
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
SNAr as a facile method to prepare polystyrene-grafted conjugated copolymers with enhanced photoluminescence properties†
Martina
Rimmele
 a,
Adam V.
Marsh
b,
Charlotte L.
Rapley
a,
Ashraf
Al-Amoudi
c and
Martin
Heeney
*ab
a,
Adam V.
Marsh
b,
Charlotte L.
Rapley
a,
Ashraf
Al-Amoudi
c and
Martin
Heeney
*ab
aDepartment of Chemistry and Centre for Processable Electronics, Imperial College London, London, W12 0BZ, UK. E-mail: m.heeney@imperial.ac.uk; martin.heeney@kaust.edu.sa
bKing Abdullah University of Science and Technology (KAUST), KAUST Solar Centre (KSC), Physical Sciences and Engineering Division (PSE), Thuwal, 23955-6900 Saudi Arabia
cKing Abdullah University of Science and Technology (KAUST), Imaging and Characterization Core Labs, Electron Microscopy Lab, Thuwal, 23955-6900 Saudi Arabia
First published on 28th May 2024
Abstract
The preparation of graft copolymers remains a challenge, especially for conjugated materials, due to the limited number of reactions available for backbone modification. Herein, we report a reliable method to conjugated graft copolymers (also known as rod-graft-coil copolymers) starting from different well studied polymers in a one-step post-polymerisation reaction. The starting polymers all contain a commonly used benzo[c][1,2,5]thiadiazole unit, which includes a fluorine atom as a functional handle for replacement via a nucleophilic aromatic substitution (SNAr) reaction. We functionalised the polymers in a post-polymerisation approach with secondary thiol-terminated polystyrene (PS), readily available from the RAFT polymerisation. Furthermore, we prepared stable, spherical, emissive nanoparticles from the resulting graft copolymers as shown by dynamic light scattering (DLS) and transmission electron microscopy (TEM). The optical properties of the parent and the graft copolymers were studied in solution and solid state, as well as in nanoparticles (NPs). PLQY studies revealed, that the emission was enhanced upon grafting of PS in the NPs. This approach offers the preparation of graft copolymers in one additional step after polymerisation only, and moreover enhances the emissive properties of the resulting graft copolymers in the solid state which is promising in the field of biologically benign fluorescent nanoprobes.
Introduction
Graft copolymers are a fascinating class of materials characterised by the attachment of polymer chains (grafts) onto the backbone of another polymer. Through grafting, the combination of different polymers enables the synthesis of a unique material that capitalises on the individual functionality of each polymer component, resulting in a diverse range of properties and applications.Methods of synthesising graft polymers can be divided into three processes: grafting-from, grafting-through, and grafting-onto. Grafting-from involves attaching a polymerisable monomer or initiator to the parent polymer's backbone, followed by subsequent polymerisation; grafting-through uses a preformed polymer with a reactive end group for subsequent polymerisation (sometimes called the macromonomer approach); while grafting-onto is the only method that bonds pre-formed polymer chains onto the parent polymer's surface. The latter has a distinct advantage over the other processes in that it allows for the incorporation of well-defined polymer segments, resulting in precise control over the grafting density and distribution. Such grafting-onto strategies have found recent application in the field of conjugated polymers.1–4
Conjugated polymers have been widely studied due to their unique properties, which make them promising material in various applications, such as organic photovoltaics (OPVs),5,6 organic light emitting diodes (OLEDs),7 and organic photodetectors (OPDs),8 amongst others. Their attraction in these applications stems from their highly tunable electronic and optical properties, low-cost processing, flexibility, low weight and biocompatibility.9,10 Despite the enormous interest in conjugated polymers, reports of graft copolymers, in which non-conjugated polymers are attached to a conjugated backbone (sometimes called rod-coil graft copolymers) are rare.11–15 The alternative arrangement, in which conjugated polymers are grafted to a non-conjugated polymer is also uncommon.16 Often this latter scenario uses the grafting through or macromonomer approach, in which a reactive end-group on a conjugated polymer is subsequently polymerised by techniques such as controlled radical polymerisation. Many reports utilise poly(3-hexyl)thiophene (P3HT) as the macromonomer, since it can be prepared with reactive end-groups via the well-controlled catalyst transfer polymerisation.1,17
In the case of conjugated polymers with non-conjugated grafts, the ‘grafting to’ approach has been the most explored since the advent of click chemistry.18 Thus, conjugated polymers can be prepared with sidechains containing reactive functionality, most often azide or alkyne groups, and non-conjugated polymers with complimentary reactive ends attached. For example, P3HT containing an alkylazide sidechain can be grafted with poly(vinylpyridine) containing an alkyne end-group under copper-catalyzed azide–alkyne cycloaddition (CuAAC) conditions.19 The resulting graft was used as a compatibiliser to improve the thermal stability of bulk-heterojunction OPVs. Similar approaches were used to graft the same polymer to an emissive polyfluorene backbone,20 or graft poly(ethyleneglycol) (PEG) to a low band gap polymer.21 One interesting application has been the grafting of PEG and poly(methyl methacrylate) (PMMA) to an azide functionalised P3HT.22 Nanoparticles (NPs) of all three polymers were formed, as potential low toxicity, fluorescent nanoprobes.23,24 Whilst the parent P3HT based NPs were poorly emissive, due to the formation of aggregates that quenched excitons in the aggregated state, NPs of the grafts with PEG or PMMA demonstrated up to two orders of magnitude higher photoluminescent quantum yields.
