
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoregular polymerization of phenylacetylene using alkynyl and methyl rhodium(I) complexes with functionalized phosphine ligands: linear vs. branched poly(phenylacetylene)s†
Marta
Angoy
a,
M. Victoria
Jiménez
 *a,
Eugenio
Vispe
a,
Stepan
Podzimek
bcd and
Jesús J.
Pérez-Torrente
*a
*a,
Eugenio
Vispe
a,
Stepan
Podzimek
bcd and
Jesús J.
Pérez-Torrente
*a
aDepartamento de Química Inorgánica, Instituto de Síntesis Química y Catálisis Homogénea–ISQCH, Universidad de Zaragoza–CSIC, Facultad de Ciencias, C/Pedro Cerbuna, 12, 50009 Zaragoza, Spain. E-mail: perez@unizar.es; vjimenez@unizar.es
bInstitute of Chemistry and Technology of Macromolecular Materials, Faculty of Chemical Technology, University of Pardubice, Pardubice, Czech Republic
cWaters|Wyatt Technology, D-56307 Dernbach, Germany
dSYNPO, 53207 Pardubice, Czech Republic
First published on 8th July 2024
Abstract
The alkynyl [Rh(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C-Ph)(nbd){Ph2P(CH2)3Z}2] and [Rh(CC-Ph)(cod){Ph2P(CH2)3Z}] (cod = 1,5-cyclooctadiene, nbd = 2,5-norbornadiene) and methyl complexes [Rh(CH3)(cod){Ph2P(CH2)3Z}] featuring functionalized phosphine ligands (Z = NMe2, OEt) have been prepared. These complexes efficiently catalyze the polymerization of phenylacetylene in the absence of a base to afford stereoregular poly(phenylacetylene)s with high molar masses. Polymer characterization by SEC-MALS and A4F-MALS revealed a bimodal molar mass distribution due to the presence of a high molar mass polymer fraction. The diene ligand in the alkynyl complexes influences the morphology of the polymers. The poly(phenylacetylene)s prepared with the square-planar alkynyl complexes [Rh(CC-Ph)(cod){Ph2P(CH2)3Z}] having cod as the diene are linear, whereas the catalysts [Rh(CC-Ph)(nbd){Ph2P(CH2)3Z}2] with nbd as the diene afford PPAs with a fraction of high molar mass branched polymer. However, neither the catalytic performance nor the PPA morphology is affected by the donor function of the functionalized phosphine ligand (–NMe2 or –OEt). The alkynyl complexes having nbd as the diene ligand, [Rh(CC-Ph)(nbd){Ph2P(CH2)3Z}2], are much more active than the square-planar alkynyl and methyl complexes having cod as the diene. These catalysts polymerize phenylacetylene at a faster rate than the catalyst [Rh(CC-Ph)(nbd)(PPh3)2] bearing a non-functionalized phosphine ligand and afford polymers with a much higher molecular weight.
C-Ph)(nbd){Ph2P(CH2)3Z}2] and [Rh(CC-Ph)(cod){Ph2P(CH2)3Z}] (cod = 1,5-cyclooctadiene, nbd = 2,5-norbornadiene) and methyl complexes [Rh(CH3)(cod){Ph2P(CH2)3Z}] featuring functionalized phosphine ligands (Z = NMe2, OEt) have been prepared. These complexes efficiently catalyze the polymerization of phenylacetylene in the absence of a base to afford stereoregular poly(phenylacetylene)s with high molar masses. Polymer characterization by SEC-MALS and A4F-MALS revealed a bimodal molar mass distribution due to the presence of a high molar mass polymer fraction. The diene ligand in the alkynyl complexes influences the morphology of the polymers. The poly(phenylacetylene)s prepared with the square-planar alkynyl complexes [Rh(CC-Ph)(cod){Ph2P(CH2)3Z}] having cod as the diene are linear, whereas the catalysts [Rh(CC-Ph)(nbd){Ph2P(CH2)3Z}2] with nbd as the diene afford PPAs with a fraction of high molar mass branched polymer. However, neither the catalytic performance nor the PPA morphology is affected by the donor function of the functionalized phosphine ligand (–NMe2 or –OEt). The alkynyl complexes having nbd as the diene ligand, [Rh(CC-Ph)(nbd){Ph2P(CH2)3Z}2], are much more active than the square-planar alkynyl and methyl complexes having cod as the diene. These catalysts polymerize phenylacetylene at a faster rate than the catalyst [Rh(CC-Ph)(nbd)(PPh3)2] bearing a non-functionalized phosphine ligand and afford polymers with a much higher molecular weight.
Introduction
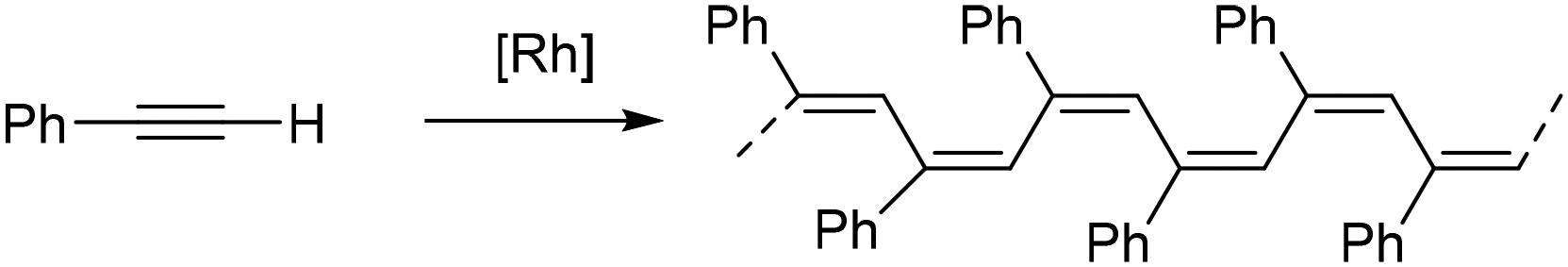
The polymerization of terminal alkynes such as phenylacetylene (PA) and its derivatives is one of the most studied chain polymerization reactions for accessing π-conjugated polymers.1 Unlike poly(acetylene), which has poor solubility and stability, poly(phenylacetylene) (PPA) is stable in air and soluble in organic solvents and has good processability.2 Interestingly, functional materials with applications in electronics, photoelectronics, optics and membrane separation have been prepared by introducing appropriate functionalized substituents into the phenyl ring of phenylacetylene.3–6 In addition, a wide range of supramolecular assemblies derived from PPAs have been reported. These include nanotubes, fibers, gels, liquid crystals and composites.7 The polymerization of substituted phenylacetylenes can be performed using a wide range of transition metal-based catalysts.8,9 In contrast to early-transition metal catalysts, which generally require the presence of a co-catalyst, well-defined complexes of late-transition metals have shown high activity and good stability in the presence of air and moisture.10 In particular, rhodium(I) catalysts have been widely used for the synthesis of PPAs because they are highly tolerant to many of the heteroatoms in alkyne-functionalized monomers and typically provide highly stereoregular polymers with a cis-transoidal configuration through a coordination-insertion mechanism.11,12Recently, considerable attention has been paid to the design of rhodium(I) catalysts for the living polymerization of alkyne-based monomers.13 Living polymerization allows the control of the polymer molecular weight, the modification of polymer chain ends, and the synthesis of block polymers with well-defined architectures.14–17 In this context, molecular Rh–alkynyl, Rh–vinyl18–20 and Rh–aryl21–23 complexes enable the controlled polymerization of PA derivatives to give highly stereoregular polymers with narrow molecular weight distributions and very high initiation efficiencies. The first controlled stereospecific polymerization of PAs using a well-characterized group 9 metal complex was reported by Noyori and co-workers using the pentacoordinated alkynyl rhodium(I) catalyst [Rh(CCPh)(nbd)(PPh3)2] and DMAP (4-(dimethylamino)pyridine) as the co-catalyst.24 PA polymerization using this catalytic system under different conditions yielded PPAs with low dispersities and initiation efficiencies in the range of 33–56%. The role of DMAP was shown to be critical in achieving a controlled polymerization to yield materials with lower molecular weights and narrower dispersities. Following that report, Noyori et al. showed a controlled PA polymerization using the related tetracoordinated compound [Rh(CCPh)(nbd)(PPh3)]. This catalyst exhibited improved catalytic performance affording a stereoregular PPA with very low dispersity and an initiation efficiency up to 72%. Unfortunately, this catalyst could not be isolated but was generated in situ from the multicomponent catalytic system [Rh(μ-OMe)(nbd)]2/PPh3/DMAP or [Rh(μ-Cl)(nbd)]2/NaOMe/PPh3/DMAP in the presence of PA.25,26
We have developed an alternative strategy for the design of efficient rhodium(I) PA polymerization catalysts based on the use of functionalized ligands with the ability to act as an internal base. Rhodium(I) catalysts with N-functionalized phosphine and N-heterocyclic carbene (NHC) ligands efficiently catalyzed PA polymerization leading to very high molecular weight stereoregular PPAs with a cis-transoidal configuration and moderate dispersities, albeit with low initiation efficiencies. For example, cationic rhodium(I) complexes based on amino-functionalized phosphine ligands, such as [Rh(nbd){κ2P,N-Ph2P(CH2)3NR2}]+ (R = H, Me),27 or neutral complexes with amino-functionalized NHC ligands, such as [RhX(nbd){κC-MeIm(CH2)3NMe2}] (X = Cl, Br),28 yielded stereoregular megadalton PPAs (Mn in the range of 1.1–1.5 × 106).29 Interestingly, characterization of the PPAs prepared with some of our catalysts by size exclusion chromatography (SEC) and asymmetric flow field flow fractionation (A4F) using a multiangle light-scattering detector (MALS) revealed that some samples contained a mixture of linear and branched polymers with high molecular weights.28,30
Mechanistic studies on PA polymerization with the catalyst [Rh(cod){κ2P,N-Ph2P(CH2)3NMe2}]+ allowed us to observe a cationic alkynyl species resulting from the intramolecular proton transfer from an η2-alkyne ligand to the coordinated-NMe2 group, which acts as an internal base. The presence of an external base, such as DMAP, promoted the deprotonation process leading to the formation of a related neutral alkynyl species and [HDMAP]+ (Fig. 1).27 The use of DMAP as a co-catalyst usually has a positive effect on the performance of the catalyst, resulting in an increase in both the activity and the initiation efficiency, and a decrease in the dispersity. For instance, the catalytic system [Rh(tfb){κ2P,N-PPh2(CH2)3NMe2}]+/DMAP polymerized PA in a quasi-living manner producing a PPA with a dispersity of 1.20, but with an initiation efficiency of only 10%.27 The key role of alkynyl rhodium species as PA polymerization catalysts was also demonstrated by mechanistic studies on the catalytic system [Rh(cod)(κ2P,N-Ph2PPy)]+/iPrNH2, which identified the neutral alkynyl compound [Rh(CCPh)(cod){κP-Ph2PPy}] as the active species.31
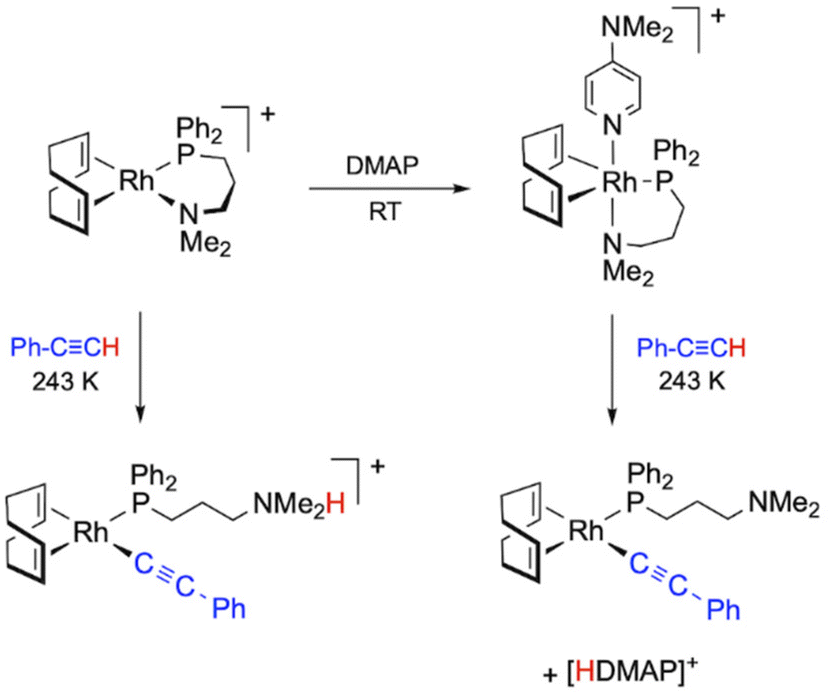 | ||
| Fig. 1 Formation of Rh–alkynyl species from the complex [Rh(cod){κ2P,N-Ph2P(CH2)3NMe2}]+ and PA in the absence of a base and in the presence of DMAP. | ||
Inspired by these results, and in order to improve the catalytic performance of our catalytic systems based on functionalized phosphine ligands, we consider the potential of rhodium(I) alkynyl initiators. On the one hand, the presence of an alkynyl ligand in the initiators could increase the initiation efficiency and, on the other hand, the donor function in the ligand, which is no longer needed in the initiation step to form the alkynyl species, could play a role similar to that of the base co-catalyst.
We report herein on the synthesis of a series of neutral alkynyl and methyl complexes bearing functionalized phosphine ligands with groups of very different basicity and coordination ability, Ph2P(CH2)3Z (Z = NMe2 or OEt), and their performance as PA polymerization catalysts. The characterization of the PPAs by SEC-MALS and A4F-MALS has shown that some of the samples contain a fraction of high molar mass (MM) branched polymer.
