
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceScalable design of uniform oligourethanes for impact study of chain length, sequence and end groups on thermal properties†
Jens
Van Hoorde
 ,
Nezha
Badi
* and
Filip E.
Du Prez
*
,
Nezha
Badi
* and
Filip E.
Du Prez
*
Polymer Chemistry Research Group, Centre of Macromolecular Chemistry (CMaC), Department of Organic and Macromolecular Chemistry, Faculty of Sciences, Ghent University, Krijgslaan 281 S4, 9000 Ghent, Belgium. E-mail: Filip.DuPrez@UGent.be; Nezha.Badi@UGent.be
First published on 10th October 2024
Abstract
The full potential of sequence-defined macromolecules remains unexplored, hindered by the difficulty of synthesizing sufficient amounts for the investigation of the properties of such uniform structures and their derived materials. Herein, we report the bidirectional synthesis and thermal behavior analysis of sequence-defined oligourethanes. The synthesis was conducted on a large scale (up to 50 grams) using a straightforward protocol, yielding uniform macromolecules as validated by NMR, ESI-MS and SEC. With this approach, a library of uniform oligourethanes (up to the octamers) was produced using two structural units: a hydrogen-bonding carbamate and a methyl-substituted alternative structure. By varying the chain length, monomer sequence and functionality, we were able to perform a systematic study of the impact of hydrogen bonding on the thermal properties of polyurethanes. Thermal analysis of the discrete oligomers using DSC revealed that both the molecular weight and microstructure significantly affect the glass transition and melting temperatures. TGA measurements also revealed differences in the thermal stability of the oligomers, underscoring the significance of the primary structure of polyurethanes. Additionally, the influence of the terminal groups on the degradation pathway was assessed via pyrolysis-GC-MS, which specifically highlighted the increased thermal stability in the absence of hydroxyl end groups. This work shows the interest of using sequence-defined synthetic macromolecules for the elucidation of structure–property relationships and thereby facilitates their fine-tuning towards specific material applications.
Introduction
Hydrogen bonds are an important class of non-covalent interactions that can greatly impact the performance of a polymeric material. A prime example of this is spider silk, one of nature's most resilient protein fibers, which possesses an exceptional strength while maintaining high deformation.1,2 This extraordinary behavior arises from the hierarchical structure of the material, which comprises glycine-containing amorphous regions and alanine-rich domains. The aggregation of the latter into crystalline regions adds mechanical strength and hardness through physical crosslinks, a phenomenon that is responsible for the high strength of polyurethanes (PUs), one of the most widely used commodity plastics.3 The application of PU-based materials ranges from soft foams and mattresses to thermal insulation for buildings, leading to an estimated annual consumption of about 20 million metric tons.4 The popularity of PUs is driven by their superior and variable mechanical properties compared to alternative polymers, a characteristic that is primarily linked to their strong structure–property relationships.3,5–7 Indeed, within their structure, the segregation of the hard segments, which are crystalline regions formed by the strong hydrogen bonds between carbamate and urea groups, plays a crucial role in influencing the material's behavior. Given the vital role of these non-covalent interactions in dictating material properties, numerous efforts have been dedicated to the study of their thermal and material properties.8–13 However, due to the inherent dispersity resulting from both the applied polyols and the step-growth polymerization process, extracting exact fundamental information on the structure–property relationship proves challenging.Recently, the issue of dispersity has been addressed through the introduction of sequence-defined macromolecules in polymer science.14–16 Aiming to mimic nature's biopolymers, monodisperse macromolecules with a defined monomer order were developed, targeting applications where the primary structure is of utmost importance, such as catalysis, self-assembly and data-storage.17–26 Such uniform polymers allow for a thorough examination of structure–property relationships, which cannot be properly achieved by using conventional polymers or polymers derived from reversible-deactivation radical polymerizations (RDRPs), due to their dispersity. Several research groups have already demonstrated this potential in their studies.19,27–30 For example, the group of Alabi investigated the duplex formation of sequence-defined oligomers and found that the assembly is highly dependent on the positioning and valency of the complementary hydrogen binding partners.27 Our own research group has also provided an example where the structure-dependent binding affinity has been clearly demonstrated for uniform antibody-recruiting macromolecules.28
The synthesis of these discrete polymers involves iterative protocols, progressively expanding the polymer chain by one unit after each cycle. Throughout these iterations, it is essential to ensure that all steps reach complete conversion to prevent the emergence of dispersity or irregularities in the sequence, which is often achieved by choosing efficient chemistries and using an excess of reagents. Since the remaining reagents may disrupt subsequent couplings, it is imperative to conduct a work-up after each step. The introduction of solid supports significantly simplifies this purification, requiring only a straightforward filtration and washing step to remove all waste products effectively.31,32 However, some drawbacks of this approach include the high cost of the resins, their restricted loading capacity, and the need for a large excess of often expensive reagents, which hamper the scalability of the synthesis. Transitioning from a solid to a soluble support addresses several of these challenges by reducing the excess reagents and costs related to the support, while simultaneously providing more homogeneous reaction conditions. Furthermore, this approach also allows for direct characterization without the need for chain cleavage, and the purification is limited to precipitation.33,34 Nevertheless, a supportless synthesis remains the most advantageous approach due to its increased atom efficiency and the elimination of the often cumbersome support synthesis. Recently, this strategy was exploited by our group and others for the synthesis of sequence-defined oligourethanes.17,35–37
In this work, the approach has been adapted to synthesize these uniform macromolecules in a scalable manner from aminoethanol and using a coupling agent. Not only has the protocol been modified to allow a bidirectional growth of the oligomer chain but the purification is also limited to a single extraction or precipitation step (Fig. 1). Two structural units have been selected in this study, i.e. one containing a regular N–H carbamate unit with hydrogen bonding capacity, and one methyl-substituted counterpart that lacks this capability. A detailed exploration of the structure–property relationships of a series of oligomers with varying chain length, sequences, and end-group functionalities was performed to provide an in-depth understanding for the precise impact of hydrogen bonding on polyurethane thermal characteristics. By exploring the interplay between molecular design and thermal properties, this study paves the way for tailoring material performance to specific application requirements.