Whilst these reports have demonstrated the utility of the graft approach, they can be complex to implement synthetically. For example, in the case of P3HT grafts, the polythiophene is first prepared with alkylbromide sidechains, followed by a post-polymerisation reaction to convert the alkylbromide to azide, and a second post-polymerisation reaction to couple the azide to alkyne terminated polymer (which also need to be prepared). Two post-polymerisation steps increases the chance to introduce undesired defects and miscouplings. Thus new, simpler synthetic approaches to such rod-coil graft copolymers are highly desirable.25
One potential way for the functionalisation of conjugated polymer backbones is a nucleophilic aromatic substitution (SNAr) reaction directly on the conjugated backbone. Our group has previously demonstrated the direct functionalisation of conjugated polymers by halide displacement from fluorinated, electron deficient monomers such as benzo[c][1,2,5]thiadiazole units within a conjugated polymer backbone. A variety of primary thiols and alcohols were used as nucleophiles to substitute the backbone in quantitative or near-quantitative yield.26,27 Promisingly, graft copolymers could be readily prepared by the reaction of PEG (up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) with a hydroxyl end-group. This graft-onto strategy induced remarkable changes in the properties of the polymers, such as enabling the PEGylated poly(dioctylfluorene-co-benzothiadiazole) (F8BT-PEG) to dissolve in protic solvents including water.28 F8BT-PEG derivatives have also been shown to form in vivo stable semiconducting polymer nanoparticles (NPs) through simple nanoprecipitation techniques, with potential applications in cancer theranostics where the conjugated polymer is encapsulated within the benign PEG host material.29 Similar reactions have also been reported on an electron deficient perfluorinated benzene within conjugated polymer systems.30–32 Given the simplicity of this approach, we were keen to investigate if other graft copolymers could be prepared, for example by the coupling of secondary thiols to the conjugated backbone. Such thiols are readily available in well-controlled manner via the reversible addition and fragmentation chain transfer (RAFT) technique, followed by the facile conversion of the thiocarbonylthio end-group to a secondary thiol.33,34 Given the large variety of such RAFT copolymers that can be readily prepared from different monomers, direct coupling of the secondary thiol end-group to the conjugated backbone offers a potentially simple route to a range of rod-coil copolymers. However, given the additional steric hindrance associated with a secondary thiol end-group, its ability to participate in SNAr onto the conjugated backbone was unclear at the outset of this work.
000 g mol−1) with a hydroxyl end-group. This graft-onto strategy induced remarkable changes in the properties of the polymers, such as enabling the PEGylated poly(dioctylfluorene-co-benzothiadiazole) (F8BT-PEG) to dissolve in protic solvents including water.28 F8BT-PEG derivatives have also been shown to form in vivo stable semiconducting polymer nanoparticles (NPs) through simple nanoprecipitation techniques, with potential applications in cancer theranostics where the conjugated polymer is encapsulated within the benign PEG host material.29 Similar reactions have also been reported on an electron deficient perfluorinated benzene within conjugated polymer systems.30–32 Given the simplicity of this approach, we were keen to investigate if other graft copolymers could be prepared, for example by the coupling of secondary thiols to the conjugated backbone. Such thiols are readily available in well-controlled manner via the reversible addition and fragmentation chain transfer (RAFT) technique, followed by the facile conversion of the thiocarbonylthio end-group to a secondary thiol.33,34 Given the large variety of such RAFT copolymers that can be readily prepared from different monomers, direct coupling of the secondary thiol end-group to the conjugated backbone offers a potentially simple route to a range of rod-coil copolymers. However, given the additional steric hindrance associated with a secondary thiol end-group, its ability to participate in SNAr onto the conjugated backbone was unclear at the outset of this work.
Herein, we present an efficient approach for the preparation of polystyrene-grafted conjugated copolymers using a SNAr based method. The method couples a secondary thiol-terminated polystyrene to three different conjugated backbones with different graft densities. Moreover, we demonstrate the subsequent preparation of stable, uniform, spherical, and emissive NPs from these grafted polymers using a nanoprecipitation method, with no requirement to add additional surfactants or other additives. Significant suppression of aggregation-caused quenching (ACQ) in aggregated states was observed upon grafting, leading to an increased photoluminescence quantum yield (PLQY) for the grafted, relative to the un-grafted, polymers with promise for application as fluorescent probes in biological systems.
Synthesis
We chose three different conjugated polymers for this investigation, which were all prepared according to different protocols, but all contain an electron deficient fluorinated benzo[c][1,2,5]thiadiazole derivative (fBT) copolymerised with a more electron-rich comonomer. F8BT (poly(dioctylfluorene-co-benzothiadiazole)) is a well-studied highly emissive conjugated polymer,35–37 and the analogue with a fBT unit instead of the BT is also known.38 We prepared F8fBT polymer from two commercially available monomers, 9,9-dioctyl-9H-fluorene-2,7-diboronic acid bis (pinacol) ester and 4,7-dibromo-5-fluorobenzo[c][1,2,5]thiadiazole, in a Suzuki cross-coupling polymerisation. Another thoroughly studied polymer, poly(indacenodithiophene-co-benzothiadiazole) (IDTBT), was chosen as a second candidate for our study and was prepared following a recently published protocol using C–H activation conditions, again replacing the BT unit with fBT.39–41 C16-IDT was copolymerised with 4,7-dibromo-5-fluoro benzo[c][1,2,5]thiadiazole under direct (hetero)arylation polymerisation (DHAP) conditions to give IDTfBT. Our recently reported FO6-T was chosen as the third, more challenging candidate.42 In this case, the benzo[c][1,2,5]thiadiazole unit also contains an electron donating alkoxy substituent adjacent to the fluorine. Electron donating groups reduce the rate of reactivity for SNAr, and in this case, also increase the steric shielding around the reactive site. Thus, we were excited to investigate if reactivity could still be observed. FO6-T was prepared from commercially available 2,5-bis(trimethylstannyl)thiophene and a 4,7-dibromo-5-fluoro-6-((2-hexyldecyl)oxy)benzo[c][1,2,5]thiadiazole in a Stille polymerisation (see details in the ESI†).All three conjugated polymers were purified using Soxhlet extraction with methanol, acetone and hexane to remove lower molecular weight fractions and finally extracted in chloroform and precipitated. Their molecular weights were analysed by gel permeation chromatography (GPC) in 1,2,4-trichlorobenzene (1,2,4-TCB) at 150 °C versus polystyrene standards (see Table 1 and Fig. S.1†). All polymers exhibited monomodal distributions, with molecular weight comparable to those previously reported (F8fBT: Mn = 44.8 kDa; IDTfBT: Mn = 15.9 kDa; FO6-T: Mn = 24.4 kDa).
| Sample | M n (kDa) | M w (kDa) | Đ |
|---|---|---|---|
| F8fBT | 44.8 | 133 | 2.99 |
| PS-F8BT | 98.2 | 222 | 2.26 |
| IDTfBT | 15.9 | 35.8 | 2.26 |
| PS-IDTBT | 34.4 | 51.0 | 1.48 |
| FO6-T | 24.4 | 69.4 | 2.85 |
| PS-FO6-T | 40.2 | 73.7 | 1.83 |
To date, only primary thiols have been utilised in post-polymerisation SNAr reactions. To establish that secondary thiols could undergo SNAr smoothly, we initially investigated the reaction of 2-butanethiol with F8fBT. After heating in the microwave for 1.5 hours in presence of potassium carbonate, gratifyingly the resulting polymer 2S-F8BT was found to have undergone complete fluorine substitution. We then moved onto the more challenging coupling of the thiol terminated PS (PS-SH), using a narrow dispersity (Đ of ≤1.1) sample with a Mn of 5000 g mol−1. As discussed above, a wide range of thiol terminated polymers can be readily prepared by RAFT techniques,34 although in this case commercially available PS-SH was chosen for convenience. An additional attraction of the polystyrene grafts was that simple blending of fluorescent conjugated polymers with PS has been shown to improve the emissive performance in OLED devices, due to a reduction in trap density.43–45 Moreover, while it can be difficult to ensure good mixing of polymer blends due to the low entropy of mixing polymers, we envisioned that grafting PS to conjugated polymer backbones would ensure good miscibility.
