
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-Heterocyclic carbene-initiated epoxide/anhydride ring-opening copolymerization: effective and selective organoinitiators for the production of various polyesters†
Gaël
Printz
a,
Redoy Gazi
Shuvo
a,
Antoine
Schweizer
a,
Dmytro
Ryzhakov
b,
Christophe
Gourlaouen
 c,
Lielou
Lantigner
b,
Béatrice
Jacques
a,
Samir
Messaoudi
b,
Franck
Le Bideau
*b and
Samuel
Dagorne
*a
c,
Lielou
Lantigner
b,
Béatrice
Jacques
a,
Samir
Messaoudi
b,
Franck
Le Bideau
*b and
Samuel
Dagorne
*a
aInstitut de Chimie, CNRS – Université de Strasbourg, Strasbourg, France. E-mail: dagorne@unistra.fr
bUniversité Paris-Saclay, CNRS, BioCIS 91400, Orsay, France. E-mail: franck.lebideau@universite-paris-saclay.fr
cLaboratoire de Modélisation et Simulations Moléculaires, Université de Strasbourg, UMR 7140, F-67000 Strasbourg, France
First published on 10th September 2024
Abstract
The present report describes the first use of unprotected and “free” N-heterocyclic carbenes (NHCs), such as IMes, IPr and air-stable Cl-IPr carbenes, as single component initiators for the alternating ring-opening co-polymerization (ROCOP) of cyclic anhydrides (phthalic anhydride, PA; succinic anhydride, SA) and various epoxides to efficiently and selectively access the corresponding polyesters in a well-defined manner and as metal-free materials. In the case of ROCOP runs with PA as the anhydride source, control experiments are in line with an initiation via ring-opening of PA by the NHC moiety, as established with the synthesis and structural characterization of the IMes-PA ring-opened product 1. The present NHC systems were further exploited as ROCOP initiators of PA and bio-sourced epoxides such as eugenyl glycidyl ether (EGE) and safrole glycidylether (SO) yielding the production of regioregular p(PA-alt-EGE) polyester, as well as p(PA-alt-SO), a novel polyester material.
Introduction
The production of sustainable materials is currently of key importance for society for replacement of petroleum-based materials.1 In this regard, aliphatic polyesters are promising polymers as they may incorporate sustainable and/or bio-sourced monomers and be readily degraded to non-toxic and re-usable components.2 Industrially, polyesters are typically produced by polycondensation of diacids and diols and ring-opening polymerization (ROP) of cyclic esters. For instance, the production of a biodegradable polyester of current importance like polylactide (PLA) is industrially performed by ring-opening polymerization (ROP) of lactide using a Sn(II)-based initiator.3Over the past fifteen years, the alternating ring-opening co-polymerization (ROCOP) of epoxides and anhydrides has become a powerful approach to access structurally diverse polyesters in a controlled manner using a variety of well-defined metal complexes as ROCOP initiators.4 Notably, Co-, Cr-, Zn- and Al-based complexes when operating in the presence of a co-catalyst (typically a halide salt) were shown to selectively produce polyesters via ROCOP.4a,5 More recently, due to the potential toxicity of some metal salts,6 organocatalyzed and/or initiated epoxide/anhydride ROCOP has emerged as an effective and attractive tool to access well-defined metal-free polyesters materials.7 Though the field is only emerging, a number of two-component catalytic systems, most notably those involving boron-based Lewis acidic organocatalysts in combination with a halide source acting as a nucleophile/co-catalyst, were demonstrated to display high activity and ester selectivity in epoxide/anhydride ROCOP.8 Other two-component organocatalysts, including several ingeniously designed bifunctional borane-ammonium catalysts, organic superbases combined with alcohol/Brønsted acid sources, were also shown to mediate epoxide/anhydride ROCOP.9 In contrast, ROCOPs initiated by a single component/nucleophile, most frequently halide salts and various amines,7,10 remain less studied since typically thought to exhibit lower catalytic performance. In the area, a recent report showed that carbon(0) species of the type carbodicarbene may efficiently initiate epoxide/anhydride ROCOP to produce polyesters.11
Due to their exceptional steric/electronic properties and ready synthesis,12 N-heterocyclic carbenes (NHCs) are ubiquitous organic initiators able to mediate various ROP processes. Thus, acting as strong nucleophiles, they are well-established ROP initiators of various cyclic esters, and efficiently mediate the homo-polymerization of epoxides.13 The use of NHCs as single component initiators of the alternating epoxides and anhydrides ROCOP remain little explored. In that regard, Buchmeiser showed that latent NHC-metal/NHC-CO2 adducts, which may be seen as “protected” NHC sources, are excellent epoxide/anhydride ROCOP initiators for a broad scope of monomers.14 Herein, we report that unprotected, “free” NHCs such as classical IMes, IPr and air-stable Cl-IPr carbenes (Fig. 1) may act as single component initiators for the effective and selective ROCOP of various epoxides (including bio-sourced derivatives) and anhydrides.