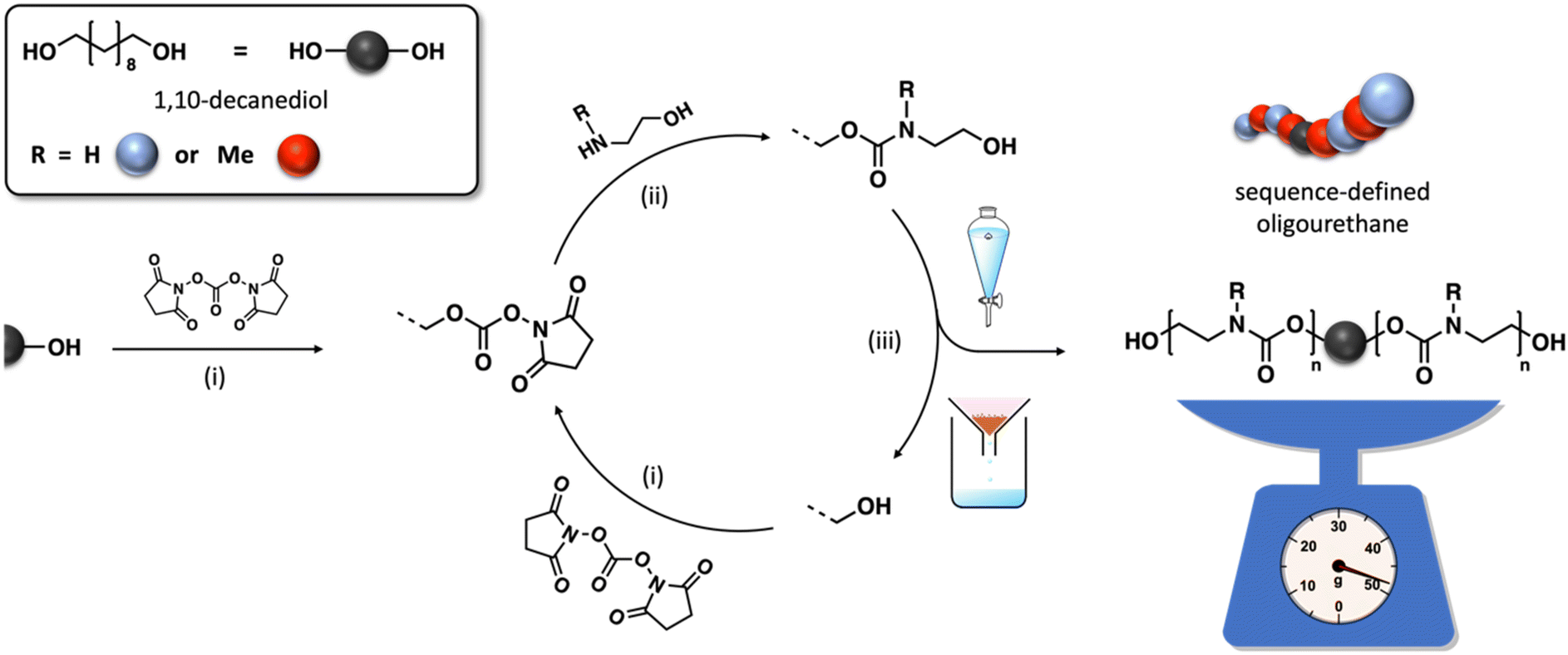 | ||
| Fig. 1 General synthesis protocol for the synthesis of uniform oligourethanes consisting of (i) an activation of the terminal alcohol groups to a carbonate, (ii) the subsequent chemoselective coupling with an aminoethanol and (iii) the purification with either an extraction or a precipitation. | ||
Results and discussion
Synthesis of oligourethanes with distinct primary structure
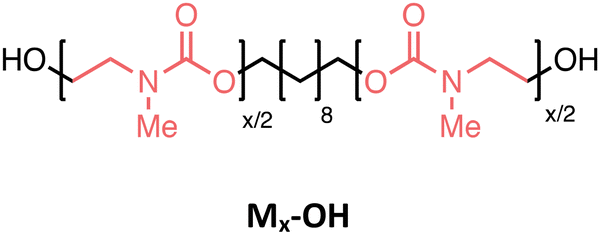
Several oligourethanes with distinct primary structures were synthesized using the protocol outlined in Fig. 1. By selecting 1,10-decanediol as the initiator, bidirectional growth of the oligomer chain was achieved, which accelerates the oligomerization process and opens up possibilities for telechelic oligomer synthesis. Furthermore, this procedure facilitates the purification of the oligomers by increasing their hydrophobicity thanks to the long aliphatic chain. The iterative protocol, carried out in acetonitrile (ACN) as the solvent, starts by converting the terminal hydroxyl groups of the initiator into N-hydroxysuccinimide (NHS)-containing carbonates by making use of disuccinimidyl carbonate (DSuC) in slight excess (i.e., 1.25 equiv. per OH group). Subsequently, an alkanolamine is introduced that chemoselectively forms carbamate bonds, while simultaneously regenerating terminal alcohol moieties. In this step, the choice of primary or secondary amine determines whether the incorporated monomer can form hydrogen bonding interactions or not. Given that some DSuC remains from the first step, an intermediate work-up would be required to prevent side reactions. However, this additional step can be avoided by introducing the alkanolamine in excess (i.e., 4 equivalents) in the second step. This excess leads to the quenching of the remaining DSuC without causing side reactions that could interfere with the growing sequence. Using this initiator and 2-(methylamino)ethan-1-ol, four OH-endcapped methylated oligourethanes (Mx-OH) with different molecular weights (see Table 1) were synthesized: a dimer (M2-OH), a tetramer (M4-OH), a hexamer (M6-OH) and an octamer (M8-OH). All these products were obtained on a multigram scale (23 g of M2-OH, 33 g of M4-OH, 42 g of M6-OH, and 50 g of M8-OH) with high yields (95% per step), and their purity was confirmed by ESI-MS, SEC and NMR (Fig. S1–S26†). To demonstrate that the protocol can be performed on larger scale, the synthesis of M8-OH was successfully repeated on a 50 gram scale.| Sequence-defined oligourethane | x = |
m/zth![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
m/zexpb |
T
gc (°C) |
T
md (°C) |
ΔHfuse (kJ mol−1) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Theoretical monoisotopic m/z. b Determined monoisotopic m/z via orbitrap-MS measurements. c Glass transition temperature determined via the second heating run in DSC. d Melting temperature determined via the first heating run in DSC. e Enthalpy of fusion integrated from the first heating run in DSC. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
2 | 377.2646 | 377.2641 | −66 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 579.3600 | 579.3602 | −44 | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 781.4553 | 781.4552 | −32 | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 983.5507 | 983.5529 | −24 | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
2 | 349.2333 | 349.2331 | — | 120 | 70 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 523.2974 | 523.2977 | — | 136 | 107 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 697.3614 | 697.3612 | — | 147 | 130 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 871.4255 | 871.4235 | — | 150 | 151 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The oligomers were purified by means of an aqueous extraction, successfully removing all water-soluble side products. This process is made possible by the non-solubility of the methyl-substituted oligourethanes in water and their solubility in non-miscible organic solvents, such as dichloromethane (DCM), improved by the long aliphatic chain of decanediol. Earlier attempts with ethanediol resulted in a more challenging extraction and low yields after purification (<50%, data not shown).
