
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent-dependent valence tautomerism and polarization switching in a heterodinuclear [CrCo] complex†
Wenwei
Zheng
,
Xiaopeng
Zhang
,
Qirui
Shui
,
Tatsuki
Fukuyama
,
Wen-huang
Xu
,
Yu-bo
Huang
,
Tianchi
Ji
,
Ziqi
Zhou
,
Mikoto
Uematsu
,
Sheng-Qun
Su
,
Shinji
Kanegawa
 ,
Shu-Qi
Wu
* and
Osamu
Sato
*
,
Shu-Qi
Wu
* and
Osamu
Sato
*
Institute for Materials Chemistry and Engineering & IRCCS, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: sato@cm.kyushu-u.ac.jp
First published on 17th May 2024
Abstract
Macroscopic polarization switching via directional electron transfer has attracted great attention owing to its potential application in data storage devices, sensors, and energy conversion. However, the acquisition of the desired polar crystals is still a huge challenge to realize polarization switching. Here, a heterometallic [CrCo] complex with an enantiopure ligand was successfully obtained by a stepwise synthetic route, which effectively avoids the contamination caused by the lack of recognition of chiral ligands. Both compound 1·sol and its desolvated form, 1, are packed in the polar P21 space group. These forms exhibit valence tautomerism to varying degrees and interconversion to each other through single-crystal-to-single-crystal transformation, which enables solvent-dependent polarization-switching behavior. Moreover, both forms show trapping of the photoinduced electron-transferred states upon light irradiation. These findings highlight the effectiveness of using enantiopure ligands in constructing polar crystals with versatile polarization-switching behavior.
Introduction
Polar materials responsive to external stimuli, such as temperature, light, and electric field, offer potential applications in next-generation molecular memory devices.1–7 Ferroelectric materials where spontaneous polarization could be reversed by the application of an electric field have been the most intensively investigated polarization-switching materials. However, as a collective phenomenon in its nature, the rational design of ferroelectrics with the desired performance usually represents a formidable challenge from a chemical perspective.8–12 To diversify polarization-switching materials, a bottom-up approach, utilizing dynamic molecules with switchability between states and different electric dipole moments, has been proposed for non-ferroelectric systems to realize polarization switching. In this regard, valence tautomeric (VT) molecules are ideal structural motifs exhibiting variable electronic configurations responsive to temperature, pressure, and external fields.13–48 Furthermore, for crystalline materials with switchable macroscopic polarization, it is imperative that the target molecules crystallize in polar space groups so that their dipole moments will not be canceled. To date, there have been only a few non-ferroelectric molecular crystals exhibiting macroscopic polarization switching via electron transfer owing to the difficulties in crystal engineering to align molecules in polar space groups.14,18,49–51 The acquisition of such crystals still largely relies on serendipity. Hence, it is still full of challenges in synthesizing polar crystals through an appropriate molecular design to enhance the probability of arranging target molecules in a polar space group.Recently, a series of dinuclear VT complexes with racemic ligands in a polar space group were synthesized via a chiral-assisted approach, which relies on the preference of left-handed chiral ligands to interact with their right-handed counterparts, resulting in a polar crystal structure.3,5,46,47 A natural extension would be the introduction of enantiopure ligands to enhance the possibility of obtaining polar crystals. Indeed, subsequent research in the case of mononuclear cobalt complexes has demonstrated its efficacy in diversifying polar crystals with VT behavior.2,40,52 However, the previously used crystallization strategy is invalid for dinuclear systems with homochiral ligands due to the lack of complementary interactions. Therefore, the [CoIICoIII] complex with an enantiopure SS-cth ligand was first investigated (cth = (7S,14S)-5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraazacyclotetradecane) to aid in our initial screening. Although it is in the high-spin (HS) state in the temperature range between 2 and 400 K, the complex indeed crystallizes in the polar P21 space group with a regular molecular arrangement, where the CoII sites are well distinguished from the CoIII sites crystallographically. This is a crucial factor for dictating directional electron transfer aiming at polarization-switching behavior. Given the fact that the strong coupling between chromium and dhbq proves beneficial for stabilizing a low-spin trivalent cobalt state,53 thereby enhancing the VT transition temperature in the corresponding racemic system, a site-selective substitution with CrIII ions for CoIII sites would possibly lead to the desired VT behavior. Consequently, we synthesized the [CrCo] complex with the enantiopure SS-cth ligand. By careful design of synthetic procedures by first incorporating the more inert Cr3+ sites, we successfully isolated and obtained the pure heterometallic dinuclear [CrCo] complex [Cr(SS-cth)(μ-dhbq)Co(SS-cth)](PF6)3·2.5H2O·0.5MeOH ((denoted as compound 1·sol), H2dhbq = 2,5-dihydroxy-1,4-benzoquinone). Single-crystal structural analysis revealed that 1·sol is isostructural to the above-mentioned [CoIICoIII] complex. Magnetometry and spectroscopic studies showed that 1·sol exhibits an incomplete VT transition between the Co ion and the dhbq ligand. Polarization switching from such an intramolecular electron transfer process could be directly probed by pyroelectric measurements. Upon heating, the crystals still retain their integrity without changing the space group. Interestingly, detailed structural analysis and magnetometry revealed that the desolvated complex, 1, exhibits a much more complete VT behavior. When exposed to water–methanol vapor, complex 1 can re-absorb the solvent molecules, recovering to its solvated form, 1·sol, with incomplete VT. Such solvent-induced switching of physical behavior could be repeatedly observed.