For each conjugated polymer, we reacted 1.2 equivalents of PS-SH in the presence of excess K2CO3 under heating at 120 °C for 3 days (see Scheme 1). Note that whilst the molar excess, based on the repeat unit mass, is small, the mass difference of each reagent is large (see ESI†). The crude products were precipitated in methanol and purified using Soxhlet extraction with methanol and acetone (24 h each) to remove excess thiol, and finally extracted into chloroform. The resulting polymers PS-F8BT, PS-IDTBT and PS-FO6-T were then characterised with GPC and NMR spectroscopy.
 | ||
| Scheme 1 Synthesis of the PS-grafted target polymers PS-F8BT, PS-IDTBT, and PS-FO6-T from the fluorinated starting polymers via a SNAr approach. | ||
The molecular weights of the graft copolymers were analysed using GPC in 1,2,4-TCB (Table 1, with more details in the ESI†). While PS-IDTBT exhibited a monomodal peak, both PS-F8BT and PS-FO6-T exhibited traces of unreacted PS-SH (and is oxidised dimer PS-SS-PS), which was incompletely removed by washing due to the large physical excess (Fig. S.2b†). The excess PS-SH could be removed from PS-FO6-T by preparative GPC (Fig. S.3†). Whilst the GPC confirmed an increase in weight, a higher value might have been expected given the size of the graft. Here we note that GPC analysis of graft copolymers against linear polystyrene standards is unrepresentative, given that the more densely packed conformations of grafts typically give smaller hydrodynamic radii compared to linear polymers. Similar observations have been observed in P3HT graft copolymers.22
1H-NMR of the grafts showed the occurrence of new signals corresponding to the PS. However, as the ratio of grafted PS to the respective backbone polymers is high, the backbone conjugated polymer signals are largely not visible in the 1H-NMR spectra. Additional proof of complete fluorine displacement comes from the absence of a signal in the 19F-NMR spectra of the three graft polymers. To further confirm the grafting of the PS to the conjugated polymer backbones, we used diffusion ordered NMR spectroscopy (DOSY), comparing the graft copolymers to a blend of both polymers (Fig. S11–13†). The 2D DOSY spectra plot chemical shift versus diffusion coefficients for individual species and are therefore able to determine the number of polymeric species present in a sample. For the samples with F8fBT, IDTfBT and FO6-T blended with PS-SH we observed two diffusion peaks, indicating that there are two separate polymers diffusing at different rates in each sample. For the graft copolymers the DOSY spectra display a single diffusion peak in all cases, providing strong evidence for the coupling of the PS to the conjugated backbone. The diffusion coefficient decreased compared to the starting polymers, due to the higher molecular weight of the grafted polymers.46
Further evidence for the success of the grafting approach was obtained by study of the material properties of grafted and parent polymers. Contact angle measurements using solvents of varying polarity (water, ethylene glycol and benzylic alcohol) showed that, upon grafting of PS, the wetting of the polymer films changed markedly (Table S.1†). Due to the decrease in the polarity that stems from the functionalisation with apolar PS, the contact angles decreased for ethylene glycol and benzylic alcohol. The thermal properties of the parent polymers and the respective graft copolymers were also studied, using thermogravimetric analysis (TGA) (Fig. S.14†) and differential scanning calorimetry (DSC) (Fig. S.15 and S.16†). TGA revealed good stability for the graft copolymers up to 328 °C for PS-FO6-T, 344 °C for PS-IDTBT and 337 °C for PS-F8BT, at which point a 5% weight loss was observed (Td). Comparing to the Td of the parent polymers, no clear trend in terms of changes in this metric were observed; for F8fBT, the Td was determined to be 328 °C, hence the Td was increased upon grafting, with similar results obtained for FO6-T (Td of 305 °C at 5% weight loss, slightly lower than the graft copolymer), but in the case of IDTfBT, the Td was found to be 403 °C, corresponding to a lowering of the Td upon grafting with PS.
DSC curves showed a glass transition temperature (Tg; determined using the derivative of the heat flow) of 61 °C for PS-F8BT, 59 °C for PS-IDTBT, and 58 °C for PS-FO6-T. We attribute this transition to the PS grafts, since none of the parent polymers show any features in the DSC traces up to 300 °C. This is in agreement with previous reports for F8fBT and FO6-T, as well as non-fluorinated IDTBT, and is generally characteristic for rigid rod conjugated polymers.38,47 The observed Tg transitions for graft copolymers are lower compared to the literature value of around 80 °C for pure PS samples with a Mn around 5000 g mol−1.48 Overall, these data collectively demonstrate the efficiency of the SNAr-based grafting approach, and the ability of PS-grafting to tune the material properties of conjugated polymers.
