
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Water-based polymer colloids with a branched chain architecture as low-gel pressure-sensitive adhesives†
Emily M.
Brogden
 and
Stefan A. F.
Bon
*
and
Stefan A. F.
Bon
*
Department of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: s.bon@warwick.ac.uk; Web: https://bonlab.info
First published on 19th June 2024
Abstract
The performance of water-based acrylic pressure-sensitive adhesives, in which the latex particles had minimal gel content of typically less than a few percent and the polymer chains had a branched architecture, were investigated. A series of semi-batch emulsion copolymerizations of 2-octyl acrylate, isobornyl acrylate and acrylic acid was carried out in the presence of ethylene glycol dimethacrylate as crosslinker. The molecular weight distributions and branched chain topology were regulated by using 2-ethylhexyl thioglycolate as a chain-transfer agent. The monomers and chain-transfer agent were selected as these were already available as bio-based feedstocks, or had a realistic potential to become available from non-petrochemical resources. Adhesive films cast from the polymer dispersions demonstrated good peel, shear strength and tack adhesion energies. This is attributed to polymer chain branching, which unlocks a broader window for the design rules for pressure-sensitive adhesives. Detailed rheological studies of the viscoelastic materials were conducted to support the adhesive test results.
1. Introduction
A pressure-sensitive adhesive (PSA) is a viscoelastic material, typically characterized by its adhesion to a variety of substrates upon application of light pressure for a short amount of time.1 One commercially important class of PSAs is made from low glass transition temperature, Tg, poly(acrylates).2 Two frequently used monomers are n-butyl acrylate and 2-ethylhexyl acrylate as the resulting polymers are soft and tacky at room temperature. Often, small amounts (1–15 wt%) of a higher Tg monomer, such as styrene, are also incorporated to improve the cohesive strength of the system, which enables good fibril formation upon debonding, typical of a PSA.3 It is also common to find the incorporation of functional monomers, such as (meth)acrylic acid, to aid wettability and enhance peel and shear strength.4–6 Although an increasing number of these or similar monomers have non-petroleum-based synthesis routes such as the microbial fermentation to produce n-butanol from biomass, which is then esterified with acrylic acid to produce butyl acrylate, they are typically not widely used in industry yet due to their current high costs.7 The sustainability and circularity drives will, no doubt, change this. Ideally, virgin petroleum-based monomers will soon be replaced by monomers made by more sustainable synthetic routes, from a combination of biomass, carbon dioxide, and polymer waste feedstocks, to negate the irregularities in the supply and price fluctuations of petroleum. Besides addressing the underlying chemistry of PSAs, one can also tackle application product design, for example by developing linerless adhesive labels.8Adhesives made from natural plant-based materials have been used for thousands of years. Some of the earliest examples were as simple as burning birch bark to produce birch-bark tar.9 However, often these simple adhesives are susceptible to environmental damage and degradation and do not provide the mechanical strength required for many of today's applications. Slowly, more complex adhesives were developed using composite materials such as combining plant gum and natural iron oxideto improve strength and water resistance.10 An excellent example of an early PSA, credited to Henry Day in 1845 (US patent 3965), was made using a composite of natural rubber, pine gum and a yellow lead oxide applied to pieces of fabric to create surgical tape.11 The problems with many of these early examples were the variability in the quality of the plant-based feedstocks as well as the synthesis and production methods not being sustainable on the scale required for today's consumers. Therefore, synthetic alternatives were desirable and this was achieved mostly by the introduction of poly(acrylate)-based polymers in the 1940s. It is now imperative to maintain the production and properties of the PSAs produced but switch back to non-petroleum-based chemicals. Researchers as early as 1975 were already considering the sustainability of PSA production, suggesting aqueous dispersions based on polymers of acrylic esters, natural or synthetic rubber and resins to replace solvent-based PSAs.12 The term ‘bio-based’ PSAs remained in the background before the turn of the 21st century but some notable plant-based research included the use of tannins from tree bark.13 However, the deforestation required to commercially produce enough for todays market is not desirable. Rosin, extracted from pine tree oils or freshly cut bole has also been used in plant-based adhesives.14 For example, a heat-activated PSA was developed from glycerol esters of rosin acids as well as blending alkyl acrylate–acrylic acid copolymers with rosin derivates.15
With the increasing pressure to use more sustainable polymerization techniques than traditional bulk and solution radical polymerization, many researchers have turned to utilizing emulsion polymerization to produce PSAs as it is a waterborne polymerization technique affording higher molecular weights than the equivalent homogeneous radical polymerizations.12,16,17 Recently, bio-sourced monomers have gained popularity to reduce the reliance on petrochemical feedstocks in the production of PSAs. For instance, Molina-Gutiérrez and coworkers incorporated eugenol-derived methacrylates, sourced from clove oil or lignin depolymerization, into typical emulsion polymerization recipes to obtain latexes with high solids contents, 50%, and glass transition temperatures between −32 and −28 °C.18 They observed improved performance in tack and peel compared to commercially available products. One of the most promising replacements for the low Tg acrylic monomers is 2-octyl acrylate (2-OA), derived from castor oil. A common synthetic route includes reacting 2-octanol with acrylic acid in water in the presence of an acid catalyst or ethyl titinate.19,20 Acrylic acid can also be obtained from bio-based feedstocks, for example through dehydration of 3-hydroxypropionic acid or lactic acid. Badía and coworkers have demonstrated the feasibility of producing latexes using 2-OA as the soft component which are comparable to PSAs made from oil-based polymers.21–23 However, they also reported that improved adhesive performance was obtained when some fraction of the 2-OA was replaced with the petroleum-based 2-ethylhexyl acrylate, creating a trade-off between high proportions of bio-based reagents and material properties.24 A popular replacement for the high Tg components, is isobornyl acrylate or isobornyl methacrylate (IBoA and IBOMA respectively), which is derived from pine resin.25,26 Typically it can be synthesized in a 2 stage one-pot process where isobornyl acetate and methanol are reacted to form isoborneol.27 Following this, a transesterification reaction takes place with (meth)acrylic methyl ester to produce the desired product. Typically, it has been incorporated between 5 and 15 wt% based on total monomer content and commonly in conjuction with 2-OA to yield bio-based coatings and PSAs.23,24 In this work we utilize a combination of 2-OA and IBOA with acrylic acid (AA), to produce bio-based latexes for PSA applications.
A prevalent finding in high-performance PSAs made from water-based polymer dispersions and characterized by high tack and shear properties, indicates that the gel content typically ranges from 50% to 70%.2,28,29 In general, a high gel fraction is attributed to increased shear resistance but decreased tack. Whilst some quote the sol-to-gel ratio as one of the most important factors controlling adhesive performance, others have indicated that the microstructure of the gel has a greater effect.29,30 For instance, Lopez and coworkers investigated a hybrid waterborne polyurethane-acrylic system with a variety of diol crosslinkers.31 They found different diols had a small effect on the overall gel content but a large effect on the gel network morphology producing a range of crosslinking densities which had a profound effect on the adhesive performance. Additionally, Tobing and coworkers also placed a large importance on the network morphology of the gel fraction and even attributed the common observation that the poorer performance, often observed with acrylic emulsion PSAs, is due to the discrete network morphology from individual particles compared to the continuous morphology seen in solution-based acrylic PSAs.16
One can question what defines the gel content in a polymer latex, in other words, what constitutes a ‘gel’ or a polymer solution (‘sol’). The maximum dimensions of crosslinked material are on the order of the size of the particles, as the polymer chains are geometrically confined within this volume. However, it is perfectly feasible that smaller fragments of crosslinked material exist. If one would try to dissolve the latex particles in a good solvent for the polymer, then at one particular point a decision needs to be made on what is a dispersed ‘gel’ and what are dissolved polymer chains, ‘sol’. These dissolved polymer chains will have a branched chain architecture, but at the same time can have some crosslinked loops. Common in high-performance waterborne acrylic PSAs is to have a considerable amount of gel content to balance the adhesive properties. Here, we challenge this approach and hypothesize that we can obtain water-based poly(acrylate) dispersion PSAs with desirable adhesive performance that have little to no gel content. If true, and branched polymer chain architectures can compete in performance, this would open up a broader PSA sustainability and circularity design window.
Branching has previously been shown to be beneficial for adhesion as it causes strain hardening as the polymers do not disentangle easily leading to a large increase in extensional viscosity at high rates of extension. However, too much branching prevents polymer–polymer interdiffusion during film formation and decreases cohesion. Recently, Hirth and coworkers showed that moderate gel contents and branching resulted in optimized tack adhesion energy.32 Additionally, Li and coworkers produced highly branched adhesive resins via polycondensation reactions but noted the difficulty to control the branching structure in these types of polymerization reactions.33 González and coworkers were able to produce highly branched polymers using emulsion polymerization which exhibited good shear strength, however the gel content remained high, around 70%.34 Callies and coworkers performed an interesting study where they synthesized low molecular weight PSAs from butyl acrylate and glycidyl methacrylate.35 They produced low molecular weight linear polymers which had some hydrogen bonding moieties. It was only after post-crosslinking these linear segments with a reaction between a diamine and the epoxy groups of the polymer that good adhesive properties were observed. As usual, it is difficult to distinguish the cause of the improvement between increasing the molecular weight and increased chain branching of the crosslinked product. Finally, Fang and coworkers studied the effects of side chain length of adhesion properties between fluorinated acrylic PSAs and PTFE.36 They found that the longer aliphatic side chains resulted in increased peel adhesion.
