is a further abbreviation of DMAEMA. b The experimental degrees of polymerisation (DP) of the polymers, after precipitation, were calculated using the Mn and the experimental compositions obtained by SEC and 1H NMR analysis, respectively. c The number average molar mass (Mn) of the polymers was determined through SEC analysis after precipitation. d The composition of each component within the terpolymer in % w/w was calculated through 1H NMR analysis. e The pKa values of the final terpolymer products were determined by potentiometric titrations. | ||||||
| C1 | DMA32 | 5724 | 4400 | 1.19 | 100–0–0 | NA |
| DMA32-b-MMA25 | 8227 | 7000 | 1.20 | 67–33–0 | NA | |
| DMA32-b-MMA25-b-NMEP20 |
12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 171 171
|
11700
|
1.28 | 44–22–34 | 5.4/7.5 | |
| C2 | MMA22 | 2724 | 2600 | 1.19 | 100–0–0 | NA |
| MMA22-b-DMA31 | 8227 | 7600 | 1.19 | 31–69–0 | NA | |
| MMA22-b-DMA31-b-NMEP18 |
12171
|
10800
|
1.30 | 21–46–33 | 5.4/7.6 | |
| C3 | MMA21 | 2724 | 2600 | 1.19 | 100–0–0 | NA |
| MMA21-b-NMEP18 | 6669 | 5200 | 1.25 | 37–63–0 | NA | |
| MMA21-b-NMEP18-b-DMA34 |
12171
|
11200
|
1.24 | 19–32–49 | 5.3/7.5 | |
| C4 | MMA21-co-NMEP16-co-DMA27 |
12171
|
9900 | 1.18 | 24–29–47 | 5.2/7.4 |
In addition to their thermoresponsive nature, all synthesised polymers C1–C4 exhibit pH-responsiveness due to the tertiary amines present in both NMEP and DMAEMA components. Their pKa values were determined through potentiometric titrations on 1% w/w aqueous polymer solutions. It is anticipated that the terpolymer components would display two distinct pKa values corresponding to NMEP (∼5.4) and DMAEMA (∼7.5) respectively. The titration curve for the triblock terpolymer C1, along with the measured pKa, is detailed in Fig. S6.† Similarly, the half-equivalent points of acid dissociation for C2–C4 were determined, with the findings summarised in Table 1. The measurements reveal that the pKa values of the terpolymers, despite their varying architectures, are broadly consistent, with any deviations falling within the method's error margin. The pKa of the statistical architecture was slightly lower, a result that aligns with findings from previous studies.54,55
3.2 Cloud point temperature (Tcp) measurements
Chemically, the NMEP units exhibit thermoresponsive properties arising from the combined effects of the pendant pyrrolidone groups and the apolar methacrylate backbone. Similarly, the thermoresponsive ability of DMAEMA units is attributed to the tertiary amine group on the side chain and the methacrylate backbone. However, DMAEMA's side chain is less polar than that of NMEP, leading to reduced solubility and a lower LCST in polymers with similar MM. This difference between NMEP and DMAEMA has been validated through research by Sun et al.,59 Cunningham et al.,39 and Billingham's group.60Building upon the understanding of NMEP and DMAEMA, this study determined the Tcps of NMEP-containing triblock terpolymers in DI water. Initially, a 1% w/w polymer solution was prepared, and the initial pH was recorded following its complete dissolution—a crucial step for accurate Tcp determination. This helps mitigate errors arising from pH variation or ionic impurities, enabling comparisons of Tcps under different pH conditions and ensuring the precision of measurements. Notably, polymers C1, C2, and C3 exhibited initial pH values of 8.4, 8.0, and 8.7, respectively. Despite being dissolved in the same solvent and sharing similar polymer compositions, these polymers showed varying pH levels, likely due to their distinct architectures, which could influence the Tcp.
Upon 10% protonation, none of the block terpolymers exhibited a Tcp due to the increased hydrophilicity of the polymers. Protonation leads to the polymer micelles becoming positively charged at the tertiary amine groups in the NMEP and DMAEMA side chains, which in turn induces electrostatic repulsion between adjacent micelles. Such repulsion prompts the micelles to swell and maintain colloidal stability in water, a phenomenon extensively documented in previous studies.61–65 However, the statistical terpolymer C4 was an exception; it only became soluble after 10% protonation, which makes the polymer more hydrophilic, thus enabling the measurement of its Tcp at 25 °C, as detailed in Table 2. The reduced solubility of the statistical terpolymer compared to their block-based counterparts, due to its inability to form micelles, is well documented in previous studies.54,55,66
| No. | Experimental polymer structurea | Architecture | Cloud point temperature, Tcpb ( °C) |
||
|---|---|---|---|---|---|
| 0% H+ in H2O | 10% H+ in H2O | 0% H+ in D2O | |||
| a Abbreviations: 2-(dimethylamino)ethyl methacrylate (DMA), methyl methacrylate (MMA), N-(2-(methacryloyloxy)ethyl) pyrrolidone (NMEP). Note that DMA is a further abbreviation of DMAEMA. b The cloud point temperature (Tcp) was determined by turbidimetry. c The determination of Tcp for the statistical terpolymer was not feasible as it is insoluble at 0% protonation. | |||||
| C1 | DMA32-b-MMA25-b-NMEP20 | ABC | 45 | No Tcp | 37 |
| C2 | MMA22-b-DMA31-b-NMEP18 | BAC | 45 | No Tcp | 36 |
| C3 | MMA21-b-NMEP18-b-DMA34 | BCA | 35 | No Tcp | 29 |
| C4 | MMA21-co-NMEP16-co-DMA27 | Statistical | NSc | 25 | NSc |
3.3 Temperature variation 1H NMR
 | ||
| Fig. 2 Transmittance plotted as a function of solution temperature for the Tcp determination of polymers C1, C2 and C3 (from left to right) in both D2O and H2O. | ||
Sun et al. attributed the lower Tcp in D2O to the absence of hydrogen bonding donors in PNMEP, coupled with weaker van der Waals interactions such as hydrophobic associations of apolar backbones and spacers.59 Extending this concept to our study, which also involves comonomers NMEP and DMAEMA—similar to NMEP—lacking hydrogen bonding donors on their side chains, suggests that the interactions of such polymers with D2O inherently lack hydrogen bonding and result in a lower Tcp. This phenomenon has also been reported by Cremer's group in elastin-like peptides (ELPs),68 which exhibit a more stabilised collapsed state in D2O than in H2O, thus displaying a lower Tcp.
