
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of a sulfonamide functionalized poly(styrene oxide) and illustration of a potential post-polymerization strategy†
Marina
Wittig
ab,
Philipp
Pfändner
ab and
Bernhard
Rieger
 *a
*a
aTechnical University of Munich, WACKER Chair of Macromolecular Chemistry, Department of Chemistry, D 85748 Garching, Germany. E-mail: rieger@tum.de; Tel: +49 (0)89 289 13571
bTUMint.Energy Research GmbH, Department of Chemistry, D 85748 Garching, Germany. Tel: +49 (0)89 289 54448
First published on 10th July 2024
Abstract
In the first part of our work, we demonstrate a design concept for the functionalization of styrene oxide (SO) with a sulfonamide protecting group. A (1S,2S)-(+)-[1,2-cyclohexanediamino-N,N′-bis(3,5-di-t-butylsalicylidene)]-chromium(III)chloride ((salen)Cr(III)) catalyst polymerizes the end grouped epoxide via ring-opening-polymerization (ROP) into the respective polyether backbone. Absolute molecular weights of the resulting homopolymers range between 14.2 to 113 kg mol−1 with a polymer stability up to 300 °C and a glass transition temperature (Tg) of around 68–73 °C. The synthesis is completed by showing a possible post-polymerization modification of the functionalized poly(styrene oxide) (PSO). By adding the polymer to a lithium methoxide solution, a new reactive group in the form of a free sulfonate moiety can be generated. This method enables the transition towards a lithium sulfonated PSO that shows a thermal stability up to 300 °C and a Tg in the range of 18–20 °C.
Introduction
Poly(ethylene oxide) (PEO) and poly(propylene oxide) (PPO) are well known in the context of medical applications, food additives, as well as biocompatible applications. A facilitated access to their monomers is ensured by easily oxidizing the respective alkenes in large quantities.1 The ring-opening-polymerization (ROP) of ethylene oxide (EO) or propylene oxide (PO) can be described with a variety of catalytic systems.2 In many cases, the mechanism is based on living type polymerizations using different anionic or coordination catalyst systems like potassium tert-butoxide,3 trialkylaluminum (e.g. i-Bu3Al),4 or aluminum porphyrins coupled with quaternary organic salts.5A related epoxide with increased steric requirements in comparison to EO and PO is styrene oxide (SO). It can conventionally be synthesized employing organic peracids on styrene or applying a base when styrene chlorohydrin is used as raw material.6 Based on reactivity differences and electronic effects, established cationic or anionic catalysts that work in the ROP of EO or PO often show only oligomer formation or low molecular weights in the case of SO.7–9 To avoid harsh reaction conditions like the elongation of polymerization time, elevated temperatures, and ROPs under vacuum, organobases can be used to increase the molecular weights of poly(styrene oxide) (PSO).7,10
This work depicts a detailed monomer synthesis and polymerization protocol of our recently designed sulfonamide functionalized PSO. The functional group is selected in a way that restrictions related to functional group tolerance during ROP are bypassed. Post-polymerization modification enables the sulfonamide transformation towards a lithium sulfonate functionalized PSO. This specific setting should enable the establishment of an ionic polymer capable of conducting lithium cations. The polyether backbone has the potential to act as lithium dissolving matrix accompanied by immobilized anionic groups bearing charge carriers.
Results and discussion
The monomer synthesis of 1-((4-(oxiran-2-yl)phenyl)sulfonyl)-1H-pyrrole (1), the catalytically supported ROP towards poly(1-((4-(oxiran-2-yl)phenyl)sulfonyl)-1H-pyrrole (2) and the subsequent polymer-analogous transformation leading to poly(lithium 4-oxiran-2-yl)benzene sulfonate) (3) are depicted in Scheme 1. The whole synthetic pathway is monitored by nuclear magnetic resonance (NMR) spectroscopy, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC).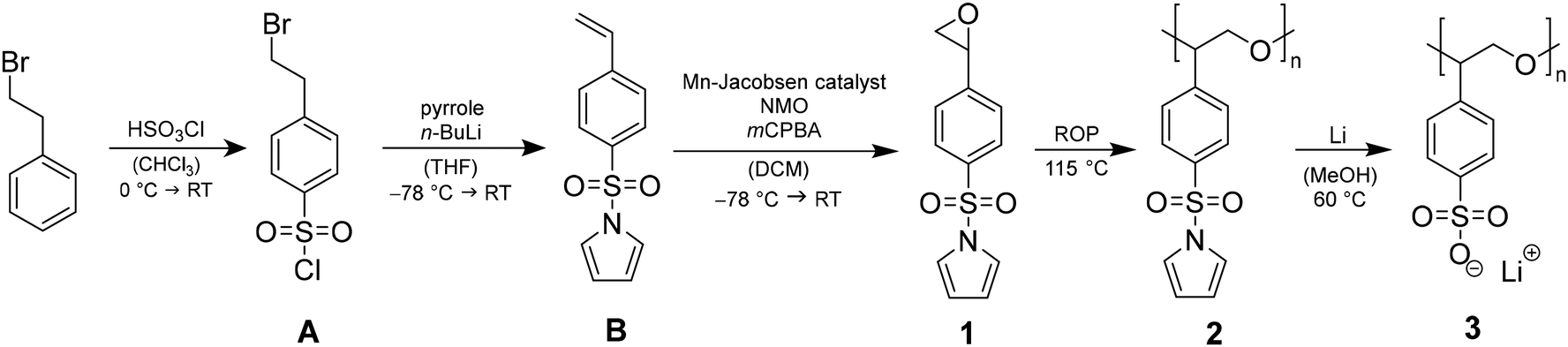 | ||
| Scheme 1 Three-step monomer synthesis including 1-((4-(oxiran-2-yl)phenyl)sulfonyl)-1H-pyrrole (1), catalytically supported anionic ROP of 1 to poly(1-((4-(oxiran-2-yl)phenyl)sulfonyl)-1H-pyrrole) (2) and cleavage of sulfonamide to obtain the final homopolymer poly(lithium 4-oxiran-2-yl)benzene sulfonate (3). | ||
Monomer synthesis