Results and discussion
Synthesis of alkynyl and methyl rhodium(I) complexes [Rh(R)(diene){Ph2P(CH2)3Z}n] (R = C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) C-Ph, Me; diene = nbd, cod; Z = NMe2, OEt; n = 1, 2)
C-Ph, Me; diene = nbd, cod; Z = NMe2, OEt; n = 1, 2)
The synthesis of alkynyl rhodium complexes [Rh(CC-Ph)(diene){Ph2P(CH2)3Z}n] was carried out following a one-pot procedure. The reaction of [Rh(μ-OMe)(nbd)]2 with the corresponding functionalized phosphine, Ph2P(CH2)3Z (Z = NMe2, OEt), and phenylacetylene (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:2.1 molar ratio) in THF afforded the pentacoordinated compounds [Rh(CC-Ph)(diene){Ph2P(CH2)3Z}2] (1, Z = NMe2; 2, Z = OEt) containing two functionalized phosphine ligands, which were isolated as dark brown solids in about 60% yield. These compounds were also obtained in lower yields when the reactions were carried out with a P/Rh ratio of 1 suggesting that the putative square planar complexes [Rh(CC-Ph)(nbd){Ph2P(CH2)3Z}] are not accessible by this procedure. In sharp contrast, the one-pot reaction of [Rh(μ-OMe)(cod)]2, Ph2P(CH2)3Z (Z = NMe2, OEt) and phenylacetylene (1:2:2.1) in THF resulted in the formation of alkynyl square planar complexes [Rh(CCPh)(cod){Ph2P(CH2)3Z}] (3, Z = NMe2; 4, Z = OEt) which were isolated as yellow brown solids in moderate yields (Chart 1).
:4:2.1 molar ratio) in THF afforded the pentacoordinated compounds [Rh(CC-Ph)(diene){Ph2P(CH2)3Z}2] (1, Z = NMe2; 2, Z = OEt) containing two functionalized phosphine ligands, which were isolated as dark brown solids in about 60% yield. These compounds were also obtained in lower yields when the reactions were carried out with a P/Rh ratio of 1 suggesting that the putative square planar complexes [Rh(CC-Ph)(nbd){Ph2P(CH2)3Z}] are not accessible by this procedure. In sharp contrast, the one-pot reaction of [Rh(μ-OMe)(cod)]2, Ph2P(CH2)3Z (Z = NMe2, OEt) and phenylacetylene (1:2:2.1) in THF resulted in the formation of alkynyl square planar complexes [Rh(CCPh)(cod){Ph2P(CH2)3Z}] (3, Z = NMe2; 4, Z = OEt) which were isolated as yellow brown solids in moderate yields (Chart 1).
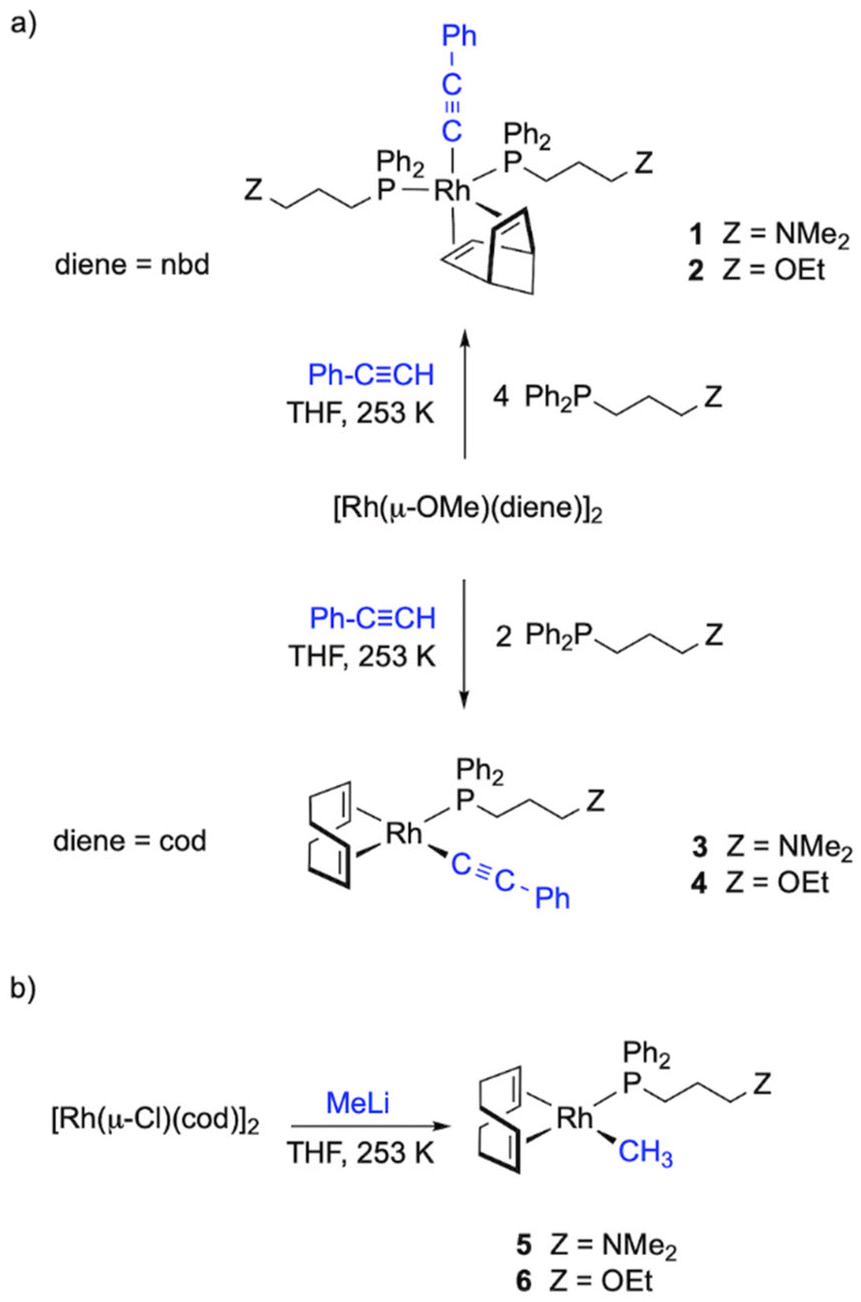 | ||
| Chart 1 Alkynyl and methyl rhodium(I) phenylacetylene polymerization catalysts. | ||
The alkynyl rhodium(I) complexes were characterized by elemental analysis, mass spectrometry and NMR spectroscopy. The 31P{1H} NMR spectra of compounds 1 and 2 recorded at room temperature in THF-d8 showed a broad resonance which was resolved into a doublet at δ ≈ 13 ppm (JRh–P ≈ 120 Hz) at 253 K. However, a broad resonance for the four = CH protons of the nbd ligand was observed in the 1H NMR spectra at 253 K, showing the fluxional behavior typical of pentacoordinated rhodium(I) and iridium complexes.32 Compounds 3 and 4 were also found to be dynamic although the fluxional behavior stopped at 233 K in CD2Cl2. Thus, the 31P{1H} NMR spectra showed a doublet at δ ≈ 26 ppm (JRh–P ≈ 160 Hz) and the 1H NMR spectra showed two resonances for the four olefin protons ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH of the cod ligand, which is consistent with a square planar structure of Cs symmetry with the more shielded resonance corresponding to the protons trans to the phosphine ligand. The 13C{1H} NMR spectra of complexes 1–4 at low temperature showed two resonances around δ ≈ 120–110 ppm for the Cα and Cβ carbon atoms of the alkynyl ligand with a multiplicity that is consistent with the number of functionalized phosphine ligands in both types of complexes (Fig. 2). Although the olefinic CH carbons of the diene ligands were not observed in the 13C{1H} NMR spectra of 1 and 2 at 253 K due to their dynamic behavior, the spectra of 3 and 4 at 233 K showed two resonances at δ ≈ 97 (dd, JC–Rh ≈ 12 Hz and JC-P ≈ 7 Hz) and 85 ppm (d, JC–Rh ≈ 8 Hz) with the expected multiplicity for a square planar structure.
CH of the cod ligand, which is consistent with a square planar structure of Cs symmetry with the more shielded resonance corresponding to the protons trans to the phosphine ligand. The 13C{1H} NMR spectra of complexes 1–4 at low temperature showed two resonances around δ ≈ 120–110 ppm for the Cα and Cβ carbon atoms of the alkynyl ligand with a multiplicity that is consistent with the number of functionalized phosphine ligands in both types of complexes (Fig. 2). Although the olefinic CH carbons of the diene ligands were not observed in the 13C{1H} NMR spectra of 1 and 2 at 253 K due to their dynamic behavior, the spectra of 3 and 4 at 233 K showed two resonances at δ ≈ 97 (dd, JC–Rh ≈ 12 Hz and JC-P ≈ 7 Hz) and 85 ppm (d, JC–Rh ≈ 8 Hz) with the expected multiplicity for a square planar structure.
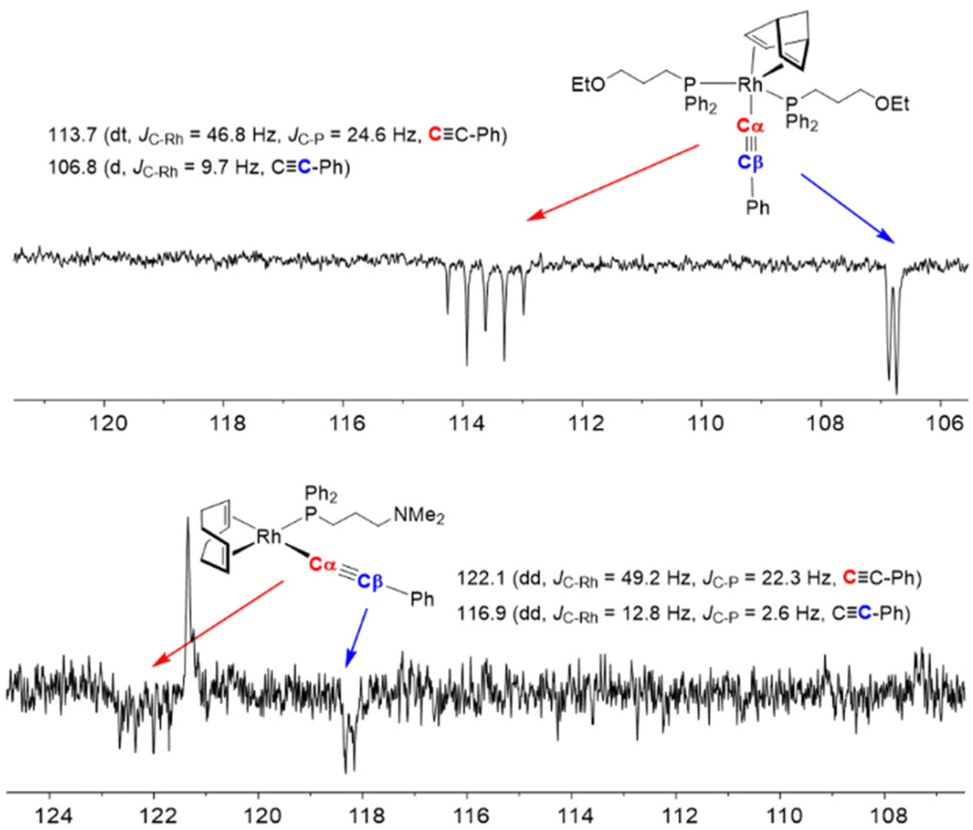 | ||
| Fig. 2 Region of the 13C{1H} NMR spectra of 2 and 3 showing the Cα and Cβ carbon resonances of the alkynyl ligand. | ||
The reaction of [Rh(μ-Cl)(cod)]2 with the functionalized phosphines Ph2P(CH2)3Z (Z = NMe2, OEt) and MeLi (1:2:2 molar ratio) in diethyl ether at 253 K gave dark orange solutions from which the square planar methyl complexes [Rh(CH3)(cod){Ph2P(CH2)3Z}] (5, Z = NMe2; 6, Z = OEt) were isolated as orange solids in moderate yields (Chart 1). Attempts to prepare related complexes with nbd as the diene ligand using this method were unsuccessful. The methyl ligand in both complexes was observed as a high field doublet at δ ≈ 0.5 ppm (JRh–H ≈ 1.4 Hz) in the 1H NMR spectra and as a doublet at δ ≈ 5 ppm (JC–Rh ≈ 24 Hz) in the 13C{1H} spectra in C6D6. In addition, the spectra showed two well-separated resonances for the CH of the cod ligand which is consistent with the expected square planar structure of Cs symmetry.
The κ1P coordination mode of the functionalized phosphine ligands in compounds 1–6 was inferred from the 13C{1H} NMR spectra in which the resonances of –NMe2 and –OEt fragments were found to be shielded by 5–6 ppm compared to that of complexes [Rh(nbd){κ2P,N-Ph2P(CH2)3NMe2}]+ and [Rh(cod){κ2P,N-Ph2P(CH2)3OEt}]+ in which the ligands exhibit a bidentate coordination mode.33
Polymerization of PA using alkynyl and methyl rhodium(I) complexes
Complexes [Rh(CC-Ph)(diene){Ph2P(CH2)3Z}n] (diene = nbd, n = 2; cod, n = 1) (1–4) and [Rh(CH3)(cod){Ph2P(CH2)3Z}] (5 and 6) (Z = NMe2, OEt) were found to be efficient initiators for PA polymerization (Table 1). The polymerization reactions were carried out in THF at 20 °C in the absence of light with a [PA]o/[Rh] ratio of 100. The PPAs were isolated as soluble yellow-orange solids in practically quantitative yields. The 1H NMR spectra showed a sharp signal at δ 5.85 ppm for the vinyl protons and six characteristic resonances in the 13C{1H} NMR spectra in CD2Cl2 suggesting a highly stereoregular structure with a cis-transoidal configuration. Indeed, a cis-content ≥ 95% was determined by NMR.34,35 The PPAs were characterized by size exclusion chromatography (SEC) and asymmetric flow field flow fractionation (A4F) using multi-angle light scattering (MALS) and differential refractive index (DRI) detectors (Table 1).
