
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot preparation of nonconventional luminescent polymer gels driven by polymerization†
Nan
Jiang
 a,
Xin
Pu
b,
Ke-Xin
Li
a,
Chang-Yi
Zhu
a,
Yu-Wei
Sun
a,
Yan-Hong
Xu
*a,
You-Liang
Zhu
*b and
Martin R.
Bryce
*c
a,
Xin
Pu
b,
Ke-Xin
Li
a,
Chang-Yi
Zhu
a,
Yu-Wei
Sun
a,
Yan-Hong
Xu
*a,
You-Liang
Zhu
*b and
Martin R.
Bryce
*c
aKey Laboratory of Preparation and Applications of Environmental Friendly Materials, Key Laboratory of Functional Materials Physics and Chemistry of the Ministry of Education, Jilin Normal University, Changchun 130103, China. E-mail: xuyh198@163.com
bState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun 130012, China. E-mail: youliangzhu@jlu.edu.cn
cDepartment of Chemistry, Durham University, Durham, DH1 3LE, UK. E-mail: m.r.bryce@durham.ac.uk
First published on 24th September 2024
Abstract
Nonconventional chromophores are good candidates for preparing luminous gels because their luminescence is usually enhanced in the aggregated state. In this work, a simple one-pot strategy for polymerization-induced gelation of polymer fluorescent gels was developed, and a self physically crosslinked luminous gel PUHG based on a non-conjugated/nonconventional luminous polyurethane derivative was obtained. Detailed experimental and theoretical studies probed the physical properties and luminescence principles of PUHG's aggregated state. Molecular dynamics simulations suggest that abundant non-covalent interactions and physical entanglement between polyurethane chains are the main driving forces of gel formation and the source of luminescence. PUHG displays stable photophysical properties, environmental tolerance, good adhesion properties and processability, leading to the validation of patterning applications of PUHG on different organic/inorganic substrates. This work broadens the application range of nonconventional luminous polymers and provides a simple route for large-scale preparation of fluorescent gel soft materials.
Introduction
Gels are very promising flexible materials with the advantages of flexible and tuneable physical and chemical properties, adjustable mechanical properties and viscoelasticity, and they have excellent application prospects in fields such as biomedical engineering, agriculture, food industry, soft robotics, etc.1–5 Therefore, new luminous gel soft materials, which combine the benefits of luminescence and gel properties, can play a vital role in high-tech applications where they are receiving increased attention.6–9 However, the development of such materials is still relatively slow. One of the major challenges is the aggregation-caused quenching (ACQ) that occurs when conventional chromophores are aggregated in the solid state or in high concentrations in solutions.10–13 Gels assume solid and liquid characteristics concurrently, and a large part of a gel consists of a network of partially aggregated states, giving the properties of a quasi-solid.14 The confined space inside gels will inevitably cause conventional aromatic or π-conjugated chromophores to accumulate and interact strongly, which sometimes leads to the ACQ effect.15 This has greatly limited the development of fluorescent gels with broad application prospects, and brings a lot of adverse effects into their production.The development of nonconventional luminescent materials (NCLMs) has overcome the ACQ dilemma faced by conventional luminescent materials.16–20 Crucially, the emission from NCLMs does not rely on chromophores with large π-conjugated structures. Instead, the emission comes from the spatial electron delocalization formed by the mutual aggregation of electron-rich or lone pair electron groups such as hydroxyl, ester, carbonyl, amide and so on, and the ensuing conformational solidification.21–25 The spatial electron aggregation principle of NCLMs is partly consistent with the semi-aggregation property of gels. In addition, NCLMs have the advantages of simple synthesis, low cost, easy structural regulation, good processability and large-scale production,26–30 which make them excellent candidates for the preparation of fluorescent gels. The polymerization-induced emission strategy31–33 is important and very relevant to the present work.