Nanoparticle synthesis and characterisation
To explore the potential of these systems as fluorescent nanoprobes, we first attempted to form stable NPs. The NPs were prepared with all three graft copolymers and parent polymers using a nanoprecipitation method.49 Briefly, 0.1 mg mL−1 solutions of the respective polymer in THF were injected into water while applying sonication. THF was removed by heating to 60 °C for 1 h while N2 was bubbled through the solution. The resulting NP suspensions were analysed using dynamic light scattering (DLS) to determine the size distribution of the particles and cryogenic transmission electron microscopy (cryo-TEM) to determine the shape and form of the particles. DLS analysis of the graft copolymers is shown in Fig. 1a, and the analysis of PS-F8BT NPs showed that narrow size distributions were obtained, with a Z-average of 41 nm. PS-IDTBT formed NPs of a broader distribution and a Z-average of 58 nm, possibly due to be more rigid conjugated backbone. The size distribution for NPs made of PS-FO6-T was very similar to PS-F8BT NPs and exhibited a Z-average of 49 nm. Overall, the size of the obtained NPs was in the same regime of 40–60 nm. Comparing the graft copolymer NPs to their parent polymers (Fig. 1b), it is apparent that the size decreases upon grafting of the PS onto the polymer backbone; for all three parent polymers the sizes are around 20 nm larger than for the respective graft copolymer NPs, with Z-averages of 63 nm for F8fBT, 81 nm for IDTfBT, and 74 nm for FO6-T. The decrease in size of the nanoparticles can tentatively be attributed to decreased interactions between conjugated backbones and hence less aggregation due to the PS grafting. DLS of IDTfBT NPs also shows the formation of some large aggregates, which are not observed in the graft NPs.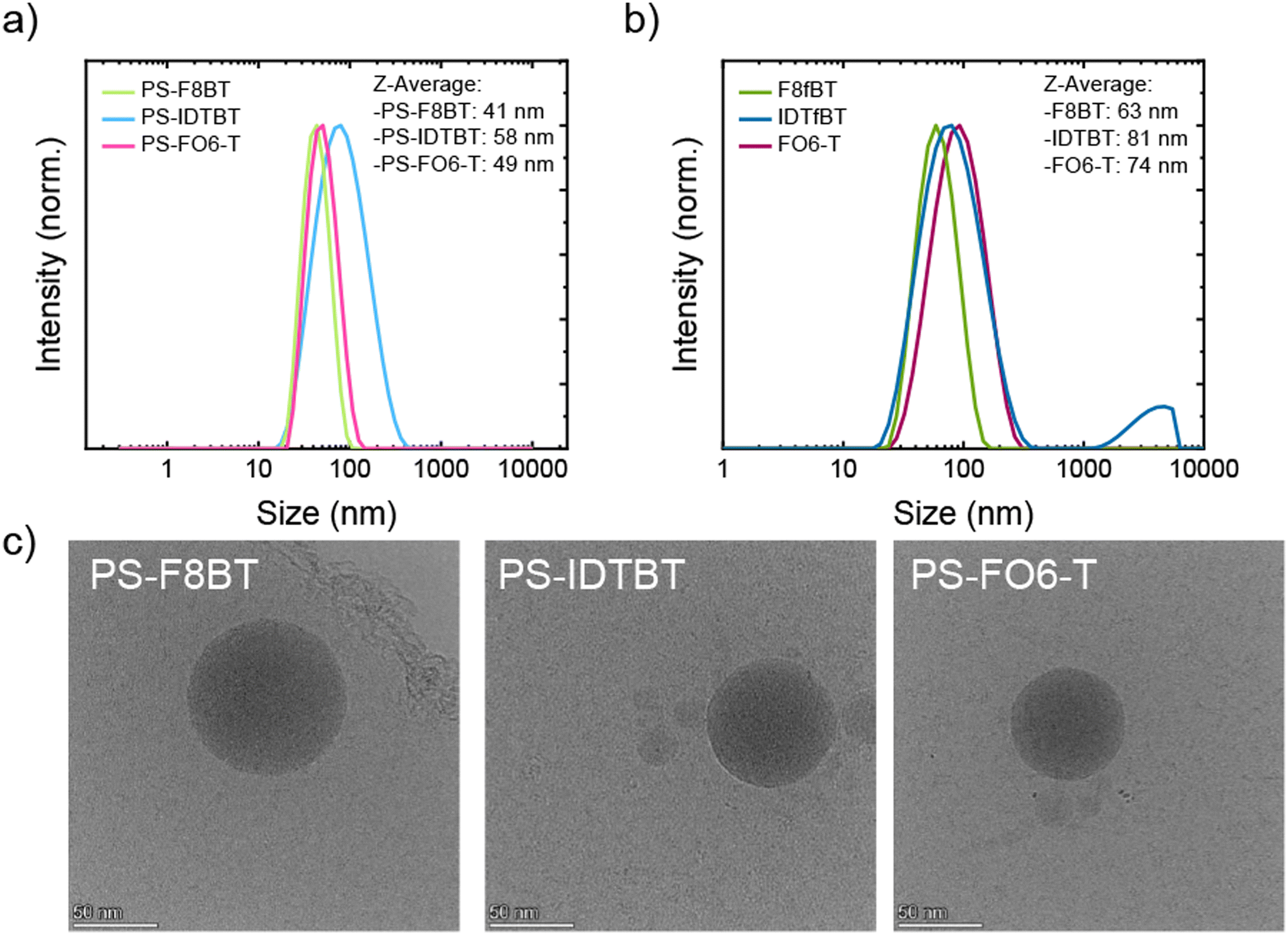 | ||
| Fig. 1 DLS traces and Z-average analyses of the NPs prepared from (a) graft copolymers, (b) parent polymers, and (c) cryogenic transmission electron microscopy (cryo-TEM) images of graft copolymer NPs. | ||
To investigate the stability of the NPs over time, NP suspensions were prepared from the graft copolymers and analysed over periods of up to 14 days. The DLS results showed that the particles were stable in suspension over these time periods, with no observed changes in the size distribution (Fig. S.17a–c†). These results were consistent with the stability of the parent polymer nanoparticles, which also showed no change of Z-average upon aging (Fig. S.17d†).
To study the shape and confirm the size of the NPs, cryo-TEM experiments were performed. The selected images in Fig. 1c demonstrate the overall trend in the cryo-TEM observations that the particle sizes were indeed around 50 nm in the case of all grafted polymers. Furthermore, the images show that spherical, uniform, and morphologically homogeneous particles were prepared with this method.