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Cat. | t (min) | Conv.b (%) | Anal. | M w (g mol−1) | Đ | IEd (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reaction conditions: [PA]o = 0.25 M, [PA]o/[Rh] = 100, in THF at 293 K. b Determined by GC (octane as an internal standard). c Đ = dispersity (Mw/Mn, where Mn = number-average molecular weight). d Initiation efficiency, IE = Mtheor/Mn × 100, where Mtheor = [PA]o/[Rh] × MWPA × polymer yield. e Bimodal distribution of MM. Data for the low MM polymer. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 1 | 10 | 100 | SEC | 7.88 × 105e |
1.17 | 1.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 1 | 10 | 100 | A4F | 1.85 × 106e |
2.81 | 1.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 2 | 20 | 100 | SEC | 7.40 × 105e |
1.51 | 2.1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 2 | 20 | 100 | A4F | 1.44 × 106e |
3.03 | 2.1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 3 | 40 | 100 | SEC | 2.76 × 105e |
2.03 | 7.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 3 | 40 | 100 | A4F | 3.08 × 105e |
2.22 | 7.4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 4 | 60 | 100 | SEC | 2.56 × 105e |
2.02 | 8.0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 4 | 60 | 100 | A4F | 2.84 × 105e |
2.05 | 7.4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | 5 | 60 | 100 | SEC | 2.68 × 105e |
1.55 | 5.9 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | 6 | 60 | 100 | SEC | 2.56 × 105e |
1.74 | 6.9 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The polymerization reactions were very fast, with 100% conversions in relatively short reaction times (10–60 min), resulting in high molecular weight PPAs with variable dispersity. The alkynyl complexes [Rh(CC-Ph)(nbd){Ph2P(CH2)3NMe2}2] (1) and [Rh(CC-Ph)(nbd){Ph2P(CH2)3OEt}2] (2) were found to be the most active catalysts with full PA conversion in just 10 and 20 min, respectively, affording PPAs with Mw ≈ 7.5 × 105 and low dispersities of 1.17 and 1.51, respectively (entries 1 and 3). However, PA polymerization with catalysts [Rh(CC-Ph)(cod){Ph2P(CH2)3NMe2}] (3) and [Rh(CC-Ph)(cod){Ph2P(CH2)3OEt}] (4) was slower, requiring 40 and 60 min respectively for full conversion, affording PPAs with Mw ≈ 2.6 × 105 and slightly higher dispersities (entries 5 and 7). The higher activity of the catalysts having nbd as the diene ligand is consistent with its smaller size and higher π-acceptor capacity compared to cod, which facilitates both PA coordination and insertion into the Rh–C bond of the polymer chain, and is in agreement with previous observations with related rhodium initiators.36,37 In contrast, neither the MM nor the dispersity of the polymer obtained with each type of catalyst was significantly affected by the functional group of the phosphine ligand (–NMe2 or –OEt). Furthermore, catalysts of the same type showed similar initiation efficiencies, suggesting that the functionalized phosphine does not play a relevant role in the initiation step and that neutral alkynyl complexes are likely to be the initiating species in the PA polymerization. Finally, methyl complexes [Rh(CH3)(cod){Ph2P(CH2)3NMe2}] (5) and [Rh(CH3)(cod){Ph2P(CH2)3OEt}] (6) also showed good catalytic performance, with activities, Mw and dispersities similar to those obtained with 4 (entries 9 and 10).
SEC-MALS analysis of the polymers revealed an unresolved bimodal molar mass distribution as observed in the SEC chromatogram of a PPA sample prepared with catalyst 1 as a representative example (Fig. 3a). The shoulder to the left of the main peak in the light scattering detector response indicates a higher MM polymer fraction, albeit at a low concentration according to the DRI response which is sensitive to the polymer concentration. Irregular (non-linear) increases in MM were observed in the plot of MM vs. elution volume in both the main and secondary peaks, which may be due to the characteristic co-elution effect in SEC.38 Thus, the late SEC elution consists of species with different molar masses. This indicates that a significant number of high MM species are eluted later than expected, either because of the presence of branches or because the high molar mass species are retained on the stationary phase, or a combination of both. The log–log plot of the radius of gyration (rg) vs. the molar mass (conformation plot) from the SEC-MALS data showed two regions with slightly different slopes that turn almost flat at very high MM (Fig. 3b) which also suggests the presence of different PPA species, likely as a result of the combination of linear and branched species. The first region, with a slope of m = 0.52, collects species of MM up to Mw = 1.16 × 106, and a second region, with a slope of m = 0.48, includes species with very high molar masses, up to Mw = 3.22 × 106. Although both fractions appear to be linear in the ranges where the light scattering and differential refractometer detectors measure reliably, the slope of the second fraction (m = 0.48) is well below the reference value of 0.58 typical of linear PPA polymers.39 Analysis of this sample by A4F-MALS also confirmed a bimodal molecular weight distribution and the presence of a branched polymer. The conformation plot obtained from the A4F-MALS data also showed two regions with different slopes, with a very significant deviation from linearity in the higher molar mass region, suggesting the presence of a fraction of very high MM branched polymer (Fig. 3d). It should be noted that the higher exclusion limit and the absence of a stationary phase in A4F avoid the effects of column interaction with the polymer and allow better separation and therefore more accurate MM and rg distributions.40
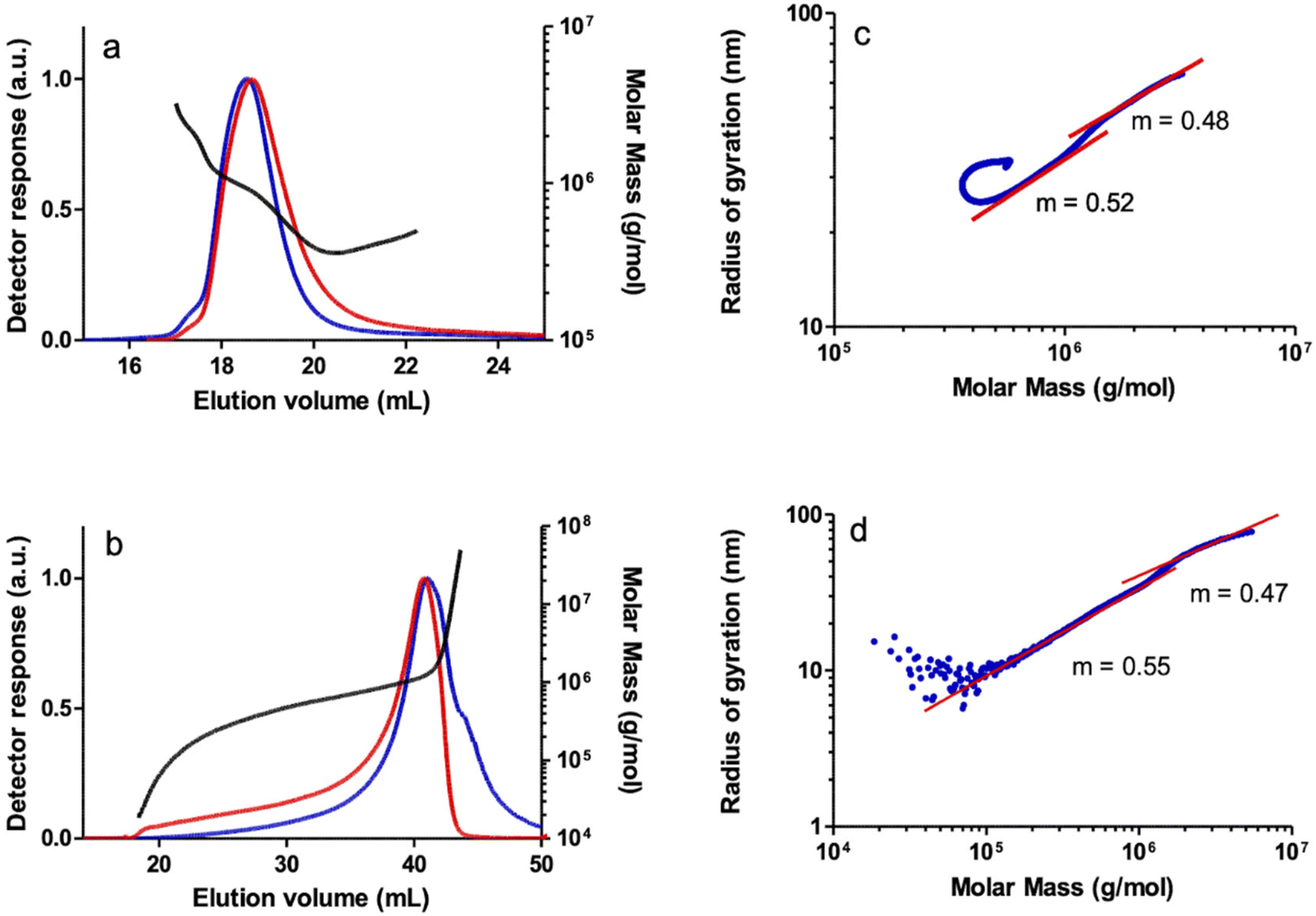 | ||
| Fig. 3 SEC chromatograms: light scattering detector response (90 degrees) (blue) and differential refractometer response (red), and MM (molar mass) vs. elution volume plot for a PPA sample prepared with the catalyst [Rh(CC-Ph)(nbd){Ph2P(CH2)3NMe2}2] (1) in THF analyzed by: (a) SEC-MALS and (b) A4F-MALS. Log–log plot of the radius of gyration (rg) vs. MM (blue) and linear fit (red) of the same sample analyzed by: (c) SEC-MALS and (d) A4F-MALS. | ||
The SEC-MALS analysis of a PPA sample obtained with catalyst 2 also showed a bimodal molar mass distribution, although in this case the shoulder corresponding to a higher MM fraction practically overlapped the broad main peak. To a lesser extent, a non-linear increase in MM versus elution volume was also observed in the central part of the main peak, probably resulting from the co-elution effect in SEC of linear and branched high MM species discussed above. The log–log plot of rgvs. MM also showed two regions with different slopes. The small slope of the high MM region (m = 0.38), well below the reference value for a linear polymer, is indicative of the presence of branched species in the very high MM region. Indeed, the log–log plot of rgvs. MM showed an appreciable deviation from the linear behavior in the high MM region which is compatible with the presence of a fraction of high MM branched polymer which was also confirmed by the A4F-MALS analysis of the polymer (see the ESI†). The polymers prepared with catalysts 3 and 4 were also found to show bimodal behavior at very high MM as shown by the MALS detector, but the contribution of these species is negligible since there is no quantifiable amount as shown by the DRI detector. However, both polymers appear to be linear, which was confirmed by the linear relationship of the log–log plot of rgvs. MM in the high molar mass region with both SEC-MALS and A4F-MALS (Fig. 4).
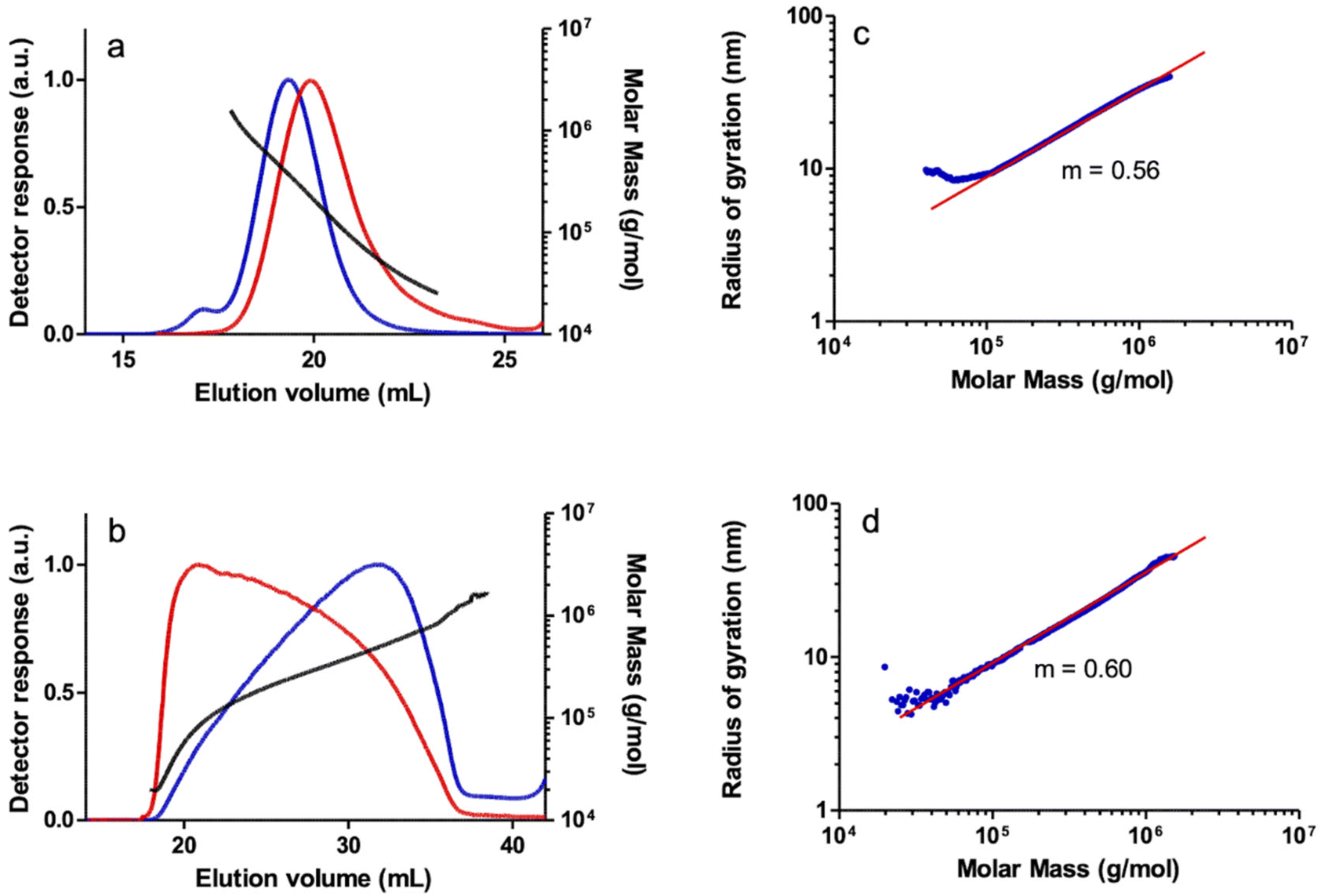 | ||
| Fig. 4 SEC chromatograms: light scattering detector response (90 degrees) (blue) and differential refractometer response (red), and MM (molar mass) vs. elution volume plot for a PPA sample prepared with the catalyst [Rh(CC-Ph)(cod){Ph2P(CH2)3OEt}] (4) in THF analyzed by: (a) SEC-MALS and (b) A4F-MALS. Log–log plot of the radius of gyration (rg) vs. MM (blue) and linear fit (red) of the same sample analyzed by: (c) SEC-MALS and (d) A4F-MALS. | ||
In general, the Mw of the PPAs obtained with catalysts 1–4, as determined by A4F-MALS, are higher than those determined by SEC-MALS and also show higher dispersities (Table 2, entries 2, 4, 6 and 8). The lower MM determined by SEC-MALS may indicate shear degradation and/or removal of very high molecular weight macromolecules from the sample by interaction with the stationary phase (filtration effect of SEC columns).40 The differences between the Mw determined by SEC and A4F are very noticeable in the case of the polymer samples prepared with catalysts 1 and 2. These polymers are susceptible to the negative effects of the SEC technique due to the presence of a high MM branched polymer. In contrast, a moderate increase in the Mw determined by A4F compared to SEC was observed in the PPAs prepared with catalysts 3 and 4, which is consistent with the linearity of these samples.