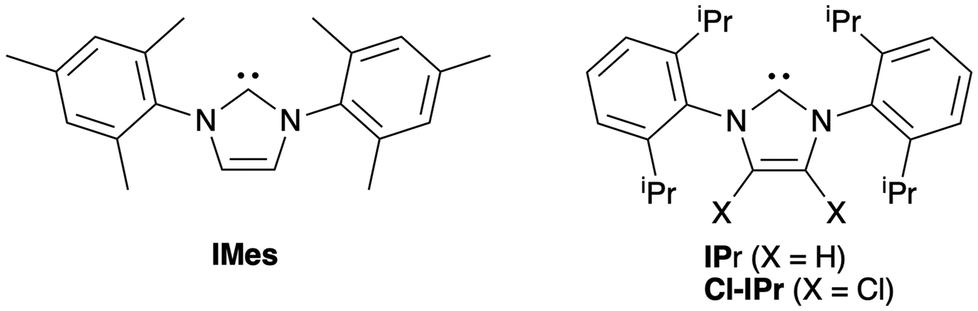 | ||
| Fig. 1 NHC initiators used for epoxide/anhydride ROCOP. | ||
Results and discussion
ROCOP tests were first performed with CHO and PA as monomers using IPr and IMes carbenes as initiators, and all results are compiled in Table 1. For initial tests, a low monomer feed was selected (60/70/1 PA/CHO/NHC ratio) to first probe/validate the possibility of NHC-mediated ROCOP. Thus, carbene IPr was satisfyingly found to effectively mediate PA/CHO ROCOP to afford quantitatively and selectively well-defined and monodisperse p(PA-CHO) polyester after 16 h at 90 °C (Mn = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 g mol−1, Đ = 1.05, >98% ester linkage; entry 1, Table 1).
200 g mol−1, Đ = 1.05, >98% ester linkage; entry 1, Table 1).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | NHC | epoxide | PA/epoxide/NHC ratio | Time (h) | Conv PAb (%) | Ester linkagec (%) |
M
n
d (kg mol−1) |
Đ |
| a Conditions (unless indicated otherwise): neat conditions (no solvent), 90 °C. b Determined by 1H NMR of the crude polymer mixture (integration of the polyester signals relative to those of unreacted PA). c Determined from 1H NMR by integration of CH-ester signals (typically in the 4.0–5.5 ppm region) vs. CH-O polyether resonances (typically in the 3.0–3.5 ppm region). d Determined by GPC analysis (THF, room temperature) using polystyrene standards. e Crude IPr was generated by reaction of [IPr-H][BF4] with 0.9 equiv. K[N(SiMe3)2] and the crude mixture was only washed with pentane prior to ROCOP. f ROCOP performed at 70 °C. g The GPC trace is shouldered to the lower masses, possibly due to the presence of impurities leading to transesterification side reactions. h Performed under air (closed vial). | ||||||||
| 1 | IPr | CHO | 60:70:1 |
16 | 95 | >98 | 10.2 | 1.05 |
| 2 | IPr | CHO | 60:70:1 |
6 | 95 | >98 | 13.4 | 1.13 |
| 3 | IMes | CHO | 60:70:1 |
6 | 80 | >98 | 13.0 | 1.19 |
| 4 | IPr (crude)e | CHO | 60:70:1 |
6 | 73 | >98 | 10.1 | 1.23 |
| 5 | IPrf | CHO | 60:70:1 |
6 | 0 | — | — | — |
| 6 | IMesf | CHO | 60:70:1 |
6 | 0 | — | — | — |
| 7 | [PPN]Cl | CHO | 60:70:1 |
6 | <20 | — | — | — |
| 8 | IPr | CHO | 250:300:1 |
4 | 72 | >98 | 26.7 | 1.11 |
| 9 | IPr | CHO | 250:300:1 |
6 | 100 | >98 | 32.0 | 1.12 |
| 10 | Cl-IPr | CHO | 250:300:1 |
6 | 100 | >98 | 25.2 | 1.20 |
| 11 | Cl-IPrh | CHO | 250:300:1 |
16 | 53 | >98 | 13.4 | 1.37 |
| 12 | IMes-PA (1) | CHO | 250:300:1 |
16 | 97 | >98 | 29.7g | 1.17g |
| 13 | IPr | PO | 250:300:1 |
16 | 80 | >98 | 35.0 | 1.21 |
| 14 | IMes | PO | 250:300:1 |
16 | 83 | >98 | 33.0 | 1.21 |
| 15 | Cl-IPr | PO | 250:300:1 |
16 | 89 | >98 | 29.2 | 1.22 |

Optimization of the reaction showed that the ROCOP is complete within 6 h at 90 °C (entry 2, Table 1). Polymerization also proceeds efficiently when the less sterically hindered IMes carbene was used as initiator leading to the selective formation of p(PA-CHO) polyester material (entry 3, Table 1). It may also be noted that in situ generated IPr (from deprotonation of salt [IPr-H][BF4] by K[N(SiMe3)2], with a subsequent pentane wash) also mediates PA/CHO ROCOP with the selective production of polyester, though with lower activity relative to pure IPr (entry 2 vs. 4, Table 1). For both IPr and IMes, lowering the reaction temperature to 70 °C with an identical monomer/initiator feed ratio led to no reaction after 6 h (entries 5 and 6, Table 1). Overall, these initial results show that carbenes IMes and IPr may behave as suitable and effective nucleophiles for epoxide/anhydride ROCOP for polyester production. As a comparison, the use of a halide salt such as [PPN]Cl as an initiator under identical ROCOP conditions (60/70/1 PA/CHO/[PPN]Cl ratio, 90 °C, 16 h) only allowed low monomer conversion (<20%) and no well-defined p(PA-CHO) material could be isolated (entry 7, Table 1).
