
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymers with quadruple hydrogen-bonding end groups: controlling molecular weight using a small molecule photoswitch†
Eleanor M.
Hilton‡
ab,
Yasmeen
Jhons
b,
Nicholas J.
Warren
 *bc and
Andrew J.
Wilson
*ad
*bc and
Andrew J.
Wilson
*ad
aSchool of Chemistry, University of Leeds, Woodhouse Lane, Leeds LS2 9JT, UK
bSchool of Chemical and Process Engineering, University of Leeds, Woodhouse, Lane, Leeds LS2 9JT, UK
cSchool of Chemical, Materials and Biological Engineering, University of Sheffield, Mappin Street, Sheffield, S1 3JD, UK. E-mail: n.warren@sheffield.ac.uk
dSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK. E-mail: a.j.wilson.1@bham.ac.uk
First published on 24th September 2024
Abstract
The molecular weight and topology of polymer chains is typically defined during their synthesis, after which these parameters remain fixed. This limitation has motivated efforts to develop reversibly reconfigurable polymers, which offer opportunities for advanced applications and/or efficient reprocessing. Herein, we report the preparation of a library of poly(methyl acrylate) (PMA) homopolymers and a poly(methyl methacrylate) (PMMA) homopolymer bearing pendant hydrogen bonding motifs (HBMs) which have the potential to generate reconfigurable materials. This approach includes the synthesis of alkyne-functional HBMs which were successfully coupled to an azide-functionalised RAFT agent via alkyne/azide “click” chemistry. The resulting ureidopyrimidinone (UPy) and amidonaphthyridine (NAP) RAFT agents could successfully be used to prepare PMA, but the attempt using the diamidonaphthyridine (DAN) RAFT agent was unsuccessful. In this example, a post-polymerisation technique was demonstrated as a viable alternative (in this case with a PMMA homopolymer). To demonstrate proof-of-concept that this can be achieved using a supramolecular approach: a small molecule photoswitch comprising ditopic azobenzene linked ureidopyrimidinone (UPy) was shown to effect reconfiguration of polymer molecular weight of DAN-functionalised PMMA under visible/UV light irradiation. In the cis (Z) photostationary state, intramolecular UPy·UPy homodimerization within the photoswitch was preferred, whereas in the trans (E) configuration, the need for UPy to satisfy its hydrogen-bonding requirements resulted in intermolecular DAN·UPy heterodimerization with the DAN motif of poly(methyl-methacrylate) resulting in a doubling of its molecular weight as observed by 1H DOSY NMR spectroscopy.
Introduction
Supramolecular polymers have had significant impact in materials research,1,2 spanning a range of applications3 that include: self-healing,4,5 adhesives,6–8 biomaterials,9,10 actuating and memory materials.11–13 Hydrogen-bond assembled polymers14 have proven attractive given the availability of a range of hydrogen-bonding motifs (HBMs);14,15 when hydrogen-bond donor and acceptor groups are combined to give patterns, this leads to sequence selective recognition modules with a range of dimerization affinities14 that directly influence macromolecular behaviour.16–18 In solution, polymers assembled through main chain hydrogen-bonds require high affinity HBMs,19 whereas weaker affinities can be accommodated where hydrogen-bonds between side chains are used for assembly20–22 or hydrogen-bonds are used for crosslinking. A specific category amongst the former class, are those which involve assembly via a single HBM at the chain end of the polymer and which have been used to prepare e.g. diblock co-polymers23 which phase separate.24 Indeed, the topology of a polymer plays a key role in determining its shape, which in turn must be considered for different applications, however, polymer topology is usually static once synthesis is complete and has motivated efforts to generate reconfigurable polymers.25 Thus, the ability to readily reconfigure polymers assembled through HBMs at the chains ends would be desirable, but is not yet readily achievable. Amongst available stimuli e.g. temperature, pH, redox control and light, light is attractive.26–29 Indeed, a number of light responsive hydrogen-bond assembled supramolecular “condensation” and side-chain functionalized polymers have been described.30–36 We recently reported on the photoswitchable hydrogen-bond mediated assembly of ditopic azobenzene linked HBMs;37 the cis (Z) and trans (E) photostationary states were shown to be biased toward cyclic or extended conformations leading to different supramolecular assemblies based on the concentration dependent ring-chain equilibrium and varying hydrogen-bond dimerization affinity of the HBMs.In this manuscript we synthesise a series of poly(methyl acrylate) and poly(methyl methacrylate) (PMA and PMMA) homopolymers, end-functionalised with HBMs using alkyne/azide “click” chemistry. This was facilitated using a commercially available azide functional RAFT agent, combined with alkyne functionalized HBMs reported in our previous work.37 To develop a photoresponsive reconfigurable polymer, PMMA bearing a single terminal DAN HBM38 was mixed with a ditopic azobenzene linked ureidopyrimidine (UPy) HBM.39 By switching between E and Z photostationary states using visible light at a concentration below the supramolecular polymerization regime of the ring-chain equilibria for the ditopic azobenzene linked UPy HBM, we show that the molecular weight of the polymer bearing a single terminal diamidonapthyridine (DAN) HBM can be reversibly doubled through multiple cycles of photoswitching.