Optical characterisation
To examine the effects of the grafting of PS to the backbone of the conjugated polymers, we studied their absorption and photoluminescence (PL) in solutions in CHCl3 in comparison to blends of the parent polymers and PS-SH. For PS-F8BT the characteristic occurrence of an additional absorption peak at around 370 nm can be observed in comparison to starting polymer F8fBT, in addition to a change of the ratio of the relative intensities of the long wavelength intramolecular charge transfer (ICT) band to the high-energy absorption band (see Fig. 2a).26 These finding are very similar to our investigation into the substitution of F8fBT with 1-octanethiol, and provide further support for the grafting of the PS to the conjugated backbone.27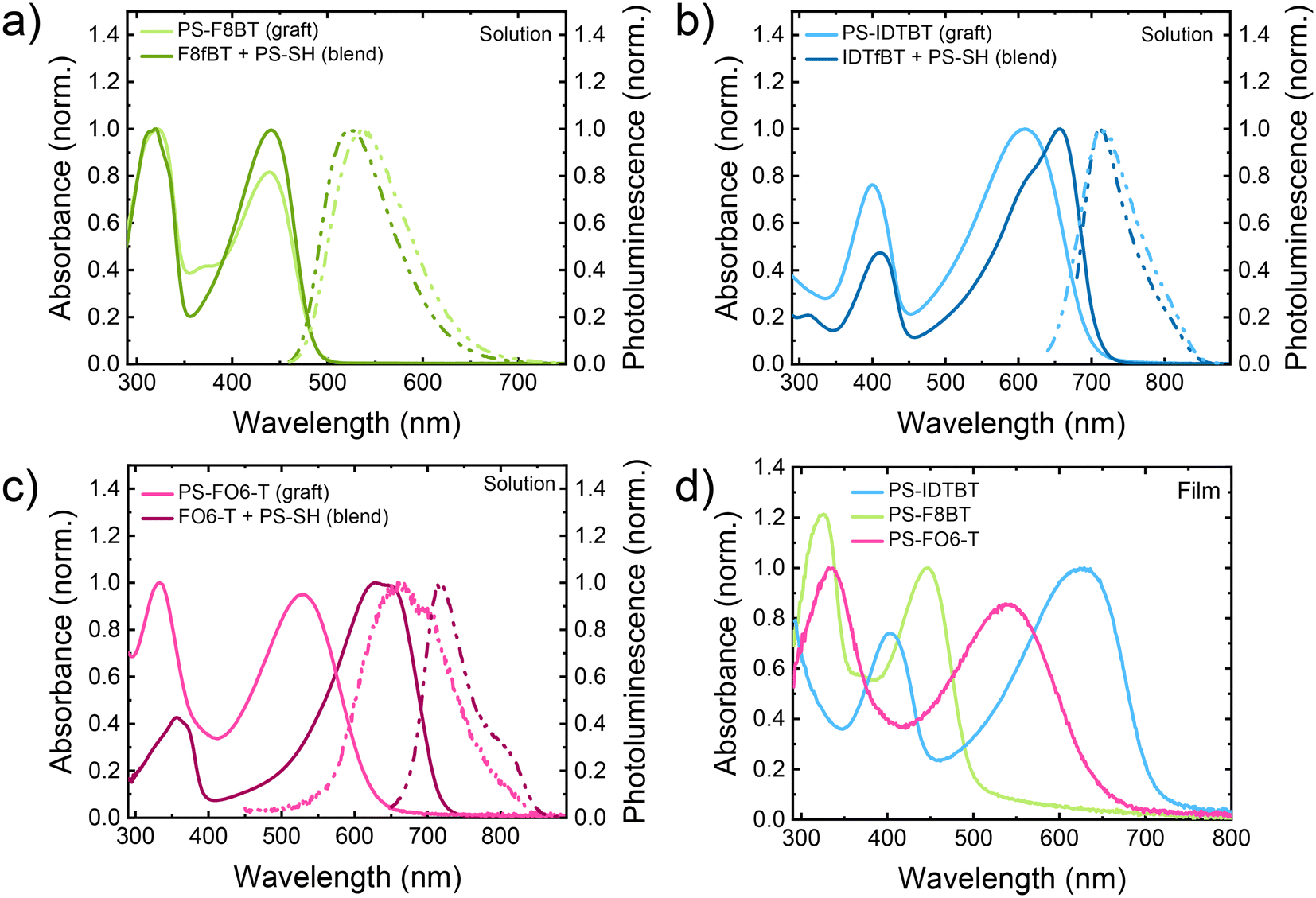 | ||
| Fig. 2 (a–c) UV-Vis (solid curves) and PL spectra (dot dash curves) of the graft copolymers in comparison to the blended parent polymers with PS-SH in solution (CHCl3). For the PL studies, PS-F8BT was excited at 440 nm, PS-IDTBT at 611 nm, and PS-FO6-T at 333 nm. (d) UV-vis spectra of thin films spun cast from graft copolymer solutions. | ||
For PS-IDTBT a significant blue shift of the main absorption peak can be observed, from 657 nm for IDTfBT to 609 nm for PS-IDTBT, along with a change in the relative intensities for the lower energy ICT band compared to the higher energy band (Fig. 2b). The widening of the polymer band gap and reduction in ICT intensity has previously been attributed to steric backbone twisting effect due to the presence of sulfur from the thioether group.27,50 Similar trends were observed for PS-FO6-T (Fig. 2c), although here the blue shift was larger, possibly due to a more pronounced steric effect due to the presence of two substituents on the BT group. The absence of a shift for F8fBT suggests the conjugated backbone is already twisted, as previously noted,35–38 relative to the more planar IDTFBT and FO6-T. The photoluminescence spectra of the grafts and the blends are shown in Fig. 2, and results summarised in Table 2. For PS-IDTBT there was no shift of the λPL,max (in solution) after the grafting, for PS-F8BT there was a small shift of 17 nm upon grafting (in agreement with the previous reports on thioether grafting), and for PS-FO6-T was a significant shift of 54 nm after grafting. The Stokes shifts of all polymers increased upon functionalisation with PS, with values of 0.494, 0.299, and 0.467 eV for PS-F8BT, PS-IDTBT, and PS-FO6-T, respectively. The observed increases in Stokes shift for the functionalised polymers can be attributed to a combination of steric effects, principally from the replacement of fluorine with the significantly larger sulfur, as well as the change from an electron withdrawing group to a slightly electron donating group.27
| λ abs,max (nm) | λ onset (nm) | λ PL,max (nm) | Stokes Shift (eV) |
E
g,opta (eV) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Polymer | Sol. | Film | Sol. | Film | Sol. | Sol. | Sol. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Estimated from the onset of absorption from the measured UV-vis spectra where E = hc/λ. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| F8fBT | 319, 441 | 455 | 486 | 514 | 521 | 0.432 | 2.55 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PS-F8BT | 322, 443 | 326 | 486 | 504 | 538 | 0.494 | 2.55 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IDTfBT | 657 | 680 | 712 | 722 | 713 | 0.148 | 1.74 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PS-IDTBT | 609 | 629 | 699 | 712 | 714 | 0.299 | 1.77 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FO6-T | 625 | 665 | 723 | 737 | 713 | 0.245 | 1.72 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PS-FO6-T | 528 | 651 | 633 | 785 | 659 | 0.467 | 1.96 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The graft copolymers were further studied in thin films on glass substrates spun cast from CHCl3 solutions (Fig. 2). For all PS-graft copolymers, the absorption maxima were red shifted compared to solution maxima, which is commonly observed for conjugated polymers and can be attributed to more planar structures in the solid state.51 The shapes of the curves did not, however, change significantly, although some broadening can be observed. Interestingly, the optical band gap (calculated from the onset of the absorption from the solution) for F8fBT and IDTfBT-based polymers showed no significant change upon grafting of the PS. In the case of FO6-T however, a large change was observed, increasing the gap significantly from 1.72 eV to 1.96 eV, which we attribute to significant twisting of the backbone upon grafting of the PS likely due to the higher grafting density.