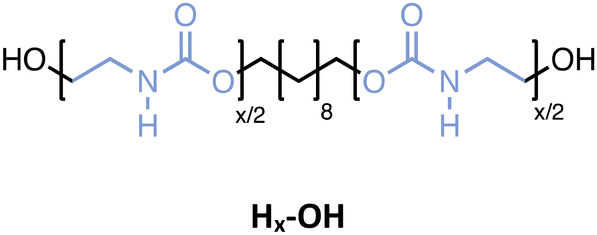
In parallel, four monodisperse oligomers (Hx-OH), having similar structures but without methyl-substituents, have been prepared using 2-aminoethan-1-ol this time. While the Mx-OH oligomers containing substituted carbamates could be isolated using extraction, the Hx-OH oligomers were insoluble in any organic solvent. Remarkably, polar solvents like methanol, ethanol, isopropanol, and water did not allow solubilization of those structures, whereas only dimethyl sulfoxide (DMSO) and dimethyl formamide effectively disrupted the non-covalent interactions, thereby dissolving the product. As a consequence, the solvent used in the protocol was switched from ACN to DMSO and the work-up approach adjusted accordingly. For the latter, the entire reaction mixture was added to water and stirred vigorously to dissolve all byproducts, with the oligourethane remaining as a white powder that could be filtered off. Because filtration is a straightforward and scalable work-up method, this alternative procedure does not compromise any efficiency compared to the conventional extraction process. This was demonstrated by synthesizing a similar series of oligourethanes, including a dimer (H2-OH), tetramer (H4-OH), hexamer (H6-OH) and octamer (H8-OH), in high yields per step (i.e., 97%). Moreover, the monodisperse octamer synthesis was performed on a 10 gram scale and the purity of these oligomers has been confirmed again by ESI-MS, SEC and NMR (see Fig. S1–S26†).
Since these sequence-defined oligourethanes vary in chain length, further investigations could be conducted on the effect of this parameter on their thermal properties including glass transition temperature (Tg), thermal degradation and melting behavior.
Influence of the length