Results and discussion
It was anticipated that the most efficient route for the preparation of PMA end-functionalised with HBMs was via either a pre-polymerisation functionalisation or post-polymerisation functionalisation. The pre-polymerisation approach, by employing HBM-functional RAFT agents was initially preferred on the basis that all chains bear the HBM. Although this would be possible with the post-polymerization approach, there is the potential for incomplete reaction, which is likely to be exacerbated by the steric hindrance of the long polymer chains.For the pre-polymerisation strategy, we identified a series of alkyne-functional HBMs from our previous work which could be reacted with an azide-functionalized RAFT agent via an alkyne/azide click reaction (Scheme 1). This resulted in a series of CTAs where alkynes 1, 2 and 3 were linked to azide functionalised RAFT agent 4 by a triazole ring. Using this approach amidonaphthyridine (NAP)-RAFT agent 5, UPy-RAFT agent 6 and DAN-RAFT agent 7 were all successfully synthesised in reasonable yields. The ability of 5–6 to serve as chain transfer agents was then assessed for the RAFT polymerization of MA (Scheme 2).
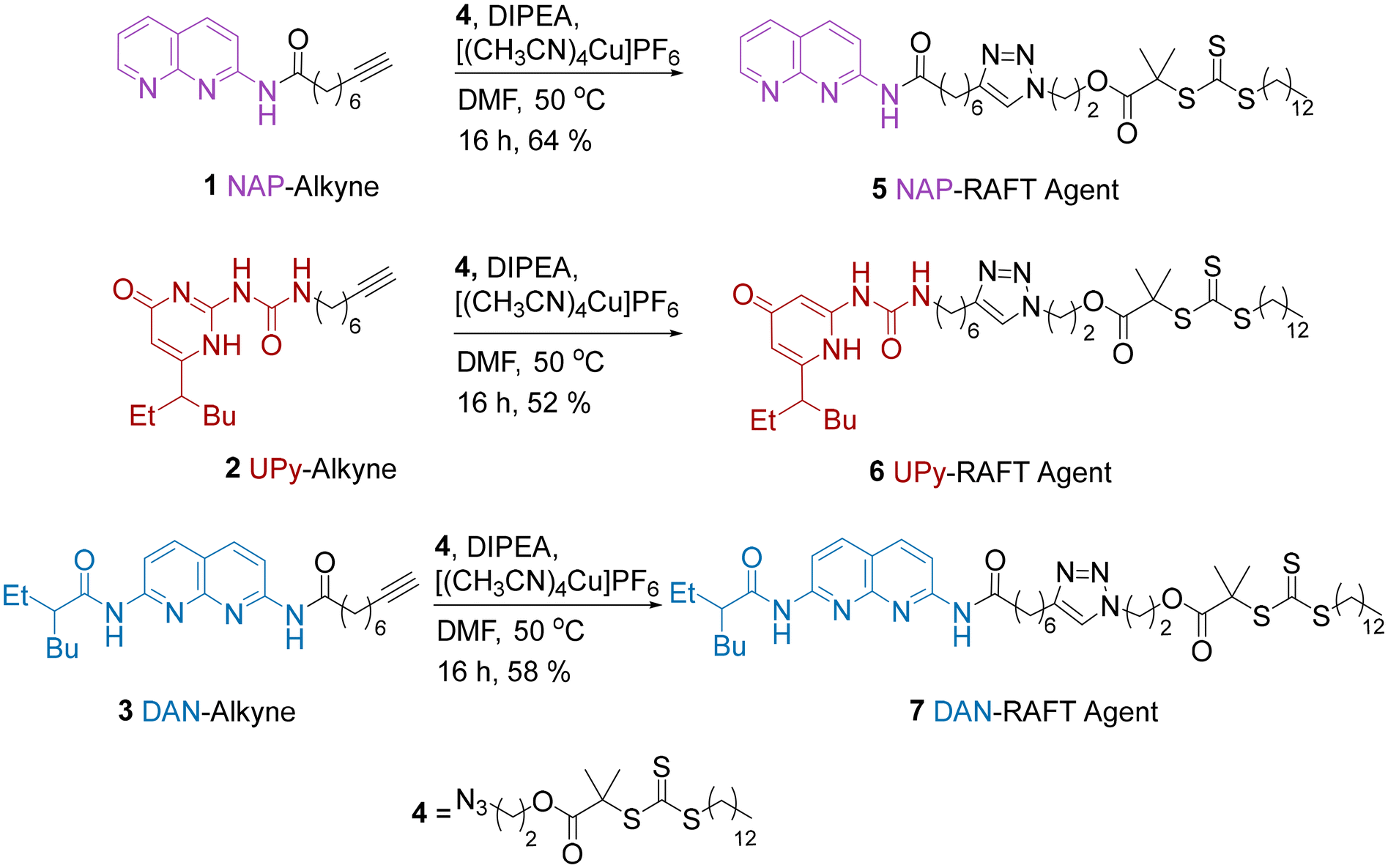 | ||
| Scheme 1 Click chemistry syntheses of NAP-RAFT agent 5, UPy-RAFT agent 6 and DAN-RAFT agent 7. | ||
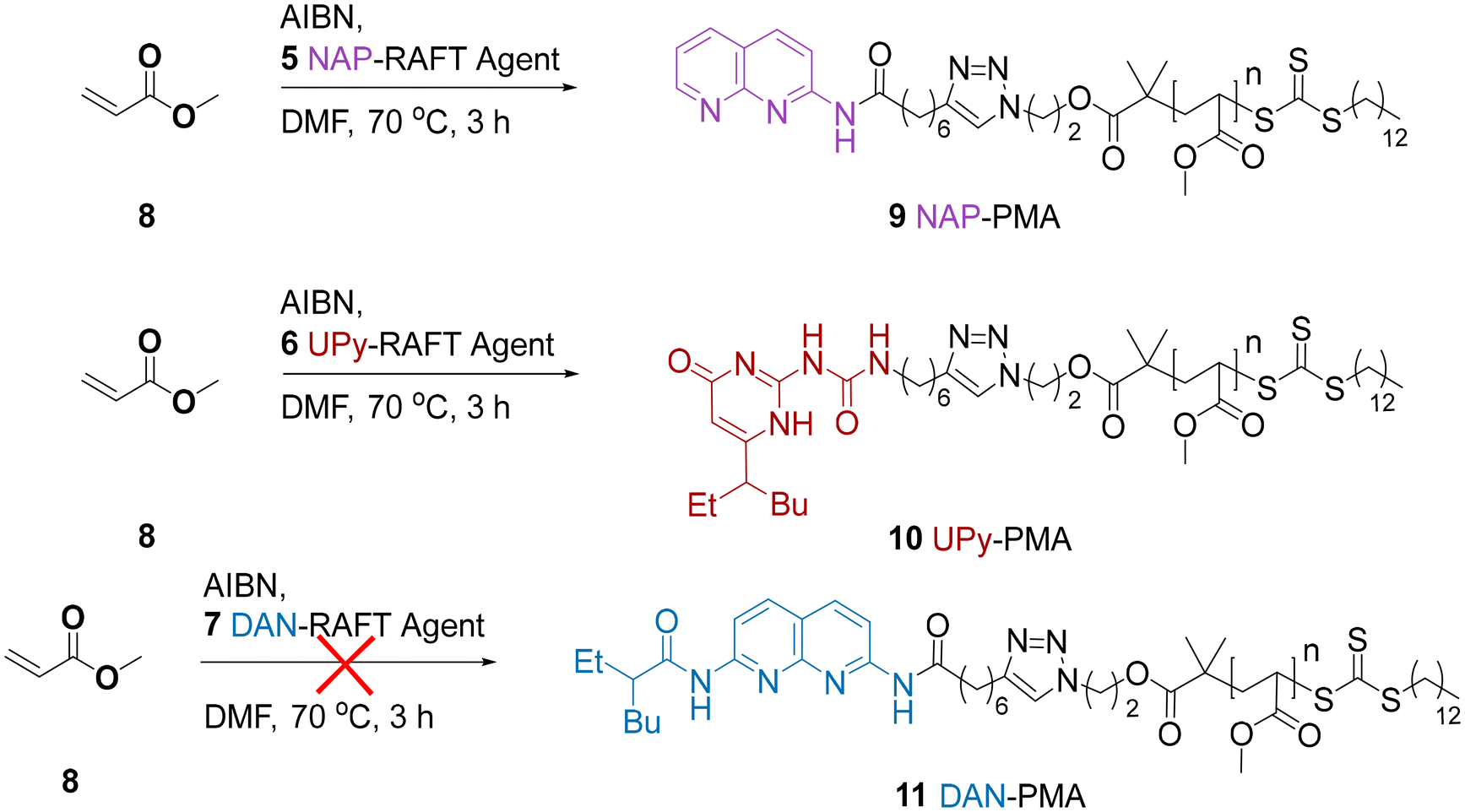 | ||
| Scheme 2 RAFT polymerisations of MA 8 using NAP-RAFT agent 5, UPy-RAFT agent 6, and DAN-RAFT agent 7. | ||
GPC and 1H NMR analysis conducted on the resulting products (Table 1 and Fig. 1) indicated that the NAP-RAFT agent 5 produced reasonably low dispersity polymer 9 (Đ = 1.19) but monomer conversion was only 31% after three hours. This suggested that the addition of the NAP HBM had an adverse impact on polymerisation. More promising was the UPy-RAFT agent 6, which afforded reasonably low dispersity polymers (Đ = 1.18) alongside a much more respectable monomer conversion of 84%. However, the resulting UPy functionalized polymer 10, was considered less desirable for reconfiguring, given the self-dimerization of the UPy motif. Unfortunately, the DAN-RAFT agent 7 failed to control the MA polymerization, with negligible conversion indicated by NMR.