Fluorescent gels have many advantages. From the viewpoint of processability they have excellent plasticity, and due to their high stability in the aggregation state they can be processed into structures with a specific shape and size by injection molding and other methods which are suitable for the laboratory or commercial production. Gel-state materials are generally easy to handle because they do not spill, scatter or fly like powders, and are not prone to leak like solutions.34–39 At present, the acquisition of many fluorescent gels relies on embedding chromophores into the three-dimensional network of the gel, which relaxes the threshold for the preparation of luminous gels.40–43 However, these doped gels face many problems. For example, chromophore molecules may interact with other gel components, affecting the stability and controllability of the gel structure. The preparation of composite gels may require more complex synthesis and preparation methods, involving a multi-step process, increasing the difficulty of the experiments, especially for commercial scale-up.44 Therefore, research on luminous gels should focus on developing single molecules that can be used as gelators and chromophores concurrently. Polymer gels typically have the advantage of increased mechanical strength compared to supramolecular gels that are formed from small molecules.45
In this study, a fluorescent self physically crosslinked polymer gel was obtained via a simple one-pot reaction. Compared with conventional methods, the one-pot protocol needs fewer steps, thus simplifying the preparation process. It can use raw materials more efficiently, save resources and costs, and help reduce the chemical waste generated, which is in line with the principles of green chemistry. Moreover, self-crosslinked gel preparation often does not require external additives or catalysts, reducing dependence on external substances.
Results and discussion
Synthesis
Through a simple one-pot reaction of heptaethylene glycol, poly(ethylene glycol) monomethyl ether and trimethylhexamethylene diisocyanate (Fig. 1a) the self-crosslinked polyurethane derivative fluorescent gel (named PUHG, Fig. 1a and Table S1†) was prepared. The 1H nuclear magnetic resonance (NMR) spectrum showed that PUHG has a clear fine structure (Fig. S1†). The FT-IR spectrum showed the N–H stretching vibration absorption peak at 3340 cm−1, C–H asymmetric and symmetrical vibration peaks at 2881 and 2955 cm−1, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration absorption peak at 1643 cm−1; is the stretching vibration absorption peak of C–O–C is at 1105 cm−1. The combination of the characteristic NH and CO peaks of the carbamate [–NHC(O)O–] functional group confirmed the successful synthesis of the polyurethane derivative (Fig. S2†). X-ray diffraction (XRD) data (Fig. S3†) show that a wide diffused diffraction peak unique to polyurethane derivatives appears at the diffraction angle of 18°. This semi-crystalline structure can enhance the physical binding of materials, which is conducive to their luminescence.46 The average molecular weight and polydispersion index of PUHG were 8333 and 4.37, respectively, obtained by gel permeation chromatography (GPC).
O stretching vibration absorption peak at 1643 cm−1; is the stretching vibration absorption peak of C–O–C is at 1105 cm−1. The combination of the characteristic NH and CO peaks of the carbamate [–NHC(O)O–] functional group confirmed the successful synthesis of the polyurethane derivative (Fig. S2†). X-ray diffraction (XRD) data (Fig. S3†) show that a wide diffused diffraction peak unique to polyurethane derivatives appears at the diffraction angle of 18°. This semi-crystalline structure can enhance the physical binding of materials, which is conducive to their luminescence.46 The average molecular weight and polydispersion index of PUHG were 8333 and 4.37, respectively, obtained by gel permeation chromatography (GPC).