Monomer 1 consists of a terminal epoxide bound to an aromatic ring that is functionalized with a protected sulfonyl group in the form of a sulfonamide. The synthesis pathway is built up of three steps in which the first step is known as a classical chlorosulfonation in the para position of the alkyl substituent.11In the second step, two reactions take place, starting with n-butyllithium (n-BuLi) deprotonating pyrrole and activating the secondary amine as a nucleophile and a base. Via SN2, the chloride is cleaved of and substituted to form a sulfonamide bond.12 Due to the excess of deprotonated pyrrole in the solution, an elimination leads to the release of hydrogen bromide and thus, the generation of a vinylic double bond. The structure after this synthesis step bears styrene as the main structural motif with sulfonamide in its function as protecting group. Literature attempts often modify the sulfonyl group towards a larger anionic unit like trifluoromethanesulfonyl imide (TFSI) and radically polymerize the terminal double bond.13,14 In contrast, our approach is to convert the double bond in an oxygen transfer reaction into an epoxide to get to SO as the main core. First, epoxidation strategies with meta-chloroperoxybenzoic acid (mCPBA), hydrogen peroxide (H2O2), sodium hypochlorite (NaOCl) or dimethyldioxirane (DMDO) show no or only minimal conversion (see ESI S1†). This can be potentially ascribed to the electron deficiency of the double bond because both the aromatic ring and the sulfonamide exhibit electron withdrawing effects. In this case, transition metal catalysts like (R,R)-(−)-N,N′-bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexanediaminomanganese(III) chloride ((salen)Mn(III) catalyst), also known as Jacobsen catalyst, can promote the epoxidation.15 The chiral environment of the catalyst in combination with an excess of N-methylmorpholine-N-oxide (NMO) as co-catalyst and low temperature enables the enantioselective oxo transfer with an enantiomeric excess (ee) up to 91% in relation to styrene.15 Utilization of this catalytically supported epoxidation route leads to 70% conversion and 60–65% yield of 1. The execution at −78 °C prevents the isomerization to phenylacetaldehyde like in the reaction with DMDO (see ESI S2†). The structure of 1 is confirmed by 1H and 13C-NMR spectra (see ESI S3†). DSC measurements observe a Tm at 108 °C (see ESI S4†). Prior to polymerization, 1 is sublimated three times to exclude water as chain transfer agent.
Polymerization
For catalyst screening, polymerizations are applied in small scales in solution and bulk. Anionic ROP with n-BuLi or the implementation of titanium(IV)isopropoxide, diethylzinc, Lewis acidic β-diiminate (BDI)CF3–Zn–(SiMe3)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16 or tin(II) 2-ethylhexanoate do not show polymerization or minor oligomerization. Due to the decreased nucleophilicity of the alkoxide chain end induced by the electron-withdrawing effect of the phenyl ring, the polymerization behaviour of smaller and well-known monomers EO or PO cannot be directly transferred to SO.10 Nevertheless, there is the possibility of a living type, metal-free ROP with 1-tert-butyl-4,4,4-tris(dimethylamino)-2,2-bis[tris(dimethylamino)phosphor-anylidenamino]-2Λ5,4Λ5-cat-enadi(phosphazene) (t-Bu-P4). In contrast to alkali metal compounds, the phosphazene base possesses high basicity in combination with low nucleophilicity. The hereby controlled ROP of SO with 3-phenyl-1-propanol as initiator produces PSO with precise molecular masses up to 21.8 kg mol−1 and narrow dispersities (Đ) < 1.14 at room temperature.7 The performance of this organobase can however not be transferred to 1 which may be explained by an oxophilic attachment of the phosphorus compound to the spatially approachable sulfone moiety. If 1 is slightly modified and synthesized with the free sulfonyl group in its salt form, no polymerization is observed at all. These results strongly indicate the necessity of the pyrrole protecting group to bypass most of the polymerization restrictions based on functional group tolerance of the catalyst. The group of Coates et al. depicted elaborated catalytically supported ROPs based on a variety of transition metal complexes.2 Surrounded by larger salen ligands, especially cobalt and chromium organometallic catalysts effectively promote the ROP of epoxides like EO, PO, SO, and cyclohexene oxide (CO). The catalyst efficiency depends on the choice of initiator and the ratio of catalyst to cocatalyst.17 Aside from that, Jacobsen et al. demonstrated with their (salen)Mn(III) catalysts that the central atom needs to show coordination to the epoxide to an extent that it can display its Lewis acidic properties.18 After this screening, (1S,2S)-(+)-[1,2-cyclohexanediamino-N,N′-bis(3,5-di-t-butylsalicylidene)]-chromium(III)chloride ((salen) Cr(III) catalyst) (see Fig. 1) was found to be the best working catalyst for the anionic ROP of 1.