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Cat. | t (min) | Conv.b (%) | Anal. | M w (g mol−1) | Đ | IEd (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reaction conditions: [PA]o = 0.25 M, [PA]o/[Rh] = 100, [DMAP]/[Rh] = 1 in THF at 293 K. b Determined by GC (octane as an internal standard). c Đ = dispersity (Mw/Mn, where Mw = weight-average molecular weight and Mn = number-average molecular weight). d Initiation efficiency, IE = Mtheor/Mn × 100, where Mtheor = [PA]o/[Rh] × MWPA × polymer yield. e Bimodal distribution of MM. Data for the low MM polymer. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 1 | 10 | 100 | SEC | 3.11 × 105e |
1.13 | 3.7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 2 | 20 | 100 | SEC | 3.18 × 105e |
1.22 | 3.9 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 3 | 50 | 100 | SEC | 2.58 × 105e |
2.08 | 8.2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 4 | 40 | 100 | SEC | 2.84 × 105e |
1.68 | 6.0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The PPAs prepared with the methyl rhodium(I) catalysts 5 and 6 also showed bimodal distributions of MM in SEC-MALS although the concentration of the higher MM polymer fraction is too small to be observed using the DRI detector. Although a regular increase in MM was observed in the plot of MM vs. elution volume in the main peak of the PPA prepared with catalyst 5, the plot shows an increase in MM in the high-MM region at short elution volumes which suggests the presence of a branched material. In addition, the log–log plot of rgvs. MM showed a significant deviation from the linear behavior in the high MM region, consistent with the presence of a fraction of high MM branched polymer (Fig. 5). In contrast, the PPA prepared with catalyst 6 showed a linear relationship of the log–log plot of rgvs. MM, consistent with a linear polymer (see the ESI†).
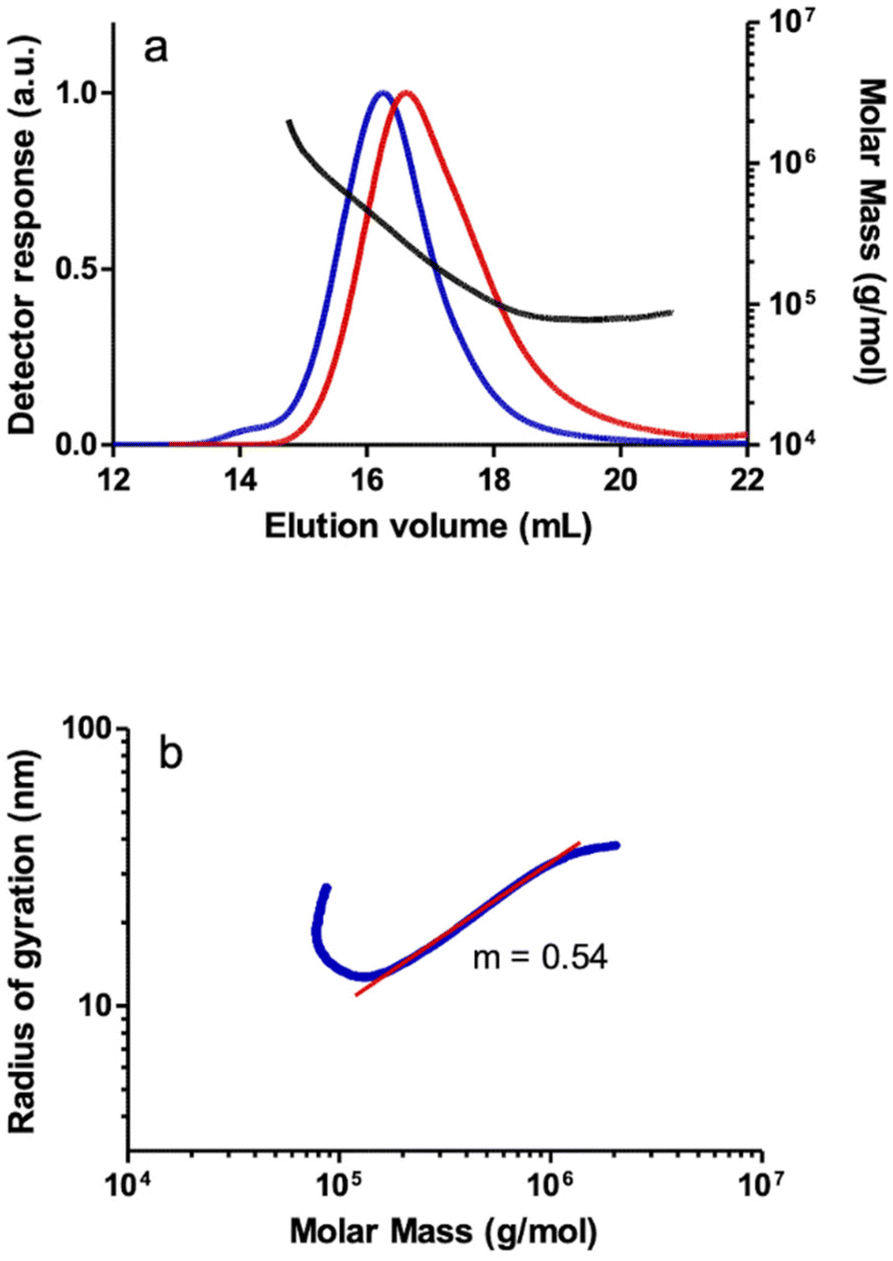 | ||
| Fig. 5 (a) SEC chromatograms: light scattering detector response (90 degrees) (blue) and differential refractometer response (red), and MM (molar mass) vs. elution volume plot for a PPA sample prepared with the catalyst [Rh(CH3)(cod){Ph2P(CH2)3NMe2}] (5) in THF analyzed SEC-MALS and (b) log–log plot of the radius of gyration (rg) vs. MM of the same sample analyzed by SEC-MALS (blue) and linear fit (red). | ||
It is well known that many rhodium complexes in combination with an external base, such as triethylamine or 4-(dimethylamino)pyridine (DMAP), efficiently catalyze the polymerization of PA derivatives.41–43 Considering these results, we decided to study the influence of the external base DMAP on the polymerization of PA with initiators [Rh(CC-Ph)(nbd){Ph2P(CH2)3L}2] (1 and 2) and [Rh(CC-Ph)(cod){Ph2P(CH2)3L}] (3 and 4) (L = NMe2, OEt). The performance of these catalysts in the presence of DMAP as a co-catalyst, using a [DMAP]/Rh ratio of 1, is shown in Table 2.
The activity of alkynyl initiators 1 and 2 was not affected by the use of DMAP as a co-catalyst. However, a significant decrease in the Mw of the PPAs, with a corresponding increase in the initiation efficiencies, and a decrease in the dispersity were observed. The narrow molecular weight distributions of the PPAs prepared with catalysts 1 and 1/DMAP, having Đ values of 1.17 and 1.13, are consistent with the quasi-living nature of the PA polymerization.27 The morphology of the PPAs prepared with catalysts 1/DMAP and 2/DMAP is very similar to that of the PPAs prepared in the absence of DMAP. In particular, the DMAP co-catalyst did not affect either the bimodality of the molecular weight distribution or the branching. The same trend was observed with the catalyst 3/DMAP although the decrease in Mw was less pronounced and the dispersity was maintained. In contrast, 4/DMAP showed an increase in catalytic activity and a slight increase in Mw with an improvement in dispersity. Interestingly, although the bimodality of the PPA prepared with 3/DMAP was not seen, the morphology of the PPAs prepared with catalysts 3/DMAP and 4/DMAP was maintained and is consistent with the presence of a linear polymer (see the ESI†).
The polymerization reactions with catalysts 1 and 2 were faster than those with the related catalyst [Rh(CC-Ph)(nbd)(PPh3)2] based on non-functionalized phosphine ligands reported by Noyori et al. and gave polymers with much higher MM.24,26 Also, the catalytic systems 1/DMAP and 2/DMAP polymerized PA at a faster rate than [Rh(CC-Ph)(nbd)(PPh3)2]/DMAP (1:10) affording PPAs with higher molar masses. Moreover, the PA polymerization using the catalyst [Rh(CC-Ph)(cod)(PPh3)], prepared in situ, in the presence of DMAP is much slower than that using 3/DMAP and 4/DMAP affording a PPA with lower MM (Mn of 82.500) and much higher dispersity (Đ of 3.77).25
PA polymerization mechanism when using alkynyl and methyl rhodium(I) complexes
The most likely polymerization mechanism when using initiators [Rh(CC-Ph)(nbd){Ph2P(CH2)3Z}2] (1 and 2) which explains the marginal influence of the donor set of the functionalized phosphine ligand on the catalytic performance is shown in Fig. 6. Both initiators are pentacoordinated Rh(I) species with 18 valence electrons, so dissociation of a phosphine ligand is required for the polymerization reaction to occur. Then, η2-coordination of PA to the resulting square planar species, [Rh(CC-Ph)(nbd){Ph2P(CH2)3Z}] (Z = NMe2, OEt), followed by 2,1-insertion of PA into the Rh–alkynyl bond should afford a Rh–alkenyl species responsible for the propagation step through successive PA coordination-insertion events with 2,1-regioselectivity. In this respect, the involvement of the donor function of the functionalized phosphine ligand (–NMe2 or –OEt) in the coordination of phenylacetylene could be envisaged.
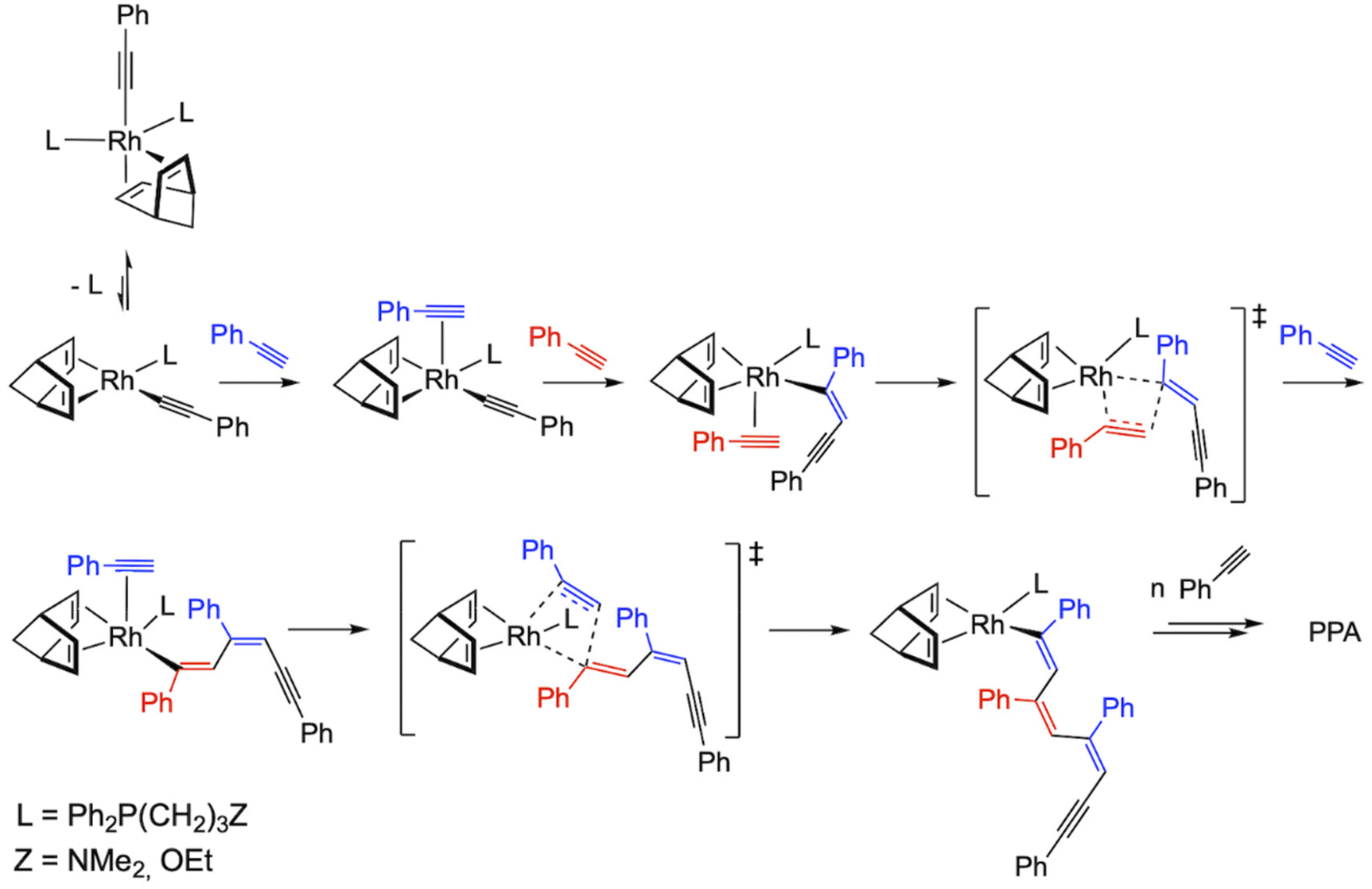 | ||
| Fig. 6 Plausible mechanism for the polymerization of PA using the catalysts [Rh(CC-Ph)(nbd){Ph2P(CH2)3Z}2] (1 and 2). | ||
It is worth noting that alkynyl species have been suggested as the initiating species likely to be involved in the generation of stable rhodium–vinyl propagating species by PA insertion into the Rh–alkynyl bond.42,44,45
Theoretical studies reported by Morokuma et al.46 have demonstrated the key role of alkynyl species as PA polymerization initiators showing that the energy barrier for the PA insertion into the Rh–alkynyl bond of [Rh(nbd)(CC-Ph)(PA)] (initiation step) is almost 4 kcal mol−1 higher than the barrier for the insertion into the Rh–alkenyl bond (propagation step), which could explain the low initiation efficiencies observed. However, the low concentration of the square-planar species [Rh(CC-Ph)(nbd){Ph2P(CH2)3Z}] in equilibrium with the pentacoordinated catalyst precursor could also contribute to the low initiation efficiencies. On the other hand, this mechanistic proposal should also be operative for the square-planar catalysts [Rh(CC-Ph)(cod){Ph2P(CH2)3Z}] (3 and 4). The higher initiation efficiencies calculated for both catalysts (7.5 and 8.0, respectively) are consistent with the absence of a pre-equilibrium step.