Starting with the methylated carbamate series (Mx-OH), SEC analysis shows narrow peaks, as expected from the uniformity of the macromolecules, along with a clear shift that demonstrates the consistent increase in molecular weight with each monomer addition (Fig. 2). When examining their thermal behavior using differential scanning calorimetry (DSC), only a single thermal transition is observed, confirming their amorphous state. This Tg increase can be correlated to the polymer's molecular weight via the Flory–Fox equation (eqn (1)).38 This equation includes two polymer specific parameters: Tg,∞, which denotes the highest possible Tg achievable at a theoretical infinite molecular weight, and K, an empirical parameter related to the free volume present in the polymer sample. Fitting the values to this equation yields a Tg,∞ of 1 °C and a K of 25322 kg mol−1, supported by a high R2 value, demonstrating the suitability of sequence-defined polymers for elucidating structure–property relationships.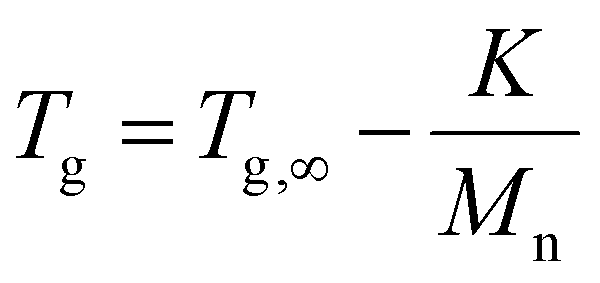 | (1) |
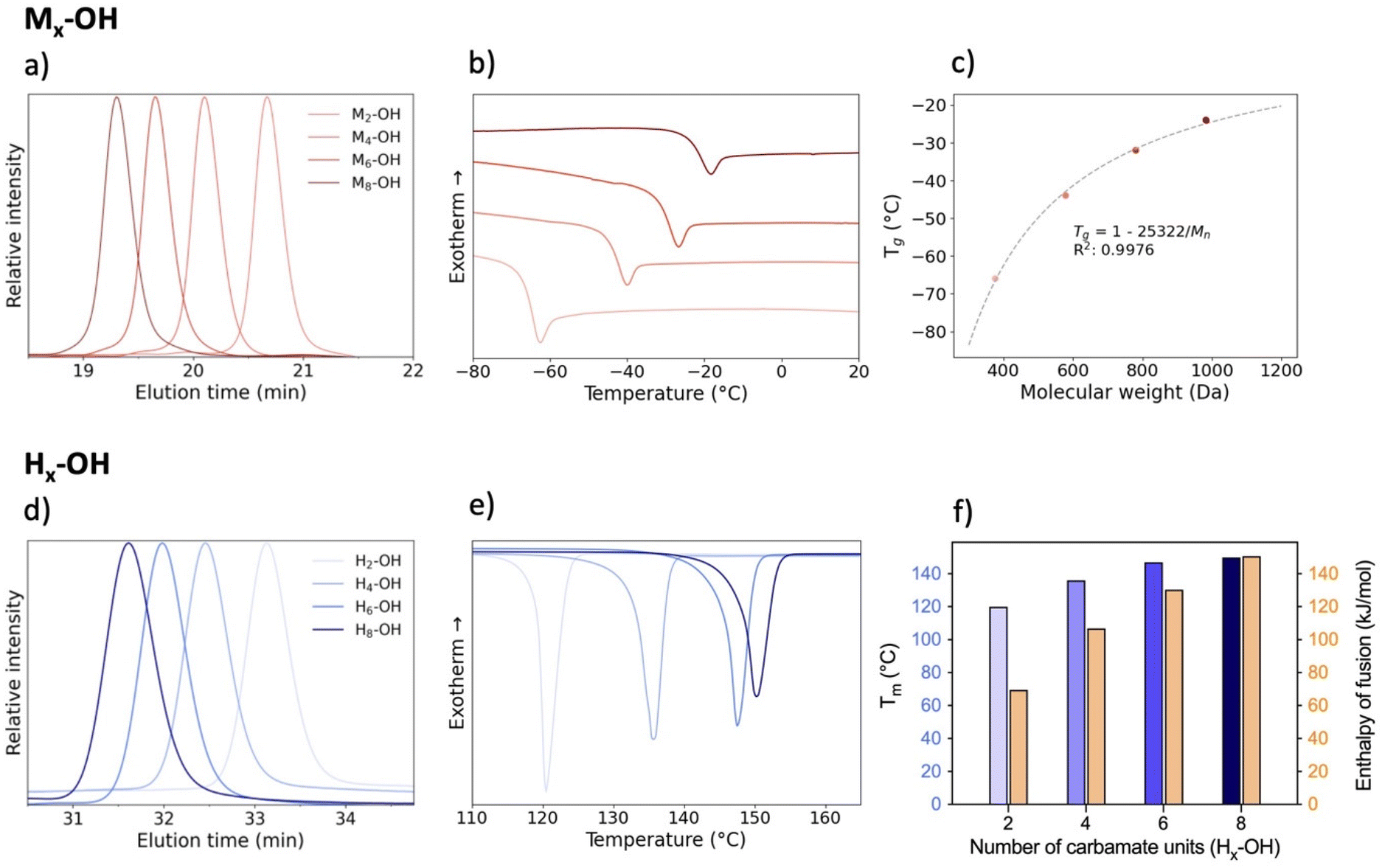 | ||
| Fig. 2 Characterization of the Mx-OH (top) and Hx-OH (bottom) oligomers. (a and d) SEC traces of the various macromolecules. (b and e) DSC curves showing the evolution of Tg or the melting temperature when varying the chain length for oligomers Mx-OH or Hx-OH, respectively. (c) Flory–Fox fitting showing the relationship between the Tg and the molecular weight. (f) Bar plots of the melting temperatures and enthalpy of fusion. | ||
The oligomers with hydrogen bonding moieties (Hx-OH) exhibit no Tg in the DSC-analysis due to their high degree of crystallinity. On the other hand, the melting peak gives information about the melting point and energy required to put the white powders into their liquid phase. As expected, extending the chain length and, consequently, increasing the number of hydrogen bonds results in a higher melting point. Furthermore, the enthalpy of fusion (ΔHfus) increases with the chain length, with a value of about 70 kJ mol−1 for H2-OH up to about 150 kJ mol−1 for the octamer H8-OH. This clearly shows that for such low mass oligourethanes, the amount of N–H containing carbamates has a significant impact on the crystallinity of the polymers and consequently on their thermal behavior.