However, this mode of interaction stands in contrast to that observed in polymers like PNIPAM, which exhibit a higher Tcp in D2O compared to H2O. This is likely attributed to PNIPAM's stronger hydrogen bonding with D2O molecules, resulting in higher bond strength and an increased enthalpic cost to break these hydrogen bonds.69–71 This difference is possibly due to the fact that NIPAM units have a secondary amine with a hydrogen next to the nitrogen, free to form hydrogen bonds, while both NMEP and DMAEMA units have a tertiary amine with no free hydrogen to form hydrogen bonds.
Fig. 3 reveals that at 25 °C, all three terpolymers display clear and detectable proton resonance signals in D2O, confirming their good solubility at room temperature and near their respective Tcp. Notably, a significant decrease in these proton resonance signals was observed upon reaching their Tcp in D2O, suggesting reduced chain mobility at elevated temperatures.
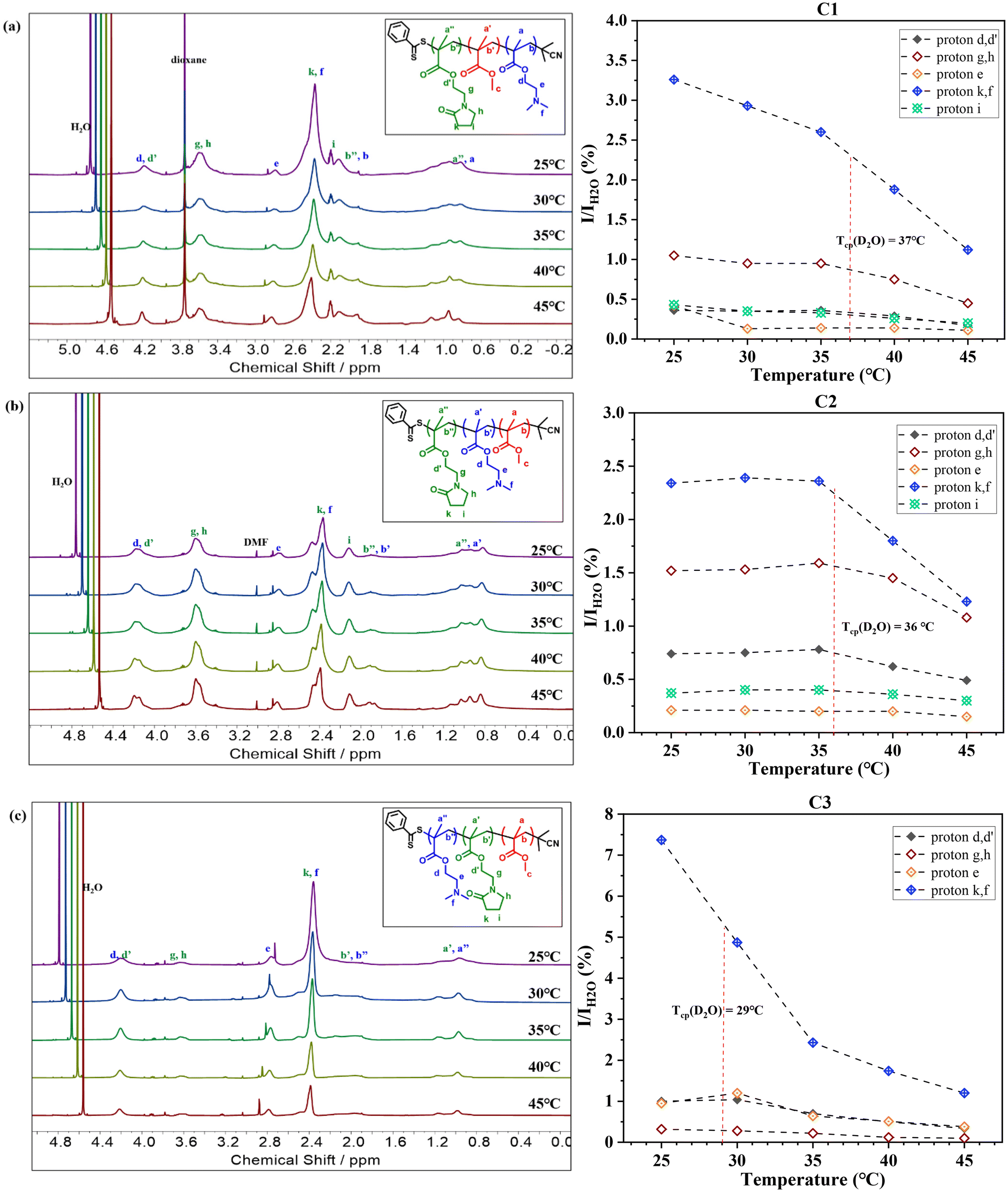 | ||
| Fig. 3 Temperature-variation 1H NMR spectra and corresponding proton integration ratios for a 10.0 mg mL−1 sample of (a) C1, DMA32-b-MMA25-b-NMEP20 (b) C2, MMA22-b-DMA31-b-NMEP18 and (c) C3, MMA21-b-NMEP18-b-DMA34 in D2O to that of H2O (I/IH2O), plotted as a function of solution temperature. | ||
Further analysis involved calculating the relative integral ratios of protons from NMEP and DMAEMA to the integral of H2O in each terpolymer, as illustrated in Fig. 3. This analysis validated the reduction in proton integrals for all three terpolymers above the Tcp. It was observed that the sharpest drop in proton integrals occurs in the peak with a chemical shift around 2.25–2.65 ppm. This drop is attributed to both the protons in the pyrrolidone ring of NMEP (2H, NCOCH2CH2CH2, δ = 2.45 ppm) and the protons of methyl groups in the tertiary amine of DMAEMA (6H, –N(CH3)2, δ = 2.35 ppm).
While most proton resonance signals identifiable in CDCl3 were also detectable in D2O, C3 (MMA21-b-NMEP18-b-DMAEMA34) was an exception. As shown in Fig. 3(c), this terpolymer exhibited markedly weak signals for NMEP protons, particularly protons g and h, and a complete absence of proton i, in contrast to the other terpolymers. Notably, in C3, the only clearly detectable NMEP proton peaks—d and d′, k and f, a′ and a′′, b′ and b′′—are attributed simultaneously to both NMEP and DMAEMA. This suggests that the detectability of these peaks may be primarily due to DMAEMA's influence, rather than NMEP's.
