
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lewis acid ionic liquid catalysed synthesis of bioderived surfactants from β-pinene†
Philippa L.
Jacob
 a,
Fabricio
Machado
ab,
Graham A.
Rance
c,
Gary
Walker
d,
Vincenzo
Taresco
a,
Daniel J.
Keddie
a and
Steven M.
Howdle
*a
a,
Fabricio
Machado
ab,
Graham A.
Rance
c,
Gary
Walker
d,
Vincenzo
Taresco
a,
Daniel J.
Keddie
a and
Steven M.
Howdle
*a
aSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: steve.howdle@nottingham.ac.uk
bUniversity of Brasília, Institute of Chemistry, Campus Universitário Darcy Ribeiro, Brasília, DF 70910-900, Brazil
cNanoscale and Microscale Research Centre (nmRC), University of Nottingham, Nottingham, NG7 2QL, UK
dLubrizol Ltd., Hazelwood, Derby DE56 4AN, UK
First published on 7th October 2024
Abstract
Cationic polymerisation of β-pinene (βP) via earth abundant catalysis has been investigated as a route to low molar mass poly(β-pinene) (PBP) for surfactant applications. As a ‘greener’ alternative to the often hazardous and poorly abundant Lewis acid catalysts reported for the cationic polymerisation of βP, imidazolium-based Lewis acid ionic liquids have been used as catalysts for the polymerisation, yielding polymers of up to Mn = 2560 g mol−1. Iron(III) chloride (FeCl3) proved to be an effective catalyst for the transformation in a scaled-up, industrially applicable polymerisation resulting in polymers of slightly higher molar mass (Mn = 5680 g mol−1). Supercritical carbon dioxide (scCO2) proved to be an effective solvent for the purification of the polymers on a large scale, efficiently removing unreacted monomer and solvent. The unsaturated nature of the polymer has been exploited via post-polymerisation functionalisation reactions (epoxidation/hydrolysis and radical thiol–ene), endowing the polymers with hydrophilic groups. The functionalised PBPs were fully characterised, demonstrating variations in thermal properties compared to the unfunctionalised polymer. Finally, with careful balancing of the amphiphilicity, the functionalised polymers were shown to stabilise oil/water emulsions for up to two weeks, demonstrating the potential of these bioderived materials in several surfactant applications.
Introduction
Polymeric surfactants find applications in a wide range of industries and sectors, including lubricant additives and personal care as well as paints and coatings.1,2 Their highly tuneable chemistry renders them well-suited to a host of applications. However, many of the most commonly used polymeric surfactants are fossil fuel derived. With increasing regulatory pressure on the chemical industry to work towards net-zero goals, there is a need to transition towards more sustainable synthesis and the use of more sustainable feedstocks.3There are countless examples of polymeric surfactants derived from fossil fuel-based feedstocks as well as several examples of the synthesis of commonly used monomers from bioderived feedstocks. However, as proposed by Hillmyer, there are two approaches to the development of more sustainable polymers: (1) new, bioderived synthetic routes to the most commonly used monomers; and (2) conversion of renewable feedstocks into novel materials.4 Considering these approaches, there is a drive towards the use of widely available, bioderived molecules for the development of new materials.5 Terpenes present an attractive alternative to some of the more traditionally used monomers in the synthesis of polymeric surfactants. These molecules are capable of undergoing polymerisation whilst maintaining modifiable functional groups, namely alkenes, along the polymer backbone, useful for post-polymerisation functionalisation.6–10
Of particular interest in the research presented here is β-pinene (βP), found abundantly in turpentine. The global production of turpentine in 2019 was reported to be 316 kilotons, and 185 kt of this was crude sulfate turpentine (CST), a side-product of the Kraft process.2 Importantly, the use of turpentine as a monomer source does not compete with land use for food production.11
βP, a major constituent of turpentine, accounts for up to 60 wt% of turpentine depending on the type and origin of the pine tree.12 The cationic polymerisation of βP is well reported and was first published by Roberts and Day in 1950.13 This early work demonstrated the cationic polymerisation of βP using a range of Lewis acids, yielding low molar mass oligomeric poly(β-pinene) (PBP). Since then, there have been considerable developments in the polymerisation of βP with a diverse array of catalytic systems ranging from heteropolyacids to Schiff-base nickel complexes to rare earth metal catalysts.13–23
Imidazolium-based Lewis acid ionic liquids (LA-ILs) have been shown to be efficient catalysts for the cationic polymerisation of styrene as well as α-pinene.24–28 ILs also exhibit advantageous properties including low toxicity and potential recyclability.29 However, their applicability as catalysts for the cationic polymerisation of βP has not yet been investigated. In the polymerisation of α-pinene, low molar mass oligomers were obtained when the LA-ILs were used in combination with the Lewis acid SbCl3.24 Meanwhile, the LA-IL catalysed polymerisation of styrene has been shown to yield high molar mass polystyrene with reported Mn values of greater than 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1.28 Notably, in this work, iron-based imidazolium LA-ILs were shown to be particularly active towards the cationic polymerisation of styrene, particularly 1-n-butyl-3-methylimidazolium heptachlorodiferrate ([bmim]-Fe2Cl7).25,27,28,30
000 g mol−1.28 Notably, in this work, iron-based imidazolium LA-ILs were shown to be particularly active towards the cationic polymerisation of styrene, particularly 1-n-butyl-3-methylimidazolium heptachlorodiferrate ([bmim]-Fe2Cl7).25,27,28,30
Despite the wide-ranging studies on the cationic polymerisation of βP, AlCl3 in combination with adventitious water remains the industrial catalyst of choice in the synthesis of polyterpene resins.31 Whilst AlCl3 is an earth abundant catalyst, it is known to react violently with water liberating HCl gas.
There are few examples of the exploitation of the unsaturation of PBP in the synthesis of polymeric surfactants via post-polymerisation functionalisation. There are examples of the hydrogenation, doping and end-functionalisation PBP,32–34 however, the exploitation of the unsaturated repeat units of PBP is relatively unexplored as a post-polymerisation functionalisation strategy.
Herein, we demonstrate the use of earth abundant, iron-based catalysts in the polymerisation of bioderived βP. We describe the use of imidazolium-based Lewis acid ionic liquids (LA-ILs) as catalysts for the synthesis of low molar mass PBP as well as the use of iron chloride as an industrially viable alternative for a scaled-up synthesis. Supercritical carbon dioxide (scCO2) extraction has been used as a more efficient method for polymer purification.
Generally, polymers are known to be poorly soluble, but are known to swell and become plasticised in scCO2. Therefore, the removal of small molecules from polymers via scCO2 extraction is a facile route to pure materials. The plasticisation effect of scCO2 on polymers renders them more flexible, allowing scCO2 to penetrate the polymer matrix and solubilise small molecules trapped within. This very efficient route to polymer purification is also tuneable; the density of scCO2 can be easily manipulated to facilitate specific solubilisation requirements.35 Additionally, the use of scCO2 facilitates the avoidance of large volumes of traditional solvents that are conventionally used in polymer purification.36,37 scCO2 is non-flammable, non-toxic, inexpensive and readily available as an industrial side-product, and is therefore considered a ‘greener’ solvent.38
We also demonstrate the successful modification of PBP in the synthesis of bioderived polymeric surfactants and show that the post-polymerisation functionalisation yields polymeric surfactants capable of stabilising an oil/water emulsion.