Influence of sequence and end group functionality on the thermal behavior of sequence-defined oligourethanes
These first experimental results raised the question whether the transitions of such oligourethanes could be precisely controlled when combining regular carbamates with substituted carbamates. Specifically, we aimed to assess the influence of the sequence on the resulting material properties. To study the impact of monomer arrangement, the library of sequence-defined oligourethanes was further expanded (see Fig. 3) to include both a blocky structure (B8-OH), featuring a substituted urethane domain in the center, flanked by N–H urethane segments, and an alternating structure (A8-OH). Since the synthesis of these copolymers did not result in any solubility issues thanks to the insertion of a methyl-substituted monomer as the first unit, the protocol was carried out in a manner similar to the M8-OH octamer (see Experimental section). Even though the molecular weights and number of non-substituted carbamates are identical, the varying synergetic effects exerted by these hydrogen bonding groups can lead to differences in thermal behavior. Furthermore, the influence of the terminal groups is also examined by replacing the OH end groups of the oligomers with alkene moieties. The latter, chosen for the potential introduction of such oligomers into crosslinked structures, is achieved by exchanging the last added alkanolamine with the corresponding allylamine. This procedure gives rise to four new oligomers, H8-ene, M8-ene, B8-ene and A8-ene.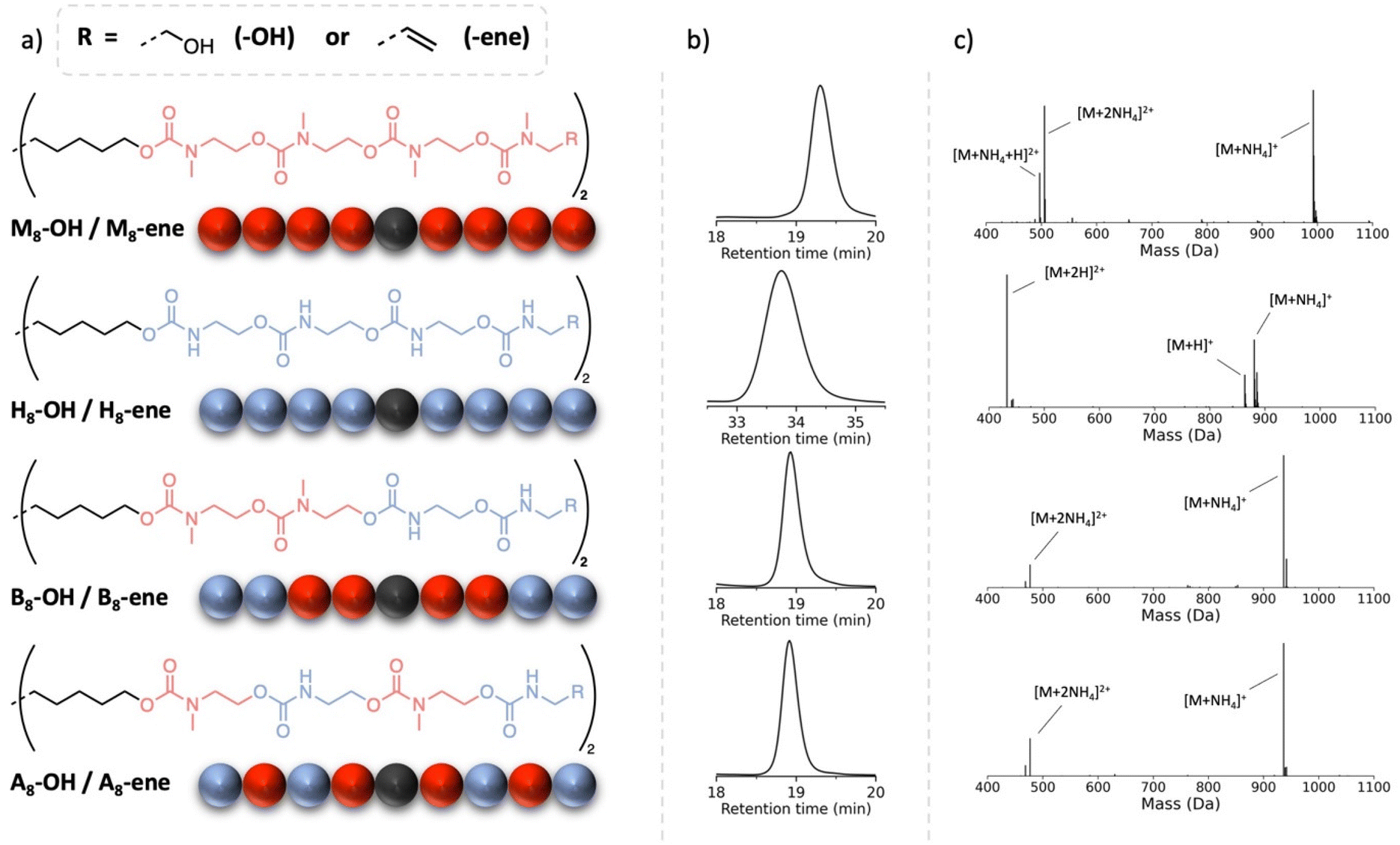 | ||
| Fig. 3 (a) The different structures synthesized, from top to bottom: methylated (M8-OH/M8-ene), non-methyl substituted (H8-OH/H8-ene), block (B8-OH/B8-ene) and alternating (A8-OH/A8-ene) oligomers with hydroxyls (–OH) or alkene (–ene) end groups. (b) SEC traces and (c) ESI-MS of the corresponding alkene-terminated oligomers. Note that the second SEC measurement has been performed in dimethylacetamide due to a solubility issue. This SEC system utilizes a column calibrated for higher molecular weight polymers, resulting in the oligomers having a later retention time compared to the other measurements. | ||
All eight oligomers were analyzed using DSC (see Fig. 4a and Table 2), with Tg values obtained from the second heating run. On the other hand, Tm along with melting enthalpy values – if present – were recorded from the first heating run because of a very slow recrystallization process that could not be fully achieved, even with very low cooling rates. With the exception of the highly crystalline, non-methyl-substituted oligourethanes (H8-OH and H8-ene), all oligomers demonstrate a clear Tg transition. Efforts have been made to enable the identification of the Tg of H8-OH and H8-ene, such as annealing these products and performing dynamic mechanical analysis (DMA), yet no results have been obtained. Focusing first on the influence of the end group, varying effects on the Tg can be observed. In purely methyl-substituted oligomers, terminal hydroxyl groups slightly raise the Tg from −34 °C (M8-ene) to −24 °C (M8-OH), whereas in the block and alternating copolymers, these OH groups lead to a decrease of the Tg (e.g., Tg = −42 °C for B8-OHvs. Tg = −13 °C for B8-ene). A potential explanation is that the OH groups in M8-OH introduce additional intermolecular interactions, increasing the Tg, while the hydroxyl groups in the copolymers (B8-OH and A8-OH) lower the Tg by interfering with the existing hydrogen bonds. The sequence also has a pronounced effect on the Tg, as is clearly demonstrated by the differences between the ene-terminated oligomers. Indeed, it was found that B8-OH and A8-OH possess similar Tg values (−11 °C and −13 °C, respectively), whereas M8-OH exhibits a significant decrease in its Tg value (−34 °C). This highlights the Tg-lowering effect of carbamate substitution (see Fig. 4a), as those substituted moieties do not reduce the molecular chain mobility in a significant way.