This observation highlights the distinct molecular architecture of C3, which forms a core–shell-corona structure with a permanently hydrophobic MMA core, a partially desolvated NMEP inner shell, and a thick and well-solvated DMAEMA corona. Consequently, NMEP exhibits a hydrophobic character and collapses onto the MMA hydrophobic core when positioned as the middle block. This type of core–shell-corona micelle structure, featuring a pH- or thermo-responsive shell to promote pH- or temperature-induced micellization, has been studied under 1H NMR analysis in D2O and is well-documented in the literature.72–75
This architectural influence also sheds light on the unconventional Tcp trends observed in these triblock terpolymers. Notably, C1 and C2, despite having different architectures, exhibit surprisingly similar Tcp values. In contrast, C2 and C3, both incorporating hydrophobic MMA as their outer block, demonstrate distinctly different Tcp values. This variation can be attributed to the terpolymers’ differing degrees of hydration/solvation of the blocks, influenced by their structural differences, as evidenced by the change in intensity of the signals for different terpolymer architectures under 1H NMR analysis in D2O. Specifically, in C3, the positioning of NMEP as the middle block contributes to its more solvophobic properties, resulting in much attenuated signals under 1H NMR, which in turn affects its thermoresponsive ability. Consequently, the Tcp of C3 is predominantly influenced by the thermoresponsive behaviour of DMAEMA at the outer corona, unlike in C1 and C2, where it is affected by the mutual impact of both NMEP and DMAEMA blocks, resulting in a lower Tcp compared to C1 and C2.
3.4 Self-assembly behaviour
| No. | Experimental polymer structurea | Architecture | Hydrodynamic diameter (±0.5 nm) | ||
|---|---|---|---|---|---|
| Theoreticalb | By intensity | By number | |||
| a Abbreviations: 2-(dimethylamino)ethyl methacrylate (DMA), methyl methacrylate (MMA), N-(2-(methacryloyloxy)ethyl) pyrrolidone (NMEP). Note that DMA is a further abbreviation of DMAEMA. b The theoretical hydrodynamic diameter was calculated by assuming the formation of either a classical core–shell micelle or a polymer coil. Specifically, the following equations were used: (i) for the ABC architecture, where the hydrophobic MMA is positioned as the middle block, the theoretical dh is calculated as dh (nm) = (DPMMA + 2 × DPDMAEMA) × 0.254 nm; (ii) for the BAC and BCA architectures, where the hydrophobic MMA forms a distinct block at the end of the polymer chain, the theoretical dh is calculated as dh (nm) = [DPMMA + 2 × (DPDMAEMA + DPNMEP)] × 0.254 nm; (iii) for the statistical terpolymer, the theoretical dh is calculated as 〈dg2〉1/2 = 2 × [2 × 2.20 × (DPNMEP + DPDMAEMA + DPMMA)/3]1/2 × 0.154 nm, assuming the formation of a random polymer coil. Here, the experimental degrees of polymerisation were used, calculated by multiplying the actual molar mass obtained from SEC with the actual composition determined by 1H NMR. c The hydrodynamic diameter and Tcps of the statistical terpolymer were measured under 10% H+ protonation, as this polymer was insoluble in aqueous solutions without any protonation. Notably, the hydrodynamic diameter of the statistical terpolymer was measured at 10 °C, instead of room temperature (25 °C), due to its cloud point being close to room temperature, which could affect the measurement. | |||||
| C1 | DMA32-b-MMA25-b-NMEP20 | ABC | 22.6 | 15.7 | 6.3 |
| C2 | MMA22-b-DMA31-b-NMEP18 | BAC | 30.5 | 27.4 | 11.7 |
| C3 | MMA21-b-NMEP18-b-DMA34 | BCA | 31.8 | 18.2 | 7.5 |
| C4 | MMA21-co-NMEP16-co-DMA27 | Statistical | 3.0 | 4.2c | 2.3c |
Therefore, the model predicts that C1, characterised by its ABC structure and a central hydrophobic block, forms smaller and structurally distinct micelles compared to C2 and C3, which have hydrophobic MMA at the end of their chains. As depicted in the configuration schematics in Fig. 4, C1 with the centrally located MMA likely contributes to a hydrophilic corona with a thickness comparable to DMAEMA. Conversely, C2 and C3, with terminal MMA, are expected to form micelles with thicker and more extended hydrophilic shells consisting of both DMAEMA and NMEP.
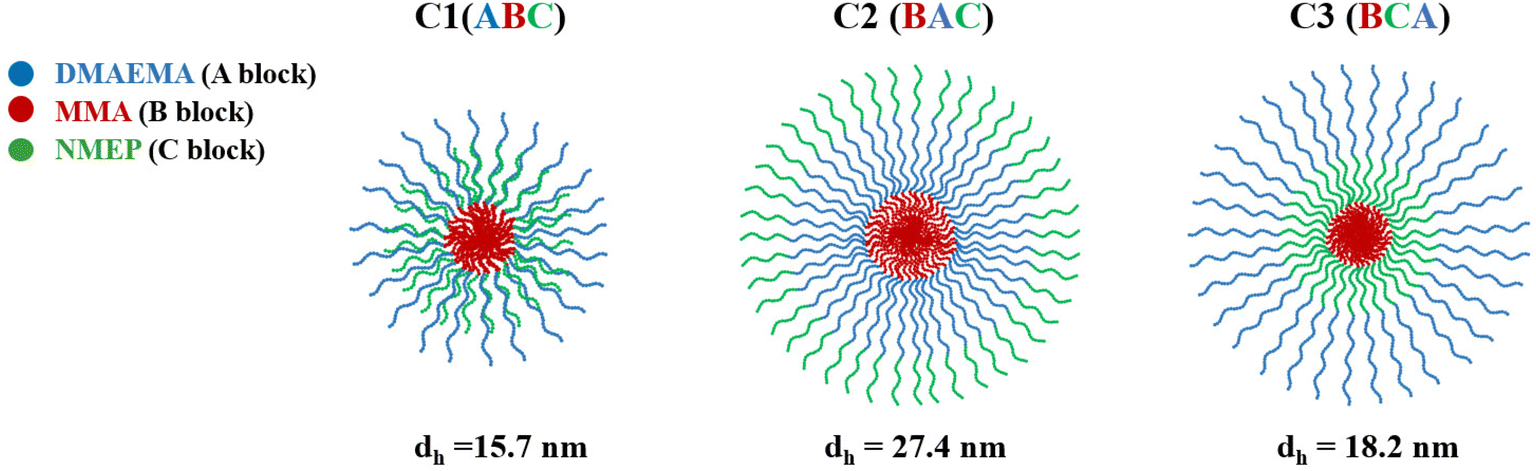 | ||
| Fig. 4 Schematic representation of the proposed micelle configuration and the hydrodynamic diameters as measured by DLS for C1, C2, and C3. | ||
Empirical DLS data supports the theoretical predictions, with C1 showing the smallest diameter compared to the other two. Specifically, the DLS results indicate a descending size order of C2, C3, and C1, with C2 being approximately 1.5 to 2 times larger than the others. The measured micelle diameters are consistently smaller than the theoretically predicted values. This discrepancy between empirical and theoretical values was expected and has been previously reported,54,55,66 attributed to the model assuming fully stretched polymer chains.
