
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pyramidal inversion in the solid state†
Robin
Turnbull
 *a,
Javier Gonzalez
Platas
b,
Alfonso
Muñoz
c,
Josu
Sánchez-Martín
a,
M.
Jasmin
d,
Gaston
Garbarino
e,
Daniel
Errandonea
a and
Akun
Liang
*a
*a,
Javier Gonzalez
Platas
b,
Alfonso
Muñoz
c,
Josu
Sánchez-Martín
a,
M.
Jasmin
d,
Gaston
Garbarino
e,
Daniel
Errandonea
a and
Akun
Liang
*a
aDepartamento de Física Aplicada-ICMUV-MALTA Consolider Team, Universitat de Valencia, 46100 Valencia, Spain. E-mail: robin.turnbull@uv.es; aliang@ed.ac.uk
bDepartmento de Física, Instituto Universitario de Estudios Avanzados en Física Atómica, Molecular y Fotónica (IUDEA) and MALTA Consolider Team, Universidad de La Laguna, E-38206 La Laguna, Spain
cDepartamento de Física, Instituto de Materiales y Nanotecnología, MALTA Consolider Team, Universidad de La Laguna, E-38206 La Laguna, Spain
dTKM College of Arts and Science, 691005, Kerala, India
eEuropean Synchrotron Radiation Facility, 71 Avenue des Martyrs, 38000 Grenoble, France
First published on 26th July 2024
Abstract
Pyramidal inversion is a stereochemical phenomenon that describes the interconversion between two equivalent pyramidal configurations of the same chemical species. Using the IO3 molecule as a prototypical trigonal pyramidal unit, pyramidal inversion has been observed in the solid state by applying hydrostatic pressure to crystals of barium di-iodate monohydrate, Ba(IO3)2·H2O, without chemical reaction or the introduction/elimination of guest molecules. The pyramidal inversion was identified by high-pressure single-crystal synchrotron X-ray diffraction from the appearance of charge density on the unoccupied side of the IO3 pyramid at pressures above 5 GPa. The percentage of inverted pyramids increases with pressure, from 2.5% at 5.10(5) GPa to 17.5% at 14.84(5) GPa. The energetic competition between the original and inverted IO3 pyramids as a function of pressure is investigated by density functional theory calculations, finding the two configurations to be very close in energy. Factors contributing to the observation of pyramidal inversion in barium iodate monohydrate are discussed and it is suggested that hydrogen bonding due to the presence of water may play a significant role.
1. Introduction
A tri-coordinated atom can be positioned outside of the plane defined by its three bonded atoms, and it may exist on either side of the plane, thereby making a trigonal pyramid. Pyramidal inversion, as described in ref. 1–3, is the phenomenon of interconversion between these configurations, and is observable in many molecules, including the well-known example of ammonia (NH3)4 and its analogs.5 Pyramidal inversion has traditionally been observed in the fluid state (gas/liquid/solvent). To the best of the authors’ knowledge, this study represents the first observation of pyramidal inversion in a system composed of a single chemical species in the solid state. Here, the inversion of IO3 pyramids in barium iodate monohydrate, Ba(IO3)2·H2O, is driven solely by an increase in the thermodynamic variable of pressure. No other chemical species are involved. Recently, the inversion of a VO5 square pyramidal unit, which forms a constituent of a larger dodecavanadate shell, was observed in the solid state, however, differing from the current work, the VO5 inversion was driven by the capture/elimination of guest gas molecules.6 In the current work we observe that the statistical distribution of original and inverted IO3 pyramids shifts towards inverted pyramids with increasing pressure, with the percentage of inverted pyramids increasing from 2.5% at 5.10(5) GPa to 17.5% at 14.84(5) GPa. Below 5.1 GPa the inversion is not observed, suggesting the possible existence of a critical pressure.The properties of metal iodate materials under high-pressure conditions have been increasingly attracting attention over recent years due to their piezoelectric and nonlinear optical properties7 which originate from the relative orientations of the IO3 pyramids in the crystal structure, in particular the relative orientations of the iodine lone electron pairs.8 However, previous studies have solely focused on anhydrous metal iodate structures (with the exception of ref. 9), disregarding the fact that the IO3 ion is strongly hydratable10 and that naturally occurring iodates can be hydrated.11 The inclusion of H2O may play an important role in facilitating the inversion of the IO3 pyramids in Ba(IO3)2·H2O which was not observed in the only other previously studied hydrated metal iodate, Ca(IO3)2·H2O,9 although it should be noted that, in contrast to the present work, Ca(IO3)2·H2O was not studied by single-crystal X-ray diffraction. The crystal structures of barium and calcium bi-iodate monohydrate are also different, suggesting that the specific crystal structure of Ba(IO3)2·H2O facilitates the pyramidal inversion.