Over the last two decades, PSA performance has been improved by incorporating a delicate balance of crosslinker and chain transfer agent to control the sol-to-gel ratio and maintain high sol molecular weights.29,37–40 In almost all cases the gel content remains above 20% to give good adhesive properties. Liu and coworkers have reported achieving low gel contents and branched polymer architectures from emulsion polymerization of petroleum-based butyl acrylate using this approach but no adhesive testing was performed.41 In the following work, the crosslinker ethylene glycol dimethacrylate (EGDMA), and the conventional chain transfer agent 2-ethylhexyl thioglycolate (2-EHTG) are used in an attempt to minimise gel content but maintain a high enough molecular weight to demonstrate good adhesive properties. Both EGDMA and 2-EHTG have a realistic potential to become available from non-petrochemical resources. Previously, Agirre and coworkers have demonstrated that at low incorporations of EGDMA (less than 1 mol% with respect to monomer) there is limited effect on gel content and sol molecular weight, possibly due to its extensive primary cyclization.37 This work will investigate EGDMAs effect at substantially higher incorporations.
2. Experimental
2.1. Materials
Aluminium oxide activated (neutral, Brockmann I), Brij L23 solution (30% (w/v) in H2O), ammonium persulfate (reagent grade 98%), isobornyl methacrylate (technical grade), 2-ethylhexyl thioglycolate (≥95.0%), ethylene glycol dimethacrylate (98%, contains 90–110 ppm monomethyl ether hydroquinone as inhibitor), acrylic acid (stabilised with hydroquinone monomethyl ether for synthesis), tetrahydrofuran (inhibitor-free, for HPLC, ≥99.9%), and ethylene glycol (ReagentPlus ≥99%) were purchased from Sigma Aldrich. Sodium hydrogen carbonate (99%) was purchased from Alfa Aesar. Aluminium oxide activated (basic, Brockmann I) was purchased from Honeywell Fluka. Potassium hydroxide pellets were purchased from Chemiphase. Lakeland PAE 136 was a gift from Lakeland Laboratories. 2-Octyl acrylate was kindly donated by Covestro.Acrylic acid was filtered through activated aluminium oxide (neutral) before use. Isobornyl methacrylate, ethylene glycol dimethacrylate and 2-octyl acrylate were filtered through activated aluminium oxide (basic) before use. Size exclusion chromatography samples were run in tetrahydrofuran with 0.01% butylated hydroxytoluene. Mylar®A polyethylene terephthalate (PET), 50 μm thickness, was purchased from UK Insulations in rolls of 30 mm width and used as a film formation substrate after chemical modification, detailed in section 2.2.8. All other chemicals were used as purchased with no further purification. Deionized water was used in all reactions and analyses.
2.2. Methods
All reactions proceeded in the following way, where exact amounts of each chemical used for each reaction can be found in Tables S1 and S2.† Lakeland PAE 136 (0.35 g) in water (112.32 g) was added to the reactor. This was degassed, via nitrogen bubbling, for 30 min, together with each of the following in three separate round bottom flasks: monomer mixture (2-octyl acrylate, isobornyl acrylate and acrylic acid, 92.6%, 5.0% and 2.4% mol% respectively, with ethylene glycol dimethacrylate and 2-ethylhexyl thioglycolate), surfactant mixture (Brij L23 solution 30% w/v (9.28 g), Lakeland PAE 136 (0.33 g), sodium hydrogen carbonate (0.78 g) and water (6.74 g)) and initiator mixture (ammonium persulfate (0.24 g) in water (11.52 g)). For reactions where the ethylene glycol dimethacrylate was only incorporated during the feeding stage, the monomer mixture was prepared by adding all the other reagents, removing 10 mL of the mixture to a separate flask, which was also degassed, and adding the crosslinker to the remaining mixture to create the monomer feed solution.
Once degassed, the monomer charge (8 mL) was added to the reaction vessel using a degased syringe. The initiator charge (9.5 mL) was added at t = 0. Feed 1 (monomer, 25.962 mL h−1) and feed 2 (aqueous surfactant solution, 4.10 mL h−1) began at t = 30 min and fed for 3 h. The reaction mixture was stirred throughout at 225 rpm and heated via inbuilt water jacket at 70 °C. Samples were taken throughout the reaction to monitor the overall monomer conversion and particle size. The total reaction time was 5 h and 30 min, at which time the latex was removed from the vessel, whilst still hot and filtered through a mesh with a pore-size of 200 μm. All latexes were coagulum-free, had a final monomer conversion ≥99% calculated using gravimetry, average hydrodynamic diameters, dz, between 140 and 200 nm with polydispersity indexes, PDI, generally below 10%. The final solids contents of the latexes were between 37 and 40 wt% and the glass transition temperatures, Tg, measured by dynamic scanning calorimetry varied. For analyses from individual latexes see Table 1.
| Latex | SC/% | T g/°C | M coag/% | d z /nm | PDI/% | p M,cum/% | gel0.22 μm/% | M n,sol/g mol−1 | M w,sol/g mol−1 | Đ/— |
|---|---|---|---|---|---|---|---|---|---|---|
| B_X04_C4 | 40.12 | −58 | <1 | 154 | 3.7 | >99.9 | 0.87 ± 0.65 | 3128 | 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 666 666 |
4.0 |
| B_X07_C4 | 39.38 | −43 | <1 | 172 | 9.6 | >99.9 | 3.44 ± 4.63 | 4211 | 14681 |
3.5 |
| B_X16_C4 | 38.73 | −31 | <1 | 151 | 6.1 | >99.9 | 10.53 ± 3.82 | 4346 | 37710 |
8.7 |
| F_X00_C4 | 37.71 | −57 | 0 | 182 | 8.1 | 99.7 | 0.63 ± 0.11 | 2739 | 5932 | 2.2 |
| F_X08_C4 | 39.46 | −46 | <1 | 155 | 5.9 | >99.9 | 1.08 ± 0.12 | 1848 | 10756 |
5.8 |
| F_X11_C4 | 40.45 | −43 | 0 | 167 | 10.4 | >99.9 | 0.32 ± 0.29 | 4669 | 19908 |
4.3 |
| F_X13_C4 | 39.58 | −38 | <1 | 164 | 5.1 | >99.9 | 0.31 ± 0.21 | 5235 | 24567 |
4.7 |
| F_X18_C4 | 39.95 | −33 | <1 | 168 | 7.9 | >99.9 | 0.99 ± 0.31 | 2350 | 42637 |
18.1 |
| F_X00_C2 | 37.43 | −55 | <1 | 165 | 3.8 | 99.9 | 0.45 ± 0.20 | 4453 | 11713 |
2.6 |
| F_X08_C2 | 38.22 | −43 | <1 | 152 | 1.6 | >99.9 | 0.98 ± 0.04 | 4115 | 23214 |
5.6 |
| F_X11_C2 | 38.97 | −39 | 0 | 154 | 5.2 | >99.9 | 1.02 ± 0.55 | 7079 | 42737 |
6.0 |
| F_X13_C2 | 37.70 | −29 | <1 | 157 | 4.1 | >99.9 | 5.13 ± 5.16 | 5481 | 41917 |
7.6 |
| F_X18_C2 | 38.81 | −27 | <1 | 163 | 6.1 | >99.9 | 2.44 ± 0.90 | 3581 | 43460 |
12.1 |
| F_X13_C1 | 38.77 | −32 | <1 | 152 | 5.2 | >99.9 | 2.98 ± 2.52 | 8935 | 80087 |
9.0 |
| F_X13_C0.5 | 38.23 | −25 | 0 | 153 | 3.4 | >99.9 | 1.67 ± 0.65 | 8929 | 70488 |
7.9 |
The number of particles per litre of water, Np, at each sample time was calculated using eqn (1) to check for coagulation, characterized by a decreasing Np and increasing dz, or secondary nucleation, characterized by an increasing Np and decreasing dz. mmon refers to the mass of monomer added to the reactor overall at the sampling time, pM,cum refers to the instantaneous monomer conversion at the sampling time, ρpol is the average polymer density, calculated using a weighted mass average, and VH2O is the volume of water in the reaction vessel at the sampling time. The Np for each latex as a function of reaction time can be seen in Fig. S1e, S2e, S3e and S4e.†
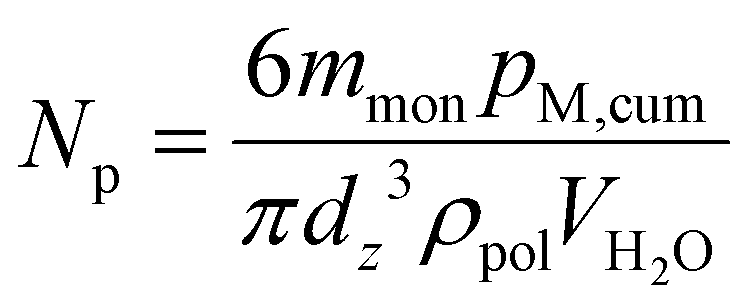 | (1) |
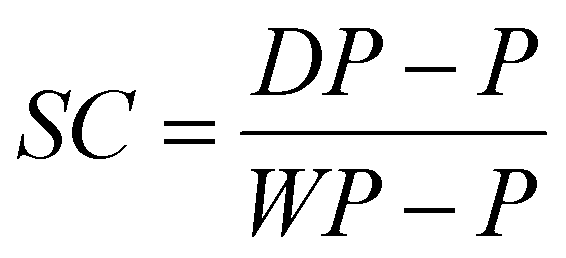 | (2) |
The SC was used to calculate the instantaneous conversion, pm,inst, at each time point using eqn (3), where Mt,sol is the mass of all solid components, not including polymer, Mt,tot is the mass of all components and Mt,mon is the cumulative mass of monomer at the time of sampling.