This size sequence is also consistent with our group's earlier studies on amphiphilic triblock terpolymer systems based on comonomers with slight variations, namely oligo(ethylene glycol) methyl methacrylate (OEGMA, average Mn = 232.27 or 300 g mol−1).55,66 Similar to our current findings, those studies reported a size sequence of BAC, BCA, and ABC, with ‘A’ denoting DMAEMA, ‘B’ denoting butyl methacrylate, and ‘C’ denoting OEGMA instead of NMEP.
Meanwhile, the size of the statistical polymer was measured by DLS at 10% protonation, since it is insoluble at 0% protonation. The theoretical dh for the statistical terpolymer is calculated as 〈dg2〉1/2 = 2 × [2 × 2.20 × (DPNMEP + DPDMAEMA + DPMMA)/3]1/2 × 0.154 nm, assuming the formation of a random polymer coil.50 The experimental hydrodynamic diameter was measured to be 4.2 nm, which is close to the theoretically predicted value, and also aligns with our expectation that this polymer cannot self-assemble into micelles but mainly exists as coils.
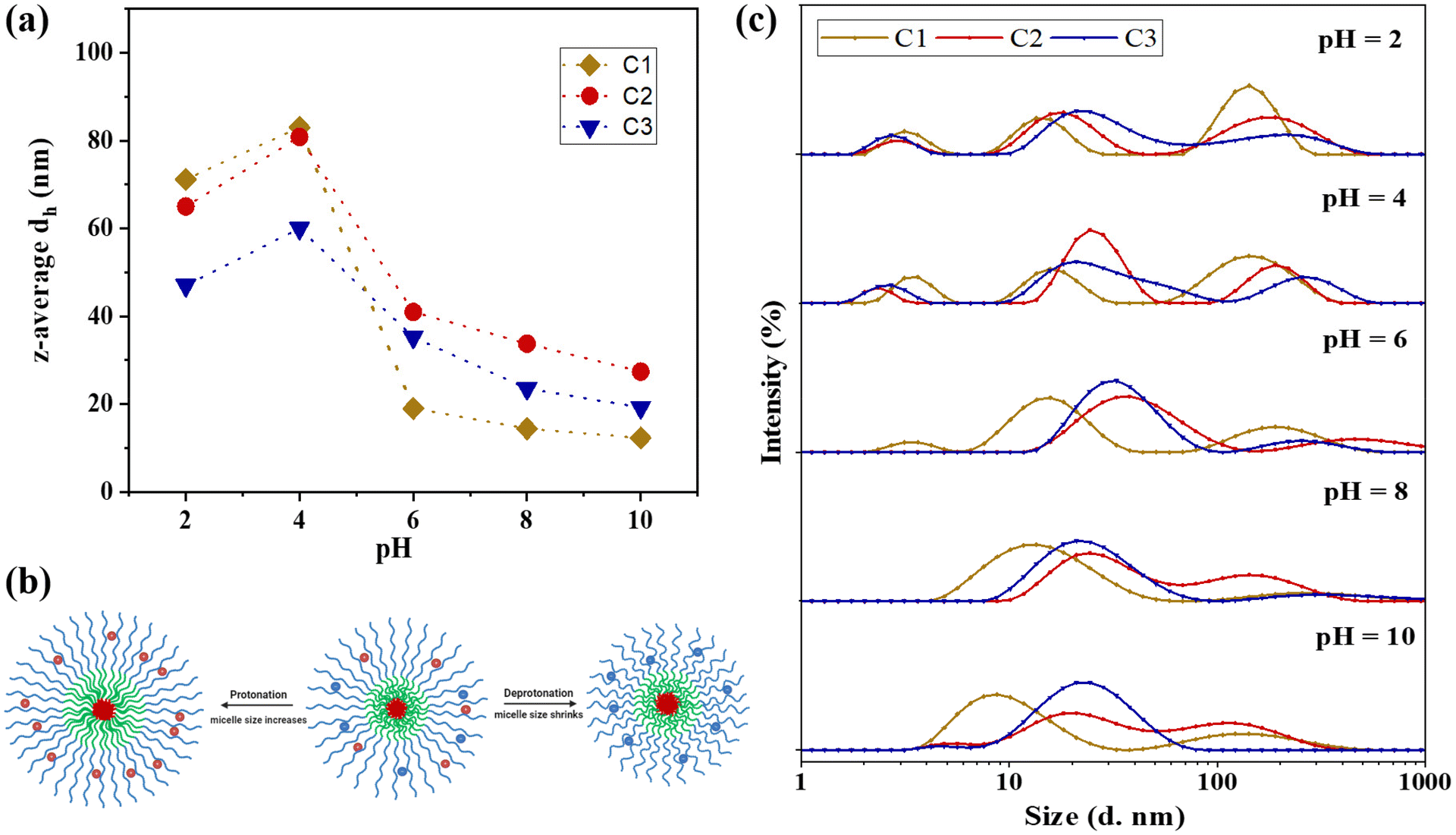 | ||
| Fig. 5 (a) Changes in the z-average dh against different pH values for C1, C2, and C3. (b) Schematic demonstrating the impact of protonation on the terpolymer (C3) and its sizes. (c) Particle size distribution by intensity under varying pH levels: 2, 4, 6, 8, and 10. | ||
Fig. 5(c) demonstrates that upon increasing the pH from 8.0 to 10.0 through titration with OH−, the z-average particle sizes remain largely unchanged, exhibiting only a slight decrease. This decrease is attributed to the deprotonation of the corona at pH levels exceeding the pKa values of both pH-responsive blocks, resulting in more compact micelle structures and a consequent reduction in hydrodynamic diameter.
Conversely, titration of the polymer sample with H+ to decrease the pH from 8.0 to 2.0 leads to protonation of the tertiary amine groups (R3N) in both NMEP and DMAEMA comonomers, converting them to their positively charged forms (R3NH+). This protonation significantly impacts the overall morphology and size of the micelles. Specifically, as the pH decreases from 8.0 to 6.0, a gradual increase in particle size is observed due to the increased protonation, causing the polymer to transition from a hydrophobic to a hydrophilic state and resulting in the formation of swollen micellar structures.