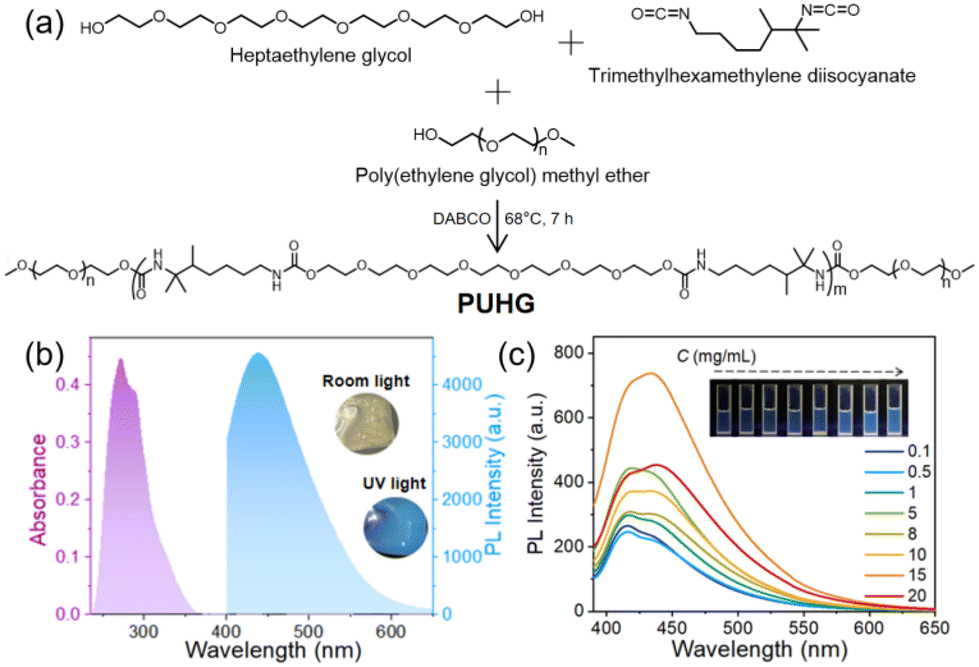 | ||
| Fig. 1 (a) Synthetic route to the polyurethane derivative PUHG. (b) Absorption and emission (photoluminescence, PL) spectra of PUHG in the gel state. Insert: Photograph of PUHG glowing under room light and 365 nm UV light. (c) Emission of PUHG/DMSO solutions with different concentrations. λex = 365 nm; Inset: Corresponding photographs taken under 365 nm UV light. | ||
Due to the chemical structure of the diol monomer (heptaethylene glycol), PUHG has a relatively flexible linear structure and contains a large number of O and N heteroatoms, which is very conducive to enhancing emission through the overlap of lone pairs of electrons. PUHG appeared as a transparent gel-like object under room light and as a bright blue, fluorescent gel under a 365 nm UV lamp (Fig. 1b). In comparison, the optical and mechanical properties of heptaethylene glycol monomer are mediocre (Fig. S4†). Therefore, we project a chain-extension type of addition polymerization reaction, for amplifying the effect of polymerization on the material's properties.
Photophysical characterization and mechanical property
The absorption and emission peak of PUHG is at λmax 280 and 438 nm, respectively, showing a large Stokes shift of >150 nm (Fig. 1b and S4†), which can reduce the re-excitation caused by light. PUHG exhibits the common excitation-dependent properties of non-conjugated polymers (Fig. S5†).26 The optical properties, including the excited state lifetime (6.18 ns) and quantum yield (0.8%) are listed in Table S1 in ESI.† To understand the luminous mechanism of PUHG in detail PUHG/DMSO solutions at different concentrations were monitored. Both the emission and absorption showed a concentration-dependent enhancement as the solution concentration gradually increased from 0.1 mg mL−1 to 15 mg mL−1 (Fig. 1c and S6†). This is because the aggregation behavior of PUHG chains leads to enhanced electron delocalization. However, with the further increase in concentration, PUHG chains gather randomly, and more heterogeneous electron delocalization is generated in the aggregates. At 20 mg mL−1, PUHG's emission shows a distinct change, slightly broadening and red-shifting, yet the strength is reduced as observed previously with different aggregating fluorophores.47–49The illustration in Fig. 1c is the corresponding luminescent enhanced photograph. In addition, PUHG maintained good excitation-dependent emission properties even in dilute solutions as low as 0.1 mg mL−1 (Fig. S7†).
Heptaethylene glycol monomer and the product PUHG were observed by scanning electron microscopy (SEM) to explore the relationship between the properties of the compounds and their microscopic aggregation features (Fig. 2). Quantified heptaethylene glycol monomer and PUHG were dispersed in ethanol (20 mg mL−1). The two samples were sufficiently dispersed by the vortex instrument to observe clearly their micromorphology. At the same magnification, heptaethylene glycol monomer showed a very dispersed blocky structure (Fig. 2a). In contrast, PUHG showed a slightly folded and ruptured membrane structure (Fig. 2b). In addition, thicker nanowire entanglements were observed (Fig. 2c and d). The above results show that the material's microstructure changed considerably after only one step of chain extension addition polymerization, which further affects the macroscopic emission properties.