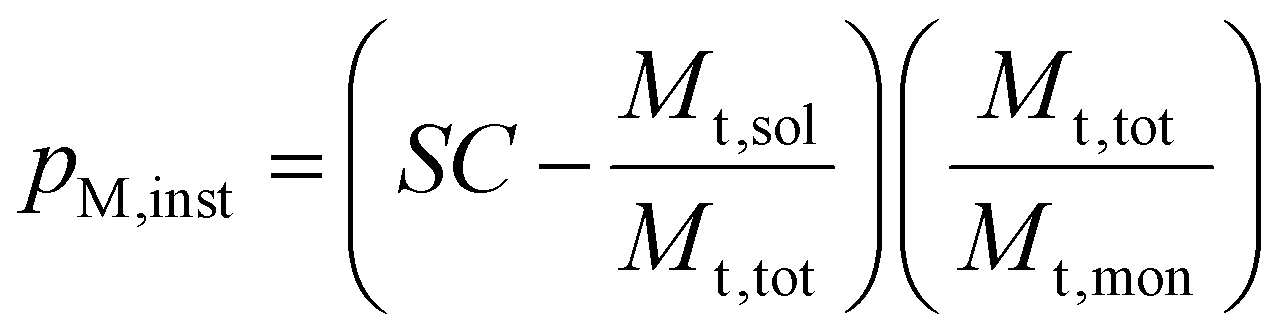 | (3) |
Using pM,inst, the cumulative conversion, pM,cum, was then calculated using eqn (4), where Mmon is the total mass of the monomer used for the reaction.
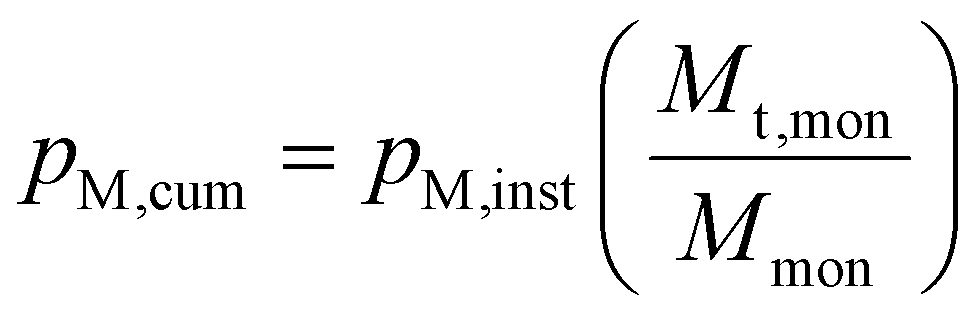 | (4) |
M were produced using Agilent GPC/SEC software using dn/dc = 0.068 and two light scattering detectors (90 and 15°). The linear region of the molecular weight against elution time was selected below 15.4 min elution time, to remove the effect of surfactant contamination, and encompassed the majority of the peak. A discussion of why these regions were picked can be found in the Results and Discussion. The Mark–Houwink parameter α was determined by the software as the gradient of the Mark–Houwink plot and is used as a value to compare the extent of branching between each latex, see Fig. 2b, c, 3b, c and 4b, c.
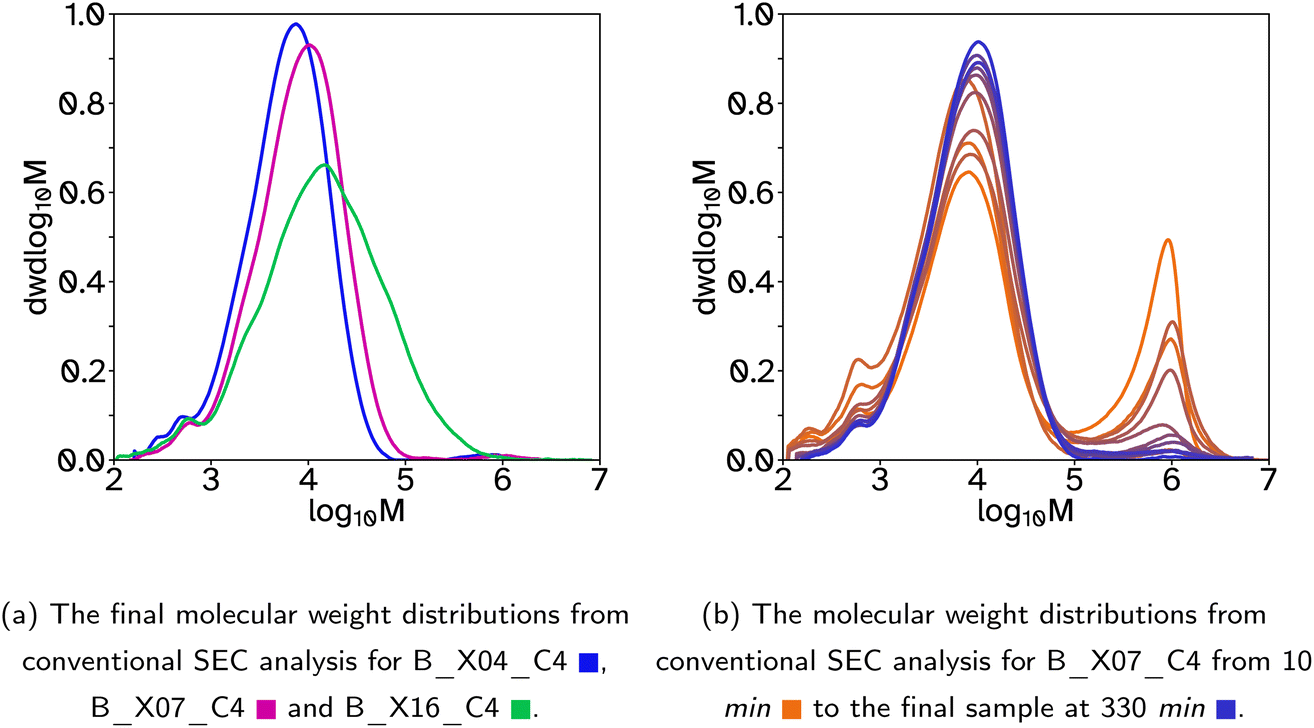 | ||
| Fig. 1 Molecular weight distributions for latexes where the crosslinker was included in both the batch and feed stages of the emulsion polymerization. | ||
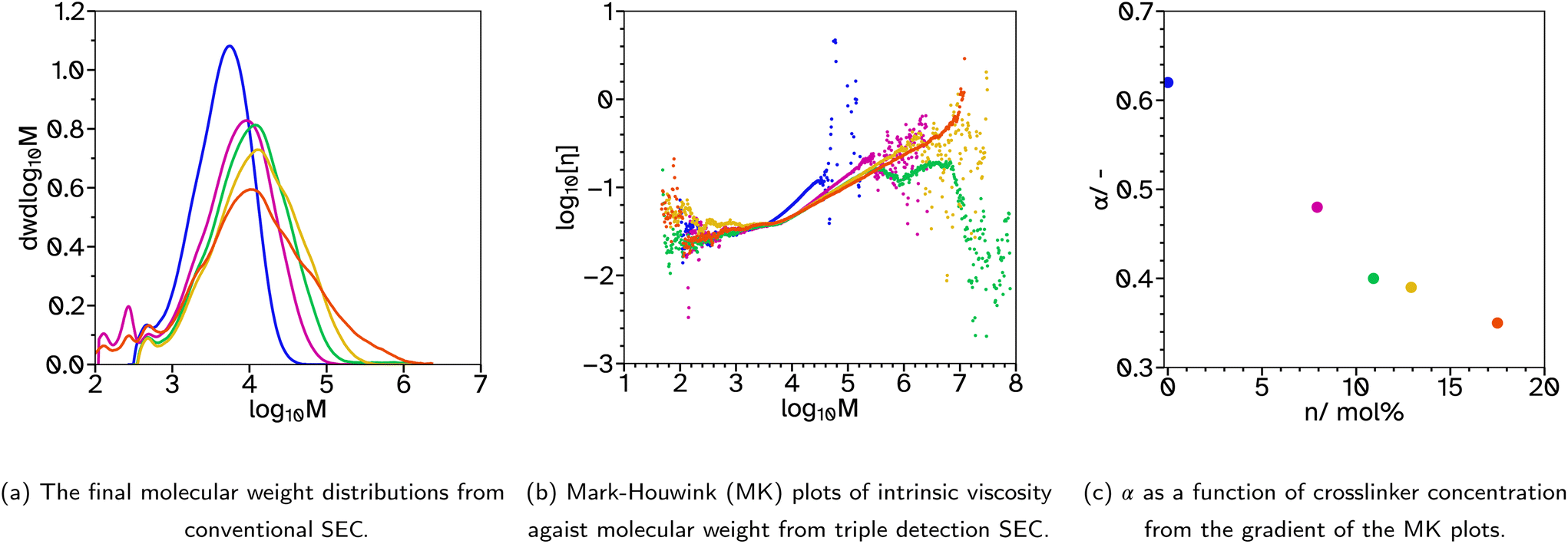 | ||
Fig. 2 SEC analysis for F_Xn_C4, where n = 0  , 8 , 8  , 11 , 11  , 13 , 13  and 18 and 18  . . | ||
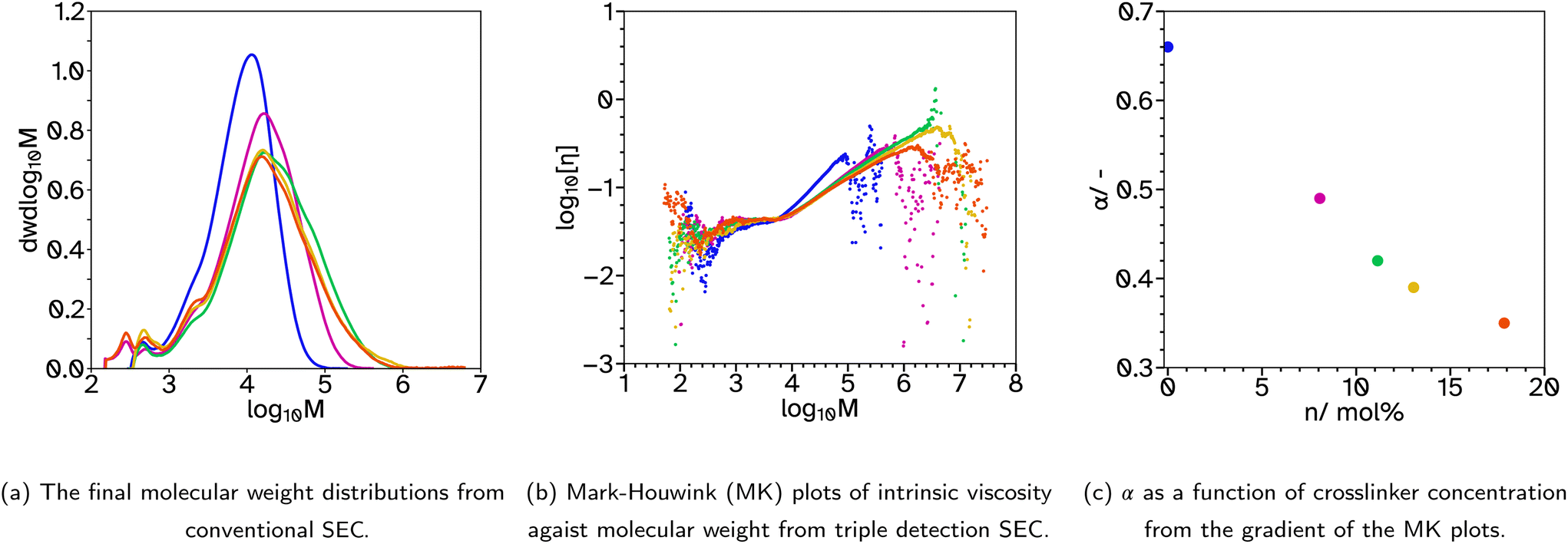 | ||
Fig. 3 SEC analysis for F_Xn_C2, where n = 0  , 8 , 8  , 11 , 11  , 13 , 13  and 18 and 18  . . | ||
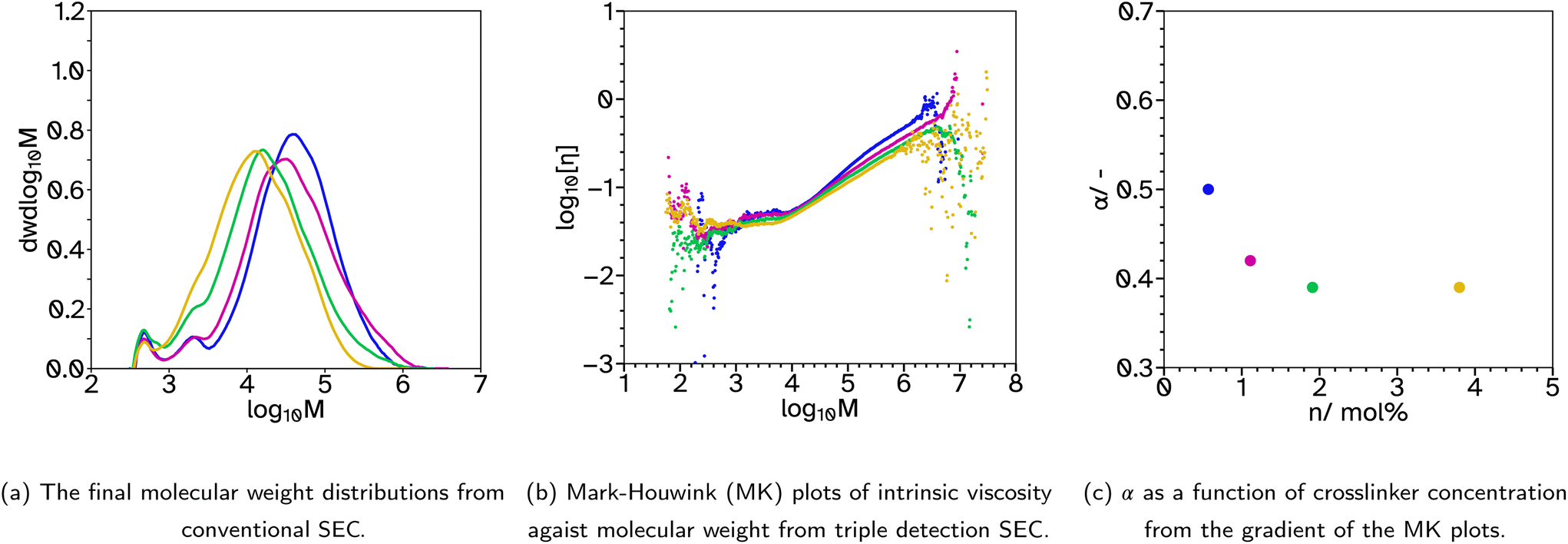 | ||
Fig. 4 SEC analysis for F_X13_Cn, where n = 0.5  , 1 , 1  , 2 , 2  and 4 and 4  . . | ||
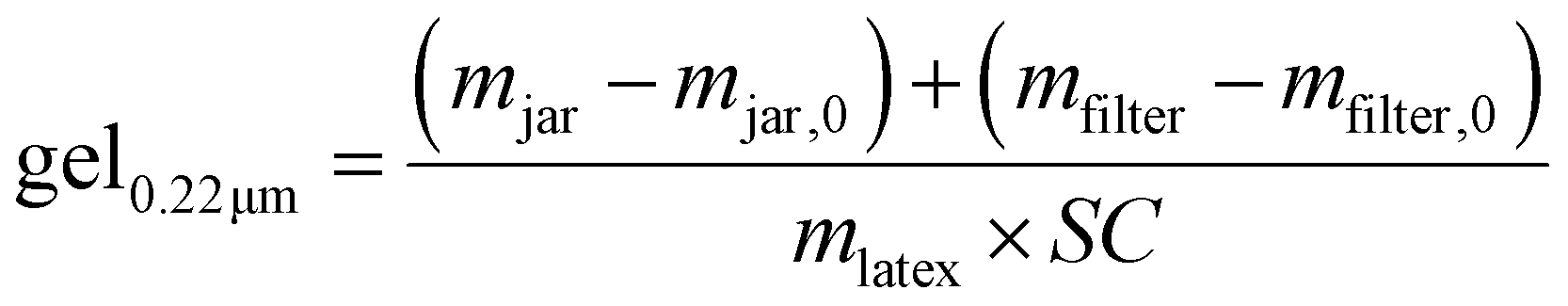 | (5) |
Chemical modification of substrate. All the latexes were too hydrophilic to be cast directly onto mylar®A PET as they exhibited dewetting behaviour. To achieve a homogeneous film the PET substrate was modified using a method similar to that reported by Kim and coworkers.42