The selective formation of polyesters in the NHC-mediated ROCOP was confirmed by NMR data and mass spectrometric MALDI-TOF analysis. 1H and 13C NMR data of the crude and isolated materials agree in all cases with the exclusive presence of ester linkages, and thus the absence of any polyether material that would arise from CHO homopolymerization by the NHC initiator. In addition, 1H DOSY NMR data of the material produced from a 60/70/1 PA/CHO/IPr run only display signals correlating with one diffusion coefficient value, in line with the presence of one monodisperse copolymer (see ESI†). MALDI-TOF data for the ROCOP runs with IMes and IPr both display peaks alternatively spaced by CHO (98 u.a.) and PA (148 u.a.), consistent with an alternating p(PA-alt-CHO) polyester, as shown in Fig. 2 in the case of a PA/CHO ROCOP initiated by IPr. These data also agree with NHC and OH end groups at the α and ω chain ends, respectively, confirming that the NHC moiety acts as the actual ROCOP initiator and in line with a zwitterionic polymer chain growth. Taking the example of a PA/CHO ROCOP initiated by IPr (Fig. 2), a closer inspection of the MALDI-TOF peaks sequence (and associated mass values) is consistent with a polymer chain of the type [IPr-PA-p(CHO-alt-PA)-H]+, in line with a ring-opening of PA by IPr as the initiating step. ROCOP initiation through ring-opening of PA (instead of the epoxide source) contrasts with documented and well-established ROCOP processes initiated by various nucleophiles, most notably halide salts, for which the halide source typically ring-opens the epoxide as the initiating step.7,10,15 To gain additional insight, stoichiometric control experiments were performed. Thus, the NMR scale reaction of one equiv. of IMes with a 1/1 PA/CHO mixture (CD2Cl2, RT) led to the immediate (<15 min) and quantitative formation of zwitterion IMes-PA (1, Scheme 1), arising from a selective nucleophilic ring-opening of PA by IMes, along with unreacted CHO, according to 1H and 13C NMR data. Species 1 was also prepared through an independent synthesis by a 1/1 PA/IMes reaction leading to its isolation in 79% yield.
 | ||
| Fig. 2 MALDI-TOF mass spectrum (zoom of a region) for isolated poly(PA-alt-CHO) formed from the ROCOP of PA and CHO initiated by IPr. Conditions: 60/70/1 (PA/CHO/IPr), no solvent, 90 °C, 16 h (95% conversion of PA). See the ESI† for the full MALDI-TOF spectrum. | ||
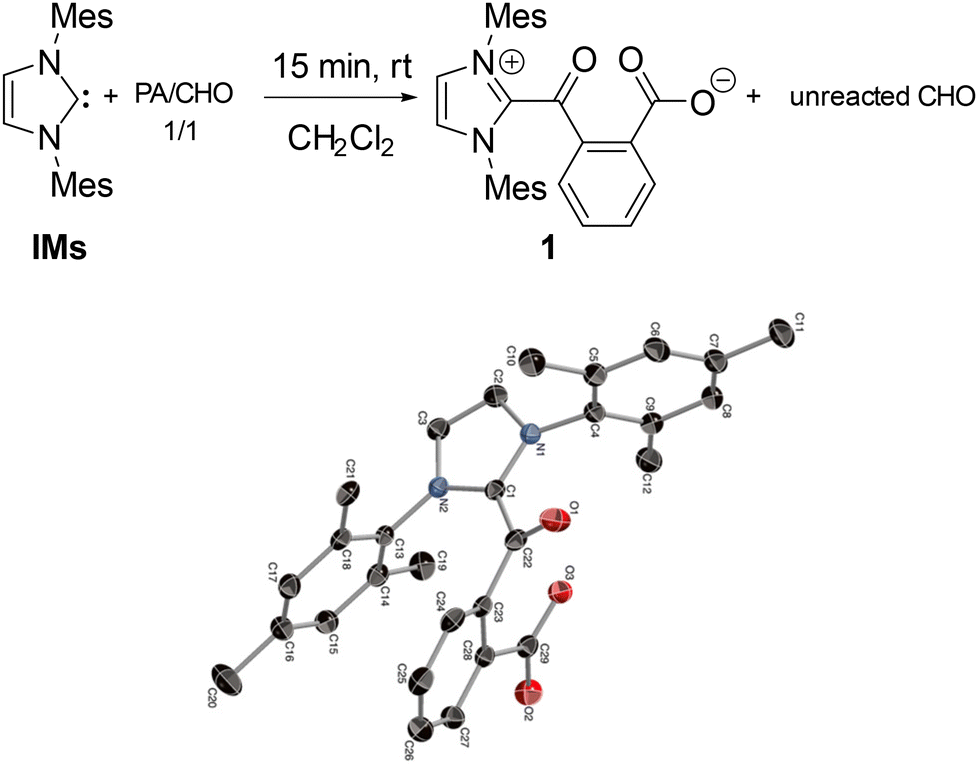 | ||