16 or tin(II) 2-ethylhexanoate do not show polymerization or minor oligomerization. Due to the decreased nucleophilicity of the alkoxide chain end induced by the electron-withdrawing effect of the phenyl ring, the polymerization behaviour of smaller and well-known monomers EO or PO cannot be directly transferred to SO.10 Nevertheless, there is the possibility of a living type, metal-free ROP with 1-tert-butyl-4,4,4-tris(dimethylamino)-2,2-bis[tris(dimethylamino)phosphor-anylidenamino]-2Λ5,4Λ5-cat-enadi(phosphazene) (t-Bu-P4). In contrast to alkali metal compounds, the phosphazene base possesses high basicity in combination with low nucleophilicity. The hereby controlled ROP of SO with 3-phenyl-1-propanol as initiator produces PSO with precise molecular masses up to 21.8 kg mol−1 and narrow dispersities (Đ) < 1.14 at room temperature.7 The performance of this organobase can however not be transferred to 1 which may be explained by an oxophilic attachment of the phosphorus compound to the spatially approachable sulfone moiety. If 1 is slightly modified and synthesized with the free sulfonyl group in its salt form, no polymerization is observed at all. These results strongly indicate the necessity of the pyrrole protecting group to bypass most of the polymerization restrictions based on functional group tolerance of the catalyst. The group of Coates et al. depicted elaborated catalytically supported ROPs based on a variety of transition metal complexes.2 Surrounded by larger salen ligands, especially cobalt and chromium organometallic catalysts effectively promote the ROP of epoxides like EO, PO, SO, and cyclohexene oxide (CO). The catalyst efficiency depends on the choice of initiator and the ratio of catalyst to cocatalyst.17 Aside from that, Jacobsen et al. demonstrated with their (salen)Mn(III) catalysts that the central atom needs to show coordination to the epoxide to an extent that it can display its Lewis acidic properties.18 After this screening, (1S,2S)-(+)-[1,2-cyclohexanediamino-N,N′-bis(3,5-di-t-butylsalicylidene)]-chromium(III)chloride ((salen) Cr(III) catalyst) (see Fig. 1) was found to be the best working catalyst for the anionic ROP of 1.
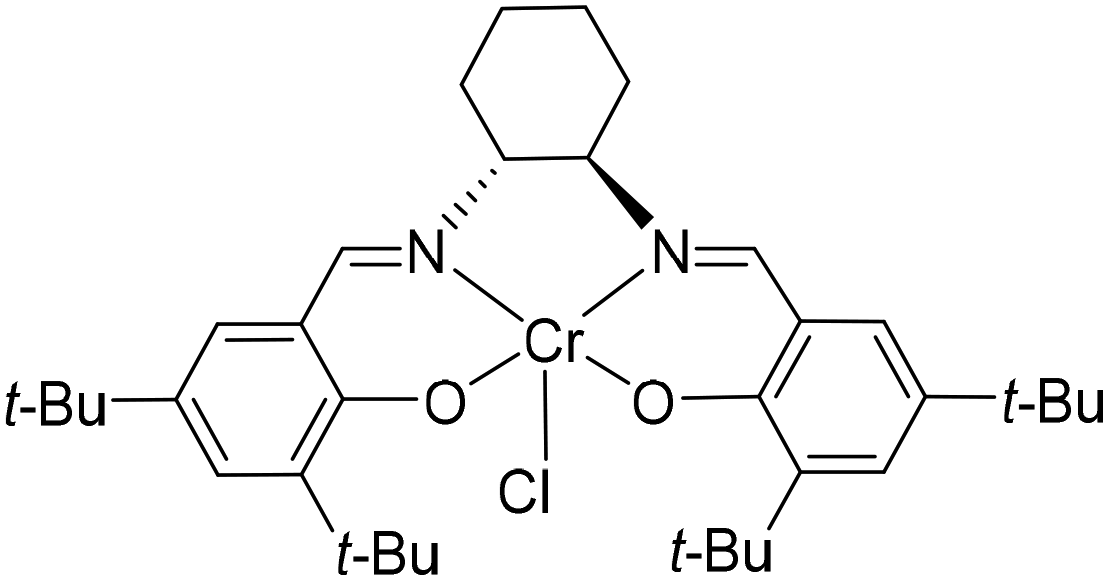 | ||
| Fig. 1 Structure of (salen)Cr(III) catalyst used in the anionic ROP of 1 to establish the polyether backbone of 2. | ||
In Table 1, the gel permeation chromatography (GPC) data (see ESI S5†) as well as the thermal behaviour of the respective polymers 2 determined via TGA and DSC are presented.