 | ||
| Fig. 2 SEM images of 20 mg mL−1 (a) heptaethylene glycol monomer, and PUHG (b–d) dispersed in ethanol. | ||
To better understand the effects of polymerization and aggregation behavior, PUHG solutions with different concentrations were selected for quantitative SEM measurements and dynamic light scattering (DLS) monitoring (Fig. 3 and S8†). At the same magnification, 0.5 mg mL−1 samples showed sparse nanosphere structures (Fig. 3a). When the sample concentration increased successively, the microstructure showed an increasing trend of aggregation, until at 20 mg mL−1 aggregation and large nano-aggregates appear (Fig. 3b–d and S8†) and the polydispersity index increases with increasing concentration (Fig. S8†). Combined with the emission profile in Fig. 1c, we can infer that in the low concentration range, the aggregation behavior enhances the emission intensity. In the high concentration range, the aggregation behavior causes the emission wavelength to redshift. This property is commonly observed with non-conjugated polymers.16,17,26 Compared with the conventional aggregation-induced luminescence induced by molecular motion restriction, the nontraditional intrinsic luminescence (NTIL) of non-conjugated polymers is realized through the spatial electron conjugation of nonconventional chromophores. Hence, the increased concentration has a more significant influence on their NTIL emission behavior.
 | ||
| Fig. 3 SEM images of (a) 0.5 mg mL−1, (b) 5 mg mL−1, (c) 8 mg mL−1, (d) 20 mg mL−1 of PUHG/ethanol solutions. | ||
The theoretical calculations of PUHG system were conducted using Compass III force field. PUHG molecules with two repeating units were initially structurally optimized, annealed, and then molecular dynamics simulation with a simulation time of 5000 ps was implemented (Fig. S9–S12†).50–54 Various non-covalent interaction forces and chain-to-chain interactions were calculated via radial distribution functions. As shown in Fig. 4a, there are abundant N–CO, N–C–O, N–C(O)O, CO–C–O and inter-chain interactions within the PUHG system. These interactions make PUHG prone to inter- and intra-chain aggregation behavior, which is conducive to the through-space communication, conjugation of electrons and physical (non-covalent) crosslinking. These effects should be responsible for the gelation and luminescence of PUHG. A statistical analysis of the trend of the non-short interactions in the PUHG system over time shows that their energies reach equilibrium within 100 ps and then change little over time (Fig. S10†). The calculated cut-off distance of the hydrogen bond in the equilibrium structure was 3.43 Å (Fig. S11†), and the bond angle of the formed hydrogen bond was defined as 90–180 degrees, which was used as the criterion of the hydrogen bond for subsequent statistics on the number of hydrogen bonds. The statistical results show that with the change of time, the number of hydrogen bonds in the whole system is in dynamic equilibrium (Fig. 4b and S12†). The above results prove that there are many strong and stable non-covalent interactions in the system, which provides a solid molecular foundation for the stability and function of PUHG. These tight non-covalent interaction networks not only enhance the intermolecular binding force, but also make PUHG molecular chains tend to entwine and aggregate with each other, and finally bring enhanced photoluminescence and self-gelation properties. To characterize the dynamic rheology of PUHG at different time scales, the strain was set at 1%, and the angular frequency (ω) was 100–0.1 rad s−1. The change curves of elastic modulus (G′) and viscosity modulus (G′′) at different frequencies were obtained by a frequency scanning test. As shown in Fig. 4d, G′ > G′′, and G′ and G′′ have no intersection, and G′ is not affected by frequency changes, indicating that PUHG is a complete gel sample.55