Results and discussion
Three heterometallic dinuclear complexes [MCr] (M = Co, Zn, Ni) featuring the enantiopure ligand SS-cth or RR-cth were synthesized according to one-pot and stepwise routes. In the former, enantiomeric complexes ([M(AcO)(SS-cth)]PF6 and [CrCl2(SS-cth)]Cl) were obtained separately and mixed in a methanol solution of dhbq, resulting in a crude complex [Cr(SS-cth)(μ-dhbq)(M(SS-cth))](PF6)3. In the latter, the [Cr(SS-cth)(μ-dhbq)]PF6 precursor was first isolated and then mixed with the corresponding complex motif to afford heterometallic [CrM] complexes, exemplified by [CrCo] (Fig. 1). The characterization of complexes using mass spectra confirmed that the stepwise method effectively yields pure heterometallic complexes. The crude samples obtained from the two methods were dissolved in MeOH and mass spectral measurements were subsequently conducted. By the one-pot route, the obtained spectrum shows three distinct types of molecular peaks for [1(PF6)2]+ (m/z = 1107.9) through conventional methods, including [(CrCr)(PF6)2]+, [(CrCo)(PF6)2]+, and [(CoCo)(PF6)2]+. Notably, alongside the predominant [CrCo] peaks, the intensity of peaks attributed to [CoCo] is remarkably strong, indicating a discernible random occupation of the two coordination sites during synthesis (Fig. 2a). In contrast, the spectrum of [1(PF6)2]+ obtained using the stepwise method reveals nearly all molecular peaks contributed by [CrCo] (Fig. 2b), consistent with calculated results (Fig. S1†). Moreover, the mass spectra of reference complexes, [NiCr] and [ZnCr], also validate the universality of the stepwise synthetic route (Fig. S2 and S3†). These findings strongly support the feasibility of the new strategy for synthesizing pure heterometallic complexes with an enantiopure ligand.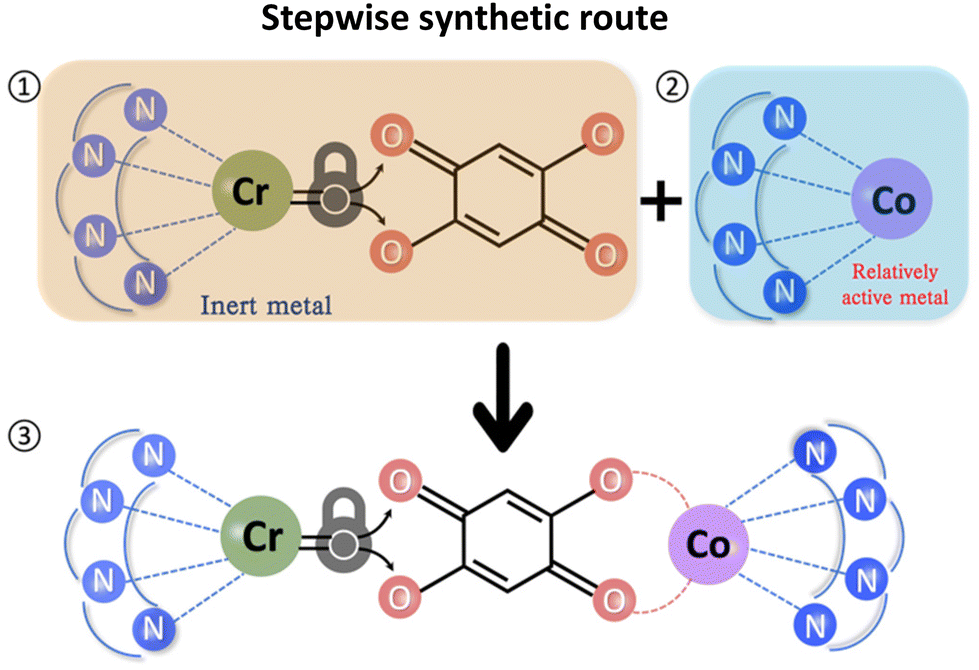 | ||
| Fig. 1 Schematic illustration of the stepwise synthetic strategy. | ||
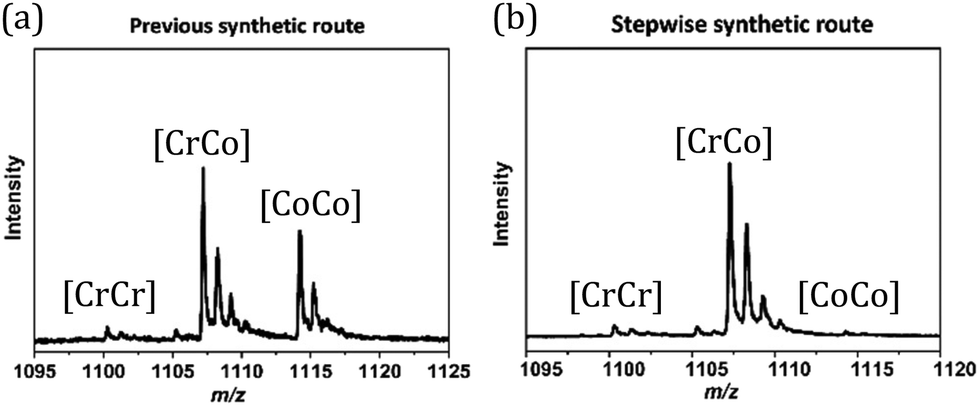 | ||
| Fig. 2 (a) Mass spectra of complex 1 synthesized through one-pot and (b) stepwise routes. | ||
Temperature-dependent single-crystal X-ray diffraction of 1·sol and 1 was performed at 100, 150, 200, and 375 K. The structural analyses revealed that both 1·sol and 1 crystallize in the polar P21 space group throughout the studied temperature range. The asymmetric unit of 1·sol involves one heterodinuclear molecular ion comprising [Co(SS-cth)] and [Cr(SS-cth)] motifs bridged by a dhbq ligand, three PF6− anions, two and a half water molecules, and a half-occupied methanol molecule. The composition of the co-crystallized solvent is further supported by thermogravimetric analysis (TGA) and elemental analysis (Fig. S4†). Interestingly, different from the eclipsed conformation in the previously reported heterochiral [CrCo] complex, the two SS-cth ligands in the complex motif of 1·sol adopted a slightly staggered conformation. Such a structural distortion may contribute to different VT transition behavior. At 100 K, 1·sol exhibited the average Co–O and Co–N bond lengths of 2.069 and 2.149 Å, indicative of the dominant presence of the Co2+ HS ions, while the average bond lengths of Cr–O and Cr–N bonds are 1.940 and 2.086 Å, characteristic of the Cr3+ ions. The evident differences in metal–ligand bond lengths clearly show that the Co and Cr sites are well distinguished rather than a random occupation. By the 21-screw rotation, the [CrCo] molecular motif is regularly packed, with a uniform projection angle (70.31(7)°) along the crystallographic b-axis, a critical factor for the manifestation of polarization switching (Fig. 3, upper). Upon heating above room temperature, the sample started to diffract badly until 375 K, where all the solvent molecules were removed according to the TGA result. Crystal structural analysis also confirmed the absence of solvent molecules in the lattice; thus, the diffraction data correspond to its desolvated form, 1. Notably, the bond lengths around the Co ions remained consistent with those typical for Co2+ ions. However, when the temperature was reduced back to 100 K, a significant contraction in cell volume by 184 Å3 was observed. Simultaneously, the average bond lengths around the Co ions contracted to 1.910 Å and 2.041 Å for Co–O and Co–N bonds, respectively, indicative of Co3+ LS ions. Furthermore, the observed contraction in the specific C–C bonds (C2–C3; C5–C6), accompanied by the elongation in the C–O bonds located on both sides of the bridging ligand, further confirms the occurrence of dhsq3− in a low-temperature range after desolvation (Fig. S5a–c†). These significant changes in bond lengths at the Co site clearly indicate a more pronounced VT behavior in complex 1, transitioning to [Cr3+-dhsq3−-Co3+LS] upon cooling. Additionally, the projection angle between the [CrCo] direction and the polar b-axis also alters to 85.66(1)° at 375 K and 85.35(8)° at 100 K after desolvation (Fig. 3, lower; S6†). Comparing the extent of VT transition and the changes in molecular arrangements before and after desolvation, we anticipate a solvent-tuned polarization change at the single-crystal level.