Experimental
Materials
All chemicals were used as purchased without further purification unless otherwise stated. N-Methylimidazole (99%) was purchased from Alfa Aesar. 1-Chlorobutane (99%, anhydrous) and β-pinene (βP) (≥97%) were purchased from Sigma-Aldrich. Acetonitrile (≥99%), toluene (laboratory reagent grade), tetrahydrofuran (THF) (laboratory reagent grade) and iron(III) chloride (98%, anhydrous) were purchased from Fisher Scientific.Characterisation
Nuclear magnetic resonance (NMR) spectroscopy was performed using a Bruker DPX 400 MHz spectrometer operating at 400 MHz (1H) and 101 MHz (13C), assigning chemical shifts in parts per million (ppm) referenced to residual solvent. NMR samples were dissolved in acetone-d6 or CDCl3. Size exclusion chromatography (SEC) was performed in THF (HPLC grade, Fisher Scientific) as the eluent at 40 °C using two Agilent PL-gel mixed-D columns in series, an injection loop of 50 μL, and a flow rate of 1 mL min−1. A differential refractometer (DRI) was used for the detection of samples (solution containing approximately 3 mg mL−1 of polymer in THF, filtered through 0.22 μm Teflon filter). The system was calibrated using low molar-mass dispersity poly(methyl methacrylate) standards with average molar mass in the range from 540 to 1.02 × 106 g mol−1. Differential scanning calorimetry (DSC) was performed on a TA-Q2000 (TA instruments), which was calibrated with indium and sapphire standards under N2 flow (50 mL min−1). The sample (5–10 mg) was weighed into a T-zero sample pan (TA instruments) with a reference T-zero pan remaining empty. Samples were heated at a rate of 10 °C min−1, at the stated temperature ranges. To remove any thermal history, two heating cycles were recorded and the second heating cycle was used to determine the glass transition temperature (Tg). Dynamic mechanical analysis (DMA) was performed using a Triton technologies DMA using a powder pocket in single cantilever bending mode. Approximately 20 mg of polymer was loaded into a powder pocket and analysed at the stated temperatures, ranging between 18 and 200 °C depending on the region of interest. Samples were measured at 1 and 10 Hz at a heating rate of 5 °C min−1. The Tg was recorded as the peak temperature in the tanδ trace. tanδ is defined as the ratio of the loss modulus to the storage modulus and is a measure of energy dissipation in a material.39 Thermogravimetric analysis (TGA) was carried out using a TGA Q500 thermogravimetric analyser (TA Instruments). Analyses were performed from 40 to 800 °C, at a heating rate of 10 °C min−1 under a flow of air. Infrared (IR) spectra were recorded on a Bruker Alpha FTIR instrument using solid polymers with an attenuated total reflectance (ATR) module. Raman spectroscopy was performed using a HORIBA LabRAM HR Raman microscope equipped with a 785 nm laser (at ∼20 mW power), a 300 lines per mm diffraction grating and a 100× objective. The sample was prepared and measured in an NMR tube. Spectra were processed by baseline subtraction and normalisation to the intensity of the spectral maximum. Matrix-assisted laser desorption ionisation-time of flight mass spectrometry (MALDI-ToF MS) was conducted using a Bruker Autoflex Max spectrometer. The spectra were collected on low molar mass polymers (Mn < 10000 g mol−1) in positive, reflective mode. trans-2-[3-(4-tert-Butylphenyl)-2-methyl-2-propenylidene] malononitrile (DCTB) in acetonitrile was used as matrix and sodium trifluoroacetate as a cationisation salt for PBP. No salt was required for the acquisition of the EPBP mass spectrum. Both polymers were dissolved in THF during the sample preparation. Electrospray ionisation mass spectrometry (ESI-MS) was conducted using a Bruker ESI-TOF MicroTOF II by electrospray ionisation.
Synthetic procedures
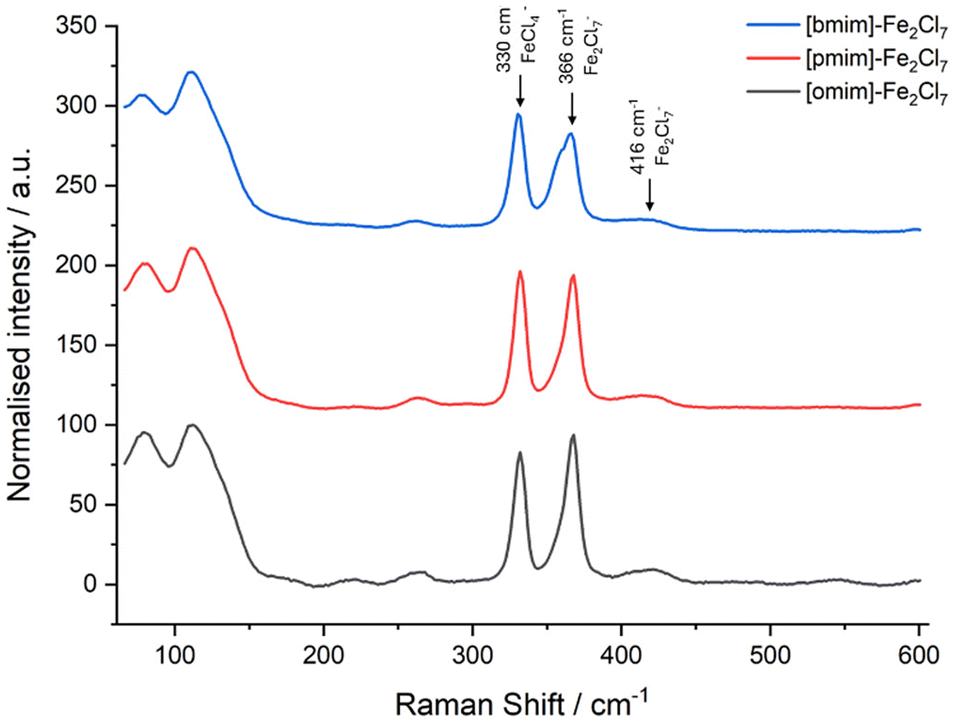 | ||
| Fig. 1 Raman spectra of the iron-based Lewis acid ionic liquids [bmim]-Fe2Cl7 (blue), [pmim]-Fe2Cl7 (red), and [omim]-Fe2Cl7 (grey). Spectra have been shifted on the Y-axis for clarity. | ||
| Entry | Solventa | Dielectric constant (T (°C)) | Temperatureb (°C) |
M
nc (g mol−1) |
Đ | Conversiond (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
a Polymerisations carried out for 18 h, [βP] = 3.2 M (except when done in bulk), [2a]:[βP] = 1:30.
b RT (room temperature) was ∼7 °C, except for DCM and bulk examples (entries 1 and 6) where it was ∼18 °C.
c From SEC analysis (THF eluent, PMMA standards).
d Conversion was determined by 1H NMR spectroscopy.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | Bulk | — | 0–RT | 890 | 2.5 | 48 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | n-Heptane | 1.92 (20)43 | 0–RT | 1160 | 1.8 | 55 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | Toluene | 2.40 (25)43 | 0–RT | 1620 | 2.7 | 83 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | EtOAc | 6.02 (20)43 | 0–RT | 600 | 1.7 | 23 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 2-MeTHF | 6.97 (25)44 | 0–RT | 510 | 2.0 | 22 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | DCM | 9.08 (20)43 | 0–RT | 2220 | 2.4 | >95 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Entrya | LA IL | [LA-IL]:[βP] |
M
nb,e (g mol−1) |
Đ , | Conversionc,e (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Polymerisations carried out at 18 °C, 18 h at 3.2 M βP in toluene (unless otherwise stated). b From SEC analysis (THF eluent, PMMA standards). c Conversion determined by 1H NMR spectroscopy. d Duplicate data of Table 1, entry 3 included for reference. e % Conversion, Mn and Đ are averages of three measurements. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1d | 3a [bmim]-Fe2Cl7 | 1:30 |
1620 | 2.7 | 83 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 3b [pmim]-Fe2Cl7 | 1:30 |
2430 | 2.1 | 82 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 3c [omim]-Fe2Cl7 | 1:30 |
2130 | 2.2 | 87 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 3a [bmim]-Fe2Cl7 | 1:80 |
2230 | 1.8 | 27 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 3b [pmim]-Fe2Cl7 | 1:80 |
2560 | 1.8 | 21 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 3c [omim]-Fe2Cl7 | 1:80 |
1860 | 2.1 | 37 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 3a [bmim]-Fe2Cl7 | 1:140 |
1230 | 1.7 | 8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 3b [pmim]-Fe2Cl7 | 1:140 |
1820 | 1.6 | 6 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | 3c [omim]-Fe2Cl7 | 1:140 |
1600 | 1.6 | 13 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Entrya | FeCl3:βP (molar ratio) |
Conversionb (%) |
M
nc,d (g mol−1) |
Đ , | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Reactions carried out in toluene, 0–18 °C, 24 h. b Determined by 1H NMR spectroscopy. c From SEC analysis (THF eluent, PMMA standards). d Values in parentheses indicate the measured values after purification by scCO2 extraction. e Reaction caried out on 2 mL scale (βP). f Reaction scaled up to 40 mL (β pinene) and run for 24 h. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1e | 1:30 |
>95 | 1310 | 4.5 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2e | 1:80 |
>95 | 2210 | 3.2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3e | 1:140 |
15 | 1080 | 3.3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4f | 1:30 |
>95 | 1630 (1410) | 5.7 (1.7) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5f | 1:100 |
>95 | 5680 (6330) | 2.4 (1.9) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
βP was filtered through a basic alumina column and degassed for 30 minutes prior to polymerisation. [bmim]-Fe2Cl7 (0.212 g, 0.423 mmol) (1:30 IL:βP molar ratio) was weighed into an oven dried glass vial equipped with a magnetic stirrer bar. The vial was sealed with a suba-seal and underwent three vacuum/argon cycles. Anhydrous toluene (4 mL) was added to the vial and stirred at 500 rpm on ice to disperse the catalyst. βP (2.00 mL, 12.7 mmol) was added to the vial and stirred on ice. The vial was then allowed to warm to room temperature over 24 h. After 24 h, the reaction was stopped by the addition of a few drops of 0.1 M sodium hydroxide solution, or by the addition of activated charcoal. The polymer solution was washed with deionised water (×3) and the solvent was removed under reduced pressure.