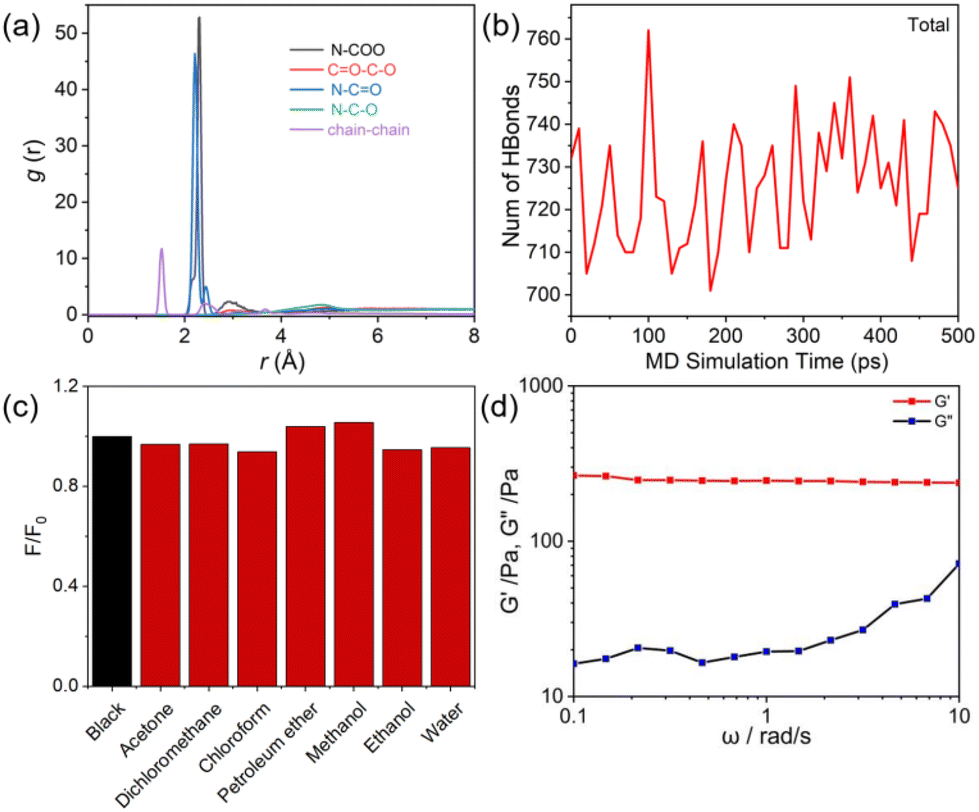 | ||
| Fig. 4 (a) Radial distribution function of PUHG by molecular dynamics simulation. (b) The number of total hydrogen bonds over time. (c) Fluorescence changes of pure PUHG (black) and PUHG treated with different solvents (red). (d) The elastic modulus (G′) and viscosity modulus (G′′) at different frequencies of PUHG. | ||
PUHG application
The environmental tolerance of fluorescent gels in practical applications is also an important index. After 30 min infiltration of PUHG with acetone, dichloromethane, chloroform, petroleum ether, methanol, ethanol and water, PUHG still maintained excellent luminescence (Fig. 4c), which indicated that PUHG has good environmental tolerance and could be used in practical applications. Therefore, as a proof-of-concept, we tested the adhesion of PUHG on different organic/inorganic substrates, including paper, glass, plastic, wood and tinfoil. PUHG exhibited effective adhesion on these substrates. In addition, thanks to the flexibility and deformability of PUHG, it can be molded into any shape which is retained after the mold is removed (Fig. 5). These results prove the excellent processability of PUHG. | ||
| Fig. 5 Fluorescent patterns of PUHG adhering to paper, glass, plastic, wood and tinfoil under daylight and an ultraviolet lamp. | ||
Conclusions
In conclusion, fluorescent gels materials have great potential for applications and for fundamental research because of their excellent photophysical properties, adhesion and plasticity. They are rising stars in luminous materials. However, due to the tight internal structure of gel materials, conventional chromophores usually face the dilemma of luminescence quenching after aggregation, so the development of such materials has been minimal. In this work, we developed a polymerization-induced self physically crosslinked polymer gel (PUHG) based on a nonconventional luminescent polyurethane derivative via a one-pot reaction. The ultra-flexible alkoxy chain favors non-covalent interactions and physical entanglement which are responsible for PUHG's gelation and luminescence behavior. Due to its excellent adhesion, processability, and fluorescence stability, PUHG was successfully applied to patterning on different substrates. This work provides a universal strategy for the facile, rapid and large-scale preparation of luminous gels.Author contributions
Nan Jiang: Investigation, data curation, formal analysis, visualization, writing original draft; Ke-Xin Li, Chang-Yi Zhu: data curation, formal analysis, visualization. Yu-Wei Sun: validation; Xin Pu and You-Liang Zhu: software; Yan-Hong Xu: conceptualization, funding acquisition, project administration, resources; Martin R. Bryce: project administration, resources, supervision, writing-review & editing.Data availability
The data associated with this article are available in the manuscript and ESI files.† Additional data can be obtained upon request to the authors.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The work was funded by the Science and Technology Development Program of Jilin Province (YDZJ202301ZYTS305, YDZJ202201ZYTS368); Industrial Technology Research and Development Program of Jilin Province (Grant No. 2022C039-1); we thank Dr Yang at Shenzhen HUASUAN Tech. Co., Ltd, for insightful discussions and suggestions. M. R. B. thanks EPSRC Grant EP/L02621X/1 for funding.References
- L. Lan, J. Ping, H. Li, C. Wang, G. Li, J. Song and Y. Ying, Adv. Mater., 2024, 2401151 CrossRef PubMed.