:10, 1:50, 1:100, 1:200) in bulk at 115 °C for 3 days yielding the respective polymers 2
| Cat:1a |
X
1
b [%] |
M
n,theoc [kg mol−1] |
M
n,absd [kg mol−1] |
Đ [—] |
I
ee [%] |
T
df [°C] |
T
gg [°C] |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
a Initial catalyst to monomer ratio.
b Conversion of monomer determined by isolated yield of polymer.
c Theoretical molecular weight Mn,theo = [Cat:1] × X1 × 249.28 g mol−1.
d Absolute molecular weight determination and dispersity of the homopolymer in DMF (30 °C, with 25 mmol L−1 LiBr, triple detection, dn/dc = 0.152 mL g−1).
e Initiator efficiencies = Mn,theo/Mn,abs.
f Onset decomposition temperatures (Tds) of first decomposition step determined via TGA measurements.
g
T
g determined via DSC measurements. Each experiment was performed at least in triplicates (standard deviations are depicted).
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1:10 |
96 | 2.50 | 14.2 ± 0.04 | 2.6 ± 0.01 | 18 ± 0.06 | 289 ± 0.47 | 69.9 ± 0.26 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1:50 |
93 | 11.8 | 36.5 ± 0.03 | 1.9 ± 0.01 | 32 ± 0.04 | 302 ± 0.82 | 68.5 ± 0.31 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1:100 |
97 | 24.1 | 67.3 ± 0.03 | 1.3 ± 0.01 | 36 ± 0.05 | 294 ± 0.56 | 69.5 ± 0.22 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1:200 |
96 | 50.8 | 113 ± 0.05 | 2.4 ± 0.01 | 43 ± 0.05 | 310 ± 0.87 | 72.6 ± 0.27 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
For homopolymerization, the (salen)Cr(III) catalyst and 1 are mixed in dry state under inert conditions and heated up to 115 °C to exceed the Tm of the monomer. To prevent solidification of the mixture, polymerization in toluene or dichloromethane (DCM) is investigated. Despite the better mixing behavior, only slight amounts of polymer formation is observed, probably due to reduced activity of the (salen)Cr(III) catalyst in solution. Bulk polymerizations with varying catalyst to monomer ratios (1:10, 1:50, 1:100, 1:200) yield a solid mixture which is soluble in dichloromethane. The respective polymers are isolated through precipitation in pentane, allowing separation from the catalyst and unconverted monomer. The last step is drying in a vacuum at 50 °C for 24 h to yield 93–97% of polymer in the form of a powder. The structures are confirmed by 1H-NMR (see ESI S6†). Energy dispersive X-ray analysis (EDX) measurements (see ESI S7†) prove the absence of chromium catalyst residues. GPC analysis show molecular weights of 14.2–113 kg mol−1, which exceed the chain lengths of unsubstituted PSO using an organobase in a living type polymerization (Mn = 5.20–21.8 kg mol−1).7 Dispersities vary between 1.3 to 2.6 quite significantly what can be traced back to inhomogeneities evoked by unpropitious stirring and increasing viscosity applying bulk conditions. Literature attempts underline, that using a more controlled set up like living type solution polymerization of SO can drastically narrow the respective dispersities (Đ = 1.06–1.14 for different catalyst to monomer ratios).7 Initiator efficiencies range from 18–43% and cannot be increased with additional sublimation steps of 1. Due to the overlaying signals of the epoxide and the aliphatic polyether backbone signals in the 1H-NMR spectra, conversion is determined gravimetrically. Concerning the ring-opening mechanism, 1 can be opened in two positions, both yielding a polyether backbone. As it is seen for catalytically supported ROP of SO, the attack of a nucleophile at the methylene or the methine position can be influenced by a variety of parameters like temperature, electronic properties of the transition metal centre, cocatalyst addition, or steric reasons.19,20 Since 1 consists of an aromatic ring and a sulfone moiety, which decrease electron density of the methine position, the nucleophilic attack is supposed to be favoured at this more electrophilic position. Mechanistic studies via (HT)-NMR are subject to ongoing studies. TGA measurements of 2 show that the Tds of the different catalyst to monomer ratios are all observed in the same range, with all compositions being stable up to at least 290 °C (see Fig. 2A) like it is also seen for commercial poly(styrene) (PS) (Tds between 300–330 °C).21 The first decomposition step between 290–310 °C shows 52% weight loss which can possibly be attributed to the cleavage of the sulfonamide from the aromatic ring. The second transition at elevated temperatures indicates a decomposition of the polyether backbone and aromatic ring. Fig. 2B comparatively displays the DSC traces for the tested catalyst to monomer ratios depicting the second heating cycle. Based on the absence of a Tm in the whole temperature profile ranging from −100–250 °C, it can be concluded that the polymer is fully amorphous. In comparison, syndiotatic PS is in contrast a semicrystalline polymer with a Tg between 90–95 °C (depending on the molecular weight) and a Tm around 260–270 °C.22 Aside from that, the Tgs of 2 range from 68–73 °C implying that the length of the polymer chains is not decisive for the thermal transition.