The morphology of the polymers is determined by both the diene ligand and the type of functionalized phosphine ligand. The PPAs obtained with the square-planar complexes [Rh(CC-Ph)(cod){Ph2P(CH2)3Z}] (3 and 4) containing the cod ligand are linear, whereas the pentacoordinated complexes [Rh(CC-Ph)(nbd){Ph2P(CH2)3Z}2] (1 and 2) bearing the nbd ligand gave PPA samples showing the presence of a fraction of high MM branched polymer. In contrast, the PPA prepared with the methyl initiator [Rh(CH3)(cod){Ph2P(CH2)3OEt}] (6) is linear whereas that prepared with [Rh(CH3)(cod){Ph2P(CH2)3NMe2}] (5) is branched.
The factors influencing the formation of branched materials as well as the mechanism of branch formation in these materials are still the subject of debate. In principle, two possible branching mechanisms could be considered: (i) terminal branching by copolymerization with macromonomers having a reactive end group and (ii) formal chain transfer to the polymer.30 It may be possible that chain transfer to PA via protonolysis of the chain end will result in a polymer with a vinylene or vinylidene reactive end group, depending on the regiochemistry of the last PA insertion, with regeneration of the alkynyl initiator. In the latter case, the macromonomer might be more susceptible to co-polymerization leading to terminal branching (see Fig. S47 in the ESI†). On the other hand, formal chain transfer to the polymer likely involves Rh–H species which could be formed by formal PA oxidative addition followed by reductive elimination of the enyne-terminated polymer. This Rh–H species can react with a CC bond inside any macromolecule to form a π-allyl complex which could propagate further, via the σ-allyl species, leading to the formation of branches (see Fig. S48 in the ESI†). On the other hand, in contrast to an Rh–H species, a concerted transfer of a rhodium complex from the end of one growing chain to an internal CC bond of another polymer chain is unlikely to occur for steric reasons.
The branching frequency of the PPA sample prepared with catalyst 1 was calculated from the A4F data using the Zimm–Stockmayer equation, assuming trifunctional branching and monodisperse slices.47,48 The average branching frequencies per molecule and per 1000 repeat units were 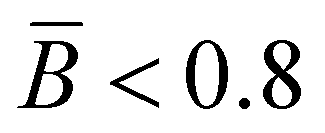 and
and 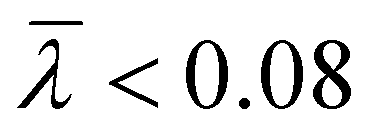 , respectively. The overlay of the cumulative molar mass distribution and branch units per macromolecule plots is shown in Fig. 7. As can be seen in the figure, approximately 50% of the polymer mass is linear and an average of less than 0.8 branches/molecule is determined for the remaining polymer, probably due to the co-elution of a mixture of linear and branched polymers. Similar calculations for the PPA sample prepared with catalyst 2 gave values of
, respectively. The overlay of the cumulative molar mass distribution and branch units per macromolecule plots is shown in Fig. 7. As can be seen in the figure, approximately 50% of the polymer mass is linear and an average of less than 0.8 branches/molecule is determined for the remaining polymer, probably due to the co-elution of a mixture of linear and branched polymers. Similar calculations for the PPA sample prepared with catalyst 2 gave values of 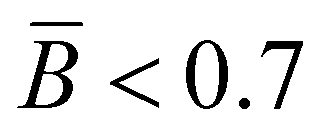 and
and 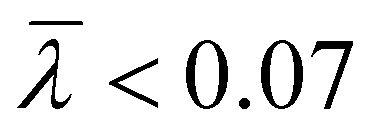 (see the ESI†). This level of branching is consistent with terminal branching via copolymerization of unsaturated macromonomers, although chain transfer to the polymer cannot be ruled out.
(see the ESI†). This level of branching is consistent with terminal branching via copolymerization of unsaturated macromonomers, although chain transfer to the polymer cannot be ruled out.
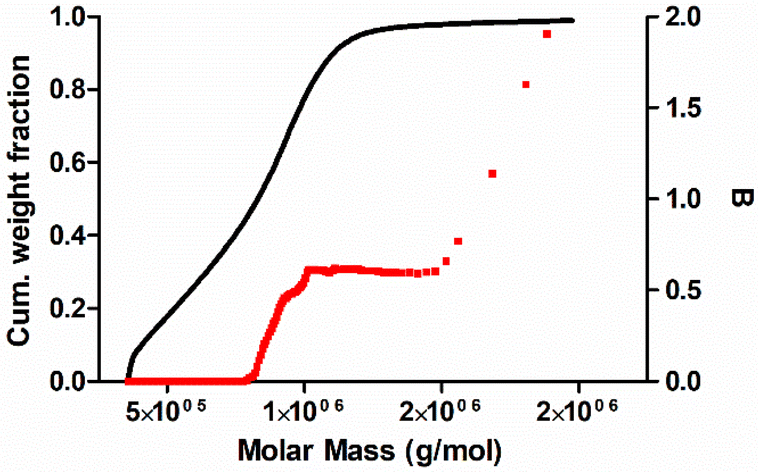 | ||
| Fig. 7 Cumulative molar mass distribution (black line) and branch units per macromolecule (B) (red dots) vs. MM determined by A4F-MALS for a PPA sample prepared with the catalyst [Rh(CC-Ph)(nbd){Ph2P(CH2)3NMe2}2] (1). | ||
The mechanism of polymerization using methyl rhodium(I) complexes is partly related to that described for alkynyl initiators. Complexes [Rh(CH3)(cod){Ph2P(CH2)3Z}] (5 and 6) are square-planar Rh(I) species with 16 valence electrons which can act directly as polymerization initiators. Thus, the η2-coordination of PA and subsequent insertion into the Rh–Me bond should generate a Rh–alkenyl species that could be responsible for the propagation process. However, the insertion of an alkyne into an M–Me bond is rare and has only been observed in palladium chemistry with activated alkynes.49 Thus, the methyl rhodium(I) compounds are likely to react with PA to give the corresponding alkynyl complexes with the release of methane.26 The transformation of the methyl complexes into alkynyl derivatives can take place via two different pathways (Fig. 8). First, PA must coordinate to the square-planar complexes [Rh(CH3)(cod){Ph2P(CH2)3Z}] to form the penta-coordinated species [Rh(PA)(CH3)(cod){Ph2P(CH2)3Z}]. The increased acidity of the alkyne resulting from its coordination to the metal center can then facilitate the transfer of the CH proton to the methyl ligand, which is eliminated as methane, resulting in the coordination of the alkynyl fragment (path A). However, oxidative addition of PA and subsequent reductive elimination with the release of methane can also occur after coordination of PA (path B).50 It is worth noting that hydrido-alkynyl Rh(III) complexes resulting from the oxidative addition of PA were isolated as intermediates in the η2-alkyne to vinylidene isomerization.51
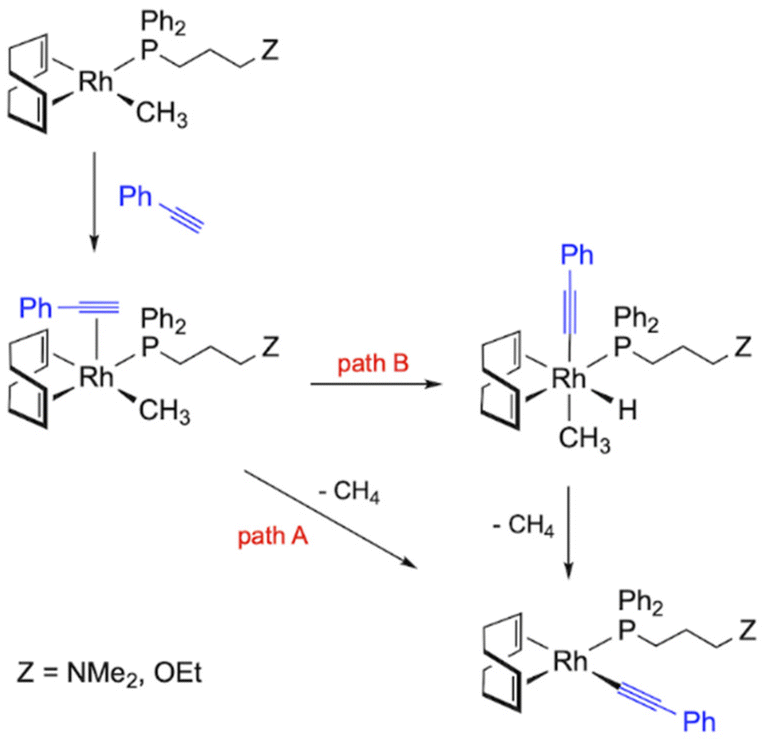 | ||
| Fig. 8 Reaction pathways leading to the formation of alkynyl initiators using the catalysts [Rh(CH3)(cod){Ph2P(CH2)3Z}] (5 and 6). | ||
Finally, for comparative purposes, the performance of related cationic complexes [Rh(cod){κ2P,N-Ph2P(CH2)3NMe2}]BF4 (7) and [Rh(cod){κ2P,O-Ph2P(CH2)3OEt}]BF4 (8), with the functionalized phosphine ligand coordinated in a bidentate fashion, in PA polymerization was studied (Table 3).
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Cat. | t (min) | Conv.b (%) | Anal. | M w (g mol−1) | Đ | IEd (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
a Reaction conditions: [PA]o = 0.25 M, [PA]o/[Rh] = 100, [DMAP]/[Rh] = 1 in THF at 293 K.
b Determined by GC (octane as an internal standard).
c
Đ = dispersity (Mw/Mn, where Mw = weight-average molecular weight and Mn = number-average molecular weight).
d Initiation efficiency, IE = Mtheor/Mn × 100, where Mtheor = [PA]o/[Rh] × MWPA × polymer yield.
e Ref. 27.
f Bimodal distribution of MM. Data for the low MM polymer.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 7 | 120 | 100 | SEC | 2.38 × 105f |
1.79 | 7.7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 8 | 150 | 90 | SEC | 1.37 × 105 | 1.90 | 11.9 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
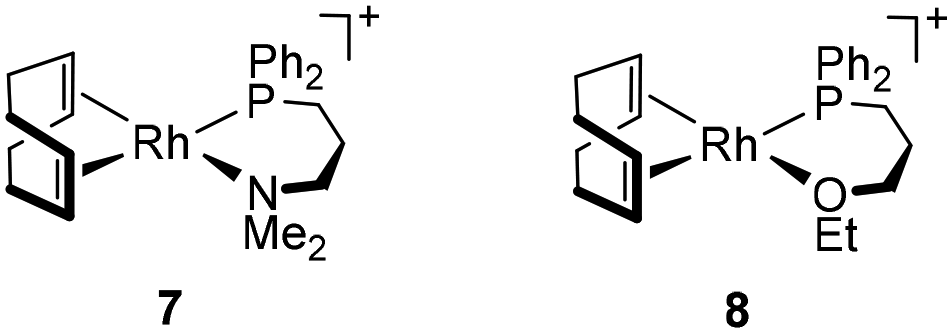
The alkynyl initiators [Rh(CC-Ph)(cod){Ph2P(CH2)3Z}] (Z = NMe2, 3; OEt, 4) were more active than the related cationic initiators 7 and 8 affording PPAs with slightly higher Mw and Đ, although no significant differences in the initiation efficiencies were observed. In terms of the morphology of the PPAs, catalyst 8 produced a linear PPA similar to catalyst 4. In contrast to catalyst 3, which gave a linear PPA, catalyst 7 produced a branched PPA. The higher activity of the alkynyl initiators 3 and 4 compared to the related cationic compounds 7 and 8 suggests the involvement of different propagating species, probably [Rh(PPA)(cod){κP-Ph2P(CH2)3Z}] and [Rh(PPA)(cod){κP-Ph2P(CH2)3ZH}]+ (PPA = polymer chain), respectively, with the neutral species being more active. This result is in line with that observed for related rhodium(I) complexes with N-functionalized N-heterocyclic carbene ligands.28
Conclusions
The alkynyl [Rh(CC-Ph)(nbd){Ph2P(CH2)3Z}2] and [Rh(CC-Ph)(cod){Ph2P(CH2)3Z}] and methyl complexes [Rh(CH3)(cod){Ph2P(CH2)3Z}] featuring functionalized phosphine ligands (Z = NMe2, OEt) efficiently catalyze the polymerization of phenylacetylene in the absence of a base to afford stereoregular PPAs with high molar masses (Mw ≈ 5 × 105). The PPAs prepared with these catalysts show a bimodal molar mass distribution due to the presence of a high molar mass polymer fraction. The alkynyl complexes [Rh(CC-Ph)(nbd){Ph2P(CH2)3Z}2], with nbd as the diene ligand, are considerably more active than the square planar alkynyl and methyl complexes having cod as the diene. The diene ligand in the alkynyl complexes also influences the morphology of the PPAs. The PPAs prepared with the square-planar initiators [Rh(CC-Ph)(cod){Ph2P(CH2)3Z}] are essentially linear, whereas the pentacoordinated initiators with the nbd ligand, [Rh(CC-Ph)(nbd){Ph2P(CH2)3Z}2], afford PPAs showing the presence of a fraction of high MM branched polymer. However, the donor function of the functionalized phosphine ligand (–NMe2 or –OEt) has little effect on either the catalytic activity or the PPA properties, except in the case of methyl initiators [Rh(CH3)(cod){Ph2P(CH2)3Z}], since the PPA obtained with the catalyst having the –OEt function is linear, whereas the –NEt2 function promotes branching.
In general, the alkynyl initiators [Rh(CC-Ph)(nbd){Ph2P(CH2)3Z}2] polymerize PA at a faster rate than the catalyst [Rh(CC-Ph)(nbd)(PPh3)2] bearing a non-functionalized phosphine ligand and afford polymers with much higher MM. However, none of the catalysts performs a stereospecific living polymerization, suggesting that, although the functionalized phosphine does not play a relevant role in the initiation step, it significantly increases the chain growth rate, which is consistent with the observed low initiation efficiency. PA polymerization in the presence of an external base as a co-catalyst, such as 4-(dimethylamino)pyridine, only has a positive effect on the performance of catalysts [Rh(CC-Ph)(nbd){Ph2P(CH2)3Z}2], resulting in a decrease in Mw and dispersity while maintaining the activity. However, neither bimodality nor branching is suppressed. On the other hand, the alkynyl catalysts [Rh(CC-Ph)(cod){κP-Ph2P(CH2)3Z}] (Z = NMe2, OEt) are much more active than the related cationic catalysts with the functionalized phosphine ligand coordinated in a bidentate fashion, [Rh(cod){κ2P,Z-Ph2P(CH2)3Z}]+ (Z = NMe2, OEt) suggesting the involvement of different propagating species.