| Scheme 1 Reaction of carbene IMes with a 1/1 PA/CHO mixture and molecular structure (ORTEP view) of ring-opened product 1. | ||
In addition, the molecular structure of 1 was further established through X-ray crystallographic analysis (Scheme 1), confirming the ring-opening reaction and the zwitterionic nature of 1. Though stable enough to be isolated, species 1 exhibits a limited stability in CD2Cl2 (room temperature) overtime and slowly decomposes to unidentified products over the course of several days (t1/2 ≈ 4 days) as monitored by 1H NMR. An identical ring opening reactivity was observed upon mixing IPr or IPr-Cl with 1 equiv. of PA, with the fast and complete formation of the corresponding ring opening products (CD2Cl2, 15 min, RT) according to 1H NMR data, thus indicating that all three studied NHCs allow fast and complete PA ring opening. However, in the case of the more sterically bulky IPr and IPr-Cl, the ring opened products are highly unstable (CD2Cl2, room temperature) and decompose within a few hours to unknown species, precluding their isolation in a pure form.
DFT calculations were also performed to rationalize the observed higher reactivity of IMes with PA (vs. CHO) leading to 1 (with A, B, C, D and E as models of IMes, PA, CHO, 1, and IMes-CHO ring-opened product, respectively; Fig. 3). As depicted in Fig. 3, the nucleophilic attack by A of a PA C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety proceeds via a concerted low energy transition state (TS) and thus a low energy barrier (ΔΔG = 5.1 kcal mol−1) to afford the ring-opened product D, which is in line with the experimentally observed fast formation of 1 at room temperature. In contrast, the energy barrier for a nucleophilic attack of model carbene A on CHO model C to form E was computed to occur with a much higher energy transition state (TS′, ΔG = 26.1 kcal mol−1), rationalizing the exclusive ring-opening of PA by IMes at room temperature.
O moiety proceeds via a concerted low energy transition state (TS) and thus a low energy barrier (ΔΔG = 5.1 kcal mol−1) to afford the ring-opened product D, which is in line with the experimentally observed fast formation of 1 at room temperature. In contrast, the energy barrier for a nucleophilic attack of model carbene A on CHO model C to form E was computed to occur with a much higher energy transition state (TS′, ΔG = 26.1 kcal mol−1), rationalizing the exclusive ring-opening of PA by IMes at room temperature.