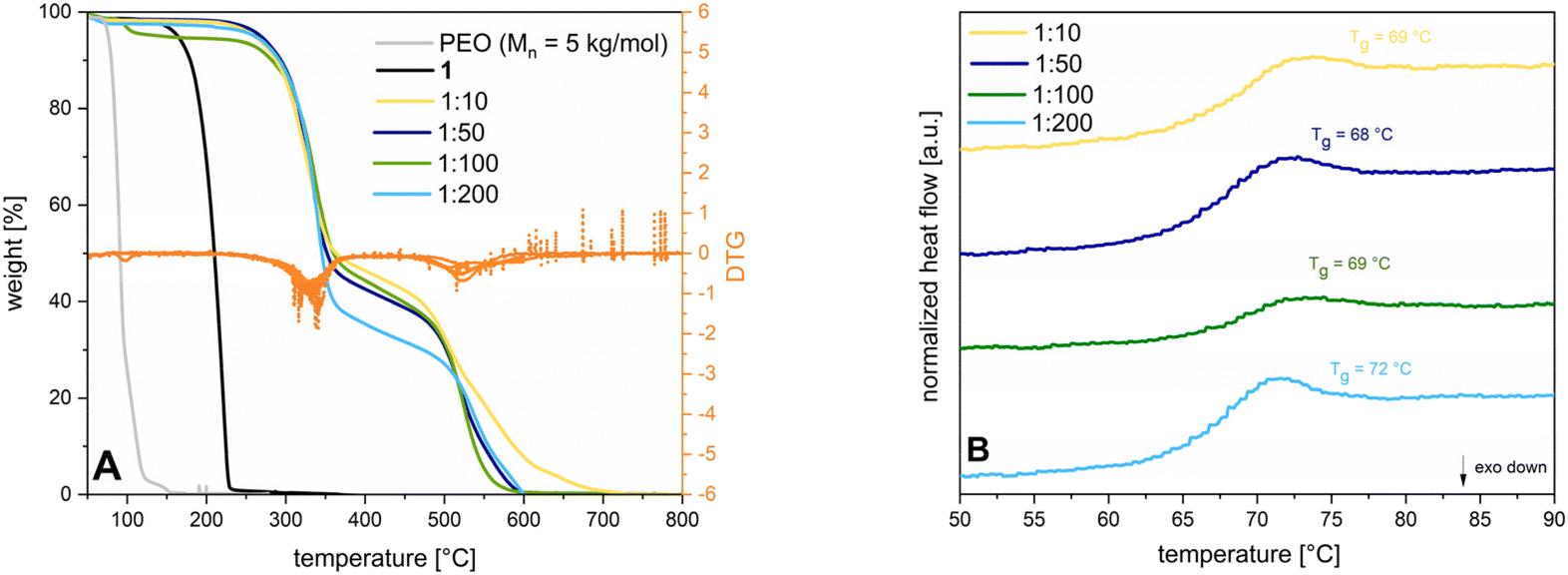 | ||
| Fig. 2 (A) TGA measurements of 2 for different catalyst to monomer ratios (1:10 (yellow line), 1:50 (dark blue line), 1:100 (green line). 1:200 (light blue line)) in comparison to PEO (Mn = 5 kg mol−1; grey line) and 1 (black line); orange lines depict the DTG of the respective catalyst to monomer ratios. Every sample is heated up from 25–1000 °C with a heating rate of 10 K min−1 under synthetic air; (B) DSC measurements of 2 for different catalyst to monomer ratios (1:10 (yellow line), 1:50 (dark blue line), 1:100 (green line), 1:200 (light blue line)) depicting the second heating cycle in an interval of 50–90 °C at a heating rate of 10 K min−1 in a non-hermetic set up. | ||
Post-polymerization modification
After successful synthesis and characterization of 2, post-polymerization modification focusses the cleavage of the sulfonamide and the generation of a lithium sulfonate bound to the PSO structure motif (see Scheme 1). Literature suggests acidic conditions like the addition of HCl, HBr, or H2SO4 as simple methods to break the sulfonamide bond.23 However, acidic treatment of the ring opened monomer 1 leads to polymer decomposition indicated by unattributable signals in the 1H-NMR spectrum. Instead of adding proton sources to the polyether backbone, the usage of nucleophiles or bases is forced. During deprotection experiments, the polymer and the deprotection reagent are dissolved separately, combined and heated up under reflux. Based on the chemical shift of the free pyrrole proton signals in the 1H-NMR, the degree of post-polymerization modification can be determined by continuously taking samples (0.1 mL of reaction mixture + 0.4 mL of NMR-solvent). KOH as alkali metal hydroxide is known to efficiently break the bond between sulfur and nitrogen in sulfonamides, yielding the sulfonic acid and amine.24 The resulting potassium salt can consequently be converted to the lithium salt in a salt metathesis reaction (see Scheme 2).25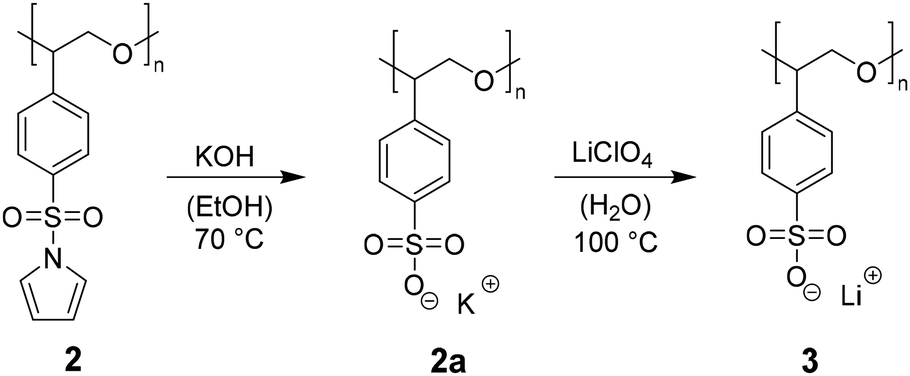 | ||
| Scheme 2 Synthesis protocol for the deprotection of 2 using a two-step approach; step 1: addition of KOH (10 eq. per monomer unit) in EtOH, 70 °C, reflux for 3 d to receive 2a; step 2: salt metathesis using LiClO4 (10 eq. per monomer unit) in H2O at 100 °C for 5 days. | ||