Experimental section
Synthesis
All experiments were carried out under an atmosphere of argon using Schlenk techniques or a glovebox. Solvents were distilled immediately prior to use with appropriate drying agents or obtained from a solvent purification system (Innovative Technologies). CD2Cl2, C6D6 and THF-d8 (Euriso-top) were dried using activated molecular sieves. The starting materials [Rh(μ-Cl)(cod)]252 and [Rh(μ-OMe)(diene)]2 (diene = cod,53 nbd54) were prepared following the reported methods. The functionalized phosphines Ph2P(CH2)3NMe2 and Ph2P(CH2)3OEt were prepared according to the procedure described in the literature.55 The compound [Rh(cod){κ2P,O-Ph2P(CH2)3OEt}][BF4] (8) was prepared as described previously.33 MeLi (1.6 M in diethyl ether) was purchased from Aldrich and used as received. Phenylacetylene (Aldrich) was purified by vacuum distillation from CaH2 and stored over molecular sieves.Scientific equipment
C, H and N analyses were carried out using a PerkinElmer 2400 Series II CHNS/O analyzer. 1H, 13C{1H} and 31P{1H} NMR spectra were recorded on a Bruker Avance 300 (300.1276 MHz) or Bruker Avance 400 (400.1625 MHz) spectrometer. NMR chemical shifts are reported in ppm relative to tetramethylsilane and referenced to partially deuterated solvent resonances. Coupling constants (J) are given in hertz. Spectral assignments were achieved by a combination of 1H–1H COSY, 13C{1H}-APT and 1H–13C HSQC experiments. Mass spectra were recorded on a Bruker MicroTof-Q spectrometer using electrospray ionization-mass spectroscopy (ESI-MS) and sodium formate as a reference. Infrared spectra were recorded on a PerkinElmer Spectrum 100 FT-IR spectrometer equipped with a Universal Attenuated Total Reflectance (UATR) accessory, which allows the observation of the electromagnetic spectrum over the 4000–250 cm−1 region.SEC-MALS analyses of PPAs
The absolute molecular weight averages (Mn and Mw), dispersity (Đ, Mw/Mn) and molecular weight distribution were determined by SEC-MALS at the Chromatography and Spectroscopy Service of the ISQCH. SEC-MALS analyses were carried out using a Waters 2695 instrument, equipped with three PL-Gel Mixed B LS columns fitted to a MALS detector (MiniDawn Treos, Wyatt) and a differential refractive index (DRI) detector (Optilab Rex, Wyatt). The polymer solutions in THF (≈2.0 mg mL−1) were filtered through a 0.45 μm PTFE membrane filter before being injected into the GPC systems. The analyses were carried out immediately after the dissolution of the polymer sample in THF to minimize sample degradation.56,57 Data analysis was performed using ASTRA software from Wyatt. The samples were eluted at 25 °C with THF at a flow rate of 1.0 mL min−1. The reported dn/dc value of 0.2864 mL g−1 determined at 633 nm for atactic PPA56 was used which resulted in calculated mass recoveries that were in reasonable agreement with the theoretical values.40A4F-MALS analyses of PPAs
Samples were characterized by asymmetric flow field flow fractionation (A4F) coupled with a multi-angle light scattering (MALS) photometer at SYNPO Pardubice. The instrumental setup consisted of a Wyatt Technology Corporation A4F system Eclipse 3+, a MALS photometer EOS (Wyatt Technology Corporation) and a refractive index (RI) detector 410 (Waters). The measurements were carried out in THF using a 5 kDa regenerated cellulose membrane, a long channel with a wide 350 μm spacer. The samples were injected as 0.1 or 0.2% w/v solutions in THF in a volume of 100 μL. The analyses were performed shortly after the dissolution to minimize sample degradation. The detector flow rate was 1 mL min−1; the separation was achieved using the following cross flow gradient: 3 mL min−1 for 5 min, then linear decay to 0.1 mL min−1 over 20 min, plus 45 min at 0.1 mL min−1. The measurements were carried out at room temperature.Synthesis of [Rh(CC-Ph)(diene){Ph2P(CH2)3Z}n] (Z = NMe2, OEt) (1–4)
CH (0.44 mmol) were sequentially added to a solution of the corresponding complex [Rh(μ-OMe)(diene)]2 (0.2 mmol) in THF (10 mL) at 253 K to give red solutions which were stirred at 273 K for 1 h. The solutions were concentrated under vacuum and filtered through a cannula. The clear solutions were brought to dryness under vacuum and the residue was washed with diethyl ether (3 mL). The red oily residues were disaggregated by stirring with cold hexane to give the compounds as red solids which were isolated by decantation, washed with cold hexane (3 × 2 mL) and dried under vacuum.
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) C-Ph)(nbd){Ph2P(CH2)3NMe2}2] (1).
[Rh(μ-OMe)(nbd)]2 (100 mg, 0.221 mmol), Ph2P(CH2)3NMe2 (240 mg, 0.884 mmol), and Ph-CCH (54 μL, 0.486 mmol). Yield: 64%. Analysis calcd for C49H57N2P2Rh: C, 70.16; H, 6.85; N, 3.34. Found: C, 69.90; H, 6.83; N, 3.34. MS (ESI+, CH3CN, m/z, %): 466.1 ([Rh(nbd){Ph2P(CH2)3NMe2}]+, 100), 739.1 ([M − CC-Ph + 2H]+, 90). IR (ATR, cm−1): 2088 (w, CC). 1H NMR (THF-d8, 253 K): δ 7.55 (m, 4H, Ph), 7.35–7.26 (m, 18H; 16H Ph, 2H C-Ph), 7.12 (t, JH–H = 7.6, 2H, C-Ph), 6.97 (t, JH–H = 7.3, 1H, C-Ph), 3.07 (br, 4H, CH nbd), 3.00 (br, 2H, CH nbd), 2.79 (m, 2H, CH2N), 2.22 (m, 2H, CH2N), 2.09 (m, 4H, CH2P), 2.01 (s, 12H, NMe2), 1.49 (m, 4H, CH2), 0.90 (m, 2H, CH2 nbd). 31P{1H} NMR (THF-d8, 253 K): δ 13.87 (d, JRh–P = 120.9). 13C{1H} NMR (THF-d8, 253 K): δ 139.6 (Ci, C-Ph), 139.4 (m, Ci), 133.7 (m, Co), 131.6 (Co, C-Ph), 129.3 (d, JC–P = 6.1, Cp), 128.8 (m, Cm), 128.5 (Cp, C-Ph), 124.5 (Cm, C-Ph), 113.7 (dt, JC–Rh = 47.82, JC–P = 24.58, CC-Ph), 107.0 (d, JC–Rh = 10.1, CC-Ph), 61.6 (app t, JC–Rh ≈ JC–P = 5.8, CH2N), 60.7 (CH2 nbd), 47.3 (CH nbd), 45.8 (NMe2), 28.7 (app t, JC–Rh ≈ JC–P = 10.4, CH2P), 23.7 (CH2) (CH nbd not observed).
C-Ph)(nbd){Ph2P(CH2)3OEt}2] (2).
[Rh(μ-OMe)(nbd)]2 (100 mg, 0.221 mmol), Ph2P(CH2)3OEt (241 mg, 0.885 mmol), and Ph-CCH (54 μL, 0.486 mmol) in THF. Yield: 58%. Analysis calcd for C49H55O2P2Rh: C, 70.00; H, 6.59. Found: C, 69.64; H, 6.57. MS (ESI+, CH3CN, m/z, %): 741.1 ([M − CC-Ph + 2H]+, 100). IR (ATR, cm−1): 2089 (w, CC). 1H NMR (THF-d8, 253 K): 7.53 (m, 4H, Ph), 7.51–7.44 (m, 16H, Ph), 7.19 (m, 2H, C-Ph), 7.13 (t, JH–H = 7.5, 2H, C-Ph), 6.99 (t, JH–H = 7.2, 1H, C-Ph), 3.30 (q, JH–H = 6.8, 4H, CH2 OEt), 3.24 (m, 4H, CH2O), 3.07 (br, 4H, CH nbd), 3.03 (br, 2H, CH nbd), 2.75 (t, JH–H = 13.6, 2H, CH2P), 2.20 (t, JH–H = 13.6, 2H, CH2P), 1.59 (m, 4H, CH2), 1.09 (t, JH–H = 6.8, 6H, CH3 OEt), 0.87 (s, 2H, CH2 nbd). 31P{1H} NMR (253K, THF-d8): δ 16.11 (d, JRh–P = 121.3). 13C{1H} NMR (THF-d8, 253 K): δ 138.6 (dd, JC–P = 21.1, JC–Rh = 10.0, Ci), 133.0 (m, JC–P = 9.8, Co), 131.5 (Co, C-Ph), 130.9 (Ci, C-Ph), 129.4 (d, JC–P = 8.8, Cp), 128.9 (m, Cm), 128.6 (Cp, C-Ph), 124.6 (Cm, C-Ph), 113.7 (dt, JC–Rh = 46.8, JC–P = 24.6, CC-Ph), 106.8 (d, JC–Rh = 9.7, CC-Ph), 72.3 (t, JC–P = 6.4, CH2O), 66.8 (CH2 OEt), 60.7 (CH2 nbd), 47.2 (CH nbd), 27.5 (app t, JC–Rh ≈ JC–P = 11.3, CH2P), 25.9 (CH2), 16.0 (CH3 OEt) (CH nbd not observed).
C-Ph)(cod){Ph2P(CH2)3NMe2}] (3).
[Rh(μ-OMe)(cod)]2 (100 mg, 0.207 mmol), Ph2P(CH2)3NMe2 (112.1 mg, 0.413 mmol), and Ph-CCH (49.9 μL, 0.454 mmol). Yield: 42%. Analysis calcd for C33H39NPRh: C, 67.92; H, 6.74; N, 2.40. Found: C, 67.77; H, 6.65; N, 2.45. MS (ESI+, THF, m/z, %): 482.1 ([M − CC-Ph]+, 100). IR (ATR, cm−1): 2077 (w, CC). 1H NMR (CD2Cl2, 233 K): 7.73–6.95 (m, 15H; 10H Ph, 5H C-Ph), 5.48 (br, 2H, CH cod), 3.57 (br, 2H, CH cod), 2.48–1.97 (m, 12H; 8H CH2 cod, 2H CH2N, 2H CH2P), 2.05 (s, 6H, NMe2), 1.95 (m, 2H, CH2). 31P{1H} NMR (298 K, C6D6): δ 26.09 (d, JRh–P = 149.6). 13C{1H} NMR (CD2Cl2, 233 K): δ 134.6 (d, JC–P = 38.0, Ci), 133.5 (d, JC–P = 10.8, Co), 132.7 (Ci, C-Ph), 130.6 (Co, C-Ph), 130.6 (Cp), 128.2 (d, J = 9.2, Cm), 127.9 (Cp, C-Ph), 124.9 (Cm, C-Ph), 122.1 (dd, JC–Rh = 49.2, JC–P = 22.3, CC-Ph), 116.9 (dd, JC–Rh = 12.8, JC–P = 2.6, CC-Ph), 97.2 (dd, JC–Rh = 11.4, JC–P = 7.5, CH2 cod), 84.9 (d, JC–Rh = 8.3, CH2 cod), 60.7 (d, JC–P = 15.5, CH2N), 45.4 (NMe2), 31.3, 30.6 (CH2 cod), 27.3 (d, JC–P = 27.3, CH2P), 24.5 (CH2).
C-Ph)(cod){Ph2P(CH2)3OEt}] (4).
[Rh(μ-OMe)(cod)]2 (100 mg, 0.207 mmol), Ph2P(CH2)3OEt (113 mg, 0.415 mmol), and Ph-CCH (49.9 μL, 0.454 mmol). Yield: 46%. Satisfactory elemental analysis could not be obtained. MS (ESI+, THF, m/z, %): 583.1 ([M]+, 8), 483.1 ([M − CCC6H5]+, 100). IR (ATR, cm−1): 2076 (w, CC). 1H NMR (THF-d8, 233 K): δ 7.69–7.41 (m, 15H; 10H Ph, 5H C-Ph), 5.40 (br, 2H, CH cod), 3.41 (m, 4H, CH2O, CH2 Et), 2.90 (br, 2H, CH cod), 2.59 (m, 2H, CH2), 2.30–1.86 (m, 10H; 8H CH2 cod, 2H CH2P), 1.16 (t, 3H, JH–H = 6.8, CH3 Et). 31P{1H} NMR (THF-d8, 233 K): δ 25.59 (d, JRh–P = 149.0). 13C{1H} NMR (THF-d8, 233 K): δ 134.9 (d, JC–P = 10.3, Co), 133.6 (d, J = 37.9, Ci), 132.2 (Co, C-Ph), 131.0 (Cp), 130.1 (Ci, C-Ph), 129.9 (Cp, C-Ph), 129.1 (d, JC–P = 9.1, Cm), 1226.3 (Cm, C-Ph), 119.3 (dd, JC–Rh = 48.9, JC–P = 22.8, CC-Ph), 115.2 (d, JC–Rh = 12.1, CC-Ph), 104.9 (br, CH cod), 72.1 (d, JC–P = 16.1, CH2O), 70.4 (d, JC–Rh = 8.4, CH cod), 64.2 (CH2 OEt), 339 (CH2 cod), 29.6 (CH2 cod), 24.0 (d, JC–P = 28.7, CH2P), 22.2 (d, JC–P = 3.6, CH2), 16.0 (CH3 OEt).
C-Ph)(nbd){Ph2P(CH2)3NMe2}2] (1).