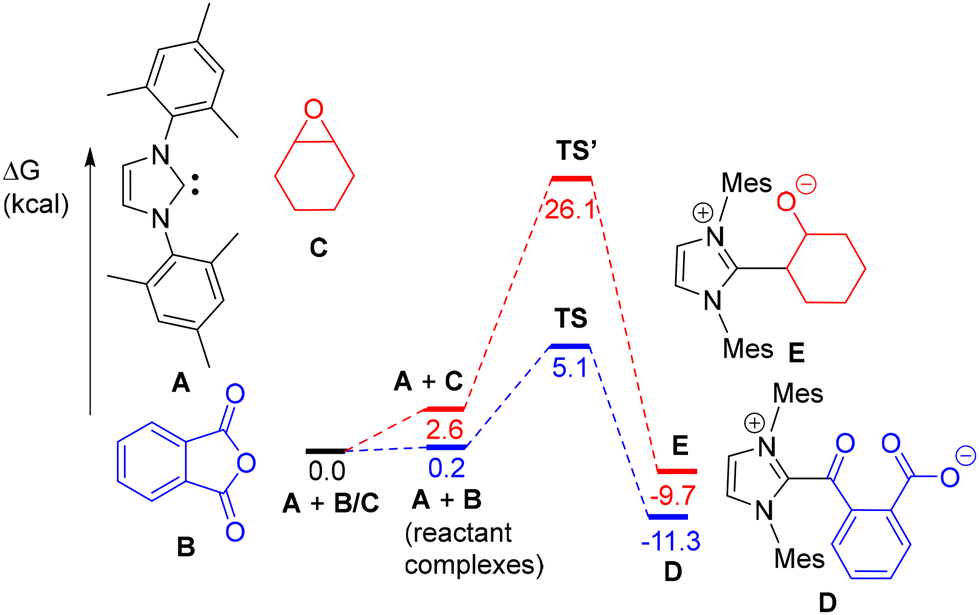 | ||
| Fig. 3 DFT-estimated reactions (GAUSSIAN 16/ωB97XD) of IMes model A with PA (B) and CHO (C) to form the ring-opened products D and E, respectively. | ||
Next, we further studied the present NHC-initiated ROCOP to probe its scope/performances (entries 8–15, Table 1). Increasing monomer feed to a 250/300/1 PA/CHO/NHC ratio did not alter ROCOP performances, leading to the quantitative and selective formation of p(PA-alt-CHO) with a higher number average molecular mass (Mn = 32.0 kg mol−1, entry 9, Table 1). Air-stable Cl-IPr (less nucleophilic than IPr and IMes) may also effectively be used as ROCOP initiator with the quantitative production of p(PA-alt-CHO) after 16 h at 90 °C (entry 10, Table 1). When performed under air (16 h, 90 °C), ROCOP with IPr-Cl also selectively produced polyester, albeit with a significantly lower catalytic activity than under inert atmosphere (entry 10 vs. 11, Table 1). The ring-opened product 1 logically performs similarly to the NHC systems with the isolation of a polyester material (Mn = 29.1 kg mol−1, Đ = 1.17; entry 12, Table 1). In addition, using PO as an epoxide source, all three NHCs initiators also mediate PA/PO ROCOP (250/300/1, 16 h, 90 °C), with high PA conversion (80 to 89%) to exclusively afford regioirregular p(PA-alt-PO) polyesters (Mn = 29.2–35 kg mol−1, Đ = 1.21–1.22; entries 13–15, Table 1).
The present NHC systems were also tested in SA/CHO (SA: succinic anhydride) and SA/PO ROCOP and all results are compiled in Table S2 (ESI†). As deduced from GPC and NMR data, IMes, IPr and IPrCl carbenes polymerize a 60/70 or 250/300 SA/CHO mixture to produce p(SA-alt-CHO) polyester (Mn = 1.5 to 5.8 kg mol−1, Đ = 1.32 to 1.79) with a good ester linkage selectivity (80 to 88%) and high SA conversion (82 to 86%) after 16 h at 90 °C under bulk conditions. Thus, going from PA to SA as anhydride source decreased ester linkage selectivity and only afforded lower molecular weight p(SA-alt-CHO) materials, as frequently observed.4b,7 This can be ascribed to the lower reactivity of the less ring-strained SA (vs. PA), resulting in a decreased ROCOP activity as well as a possible competition with CHO homopolymerization (leading to polyether segments) and thus a decreased ester linkage selectivity. However, a SA/PO ROCOP test with IPr as initiator (250/300/1 SA/PO/IPr ratio) exclusively led the high yield production of well-defined p(SA-alt-PO) polyester (96% SA conversion, ester linkage >98%, Mn = 5.92 kg mol−1, Đ = 1.82). Since PO is a significantly less reactive monomer than CHO towards ring-opening due to lower ring strain, PO homopolymerization may not compete with the PO/SA ROCOP process, thus the observed higher ester linkage selectivity in PO/SA vs. CHO/SA ROCOP.