The same general procedure was followed for the polymerisation of βP using FeCl3, with amounts of FeCl3 altered as per Table 2. Solutions containing FeCl3 were sonicated prior to the addition of the monomer to ensure good dispersion of the initiator.
Purification A (precipitation). The polymer solution was concentrated by removal of solvent in vacuo and then added dropwise to ice-cold methanol (1
:4 ratio of toluene:methanol). Upon addition of the solution to methanol, the polymer precipitated out of solution. The precipitated polymer was stored overnight at −20 °C and subsequently purified by centrifugation at 4000 rpm. The supernatant was removed, and the polymer was dried in vacuo.
Purification B (scCO2 extraction). The polymer/monomer solution (20 mL) was loaded into a 60 mL autoclave equipped with an overhead stirrer. The reaction vessel was heated to 50 °C and pressurised to 207 bar with CO2 whilst stirring at 300 rpm. The reaction mixture was left to solubilise for approximately 2 h. After this time, the solvent and unreacted monomer were extracted using supercritical CO2 for 40 minutes by maintaining a constant flow of CO2 into and out of the autoclave at the stated pressure. After 40 minutes, the reaction vessel was left at 207 bar for another hour to solubilise any remaining solvent or monomer and was extracted again for 30 minutes. After this time, the autoclave was left to cool, then vented. The product was a pale yellow/white powder.
PBP (20.00 g, 0.1470 mol w.r.t repeating monomer unit, 136 g mol−1) was weighed into a round bottom flask equipped with a magnetic stirrer bar. The polymer was dissolved in dichloromethane (200 mL) and stirred over ice for 20 minutes. Once cool, meta-chloroperoxybenzoic acid (mCPBA) (15.22 g, 88.27 mmol, 0.6 equiv. w.r.t repeating monomer unit) dissolved in DCM was added to the reaction mixture dropwise whilst stirring over ice. The reaction was stirred for 2 h over ice following the addition and was then allowed to warm to room temperature and was stirred for a further 22 h. After this time, the reaction was filtered, and mixture washed with Na2CO3 (×1) then deionised water (×2). The solvent was then removed under reduced pressure. The polymer was redissolved in THF and was precipitated into cold methanol. The precipitated polymer was collected by centrifugation and dried under reduced pressure. 1H NMR spectroscopy confirmed the successful epoxidation of PBP with the proton adjacent to the epoxide visible between 2.71 and 3.78 ppm. A corresponding decrease in the intensity of the alkene proton between 5.12 and 5.90 ppm was also observed.
Epoxidised poly(β-pinene) (EPBP-80) (2.00 g, 13.25 mmol w.r.t. repeating unit, 152 g mol−1) was dissolved in toluene (50 mL). Deionised water (30 mL) and p-toluenesulfonic acid (10 mol% w.r.t polyepoxide) (1.325 mmol, 0.2281 g) were added to the reaction mixture. The reaction was heated at 117 °C whilst stirring at 700 rpm for 24 hours. After 24 hours, the reaction was neutralised using NaOH and the product washed with DI water (×2). The product was dried using MgSO4 and the solvent was removed under reduced pressure. 1H NMR spectroscopy was used to confirm hydrolysis of epoxides in the polymer.
Results and discussion
To investigate the potential of PBP as the basis for polymeric surfactants, we targeted the synthesis of low molar mass PBP, using iron-based Lewis acid ionic liquid (LA-IL) catalysts as a greener alternative to conventional cationic polymerisation catalysts.Lewis acid ionic liquid synthesis
Imidazolium-based Lewis acid ionic liquids (LA-ILs) were synthesised over two steps, following adaption of a previously published method (see Scheme 1).26 Initially, reaction of 1-methylimidazole (MeIm) with a range of alkyl chlorides (1a–c), with differing alkyl chain length, in acetonitrile (MeCN) gave the precursor ILs 2a–2c as viscous liquids which were purified via liquid–liquid extraction. The structures 2a–2c were confirmed by 1H NMR spectroscopy and mass spectrometry. Subsequent reaction of the ILs 2a–2c with anhydrous iron(III) chloride (FeCl3) delivered the desired LA-ILs 3a–3c as dark coloured viscous liquids. Importantly, the presence of the Fe2Cl7− ion (with C2 symmetry) in the LA-ILs 3a–c was confirmed by Raman spectroscopy, with characteristic peaks at 367 (A mode) and 416 cm−1 (combination of two A and two B modes) observed (see Fig. 1), consistent with previous reports.41 FeCl4− (of Td symmetry), which is known to coexist with Fe2Cl7−, was also identified in the Raman spectrum at 330 cm−1 (A1 mode).41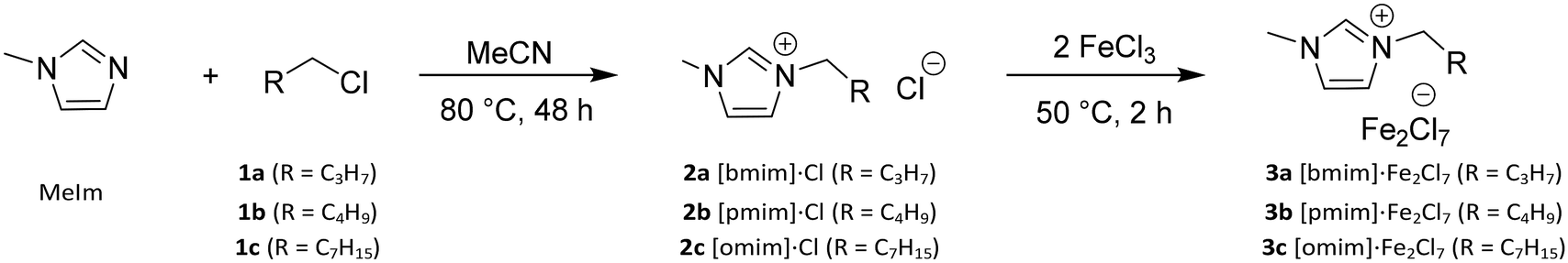 | ||
| Scheme 1 Synthesis of ionic liquids 2a–e, and iron-based Lewis acidic ionic liquids 3a–e. | ||
Polymerisation of βP using iron-based Lewis acid ionic liquids
The butyl-functional LA-IL, 1-butyl-3-methylimidazolium heptachlorodiferrate ([bmim]-Fe2Cl7) 3a has been previously shown in literature to efficiently catalyse the cationic polymerisation of styrene.25–28,30 Meanwhile, the catalytic activity of [bmim]-FeCl4 has previously been studied, showing no catalytic activity towards cationic polymerisation.28 Therefore, this [bmim]-Fe2Cl7 was initially screened as a catalyst to promote the polymerisation of βP; βP polymerises cationically, via a ring-opening mechanism which relieves ring strain of the four-membered ring (see Scheme 2(a)). Termination via proton (chain) transfer gives a mixture of endo and exo alkene end-groups (see Scheme 2(b)).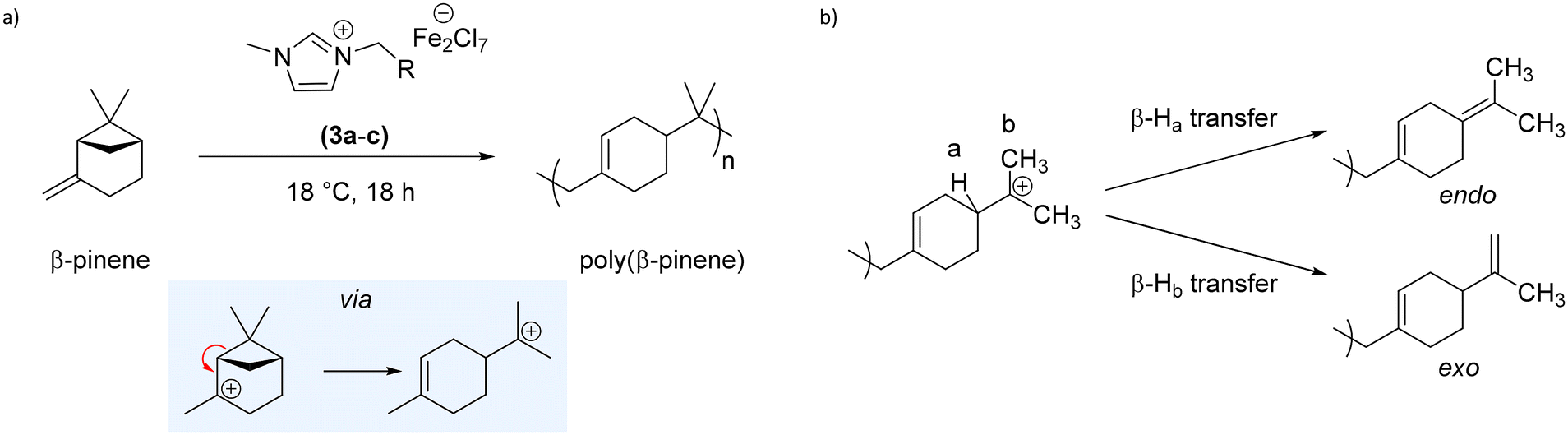 | ||
| Scheme 2 (a) Synthesis PBP using iron-based Lewis acidic ionic liquids 3a–e, and (b) pathways for the formation of endo or exo alkene end-groups via proton transfer. | ||
Effect of solvent on Lewis acid ionic liquids on the cationic polymerisation of βP
Initially we attempted polymerisation of βP, using [bmim]-Fe2Cl73a as catalyst, in solvent free conditions (bulk) at room temperature, where conversion was limited to 48% and molar mass was fairly low (890 g mol−1) (see Table 1, entry 1).This is attributed to sub-optimal dispersion of the LA-IL in the monomer due to poor solvency of βP, resulting in polymerisation confined locally to LA-IL rich regions in the reaction mixture; βP is a relatively poor solvent for the LA-IL 3a.