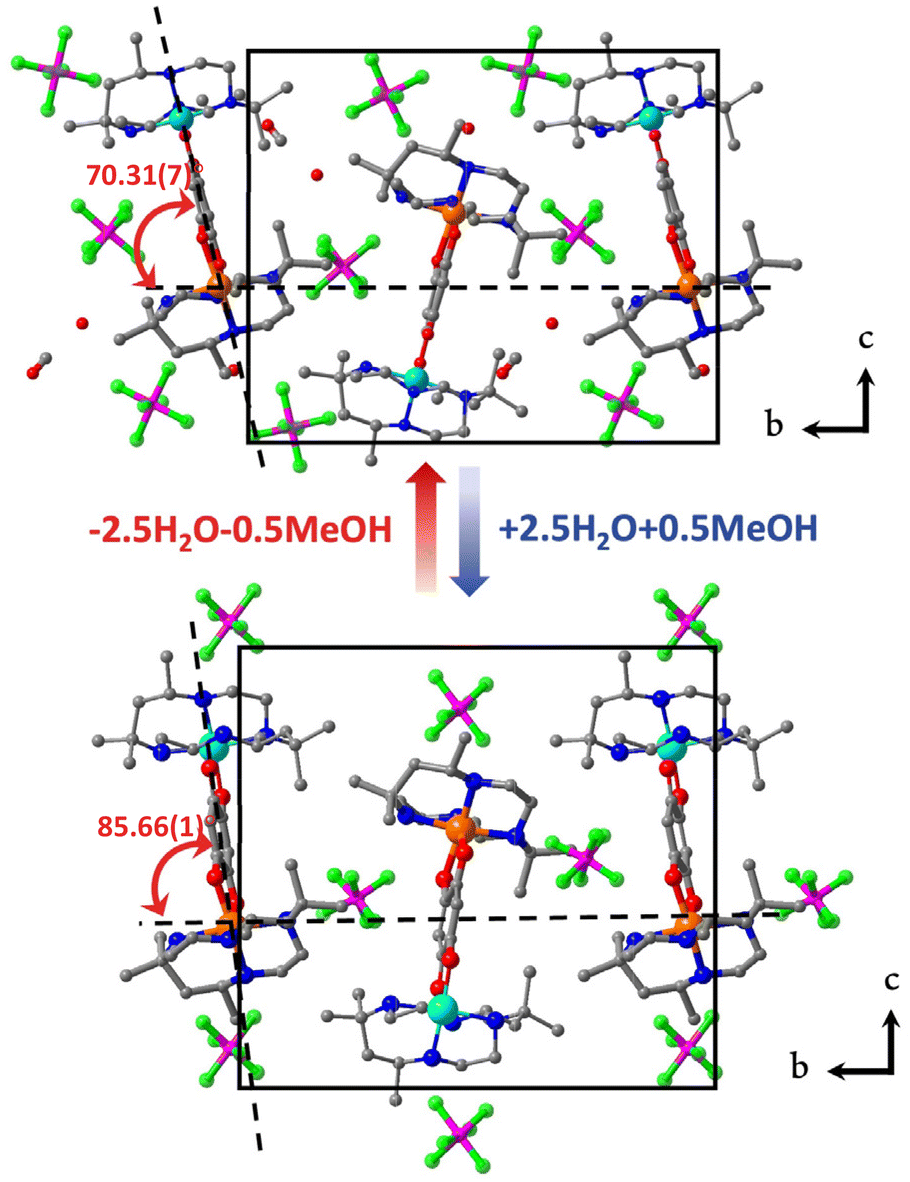 | ||
| Fig. 3 Crystal structures of compound 1·sol (upper) and 1 (lower) viewed along the a-axis. Co: orange, Cr: pale blue, O: red, C: dark gray, P: deep pink, and F: green; H atoms are omitted for clarity. | ||
The variable-temperature powder X-ray diffraction (PXRD) technique was employed to confirm the reversibility of the crystal structure during the in situ desolvation–reabsorption process (Fig. 4). To prevent the escape of the removed solvent molecules, the powder sample of 1·sol was carefully sealed using aluminum foil, and measurements were performed by following the sequence of 230–400–230 K. The PXRD patterns of 1·sol at 230 K matched well with the simulated one from single-crystal data. No obvious change in these reflections was observed until the temperature reached 326 K. When the temperature was increased further to 376 K, the PXRD patterns showed a significant and complete change of reflections at 2θ = 10.0°, 11.4°, 12.3°, and 20.5°. This change was consistent with the calculated result of the desolvated complex 1 at 375 K, indicating that compound 1 was generated in situ. The powder patterns remained unchanged until 400 K. However, when cooled down again, the diffraction pattern did not match the simulated result of compound 1 at low temperature but reverted to that of the pristine compound, 1·sol. These results clearly certify that the desolvated crystal could reabsorb the trapped solvent under ambient conditions to recover to the solvated compound, 1·sol. We also performed the PXRD measurement on thermally treated compound 1. After exposure to MeOH and H2O, the diffraction pattern agreed well with that of 1·sol (Fig. S7†). This also supported the reversibility of single-crystal-to-single-crystal transformation (SCSCT).54
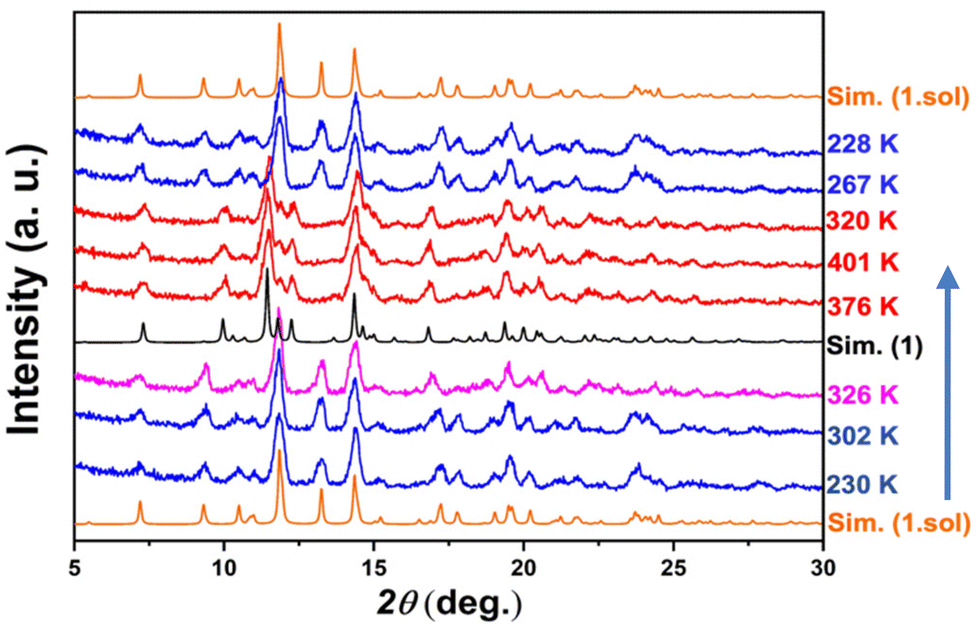 | ||
| Fig. 4 Variable-temperature powder XRD patterns in a thermal cycle of 230–400–230 K. Simulation spectra based on single-crystal XRD data at 200 (for 1·sol) and 375 K (for 1) are shown as orange and black lines. | ||
Variable-temperature magnetic susceptibility was measured on the polycrystalline sample of 1·sol. To prevent the loss of co-crystallized solvent molecules, the measurements started from 260 K under a 2 kOe magnetic field with a cooling rate of 2 K min−1 (Fig. 5). For compound 1·sol, the χmT value almost remains at 4.3 cm3 K cm−1 from 260 to 220 K, which is in good agreement with the expected value of isolated metal centers on Cr3+ (S = 3/2) and high spin Co2+ (S = 3/2) with an unquenched orbital magnetic moment. Upon further cooling, a gradual decrease in the χmT value is observed, reaching a plateau at 3.7 cm3 K cm−1 from 75 to 30 K. The VT transition temperature, defined as the peak temperature of the temperature derivative of the χmT plot, is determined to be ca. 170 K. On reheating to 260 K, the χmT value undergoes a reversible change without significant hysteresis. To explore the origin of this behavior, variable-temperature spectroscopic analyses were performed in the heating mode in this temperature range. A gradual decline in the intensity of the absorption band centered at 500 nm is observed, which is attributed to ligand–metal charge transfer from dhsq3− to Co3+ ions (Fig. S8a†). Correspondingly, the vibrational band of dhsq3− at 1470 cm−1 also shows a remarkable decrease in the same temperature range (Fig. S8b†). These findings strongly indicate that the variation in the χmT values within this temperature range is associated with