A strongly basic solution was made using ethylene glycol (1110 g) with slow addition of potassium hydroxide (278 g) and dissolved overnight. A4 sheets of PET were submerged in the solution for 2 h at room temperature. Upon removal, they were rinsed with deionized water and left to dry. A second rinse with deionized water ensured there was no basic solution remaining before using it as the film formation substrate. Water sessile drop contact angles (10 μL, Young-Laplace fitting) were measured using a Krüss DSA 100 with the modified substrate, ChemPET, to show the decrease in contact angle and improved hydrophilicity, Fig. S13.†
Latex dialysis. All latexes were dialysed before tape fabrication to remove excess surfactant. A dialysis membrane (12–14 kDa) was filled with latex (approximately 10 ml) and placed in 2 L of deionized water for 24 h where the deionized water was replaced three times at regular intervals. The solids contents were measured for the dialysed latexes using eqn (2).
Film formation and tape fabrication. Approximately 10 ml of dialysed latex was cast using an Elcometer 4340 Automatic Film Applicator with a 10 cm casting knife applicator at 5 mm s−1 with a 200 μm gap onto ChemPET at 30 °C. The films were all dried for 1.5 h before a top sheet of Mylar®A PET (not chemically modified) was applied using a cylindrical roller bar (approximately 800 g) at 5 mm s−1, which was made to attach to the Elcometer Film Applicator to minimize bubbles in the finalized tape, Fig. S14.† After 7 days, the films were cut orthogonally to the casting direction using a guillotine to obtain strips of tape with dimensions 2 cm in width and 10 cm long. Accounting for the solids content of the dialysed latexes the film heights were on average 39.5 μm ± 4.6 μm with a maximum of 44.4 μm and a minimum of 27.7 μm.