The optical properties of the NPs were studied using UV-vis absorption and PL spectroscopy and compared to NPs prepared from the parent polymers, with the results shown in Fig. 3 and Table 3. One commonality between them all, apart from broadening of the signals, is the weakened absorption upon grafting, which can be explained by the decreased percentage of conjugated polymer in the samples. From the PL spectra, strong emission of PS-F8BT particles was noted (see Fig. 3d), whereas the emission for PS-IDTBT and PS-FO6-T NPs was found to be moderate (Fig. 3e and f). Interestingly, however, the emission intensity increased upon grafting in the cases of PS-F8BT and PS-FO6-T, while for PS-IDTBT NPs it was found to be similar to the parent polymer NPs.
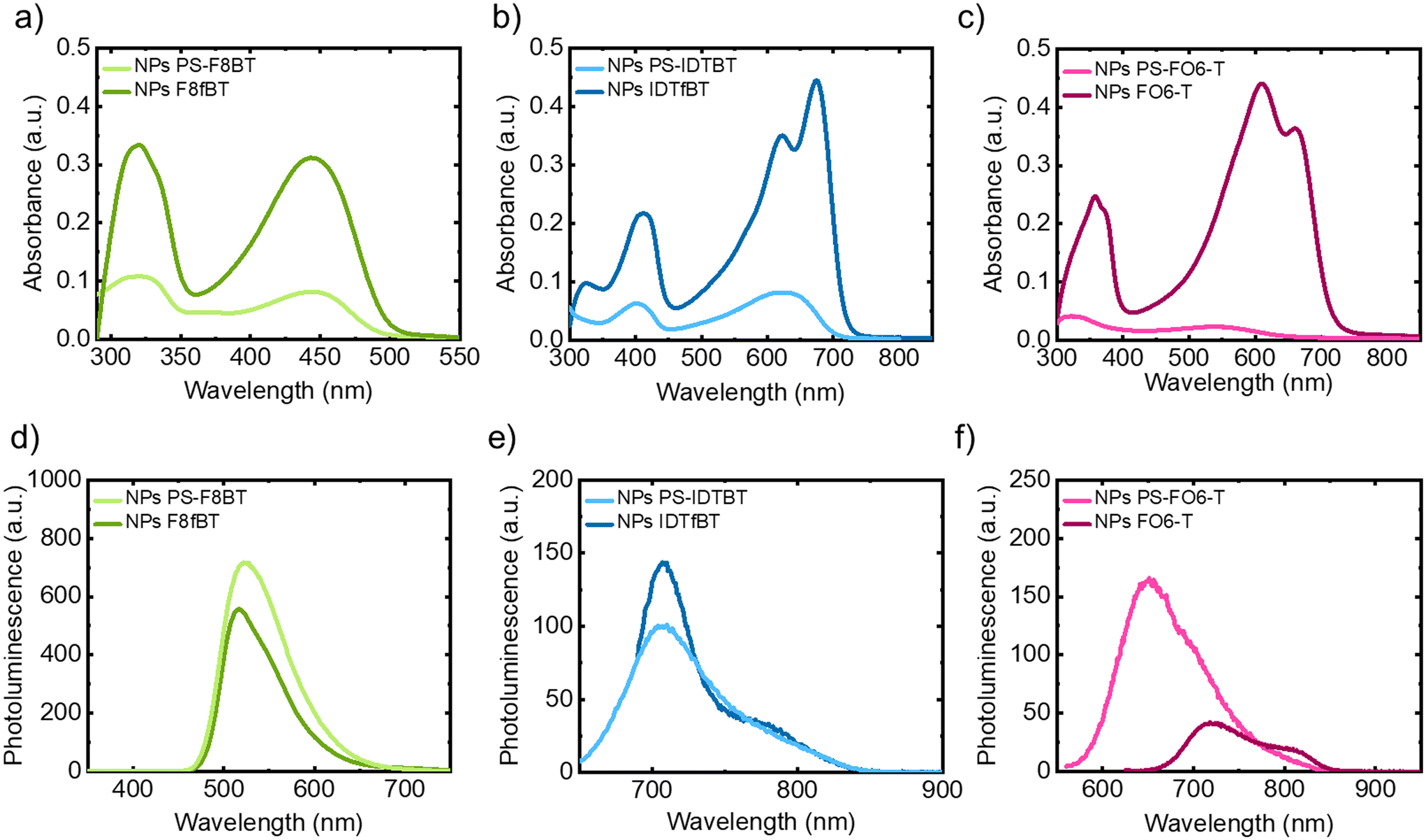 | ||
| Fig. 3 Optical characterisation of NP suspensions in water: UV-vis (a–c) and PL (d–f) spectra of graft copolymer NPs, for PL solutions were excited at λAbs,max: (a) and (d) F8fBT and PS-F8BT NPs, (b) and (e) IDTfBT and PS-IDTBT and (c) and (f) FO6-T and PS-FO6-T. | ||
| Sample | λ abs,max (nm) | λ PL,max (nm) |
|---|---|---|
| F8fBT | 320 | 517 |
| PS-F8BT | 321 | 522 |
| IDTfBT | 675 | 707 |
| PS-IDTBT | 625 | 710 |
| FO6-T | 611 | 718 |
| PS-FO6-T | 322 | 651 |
The CIE colour spaces of the three graft copolymer NP suspensions are shown in the ESI (Fig. S.18–20†). PS-F8BT shows yellow-green emission (x = 0.41, y = 0.56, CIE 1931), while for PS-IDTBT and PS-FO6-T NPs we observed emission mainly in the near-infrared (NIR) region of the solar spectrum, and hence the coordinates are located at the border of the colour plots (pictures of the grafted NP suspensions are shown in Fig. S.21†).