Last, to further expand the scope of the present NHC systems, carbene IPr was exploited as ROCOP initiator of PA and bio-sourced epoxides eugenyl glycidyl ether and safrole glycidylether (EGE, SO, Table 2). Epoxides EGE and SO, which are derived from natural products eugenol and safrole, respectively, were prepared according to literature procedures.16 As an initial run, IPr was found to polymerize a 70/85 PA/EGE mixture (90 °C, 5 h, 83% conv. PA) to selectively afford p(PA-alt-EGE) polyester with narrow dispersity (Mn = 12.1 kg mol−1, Đ = 1.20; entry 1, Table 2). Satisfyingly, the ROCOP also proceeds efficiently and with high selectivity upon increasing monomer loading to 250/300/1 (PA/EGE/IPr) but allows the production of a p(PA-alt-EGE) material with significantly higher Mn (Mn = 43.5 kg mol−1, Đ = 1.60). To our knowledge, polyester p(PA-alt-EGE) was only reported on one occasion yet using a rather toxic Cr(III)-based ROCOP catalyst, and was only characterized by 1H NMR.171H NMR data of crude and isolated p(PA-alt-EGE) agree with the exclusive formation of ester linkage in the material (see ESI†). The 13C NMR spectrum (Fig. 4) is consistent with a highly regioregular material with >95% head-to-tail epoxide sequence (H–T), by comparison with literature data.4b,17 In particular, the 13C NMR signals for the CO groups resonate as two sharp singulets (δ 166.9 and 166.7 ppm), in line with regioregular H–T sequences. Using safrole oxide as an epoxide source, the PA/SO ROCOP initiated by IPr (250/300/1 PA/SO/IPr ratio) also proceeds with high ester selectivity (90 °C, 16 h, 85% PA conversion) to afford p(PA-alt-SO) (Mn = 45.3 kg mol−1, Đ = 1.30), a novel polyester material. All 1D and 2D 1H and 13C NMR data for p(PA-alt-SO) are consistent with the exclusive presence of ester linkage (see ESI†), though 13C NMR data suggest a regioirregular material. Also, in line with GPC data, the 1H DOSY NMR spectrum of isolated p(PA-alt-SO) only displays one diffusion coefficient value, as expected for one type of (monodisperse) polymer chains (see ESI†). In addition, MALDI-TOF mass spectrometric data of a p(PA-alt-SO) sample with a shorter chain length (produced from a 20/25/1 PA/SO/IPr ROCOP run, 1 h, 90 °C, quantitative) agree with PA-SO sequences as indicated by the presence of mass peaks equally spaced by 326.3 u.a. (sum of PA and SO molecular weights). Mass values also match well with [IPr-(PA-SO)n-K]+ ions, in line with a IPr residue at the α chain-end of the copolymer chain, as expected. DSC data for the produced p(PA-alt-EGE) and p(PA-alt-SO) polymers agree with amorphous materials with Tg values of 44 and 48 °C, respectively (see ESI†).
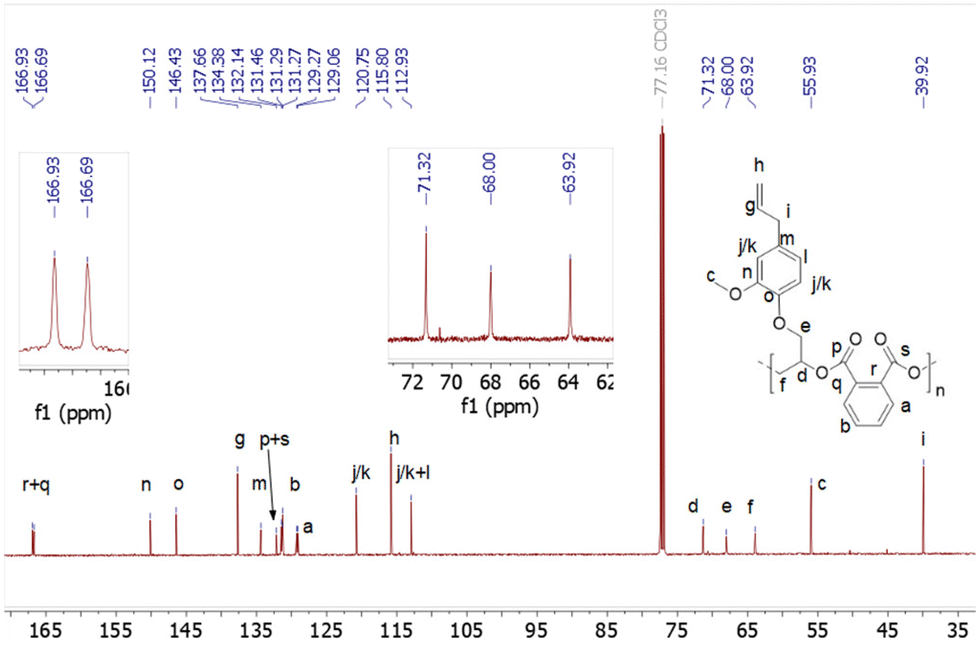 | ||
| Fig. 4 13C{1H} NMR spectrum (125 MHz, CDCl3) of isolated p(PA-alt-EGE) formed from the ROCOP of PA and EGE initiated by IPr. Conditions: 250/300/1 (PA/CHO/IPr), no solvent, 90 °C, 16 h (100% conversion of PA). | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Epoxide | PA/epoxide/IPr ratio | Time (h) | Conv PAb (%) | Ester linkagec (%) |