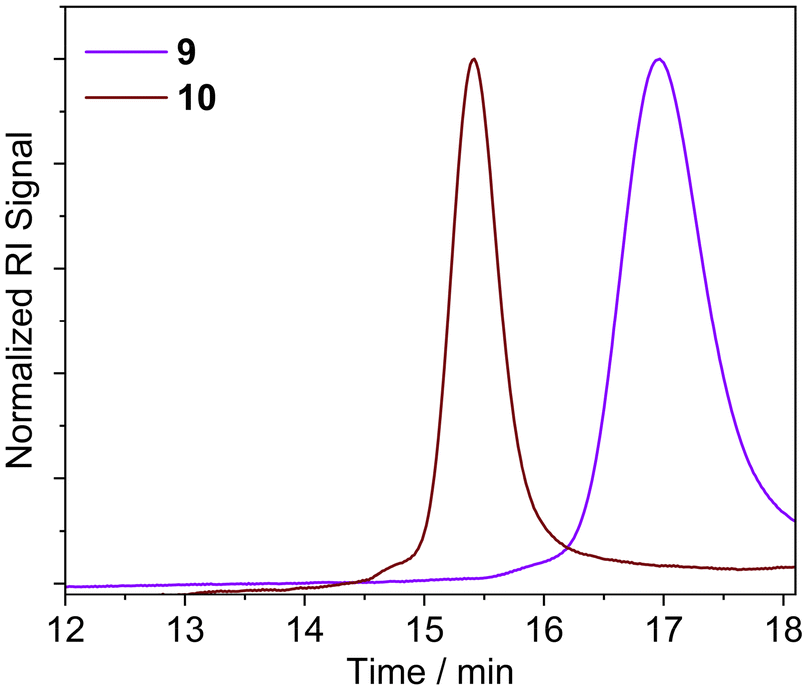 | ||
| Fig. 1 GPC traces of NAP-PMA 9; and UPy-PMA 10 obtained in DMF. | ||
| Raft agent | % MA conver. | DP | M n (NMR) (g mol−1) | M n (GPC) (g mol−1) | Đ (GPC) |
|---|---|---|---|---|---|
| 5 | 31 | 24 | 2800 | 3700 | 1.19 |
| 6 | 84 | 148 | 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 500 |
18200 |
1.18 |
| 7 | — | 3 | 1100 | — | — |
Given our objective to switch molecular weight using a small molecule, it was desirable to have a polymer functionalized with an HBM, such as DAN, that does not self-associate. Hence, we instead evaluated the use of post-polymerisation functionalization. Furthermore, we also moved to methyl methacrylate (MMA) instead of MA owing to the latter polymers being challenging to purify due to their low Tg. In principle, the behaviour of PMMA and PMA in solution should be similar enough to allow for proof-of-concept reconfiguration experiments to be explored.
The polymerization of MMA was conducted in DMF in the presence of the azide functionalised RAFT agent 4. The azide group was maintained in the resulting PMMA 13, allowing for an azide click reaction to be performed using the DAN-alkyne 3 (Scheme 3). Due to RAFT agent 4 being less suited for polymerisation of MMA, the polymers synthesised had a higher dispersity than those obtained with MA (Table 2 and Fig. 1); the trithiocarbonate RAFT was not ideal for controlling the polymerization of methacrylate monomers, resulting in a larger molar mass dispersity of 1.52 compared to values <1.20 recorded for the acrylate monomers. After functionalization with DAN, GPC (Fig. 2) indicated that the Mn increased from 24600 g mol−1 for 13 to 31200 g mol−1 for 14 On first appearance, this increase seems large for a simple end-group functionalization, but it is unsurprising given the additional purification performed after functionalisation involved a size exclusion column which would have also resulted in the removal of some of the shorter polymer chains in the sample (which also resulted in subtle reduction in molar mass dispersity). Analyses by NMR were more challenging; whilst conversion and some structural data could be obtained it was not possible to perform end group analysis due to overlapping RAFT agent and polymer proton resonances (see ESI for further details†). Nevertheless, it was deemed sufficiently modified to continue proof-of-concept studies.