Further titration of the polymer solution from pH 6.0 to pH 4.0 results in more pronounced changes in hydrodynamic diameter, indicating significant alterations in the ionisation state of the polymers. This effect is especially noticeable as the pH drops below 5.3, the pKa of the PNMEP blocks. At these lower pH levels, the corona, which is pH-responsive, becomes increasingly hydrophilic, swollen, and loose, leading to a heightened tendency to form aggregates. As the pH continues to decrease to between 4.0 and 2.0, the terpolymers become fully protonated, further enhancing their hydrophilicity. This leads to a reduction in aggregation number due to excessive protonation and electrostatic repulsions, as well as a slight dissociation of the micelles into unimer structures resulting from the dissolution of polymer chains, which contributes to a minor decrease in overall particle size. Furthermore, at this pH, there is also a higher ionic strength from adding more HCl, and the excess of Cl− are counteracting the cationic charges of the tertiary amines, thus allowing the chains to be slightly less stretched. This charge screening has been previously reported in crosslinked amino-containing gels.76 It should be noted that the change in hydrodynamic diameters within this low pH range is more pronounced for C1 and C2, compared to the less significant change for C3. This disparity can be attributed to the architectural effect: in C3, the NMEP block is located in the inner shell, sterically surrounded by the thick outer corona consisting of the DMAEMA block, which decreases the degree of hydration of NMEP, as previously discussed.
The above findings are in line with previous studies on the pH-responsive behaviour of PDMAEMA-containing polymers, which have similarly reported changes in particle sizes in response to varying pH levels.77–79
3.5 Transmission electron microscopy images
TEM images were taken for all terpolymer samples to visualize their configurations and compared with their DLS results. It is worth noting that TEM samples were prepared from polymer solutions in DI water without adjusting the pH for the triblock terpolymers. However, the sample was titrated to 10% protonation for the statistical terpolymer (C4) to ensure solubility. The TEM results confirm the formation of spherical-like micelles by the block terpolymers, as expected. In contrast, the statistical terpolymer does not exhibit a definitive shape of micelles in the TEM images. The diameter of these polymer particles was measured from TEM images with standard deviations calculated, and then compared with the hydrodynamic diameter, as shown in Fig. 6. | ||
| Fig. 6 TEM images were obtained with the samples negatively stained using 1% w/w uranyl acetate, showing C1 (ABC), C2 (BAC), C3 (BCA), and C4 (statistical) dissolved in DI water at 1% w/w. Note that C4 was prepared in a solution with 10% protonation to ensure solubility. Particle sizes in the TEM images were estimated using the measuring scales located in the left corner of each figure and were then compared with their hydrodynamic diameters measured by DLS. | ||
It is observed that both results align well with each other. Specifically, the diameter size increases from C1 to C3 to C2, consistent with observations from DLS. The diameters determined by TEM are smaller than the DLS values, as expected and observed in previous research.46,55,80 This discrepancy is attributed to different sample conditions. Specifically, TEM analysis is performed on dried samples, where the absence of water leads to the collapse of hydrophobic and hydrophilic segments of the micelles, resulting in smaller measurements. In contrast, DLS is conducted in aqueous conditions, measuring the hydrodynamic radius that includes both the micelle and its hydration shell, yielding larger size estimations. Therefore, the ‘drying effect’ in TEM removes the water that normally hydrates and swells the micelle structures. Unlike DLS, which measures the hydrodynamic radius including the solvation layer of water molecules around the polymer particle, only the solid components of the micelles are visible in TEM, making them inherently smaller.81
Overall, TEM provides a visual representation of all particles in the field of view and confirms the spherical micelles formed by the triblock terpolymers, as well as the absence of self-assembled structures in the statistical terpolymer sample. By comparing the results measured by DLS and TEM, the differences in size and conformation of the terpolymers in various architectures were verified.
3.6 Visual test and rheology
Polymer solutions with higher concentrations are more prone to thermogel formation due to their enhanced ability to entangle and create physical cross-links within a 3D network. To evaluate the polymer's gelation behaviour—the transition from a sol to a gel—a “tube inversion” test was conducted. This test determined whether the sample remained fluid upon inverting the vial. The gelation region, when present, was marked on the phase diagrams with a blue-coloured symbol, as illustrated in Fig. 7. Interestingly, two of the three polymers studied, C2 and C3, which possess BAC and BCA architectures with a hydrophobic end block instead of the ABC configuration, underwent thermal gelation transitions. This observation deviates from our previous studies on the architectural influence in different triblock terpolymer systems.54,55,84 Those studies suggested that polymers with an ABC architecture typically displayed a more well-defined sol–gel transition, characterised by a wider gelation region, a lower critical gelation concentration, and the formation of a mechanically stronger gel. However, it is important to note that in those previous studies, only one monomer was ionic, which could significantly affect the thermoresponsive sol–gel transitions observed. Regarding this study, the structure of the triblock terpolymer, featuring two ionic end blocks, might influence the formation of micelle-bridging links essential for gelation.
 | ||
| Fig. 7 Phase diagram of C1–C3, plotted using visual observation results for polymer solutions prepared in PBS (pH = 7.4) at concentrations of 1% w/w, 2% w/w, 5% w/w, 10% w/w, 15% w/w, and 20% w/w. Visual transition states are reported as: (a) runny solution in white, (b) viscous solution in red, (c) stable gel in blue, and (d) phase separation in green. | ||
The rheological behaviour of these polymers should correlate with their visual characteristics, particularly at critical transition points identified by both rheological measurements and visual inspections. The critical temperatures include the gelation temperature (Tgel) and degel temperature (Tdegel) from rheological data, as well as the gelation temperature (Tgel), syneresis temperature (Tsyn), and precipitation temperature (Tprec) from visual observations. In rheology, Tgel is the temperature at which G′ exceeds G′′, indicating the gel point, while Tdegel is the temperature at which G′′ exceeds G′ again, showing that viscous flow is more significant than elastic deformation. For visual observations, Tsyn marks the onset of gel syneresis (defined as disturbance in the gel due to internal stress),85 and Tprec indicates the start of precipitation (the complete separation into solid and aqueous phases).
Table 4 demonstrates that rheological measurements are consistent with the visual test results. Specifically, these measurements confirm C1's lack of gelation, evidenced by the absence of a crossover or notable increase between its two dynamic moduli within the examined temperature range, as shown in Fig. 8. This behaviour suggests that C1 (ABC) predominantly exhibits viscous, liquid-like characteristics. In contrast, the gelation temperatures for C2 (BAC) and C3 (BCA), as determined by both testing methods, show a minor discrepancy of 1–2 °C, which is within the acceptable experimental error margin of the techniques.