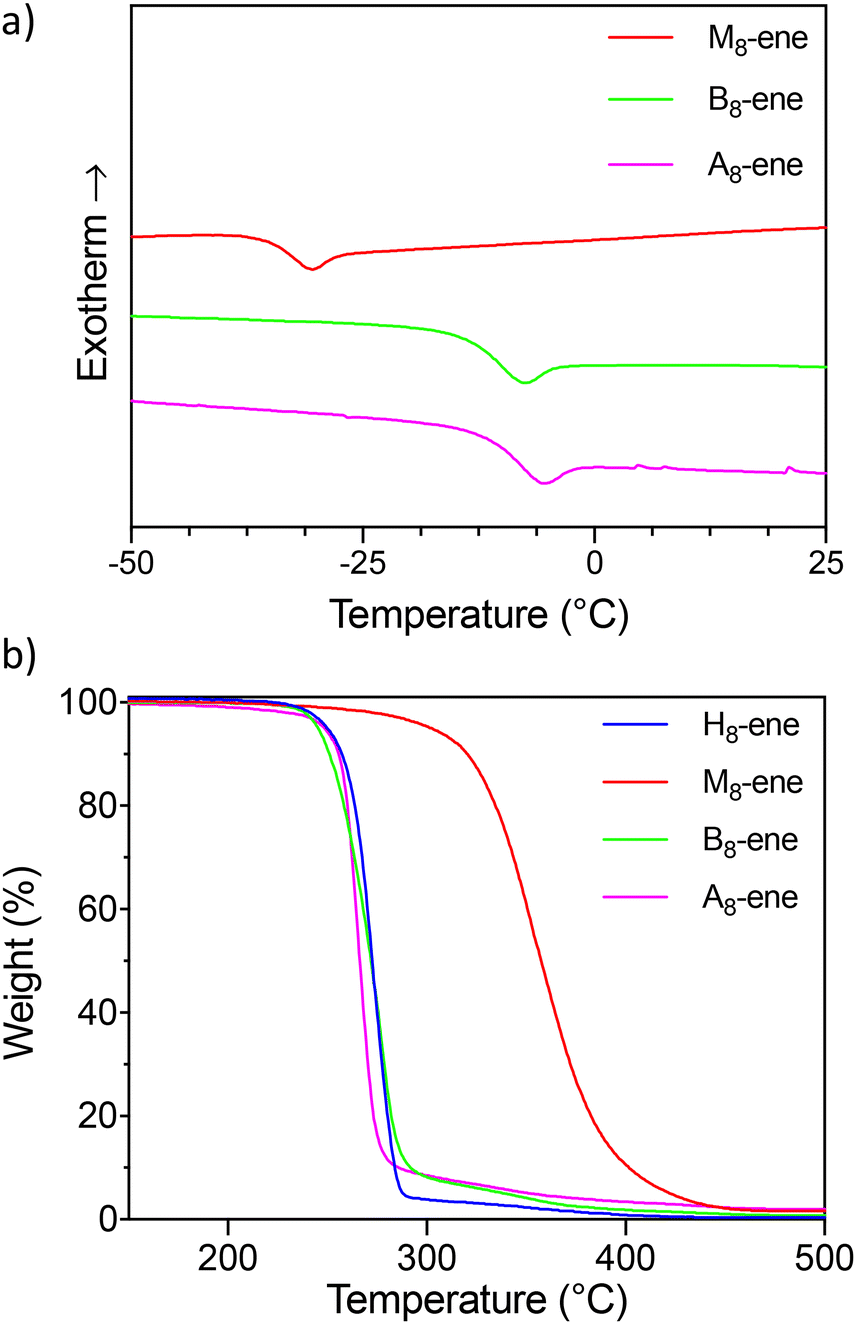 | ||
| Fig. 4 (a) DSC measurements of the alkene-terminated octamers showing their Tg. (b) TGA measurements of the alkene terminated octamers. | ||
| Sequence |
m/ztha |
m/zexpb |
Yieldc (%) |
T
gd (°C) |
T
me (°C) |
T
d5%f (°C) |
ΔHfusg (kJ mol−1) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Theoretical monoisotopic m/z. b Determined monoisotopic m/z via orbitrap-MS measurements. c Yields obtained for the entire synthesis process. d T g determined via the second heating run in DSC. e Melting temperature determined via the first heating run in DSC. f Thermal onset-temperature of 5% weight-loss measured via TGA. g Enthalpy of fusion integrated from the first heating run in DSC. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| M8-OH | 983.5507 | 983.5529 | 80.6 | −24 | — | 244 | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| M8-ene | 975.5609 | 975.5583 | 77.6 | −34 | — | 302 | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| H8-OH | 871.4255 | 871.4235 | 88.4 | — | 150 | 247 | 151 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| H8-ene | 863.4357 | 863.4333 | 85.7 | — | 158/168 | 250 | 159 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| B8-OH | 927.4881 | 927.4886 | 82.8 | −42 | — | 220 | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| B8-ene | 919.4983 | 919.4974 | 78.1 | −13 | — | 245 | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| A8-OH | 927.4881 | 927.4885 | 75.2 | −26 | — | 216 | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| A8-ene | 919.4983 | 919.4971 | 86.3 | −11 | 70 | 249 | 58 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
While H8-OH and H8-ene cannot be compared to the other oligomers in terms of their Tg values, they can be studied in terms of Tm and ΔHfus. It was anticipated that substituting the OH group with an alkene would lower the melting temperature due to reduced non-covalent interactions. However, contrary to this expectation, the diene-terminated octamer demonstrates higher values for both the melting temperature and enthalpy of fusion. While all methyl-containing oligomers are amorphous and thus do not exhibit this transition, A8-ene is an exception. It slowly crystallized from a viscous oil to a white powder with a melting temperature of 70 °C. This confirms the strong structure–property relationship in oligourethanes, which can be accurately investigated with sequence-defined macromolecules, in contrast to disperse oligomers and polymers that only allow for averaged property determination.