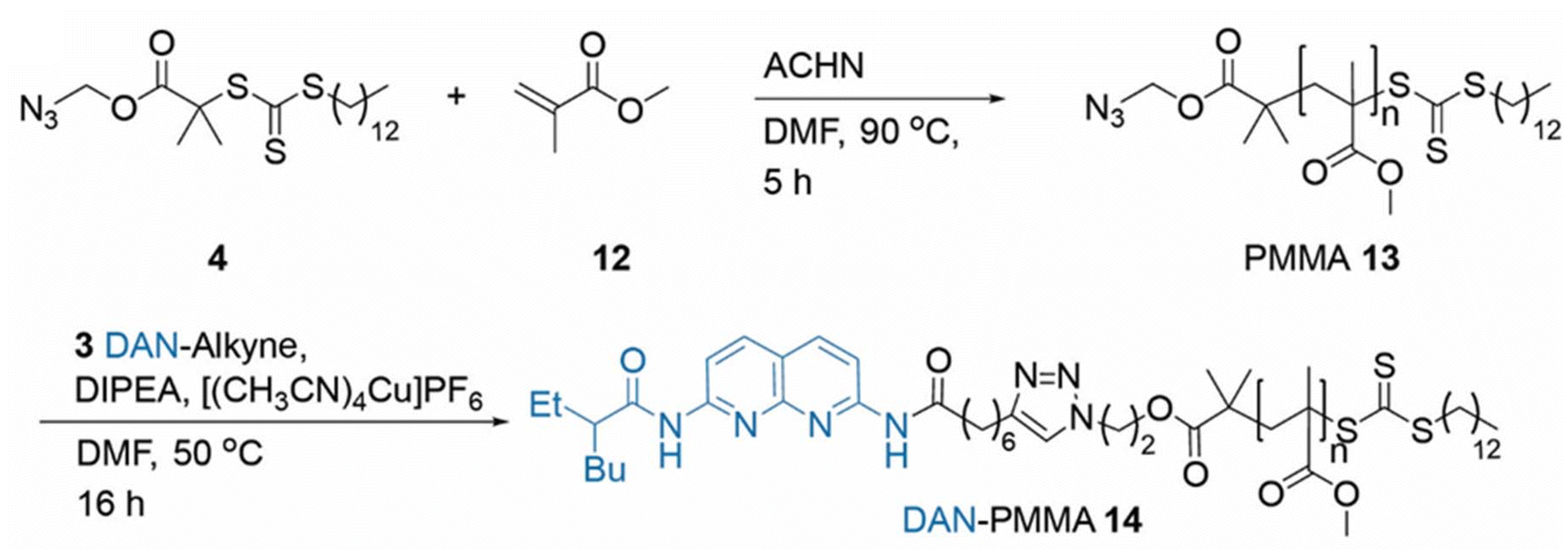 | ||
| Scheme 3 Synthesis of PMMA 13 using azide RAFT agent 4 and post-polymerisation azide click reaction producing DAN-PMMA 14. | ||
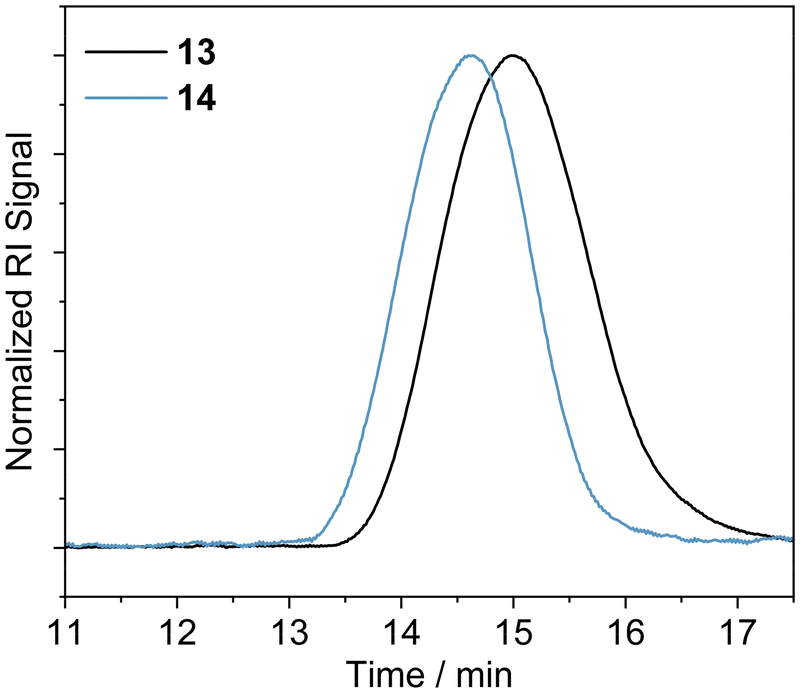 | ||
| Fig. 2 GPC traces of PMMA 13 (pre-functionalisation); and DAN-PMMA 14 (post-functionalisation) obtained in DMF. | ||
| Polmyer | % MMA conver. | M n (GPC) (g mol−1) | Đ (GPC) |
|---|---|---|---|
| 13 | 99 | 24600 |
1.52 |
| 14 | — | 31200 |
1.32 |
To develop a photoresponsive reconfigurable polymer system, DAN-PMMA polymer 14 was mixed with supramolecular synthon 15 (Fig. 3a) comprising ditopic azobenzene linked UPy species where the UPy is capable of forming quadruply hydrogen-bonded UPy·UPy homodimers through self-association or DAN·UPy heterodimers.37 We hypothesized that as a consequence of preferential intramolecular cyclization (as shown previously),37Z-15 would show limited interaction with DAN-PMMA 14, but that upon photoisomerization to give E-15, intermolecular dimerization would be suppressed in favour of chain extension permitting interaction between DAN-PMMA 14 and E-15. Such a change would be anticipated to result in a change in molecular weight of the polymer, effectively doubling the size of each chain via central hydrogen bonded linkages. This size change would result in a reduction in diffusion coefficient in the solution, meaning DOSY NMR would provide an ideal tool to assess this behaviour. We previously established a critical concentration of ∼24 mM for the ring chain equilibrium for 15 in chloroform. Therefore to limit supramolecular polymer formation by 15, samples of DAN-PMMA 14 and 15 prepared at lower concentrations (8 mM and 4 mM respectively). At a concentration of 4 mM, DOSY measurements on 15, indicate it adopts a cyclic monomer in the E form and tends towards a cyclic dimer in the Z form as previously published.37 Samples were first exposed to green light (530 nm) for 10 minutes to maximize the concentration of Z isomer (photostationary state, 530 nm = 71:29 Z:E), then DOSY NMR was performed (Fig. 3c). This process was repeated after exposure to blue light (405 nm) for 10 minutes to maximize the concentration of E isomer (photostationary state at 405 nM = 23:73, Z:E, Fig. 3b). A reduction in