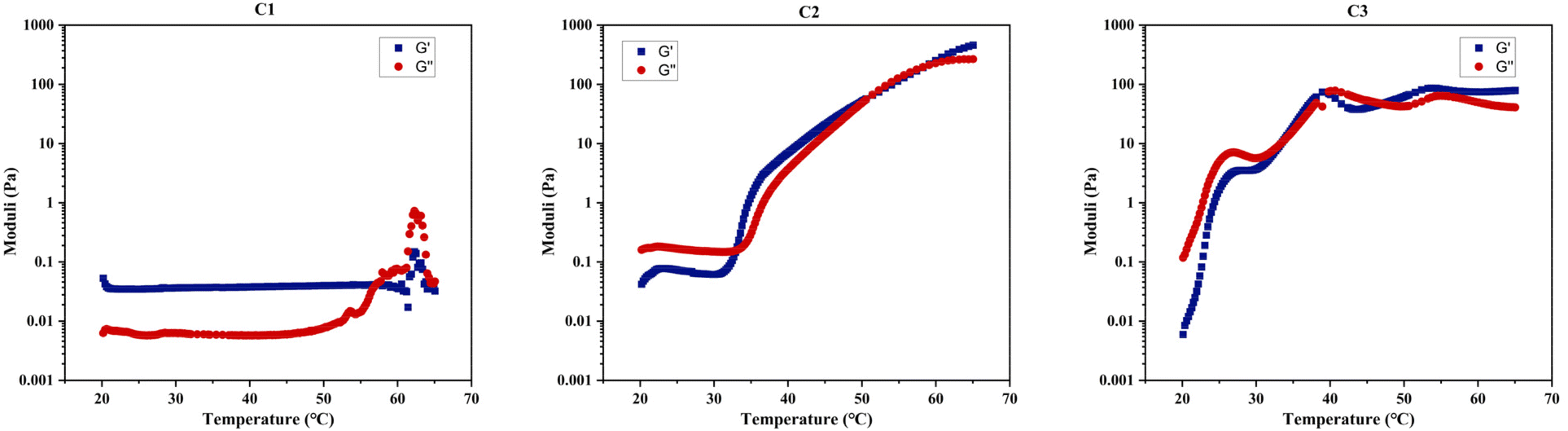 | ||
| Fig. 8 Rheological measurements during a 1 °C min−1 temperature scan from 20 to 65 °C under a constant shear strain of 1% (γ) for C1–C3 dissolved in PBS at 20% w/w. | ||
| No. | Experimental chemical structurea | Architecture | Critical temperatures (°C) | ||||
|---|---|---|---|---|---|---|---|
| Visual test (±2 °C) | Rheology (±1 °C) | ||||||
| T gel | T syn | T prec | T gel | T degel | |||
| a Abbreviations: 2-(dimethylamino)ethyl methacrylate (DMA), methyl methacrylate (MMA), N-(2-(methacryloyloxy)ethyl) pyrrolidone (NMEP). Note that DMA is a further abbreviation of DMAEMA. | |||||||
| C1 | DMA32-b-MMA25-b-NMEP20 | ABC | NA | ||||
| C2 | MMA22-b-DMA31-b-NMEP18 | BAC | 32 | 52 | 59 | 32 | 51 |
| C3 | MMA21-b-NMEP18-b-DMA34 | BCA | 31 | 41 | 48 | 33 | 40 |
A clear difference in gelation behaviour between C2 and C3 can be observed from both Table 4 and Fig. 8: C2 (BAC) has a gelation window spanning approximately 20 °C, while C3 (BCA) has a gelation window of approximately 10 °C. Additionally, C2 demonstrates a gradual increase in gel strength as the temperature rises, whereas C3 shows less variation in G′, upon examining their changes in the storage modulus with temperature for 20% w/w concentrated polymer solutions in PBS. The storage modulus is key to indicating the energy elastically stored within the gel during deformation, signifying a shift towards solid-like behaviour and a mechanically stronger network that can retain its shape when subjected to oscillatory shear deformation. Therefore, both C2 and C3 should be classified as ‘weak gels’,86,87 based on the maximum G′ values recorded for both polymers before reaching their degradation temperatures, consistently remaining below 100 Pa, despite C2 forming a stronger gel as temperature increases.
4. Conclusions
In this study, pyrrolidone-based amphiphilic thermoresponsive terpolymers were synthesised through reversible addition–fragmentation chain transfer (RAFT) polymerisation, examining four unique terpolymer architectures: ABC, BAC, BCA, and statistical. The molar masses and compositions were kept similar across the terpolymers to investigate the influence of polymer architecture on their self-assembly conformations and thermoresponsive characteristics by employing techniques such as Turbidimetry, Dynamic Light Scattering, Transmission Electron Microscopy, temperature-dependent 1H NMR spectroscopy, and rheometry. Notably, the terpolymers exhibited a lower cloud point temperature in D2O than in H2O and showed enhanced hydrophilicity at 10% protonation. While all block terpolymers formed spherical micelles whose sizes were influenced by pH changes, the statistical terpolymer did not. The arrangement of the block architecture and the position of hydrophobic groups were found to significantly influence thermoresponsive behaviour, with the BAC and BCA architectures demonstrating a clear sol–gel transition. This research highlights the vital importance of the comonomer arrangement in pyrrolidone-based terpolymers, offering significant insights for their application in the development of stimuli-responsive materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
EPSRC is acknowledged for HI's funding (EP/W034093/1).References
- A. Bordat, T. Boissenot, J. Nicolas and N. Tsapis, Adv. Drug Delivery Rev., 2019, 138, 167–192 CrossRef CAS PubMed.
- D. Roy, W. L. A. Brooks and B. S. Sumerlin, Chem. Soc. Rev., 2013, 42, 7214–7243 RSC.
- M. R. Matanović, J. Kristl and P. A. Grabnar, Int. J. Pharm., 2014, 472, 262–275 CrossRef PubMed.
- W. Tao, J. Wang, W. J. Parak, O. C. Farokhzad and J. Shi, ACS Nano, 2019, 13, 4876–4882 CrossRef CAS PubMed.
- X. Pang, Y. Jiang, Q. Xiao, A. W. Leung, H. Hua and C. Xu, J. Controlled Release, 2016, 222, 116–129 CrossRef CAS PubMed.
- E. Pantuso, G. De Filpo and F. P. Nicoletta, Adv. Opt. Mater., 2019, 7, 1900252 CrossRef.
- B. Sana, A. Finne-Wistrand and D. Pappalardo, Mater. Today Chem., 2022, 25, 100963 CrossRef CAS.
- G. Stoychev, A. Kirillova and L. Ionov, Adv. Opt. Mater., 2019, 7, 1900067 CrossRef.