Additionally, thermogravimetric analysis (TGA) was performed to gain insight into the impact of the microstructure on the thermostability of these sequence-defined oligomers. In this context, differences are anticipated due to the reversibility of a regular carbamate, which is eliminated when substituted with a methyl group.39 Indeed, when analyzing the temperatures where a 5% mass loss occurs (Td5%), clear differences between H8-ene and M8-ene are observed, indicating that methylation increases the thermal stability by 50 °C. However, this trend is not seen in the OH-terminated oligomers (Td5% of H8-OH is 247 °C and Td5% of M8-OH is 244 °C, see Fig. S28†), suggesting that also the terminal groups influence the thermal stability. Previous research studies have demonstrated that these OH-endcapped oligourethanes have a self-immolative character, undergoing a 5-exo-trig cyclization initiated by an alkaline trigger.17,37,40 Although no basic conditions are present, it is possible that the cyclization occurs under thermal conditions, which could explain the earlier observed absence of differences in thermal stability. Indeed, comparing the thermal stability of the ene-terminated oligomers with the corresponding OH-terminated alternatives, a clear trend can be observed. Across all oligomers, the Td5% values improve when the potential for backbiting is eliminated, indicating the cyclization as a predominant degradation pathway.
To validate this assumption, pyrolysis-GC-MS analysis of the ene-terminated oligomers was conducted above their respective degradation temperatures (see Tables S1–S4†). While the presence of oxazolidin-2-one can be attributed to both a dissociative intramolecular transcarbamoylation and a cyclization reaction, 3-methyl-2-oxazolidinone can only be generated through the latter. Because this is the main degradation product in M8-ene, B8-ene and A8-ene (see Tables S1–S4†), this serves as additional proof that the cyclization is indeed the predominant degradation pathway. Additionally, this implies that the thermal stability is dictated by the generation of the terminal alcohol, which is determined by the integrity of the (terminal) carbamate moieties. This translates into a substantial enhancement in the thermal stability of the M8-ene oligomer relative to the H8-ene, B8-ene, and A8-ene oligomers (see Fig. 4b), given the increased resilience of Me-substituted carbamate. Allyl isocyanate detected in the pyrolysis-GC-MS spectrum of H8-ene, B8-ene, and A8-ene provides additional confirmation of the reversibility and resulting lability of the N–H carbamate bonds in these oligomers. These findings offer valuable insights into the thermostability and degradation pathway of oligourethanes, enabling precise material design through careful selection of the design parameters.
Conclusion
First, we demonstrated that sequence-defined oligourethanes can be produced using a straightforward and scalable protocol, allowing the preparation of up to 50 g of these uniform oligomers on lab scale. Through a bidirectional process, we reduced the synthetic steps and achieved precise control over the length, microstructure, and end-functionality of the targeted macromolecules. Hence, non-substituted, substituted, blocky, and alternating octamers bearing OH- or alkene-terminated groups have been synthesized. Subsequent thermal analysis gave important insights into the parameters that dictate the thermal transitions characterizing these products, including Tg, melting temperatures and melting enthalpies. This research was specifically focused on the effect of hydrogen bonding, which is present in N–H carbamates but absent in the methyl-substituted monomers. While increasing the amount of these non-covalent interactions showed an increase in both melting temperature and enthalpy, the chain length could be used as a parameter to regulate the Tg of this type of materials. Furthermore, the arrangement of the monomers plays a crucial role in shaping these thermal properties, as illustrated by the fact that B8-ene is amorphous while A8-ene is a semi-crystalline structure. Thermal degradation was also examined, revealing no significant differences among the OH-terminated oligomers, which contradicts the expectation that methylated carbamates would be more stable due to their lack of reversibility. However, when the alcohol end group was replaced with an allyl group, the anticipated behavior emerged, showing that conventional urethanes exhibited inferior thermal stability. This indicated that cyclization, rather than carbamate breakage, is the primary degradation pathway. This conclusion was supported by pyrolysis-GC-MS measurements. The information gathered in this work offers valuable insights into the structure–property relationships of PUs, emphasizing design parameters that allow their tailor-made engineering and thus paving the way to the development of next-generation polyurethane materials.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This project received funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme 101021081 (ERC-AdG-2020, CiMaC-project). J. V. H. acknowledges the Research Foundation-Flanders (FWO) for his Ph.D. fellowship under application number 11PJ624N. The NMR expertise centre (Ghent University) is also acknowledged for providing support and access to NMR infrastructure funded by a Research Foundation Flanders (FWO I006920N) and the Bijzonder Onderzoeksfonds (BOF.BAS. 2022.0023.01). Tine Van Laere, Dr. Dave Manhaeghe and Professor Steven De Meester are acknowledged for the pyrolysis GC-MS measurements. Pieter Surmont is thanked for the orbitrap-MS measurements and Bernhard De Meyer for assistance regarding thermal analyses. The authors thank Dr. Susanne Fischer for the experimental help and both Dr. Matthieu Soete and Dr. Susanne Fischer for the fruitful discussions.References
- M. Elices, G. V. Guinea, G. R. Plaza, C. Karatzas, C. Riekel, F. Agulló-Rueda, R. Daza and J. Pérez-Rigueiro, Macromolecules, 2011, 44, 1166–1176 CrossRef CAS.
- A. H. Simmons, C. A. Michal and L. W. Jelinski, Science, 1996, 271, 84–87 CrossRef CAS PubMed.
- B. Fernández-D'Arlas and A. Eceiza, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 739–746 CrossRef.
- B. Eling, Ž. Tomović and V. Schädler, Macromol. Chem. Phys., 2020, 221, 2000114 CrossRef CAS.
- D. K. Chattopadhyay and K. V. S. N. Raju, Prog. Polym. Sci., 2007, 32, 352–418 CrossRef CAS.
- J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnam and A. R. Yuvaraj, RSC Adv., 2016, 6, 114453–114482 RSC.
- K. Kojio, S. Nozaki, A. Takahara and S. Yamasaki, J. Polym. Res., 2020, 27, 140 CrossRef CAS.
- C. M. Brunette, S. L. Hsu and W. J. MacKnight, Macromolecules, 1982, 15, 71–77 CrossRef CAS.