For cationic polymerisation the polarity of the solvent plays a critical role from a mechanistic standpoint. Polar solvents can separate ions efficiently promoting polymerisation. Non-polar solvents result in poorly separated ions which are kept together as intimate pairs, hindering polymerisation (and diminishing chain transfer). To investigate this point, a range of solvents with varying polarity were screened for the cationic polymerisation of βP. The solvents screened range in dielectric constant from 1.92 to 9.08 (see Table 1, entries 2–6). Polymerisation in n-heptane showed a marginal increase in conversion (55%) and molar mass (1160 g mol−1) (see Table 1, entry 2), when compared to bulk (see Table 1, entry 1). Switching to toluene delivered a substantial increase in conversion (83%), with higher molar mass (1620 g mol−1) (see Table 1, entry 3). Moving to the more polar solvents, ethyl acetate and 2-methyltetrahydrofuran (2-MeTHF) proved detrimental to the polymerisation, resulting in poor monomer conversion (∼20%) and low molar masses (Mn = ∼500–600 g mol−1) (see Table 1, entries 4 and 5), possibly due to higher rates of chain transfer than in the heptane and toluene cases. It is also possible that coordination of these solvents to the LA-ILs provided decent solvency, however resulted in catalyst poisoning causing poorer monomer conversion. Furthermore, these solvents are known to act as Lewis bases which may result in unwanted interactions with the catalyst or propagating cation.42 DCM, the most polar solvent tested, dispersed LA-IL significantly better than the other solvents, with the reaction proceeding vigorously to high conversion (>95%), with a significant exotherm even when cooled over ice. The polymerisation gave polymers with the highest molar mass (2220 g mol−1) of the solvents tested (see Table 1, entry 6).
Here, we attribute the increased activity of the LA-IL with βP to the increased solvency of the catalyst in DCM, giving higher molar mass polymers than the other systems. The LA-ILs were readily soluble in DCM, which can be attributed to its highly polar nature. The solvation of the LA-ILs in this solvent, combined with the polar nature of the solvent and its ability to stabilise a positive charge is thought to have contributed to the higher reactivity of this system. Overall, from the solvent screen, DCM and toluene gave the most promising results in terms of monomer conversion and molar mass. With the aim of maintaining the green credentials of the chemistry, the use of DCM in further polymerisations was avoided, in agreement with the selection of greener solvents.45 As such, all polymerisations in subsequent sections were conducted in toluene.
Effect of Lewis acid ionic liquid structure and concentration on the cationic polymerisation of βP
To investigate the effect of increased hydrophobicity of the LA-ILs, the pentyl ([pmim]-Fe2Cl7, 3b) and octyl ([omim]-Fe2Cl7, 3c) alkyl functional LA-ILs were examined as catalysts for the cationic polymerisation of βP. This was with a view to improve the polymerisation by: (i) achieving better solubility of the LA-IL in the reaction mixture, and (ii) enhancing the interaction of the LA-IL with the non-polar βP. Reactions were also performed with the butyl LA-IL [bmim]-Fe2Cl73a for comparison.Initially, the catalysts 3a–3c were screened at a LA-IL:monomer ratio ([3]:[βP]) of 1:30 and the conversion, average molar mass and molar-mass dispersity were analysed (Table 2, entries 1–3). At this relatively high LA-IL catalyst loading the conversion in all cases was high (>82%), with the more hydrophobic catalysts 3b and 3c delivering polymers with marginally higher average molar masses. Decreasing [3]:[βP] to 1:80 resulted in reduced conversion (∼20–40%), while delivering polymers of similar molar mass and dispersity to that obtained with [3]:[βP] at 1:30 (see Table 2, entries 4–6). Further reduction of [3]:[βP] to 1:140 resulted in even lower monomer conversion (∼5–15%), but gave polymers of similar molar masses as those prepared with more catalyst (see Table 2, entries 7–9).
Interestingly, across all [3]:[βP] ratios the octyl LA-IL 3c consistently gave the highest monomer conversion. This illustrates that incorporation of the longer alkyl groups is beneficial for the LA-IL catalysed polymerisation of βP. Thus, we have demonstrated the efficient polymerisation of PBP using LA-IL catalysts on a small scale where low molar mass oligomers are efficiently synthesised in mild conditions.
Scaled-up polymer synthesis using iron(III) chloride as a Lewis acidic catalyst
In the interest of an optimised scale-up of the polymerisation with a view to industrial viability, iron(III) chloride was also investigated as a catalyst for the cationic polymerisation. This earth abundant, cost-effective metal salt presents as an attractive alternative for a larger scale reaction. Here, preliminary experiments using FeCl3 at three different catalyst:monomer ratios ([FeCl3]:[βP]) were undertaken. Like the results obtained with the LA-ILs, higher catalyst concentrations delivered higher monomer conversion (see Table 3, entries 1–3). At [FeCl3]:[βP] ratios of 1:30 and 1:80 high monomer conversion is achieved (>95%). With [FeCl3]:[βP] of 1:140, the concentration of FeCl3 is insufficient, resulting in very low conversion (15%). It should be noted that these small scale FeCl3 catalysed reactions gave polymers of much higher dispersity than the analogous reactions where LA-IL catalysts were used (see Table 2).
As the [FeCl3]:[βP] ratio of 1:30 demonstrated very good conversion, these conditions were scaled up to use 40 mL of βP (in toluene). It was found that when using a catalyst concentration this high, the molar mass of the polymers was relatively low (see Table 3, entry 4). This is likely to be due to a high number of propagating cations present in the reaction mixture, resulting in the formation of many, shorter chain oligomers. Due to the small reaction scales used in this work, the stirring efficiency of the reactions may have been sub optimal. Therefore, a larger scale reaction with a lower catalyst concentration was investigated with the aim of achieving a better dispersion of catalyst as a result of more efficient stirring. With a [FeCl3]:[βP] ratio of 1:100, >95% monomer conversion is achieved while producing polymers of relatively high molar mass (5680 g mol−1) (Table 3, entry 5), possibly due to improved mixing on a larger scale. Significantly, this molar mass is much larger than that achieved for the reactions reported above. Thus, we have demonstrated that PBP can be synthesised using FeCl3 on a larger, more industrially applicable scale, however polymers of significantly higher dispersity are achieved compared to the LA-IL catalysed synthesis.