diffusion coefficient was observed after blue light irradiation, to confirm this behaviour was reversible, irradiation was performed using each wavelength again, and DOSY spectra recorded. The data obtained showed good evidence of a reversible change in diffusion coefficient upon irradiation (Fig. 3c and d). When converted to molecular weight it became clear that the species approximately doubles in size after blue light irradiation (Fig. 3d). Using the diffusion co-efficient and the Stokes Einstein relationship we estimated a hydrodynamic radius (rs) for the assembly of DAN-PMMA 14 in the presence of Z-15 ∼34 nm and (E-15) ∼43 nm.
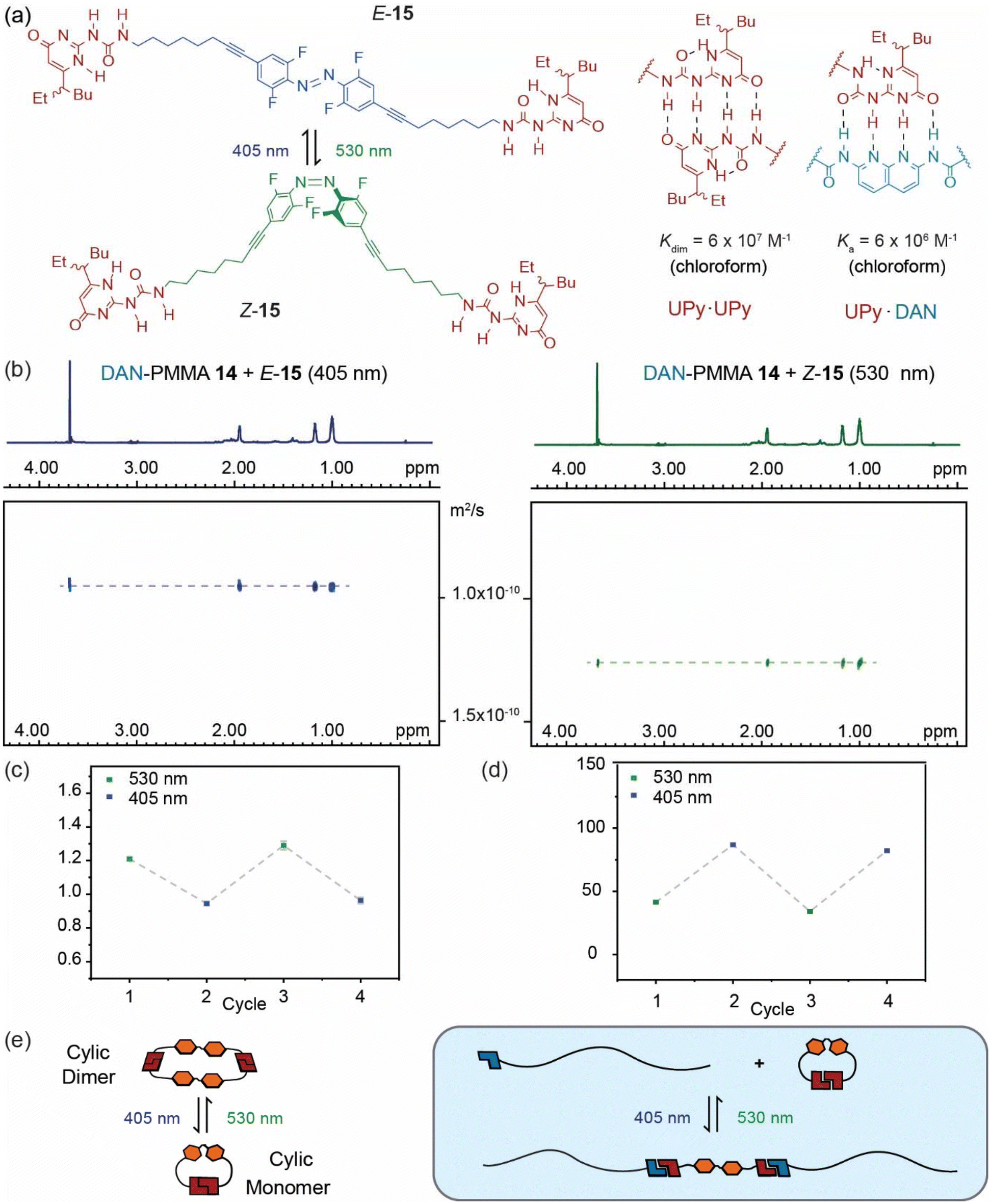 | ||
| Fig. 3 Controlling polymer weight using a hydrogen-bonding photoswitch: (a) structure of E- and Z-15 alongside structures and affinities of UPy·UPy homodimer and UPy·DAN heterodimer (b) DOSY spectra for DAN-PMMA 14 and 15 after irradiation at 405 nm (blue) and 530 nm (green); (c) diffusion coefficient of DAN-PMMA 14 and 15 after 2 cycles of green and blue light irradiation; (d) molecular weight of DAN-PMMA 14 and UPy·UPy foldamer 15 after 2 cycles of green and blue light irradiation; (e) schematic illustrating reversible rearrangement of 15 in the absence and presence of DAN-PMMA 14 in response to blue and green light. | ||
Conclusions
In this work, we successfully synthesised a range of RAFT agents by reacting alkyne functionalised NAP, UPy and DAN precursors with an azide functional RAFT agent by alkyne/azide click chemistry. The resulting NAP and UPy RAFT agents were successfully used for the RAFT polymerisation of methyl acrylate, yielding low molar mass dispersity polymers. However, neither reached 100% conversion, with the NAP only reaching 31%. Despite this, each polymer was successfully purified, resulting in polymers with terminal NAP and UPy functionalities (9 and 10). On the contrary, the attempt to polymerise MA with the DAN functionalised RAFT agent was unsuccessful, with no conversion obtained. However, in this case polymers bearing terminal HBMs could be obtained by post polymerization addition of the DAN to poly(methyl-methacrylate) to give 14.We then demonstrated that a DAN-terminated poly(methyl-methacrylate) 14 could undergo a reversible change in