[Rh(μ-OMe)(nbd)]2 (100 mg, 0.221 mmol), Ph2P(CH2)3NMe2 (240 mg, 0.884 mmol), and Ph-CCH (54 μL, 0.486 mmol). Yield: 64%. Analysis calcd for C49H57N2P2Rh: C, 70.16; H, 6.85; N, 3.34. Found: C, 69.90; H, 6.83; N, 3.34. MS (ESI+, CH3CN, m/z, %): 466.1 ([Rh(nbd){Ph2P(CH2)3NMe2}]+, 100), 739.1 ([M − CC-Ph + 2H]+, 90). IR (ATR, cm−1): 2088 (w, CC). 1H NMR (THF-d8, 253 K): δ 7.55 (m, 4H, Ph), 7.35–7.26 (m, 18H; 16H Ph, 2H C-Ph), 7.12 (t, JH–H = 7.6, 2H, C-Ph), 6.97 (t, JH–H = 7.3, 1H, C-Ph), 3.07 (br, 4H, CH nbd), 3.00 (br, 2H, CH nbd), 2.79 (m, 2H, CH2N), 2.22 (m, 2H, CH2N), 2.09 (m, 4H, CH2P), 2.01 (s, 12H, NMe2), 1.49 (m, 4H, CH2), 0.90 (m, 2H, CH2 nbd). 31P{1H} NMR (THF-d8, 253 K): δ 13.87 (d, JRh–P = 120.9). 13C{1H} NMR (THF-d8, 253 K): δ 139.6 (Ci, C-Ph), 139.4 (m, Ci), 133.7 (m, Co), 131.6 (Co, C-Ph), 129.3 (d, JC–P = 6.1, Cp), 128.8 (m, Cm), 128.5 (Cp, C-Ph), 124.5 (Cm, C-Ph), 113.7 (dt, JC–Rh = 47.82, JC–P = 24.58, CC-Ph), 107.0 (d, JC–Rh = 10.1, CC-Ph), 61.6 (app t, JC–Rh ≈ JC–P = 5.8, CH2N), 60.7 (CH2 nbd), 47.3 (CH nbd), 45.8 (NMe2), 28.7 (app t, JC–Rh ≈ JC–P = 10.4, CH2P), 23.7 (CH2) (CH nbd not observed).
C-Ph)(nbd){Ph2P(CH2)3OEt}2] (2).
[Rh(μ-OMe)(nbd)]2 (100 mg, 0.221 mmol), Ph2P(CH2)3OEt (241 mg, 0.885 mmol), and Ph-CCH (54 μL, 0.486 mmol) in THF. Yield: 58%. Analysis calcd for C49H55O2P2Rh: C, 70.00; H, 6.59. Found: C, 69.64; H, 6.57. MS (ESI+, CH3CN, m/z, %): 741.1 ([M − CC-Ph + 2H]+, 100). IR (ATR, cm−1): 2089 (w, CC). 1H NMR (THF-d8, 253 K): 7.53 (m, 4H, Ph), 7.51–7.44 (m, 16H, Ph), 7.19 (m, 2H, C-Ph), 7.13 (t, JH–H = 7.5, 2H, C-Ph), 6.99 (t, JH–H = 7.2, 1H, C-Ph), 3.30 (q, JH–H = 6.8, 4H, CH2 OEt), 3.24 (m, 4H, CH2O), 3.07 (br, 4H, CH nbd), 3.03 (br, 2H, CH nbd), 2.75 (t, JH–H = 13.6, 2H, CH2P), 2.20 (t, JH–H = 13.6, 2H, CH2P), 1.59 (m, 4H, CH2), 1.09 (t, JH–H = 6.8, 6H, CH3 OEt), 0.87 (s, 2H, CH2 nbd). 31P{1H} NMR (253K, THF-d8): δ 16.11 (d, JRh–P = 121.3). 13C{1H} NMR (THF-d8, 253 K): δ 138.6 (dd, JC–P = 21.1, JC–Rh = 10.0, Ci), 133.0 (m, JC–P = 9.8, Co), 131.5 (Co, C-Ph), 130.9 (Ci, C-Ph), 129.4 (d, JC–P = 8.8, Cp), 128.9 (m, Cm), 128.6 (Cp, C-Ph), 124.6 (Cm, C-Ph), 113.7 (dt, JC–Rh = 46.8, JC–P = 24.6, CC-Ph), 106.8 (d, JC–Rh = 9.7, CC-Ph), 72.3 (t, JC–P = 6.4, CH2O), 66.8 (CH2 OEt), 60.7 (CH2 nbd), 47.2 (CH nbd), 27.5 (app t, JC–Rh ≈ JC–P = 11.3, CH2P), 25.9 (CH2), 16.0 (CH3 OEt) (CH nbd not observed).
C-Ph)(cod){Ph2P(CH2)3NMe2}] (3).
[Rh(μ-OMe)(cod)]2 (100 mg, 0.207 mmol), Ph2P(CH2)3NMe2 (112.1 mg, 0.413 mmol), and Ph-CCH (49.9 μL, 0.454 mmol). Yield: 42%. Analysis calcd for C33H39NPRh: C, 67.92; H, 6.74; N, 2.40. Found: C, 67.77; H, 6.65; N, 2.45. MS (ESI+, THF, m/z, %): 482.1 ([M − CC-Ph]+, 100). IR (ATR, cm−1): 2077 (w, CC). 1H NMR (CD2Cl2, 233 K): 7.73–6.95 (m, 15H; 10H Ph, 5H C-Ph), 5.48 (br, 2H, CH cod), 3.57 (br, 2H, CH cod), 2.48–1.97 (m, 12H; 8H CH2 cod, 2H CH2N, 2H CH2P), 2.05 (s, 6H, NMe2), 1.95 (m, 2H, CH2). 31P{1H} NMR (298 K, C6D6): δ 26.09 (d, JRh–P = 149.6). 13C{1H} NMR (CD2Cl2, 233 K): δ 134.6 (d, JC–P = 38.0, Ci), 133.5 (d, JC–P = 10.8, Co), 132.7 (Ci, C-Ph), 130.6 (Co, C-Ph), 130.6 (Cp), 128.2 (d, J = 9.2, Cm), 127.9 (Cp, C-Ph), 124.9 (Cm, C-Ph), 122.1 (dd, JC–Rh = 49.2, JC–P = 22.3, CC-Ph), 116.9 (dd, JC–Rh = 12.8, JC–P = 2.6, CC-Ph), 97.2 (dd, JC–Rh = 11.4, JC–P = 7.5, CH2 cod), 84.9 (d, JC–Rh = 8.3, CH2 cod), 60.7 (d, JC–P = 15.5, CH2N), 45.4 (NMe2), 31.3, 30.6 (CH2 cod), 27.3 (d, JC–P = 27.3, CH2P), 24.5 (CH2).
C-Ph)(cod){Ph2P(CH2)3OEt}] (4).
[Rh(μ-OMe)(cod)]2 (100 mg, 0.207 mmol), Ph2P(CH2)3OEt (113 mg, 0.415 mmol), and Ph-CCH (49.9 μL, 0.454 mmol). Yield: 46%. Satisfactory elemental analysis could not be obtained. MS (ESI+, THF, m/z, %): 583.1 ([M]+, 8), 483.1 ([M − CCC6H5]+, 100). IR (ATR, cm−1): 2076 (w, CC). 1H NMR (THF-d8, 233 K): δ 7.69–7.41 (m, 15H; 10H Ph, 5H C-Ph), 5.40 (br, 2H, CH cod), 3.41 (m, 4H, CH2O, CH2 Et), 2.90 (br, 2H, CH cod), 2.59 (m, 2H, CH2), 2.30–1.86 (m, 10H; 8H CH2 cod, 2H CH2P), 1.16 (t, 3H, JH–H = 6.8, CH3 Et). 31P{1H} NMR (THF-d8, 233 K): δ 25.59 (d, JRh–P = 149.0). 13C{1H} NMR (THF-d8, 233 K): δ 134.9 (d, JC–P = 10.3, Co), 133.6 (d, J = 37.9, Ci), 132.2 (Co, C-Ph), 131.0 (Cp), 130.1 (Ci, C-Ph), 129.9 (Cp, C-Ph), 129.1 (d, JC–P = 9.1, Cm), 1226.3 (Cm, C-Ph), 119.3 (dd, JC–Rh = 48.9, JC–P = 22.8, CC-Ph), 115.2 (d, JC–Rh = 12.1, CC-Ph), 104.9 (br, CH cod), 72.1 (d, JC–P = 16.1, CH2O), 70.4 (d, JC–Rh = 8.4, CH cod), 64.2 (CH2 OEt), 339 (CH2 cod), 29.6 (CH2 cod), 24.0 (d, JC–P = 28.7, CH2P), 22.2 (d, JC–P = 3.6, CH2), 16.0 (CH3 OEt).
Synthesis of [Rh(CH3)(cod){Ph2P(CH2)3Z}] (Z = NMe2, OEt) (5 and 6)
CH cod), 3.73 (br, 2H, CH cod), 2.39 (m, 2H, CH2P), 2.20 (m, 6H; 4H CH2 cod, 2H NCH2), 2.08 (s, 6H, NMe2), 2.03–1.98 (m, 6H; 4H CH2 cod, 2H CH2), 0.40 (d, JH–Rh = 1.4, 3H, CH3). 31P{1H} NMR (C6D6, 298 K): δ 28.46 (d, JRh–P = 179.2). 13C{1H} NMR (C6D6, 298 K): δ 136.8 (d, JC–P = 33.5, Ci), 133.5 (d, JC–P = 11.4, Co), 129.3 (Cm), 128.2 (d, JC–P = 8.8, Cp), 95.1 (d, JC–Rh = 8.9, CH cod), 79.9 (d, JC–Rh = 7.7, CH cod), 61.0 (d, JC–P = 14.3, CH2N), 45.6 (NMe2), 32.0 (CH2 cod), 31.2 (CH2 cod), 25.8 (d, JC–P = 23.2, CH2P), 25.4 (d, JC–P = 4.8, CH2), 4.8 (d, JC–Rh = 23.9, CH3).
CH cod), 3.84 (br, 2H, CH cod), 3.40 (m, 4H; 2H CH2 Et, 2H CH2O), 2.55 (m, 2H, CH2P), 2.38–2.03 (m, 10H, 8H CH2 cod, 2H CH2), 1.22 (t, 3H, JH–H = 7.0, CH3 Et), 0.51 (d, 3H, JH–Rh = 1.4, CH3). 31P{1H} NMR (C6D6, 298 K): δ 28.72 (br). 13C{1H} NMR (C6D6, 298 K): δ 136.8 (d, JC–P = 33.1, Ci), 133.5 (d, JC–P = 11.4, Co), 129.3 (Cm), 128.2 (d, JC–P = 8.8, Cp), 95.1 (CH cod), 80.2 (d, JC–P = 7.5, CH cod), 71.5 (d, JC–P = 14.8, CH2O), 66.26 (CH2 Et), 32.0 (CH2 cod), 31.21 (CH2 cod), 27.8 (d, JC–P = 4.2, CH2), 24.9 (d, JC–P = 23.2, CH2P), 15.5 (CH3 Et), 4.6 (d, JC–Rh = 24.1, CH3).
Phenylacetylene polymerization reactions
The polymerization reactions were carried out in round bottom flasks with efficient stirring. A typical polymerization procedure is as follows: phenylacetylene (70 μL, 0.64 mmol) was added to a THF solution (2.5 mL) of the catalysts (6.4 μmol) and the mixture was stirred at 293 K in the absence of light. The consumption of phenylacetylene was monitored by GC using octane as an internal standard. The polymer solutions were transferred into vigorously stirred cold methanol (25 mL, 273 K) using a cannula under argon. The polymers were filtered, washed with methanol (3 × 5 mL) and dried under vacuum to a constant weight. The polymers were obtained as yellow-orange solids in good yields.The NMR data of a representative PPA sample: 1H NMR (298 K, CD2Cl2): δ 6.98 (m, o-H and p-H, Ph), 6.67 (m, m-H, Ph), 5.85 (s, CH). 13C{1H} NMR (298, CD2Cl2): δ 142.9 and 139.4 (Cq), 131.8 (CH), 127.8 and 127.2 (o- and m-Ph), 126.7 (p-Ph).
Author contributions
Marta Angoy: investigation and methodology. M. Victoria Jiménez: conceptualization, supervision, and validation. Eugenio Vispe: investigation, validation, and visualization. Stepan Podzimek: investigation and validation. Jesús J. Pérez-Torrente: funding acquisition, conceptualization, supervision, writing – original draft, and writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish Ministry of Science and Innovation MCIN/AEI/10.13039/501100011033, under the Projects PID2019-103965GB-I00 and PID2022-137208NB-I00, and the Departamento de Educación, Ciencia y Universidades del Gobierno de Aragón (group E42_23R) is gratefully acknowledged.References
- R. M. Pankow and B. C. Thompson, The development of conjugated polymers as the cornerstone of organic electronics, Polymer, 2020, 207, 122874 CrossRef CAS.
- J. W. Lam and B. Z. Tang, Functional polyacetylenes, Acc. Chem. Res., 2005, 38, 745–754 CrossRef CAS.
- T. Masuda, Substituted Polyacetylenes: Synthesis, Properties, and Functions, Polym. Rev., 2017, 57, 1–14 CrossRef CAS.
- L. Liu, Y. Zang, H. Jia, T. Aoki, T. Kaneko, S. Hadano, M. Teraguchi, M. Miyata, G. Zhang and T. Namikoshi, Helix-Sense-Selective Polymerization of Achiral Phenylacetylenes and Unique Properties of the Resulting Cis-cisoidal Polymers, Polym. Rev., 2017, 57, 89–118 CrossRef CAS.
- A. Xu, T. Masuda and A. Zhang, Stimuli-Responsive Polyacetylenes and Dendronized Poly(phenylacetylene)s, Polym. Rev., 2017, 57, 138–158 CrossRef CAS.
- R. Sakai, T. Satoh and T. Kakuchi, Polyacetylenes as Colorimetric and Fluorescent Chemosensor for Anions, Polym. Rev., 2017, 57, 159–174 CrossRef CAS.
- F. Freire, E. Quiñoá and R. Riguera, Supramolecular Assemblies from Poly(phenylacetylene)s, Chem. Rev., 2016, 116, 1242–1271 CrossRef CAS PubMed.
- T. Masuda, F. Sanda and M. Shiotsuki, Polymerization of Acetylenes, in Comprehensive Organometallic Chemistry III, ed. D. M. P. Mingos and R. H. Crabtree, Elsevier, Amsterdam, Holland, 2007, 1st edn, vol. 11, pp. 557–593 Search PubMed.
- F. Sanda, M. Shiotsuki and T. Masuda, Alkyne Polymerization, in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski and M. Möller, Elsevier, 2012, vol. 3, pp. 875–954 Search PubMed.
- M. Shiotsuki, F. Sanda and T. Masuda, Polymerization of substituted acetylenes and features of the formed polymers, Polym. Chem., 2011, 2, 1044–1058 RSC.
- M. A. Casado, A. Fazal and L. A. Oro, Rhodium-Catalyzed Polymerization of Phenylacetylene and its Derivatives, Arabian J. Eng., 2013, 38, 1631–1646 CrossRef CAS.