- J. Deng, P. Wang, Z. Xu, T. Hu, D. Li, X. Wei, C. Chen, Y. Li and Y. Zhang, J. Hazard. Mater., 2024, 469, 133965 CrossRef PubMed.
- W. Cheng, X. Wu, Y. Zhang, D. Wu, L. Meng, Y. Chen and X. Tang, Trends Food Sci. Technol., 2022, 129, 244–257 CrossRef.
- S. Song, Y. Shi, L. Zhu and B. Yue, Sci. China: Chem., 2024, 67, 1865–1880 CrossRef.
- F. Chen, Y. M. Wang, W. W. Gou and X. B. Yin, Chem. Sci., 2019, 10, 1644–1650 RSC.
- Z. Li, X. Ji, H. Xie and B. Z. Tang, Adv. Mater., 2021, 33, 2100021 CrossRef CAS PubMed.
- Y. Sun, X. Le, S. Zhou and T. Chen, Adv. Mater., 2022, 34, 2201262 CrossRef CAS.
- H. Shi, S. Wu, M. Si, S. Wei, G. Lin, H. Liu, W. Xie, W. Lu and T. Chen, Adv. Mater., 2022, 34, 2107452 CrossRef CAS PubMed.
- J. Wu, Y. Wang, P. Jiang, X. Wang, X. Jia and F. Zhou, Nat. Commun., 2024, 15, 3482 CrossRef PubMed.
- R. Hu, A. Qin and B. Z. Tang, Prog. Polym. Sci., 2020, 100, 101176 CrossRef.
- J. Ma, Y. Gu, D. Ma, W. Lu and J. Qiu, Front. Chem., 2022, 10, 985578–985593 CrossRef PubMed.
- D. A. Tomalia, B. Klajnert-Maculewicz, K. A. M. Johnson, H. F. Brinkman, A. Janaszewska and D. M. Hedstrand, Prog. Polym. Sci., 2019, 90, 35–117 CrossRef.
- R. Wang, W. Yuan and X. Zhu, Chin. J. Polym. Sci., 2015, 33, 680–687 CrossRef.
- M. A. Kuzina, D. D. Kartsev, A. V. Stratonovich and P. A. Levkin, Adv. Funct. Mater., 2023, 33, 2301421 CrossRef CAS.
- M. K. Dixit, C. Mahendar and M. Dubey, Chem. – Asian J., 2022, 17, e201900559 CrossRef CAS PubMed.
- Z. Wang, H. K. Zhang, S. Q. Li, D. Y. Lei, B. Z. Tang and R. Q. Ye, Top. Curr. Chem., 2021, 379, 14–36 CrossRef CAS PubMed.
- N. Jiang, D. X. Zhu, Z. M. Su and M. R. Bryce, Mater. Chem. Front., 2021, 5, 60–75 RSC.
- W. F. Lai, Mater. Today Chem., 2022, 23, 100712–100725 CrossRef CAS.
- Y. Wang, X. Bin, X. Chen, S. Zheng, Y. Zhang and W. Z. Yuan, Macromol. Rapid Commun., 2018, 39, 1800528–1800534 CrossRef.