the partial electron transfer from the [Cr3+-dhsq3−-Co3+LS] to the [Cr3+-dhbq2−-Co2+HS] state. Upon further heating to 400 K, the χmT value reaches ca. 4.2 cm3 K cm−1 accompanied by desolvation. The desolvated sample, 1, exhibits a more pronounced change. Below 340 K, the χmT value decreased abruptly and reached the minima 1.26 cm3 K cm−1, corresponding to an almost complete transition to the [Cr3+-dhsq3−-Co3+LS] state (expected χmT value: 1.0 cm3 K cm−1). The transition temperature is estimated to be 215 K with no obvious hysteretic behavior. Similar changes could also be observed from UV-vis and IR absorption spectra. The ground powder sample adhered to a CaF2 plate (for IR) and transparent tape (for UV-vis) was heated up to 400 K. After removal of the co-crystallized solvent molecules, the samples were quickly sealed into a vacuum tube to prevent the re-absorption of water molecules from air. The spectra of 1 were recorded from 79 to 300 K. At low temperature, two strong absorption peaks were observed at around 1210 and 1470 cm−1, attributed to the C–O stretching vibration from dhsq3−. As the temperature increased, a continuous decay of these absorption peaks was observed, whereas the characteristic absorption peaks (approximately 1560 and 1270 cm−1) belonging to dhbq2− became more intense (Fig. S8c†). These results directly supported that a variation of the electronic structure occurred on the bridging ligand. The UV-vis spectra also revealed similar changes to those for the pristine compound, 1·sol, with different transition temperature domains (Fig. S8d†). Notably, when the absorption intensities of the characteristic absorption bands in IR and UV-vis spectra were plotted against temperature, a clear shift in the transition temperature range could be identified, in good agreement with the magnetic data of the solvent effect of this compound (Fig. S9a–c†).
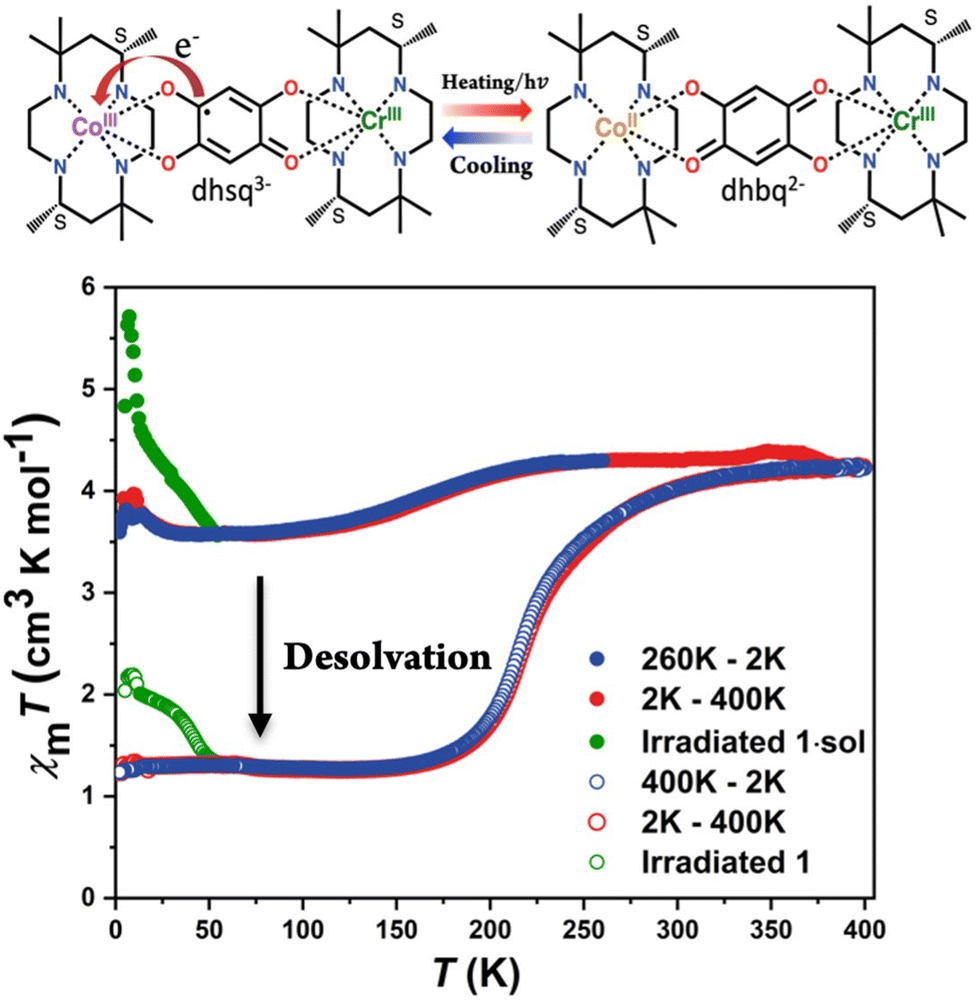 | ||
| Fig. 5 Schematic illustration of valence tautomerism in 1·sol (upper). Variable-temperature magnetic susceptibility of compound 1·sol before and after desolvation. The data were collected using the ground crystal with a sweeping rate of 2 K min−1 (lower). | ||
Additionally, the desolvated powder sample was placed above a MeOH/H2O mixture, ensuring no direct contact, and left overnight. Subsequently, the magnetic properties of the solvent-reabsorbed sample were measured under the same conditions as those for the original sample to confirm the recovery of magnetic properties upon solvent reabsorption. The results showed similar behavior, i.e., a partial and nearly complete electron transfer before and after solvent removal (Fig. S10†). Elemental analysis further corroborated the transition from compound 1 to 1·sol (Table S1†). These findings indicate that the solvent influence on the VT process of compound 1·sol can be reversibly manipulated through solvent reabsorption. Moreover, both compounds 1·sol and 1 exhibited more pronounced photoinduced valence tautomerism than the previous [CrCo] complex with racemic ligands. Compounds 1·sol and 1 were subjected to photomagnetic measurements by irradiation with green light (532 nm continuous wave laser) at 5 K. Indeed, both samples exhibited a moderate increase in magnetization after irradiation, followed by a gradual relaxation to the ground state as the temperature increased (Fig. 5). This behavior combined with thermally-induced polarization change suggests the possibility of light-induced polarization changes in these complexes.