During the treatment of 2 with KOH, a slight amount of precipitate is formed, similar to attempts in literature.261H-NMR analysis of this residue reveals the successful post-polymerization modification (see ESI S8†). After dialysis, excess salt is removed yielding <1% of 2a. The low yields can be explained with the poor solubility of 2 in EtOH, preventing an efficient interaction between the protected polymer and KOH. To bypass the two-step post-modification, a singular step cleavage of the sulfonamide is preferred. Therefore, 2 is dissolved in dry DCM and added to a solution of elemental lithium in dry MeOH.27 After refluxing the solution at 65 °C for 3 d and evaporation of the solvent, the 1H-NMR spectrum of the crude product (catalyst to monomer ratio 1:100 as model system) shows a shift of both pyrrole proton signals to 6.5 and 5.9 ppm (see Fig. 3, full 1H-NMR of purified 3 in ESI S9†). Since the benzene as well as the polyether backbone signals are preserved, a successful post-modification is assumed.
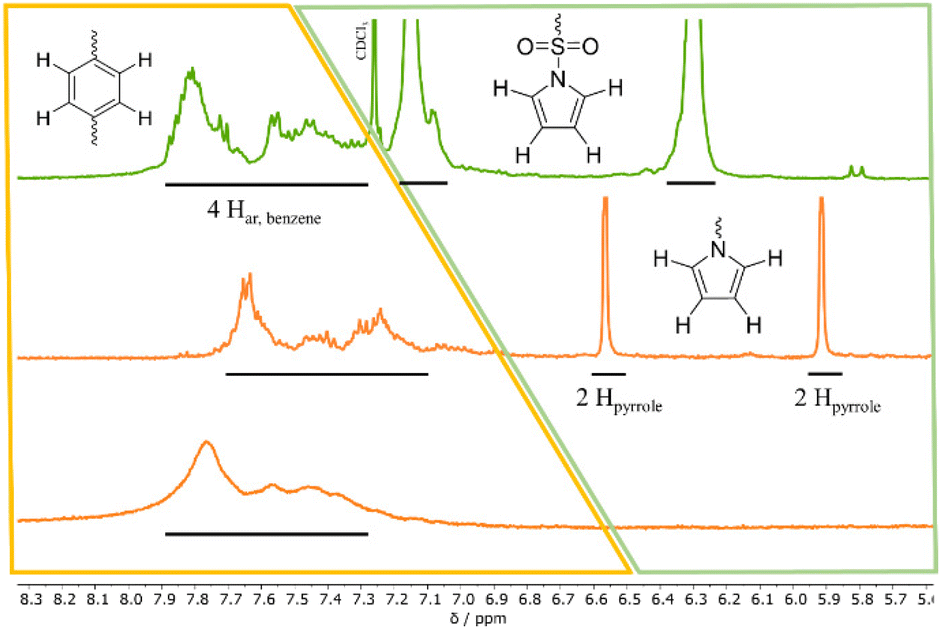 | ||
| Fig. 3
1H-NMR spectra in CDCl3 (δ = 7.26 ppm) at 400 MHz; extracts depicting the post-polymerization modification of 2 (catalyst to monomer ratio 1:100) after treatment with elemental Li (10 eq. per monomer unit) in a MeOH/DCM mixture; top (green line): 1H-NMR spectrum of 2 as reference in CDCl3; middle (orange line): 1H-NMR spectrum of crude product of 3 in D2O after the removal of solvent; bottom (orange line): 1H-NMR spectrum of 3 in D2O after dialysis; yellow areas highlight the benzene protons, green areas the pyrrole protons. | ||
The presence of lithium at the free sulfonate moiety is determined via7Li-NMR (see ESI S10†). The excessive pyrrole species is removed by dialysis, yielding 40–45% of purified 3. EDX spectra again show the absence of chromium (see ESI S11†). FTIR-spectra express the differences concerning vibration mode changes before and after deprotection (see ESI S12†). To determine the thermal properties of 3, the deprotected polymers are dried under inert conditions and sample preparation is carried out in the glovebox. The respective Tds are all located in a similar range as 2 at around 300 °C indicating the cleavage of the C–S bond to a styryloxide and SO3Li radical with subsequent PSO degradation. The cleavage of the pyrrole protecting groups results in a Tg shift from around 68–73 °C down to 18–22 °C (see Fig. 4).