molecular weight upon photoisomerization of Z-15 to E-15. This was due to the ability of Z-15 to readily cyclize through homo-dimerization of its UPy motif and the preference for E-15 to satisfy the hydrogen-bonding requirements of its UPy through intermolecular heterodimerization with the DAN motif of 14. Such behaviour represents a first step in being able to reversibly reconfigure polymer architecture and subsequent assembly; for instance, our future studies will be geared towards photoswitchable reconfigurations that lead to diblock formation and subsequent phase separation.
Author contributions
N. J. W. and A. J. W. conceived and designed the research program, E. M. H and Y. J. designed studies and performed the research. The manuscript was written by A. J. W. and edited into its final form by N. J. W. and A. J. W. with contributions from all authors.Data availability
All relevant data are included in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by The Leverhulme Trust [RPG-2019-169].References
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CrossRef CAS PubMed.
- L. R. Hart, J. L. Harries, B. W. Greenland, H. M. Colquhoun and W. Hayes, ACS Appl. Mater. Interfaces, 2015, 7, 8906–8914 CrossRef CAS PubMed.
- Y. Chen and Z. Guan, Chem. Commun., 2014, 50, 10868–10870 RSC.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC.
- S. Chen, Z. Li, Y. Wu, N. Mahmood, F. Lortie, J. Bernard, W. H. Binder and J. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202203876 CrossRef CAS.
- Y. Ahn, Y. Jang, N. Selvapalam, G. Yun and K. Kim, Angew. Chem., Int. Ed., 2013, 52, 3140–3144 CrossRef CAS PubMed.
- C. A. Anderson, A. R. Jones, E. M. Briggs, E. J. Novitsky, D. W. Kuykendall, N. R. Sottos and S. C. Zimmerman, J. Am. Chem. Soc., 2013, 135, 7288–7295 CrossRef CAS.
- Y. Liu, L. Wang, L. Zhao, Y. Zhang, Z.-T. Li and F. Huang, Chem. Soc. Rev., 2024, 53, 1592–1623 RSC.
- O. J. G. M. Goor, S. I. S. Hendrikse, P. Y. W. Dankers and E. W. Meijer, Chem. Soc. Rev., 2017, 46, 6621–6637 RSC.
- A. Goujon, G. Mariani, T. Lang, E. Moulin, M. Rawiso, E. Buhler and N. Giuseppone, J. Am. Chem. Soc., 2017, 139, 4923–4928 CrossRef CAS PubMed.
- T. Liu, S. Zou, C. Hang, J. Li, X. Di, X. Li, Q. Wu, F. Wang and P. Sun, Polym. Chem., 2020, 11, 1906–1918 RSC.