M
n
d (kg mol−1) |
Đ |
| a Conditions (unless indicated otherwise): neat conditions (no solvent), 90 °C. b Determined by 1H NMR of the crude polymer mixture (integration of the polyester signals relative to those of unreacted PA). c Determined from 1H NMR by integration of CH-ester signals (typically in the 4.0–5.5 ppm region) vs. CH–O polyether resonances (typically in the 3.0–3.5 ppm region). d Determined by GPC analysis (THF, room temperature) using polystyrene standards. | |||||||
| 1 | EGE | 70:85:1 |
5 | 83 | >98 | 12.1 | 1.20 |
| 2 | EGE | 250:300:1 |
16 | >98 | >98 | 43.5 | 1.60 |
| 3 | SO | 250:300:1 |
16 | 85 | >98 | 45.3 | 1.30 |
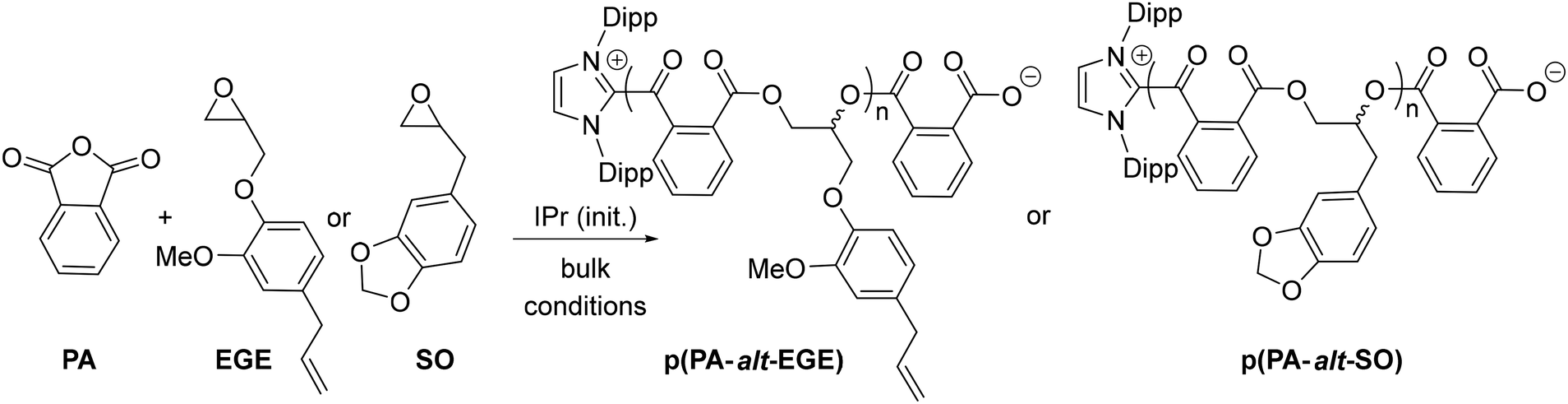
Conclusions
In conclusion, we have shown that widespread and readily available NHCs acting as single component initiators may effectively and selectively promote the alternating epoxide/anhydride ROCOP to afford well-defined polyesters, with, in all cases, a high ester linkage selectivity (from 85% to quantitative). Control experiments of the PA/CHO system in the presence of IMes, IPr and IPr-Cl suggest a fast and complete initiation proceeding via selective ring-opening of the anhydride by the NHC moiety, as confirmed by the isolation and structure determination of species 1. These simple organoinitiators were also further exploited to access polyesters materials p(PA-alt-EGE) and p(PA-alt-SO) of potential interest through ROCOP reactions of bio-sourced EGE and SO with PA. Such NHC-mediated ROCOP process allows the efficient production of various polyesters, included bio-sourced, using metal-free, non-toxic and inexpensive initiators.Author contributions
Gaël Printz, Redoy Gazi Shuvo, Antoine Schweizer: polymerization, formal analysis; Dmytro Ryzhakov, Lielou Lantigner: monomer synthesis; formal analysis. Christophe Gourlaouen: DFT calculations and analysis. Béatrice Jacques, Samir Messaoudi, Franck Le Bideau, Samuel Dagorne: supervision, conceptualization, funding acquisition, data curation; formal analysis, writing – review & editing.Data availability
The data that support the findings of this study are available in the ESI† of this article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The present work was supported by the CNRS, Université Paris-Saclay, Université de Strasbourg, project ANR (CoCaBio ANR-19-CE07-0018). In particular, the ANR is gratefully acknowledged for a doctoral fellowship to G. P. and a post-doctoral fellowship to D. R.References
- (a) M. Rabnawaz, I. Wyman, R. Auras and S. Cheng, Green Chem., 2017, 19, 4737 RSC; (b) D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733 CrossRef CAS.
- J.-G. Rosenboom, R. Langer and G. Traverso, Nat. Rev. Mater., 2022, 7, 117 CrossRef.
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841 CrossRef CAS.
- (a) R. C. Jeske, A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 11330 CrossRef CAS; (b) J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167 CrossRef CAS.