The photoluminescence quantum efficiency (PLQY) of the parent and the graft copolymers in chloroform solutions, thin films and NP suspensions were investigated and the results are summarised in Table 4. For the graft copolymers PS-F8BT, PS-IDTBT and PS-FO6-T we observed that the PLQYs in solution were 31%, 20% and 9% respectively. If these are compared to the solution PLQY of the parent polymers, the grafting has a different effect in all three cases. F8fBT shows a significantly higher PLQY compared to the graft, whereas in the case of FO6-T the grafting has a positive effect on the PLQY and for IDTfBT no changes are observed. In the aggregated states, film and NP, the PLQY decreases compared to the solutions which can be ascribed to aggregation-caused quenching (ACQ), a well-known effect for planar aromatic structures due to their inherently strong tendency to π–π stacking.52 Gratifyingly, ACQ is moderately suppressed upon grafting in the condensed states, indicated by an increase of the PLQY for graft copolymers in the films and NPs (with the exception of the films of FO6-T and PS-FO6-T, where the signals were too low to be accurately measured). This demonstrates that the emission of the polymers can be significantly enhanced by grafting of PS onto the backbones.
| Sample | Solution PLQY (%) | NP PLQY (%) | Film PLQY (%) |
|---|---|---|---|
| F8fBT | 54 | 8 | 22 |
| PS-F8BT | 31 | 20 | 27 |
| IDTBT | 20 | <1 | <1 |
| PS-IDTBT | 20 | 3 | 7 |
| FO6-T | 2 | <1 | n.m. |
| PS-FO6T | 9 | 1 | n.m. |
Conclusion
In conclusion, we have demonstrated that the secondary thiol end-group of a well-defined polystyrene can be used to graft-onto the backbone of three conjugated polymers containing an electron deficient fluorinated comonomer by SNAr. In a simple and efficient procedure, three different conjugated polymer backbones, F8fBT, IDTfBT and FO6-T were fully functionalised with a thiol terminated polystyrene to form the grafted polymers PS-F8BT, PS-IDTBT, and PS-FO6-T, respectively. The complete substitution was confirmed with a combination of DOSY and conventional NMR spectroscopy. Spherical NPs were prepared using a nanoprecipitation method with the three graft copolymers. Using DLS and cryo-TEM, the NPs were found to be comparable in size of around 50 nm diameter and be uniform in shape. Investigation of the optical properties of the graft copolymers in comparison to the parent polymers revealed that the grafting affects the λabs,max as well as the overall shape of the absorbance. Comparisons of the PL of the solution, thin film and NP suspensions of parent and graft copolymers showed that the emission intensity was quenched in solution when going from parent polymers to graft copolymers in the majority of the cases, but the opposite effect was observed in aggregated states. This was further supported by PLQY characterisation, which revealed increased emission for graft copolymers in aggregated NP suspension and film states. We believe that these promising results demonstrate that grafting by a SNAr approach is a simple and useful method for tuning the optical properties of emissive polymer backbones, which could expedite the synthesis of functional nanoprobes for use in biological imaging and sensing applications. This is particularly the case given the wide range of polymers that can be prepared with a thiol end-group by RAFT.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Peter Haycock for recording of DOSY NMR spectra. We thank the Engineering and Physics Science Research Council (EPSRC) (EP/R513052/1, EP/V048686/1 and EP/T028513/1), the Royal Society and Wolfson Foundation for financial support. We acknowledge baseline funding from the King Abdullah University of Science and Technology (KAUST).References
- C. D. Heinrich and M. Thelakkat, J. Mater. Chem. C, 2016, 4, 5370–5378 RSC.
- N. K. Obhi, D. M. Peda, E. L. Kynaston and D. S. Seferos, Macromolecules, 2018, 51, 2969–2978 CrossRef CAS.
- J. Steverlynck, J. De Winter, P. Gerbaux, R. Lazzaroni, P. Leclère and G. Koeckelberghs, Macromolecules, 2015, 48, 8789–8796 CrossRef CAS.
- P. D. Topham, A. J. Parnell and R. C. Hiorns, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1131–1156 CrossRef CAS.
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- M. Riede, D. Spoltore and K. Leo, Adv. Energy Mater., 2021, 11, 2002653 CrossRef CAS.
- O. Nuyken, S. Jungermann, V. Wiederhirn, E. Bacher and K. Meerholz, Monatsh. Chem./Chem. Monthly, 2006, 137, 811–824 CrossRef CAS.
- P. Jacoutot, A. D. Scaccabarozzi, T. Zhang, Z. Qiao, F. Aniés, M. Neophytou, H. Bristow, R. Kumar, M. Moser, A. D. Nega, A. Schiza, A. Dimitrakopoulou-Strauss, V. G. Gregoriou, T. D. Anthopoulos, M. Heeney, I. McCulloch, A. A. Bakulin, C. L. Chochos and N. Gasparini, Small, 2022, 18, 2200580 CrossRef CAS PubMed.
- L. W. T. Ng, S. W. Lee, D. W. Chang, J. M. Hodgkiss and D. Vak, Adv. Mater. Technol., 2022, 7, 2101556 CrossRef.
- G. Bernardo, T. Lopes, D. G. Lidzey and A. Mendes, Adv. Energy Mater., 2021, 11, 2100342 CrossRef CAS.
- T. Jarosz, K. Gebka and A. Stolarczyk, Molecules, 2019, 24, 3019 CrossRef CAS PubMed.
- L. T. Strover, J. Malmström and J. Travas-Sejdic, Chem. Rec., 2016, 16, 393–418 CrossRef CAS PubMed.
- D. Wu, Y. Huang, F. Xu, Y. Mai and D. Yan, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1459–1477 CrossRef CAS.
- D. Wu, F. Xu, Y. Huang, C. Chen, C. Yu, X. Feng, D. Yan and Y. Mai, Macromolecules, 2018, 51, 161–172 CrossRef CAS.
- Y. Huang, Y. Mai, X. Yang, U. Beser, J. Liu, F. Zhang, D. Yan, K. Müllen and X. Feng, J. Am. Chem. Soc., 2015, 137, 11602–11605 CrossRef CAS PubMed.
- F. Xu, J. Zhang, P. Zhang, X. Luan and Y. Mai, Mater. Chem. Front., 2019, 3, 2283–2307 RSC.
- D. van As, J. Subbiah, D. J. Jones and W. W. H. Wong, Macromol. Chem. Phys., 2016, 217, 403–413 CrossRef CAS.
- Z. Geng, J. J. Shin, Y. Xi and C. J. Hawker, J. Polym. Sci., 2021, 59, 963–1042 CrossRef CAS.
- H. J. Kim, K. Paek, H. Yang, C.-H. Cho, J.-S. Kim, W. Lee and B. J. Kim, Macromolecules, 2013, 46, 8472–8478 CrossRef CAS.
- W. Lee, J.-S. Kim, H. J. Kim, J. M. Shin, K. H. Ku, H. Yang, J. Lee, J. G. Bae, W. B. Lee and B. J. Kim, Macromolecules, 2015, 48, 5563–5569 CrossRef CAS.