- L. L. Du, B. L. Jiang, X. H. Chen, Y. Z. Wang, L. M. Zou, Y. L. Liu, Y. Y. Gong, C. Wei and W. Z. Yuan, Chin. J. Polym. Sci., 2019, 37, 409–415 CrossRef CAS.
- X. B. Zhou, W. W. Luo, H. Nie, L. G. Xu, R. R. Hu, Z. J. Zhao, A. J. Qin and B. Z. Tang, J. Mater. Chem. C, 2017, 5, 4775–4779 RSC.
- A. Pucci, R. Rausa and F. Ciardelli, Chem. Phys., 2008, 209, 900–906 CAS.
- S. X. Tang, T. J. Tang, Z. H. Zhao, T. W. Zhu, Q. Zhang, W. B. W. Hou and W. Z. Yuan, Chem. Soc. Rev., 2021, 50, 12616–12655 RSC.
- H. Lu, L. Feng, S. Li, J. Zhang, H. Lu and S. Feng, Macromolecules, 2015, 48, 476–482 CrossRef CAS.
- Q. Zhou, B. Cao, C. Zhu, S. Xu, Y. Gong, W. Z. Yuan and Y. Zhang, Small, 2016, 12, 6586–6592 CrossRef CAS.
- H. Wang, Q. Li, P. Alam, H. Bai, V. Bhalla, M. R. Bryce, M. Cao, C. Chen, S. Chen, X. Chen, Y. Chen, Z. Chen, D. Dang, D. Ding, S. Ding, Y. Duo, M. Gao, W. He, X. He, X. Hong, Y. Hong, J. J. Hu, R. Hu, X. Huang, T. D. James, X. Jiang, G. Konishi, R. T. K. Kwok, J. W. Y. Lam, C. Li, H. Li, K. Li, N. Li, W. J. Li, Y. Li, X. J. Liang, Y. Liang, B. Liu, G. Liu, X. Liu, X. Lou, X. Y. Lou, L. Luo, P. R. McGonigal, Z.-W. Mao, G. Niu, T. C. Owyong, A. Pucci, J. Qian, A. Qin, Z. Qiu, A. L. Rogach, B. Situ, K. Tanaka, Y. Tang, B. Wang, D. Wang, J. Wang, W. Wang, W. X. Wang, W. J. Wang, X. Wang, Y. F. Wang, S. Wu, Y. Wu, Y. Xiong, R. Xu, C. Yan, S. Yan, H. B. Yang, L. L. Yang, M. Yang, Y. W. Yang, J. Yoon, S. Q. Zang, J. Zhang, P. Zhang, T. Zhang, X. Zhang, X. Zhang, N. Zhao, Z. Zhao, J. Zheng, L. Zheng, Z. Zheng, M. Q. Zhu, W. H. Zhu, H. Zou and B. Z. Tang, ACS Nano, 2023, 17, 14347–14405 CrossRef CAS.
- L. L. D. Mercato, P. P. Pompa, G. Maruccio, A. D. Torre, S. Sabella, A. M. Tamburro, R. Cingolani and R. Rinaldi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18019–18024 CrossRef.
- H. Zhang, Z. Zhao, P. R. McGonigal, R. Q. Ye, S. J. Liu, J. W. Y. Lam, R. T. K. Kwok, W. Z. Yuan, J. P. Xie, A. L. Rogach and B. Z. Tang, Mater. Today, 2020, 32, 1369–7021 CrossRef.
- X. Ji, W. G. Tian, K. F. Jin, H. L. Diao, X. Huang, G. J. Song and J. Zhang, Nat. Commun., 2022, 13, 3717–3729 CrossRef CAS.
- B. Chu, H. Zhang, K. Chen, B. Liu, Q. L. Yu, C. J. Zhang, J. Sun, Q. Yang, X. H. Zhang and B. Z. Tang, J. Am. Chem. Soc., 2022, 144, 15286–15294 CrossRef CAS PubMed.
- X. L. Sun, D. M. Liu, D. Tian, X. Y. Zhang, W. Wu and W. M. Wan, Nat. Commun., 2017, 8, 1210 CrossRef PubMed.
- Y. N. Jing, S. S. Li, M. Su, H. Bao and W. M. Wan, J. Am. Chem. Soc., 2019, 141, 16839–16848 CrossRef PubMed.