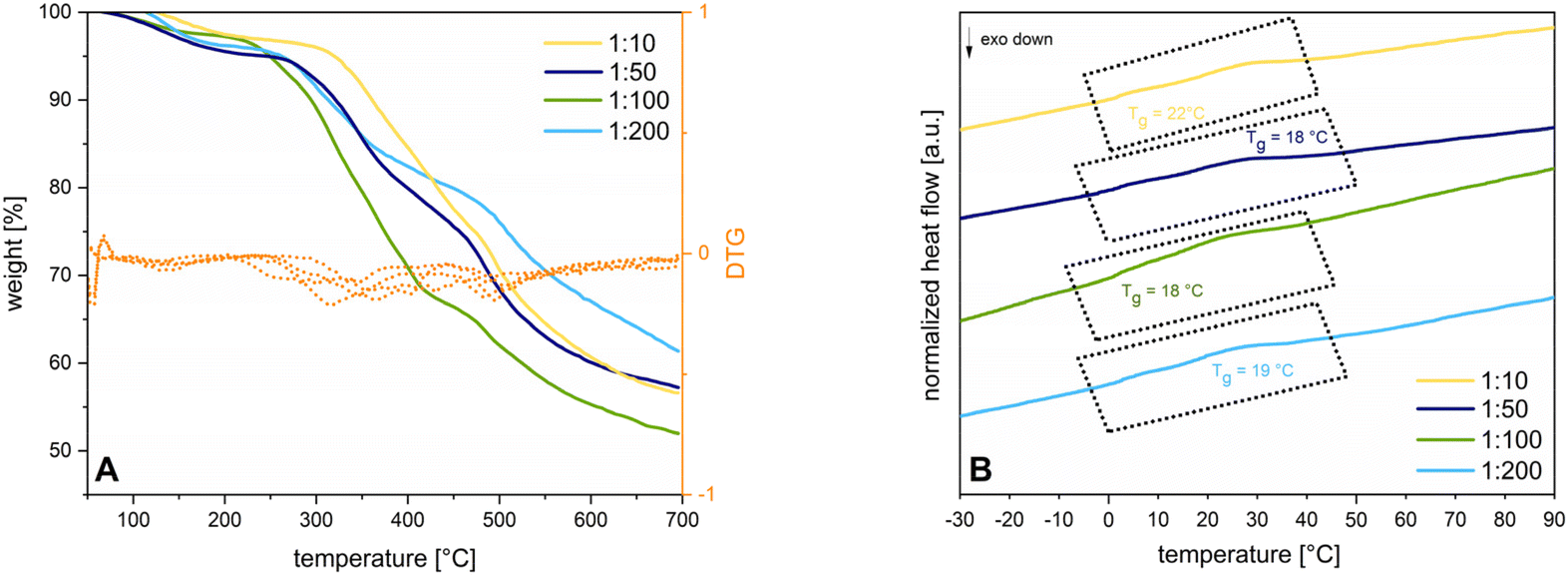 | ||
| Fig. 4 (A) TGA measurements of 3 for different catalyst to monomer ratios (1:10 (yellow line), 1:50 (dark blue line), 1:100 (green line). 1:200 (light blue line)); orange lines depict the DTG of the respective catalyst to monomer ratios. Every sample is heated up from 50–700 °C with a heating rate of 10 K min−1 under argon; (B) DSC measurements of 3 for different catalyst to monomer ratios (1:10 (yellow line), 1:50 (dark blue line), 1:100 (green line), 1:200 (light blue line)) depicting the second heating cycle in an interval of −30–90 °C at a heating rate of 10 K min−1 in a hermetic set up. | ||
Conclusions
In this work, we present a new synthetical concept towards a sulfonated PSO. The three-step monomer synthesis leads to a di-substituted benzene ring with a terminal epoxide and a sulfonamide in para position. Based on a well-designed protecting strategy, the catalytically supported ROPs with different (salen)Cr(III) catalyst to monomer ratios (1:10, 1:50; 1:100, 1:200) can be realized with yields up to 97%. Molecular weights range from 14.2 to 113 kg mol−1. Post-polymerization modification applying lithium methoxide converts the sulfonamide into a lithium sulfonate group. The sulfonated PSO shows thermal stability up to 300 °C and Tgs between 18–22 °C.
As our herein designed polymer bears one anionic charge per repetition unit and a lithium cation as counterion, its incorporation in solid state batteries as lithium single ion-conducting polymer electrolyte (SICPE) seems feasible. The enriched charge density should favour an efficient ion migration with high lithium transference numbers.28 Additionally, the generated polyether backbone offers an increased chain mobility at room temperature based on the low Tg. The application as SICPE, including a holistic electrochemical characterization, should be further investigated in future works. Furthermore, the anionic structure motif shows the potential to be optimized in the sense of a larger and more delocalized group like lithium trifluoromethanesulfonylimide (LiTFSI) to enhance ion dissociation.13
Author contributions
Marina Wittig: conceptualization, synthesis, funding acquisition, writing-original draft, visualization, data curation, formal analysis; Philipp Pfändner: writing-review and editing, data curation; Bernhard Rieger: supervision, project administration, resources, writing-review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors want to thank Anton Maier, Brigita Bratić, and Kerstin Halama for the great proofreading process. Additionally, the authors are grateful for the good discussion with the group of the WACKER chair of Macromolecular Chemistry and the TUMint.Energy Research GmbH. The authors additionally want to thank the Bavarian Ministry of Economic Affairs, Regional Development and Energy for the funding.References
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170–2243 CrossRef CAS PubMed.