| Latex | G′ (1 Hz)/MPa | F peel/N/20 mm | W shear/MPa | W adh/J m−2 |
|---|---|---|---|---|
| B_X04_C4 | — | — | — | 24.19 ± 2.73 |
| B_X07_C4 | — | — | — | 34.07 ± 2.88 |
| B_X16_C4 | — | — | — | 340.87 ± 20.29 |
| F_X00_C4 | 0.00037 | 0.08 ± 0.03 | 0.0006 ± 0.0008 | 15.89 ± 1.63 |
| F_X08_C4 | 0.00201 | 0.13 ± 0.01 | 0.0001 ± 0.0001 | 29.59 ± 3.33 |
| F_X11_C4 | 0.00206 | 0.25 ± 0.01 | 0.0021 ± 0.0020 | 72.20 ± 4.47 |
| F_X13_C4 | 0.00575 | 0.60 ± 0.04 | 0.0016 ± 0.0001 | 77.19 ± 2.31 |
| F_X18_C4 | 0.05189 | 8.96 ± 2.68 | 0.0365 ± 0.0035 | 554.90 ± 240.90 |
| F_X00_C2 | 0.00067 | 0.12 ± 0.03 | 0.0008 ± 0.0002 | 18.26 ± 3.31 |
| F_X08_C2 | 0.00361 | 0.40 ± 0.02 | 0.0009 ± 0.0006 | 54.75 ± 5.05 |
| F_X11_C2 | 0.00674 | 1.72 ± 0.07 | 0.0093 ± 0.0031 | 108.99 ± 6.13 |
| F_X13_C2 | 0.01207 | 9.81 ± 0.54 | 0.0191 ± 0.0022 | 965.96 ± 65.15 |
| F_X18_C2 | 0.14033 | 5.90 ± 2.68 | 0.1535 ± 0.0035 | 78.56 ± 12.52 |
| F_X13_C1 | 0.08717 | 13.45 ± 2.02 | 0.0539 ± 0.0075 | 36.62 ± 5.81 |
| F_X13_C0.5 | 0.06449 | 14.23 ± 1.31 | 0.1015 ± 0.0054 | 124.73 ± 45.09 |
For the tack test, the plate with dried sample was loaded at 25 °C with the top plate not in contact with the sample. The sample was then conditioned at 25 °C for 3 min. The upper plate was lowered at 5 μm s−1 with a terminating axial force of 0.1 N, where the gap at this stage is taken as the sample height (reported in Table S4†). The top plate then pressed into the sample for 5 s with a maximum force of 10 N. The upper plate was raised at 100 mm min−1 until the axial force returned to 0 N. The load when the top plate was lifted up was divided by the contact area to obtain stress and the displacement of the probe was divided by the initial sample height to obtain strain. The work of adhesion, Wadh, was then obtained using eqn (6).43
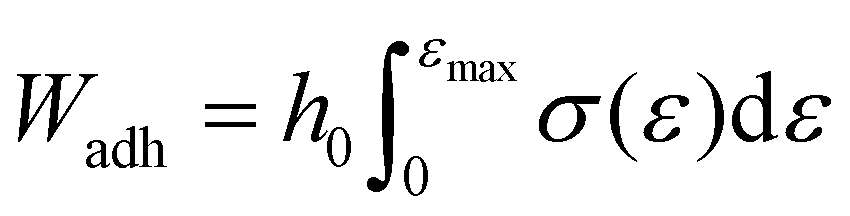 | (6) |
Each tack experiment was repeated 5 times and the average Wadh is reported with one standard deviation, Table 2. The stress–strain data for all films and their repeats can be seen in Fig. S23–S26.†
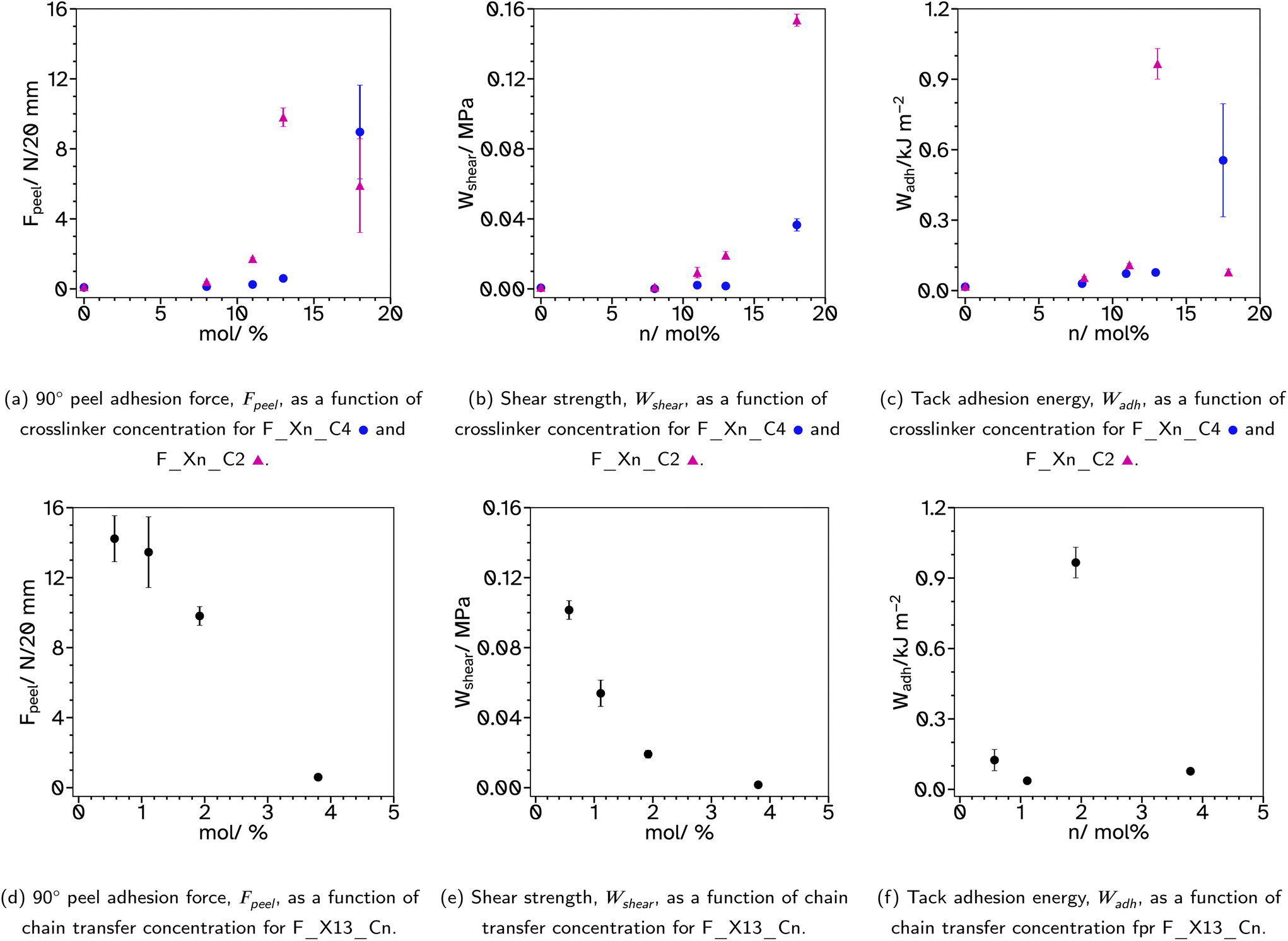 | ||
| Fig. 5 Results from typical pressure sensitive adhesive tests, 90° peel adhesion force, shear strength and tack, relating to crosslinker and chain transfer agent concentrations. | ||
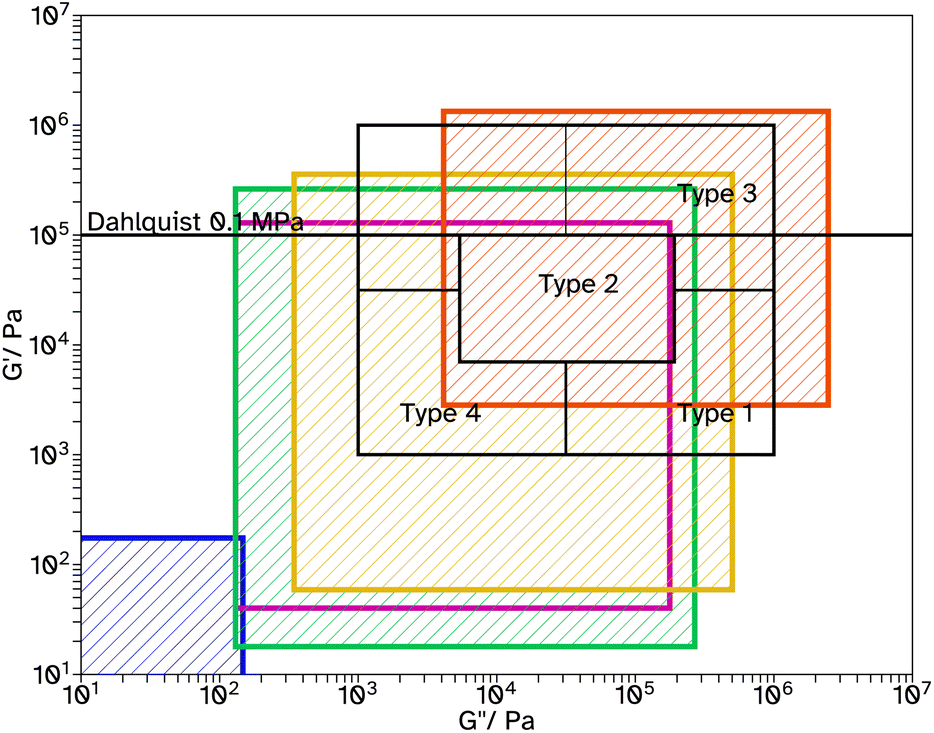 | ||
Fig. 6 Chang windows when varying crosslinker concentration in F_Xn_C4, where n = 0  , 8 , 8  , 11 , 11  , 13 , 13  and 18 and 18  . . | ||
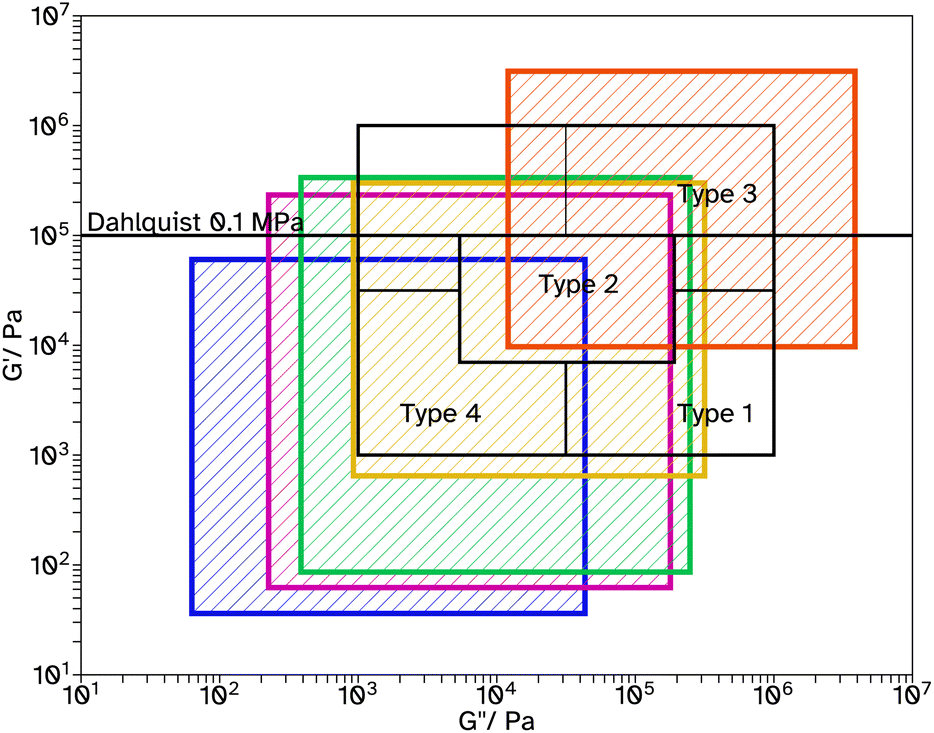 | ||
Fig. 7 Chang windows when varying crosslinker concentration in F_Xn_C2, where n = 0  , 8 , 8  , 11 , 11  , 13 , 13  and 18 and 18  . . | ||
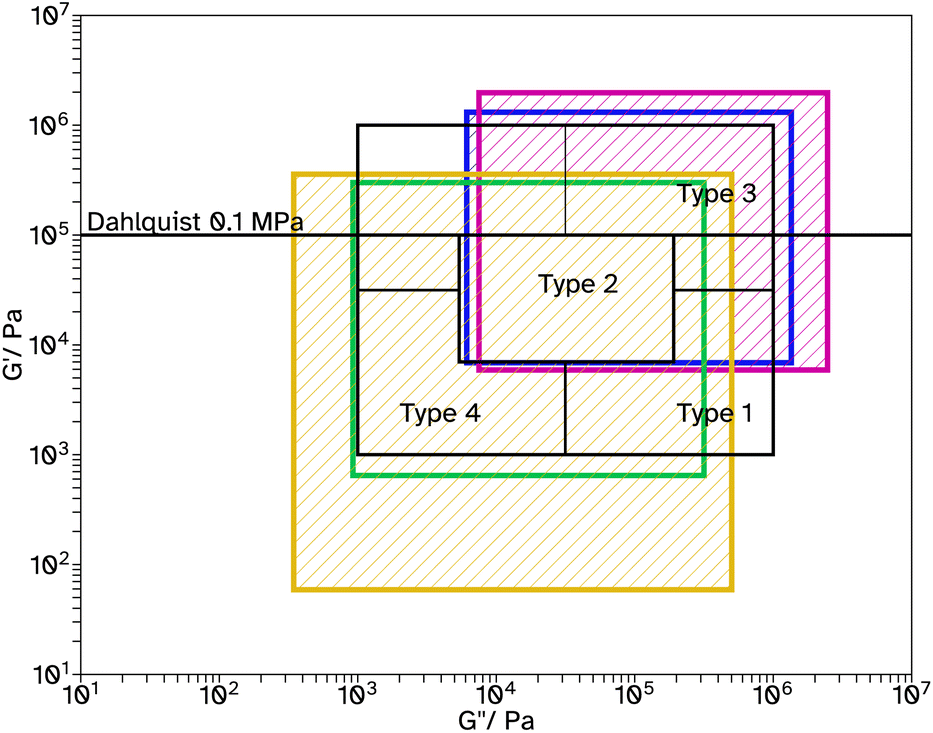 | ||
Fig. 8 Chang windows when varying crosslinker concentration in F_X13_Cn, where n = 0.5  , 1 , 1  , 2 , 2  and 4 and 4  . . | ||
3. Results and discussion
A collection of polymer latexes were produced with varying degrees of polymer chain branching and low gel contents to investigate their adhesive properties. The semi-batch emulsion polymerization reactions formed random copolymers using the following monomers; 2-octyl acylate (2-OA) to provide a low glass transition temperature, Tg, tacky component; isobornyl acrylate (IBoA) to provide a small amount of high Tg component; and acrylic acid (AA) to aid particle stabilization. In all the latexes produced the mole ratio of these monomers was kept constant to investigate only the effects of changing the crosslinker or chain transfer agent (CTA) concentrations, and therefore the polymer chain architecture and associated molecular weight distribution. All reactions produced latexes of approximately 40 wt% solids contents, SC, with <1% coagulation by mass. Additionally, all latexes were stable over time, namely the average hydrodynamic diameter, dz, remained constant. However in some cases, particularly where the Tg was lowest, there was some film formation at the top of the containers when stored between 25 and 35 °C. This could be arrested when the hot latex was removed from the reaction vessel by cooling it in an ice bath immediately. For details of the measured properties of each latex, including Tg, final dz, particle size dispersity, PDI, final monomer conversion, ρM,cum, and average molecular weights and dispersity from conventional SEC, see Table 1.Initially, a series of latexes was produced where the crosslinker, ethylene glycol dimethacrylate (EGDMA), concentration was varied and the CTA, 2 ethlyhexyl thioglycolate (2-EHTG), concentration was kept constant. These are denoted with a latex name beginning with a ‘B’ to indicate that the crosslinker was added in both the batch and feed stages of the polymerization, as opposed to starting with an ‘F’ indicating the crosslinker was only added during the feed stage. The rest of the name denotes the concentration of crosslinker and CTA with respect to the monomer solution (2-OA, IBoMA, AA, EGDMA and 2-EHTG). For example ‘B_X07_C4’, relates to a latex which has 7 mol% EGDMA and 4 mol% 2-EHTG, where the mole percentage relates to the number of molecules and not the number of vinyl groups. The instantaneous and cumulative conversions over time for these polymerizations were all almost identical, Fig. S1a and b,† indicating that the concentration of crosslinker has a negligible effect on the reaction kinetics and that the polymerization rate is controlled by the monomer feed rate and thus under monomer-starved conditions, which is a similar result to that determined by Chauvet and coworkers.29 The average dz, are also relatively similar, Table 1 and Fig. S1c and d,† with low PDI. To ensure the reactions progressed with minimal coagulation or secondary nucleation the number concentration of particles, Np, was plotted as a function of time, Fig. S1e.† During the initial batch reaction, prior to 30 min, the number of particles increases, as was expected during the particle nucleation stage. Once the feed begins, Np is stable until the end of the reaction, suggesting in all cases the reaction progresses as expected.