- J. Kolosnjaj-Tabi, L. Gibot, I. Fourquaux, M. Golzio and M. P. Rols, Adv. Drug Delivery Rev., 2019, 138, 56–67 CrossRef CAS PubMed.
- T. Manouras and M. Vamvakaki, Polym. Chem., 2016, 8, 74–96 RSC.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS PubMed.
- A. Zhang, K. Jung, A. Li, J. Liu and C. Boyer, Prog. Polym. Sci., 2019, 99, 101164 CrossRef CAS.
- N. K. Singh and D. S. Lee, J. Controlled Release, 2014, 193, 214–227 CrossRef CAS PubMed.
- Y. Sun, J. Zhou, Q. Cheng, D. Lin, Q. Jiang, A. Dong, Z. Liang and L. Deng, J. Appl. Polym. Sci., 2016, 133, 43303 CrossRef.
- L. J. Zhu, L. P. Zhu, Y. F. Zhao, B. K. Zhu and Y. Y. Xu, J. Mater. Chem. A, 2014, 2, 15566–15574 RSC.
- L. Li, Z. Xu, C. Zhang, R. Zhang, Z. Xu and A. K. Whittaker, ACS Appl. Nano Mater., 2018, 1, 5027–5034 CrossRef CAS.
- F. J. Xu, E. T. Kang and K. G. Neoh, Biomaterials, 2006, 27, 2787–2797 CrossRef CAS PubMed.
- C. N. Kotanen, D. R. Janagam, R. Idziak, L. Rhym, R. Sullivan, A. M. Wilson, T. L. Lowe and A. Guiseppi-Elie, Eur. Polym. J., 2015, 72, 438–450 CrossRef CAS.
- Y. Li, S. P. Armes, X. Jin and S. Zhu, Macromolecules, 2003, 36, 8268–8275 CrossRef CAS.
- J. I. Amalvy, E. J. Wanless, Y. Li, V. Michailidou, S. P. Armes and Y. Duccini, Langmuir, 2004, 20, 8992–8999 CrossRef CAS PubMed.
- C. De las Heras Alarcón, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- R. París and I. Quijada-Garrido, Eur. Polym. J., 2010, 46, 2156–2163 CrossRef.
- X. Zhou, X. Fan and C. He, Macromolecules, 2016, 49, 4236–4244 CrossRef CAS.
- Y. Huang, P. Yong, Y. Chen, Y. Gao, W. Xu, Y. Lv, L. Yang, R. L. Reis, R. P. Pirraco and J. Chen, RSC Adv., 2017, 7, 28711–28722 RSC.
- A. Roointan, J. Farzanfar, S. Mohammadi-Samani, A. Behzad-Behbahani and F. Farjadian, Int. J. Pharm., 2018, 552, 301–311 CrossRef CAS PubMed.
- J. Kopeĉek, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5929–5946 CrossRef PubMed.
- L. H. Hess, A. Lyuleeva, B. M. Blaschke, M. Sachsenhauser, M. Seifert, J. A. Garrido and F. Deubel, ACS Appl. Mater. Interfaces, 2014, 6, 9705–9710 CrossRef CAS PubMed.
- Z. Jiang, X. Li, G. Yang, L. Cheng, B. Cai, Y. Yang and J. Dong, Langmuir, 2012, 28, 7174–7181 CrossRef CAS PubMed.
- M. V. Risbud, A. A. Hardikar, S. V. Bhat and R. R. Bhonde, J. Controlled Release, 2000, 68, 23–30 CrossRef CAS PubMed.
- M. Kurakula and G. K. Rao, Eur. Polym. J., 2020, 136, 109919 CrossRef CAS.
- J. E. Kennedy and C. L. Higginbotham, Mater. Sci. Eng., C, 2011, 31, 246–251 CrossRef CAS.
- Q. V. Vo, T. L. B. Tram, N. T. Hoa, A. N. Au-Duong and A. Mechler, Polym. Degrad. Stab., 2023, 216, 110483 CrossRef CAS.
- G. Opiyo and J. Jin, Eur. Polym. J., 2021, 159, 110713 CrossRef CAS.
- X. Pan, X. Guo, B. Choi, A. Feng, X. Wei and S. H. Thang, Polym. Chem., 2019, 10, 2083–2090 RSC.
- P. Van De Watering, N. J. Zuidam, M. J. Van Steenbergen, O. A. G. J. Van Der Houwen, W. J. M. Underberg and W. E. Hennink, Macromolecules, 1998, 31, 8063–8068 CrossRef.
- G. M. Iskander, L. E. Baker, D. E. Wiley and T. P. Davis, Polymer, 1998, 39, 4165–4169 CrossRef CAS.
- Z. Hao, G. Li, K. Yang and Y. Cai, Macromol. Rapid Commun., 2013, 34, 411–416 CrossRef CAS PubMed.
- J. Deng, Y. Shi, W. Jiang, Y. Peng, L. Lu and Y. Cai, Macromolecules, 2008, 41, 3007–3014 CrossRef CAS.
- V. J. Cunningham, M. J. Derry, L. A. Fielding, O. M. Musa and S. P. Armes, Macromolecules, 2016, 49, 4520–4533 CrossRef CAS PubMed.
- R. R. Gibson, S. P. Armes, O. M. Musa and A. Fernyhough, Polym. Chem., 2019, 10, 1312–1323 RSC.
- L. W. Teunissen, A. R. Kuzmyn, F. S. Ruggeri, M. M. J. Smulders and H. Zuilhof, Adv. Mater. Interfaces, 2022, 9, 2101717 CrossRef CAS.
- L. W. Teunissen, M. M. J. Smulders and H. Zuilhof, Langmuir, 2023, 39, 7613–7622 CrossRef CAS PubMed.
- J. Magalhaes, R. A. Sousa, J. F. Mano, R. L. Reis, F. J. Blanco and J. S. Román, J. Biomed. Mater. Res., Part A, 2013, 101A, 157–166 CrossRef CAS PubMed.
- S. Jia, J. Fei, J. Deng, Y. Cai and J. Li, Sens. Actuators, B, 2009, 138, 244–250 CrossRef CAS.
- S. Cheng, Y. Xue, Y. Lu, X. Li and J. Dong, ACS Omega, 2017, 2, 105–112 CrossRef CAS PubMed.
- L. Wang, A. P. Constantinou, Y. Li and T. K. Georgiou, Eur. Polym. J., 2024, 207, 112810 CrossRef CAS.
- M. Elladiou and C. S. Patrickios, Polym. Chem., 2012, 3, 3228–3231 RSC.
- M. Elladiou and C. S. Patrickios, Macromolecules, 2015, 48, 7503–7512 CrossRef CAS.