Structural analysis and purification of PBP prepared via iron-based Lewis acid catalysed cationic polymerisation
In all cases directly after synthesis, the crude polymers were obtained as highly coloured materials, containing residual βP which was clearly visible by 1H NMR spectroscopy analysis (see Fig. 2A). 13C NMR spectroscopy indicated a preference for formation of the endo-alkene terminated polymers (see Scheme 2(b) upper right and Fig. S2†), in agreement with Zaitsev's rule.46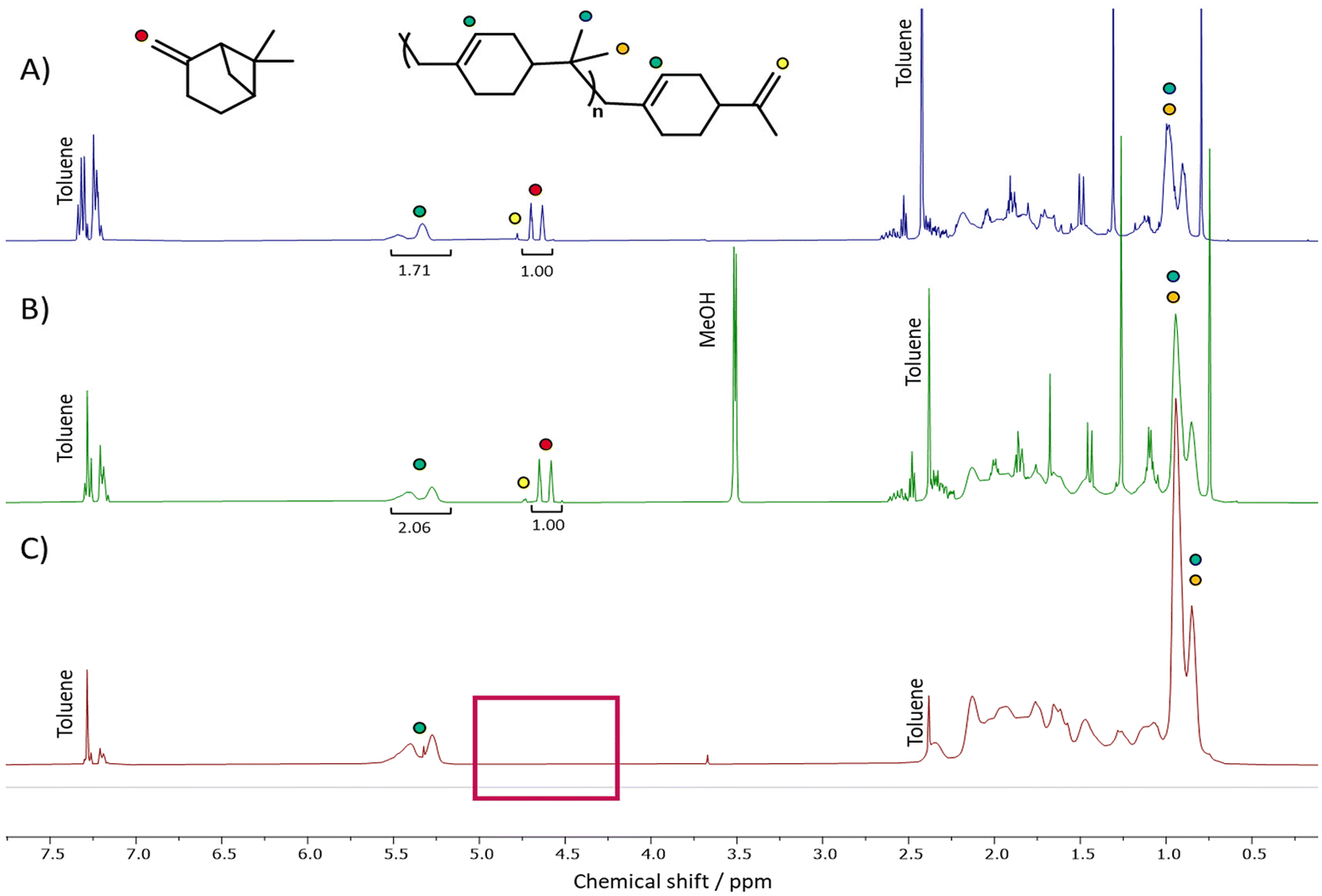 | ||
| Fig. 2 1H NMR spectra of PBP: (A) unpurified (blue trace), (B) purified by precipitation into methanol (green trace) and (C) purified by scCO2 extraction. Note that after extraction with scCO2, neither protons indicative of an exo-polymer end group nor monomer can be seen (red box). Note: Longer extraction time would have facilitated the full removal of toluene but was avoided as toluene was used as a solvent in the subsequent post-polymerisation functionalisation steps. | ||
Indeed, the absence of peaks at 108 or 149 ppm, indicative of an exo-polymer end group (Scheme 2(b) lower right),47 further support this observation. Whilst a small peak, characteristic of the exo-group was visible in the 1H NMR spectrum, the intensity of this peak is low. The endo-group, not visible by 1H NMR spectroscopy due to the tetrasubstituted alkene end group, was clearly observed in the 13C NMR spectrum. Polymer end groups were found to be comparable in the LA-IL system, however further analysis focusses on PBP from the scaled-up synthesis using FeCl3.
For all polymers, colour was removed after adsorption of the catalyst onto charcoal, giving PBP as off-white solid (Fig. S3†). Removal of the residual monomer was attempted using precipitation into an antisolvent yielding a polymer with a purity of 80% after two precipitation steps (see Fig. 2(B)). This limited removal of monomer was proposed to be the result of poor penetration of the anti-solvent into the polymer. Upon contact with the antisolvent, the polymer became solid, limiting its ability to fully mix with the antisolvent and efficiently solubilise and remove the monomer.
As a more efficient route to purify the polymers, extraction using supercritical carbon dioxide (scCO2) was investigated. Solubility testing in a view-cell demonstrated the solubility of monomeric β-pinene in scCO2 at 45 °C and 193 bar (Fig. S5†). Subsequently, scCO2 at 50 °C and 207 bar was used to remove the unreacted monomer from the polymer. A slightly higher temperature and pressure were chosen for the extraction than the conditions used in the solubility testing to account for potential pressure and temperature fluctuations that can occur during the extraction process. These conditions ensured that the extraction remained in the supercritical phase. After two, sequential extractions with scCO2,‡>95% (determined by 1H NMR spectroscopy) of the residual monomer was found to have been removed (see Fig. 2(C)), with the resultant polymers isolated as off-white dry powders.
After scCO2 extraction, SEC analysis indicated some fractionation of the samples occurred during purification; the Mn of the polymer increased, indicating that some shorter chain polymers were removed in addition to the residual βP (see entry 5, Table 3). Interestingly, the small peak at 4.70 ppm in the 1H NMR spectrum (see Fig. 2(C) and Fig. S6†), corresponding to exo- end groups, was no longer visible after extraction. This indicates the lower molar mass chains of PBP had predominantly this type of end group.
MALDI-ToF MS demonstrated the expected repeating unit of 136 g mol−1 (see Fig. 3). It also indicates the end group of the polymer does not contain a chloride atom; the peak at 1333.07 m/z corresponds to 9 repeating units of βP (136.2340 g mol−1) and one Ag+ ion (106.9046 g mol−1) from silver trifluoroacetate that was used as a cationisation agent (Fig. 3). MALDI-ToF MS analysis of PBP synthesised using [bmim]-Fe2Cl7 in a reaction that was quenched using NaOH solution also demonstrated that this polymer also does not contain chlorine (see Fig. S7†). MALDI-ToF MS also confirms that the Friedel–Crafts addition of the polymer to toluene does not occur during the polymerisation.