The molecular weight distributions were measured using size exclusion chromatography, SEC, to determine the average molecular weights and polymer architecture. In this case, where the crosslinker, EGDMA, was included in both the batch and feed stages of the reactions, a bimodal distribution occurs, with the major peak at lower molecular weights (1000–100000 g mol−1) and a minor peak at higher molecular weights (1000000 g mol−1), Fig. 1a. This can be seen best when the molecular weight distributions are shown as a function of reaction time, Fig. 1b and S9,† indicating that the high molecular weight polymer chains are made in the early stages of the reaction, particularly before the feed begins. This is due, in part, to EGDMAs slight solubility in water (1.1 g L−1 at 20 °C) compared to the more insoluble 2-EHTG (0.03 g L−1 at 23 °C) resulting in the EGDMA diffusing through the water phase and accumulating in the particles much faster than 2-EHTG. Thus creating high molecular weight, crosslinked chains before the 2-EHTG can establish its equilibrium to control the molecular weight evolution. This also explains the slightly higher gel contents measured for these reactions, Table 1. It is also more likely that higher molecular weight polymer and more branched structures will be formed at the start of the reaction due to the lower swelling capability of smaller particle sizes. This results in lower concentrations of monomer in the particles at the start of the reaction compared to the end. To avoid this bimodal distribution which lowers the molecular weight of the main peak in the distribution and complicates more complex SEC analysis, the EGDMA was removed from the batch and only added in the feed stage, allowing the 2-EHTG to establish its equilibrium during the batch stage and produce monomodal molecular weight distributions. There is an early indication that increasing the crosslinker concentration is beneficial to the adhesion properties as the tack adhesion energy, Wadh, increases, Table 2. However, the gel content also increased with the crosslinker concentration so the following work prioritized maintaining low gel contents to elucidate the impact of branching on adhesive properties.
To achieve the low gel contents sought after, the relatively high concentration of CTA, 4 mol%, was maintained but the crosslinker, EGDMA, was removed from the initial monomer charge. This produced the series F_Xn_C4 where n = 0, 8, 11, 13 and 18. As before, the kinetics and particle size evolution showed no significant changes with increased crosslinker concentrations, Fig. S2.† The molecular weight distributions no longer had a high molecular weight peak, Fig. 2a and S10,† indicating removal of the crosslinker in the batch stage prevented the very high molecular weight polymer from forming. As the EGDMA concentration increases a tail begins to appear in the high molecular weight region. This gives a good indication that there is branching or intramolecular crosslinking during the emulsion polymerizations with higher crosslinker concentrations. Measuring the gel content showed that intermolecular crosslinking was limited as its value remained below 1%, even at the highest crosslinker concentration. Previous reports have described how EGDMA, considered a highly reactive symmetrical crosslinker, has a limited effect on the gel content and sol molecular weights due to high proportions of extensive primary cyclization.37 At low incorporation, our results agree, where even 8 mol% crosslinker has a limited effect on the overall molecular weight distribution. However, above 11 mol%, there is a broadtail in the distribution suggesting some branching character. As mentioned, no considerable amounts of dissolved polymer chains of high molar mass were detected up to the exclusion limits of the columns, providing confidence in the accuracy of the low gel content values in Table 1. This was supported by no marked differences in back pressure as indication for partial column blocking, during consecutive SEC runs.
A more complex analysis of the SEC data was completed using triple detection SEC. In order to accomplish this, the refractive index increment, dn/dc, of the polymer needs to be known. For our system the dn/dc, calculated using the polymer concentrations after accounting for surfactants and gel content, was between 0.060 and 0.070. This agrees well with existing literature for poly(ethyl acrylate), poly(butyl acrylate), poly(methyl methacrylate) and poly(methyl acrylate) in THF, if a little low.45–47 In all cases dn/dc was assumed to be constant at 0.068 g mol−1. The first difficulty in the analysis is due to the overlap of the surfactant peaks with the polymer peak. There is a shoulder at low molecular weight, approximately 1200 g mol−1, which is attributed to Brij L23, Fig. S33.† This affects the calculation of molecular weight during triple detection SEC analysis as the dn/dc is significantly different, calculated as 0.055 for Brij L23, and the analysis gives higher molecular weights at high elution times. Therefore, only the linear part of the logM vs. elution time graph (Fig. 2b) was taken to avoid the contamination from surfactants at high elution times and the noisy light scattering at low elution times due to the low concentration. A Mark–Houwink logarithmic plot, eqn (7), can be used to determine the Mark–Houwink parameters, K and α. In particular, α can be determined from the gradient of the plot and indicates how compact the polymer chains are. A lower value suggests a more compact structure indicating an increase in branching in this case.
| log10[η] = log10K + αlog10M | (7) |
For F_Xn_C4, the results are reported in Fig. 2b and c. As can be seen, when the concentration of EGDMA increases the values for α drop, suggesting more branched polymer chain architectures.