Interestingly, compared to the previously reported [CrCo] complex employing a racemic ligand, the transition temperature of complex 1 is significantly lower. To understand such a discrepancy, theoretical calculations using density functional theory (DFT) were carried out at the same level of theory. Indeed, the derived energy difference of the enantiopure [CrCo] complex (0.46 kcal mol−1) is significantly lower than that of the heterochiral one (3.92 kcal mol−1) in the vacuum state. More careful inspection revealed that such a change mainly comes from the substantial stabilization (3.59 kcal mol−1) of the HS state in the enantiopure complex, which might relate to the more distorted coordination geometry in this situation. Notably, the relative stabilization of the HS electron-transferred state in the enantiopure complex also explains the improved trapping effect in the photomagnetic measurements.55
As complex 1·sol exhibits VT behavior in the P21 polar space group, it is possible to realize polarization switching at a macroscopic scale. Therefore, pyroelectric measurements were performed on the single-crystal sample of compound 1·sol to investigate the electric current attributed to the variation in dipole moments during the VT transition. The well-shaped crystal was indexed, and silver paste was attached to the (010) and (0−10) surfaces that were perpendicular to the b-axis (Fig. S11†). The pyroelectric current was recorded using a Keithley 6517B electrometer without applying any external electric/magnetic field, and the temperature was controlled using a Quantum Design magnetic property measurement system in the temperature range between 30 and 250 K with a sweeping rate of 5 K min−1. The current signal could be detected with opposite signs in the heating and cooling runs, certifying its pyroelectric nature. As temperature increases, the temperature-dependent pyroelectric coefficient of compound 1·sol exhibited a single broad peak centered at 162 and 174 K with a maximal value of ca. 2.9 nC cm−2 K−1 during cooling and heating processes, respectively. By integrating the pyroelectric current, the polarization change could be estimated as 0.27 μC cm−2, and its temperature dependence shows a perfect match with the results of magnetic measurements in thermally induced polarization.
To calculate the polarization change in 1·sol theoretically, the volume average of the molecular dipole moments (0.03 and 9.16 D for triplet and septet, respectively) along the b-axis was obtained from DFT calculations. In this way, the electron-transfer contribution was estimated as 0.17 μC cm−2, assuming that 20% of molecules (estimated from magnetic data) participate in the VT process. Besides, accompanied by the structural transition, the relative displacement between cations and anions should also contribute to polarization. This part was tentatively included by the point charge model, yielding an additional polarization change of 0.02 μC cm−2. The net polarization change is estimated as 0.19 μC cm−2, roughly consistent with the experimental value (0.27 μC cm−2) (Fig. 6). The discrepancy might result from the full negligence of the solvent orientation effects and the secondary pyroelectric effect, or inadequate consideration of the displacement contribution by an oversimplified point charge model. In any situation, it's reasonable to conclude that the observed polarization change is mainly contributed from electron transfer. As 1·sol exhibits SCSCT as confirmed by single-crystal or powder XRD, it remains interesting to investigate the solvent-induced polarization-switching behavior. Theoretically, the electron-transfer and ion-displacement contributions to polarization change could be estimated to be 0.17 and 0.00 μC cm−2, respectively. Surprisingly, this calculated value of the electron transfer-induced polarization change is similar to that of the solvated form, owing to the opposite change caused by different degrees of VT transition and change in the projection angle. Preliminary pyroelectric measurements were also performed on the in situ desolvated sample at 310 K, which shows a clear shift in the transition temperature towards a high-temperature direction, which is consistent with the results from magnetometry (Fig. S12†). However, as the large crystal pieces required for pyroelectric measurements were more fragile than those used for single-crystal XRD measurements, and the samples consistently experienced partial cracking during the desolvation treatment, it became challenging to obtain accurate pyroelectric current data with an acceptable signal-to-noise ratio. Therefore, the polarization change of the desolvated sample remains undetermined, while the solvent effect on the polarization-switching behavior is certified by the observed temperature shift. In addition, pyroelectric measurement of 1·sol was carried out under light irradiation at 5 K. A clear opposite current in comparison with the cooling process could be observed where the electron transfers from the bridging ligand to the metal. After light was switched off, the excited state gradually decayed back to the ground state as the temperature increased where an opposite peak current was detected. This experimental observation confirmed that the polarization change of 1·sol could also be stimulated by means of light irradiation (Fig. S13†).