- (a) S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459 RSC; (b) J. A. Garden, P. K. Saini and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 15078 CrossRef CAS; (c) L.-F. Hu, C.-J. Zhang, H.-L. Wu, J.-L. Yang, B. Liu, H.-Y. Duan and X.-H. Zhang, Macromolecules, 2018, 51, 3126 CrossRef CAS; (d) F. Santulli, I. D'Auria, L. Boggioni, S. Losio, M. Proverbio, C. Costabile and M. Mazzeo, Organometallics, 2020, 39, 1213 CrossRef CAS; (e) D. Wannipurage, S. D'Aniello, D. Pappalardo, L. W. Kulathungage, C. L. Ward, D. P. Anderson, S. Groysman and M. Mazzeo, Dalton Trans., 2023, 52, 8077 RSC.
- (a) S. V. Verstraeten, L. Aimo and P. I. Oteiza, Arch. Toxicol., 2008, 82, 789 CrossRef CAS PubMed; (b) H. Kozlowski, P. Kolkowska, J. Watly, K. Krzywoszynska and S. Potocki, Curr. Med. Chem., 2014, 21, 3721 CrossRef CAS.
- D. Ryzhakov, G. Printz, B. Jacques, S. Messaoudi, F. Dumas, S. Dagorne and F. Le Bideau, Polym. Chem., 2021, 12, 2932 RSC.
- (a) D. Zhang, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, J. Am. Chem. Soc., 2016, 138, 11117 CrossRef CAS; (b) C. Zhang, X. Geng, X. Zhang, Y. Gnanou and X. Feng, Prog. Polym. Sci., 2023, 136, 101644 CrossRef CAS; (c) S. Lee, W. H. Jung, M. Kim, D. J. Ahn and B.-S. Kim, ACS Macro Lett., 2023, 12, 590 CrossRef CAS.
- (a) M. N. D. Haslewood, T. J. Farmer and M. North, J. Polym. Sci., 2023, 61, 311 CrossRef CAS; (b) F. Werlinger, R. Caprile, V. Cárdenas-Toledo, B. Tarraff, Á. Mesías-Salazar, R. S. Rojas, J. Martínez, O. S. Trofymchuk and M. E. Flores, ACS Omega, 2023, 8, 21540 CrossRef CAS; (c) M. D. C. L. Cheng-Tan, A. N. Nguyen, C. T. Gordon, Z. A. Wood, Y. Manjarrez and M. E. Fieser, ACS Sustainable Chem. Eng., 2024, 12, 7246 CrossRef CAS; (d) D. Merckle, O. King and A. C. Weems, ACS Sustainable Chem. Eng., 2023, 11, 2219 CrossRef CAS; (e) J. Zhang, L. Wang, S. Liu and Z. Li, J. Polym. Sci., 2020, 58, 803 CrossRef CAS; (f) H. Li, J. Zhao and G. Zhang, ACS Macro Lett., 2017, 6, 1094 CrossRef CAS.
- Z. Hošťálek, O. Trhlíková, Z. Walterová, T. Martinez, F. Peruch, H. Cramail and J. Merna, Eur. Polym. J., 2017, 88, 433 CrossRef.
- S. Y. Jeong, M. Kim, K. Kim, H. Kim, Y. Kim, J. Seo, E. Lee and K. Son, Eur. Polym. J., 2024, 218, 113359 CrossRef CAS.
- (a) S. P. Nolan, N–Heterocyclic Carbenes, Wiley-VCH Verlag GmbH & Co. KGaA, 2014 CrossRef; (b) D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723 RSC; (c) L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz and V. César, Chem. Rev., 2011, 111, 2705 CrossRef CAS PubMed.
- (a) M. Fèvre, J. Pinaud, Y. Gnanou, J. Vignolle and D. Taton, Chem. Soc. Rev., 2013, 42, 2142 RSC; (b) W. N. Ottou, H. Sardon, D. Mecerreyes, J. Vignolle and D. Taton, Prog. Polym. Sci., 2016, 56, 64 CrossRef CAS; (c) J. Raynaud, C. Absalon, Y. Gnanou and D. Taton, Macromolecules, 2010, 43, 2814 CrossRef CAS.
- S. Naumann, M. Speiser, R. Schowner, E. Giebel and M. R. Buchmeiser, Macromolecules, 2014, 47, 4548–4556 CrossRef CAS.
- Initial ring opening of anhydride by a NHC moiety was also proposed with NHC-metal/NHC-CO2 as epoxide/anhydride ROCOP initiators: see ref. 14.
- (a) L.-F. Hu, Y. Li, B. Liu, Y.-Y. Zhang and X.-H. Zhang, RSC Adv., 2017, 7, 49490 RSC; (b) L.-C. Shen, S.-Y. Chiang, I.-T. Ho, K.-Y. Wu and W.-S. Chung, Eur. J. Org. Chem., 2012, 792 CrossRef CAS.
- B. Liu, J. Chen, N. Liu, H. Ding, X. Wu, B. Dai and I. Kim, Green Chem., 2020, 22, 5742 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2367268. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4py00778f |
| This journal is © The Royal Society of Chemistry 2024 |