The balance of crosslinker and high concentrations of CTA kept the gel content below 1% for all latexes in the series F_Xn_C4 such that the changing molecular weight distribution and architecture are the main variables. However, as the crosslinker concentration increases the Tg also increases from −57 to −33 °C. The elevated Tg's are still within the range of typically good PSAs but it should be kept in mind since adhesive properties are also often a function of Tg.
Increasing the crosslinker concentration in this series has minimal effect on the 90° peel adhesion force when lower than 13 mol% but demonstrates a drastic increase at 18 mol%, Fig. 5a. This is attributed more to the increased branched, high molecular weight polymer rather than the Tg as the Tg increases steadily as the crosslinker concentration increases which is not echoed in the Fpeel trend for this series. Typically, Fpeel and the shear strength, Wshear, are inversely proportional such that adhesives which exhibit high Fpeel show lower Wshear.44 However, in the case of the F_Xn_C4 series, increasing the crosslinker concentration increases both Fpeel and Wshear. F_X18_C4 shows peel behaviour typical of a material which is slightly too elastic as Fpeel has many spiked decreases, Fig. S16.† However, the stress–strain curve, from which Wshear is calculated, shows behaviour typical of a good PSA with a long viscoelastic plateau, Fig. S20.† The behaviour exhibited in the peel testing could be due to the poorer film formation ability of the latex which resulted in some cracks and bubbles in the tapes for this particular sample.
Tack tests of the F_Xn_C4 series, Fig. 5c, show a similar trend to the shear and peel tests. In particular, F_X18_C4 shows a high Wadh, again with an extended viscoelastic plateau, Fig. S23.† Rheological frequency sweeps were used to understand more about the adhesive behaviour, Fig. S30.† A simple comparison of the storage modulus, G′, at 1 Hz (Table 2) demonstrates that as the crosslinker concentration increases the material has a more elastic character, agreeing with the findings from the adhesive tests. All G′ at this frequency are below 0.1 MPa, suggesting the possibility of good adhesive properties according to the Dahlquist criterion. A more in-depth analysis, using Chang windows can compare the behaviour of the adhesives at low frequency, typical of bonding, where a less viscous character is desirable and high frequency, typical of debonding, where a more elastic character is preferred.44Fig. 6, shows the Chang windows for this series. When the crosslinker concentration is 0 mol% the window is not near the window typically considered good for PSAs. This suggests the material may be good at bonding, low loss modulus, G′′, but very poor at debonding as G′ is so low at high frequencies. This is confirmed in all adhesive tests with very low values for F_X00_C4. As the crosslinker concentration increases, the windows move closer to that considered typically good. Only F_X18_C4 sits mostly within the Chang window for high shear PSAs, type 1, and is the only latex in this series to demonstrate good shear and tack properties.
Typically, higher molecular weights, around 300000–400000 g mol−1, show improved adhesive properties. Therefore, a series with lower CTA concentrations was produced, F_Xn_C2 where n = 0, 8, 11, 13 and 18. For n = 0, the weight average molecular weight, Mw, was increased from 5932 to 11713 g mol−1 from conventional SEC. This is still very low for a typical PSA. Triple detection to obtain the Mark–Houwink plots, again confirms that as the EGDMA concentration increases, α decreases, suggesting more branched architectures, Fig. 3b and c. Again, the kinetics and particle size evolution during the emulsion polymerization were similar, Fig. S3† and the Tg increases as the crosslinker concentration increases, Table 1. The gel content for these latexes remained around 1% when n ≤ 11, however higher concentrations result in slightly higher gel contents, 5%, Table 1. SEC analyses again did not show polymer material of high molar mass near the exclusion limit of the column, confirming none or low gel content. Where n ≤ 13, the lower CTA concentration resulted in improved Fpeel, Wshear and Wadh compared to the equivalent latex with higher CTA concentrations. This is likely due to the increase in average molecular weight. However, when n = 18, Fpeel and Wadh decrease, likely due to the increased Tg which results in poorer wetting of the substrate. This is also seen in the rheological data as G′ at low frequencies is much higher at this crosslinker concentration, Fig. 7 and S31.† However, the decrease in peel and tack is balanced by the significant increase in Wshear. The increased average molecular weight, due to the decreased crosslinker concentration, and branched architecture, results in more entanglement points in the material and thus a greater Wshear and Wadh.
A final series was synthesized, F_X13_Cn where n = 0.5, 1, 2 and 4, to investigate the effect of varying the CTA concentration. The crosslinker concentration was maintained at 13 mol% as in previous series this concentration gave the highest Fpeel and Wadh and moderate Wshear. Again, the kinetics and particle size evolution showed similar results, Fig. S4.† In this case, the decrease in CTA concentration increased Mw from 24567 to 70488 g mol−1 from conventional SEC analysis, Fig. 4a. The Mark–Houwink plots from triple detection SEC, Fig. 4b and c, show that as the concentration of CTA is decreased, α increases and the extent of branching decreases. Interestingly, the decrease in branching results in increased Fpeel and Wshear, Fig. 5d and e. This can be attributed to the increased average molecular weight, increasing entanglement points which improves cohesion. Interestingly, Wadh does not follow the same trend as Fpeel, which is commonly observed. Only F_X13_C2 gave a high tack adhesion energy, Fig. 5f. This is likely due to the rheological behaviour of the sample, Fig. S32.†Fpeel is influenced by two factors. One, G′ at 0.01 Hz, where in this case a higher value as the CTA concentration decreases relates to poorer wetting of the substrate which should result in lower Fpeel. The second factor, higher G′ and G′′ at high frequencies resulting in stronger debonding resistance, is thus likely why Fpeel increases as CTA concentration decreases. However, the bonding frequency for tack is higher than that for peel (generally around 1 Hz) and this may explain the difference. G′ at this frequency does not follow the same trend as it does at the lower frequency and the highest tack, F_X13_C2 is likely due to the delicate balance of a lower G′ at 1 Hz which improves wetting (compared to n = 0.5 and 1) but also a longer rubbery plateau (compared to n = 4) which promotes fibril formation. Wshear is most affected by G′ at low frequencies and the difference between G′ at low and high frequencies. In this case, G′ is increasing at low frequencies and the difference between the low and high frequency values is decreasing which means using lower CTA concentrations extends the rubbery plateau which is indicative of a higher degree of entanglement due to the increased molecular weight and branching.
The most promising results from these investigations are the latexes F_X18_C4 and F_X13_C2 which show high Fpeel and Wadh and moderate Wshear. They have maintained a delicate balance of viscoelastic properties allowing for good wetting of the substrate and high cohesive forces. These results can be compared to typical PSA products on the market such as masking tape, Fpeel = 2.64 ± 0.15 N/20 mm, and cellotape, Fpeel = 8.60 ± 0.57 N/18 mm, where the experimental set-up was identical. Both the best latexes presented here surpass Fpeel for these commercial products. Additional comparisons to other literature which focuses on replacing petroleum-based chemicals with bio-based monomers reveals that they have excellent promise as PSA formulations. Many researchers have reported the use of oleo-chemicals (functionalized plant oils) to replace some parts of a typical acrylate PSA formulation. For instance, epoxidized soybean oils have been used alongside acrylic PSAs to produce UV-curable PSAs which typically exhibit Fpeel's of around 2 N/25 mm and Wshear of around 0.2 MPa.48,49 Another example uses castor oil-based urethane acrylate oligomers with an acrylic copolymer which can in one case exhibit a high peel force of 35 N/25 mm after crosslinking between the isocyanate and hydroxy groups.50,51 Other researchers who have focused on replacing the petroleum-based acrylates typically used for PSAs with bio-based acrylates such as meth(acrylate) derivatives of terpenoids like tetrahydrogeraniol acrylate and cyclademol acrylate have demonstrated maximum Fpeel values around 6.5 N/25 mm and Wadh of approximately 70 J m−1.52,53 Finally, Badía and coworkers have also produced latexes using 2-OA with much higher gel contents and molecular weights and reported maximum peel values of 8 N/25 mm and Wadh typically between 100 and 200 J m−2.21,24 These results are all highly dependent on the substrates used and the testing set-up, particularly the speed of the test so they should only be compared lightly. However, they demonstrate that the latexes presented in this work that have low gel content and a branched polymer chain architecture are promising PSA candidates.
4. Conclusions
This work demonstrates the ability to fabricate a range of viscoelastic acrylic polymer dispersions with branched polymer chain architecture by semi-batch emulsion polymerization that can exhibit a variety of adhesive properties for casted polymer films depending on the desired application. In particular, it demonstrates that high molecular weights and high gel contents are not necessary to impart particular adhesive characteristics and that these characteristics can be obtained by carefully tuning polymer chain architecture by varying the crosslinker and chain transfer agent concentrations. Our results indicate that the synthetic window for water-based polymer dispersion PSAs is broader, with a branched polymer chain architecture unlocking new options in PSA design. These findings, together with the (near future) non-petrochemical derived availability of the monomer and chain transfer agent feedstocks, will facilitate opportunities in the transition towards a more sustainable and circular PSA industry.Author contributions
Conceptualization (SAFB), Data curation (EMB, SAFB), Formal analysis (EMB, SAFB), Funding acquisition (SAFB), Investigation (EMB), Methodology (EMB, SAFB), Project administration (EMB, SAFB), Supervision (SAFB), Validation (EMB, SAFB), Visualization (EMB), Writing – original draft (EMB, SAFB), Writing – review & editing (EMB, SAFB).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Polymer Characterisation RTP, University of Warwick, for providing the use of the following equipment: Metler Toledo STARe instrument DSC, Anton Paar Litesizer 500, Agilent Infinity II MDS, Krüss DSA 100 and a Shimadzu EZ-LX universal testing machine. We thank Covestro (Netherlands) B.V. for providing the 2-OA, and Rowan Donovan and Xiaming Wu for pre-screening some 2-OA emulsion polymerization recipes during their masters projects.References
- C. Creton, MRS Bull., 2003, 28, 434–439 CrossRef CAS.