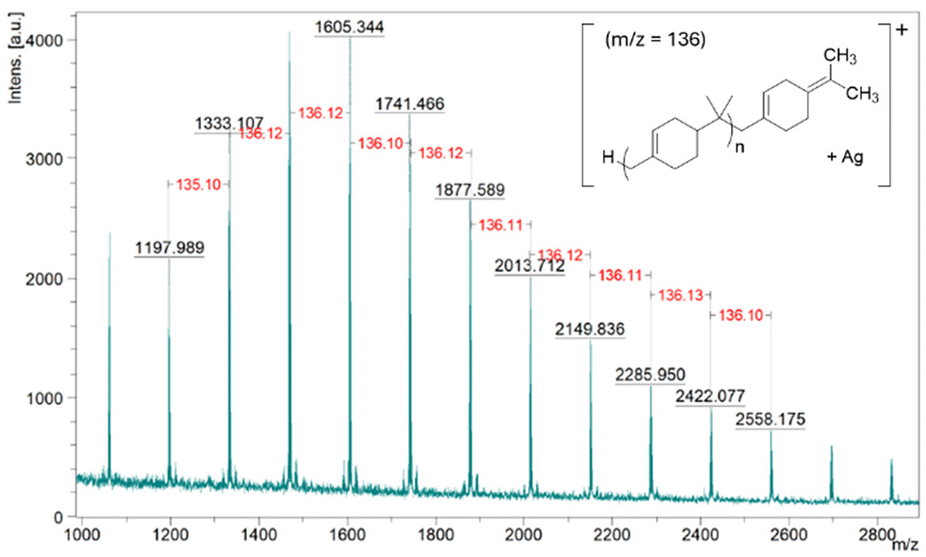 | ||
| Fig. 3 MALDI-ToF MS of PBP synthesised using FeCl3 as a catalyst. Silver trifluoroacetate was used as a cationisation agent and DCTB as a matrix. The predominant endo end group is shown, however both the exo and endo end groups are likely to be present. | ||
Post-polymerisation functionalisation
Having successfully demonstrated the small scale and scaled-up synthesis of low molar mass PBP, post-polymerisation modifications were performed to elaborate the structures towards polymeric surfactants. Strategies exploited included an epoxidation/hydrolysis, and radical thiol–ene reaction (see Scheme 3).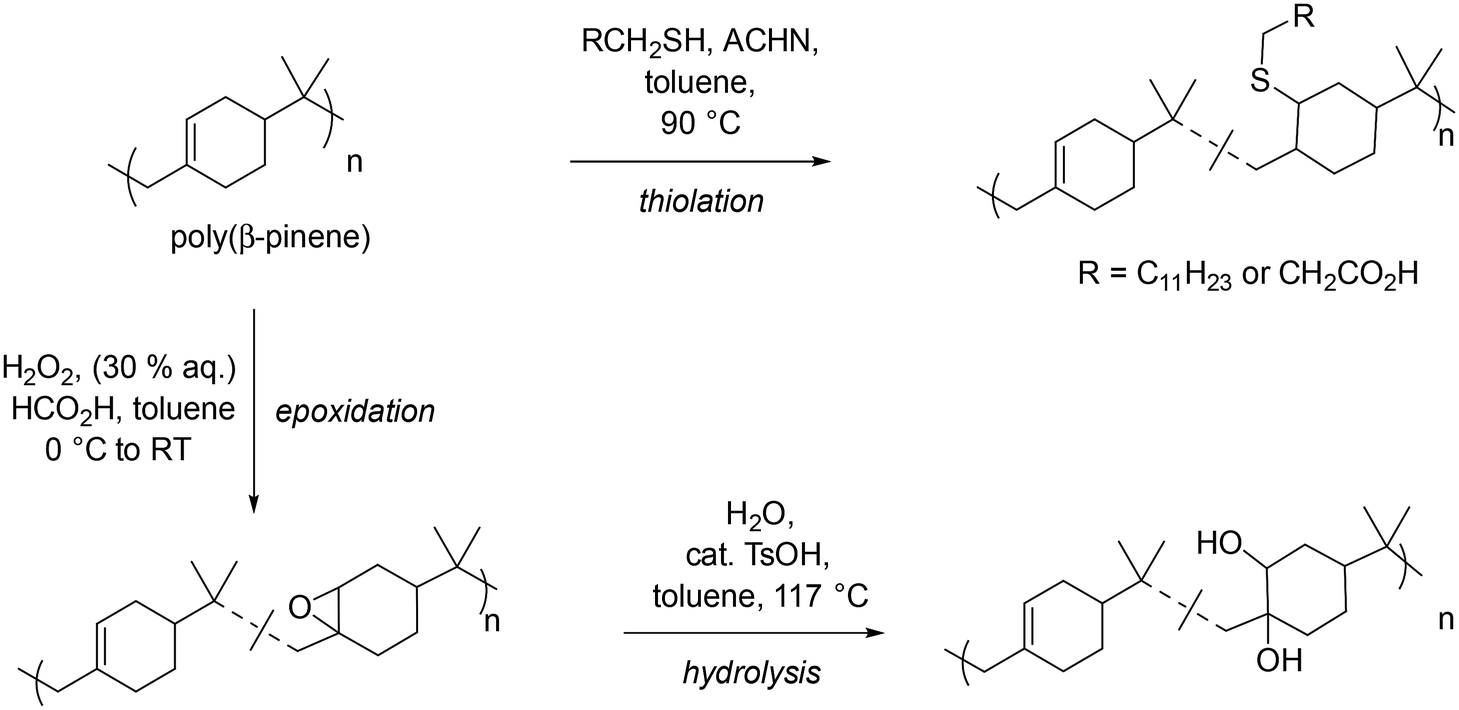 | ||
| Scheme 3 Overview of post-polymerisation modification of PBP to give a variety of functional analogues. | ||
Initially epoxidation of PBP was investigated using mCPBA as an oxidant. This procedure yielded polymers with 13 and 54% conversion of alkenes to epoxides. However, it is widely acknowledged that mCPBA offers poor atom economy, therefore, a combination of hydrogen peroxide and formic acid, which forms the oxidising agent performic acid in situ was investigated (see Scheme 3, bottom left, and Table 3 entries 1–3). In this ‘greener’ epoxidation 84% functionalisation of the alkene to epoxide was achieved. This library of functionalised polyepoxides has been characterised, highlighting the difference in thermal properties as a result of the variation in polymer functionality (Table 3 entries 1–3 respectively). Conversion of the alkenes to epoxide groups was determined by 1H NMR spectroscopy, by comparing the peak of the geminal dimethyl protons with the epoxide adjacent protons at 3.78–2.71 ppm (see Fig. S8†). MALDI-ToF MS analysis of the 84% epoxidized polymer exhibited a repeating unit of 152 m/z, indicative of one βP unit with an additional oxygen. Within the peak distributions, an m/z difference of 16 m/z was observed demonstrating that the degree of epoxidation within the polymer distribution varies (Fig. 5). A trend of increasing Tg with increasing degrees of epoxidation in both DMA and DSC analysis was observed (Table 3, entries 1–3, and Fig. S10†); all the epoxide products had higher Tgs than that of PBP (Tg = 68 °C). In the DSC analysis, it was found that in the first heating cycle, curing of the epoxide occurred, likely due to the presence of adventitious water. The epoxidized polymers were subsequently ring opened with water via an acid-catalysed (p-TsOH) hydrolysis to give polyols with varying degrees of functionality (see Scheme 3, bottom right, and Table 3, entries 4–6). Surprisingly, the ring opening reaction proved to be challenging with forcing conditions required, i.e. T > 110 °C (Scheme 3). The steric bulk associated with the geminal dimethyl groups on the repeating unit of PBP appears to limit the accessibility of the epoxide functionality for nucleophilic attack, thereby reducing its reactivity.
1H NMR spectroscopy analysis of the polyols showed extremely broad spectra which can be attributed to the many hydroxyl groups present along the polymer backbone and the large number of exchangeable protons present (see Fig. S9†). The presence of the characteristic geminal dimethyl protons demonstrates that the polymer backbone has not degraded during the reaction. IR spectroscopy further showed hydrolysis of the epoxide to an alcohol (see Fig. 4). Peaks at 3454 cm−1 and 1029 cm−1, indicative of –OH stretch and –C–OH stretches, respectively, confirm the successful hydrolysis of the epoxide groups. There was a minimal change in the Tg of the less functionalised polyols compared to less functionalised polyepoxides (see Table 3, entries 1 and 4). The more functionalised polymers saw a shift in Tg of up to approximately 20 °C when comparing the polyols to the polyepoxides (Table 3, entries 2, 3, 5 and 6). The larger shift in Tg can be attributed to the higher degree of functionality and the increase in hydrogen bonding that would be expected with a polyol compared to a polyepoxide (Table 4). Thiol–ene functionalisation was also exploited as a post-polymerisation modification of PBP with dodecanethiol and 3-mercaptopropionic acid both being successfully conjugated to the polymer via a thermally initiated radical thiol–ene reaction (see Scheme 3, top right, and Table 3, entries 7 & 8). 1H NMR spectroscopy confirmed the successful reactions as protons indicative of a thiol-adjacent –CH2 group were identified in both spectra (Fig. S11†), and this was also confirmed by HMBC analysis which showed a coupling of these protons to 13C peak at 175 ppm (Fig. S12†). Multiplicity edited HSQC analysis also corroborated the presence of the –CH2 groups (Fig. S13†). Thermal analysis of the thiol functionalised polymers indicated Tgs in line with those expected for mercaptopropionic acid (PBP-MPA) (Tg = 81 °C) and dodecanethiol (PBP-DT) (Tg = 49 °C).