- N. Li, Y. Dai, Y. Li, S. Dai, J. Strzalka, Q. Su, N. De Oliveira, Q. Zhang, P. B. J. St. Onge, S. Rondeau-Gagné, Y. Wang, X. Gu, J. Xu and S. Wang, Matter, 2021, 4, 3015–3029 CrossRef CAS.
- A. E. Masucci, M. Ghasemi, C. W. Pester and E. D. Gomez, Mater. Adv., 2023, 4, 2586–2594 RSC.
- Y.-H. Chan and P.-J. Wu, Part. Part. Syst. Charact., 2015, 32, 11–28 CrossRef CAS.
- P. Howes, M. Green, J. Levitt, K. Suhling and M. Hughes, J. Am. Chem. Soc., 2010, 132, 3989–3996 CrossRef CAS PubMed.
- M. Rimmele, F. Glöcklhofer and M. Heeney, Mater. Horiz., 2022, 9, 2678–2697 RSC.
- A. Creamer, C. S. Wood, P. D. Howes, A. Casey, S. Cong, A. V. Marsh, R. Godin, J. Panidi, T. D. Anthopoulos, C. H. Burgess, T. Wu, Z. Fei, I. Hamilton, M. A. McLachlan, M. M. Stevens and M. Heeney, Nat. Commun., 2018, 9, 3237 CrossRef PubMed.
- A. Creamer, A. Casey, A. V. Marsh, M. Shahid, M. Gao and M. Heeney, Macromolecules, 2017, 50, 2736–2746 CrossRef CAS.
- S. Cong, A. Creamer, Z. Fei, S. A. J. Hillman, C. Rapley, J. Nelson and M. Heeney, Macromol. Biosci., 2020, 20, e2000087 CrossRef PubMed.
- A. Creamer, A. L. Fiego, A. Agliano, L. Prados-Martin, H. Hogset, A. Najer, D. A. Richards, J. P. Wojciechowski, J. E. J. Foote, N. Kim, A. Monahan, J. Tang, A. Shamsabadi, L. N. C. Rochet, I. A. Thanasi, L. R. de la Ballina, C. L. Rapley, S. Turnock, E. A. Love, L. Bugeon, M. J. Dallman, M. Heeney, G. Kramer-Marek, V. Chudasama, F. Fenaroli and M. M. Stevens, Adv. Mater., 2023, e2300413, DOI:10.1002/adma.202300413.
- P. Boufflet, A. Casey, Y. Xia, P. N. Stavrinou and M. Heeney, Chem. Sci., 2017, 8, 2215–2225 RSC.
- N. Shida, K. Ninomiya, N. Takigawa, K. Imato, Y. Ooyama, I. Tomita and S. Inagi, Macromolecules, 2021, 54, 725–735 CrossRef CAS.
- K. Ninomiya, N. Shida, T. Nishikawa, T. Ishihara, H. Nishiyama, I. Tomita and S. Inagi, ACS Macro Lett., 2020, 9, 284–289 CrossRef CAS PubMed.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Polym. Int., 2011, 60, 9–25 CrossRef CAS.
- D. Kabra, L. P. Lu, M. H. Song, H. J. Snaith and R. H. Friend, Adv. Mater., 2010, 22, 3194–3198 CrossRef CAS PubMed.
- Y. Zhang and P. W. M. Blom, Appl. Phys. Lett., 2011, 98, 143504 CrossRef.
- C. L. Donley, J. Zaumseil, J. W. Andreasen, M. M. Nielsen, H. Sirringhaus, R. H. Friend and J.-S. Kim, J. Am. Chem. Soc., 2005, 127, 12890–12899 CrossRef CAS PubMed.
- W. Zhong, J. Liang, S. Hu, X.-F. Jiang, L. Ying, F. Huang, W. Yang and Y. Cao, Macromolecules, 2016, 49, 5806–5816 CrossRef CAS.
- J. F. Ponder Jr, H. Chen, A. M. T. Luci, S. Moro, M. Turano, A. L. Hobson, G. S. Collier, L. M. A. Perdigão, M. Moser, W. Zhang, G. Costantini, J. R. Reynolds and I. McCulloch, ACS Mater. Lett., 2021, 3, 1503–1512 CrossRef CAS.
- M. Nikolka, M. Hurhangee, A. Sadhanala, H. Chen, I. McCulloch and H. Sirringhaus, Adv. Electron. Mater., 2018, 4, 1700410 CrossRef.
- A. Wadsworth, H. Chen, K. J. Thorley, C. Cendra, M. Nikolka, H. Bristow, M. Moser, A. Salleo, T. D. Anthopoulos, H. Sirringhaus and I. McCulloch, J. Am. Chem. Soc., 2020, 142, 652–664 CrossRef CAS PubMed.
- M. Rimmele, Z. Qiao, J. Panidi, F. Furlan, C. Lee, W. L. Tan, C. R. McNeill, Y. Kim, N. Gasparini and M. Heeney, Mater. Horiz., 2023, 10, 4202–4212 RSC.
- J. Choi, S. Lee, M. Abbas, L. Vignau and B.-K. Ju, Curr. Appl. Phys., 2023, 45, 25–29 CrossRef.
- G. He, Y. Li, J. Liu and Y. Yang, Appl. Phys. Lett., 2002, 80, 4247–4249 CrossRef CAS.
- E. Khodabakhshi, P. W. M. Blom and J. J. Michels, Appl. Phys. Lett., 2019, 114, 093301 CrossRef.
- K. Gu, J. Onorato, S. S. Xiao, C. K. Luscombe and Y.-L. Loo, Chem. Mater., 2018, 30, 570–576 CrossRef CAS.
- Q. Wan, L. Ye and B. C. Thompson, ACS Mater. Lett., 2022, 4, 2440–2446 CrossRef CAS.
- P. Claudy, J. M. Létoffé, Y. Camberlain and J. P. Pascault, Polym. Bull., 1983, 9, 208–215 CrossRef CAS.
- Z. Tian, J. Yu, C. Wu, C. Szymanski and J. McNeill, Nanoscale, 2010, 2, 1999–2011 RSC.
- A. Casey, R. S. Ashraf, Z. Fei and M. Heeney, Macromolecules, 2014, 47, 2279–2288 CrossRef CAS.
- F. C. Spano and C. Silva, Annu. Rev. Phys. Chem., 2014, 65, 477–500 CrossRef CAS PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00492b |
| This journal is © The Royal Society of Chemistry 2024 |