- N. Ballard, Polym. Int., 2023, 73, 75–87 CrossRef.
- M. D. Gower and R. A. Shanks, Macromol. Chem. Phys., 2005, 206, 1015–1027 CrossRef CAS.
- P. K. Dhal, A. Deshpande and G. Babu, Polymer, 1982, 23, 937–939 CrossRef CAS.
- R. S. Gurney, A. Morse, E. Siband, D. Dupin, S. P. Armes and J. L. Keddie, J. Colloid Interface Sci., 2015, 448, 8–16 CrossRef CAS.
- M. Heydari, F. Sharif and M. Ebrahimi, RSC Adv., 2021, 11, 20557–20569 RSC.
- B. Schiel-Bengelsdorf, J. Montoya, S. Linder and P. Dürre, Environ. Technol., 2013, 34, 1691–1710 CrossRef CAS.
- E. M. Brogden, P. F. Wilson, S. Hindmarsh, I. Hands-Portman, A. Unsworth, E. Liarou and S. A. F. Bon, RSC Appl. Polym., 2024, 2, 248–261 RSC.
- P. Schmidt, M. Blessing, M. Rageot, R. Iovita, J. Pfleging, K. G. Nickel, L. Righetti and C. Tennie, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 17707–17711 CrossRef CAS.
- L. Wadley, T. Hodgskiss and M. Grant, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 9590–9594 CrossRef CAS.
- Handbook of adhesive technology, ed. A. Pizzi, Dekker, New York Basel, 2nd edn, 2003 Search PubMed.
- D. Buttrey and M. Rogers, J. Coated Fabr., 1975, 5, 59–73 CrossRef.
- A. Pizzi, J. Macromol. Sci.-Rev. Macromol. Chem., 1980, C18(2), 247–315 CrossRef.
- S. Maiti, S. S. Ray and A. K. Kundu, Prog. Polym. Sci., 1989, 14, 297–338 CrossRef CAS.
- F. D. Blake, US Pat, US4413080A, 3M Co, 1983 Search PubMed.
- S. D. Tobing and A. Klein, J. Appl. Polym. Sci., 2001, 79, 2230–2244 CrossRef CAS.
- M. A. Droesbeke, R. Aksakal, A. Simula, J. M. Asua and F. E. Du Prez, Prog. Polym. Sci., 2021, 117, 101396 CrossRef CAS.
- S. Molina-Gutiérrez, W. S. J. Li, R. Perrin, V. Ladmiral, R. Bongiovanni, S. Caillol and P. Lacroix-Desmazes, Biomacromolecules, 2020, 21, 4514–4521 CrossRef.
- J. L. Colby, T. A. Clem, T. D. Spawn, A. E. Hutt and W. T. Teply, EU. Pat, EP2970094A1, 3M Innovative Properties Co, 2018 Search PubMed.
- A. Riondel, C. Graire, M. Esch and R. Linemann, US Pat, US20140371482A1, Arkena France SA, 2015 Search PubMed.
- A. Badía, J. I. Santos, A. Agirre, M. J. Barandiaran and J. R. Leiza, ACS Sustainable Chem. Eng., 2019, 7, 19122–19130 CrossRef.
- A. Badía, A. Agirre, M. J. Barandiaran and J. R. Leiza, Biomacromolecules, 2020, 21, 4522–4531 CrossRef.
- A. Badía, M. Kastelijn, J. Scheerder and J. R. Leiza, Prog. Org. Coat., 2020, 147, 105708 CrossRef.
- A. Badía, J. Movellan, M. J. Barandiaran and J. R. Leiza, Ind. Eng. Chem. Res., 2018, 57, 14509–14516 CrossRef.
- L. Zhang, Y. Cao, L. Wang, L. Shao and Y. Bai, J. Appl. Polym. Sci., 2016, 133, 42886 CrossRef.
- L. Chen, Z. Bao, Z. Fu and W. Li, Colloid J., 2015, 77, 374–381 CrossRef CAS.
- J. Knebel and D. Saal, US Pat, US6479696B1, Roehm GmbH & Co KG, 2002 Search PubMed.
- J.-H. Lee, T.-H. Lee, K.-S. Shim, J.-W. Park, H.-J. Kim, Y. Kim and S. Jung, Int. J. Adhes. Adhes., 2017, 74, 137–143 CrossRef CAS.
- J. Chauvet, J. M. Asua and J. R. Leiza, Polymer, 2005, 46, 9555–9561 CrossRef CAS.
- Z. Aguirreurreta, J.-A. Dimmer, I. Willerich, J. R. Leiza and J. C. De La Cal, Int. J. Adhes. Adhes., 2016, 70, 287–296 CrossRef CAS.
- A. Lopez, E. Degrandi, E. Canetta, J. L. Keddie, C. Creton and J. M. Asua, Polymer, 2011, 52, 3021–3030 CrossRef CAS.
- C. Hirth, M. Gerst, M. Rückel, D. Botin, M. Heinz, J. C. Namyslo, J. Ruan, J. Adams and D. Johannsmann, Macromolecules, 2022, 55, 6067–6075 CrossRef CAS.
- T. Li, B. Zhang, S. Jiang, X. Zhou, G. Du, Z. Wu, M. Cao and L. Yang, ACS Sustainable Chem. Eng., 2020, 8, 5209–5216 CrossRef CAS.
- I. González, J. R. Leiza and J. M. Asua, Macromolecules, 2006, 39, 5015–5020 CrossRef.
- X. Callies, O. Herscher, C. Fonteneau, A. Robert, S. Pensec, L. Bouteiller, G. Ducouret and C. Creton, ACS Appl. Mater. Interfaces, 2016, 8, 33307–33315 CrossRef CAS.
- C. Fang and J. Xue, J. Adhes. Sci. Technol., 2022, 36, 1041–1059 CrossRef CAS.
- A. Agirre, J. Nase, E. Degrandi, C. Creton and J. M. Asua, Macromolecules, 2010, 43, 8924–8932 CrossRef CAS.
- L. Qie and M. A. Dubé, Macromol. React. Eng., 2011, 5, 117–128 CrossRef CAS.
- S. Ren and M. A. Dubé, Int. J. Adhes. Adhes., 2017, 75, 132–138 CrossRef CAS.
- M. E. Treviño and M. A. Dubé, Macromol. React. Eng., 2013, 7, 484–492 CrossRef.
- Y. Liu, J. C. Haley, K. Deng, W. Lau and M. A. Winnik, Macromolecules, 2008, 41, 4220–4225 CrossRef CAS.
- S. Kim, R. A. R. Bowen and R. N. Zare, ACS Appl. Mater. Interfaces, 2015, 7, 1925–1931 CrossRef CAS.
- F. Deplace, C. Carelli, S. Mariot, H. Retsos, A. Chateauminois, K. Ouzineb and C. Creton, J. Adhes., 2009, 85, 18–54 CrossRef CAS.
- E. P. Chang, J. Adhes., 1991, 34, 189–200 CrossRef CAS.
- T. Junkers, M. Schneider-Baumann, S. S. P. Koo, P. Castignolles and C. Barner-Kowollik, Macromolecules, 2010, 43, 10427–10434 CrossRef CAS.
- L. Couvreur, G. Piteau, P. Castignolles, M. Tonge, B. Coutin, B. Charleux and J.-P. Vairon, Macromol. Symp., 2001, 174, 197–208 CrossRef CAS.
- R. A. Cockburn, T. F. McKenna and R. A. Hutchinson, Macromol. Chem. Phys., 2010, 211, 501–509 CrossRef CAS.
- T. H. Lee, Y. I. Park, S.-H. Lee, J. Shin, S. M. Noh and J. C. Kim, Appl. Surf. Sci., 2019, 476, 276–282 CrossRef CAS.
- B. K. Ahn, S. Kraft, D. Wang and X. S. Sun, Biomacromolecules, 2011, 12, 1839–1843 CrossRef CAS.
- B. Sun, H. Wang, Y. Fan, X. Chu, S. Liu, S. Zhao and M. Zhao, Prog. Org. Coat., 2022, 163, 106680 CrossRef CAS.
- P. Hao, B. Sun, X. Chu, Y. Sun, X. Xing, S. Liu, E. Tang and X. Xu, J. Adhes. Sci. Technol., 2020, 34, 2499–2509 CrossRef CAS.
- S. Noppalit, A. Simula, L. Billon and J. M. Asua, ACS Sustainable Chem. Eng., 2019, 7, 17990–17998 CrossRef CAS.
- M. A. Droesbeke, A. Simula, J. M. Asua and F. E. Du Prez, Green Chem., 2020, 22, 4561–4569 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Exact monomer compositions for each latex (Table S1), exact emulsion polymerization recipes (Tables S2), kinetics and particle size evolutions during latex synthesis (Fig. S1–S4), DSC thermograms and analysis (Fig. S5–S8 and Table S3), SEC molecular weight distribution evolution during latex synthesis (Fig. S9–S12), contact angle measurements on chemically modified PET (Fig. S13), details on the set up of film substrate application (Fig. S14), peel (Fig. S15) and shear (Fig. S19) tests, peel adhesion data (Fig. S16–S18), shear strength stress–strain curves (Fig. S20–S22), tack adhesion energy stress–strain curves (Fig. S23–S26), rheological amplitude sweep data (Fig. S27–S29), rheological frequency sweep data (Fig. S30–S32), molecular weight distributions of the surfactants used (Fig. S33). See DOI: https://doi.org/10.1039/d4py00522h |
| This journal is © The Royal Society of Chemistry 2024 |