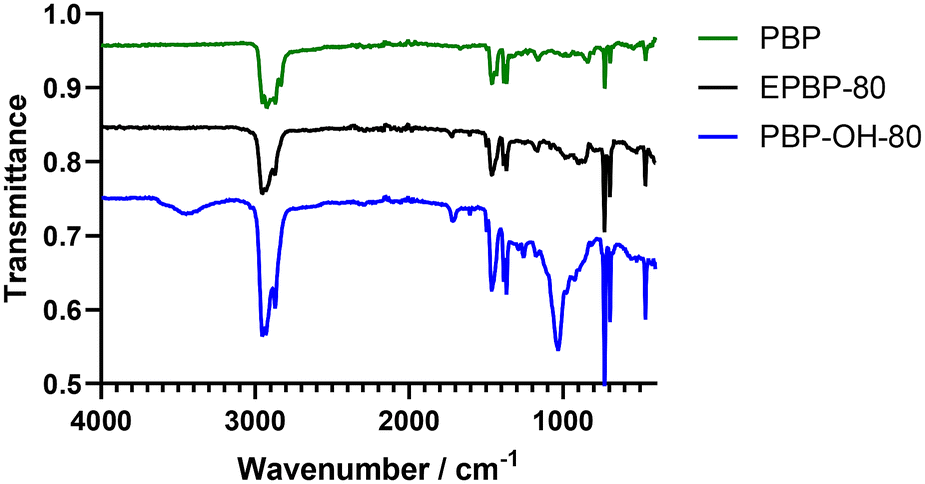 | ||
| Fig. 4 IR spectra of poly(β-pinene) (PBP) (green), poly(β-pinene) alcohol (PBP-OH-80) (blue) and poly(β-pinene) epoxide (EPBP-80) (black). | ||
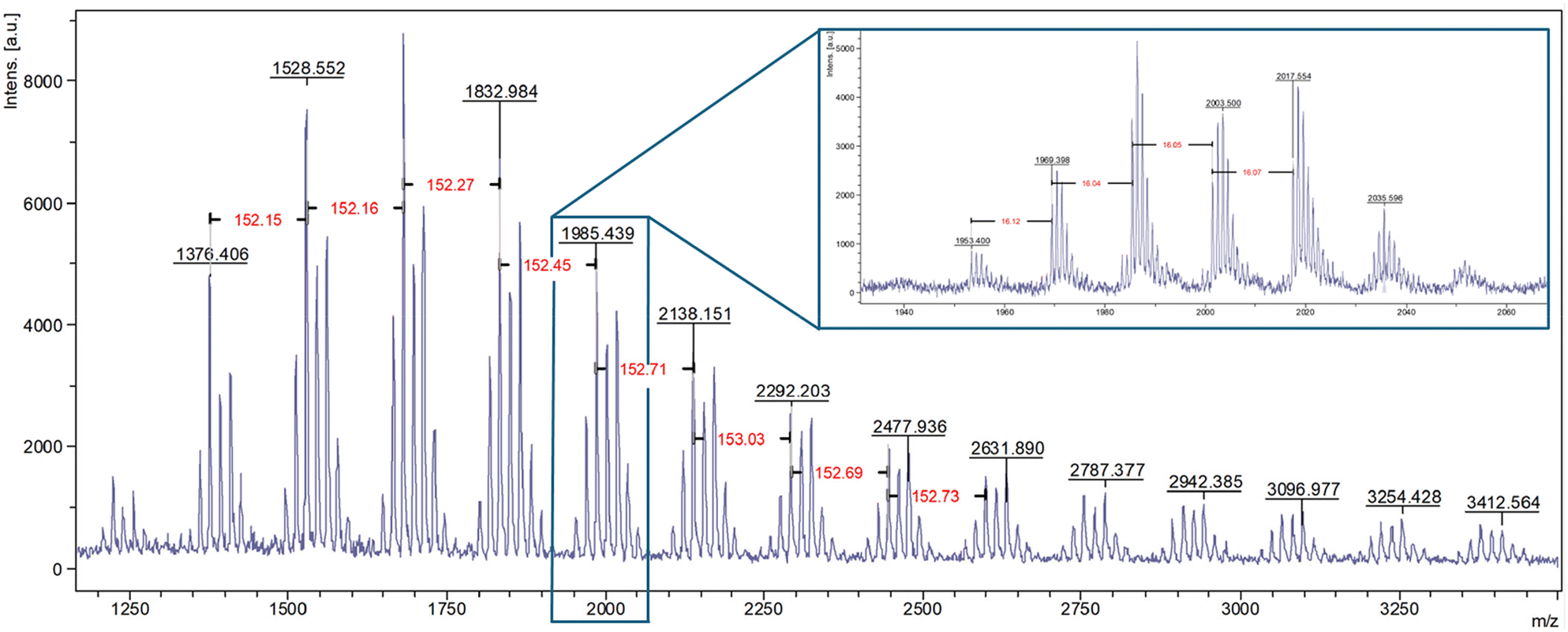 | ||
| Fig. 5 MALDI-ToF MS analysis of EPBP-80. | ||
| Entry | Post polymerisation processa | Starting material abbreviation | Product abbreviation | Product indicative structureb | Degree of functionalisationc (%) |
M
nd (g mol−1) |
Đ |
T
ge (°C) (DSC) |
T
gf (°C) (DMA) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a For reaction conditions refer to Scheme 3 and Experimental. b Percentages indicate the amount of each functionality present in the polymers. c Determined by 1H NMR analysis. d From SEC analysis (THF eluent, PMMA standards). e Determined by DSC. f Determined by DMA. g Different batches of PBP were used, which accounts for variation in molecular properties between the modified samples. h Prepared from the corresponding epoxide-functional polymer (i.e. entry 1 → 4, 2 → 5, 3 → 6). i Assuming complete hydrolysis of epoxides. j Not measured. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | Epoxidationg | PBP | EPBP-10 | P[BP87%-co-EBP13%] | 13 | 5540 | 2.0 | 78 | 92 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | PBP | EPBP-50 | P[BP46%-co-EBP54%] | 54 | 3930 | 2.2 | 130 | 121 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | PBP | EPBP-80 | P[BP16%-co-EBP84%] | 84 | 4040 | 1.8 | 126 | 131 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | Hydrolysish | EPBP-10 | PBP-OH-10 | P[BP87%-co-(PB-OH)13%] | 13i | 3560 | 2.0 | 87 | 95 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | EPBP-50 | PBP-OH-50 | P[BP46%-co-(PB-OH)54%] | 54i | 4230 | 2.1 | 103 | 159 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | EPBP-80 | PBP-OH-80 | P[BP16%-co-(PB-OH)84%] | 84i | 4650 | 1.9 | 146 | 150 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | Thiolationg | PBP | PBP-DT | P[BP68%-co-(BP-SC12)32%] | 32 | 3110 | 2.3 | 49 | —j | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | PBP | PBP-MPA | P[BP85%-co-(BP-SC2CO2H)15%] | 15 | 4620 | 1.7 | 81 | —j | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Applications testing
With the aim of synthesising polymeric surfactants, the modified polymers were tested for their ability to stabilise an oil/water emulsion with a 5 wt% loading of polymer. The stability of the emulsion was visually assessed 1 h after mixing and it was found that the highly functionalised polymers were unable to stabilise the emulsion. The highly hydrophilic nature of these polymers is thought to be the reason as these polymers will interact poorly with the oil. The less functionalised polymers (up to 50% functionalisation) were found to stabilise the emulsions well after 1 h (see Fig. 6(a)). The stability of the emulsions was visually inspected two weeks after mixing, and it was found that only the 50% epoxidized and 50% polyol polymers had successfully stabilised the emulsions after 2 weeks (see Fig. 6(b)). This demonstrates the importance of balancing the degree of functionalisation for successful surfactant preparation. The emulsions containing other, more highly functionalised additives had clearly phase separated into biphasic mixtures after the measured time. | ||
| Fig. 6 Emulsion stability study of a 1:1 mixture of oil and water with 5 wt% polymer additives after (a) 1 h, and (b) 2 weeks. | ||
Conclusions
In the work presented, it has been shown that iron-based LA-ILs are efficient catalysts for the cationic polymerisation of βP. They yield low molar mass oligomers in good conversion. These systems present a greener alternative to more typical Lewis acids used for cationic polymerisation. FeCl3 was also shown to be a viable alternative for a scaled-up polymer synthesis, however some of the molar mass control was lost, when compared to the LA-ILs systems, yielding polymers with higher dispersity. The synthesised PBP can be easily functionalised post-polymerisation to yield polyepoxides, polyols and thiol–ene adducts. Varying degrees of functionalisation were achieved whilst maintaining the green credentials of the chemistry. These polymers were shown to exhibit surfactant-like properties and in some cases can stabilise an oil/water emulsions for over two weeks. The degree of functionalisation of the polymer greatly influenced the ability of the polymer to stabilise an emulsion, highlighting the importance of fine-tuning the degree of post-polymerisation functionalisation for the intended surfactant application. It was found that over functionalisation of the polymer backbone led to poor stability of the emulsions, likely due to poor solubility of the functionalised polymers in oil. Under-functionalised polymers also demonstrated poor long-term stability of the emulsions, although short-term stability was observed. Optimal functionalisation of approximately 50% conversion of alkenes to other functionalities was shown to form the most stable emulsions, demonstrating the need for well-balanced amphiphilicity in polymeric surfactants. With the array of potential functionalisation that these PBP derivatives offer, it is envisaged that either water or oil solubility could be fine-tuned. There are a range of applications where these oligomers could serve as a chassis to deliver bespoke performance, including homecare personal care and lubricant additives.Author contributions
Philippa L. Jacob – conceptualisation, data curation, formal analysis, investigation, methodology, writing – original draft, writing – review & editing; Fabricio Machado – conceptualisation, investigation, methodology, writing – review & editing; Graham A. Rance – investigation, formal analysis, methodology, writing – review & editing; Gary Walker – project administration, supervision, writing – review & editing; Vincenzo Taresco – conceptualisation, methodology, project administration, supervision, writing – review & editing; Daniel J. Keddie – methodology, project administration, supervision, writing – original draft, writing – review & editing; Steven M. Howdle – conceptualisation, funding acquisition, project administration, supervision, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
PLJ acknowledges funding support from EPSRC Doctoral Prize [Grant Number EP/W524402/1]. PLJ also acknowledges funding support from the Engineering and Physical Sciences Research Council and SFI Centre for Doctoral Training in Sustainable Chemistry [Grant Number EP/S022236/1]. VT would like to thank the University of Nottingham for his Nottingham Research Fellowship. The authors are grateful to the Nanoscale and Microscale Research Centre (nmRC) for providing access to Raman spectroscopy facilities. We also thank the technical staff at University of Nottingham, particularly Mr Richard Wilson for specialist support with high pressure equipment.References
- P. Raffa, D. A. Wever, F. Picchioni and A. A. Broekhuis, Chem. Rev., 2015, 115, 8504–8563 CrossRef CAS.