- A. P. Constantinou, G. Patias, B. Somuncuoǧlu, T. Brock, D. W. Lester, D. M. Haddleton and T. K. Georgiou, Polym. Chem., 2021, 12, 3522–3532 RSC.
- D. Feldman and D. Feldman, JPoSL, 1984, 22, 673–673 Search PubMed.
- M. G. Arafa, R. F. El-Kased and M. M. Elmazar, Sci. Rep., 2018, 8, 1–16 CAS.
- K. Manokruang and D. S. Lee, Macromol. Biosci., 2013, 13, 1195–1203 CrossRef CAS PubMed.
- C. Gong, S. Shi, P. Dong, B. Kan, M. Gou, X. Wang, X. Li, F. Luo, X. Zhao, Y. Wei and Z. Qian, Int. J. Pharm., 2009, 365, 89–99 CrossRef CAS PubMed.
- M. A. Ward and T. K. Georgiou, Polym. Chem., 2013, 4, 1893–1902 RSC.
- A. P. Constantinou, H. Zhao, C. M. McGilvery, A. E. Porter and T. K. Georgiou, Polymers, 2017, 9, 31 CrossRef PubMed.
- J. A. Pople, I. W. Hamley, J. P. A. Fairclough, A. J. Ryan, B. U. Komanschek, A. J. Gleeson, G. E. Yu and C. Booth, Macromolecules, 1997, 30, 5721–5728 CrossRef CAS.
- G. E. Yu, H. Li, J. P. A. Fairclough, A. J. Ryan, N. McKeown, Z. Ali-Adib, C. Price and C. Booth, Langmuir, 1998, 14, 5782–5788 CrossRef CAS.
- A. Kelarakis, V. Castelletto, C. Chaibundit, J. Fundin, V. Havredaki, I. W. Hamley and C. Booth, Langmuir, 2001, 17, 4232–4239 CrossRef CAS.
- J. Sun, Y. Peng, Y. Chen, Y. Liu, J. Deng, L. Lu and Y. Cai, Macromolecules, 2010, 43, 4041–4049 CrossRef CAS.
- V. Bütün, S. P. Armes and N. C. Billingham, Polymer, 2001, 42, 5993–6008 CrossRef.
- V. J. Cunningham, Y. Ning, S. P. Armes and O. M. Musa, Polymer, 2016, 106, 189–199 CrossRef CAS.
- V. J. Cunningham, S. P. Armes and O. M. Musa, Polym. Chem., 2016, 7, 1882–1891 RSC.
- H. Lee, S. H. Son, R. Sharma and Y. Y. Won, J. Phys. Chem. B, 2011, 115, 844–860 CrossRef CAS PubMed.
- J. C. P. De Souza, A. F. Naves and F. H. Florenzano, Colloid Polym. Sci., 2012, 290, 1285–1291 CrossRef.
- D. Fournier, R. Hoogenboom, H. M. L. Thijs, R. M. Paulus and U. S. Schubert, Macromolecules, 2007, 40, 915–920 CrossRef CAS.
- M. A. Ward and T. K. Georgiou, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 775–783 CrossRef CAS.
- P. Liu, L. Xiang, Q. Tan, H. Tang and H. Zhang, Polym. Chem., 2013, 4, 1068–1076 RSC.
- Y. Cho, L. B. Sagle, S. Iimura, Y. Zhang, J. Kherb, A. Chilkoti, J. M. Scholtz and P. S. Cremer, J. Am. Chem. Soc., 2009, 131, 15188–15193 CrossRef CAS PubMed.
- P. Kujawa and F. M. Winnik, Macromolecules, 2001, 34, 4130–4135 CrossRef CAS.
- B. Sun, Y. Lin, P. Wu and H. W. Siesler, Macromolecules, 2008, 41, 1512–1520 CrossRef CAS.
- X. Wang and C. Wu, Macromolecules, 1999, 32, 4299–4301 CrossRef CAS.
- X. Jiang, G. Zhang, R. Narain and S. Liu, Langmuir, 2009, 25, 2046–2054 CrossRef CAS PubMed.
- X. Jiang, G. Zhang, R. Narain and S. Liu, Soft Matter, 2009, 5, 1530–1538 RSC.
- W. Zhang, X. Jiang, Z. He, D. Xiong, P. Zheng, Y. An and L. Shi, Polymer, 2006, 47, 8203–8209 CrossRef CAS.
- S. Liu, J. V. M. Weaver, Y. Tang, N. C. Billingham, S. P. Armes and K. Tribe, Macromolecules, 2002, 35, 6121–6131 CrossRef CAS.
- M. Vamvakaki and C. S. Patrickios, Chem. Mater., 2002, 14, 1630–1638 CrossRef CAS.
- C. Yang, J. Xiao, W. Xiao, W. Lin, J. Chen, Q. Chen, L. Zhang, C. Zhang and J. Guo, RSC Adv., 2017, 7, 27564–27573 RSC.
- H. Yang, J. Guo, R. Tong, C. Yang and J. K. Chen, Polymers, 2018, 10, 443 CrossRef PubMed.
- Y. Xing, J. Peng, K. Xu, S. Gao, X. Gui, S. Liang, L. Sun and M. Chen, Phys. Chem. Chem. Phys., 2017, 19, 23024–23033 RSC.
- C. Wu, A. Ying and S. Ren, Colloid Polym. Sci., 2013, 291, 827–834 CrossRef CAS.
- D. Laage, T. Elsaesser and J. T. Hynes, Chem. Rev., 2017, 117, 10694–10725 CrossRef CAS PubMed.
- S. Wang, G. N. Nawale, O. P. Oommen, J. Hilborn and O. P. Varghese, Polym. Chem., 2019, 10, 4322–4327 RSC.
- J. Y. Shin, Y. H. Yeo, J. E. Jeong, S. A. Park and W. H. Park, Carbohydr. Polym., 2020, 238, 116192 CrossRef CAS PubMed.
- A. P. Constantinou, L. Wang, S. Wang and T. K. Georgiou, Polym. Chem., 2023, 14, 223–247 RSC.
- M. Kunitz, J. Gen. Physiol., 1928, 12, 289–312 CrossRef CAS PubMed.
- R. Kocen, M. Gasik, A. Gantar and S. Novak, Biomed. Mater., 2017, 12, 025004 CrossRef PubMed.
- S. K. H. Gulrez, S. Al-Assaf and G. O. Phillips, Progress in molecular and environmental bioengineering-from analysis and modeling to technology applications, InTech: Rijeka, Croatia, 2011 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00405a |
| This journal is © The Royal Society of Chemistry 2024 |