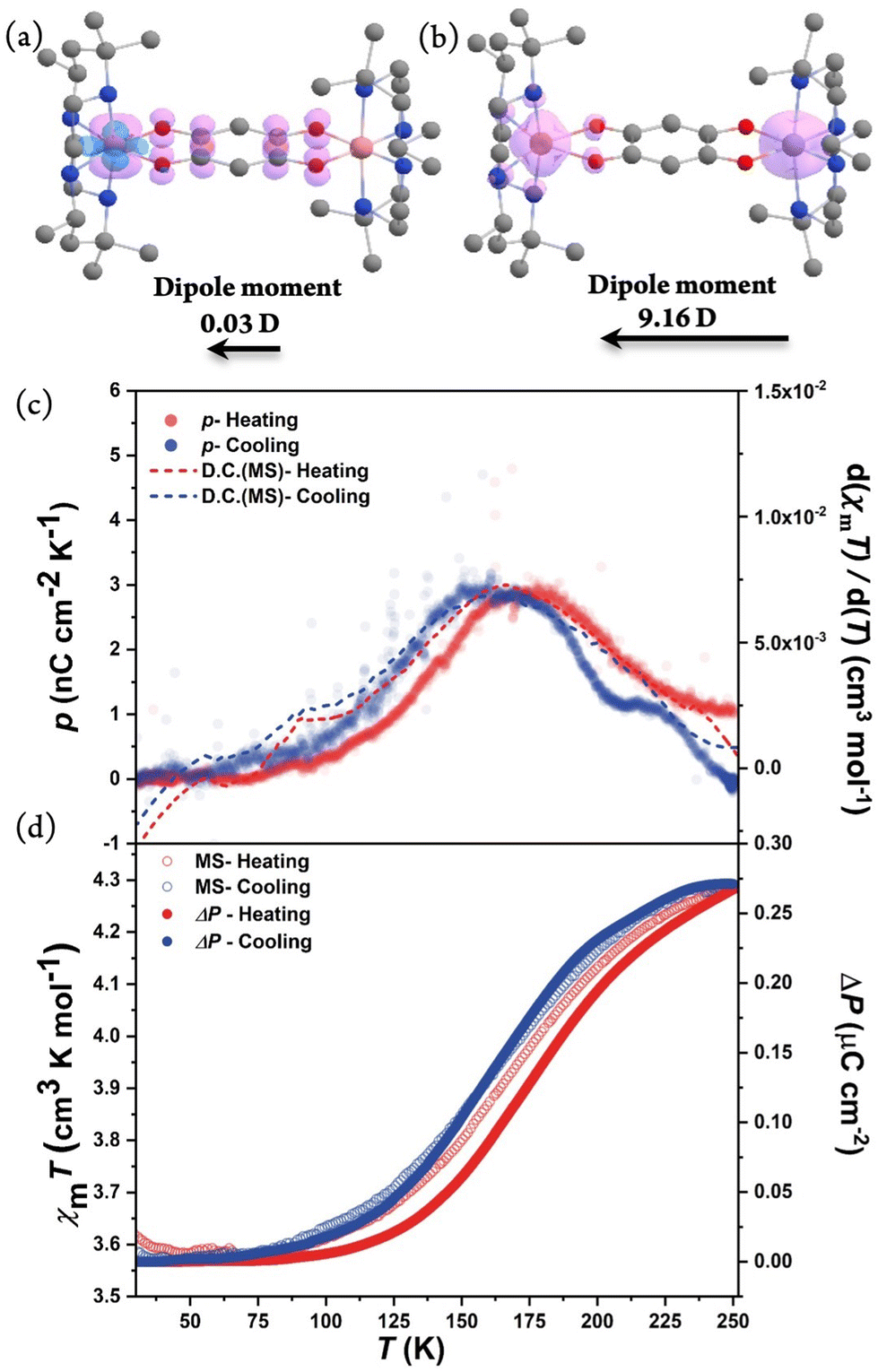 | ||
| Fig. 6 Spin density distribution shown with a contour value of 0.01 (spin Å−3). The structures were optimized based on the X-ray diffraction of 1 at (a) 100 K and (b) 375 K; (c) temperature-dependent pyroelectric coefficient of compound 1·sol from 30 to 250 K with a sweep rate of 5 K min−1. (d) Polarization change compared to magnetization of compound 1·sol. | ||
Conclusions
In conclusion, we presented a heterometallic dinuclear complex with an enantiopure ligand with regular molecular arrangements in a polar space group. The stepwise synthetic route proposed here successfully improves the purity of the desired crystals, and is applicable to the synthesis of other heterometallic dinuclear complexes with enantiopure ligands, even the achiral ones. Among the complexes we synthesized according to this approach, the less compact crystal packing of the homochiral [CrCo] complexes offered extra space to accommodate lattice solvents compared to the situation observed in previously reported [CrCo] complexes with heterochiral ligands. Thus, a new [CrCo] complex was characterized to show a solvent-dependent VT behavior via SCSCT, as demonstrated by the single-crystal structure and magnetometry and spectroscopic techniques. Moreover, pyroelectric measurements clearly confirm the polarization-switching behavior in the pristine sample. Multi-controlled polarization switching offers a good playground for potential application in multifunctional molecular devices. Our results highlight the importance of guest molecules in polarization-switching materials, which could contribute to the further study of molecular sensors by polarization.Author contributions
Wenwei Zheng: investigation, methodology, conceptualization and writing – original draft; Xiaopeng Zhang: investigation and validation; Qirui Shui, Tatsuki Fukuyama, Wen-Huang Xu, Yu-Bo Huang, Tianchi Ji, Ziqi Zhou, and Mikoto Uematsu: investigation; Sheng-Qun Su: methodology, writing – review and editing, and funding acquisition; Shinji Kanegawa: methodology, writing – review and editing, and funding acquisition; Shu-Qi Wu: formal analysis, conceptualization, writing – review and editing, and funding acquisition; Osamu Sato: supervision, project administration, writing – review and editing, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI (21K05085, 22K14694, 20H00385, 21K05086, 23H04862, 24H00466, and 24K17698). S.-Q. W. and S.-Q. S. are grateful for the support from the Iketani Science and Technology Foundation (0341056A and 0351039A).References
- O. Sato, Dynamic molecular crystals with switchable physical properties, Nat. Chem., 2016, 8, 644–656 CrossRef CAS PubMed.
- S.-Q. Wu, M. Liu, K. Gao, S. Kanegawa, Y. Horie, G. Aoyama, H. Okajima, A. Sakamoto, M. L. Baker, M. S. Huzan, P. Bencok, T. Abe, Y. Shiota, K. Yoshizawa, W. Xu, H.-Z. Kou and O. Sato, Macroscopic Polarization Change via Electron Transfer in a Valence Tautomeric Cobalt Complex, Nat. Commun., 2020, 11, 1992 CrossRef CAS PubMed.
- S. Kanegawa, Y. Shiota, S. Kang, K. Takahashi, H. Okajima, A. Sakamoto, T. Iwata, H. Kandori, K. Yoshizawa and O. Sato, Directional electron transfer in crystals of [CrCo] dinuclear complexes achieved by chirality-assisted preparative method, J. Am. Chem. Soc., 2016, 138, 14170–14173 CrossRef CAS PubMed.
- S.-Q. Su, S.-Q. Wu, Y.-B. Huang, W.-H. Xu, K.-G. Gao, A. Okazawa, H. Okajima, A. Sakamoto, S. Kanegawa and O. Sato, Photoinduced Persistent Polarization Change in a Spin Transition Crystal, Angew. Chem., Int. Ed., 2022, 61, e202208771 CrossRef CAS PubMed.
- P. Sadhukhan, S.-Q. Wu, J. I. Long, T. Nakanishi, S. Kanegawa, K. Gao, K. Yamamoto, H. Okajima, A. Sakamoto, M. L. Baker, T. Kroll, D. Sokaras, A. Okazawa, N. Kojima, Y. Shiota, K. Yoshizawa and O. Sato, Manipulating electron redistribution to achieve electronic pyroelectricity in molecular [FeCo] crystals, Nat. Commun., 2021, 12, 4836 CrossRef CAS PubMed.
- C. R. Bowen, J. Taylor, E. LeBoulbar, D. Zabek, A. Chauhan and R. Vaish, Pyroelectric materials and devices for energy harvesting applications, Energy Environ. Sci., 2014, 7, 3836–3856 RSC.
- J. F. Scott, Applications of Modern Ferroelectrics, Science, 2007, 315, 954–959 CrossRef CAS PubMed.
- K. Kobayashi, S. Horiuchi, R. Kumai, F. Kagawa, Y. Murakami and Y. Tokura, Electronic Ferroelectricity in a Molecular Crystal with Large Polarization Directing Antiparallel to Ionic Displacement, Phys. Rev. Lett., 2012, 108, 237601 CrossRef PubMed.
- S. Horiuchi, K. Kobayashi, R. Kumai, N. Minami, F. Kagawa and Y. Tokura, Quantum ferroelectricity in charge-transfer complex crystals, Nat. Commun., 2015, 6, 7469 CrossRef PubMed.
- A. S. Tayi, A. K. Shveyd, A. C. H. Sue, J. M. Szarko, B. S. Rolczynski, D. Cao, T. J. Kennedy, A. A. Sarjeant, C. L. Stern, W. F. Paxton, W. Wu, S. K. Dey, A. C. Fahrenbach, J. R. Guest, H. Mohseni, L. X. Chen, K. L. Wang, J. F. Stoddart and S. I. Stupp, Room-temperature ferroelectricity in supramolecular networks of charge-transfer complexes, Nature, 2012, 488, 485–489 CrossRef CAS PubMed.