- Q. X. Shi, H. Xiao, Y. J. Sheng, D. S. Li, M. Su, X. L. Sun, H. Bao and W. M. Wan, Polym. Chem., 2022, 13, 4524–4534 RSC.
- Y. Li, D. J. Young and X. J. Loh, Mater. Chem. Front., 2019, 3, 1489–1502 RSC.
- S. Wei, Z. Li, W. Lu, H. Liu, J. Zhang, T. Chen and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 8608–8624 CrossRef.
- S. Bhattacharya, R. S. Phatake, S. N. Barnea, N. Zerby, J. J. Zhu, R. Shikler, N. G. Lemcoff and R. Jelinek, ACS Nano, 2019, 13, 1433–1442 CrossRef PubMed.
- Z. Hu, S. Xu, H. Zhang and X. Ji, Aggregate, 2023, 4, e283 CrossRef CAS.
- X. Cong, K. Ou, J. Ma, J. Xu, Y. Liao, Y. Yang and H. Wang, Chem. Eng. J., 2024, 481, 148355 CrossRef CAS.
- J. Tavakoli, A. J. Ghahfarokhi and Y. Tang, Top. Curr. Chem., 2021, 379, 9–18 CrossRef CAS PubMed.
- P. Yu, Y. Zhao, X. Li, Z. Zhao, Y. Dong and X. Li, Mater. Today Commun., 2024, 38, 108495 CrossRef CAS.
- S. Lu, S. Wang, J. Zhao, J. Sun and X. Yang, Chem. Commun., 2018, 54, 4621–4624 RSC.
- H. Liu, S. Wei, H. Qiu, B. Zhan, Q. Liu, W. Lu, J. Zhang, T. Ngai and T. Chen, Macromol. Rapid Commun., 2020, 41, 2000123 CrossRef CAS.
- Y. Y. Chen, G. F. Gong, Y. Q. Fan, Q. Zhou, Q. P. Zhang, H. Yao, Y. M. Zhang, T. B. Wei and Q. Lin, Soft Matter, 2019, 15, 6878–6884 RSC.
- Z. Li, F. Lu and Y. Liu, J. Agric. Food Chem., 2023, 71, 10238–10249 CrossRef CAS.
- P. R. A. Chivers and D. K. Smith, Nat. Rev. Mater., 2019, 4, 463–478 CrossRef CAS.
- Y. L. Feng, N. Jiang, D. X. Zhu, Z. M. Su and M. R. Bryce, J. Mater. Chem. C, 2020, 8, 11540–11545 RSC.
- J. Zhang, H. Zou, S. Gan, B. He, J. C. Huang, C. Peng, J. W. Y. Lam, L. Zheng and B. Z. Tang, Adv. Funct. Mater., 2022, 32, 2108199 CrossRef CAS.
- Y. Qin, X. Li, S. Lu, M. Kang, Z. Zhang, Y. Gui, X. Li, D. Wang and B. Z. Tang, ACS Mater. Lett., 2023, 5, 1982–1991 CrossRef CAS.
- W. J. Shi, X. H. Yan, J. Yang, Y. F. Wei, Y. T. Huo, C. L. Su, J. Yan, D. Han and L. Niu, Anal. Chem., 2023, 95, 9646–9653 CrossRef CAS PubMed.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 4, 187–217 CrossRef CAS.
- S. W. Bunte and H. J. Sun, Phys. Chem. B, 2000, 104, 2477–2489 CrossRef.
- M. W. Deem, J. M. Newsam and S. K. Sinha, J. Phys. Chem., 1990, 94, 8356–8359 CrossRef.
- R. L. C. Akkermans, N. A. Spenley and S. H. Robertson, Mol. Simul., 2021, 47, 540–551 CrossRef.
- S. N. Ha, A. Giammona, M. Field and J. W. Brady, Carbohydr. Res., 1988, 180, 207–221 CrossRef PubMed.
- K. A. Ibrahim, M. Y. Naz, S. A. Sulaiman, A. Ghaffar, Y. Jamil and N. M. Abdel-Salam, Polymers, 2017, 9, 361 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00832d |
| This journal is © The Royal Society of Chemistry 2024 |