- S. Nozaki, S. Masuda, K. Kamitani, K. Kojio, A. Takahara, G. Kuwamura, D. Hasegawa, K. Moorthi, K. Mita and S. Yamasaki, Macromolecules, 2017, 50, 1008–1015 CrossRef CAS.
- Yu. M. Boyarchuk, L. Ya. Rappoport, V. N. Nikitin and N. P. Apukhtina, Polym. Sci. U.S.S.R., 1965, 7, 859–867 CrossRef.
- E. Yilgör, E. Burgaz, E. Yurtsever and I. Yilgör, Polymer, 2000, 41, 849–857 CrossRef.
- L. Ning, W. De-Ning and Y. Sheng-Kang, Macromolecules, 1997, 30, 4405–4409 CrossRef CAS.
- J. Qin, Y. Chen, X. Guo, Y. Huang, G. Chen, Q. Zhang, G. He, S. Zhu, X. Ruan and H. Zhu, Adv. Sci., 2024, 11, 2400255 Search PubMed.
- D. Barik and M. Porel, ACS Macro Lett., 2024, 13, 65–72 CrossRef CAS.
- X. Huang, Z. Yang, X. Yang, Z. Liang, Q. Zhong, L. Hu, Z. Huang and Z. Zhang, ACS Macro Lett., 2024, 13, 979–986 Search PubMed.
- P. Nanjan, A. Jose, L. Thurakkal and M. Porel, Macromolecules, 2020, 53, 11019–11026 CrossRef CAS.
- M. Soete, J. Van Hoorde and F. Du Prez, Polym. Chem., 2022, 13, 4178–4185 RSC.
- M. Nerantzaki, C. Husser, M. Ryckelynck and J. F. Lutz, J. Am. Chem. Soc., 2024, 146, 6456–6460 Search PubMed.
- R. Aksakal, C. Mertens, M. Soete, N. Badi and F. Du Prez, Adv. Sci., 2021, 8, 2004038 CrossRef CAS.
- Z. C. Girvin, M. K. Andrews, X. Liu and S. H. Gellman, Science, 2019, 366, 1528–1531 CrossRef CAS PubMed.
- L. Shao, D. Hu, S. Zheng, T. K. H. Trinh, W. Zhou, H. Wang, Y. Zong, C. Li and C. Chen, Angew. Chem., Int. Ed., 2024, 63, e202403263 CrossRef CAS.
- D. Karamessini, T. Simon-Yarza, S. Poyer, E. Konishcheva, L. Charles, D. Letourneur and J. Lutz, Angew. Chem., 2018, 130, 10734–10738 CrossRef.
- P. Nanjan and M. Porel, Polym. Chem., 2019, 10, 5406–5424 RSC.
- X. Huang, Z. Liang, X. Yang, M. Piao, Z. Huang and Z. Zhang, ACS Appl. Mater. Interfaces, 2024, 16, 43075–43082 CrossRef CAS PubMed.
- T. Schutz, I. Sergent, G. Obeid, L. Oswald, A. Al Ouahabi, P. N. W. Baxter, J. Clément, D. Gigmes, L. Charles and J. Lutz, Angew. Chem., Int. Ed., 2023, 62, e202310801 Search PubMed.
- S. Wegelin and M. A. R. Meier, Macromol. Chem. Phys., 2024, 225, 2300337 CrossRef CAS.
- R. K. Weigel and C. A. Alabi, Chem. Sci., 2024, 15, 9138–9146 RSC.
- R. Aksakal, C. Tonneaux, A. Uvyn, M. Fossépré, H. Turgut, N. Badi, M. Surin, B. G. De Geest and F. E. Du Prez, Chem. Sci., 2023, 14, 6572–6578 Search PubMed.
- Q. Shi, Z. Zhang and S. Liu, Angew. Chem., Int. Ed., 2024, 63, e202313370 CrossRef CAS PubMed.
- E. A. Hoff, R. K. Weigel, A. Rangamani and C. A. Alabi, ACS Polym. Au, 2023, 3, 276–283 Search PubMed.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 Search PubMed.
- J. O. Holloway, K. S. Wetzel, S. Martens, F. E. Du Prez and M. A. R. Meier, Polym. Chem., 2019, 10, 3859–3867 Search PubMed.
- I. De Franceschi, C. Mertens, N. Badi and F. Du Prez, Polym. Chem., 2022, 13, 5616–5624 Search PubMed.
- I. De Franceschi, N. Badi and F. E. Du Prez, Chem. Sci., 2024, 15, 2805–2816 RSC.
- W. Forysiak, A. Lizak and R. Szweda, ChemPhysChem, 2024, 25, e202400366 CrossRef CAS PubMed.
- E. A. Hoff, G. X. De Hoe, C. M. Mulvaney, M. A. Hillmyer and C. A. Alabi, J. Am. Chem. Soc., 2020, 142, 6729–6736 CrossRef CAS PubMed.
- S. D. Dahlhauser, P. R. Escamilla, A. N. Vandewalle, J. T. York, R. M. Rapagnani, J. S. Shei, S. A. Glass, J. N. Coronado, S. R. Moor, D. P. Saunders and E. V. Anslyn, J. Am. Chem. Soc., 2020, 142, 2744–2749 CrossRef CAS PubMed.
- T. G. Fox and P. J. Flory, J. Appl. Phys., 1950, 21, 581–591 CrossRef CAS.
- G. Montaudo, C. Puglisi, E. Scamporrino and D. Vitalini, Macromolecules, 1984, 17, 1605–1614 CrossRef CAS.
- T. Mondal, L. Charles and J. Lutz, Angew. Chem., Int. Ed., 2020, 59, 20390–20393 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details about instrumentation, synthesis and experimental procedures, NMR-, SEC-, ESI-MS- and orbitrap-MS spectra, as well as DSC and TGA thermograms. See DOI: https://doi.org/10.1039/d4py01001a |
| This journal is © The Royal Society of Chemistry 2024 |