- N. Castagnetti, M. Masino, C. Rizzoli, A. Girlando and C. Rovira, Mixed stack charge transfer crystals: Crossing the neutral-ionic borderline by chemical substitution, Phys. Rev. Mater., 2018, 2, 024602 CrossRef CAS.
- S. Tomić and M. Dressel, Ferroelectricity in molecular solids: a review of electrodynamic properties, Rep. Prog. Phys., 2015, 78, 096501 CrossRef PubMed.
- A. Caneschi and A. Dei, Valence Tautomerism in a o-Benzoquinone Adduct of a Tetraazamacrocycle Complex of Manganese, Angew. Chem., Int. Ed., 1998, 37, 3005–3007 CrossRef CAS PubMed.
- C. Carbonera, A. Dei, J.-F. Létard, C. Sangregorio and L. Sorace, Thermally and Light-Induced Valence Tautomeric Transition in a Dinuclear Cobalt–Tetraoxolene Complex, Angew. Chem., 2004, 116, 3198–3200 CrossRef.
- J. Chen, Y. Sekine, Y. Komatsumaru, S. Hayami and H. Miyasaka, Thermally Induced Valence Tautomeric Transition in a Two-Dimensional Fe-Tetraoxolene Honeycomb Network, Angew. Chem., Int. Ed., 2018, 57, 12043–12047 CrossRef CAS PubMed.
- M. G. Cowan, J. Olguín, S. Narayanaswamy, J. L. Tallon and S. Brooker, Reversible Switching of a Cobalt Complex by Thermal, Pressure, and Electrochemical Stimuli: Abrupt, Complete, Hysteretic Spin Crossover, J. Am. Chem. Soc., 2012, 134, 2892–2894 CrossRef CAS PubMed.
- Y. Gong, W.-K. Han, H.-S. Lu, Q.-T. Hu, H. Tu, P.-N. Li, X. Yan and Z.-G. Gu, Single crystal to single crystal transformation of spin-crossover coordination polymers from 3D frameworks to 2D layers, J. Mater. Chem. C, 2021, 9, 5082–5087 RSC.
- J. Tao, H. Maruyama and O. Sato, Valence Tautomeric Transitions with Thermal Hysteresis around Room Temperature and Photoinduced Effects Observed in a Cobalt−Tetraoxolene Complex, J. Am. Chem. Soc., 2006, 128, 1790–1791 CrossRef CAS PubMed.
- T. Tezgerevska, K. G. Alley and C. Boskovic, Valence tautomerism in metal complexes: Stimulated and reversible intramolecular electron transfer between metal centers and organic ligands, Coord. Chem. Rev., 2014, 268, 23–40 CrossRef CAS.
- D.-Q. Wu, D. Shao, X.-Q. Wei, F.-X. Shen, L. Shi, D. Kempe, Y.-Z. Zhang, K. R. Dunbar and X.-Y. Wang, Reversible On–Off Switching of a Single-Molecule Magnet via a Crystal-to-Crystal Chemical Transformation, J. Am. Chem. Soc., 2017, 139, 11714–11717 CrossRef CAS PubMed.
- A. Moledo Vicente Guedes, L. Sodré de Abreu, I. A. V. Maldonado, W. S. Fernandes, T. M. Cardozo, R. A. Allão Cassaro, M. Scarpellini and G. Poneti, Valence tautomerism in a cobalt–dioxolene complex containing an imidazolic ancillary ligand, RSC Adv., 2023, 13, 20050–20057 RSC.
- C. Boskovic, in Spin–Crossover Materials, 2013, pp. 203–224, DOI:10.1002/9781118519301.ch7.
- D. N. Hendrickson and C. G. Pierpont, in Spin Crossover in Transition Metal Compounds II, ed. P. Gütlich and H. A. Goodwin, Springer Berlin Heidelberg, Berlin, Heidelberg, 2004, pp. 63–95, DOI:10.1007/b95413.
- O. Sato, J. Tao and Y.-Z. Zhang, Control of Magnetic Properties through External Stimuli, Angew. Chem., Int. Ed., 2007, 46, 2152–2187 CrossRef CAS PubMed.
- O. Sato, A. Cui, R. Matsuda, J. Tao and S. Hayami, Photo-induced Valence Tautomerism in Co Complexes, Acc. Chem. Res., 2007, 40, 361–369 CrossRef CAS PubMed.
- K. K. C. T. Woods and L. Olshansky, Ligand Modifications Produce Two-Step Magnetic Switching in a Cobalt(dioxolene) Complex, Angew. Chem., Int. Ed., 2023, 62, e202311790 CrossRef PubMed.
- M. A. Hay, J. T. Janetzki, V. J. Kumar, R. W. Gable, R. Clérac, A. A. Starikova, P. J. Low and C. Boskovic, Modulation of Charge Distribution in Cobalt-α-Diimine Complexes toward Valence Tautomerism, Inorg. Chem., 2022, 61, 17609–17622 CrossRef CAS PubMed.
- G. K. Gransbury, B. N. Livesay, J. T. Janetzki, M. A. Hay, R. W. Gable, M. P. Shores, A. Starikova and C. Boskovic, Understanding the Origin of One- or Two-Step Valence Tautomeric Transitions in Bis(dioxolene)-Bridged Dinuclear Cobalt Complexes, J. Am. Chem. Soc., 2020, 142, 10692–10704 CrossRef CAS PubMed.
- G. K. Gransbury and C. Boskovic, in Encyclopedia of Inorganic and Bioinorganic Chemistry, 2021, pp. 1–24 Search PubMed.
- A. Droghetti and S. Sanvito, Electric Field Control of Valence Tautomeric Interconversion in Cobalt Dioxolene, Phys. Rev. Lett., 2011, 107, 047201 CrossRef CAS PubMed.
- T. T. Patricia, M. V. Sandra, L. Manuela, L. Andrea, F. Paolo, D. Andrea and R. Roberto, Transient infrared spectroscopy: a new approach to investigate valence tautomerism, Phys. Chem. Chem. Phys., 2012, 14, 1038–1047 RSC.
- F. V. R. Neuwahl, R. Righini and A. Dei, Femtosecond spectroscopic characterisation of the two-step valence tautomeric interconversion occurring in a cobalt–dioxolene complex, Chem. Phys. Lett., 2002, 352, 408–414 CrossRef CAS.
- D. M. Adams, B. Li, J. D. Simon and D. N. Hendrickson, Photoinduced Valence Tautomerism in Cobalt Complexes Containing Semiquinone Anion as Ligand: Dynamics of the High-Spin [CoII(3,5-dtbsq)2] to Low-Spin [CoIII(3,5-dtbsq)(3,5-dtbcat)] Interconversion, Angew. Chem., Int. Ed. Engl., 1995, 34, 1481–1483 CrossRef CAS.
- M. Wang, Z.-Y. Li, R. Ishikawa and M. Yamashita, Spin crossover and valence tautomerism conductors, Coord. Chem. Rev., 2021, 435, 213819 CrossRef CAS.
- C. Fleming, D. Chung, S. Ponce, D. J. R. Brook, J. DaRos, R. Das, A. Ozarowski and S. A. Stoian, Valence tautomerism in a cobalt-verdazyl coordination compound, Chem. Commun., 2020, 56, 4400–4403 RSC.