- C. Li, A. Iscen, H. Sai, K. Sato, N. A. Sather, S. M. Chin, Z. Álvarez, L. C. Palmer, G. C. Schatz and S. I. Stupp, Nat. Mater., 2020, 19, 900–909 CrossRef CAS PubMed.
- A. J. Wilson, Soft Matter, 2007, 3, 409–425 RSC.
- H. M. Coubrough, S. C. C. van der Lubbe, K. Hetherington, A. Minard, C. Pask, M. J. Howard, C. Fonseca Guerra and A. J. Wilson, Chem. – Eur. J., 2019, 25, 785–795 CrossRef CAS PubMed.
- H. M. Coubrough, M. Reynolds, J. A. Goodchild, S. D. A. Connell, J. Mattsson and A. J. Wilson, Polym. Chem., 2020, 11, 3593–3604 RSC.
- A. Gooch, N. S. Murphy, N. H. Thomson and A. J. Wilson, Macromolecules, 2013, 46, 9634–9641 CrossRef CAS PubMed.
- T. Park and S. C. Zimmerman, J. Am. Chem. Soc., 2006, 128, 14236–14237 CrossRef CAS PubMed.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS PubMed.
- J. M. Pollino and M. Weck, Chem. Soc. Rev., 2005, 34, 193–207 RSC.
- R. J. Thibault, P. J. Hotchkiss, M. Gray and V. M. Rotello, J. Am. Chem. Soc., 2003, 125, 11249–11252 CrossRef CAS PubMed.
- R. McHale and R. K. O'Reilly, Macromolecules, 2012, 45, 7665–7675 CrossRef CAS.
- K. E. Feldman, M. J. Kade, T. F. A. de Greef, E. W. Meijer, E. J. Kramer and C. J. Hawker, Macromolecules, 2008, 41, 4694–4700 CrossRef CAS.
- K. E. Feldman, M. J. Kade, E. W. Meijer, C. J. Hawker and E. J. Kramer, Macromolecules, 2010, 43, 5121–5127 CrossRef CAS.
- H. Sun, C. P. Kabb, Y. Dai, M. R. Hill, I. Ghiviriga, A. P. Bapat and B. S. Sumerlin, Nat. Chem., 2017, 9, 817–823 CrossRef CAS PubMed.
- F. Xu and B. L. Feringa, Adv. Mater., 2023, 35, 2204413 CrossRef CAS.
- B. J. Cafferty, R. R. Avirah, G. B. Schuster and N. V. Hud, Chem. Sci., 2014, 5, 4681–4686 RSC.
- Y. Li, T. Park, J. K. Quansah and S. C. Zimmerman, J. Am. Chem. Soc., 2011, 133, 17118–17121 CrossRef CAS.
- D. Görl, B. Soberats, S. Herbst, V. Stepanenko and F. Würthner, Chem. Sci., 2016, 7, 6786–6790 RSC.
- S.-L. Li, T. Xiao, W. Xia, X. Ding, Y. Yu, J. Jiang and L. Wang, Chem. – Eur. J., 2011, 17, 10716–10723 CrossRef CAS PubMed.
- J.-F. Xu, Y.-Z. Chen, D. Wu, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Angew. Chem., Int. Ed., 2013, 52, 9738–9742 CrossRef CAS.
- T.-G. Zhan, M.-D. Lin, J. Wei, L.-J. Liu, M.-Y. Yun, L. Wu, S.-T. Zheng, H.-H. Yin, L.-C. Kong and K.-D. Zhang, Polym. Chem., 2017, 8, 7384–7389 RSC.
- S. Tan, Y. Sha, T. Zhu, M. A. Rahman and C. Tang, Polym. Chem., 2018, 9, 5395–5401 RSC.
- L. Wei, S.-T. Han, T.-T. Jin, T.-G. Zhan, L.-J. Liu, J. Cui and K.-D. Zhang, Chem. Sci., 2021, 12, 1762–1771 RSC.
- S.-T. Han, H.-Y. Duan, L.-Y. Chen, T.-G. Zhan, L.-J. Liu, L.-C. Kong and K.-D. Zhang, Chem. – Asian J., 2021, 16, 3886–3889 CrossRef CAS PubMed.
- S.-T. Han, H.-Y. Duan, T.-G. Zhan, X.-B. Hu, L.-C. Kong and K.-D. Zhang, Chin. Chem. Lett., 2023, 34, 107639 CrossRef CAS.
- E. M. Hilton, M. A. Jinks, A. D. Burnett, N. J. Warren and A. J. Wilson, Chem. – Eur. J., 2024, 30, e202304033 CrossRef CAS.
- P. S. Corbin and S. C. Zimmerman, J. Am. Chem. Soc., 1998, 120, 9710–9711 CrossRef CAS.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, J. Am. Chem. Soc., 1998, 120, 6761–6769 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00835a |
| ‡ Current address: UCL School of Pharmacy, University College London, 29-39 Brunswick Square, London, WC1N 1AX, UK. |
| This journal is © The Royal Society of Chemistry 2024 |