- M. I. Childers, J. M. Longo, N. J. Van Zee, A. M. LaPointe and G. W. Coates, Chem. Rev., 2014, 114, 8129–8152 CrossRef CAS PubMed.
- C. C. Price and D. D. Carmelite, J. Am. Chem. Soc., 1966, 88, 4039–4044 CrossRef CAS.
- C. Billouard, S. Carlotti, P. Desbois and A. Deffieux, Macromolecules, 2004, 37, 4038–4043 CrossRef CAS.
- T. Aida, K. Sanuki and S. Inoue, Macromolecules, 1985, 18, 1049–1055 CrossRef CAS.
- M. S. Batra, R. Dwivedi and R. Prasad, ChemistrySelect, 2019, 4, 11636–11673 CrossRef CAS.
- H. Misaka, R. Sakai, T. Satoh and T. Kakuchi, Macromolecules, 2011, 44, 9099–9107 CrossRef CAS.
- R.-E. Parker and N. Isaacs, Chem. Rev., 1959, 59, 737–799 CrossRef CAS.
- L. Ge, Q. Huang, Y. Zhang and Z. Shen, Eur. Polym. J., 2000, 36, 2699–2705 CrossRef CAS.
- H. Zhang, S. Hu, J. Zhao and G. Zhang, Eur. Polym. J., 2017, 95, 693–701 CrossRef CAS.
- D. Obermayer, D. Znidar, G. Glotz, A. Stadler, D. Dallinger and C. O. Kappe, J. Org. Chem., 2016, 81, 11788–11801 CrossRef CAS PubMed.
- T. Janosik, H. Shirani, N. Wahlström, I. Malky, B. Stensland and J. Bergman, Tetrahedron, 2006, 62, 1699–1707 CrossRef CAS.
- R. Meziane, J.-P. Bonnet, M. Courty, K. Djellab and M. Armand, Electrochim. Acta, 2011, 57, 14–19 CrossRef CAS.
- R. Bouchet, S. Maria, R. Meziane, A. Aboulaich, L. Lienafa, J.-P. Bonnet, T. N. T. Phan, D. Bertin, D. Gigmes, D. Devaux, R. Denoyel and M. Armand, Nat. Mater., 2013, 12, 452–457 CrossRef CAS PubMed.
- M. Palucki, P. J. Pospisil, W. Zhang and E. N. Jacobsen, J. Am. Chem. Soc., 1994, 116, 9333–9334 CrossRef CAS.
- S. Kernbichl and B. Rieger, Polymer, 2020, 205, 122667 CrossRef CAS.
- D. J. Darensbourg, R. M. Mackiewicz, J. L. Rodgers and A. L. Phelps, Inorg. Chem., 2004, 43, 1831–1833 CrossRef CAS PubMed.
- L. E. Martinez, J. L. Leighton, D. H. Carsten and E. N. Jacobsen, J. Am. Chem. Soc., 1995, 117, 5897–5898 CrossRef CAS.
- N. D. Harrold, Y. Li and M. H. Chisholm, Macromolecules, 2013, 46, 692–698 CrossRef CAS.
- S. Abbina, V. K. Chidara and G. Du, ChemCatChem, 2017, 9, 1343–1348 CrossRef CAS.
- S. L. Malhotra, J. Hesse and L.-P. Blanchard, Polymer, 1975, 16, 81–93 CrossRef CAS.
- A. J. Pasztor, B. G. Landes and P. J. Karjala, Thermochim. Acta, 1991, 177, 187–195 CrossRef CAS.
- S. Searles and S. Nukina, Chem. Rev., 1959, 59, 1077–1103 CrossRef CAS.
- T. Ozaki, H. Yorimitsu and G. J. P. Perry, Chem. – Eur. J., 2021, 27, 15387–15391 CrossRef CAS PubMed.
- H. Hong-Bo, Z. Yi-Xuan, L. Kai, N. Jin, H. Xue-Jie, A. Michel and Z. Zhi-Bin, Chem. Lett., 2010, 39, 472–474 CrossRef.
- T. Ozaki, H. Yorimitsu and G. J. P. Perry, Tetrahedron, 2022, 117–118, 132830 CrossRef CAS.
- Z. Li, J. v. Lier and C. C. Leznoff, Can. J. Chem., 1999, 77, 138–145 CrossRef CAS.
- B. Kim, H. Kang, K. Kim, R.-Y. Wang and M. J. Park, ChemSusChem, 2020, 13, 2271–2279 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental (monomer synthesis, polymerization, post-polymerization functionalization); additional 1H/13C/7Li-NMR spectra, thermal analysis data, GPC traces, EDX data. See DOI: https://doi.org/10.1039/d4py00460d |
| This journal is © The Royal Society of Chemistry 2024 |