- J.-W. Dai, Y.-Q. Li, Z.-Y. Li, H.-T. Zhang, C. Herrmann, S. Kumagai, M. Damjanović, M. Enders, H. Nojiri, M. Morimoto, N. Hoshino, T. Akutagawa and M. Yamashita, Dual-radical-based molecular anisotropy and synergy effect of semi-conductivity and valence tautomerization in a photoswitchable coordination polymer, Natl. Sci. Rev., 2023, 10, nwad047 CrossRef CAS PubMed.
- A. Paul and S. Konar, Electronic pyroelectricity: the interplay of valence tautomerism and spin transition, J. Mater. Chem. C, 2022, 10, 4980–4984 RSC.
- G. K. Gransbury, M.-E. Boulon, S. Petrie, R. W. Gable, R. J. Mulder, L. Sorace, R. Stranger and C. Boskovic, DFT Prediction and Experimental Investigation of Valence Tautomerism in Cobalt-Dioxolene Complexes, Inorg. Chem., 2019, 58, 4230–4243 CrossRef CAS PubMed.
- A. Singh, S. Panda, S. Dey and G. K. Lahiri, Metal-to-Ligand Charge Transfer Induced Valence Tautomeric Forms of Non-Innocent 2,2′-Azobis(benzothiazole) in Ruthenium Frameworks, Angew. Chem., 2021, 133, 11306–11310 CrossRef.
- F. Cheng, S. Wu, W. Zheng, S. Su, T. Nakanishi, W. Xu, P. Sadhukhan, H. Sejima, S. Ikenaga, K. Yamamoto, K. Gao, S. Kanegawa and O. Sato, Macroscopic Polarization Change of Mononuclear Valence Tautomeric Cobalt Complexes Through the Use of Enantiopure Ligand, Chem. – Eur. J., 2022, 28, e202202161 CrossRef CAS PubMed.
- C. Metzger, R. Dolai, S. Reh, H. Kelm, M. Schmitz, B. Oelkers, M. Sawall, K. Neymeyr and H.-J. Krüger, A New Type of Valence Tautomerism in Cobalt Dioxolene Complexes – Temperature-Induced Transition from a Cobalt(III) Catecholate to a Low-Spin Cobalt(II) Semiquinonate State, Chem. – Eur. J., 2023, 29, e202300091 CrossRef CAS PubMed.
- K. S. Kumar and M. Ruben, Sublimable Spin-Crossover Complexes: From Spin-State Switching to Molecular Devices, Angew. Chem., Int. Ed., 2021, 60, 7502–7521 CrossRef CAS PubMed.
- K. Senthil Kumar and M. Ruben, Emerging trends in spin crossover (SCO) based functional materials and devices, Coord. Chem. Rev., 2017, 346, 176–205 CrossRef CAS.
- B. Li, Y.-M. Zhao, A. Kirchon, J.-D. Pang, X.-Y. Yang, G.-L. Zhuang and H.-C. Zhou, Unconventional Method for Fabricating Valence Tautomeric Materials: Integrating Redox Center within a Metal–Organic Framework, J. Am. Chem. Soc., 2019, 141, 6822–6826 CrossRef CAS PubMed.
- N. A. Vázquez-Mera, C. Roscini, J. Hernando and D. Ruiz-Molina, Liquid-Filled Valence Tautomeric Microcapsules: A Solid Material with Solution-Like Behavior, Adv. Funct. Mater., 2015, 25, 4129–4134 CrossRef.
- P. Sadhukhan, S.-Q. Wu, S. Kanegawa, S.-Q. Su, X. Zhang, T. Nakanishi, J. I. Long, K. Gao, R. Shimada, H. Okajima, A. Sakamoto, J. G. Chiappella, M. S. Huzan, T. Kroll, D. Sokaras, M. L. Baker and O. Sato, Energy conversion and storage via photoinduced polarization change in non-ferroelectric molecular [CoGa] crystals, Nat. Commun., 2023, 14, 3394 CrossRef CAS PubMed.
- X. Zhang, W.-H. Xu, W. Zheng, S.-Q. Su, Y.-B. Huang, Q. Shui, T. Ji, M. Uematsu, Q. Chen, M. Tokunaga, K. Gao, A. Okazawa, S. Kanegawa, S.-Q. Wu and O. Sato, Magnetoelectricity Enhanced by Electron Redistribution in a Spin Crossover [FeCo] Complex, J. Am. Chem. Soc., 2023, 145, 15647–15651 CrossRef CAS PubMed.
- H. Kuramochi, G. Aoyama, H. Okajima, A. Sakamoto, S. Kanegawa, O. Sato, S. Takeuchi and T. Tahara, Femtosecond Polarization Switching in the Crystal of a [CrCo] Dinuclear Complex, Angew. Chem., 2020, 132, 15999–16003 CrossRef.
- D. Kiriya, H.-C. Chang and S. Kitagawa, Molecule-Based Valence Tautomeric Bistability Synchronized with a Macroscopic Crystal-Melt Phase Transition, J. Am. Chem. Soc., 2008, 130, 5515–5522 CrossRef CAS PubMed.
- K. G. Alley, G. Poneti, J. B. Aitken, R. K. Hocking, B. Moubaraki, K. S. Murray, B. F. Abrahams, H. H. Harris, L. Sorace and C. Boskovic, A Two-Step Valence Tautomeric Transition in a Dinuclear Cobalt Complex, Inorg. Chem., 2012, 51, 3944–3946 CrossRef CAS PubMed.
- S.-Q. Su, S.-Q. Wu, S. Kanegawa, K. Yamamoto and O. Sato, Control of electronic polarization via charge ordering and electron transfer: electronic ferroelectrics and electronic pyroelectrics, Chem. Sci., 2023, 14, 10631–10643 RSC.
- W.-H. Xu, Y.-B. Huang, W.-W. Zheng, S.-Q. Su, S. Kanegawa, S.-Q. Wu and O. Sato, Photo-induced valence tautomerism and polarization switching in mononuclear cobalt complexes with an enantiopure chiral ligand, Dalton Trans., 2024, 53, 2512–2516 RSC.
- A. G. Starikov, A. A. Starikova, M. G. Chegerev, S. M. Aldoshin, A. V. Metelitsa and V. I. Minkin, Spin-State-Switching Rearrangements of Bis(dioxolene)-Bridged CrCo Complexes: A DFT Study, Eur. J. Inorg. Chem., 2021, 2021, 4113–4121 CrossRef CAS.
- S. Xue, Y. Guo and Y. Garcia, Spin crossover crystalline materials engineered via single-crystal-to-single-crystal transformations, CrystEngComm, 2021, 23, 7899–7915 RSC.
- J.-F. Létard, P. Guionneau, O. Nguyen, J. S. Costa, S. Marcén, G. Chastanet, M. Marchivie and L. Goux-Capes, A Guideline to the Design of Molecular-Based Materials with Long-Lived Photomagnetic Lifetimes, Chem. – Eur. J., 2005, 11, 4582–4589 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2320504, 2320506, 2320374, 2320375 and 2320376. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qi00836g |
| This journal is © the Partner Organisations 2024 |