- J. Sedláček and H. Balcar, Substituted Polyacetylenes Prepared with Rh Catalysts: From Linear to Network-Type Conjugated Polymers, Polym. Rev., 2017, 57, 31–51 CrossRef.
- J. Sedláček and J. Vohlídal, Controlled and living polymerizations induced with rhodium catalysts. A review, Collect. Czech. Chem. Commun., 2003, 68, 1745–1790 CrossRef.
- M. Isomura, Y. Misumi and T. Masuda, Living polymerization and block copolymerization of various ring-substituted phenylacetylenes by rhodium-based ternary catalyst, Polym. Bull., 2000, 45, 335–339 CrossRef CAS.
- N. S. L. Tan and A. B. Lowe, Polymerizations Mediated by Well-Defined Rhodium Complexes, Angew. Chem., Int. Ed., 2020, 59, 5008–5021 CrossRef CAS PubMed.
- K. Echizen, T. Taniguchi, T. Nishimura and K. Maeda, Synthesis of Stereoregular Telechelic Poly(phenylacetylene)s: Facile Terminal Chain-End Functionalization of Poly(phenylacetylene)s by Terminative Coupling with Acrylates and Acrylamides in Rhodium-Catalyzed Living Polymerization of Phenylacetylenes, J. Am. Chem. Soc., 2021, 143, 3604–3612 CrossRef CAS PubMed.
- K. Ito, T. Taniguchi, T. Nishimura and K. Maeda, Well-Controlled Living Polymerization of N-Propargylamides and Their Derivatives by Rhodium Catalysis, Angew. Chem., Int. Ed., 2022, 61, e202117234 CrossRef CAS PubMed.
- I. Saeed, M. Shiotsuki and T. Masuda, Living Polymerization of Phenylacetylene with Tetrafluorobenzobarrelene Ligand-Containing Rhodium Catalyst Systems Featuring the Synthesis of High Molecular Weight Polymer, Macromolecules, 2006, 39, 8567–8573 CrossRef CAS.
- N. Onishi, M. Shiotsuki, T. Masuda, N. Sano and F. Sanda, Polymerization of Phenylacetylenes Using Rhodium Catalysts Coordinated by Norbornadiene Linked to a Phosphino or Amino Group, Organometallics, 2013, 32, 846–853 CrossRef CAS.
- N. S. L. Tan, P. V. Simpson, G. L. Nealon, A. N. Sobolev, P. Raiteri, M. Massi, M. I. Ogden and A. B. Lowe, Rhodium(I)-α-Phenylvinylfluorenyl Complexes: Synthesis, Characterization, and Evaluation as Initiators in the Stereospecific Polymerization of Phenylacetylene, Eur. J. Inorg. Chem., 2019, 592–601 CrossRef CAS.
- N. S. L. Tan, G. L. Nealon, J. M. Lynam, A. N. Sobolev, M. R. Rowless, M. I. Ogden, M. Massi and A. B. Lowe, A (2-(naphthalen-2-yl)phenyl)rhodium(I) complex formed by a proposed intramolecular 1,4-ortho-to-ortho′ Rh metal-atom migration and its efficacy as an initiator in the controlled stereospecific polymerisation of phenylacetylene, Dalton Trans., 2019, 48, 16437–16447 RSC.
- N. S. L. Tan, G. L. Nealon, G. F. Turner, S. A. Moggach, M. I. Ogden, M. Massi and A. B. Lowe, Rh(I)(2,5-norbornadiene)(biphenyl)(tris(4-fluorophenyl)phosphine): Synthesis, Characterization, and Application as an Initiator in the Stereoregular (Co)Polymerization of Phenylacetylenes, ACS Macro Lett., 2020, 9, 56–60 CrossRef.
- T. Taniguchi, T. Yoshida, K. Echizen, K. Takayama, T. Nishimura and K. Maeda, Facile and Versatile Synthesis of End-Functionalized Poly(phenylacetylene)s: A Multicomponent Catalytic System for Well-Controlled Living Polymerization of Phenylacetylenes, Angew. Chem., Int. Ed., 2020, 59, 8670–8680 CrossRef CAS.
- Y. Kishimoto, P. Eckerle, T. Miyatake, T. Ikariya and R. Noyori, Living Polymerization of Phenylacetylenes Initiated by Rh(CCC6H5)(2,5-norbornadiene)[P(C6H5)3], J. Am. Chem. Soc., 1994, 116, 12131–12132 CrossRef CAS.
- Y. Kishimoto, T. Miyatake, T. Ikariya and R. Noyori, An Efficient Rhodium(I) Initiator for Stereospecific Living Polymerization of Phenylacetylenes, Macromolecules, 1996, 29, 5054–5055 CrossRef CAS.
- Y. Kishimoto, P. Eckerle, T. Miyatake, M. Kainosho, A. Ono, T. Ikariya and R. Noyori, Well-Controlled Polymerization of Phenylacetylenes with Organorhodium(I) Complexes: Mechanism and Structure of the Polyenes, J. Am. Chem. Soc., 1999, 121, 12035–12044 CrossRef CAS.
- M. V. Jiménez, J. J. Pérez-Torrente, M. I. Bartolomé, E. Vispe, F. J. Lahoz and L. A. Oro, Cationic rhodium complexes with hemilabile phosphine ligands as polymerization catalyst for high molecular weight stereoregular poly(phenylacetylene), Macromolecules, 2009, 42, 8146–8156 CrossRef.
- M. Angoy, M. V. Jiménez, F. J. Lahoz, E. Vispe and J. J. Pérez-Torrente, Polymerization of phenylacetylene catalyzed by rhodium(I) complexes with N-functionalized N-heterocyclic carbene ligands, Polym. Chem., 2022, 13, 1411–1421 RSC.
- M. Angoy, M. V. Jiménez, E. Vispe and J. J. Pérez-Torrente, Synthesis of megadalton stereoregular ring-substituted poly(phenylacetylene)s by a rhodium(I) catalyst with a N- functionalized hemilabile phosphine ligand, Polym. Chem., 2024, 15, 2028–2039 RSC.
- M. V. Jiménez, J. J. Pérez-Torrente, M. I. Bartolomé, E. Vispe, F. J. Lahoz and L. A. Oro, Branched Poly(phenylacetylene), Macromolecules, 2010, 43, 6278–6283 CrossRef.
- M. Angoy, M. V. Jiménez, F. J. Modrego, L. A. Oro, V. Passarelli and J. J. Pérez-Torrente, Mechanistic Investigation on the Polymerization of Phenylacetylene by 2-Diphenylphosphinopyridine Rhodium(I) Catalysts: Understanding the Role of the Cocatalyst and Alkynyl Intermediates, Organometallics, 2018, 37, 2778–2794 CrossRef CAS.
- M. Palmese, J. J. Pérez-Torrente and V. Passarelli, Cyclometalated iridium complexes based on monodentate aminophosphanes, Dalton Trans., 2022, 51, 12334–12351 RSC.
- M. V. Jiménez, M. I. Bartolomé, J. J. Pérez-Torrente, F. J. Lahoz and L. A. Oro, Rhodium(I) Complexes with Hemilabile Phosphines: Rational Design for Efficient Oxidative Amination Catalysts, ChemCatChem, 2012, 4, 1298–1310 CrossRef.
- A. Furlani, C. Napoletano, M. V. Russo and W. Feast, Stereoregular polyphenylacetylene, J. Polym. Bull., 1986, 16, 311–317 CAS.
- A. Furlani, C. Napoletano, M. V. Russo, A. Camus and N. J. Marsich, The influence of the ligands on the catalytic activity of a series of RhI complexes in reactions with phenylacetylene: Synthesis of stereoregular poly(phenyl) acetylene, Polym. Sci., Part A: Polym. Chem., 1989, 27, 75–86 CrossRef CAS.
- I. Saeed, M. Shiotsuki and T. Masuda, Effect of Diene Ligands in the Rhodium-Catalyzed Polymerization of Phenylacetylene, Macromolecules, 2006, 39, 8977–8981 CrossRef CAS.
- N. Onishi, M. Shiotsuki, F. Sanda and T. Masuda, Polymerization of Phenylacetylenes with Rhodium Zwitterionic Complexes: Enhanced Catalytic Activity by π-Acidic Diene Ligands, Macromolecules, 2009, 42, 4071–4076 CrossRef CAS.
- A. C. Makan, T. Otte and H. Pasch, Analysis of High Molar Mass Branched Polybutadienes by SEC-MALLS and AF4-MALLS, Macromolecules, 2012, 45, 5247–5259 CrossRef CAS , and references therein.
- S. Podzimek and T. Vlcek, Characterization of branched polymers by SEC coupled with a multiangle light scattering detector. II. Data processing and interpretation, J. Appl. Polym. Sci., 2001, 82, 454–460 CrossRef CAS.
- S. Podzimek, Light Scattering, Size Exclusion Chromatography and Asymmetric Flow Field Flow Fractionation, John Wiley & Sons, Hoboken, New Jersey, 2011 Search PubMed.
- M. Tabata, W. Yang and K. Yokota, 1H-NMR and UV studies of Rh complexes as a stereoregular polymerization catalysts for phenylacetylenes: Effects of ligands and solvents on its catalyst activity, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 1113–1120 CrossRef CAS.
- Y. Tian, X. Li, J. Shi, B. Tonga and Y. Dong, Monomer-induced switching of stereoselectivity and limitation of chain growth in the polymerization of amine-containing para-substituted phenylacetylenes by [Rh(norbornadiene)Cl]2, Polym. Chem., 2017, 8, 5761–5768 RSC.
- T. Tang, S. J. Lu, G. Ahumada and C. W. Bielawski, Megadalton Macromolecules Made-to-Order in Minutes: A Highly Active Nanosphere Catalys for Preparing High-Molecular Weight Polymers, Macromolecules, 2022, 55, 9943–9950 CrossRef CAS.
- M. Shiotsuki, N. Onishi, F. Sanda and T. Masuda, Living polymerization of phenylacetylenes catalyzed by cationic rhodium complexes bearing tetrafluorobenzobarrelene, Polym. J., 2011, 43, 51–57 CrossRef CAS.
- M. Angoy, M. V. Jiménez, P. García-Orduña, L. A. Oro, E. Vispe and J. J. Pérez-Torrente, Dinuclear Phosphine-Amido [Rh2(diene){μ-NH(CH2)3PPh2}2] Complexes as Efficient Catalyst Precursors for Phenylacetylene Polymerization, Organometallics, 2019, 38, 1991–2006 CrossRef CAS.
- Z. Ke, S. Abe, T. Ueno and K. Morokuma, Rh-Catalyzed Polymerization of Phenylacetylene: Theoretical Studies of the Reaction Mechanism, Regioselectivity, and Stereoregularity, J. Am. Chem. Soc., 2011, 133, 7926–7941 CrossRef CAS.
- I. Suárez and B. Coto, Determination of long chain branching in PE samples by GPC-MALS and GPC-VIS: Comparison and uncertainties, Eur. Polym. J., 2013, 49, 492–498 CrossRef.
- B. H. Zimm and W. H. Stockmayer, The Dimensions of Chain Molecules Containing Branches and Rings, J. Chem. Phys., 1949, 17, 1301–1314 CrossRef CAS.
- L. Canovese, F. Visentin, G. Chessa, P. Uguagliati, C. Santo and A. Dolmella, Insertion of Substituted Alkynes into the Pd-C Bond of Methyl and Vinyl Palladium(II) Complexes Bearing Pyridylthioethers as Ancillary Ligands. The Influence of Ligand Substituents at Pyridine and Sulfur on the Rate of Insertion, Organometallics, 2005, 24, 3297–3308 CrossRef CAS.
- V. Cîrcu, M. A. Fernandes and L. Carlton, Reaction of Bis(phosphine)(hydrotris(3,5-dimethylpyrazolyl)borato)-rhodium(I) with Phenylacetylene, p-Nitrobenzaldehyde, and Triphenyltin Hydride: Structures of [Rh(Tp*)(PPh3)2], [Rh(Tp*)(H)(C2Ph){P(4-C6H4F)3}], [Rh(Tp*)(H)(COC6H4-4-NO2)(PPh3)], and [Rh(Tp*)(H)(SnPh3)(PPh3)], Inorg. Chem., 2002, 41, 3859–3865 CrossRef.
- M. Schäfer, J. Wolf and H. Werner, Binding Two C2 Units to an Electron-Rich Transition-Metal Center: The Interplay of Alkyne(alkynyl), Bisalkynyl(hydrido), Alkynyl(vinylidene), Alkynyl(allene), Alkynyl(olefin), and Alkynyl(enyne) Rhodium Complexes, Organometallics, 2004, 23, 5713–5728 CrossRef.
- G. Giordano, R. H. Crabtree, R. M. Heintz, D. Forster and D. E. Morris, Di-μ-Chloro-Bis(η4-1,5-Cyclooctadiene) Di-rhodium(I), in Inorganic Syntheses, John Wiley & Sons, Inc, 1979, vol. 19, pp. 218–220 Search PubMed.
- R. Usón, L. A. Oro, J. A. Cabeza, H. E. Bryndza and M. P. Stepro, Dinuclear Methoxy, Cyclooctadiene, and Barrelene Complexes of Rhodium(I) and Iridium(I), in Inorganic Syntheses, John Wiley & Sons, Inc, 1985, vol. 23, pp. 126–130 Search PubMed.
- M. Falcon, E. Farnetti and N. Marsich, Stereoselective Living Polymerization of Phenylacetylene Promoted by Rhodium Catalysts with Bidentate Phosphines, J. Organomet. Chem., 2001, 629, 187–193 CrossRef CAS.
- M. V. Jiménez, J. J. Pérez-Torrente, M. I. Bartolomé and L. A. Oro, Convenient Methods for the Synthesis of a Library of Hemilabile Phosphines, Synthesis, 2009, 1916–1922 CrossRef.
- J. Sedlacek, J. Vohlidal and Z. Grubisic-Gallot, Molecular–weight Determination of Poly(phenylacetylene) by Size–exclusion Chromatography/low–angle Laser Light Scattering. Influence of Polymer Degradation, Makromol. Chem., Rapid Commun., 1993, 14, 51–53 CrossRef CAS.
- V. Percec and J. G. G. Rudick, Independent Electrocyclization and Oxidative Chain Cleavage along the Backbone of cis-Poly(phenylacetylene), Macromolecules, 2005, 38, 7241–7250 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, ESI-MS, SEC-MALS and A4F-MALS chromatograms. See DOI: https://doi.org/10.1039/d4py00497c |
| This journal is © The Royal Society of Chemistry 2024 |