- L. Canoira, D. Donoso, D. Bolonio, C. Bumharter and M. Lapuerta, Energy Fuels, 2023, 37, 15843–15854 CrossRef CAS.
- G.-J. M. Gruter, Curr. Opin. Green Sustain. Chem., 2023, 40, 100743 CrossRef CAS.
- M. A. Hillmyer, Science, 2017, 358, 868–870 CrossRef CAS PubMed.
- P. Foley, A. Kermanshahi pour, E. S. Beach and J. B. Zimmerman, Chem. Soc. Rev., 2012, 41, 1499–1518 RSC.
- L. Valencia, F. J. Enríquez-Medrano, H. R. López González, R. Handa, H. S. Caballero, R. M. Carrizales, J. L. Olivares-Romero and R. E. Díaz de León Gómez, RSC Adv., 2020, 10, 36539–36545 RSC.
- H. Chao, T. Yoo and S. Nehache, Fina Technology Inc., 2019 Search PubMed , EP3668907A1.
- P. Sahu and A. K. Bhowmick, Ind. Eng. Chem. Res., 2019, 58, 20946–20960 CrossRef CAS.
- J. Zhang, C. Aydogan, G. Patias, T. Smith, L. Al-Shok, H. Liu, A. M. Eissa and D. M. Haddleton, ACS Sustainable Chem. Eng., 2022, 10, 9654–9664 CrossRef CAS PubMed.
- A. Matic, A. Hess, D. Schanzenbach and H. Schlaad, Polym. Chem., 2020, 11, 1364–1368 RSC.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS.
- M. Gscheidmeier and H. Fleig, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2000, pp. 537–550, DOI:10.1002/14356007.a27_267.
- W. J. Roberts and A. R. Day, J. Am. Chem. Soc., 1950, 72, 1226–1230 CrossRef CAS.
- P. Yu, A.-L. Li, H. Liang and J. Lu, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3739–3746 CrossRef CAS.
- M. Akeb, A. Harrane and M. Belbachir, Green Mater., 2018, 6, 58–64 CrossRef.
- A. Moulkheir, A. Harrane and M. Belbachir, J. Appl. Polym. Sci., 2008, 109, 1476–1479 CrossRef CAS.
- A. V. Miroshnichenko, V. V. Tumanov, V. M. Menshov and W. A. Smit, J. Polym. Res., 2012, 19, 9884 CrossRef.
- H. Zhu, Z. Liu, X. An and F. Lei, React. Kinet., Mech. Catal., 2010, 100, 355–361 CAS.
- Z. Liu, S. Cao, S. Wang, W. Zeng and T. Zhang, React. Kinet., Mech. Catal., 2014, 111, 577–590 CrossRef CAS.
- Y. Karasawa, M. Kimura, A. Kanazawa, S. Kanaoka and S. Aoshima, Polym. J., 2015, 47, 152–157 CrossRef CAS.
- M. Hayatifar, F. Marchetti, G. Pampaloni, Y. Patil and A. M. R. Galletti, Catal. Today, 2012, 192, 177–182 CrossRef CAS.
- R. P. F. Guiné and J. A. A. M. Castro, J. Appl. Polym. Sci., 2001, 82, 2558–2565 CrossRef.
- J. Lu, M. Kamigaito, M. Sawamoto, T. Higashimura and Y.-X. Deng, J. Appl. Polym. Sci., 1996, 61, 1011–1016 CrossRef CAS.
- S. Liu, L. Zhou, S. Yu, C. Xie, F. Liu and Z. Song, Biomass Bioenergy, 2013, 57, 238–242 CrossRef CAS.
- R. C. Alves, T. Agner, T. S. Rodrigues, F. Machado, B. A. D. Neto, C. da Costa, P. H. H. de Arajo and C. Sayer, Eur. Polym. J., 2018, 104, 51–56 CrossRef CAS.
- G. V. S. Dutra, W. S. Neto, P. H. H. de Arajo, C. Sayer, B. A. d. S. Neto and F. Machado, Polimeros, 2021, 31, 1–14 Search PubMed.
- V. M. B. Patrocinio, T. Agner, G. V. S. Dutra, F. Machado, B. A. D. Neto, P. H. H. Arajo and C. Sayer, Macromol. React. Eng., 2019, 13, 1–7 Search PubMed.
- T. S. Rodrigues, F. Machado, P. M. Lalli, M. N. Eberlin and B. A. D. Neto, Catal. Commun., 2015, 63, 66–73 CrossRef CAS.
- S. K. Singh and A. W. Savoy, J. Mol. Liq., 2020, 297, 112038 CrossRef CAS.
- G. V. S. Dutra, T. S. Teixeira, G. A. Medeiros and P. V. a. Abdelnur, Ind. Eng. Chem. Res., 2020, 59, 21685–21699 CrossRef CAS.
- R. T. Mathers and S. P. Lewis, in Green Polymerization Methods, 2011, pp. 89–128, DOI:10.1002/9783527636167.ch5.
- K. Satoh, A. Nakahara, K. Mukunoki, H. Sugiyama, H. Saito and M. Kamigaito, Polym. Chem., 2014, 5, 3222–3230 RSC.
- P. Vippa, H. Rajagopalan and M. Thakur, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 3695–3698 CrossRef CAS.
- J. Lu, M. Kamigaito, M. Sawamoto, T. Higashimura and Y.-X. X. Deng, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 1423–1430 CrossRef CAS.
- M. v. S. Kemmere, M. Cleven, A. van Herk and J. Keurentjes, Ind. Eng. Chem. Res., 2002, 41, 2617–2622 CrossRef CAS.
- A. R. Goddard, S. Pérez-Nieto, T. M. Passos, B. Quilty, K. Carmichael, D. J. Irvine and S. M. Howdle, Green Chem., 2016, 18, 4772–4786 RSC.
- A. I. Cooper, J. Mater. Chem., 2000, 10, 207–234 RSC.
- X. Zhang, S. Heinonen and E. Levnen, RSC Advances, 2014, 4, 61137–61152 RSC.
- PerkinElmer, Dynamic Mechanical Analysis (DMA) – A Beginner's Guide, https://resources.perkinelmer.com/lab-solutions/resources/docs/app_thermaldynmechanalybasicspart1.pdf, (accessed 23rd May, 2024).
- M. Y. Abduh, M. Iqbal, F. Picchioni, R. Manurung and H. J. Heeres, J. Appl. Polym. Sci., 2015, 132, 42591 CrossRef.
- M. S. Sitze, E. R. Schreiter, E. V. Patterson and R. G. Freeman, Inorg. Chem., 2001, 40, 2298–2304 CrossRef CAS PubMed.
- R. F. Storey, C. L. Curry and L. K. Hendry, Macromolecules, 2001, 34, 5416–5432 CrossRef CAS.
- Sigmaaldrich.com, Physical properties of solvents, https://www.sigmaaldrich.com/deepweb/assets/sigmaaldrich/marketing/global/documents/614/456/labbasics_pg144.pdf, (accessed 23rd May, 2024).
- D. F. Aycock, Org. Process Res. Dev., 2007, 11, 156–159 CrossRef CAS.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. R. McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- J. J. Li, in Name Reactions, ed. J. J. Li, Springer International Publishing, Cham, 2014, pp. 650–651, DOI:10.1007/978-3-319-03979-4_296.
- N. A. Kukhta, I. V. Vasilenko and S. V. Kostjuk, Green Chem., 2011, 13, 2362 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00925h |
| ‡ For extraction with supercritical carbon dioxide (scCO2) a custom built 1 L high pressure autoclave was used (see Fig. S4†). |
| This journal is © The Royal Society of Chemistry 2024 |