The iodate ion, IO3, is the main building block of the Ba(IO3)2·H2O crystal structure and is the focus of this work. It was first observed to have a pyramidal structure in 1937 via the observation of 4 Raman active bands in iodate-containing solutions, rather than 3 bands expected of trigonal planar geometry.12 In the IO3 molecule, the iodine atom has a 5+ oxidation state. Therefore, in addition to the three covalent I–O bonds, the I atom also has a lone electron pair which repels the 3 oxygen atoms out of the planar configuration. The lone pair leads to many of the useful anisotropic phenomena associated with iodate materials, such as nonlinear optical properties, like second-harmonic generation, or ferroelectric and piezoelectric properties, making them attractive for technological applications.7
2. Experiment and calculation methods
2.1. Sample preparation
Single crystals of Ba(IO3)2·H2O were synthesised at room temperature using a single diffusion gel growth mechanism. Barium meta silicate (Ba2SiO3·9H2O) was dissolved in double distilled water, creating a gel with a density of 1.03 g cc−1. 0.5 M molarity potassium iodate was then added to the gel. Glacial acetic acid was then added to the gel raise the pH to 4.2. To prevent air bubbles in the gel medium, this acidified gel solution was poured into a test tube 20 cm long and 2.5 cm in diameter. For four days, the gel solution was left in the test tube undisturbed. 10 ml of a 0.25 M barium chloride solution was added to the gel once it had set. Sealing the test tubes helped prevent contamination. For the duration of the growth phase the tubes were not disturbed. Within a week, white, needle-shaped, transparent, high-quality single crystals of Ba(IO3)2·H2O were obtained. A single crystal of Ba(IO3)2·H2O is shown in situ in a diamond anvil cell in ESI Fig. S1.†2.2. High-pressure single-crystal X-ray diffraction
In this work three different single-crystal X-ray diffraction (SCXRD) experiments were carried out: two at the University of La Laguna (ULL); and one at the European Synchrotron Radiation Facility (ESRF). The data from the two experiments at the ULL have been combined into a single data set in all subsequent discussion and figures. All measurements were acquired at room temperature.At the ULL, SCXRD measurements were conducted using a molybdenum radiation source (λ = 0.71073 Å) with a beam diameter of 150 μm, utilizing a Rigaku SuperNOVA diffractometer equipped with an EOS charge-coupled device (CCD). High-pressure (HP) measurements involved the utilization of a Mini-Bragg diamond anvil cell featuring an 85° opening angle and anvil culets with a diameter of 500 μm, thus creating the required high-pressure environment. A stainless-steel gasket, with a central hole measuring 200 μm in diameter and 75 μm in depth, was used to contain the sample. A methanol–ethanol (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture was used as the pressure-transmitting medium (PTM). Positioned on the diffracting side of one of the diamond anvils, the sample was accompanied by a ruby sphere for use as the pressure sensor. The sample size was: 150 × 100 × 60 μm (Experiment 1)/80 × 50 × 40 μm (Experiment 2).
:1) mixture was used as the pressure-transmitting medium (PTM). Positioned on the diffracting side of one of the diamond anvils, the sample was accompanied by a ruby sphere for use as the pressure sensor. The sample size was: 150 × 100 × 60 μm (Experiment 1)/80 × 50 × 40 μm (Experiment 2).
At the ESRF, SCXRD measurements were conducted at beamline ID15B using an Eiger2 X CdTe 9M (DECTRIS) detector with an X-ray wavelength of 0.41 Å and beam size of 1 × 1 μm. The sample (50 μm × 50 μm × 50 μm) was loaded into a membrane-style diamond anvil cell with an opening angle of 60°. A 200 μm stainless steel gasket was indented to a thickness of 90 μm and a chamber of 300 μm diameter was drilled in the centre. The culet size of the diamonds was 600 μm. A ruby sphere was included in the chamber to measure the sample pressure. Helium was used as the PTM.
CrysAlisPro software was used to process all measurements. The ABSORB7 program was used for numerical absorption correction based on Gaussian integration over a multifaceted crystal model. The crystal structure was refined for each pressure, using previous results as starting points. As starting point, we solved the structure at ambient conditions, applying the novel dual-space algorithm implemented in SHELXT program.13 Fourier recycling and least-squares refinement were used for the model completion with SHELXL-2018.14 All non-hydrogen atoms were refined anisotropically, and all hydrogen atoms were placed in geometrically suitable positions and refined with isotropic thermal parameters related to the equivalent isotropic thermal parameter of the parent atom. Due to limitations of the opening angle of our diamond anvil cell, it was only possible to collect about 30–60% of the reflections present in a full dataset. Consequently, and due to the ratio between reflections and parameters to be refined, only the heaviest atoms were refined anisotropically, with the rest being refined isotropically, while the positions of the hydrogens were always calculated in the last step of the refinement procedure.
Around 3.5 GPa we observed the presence of a residual peak compatible with a positional splitting for the iodine atom, with that peak becoming more relevant as the pressure was increased. Therefore, this positional splitting was accounted for in the subsequent refinements above 4 GPa, in which the occupancy factor between both positions for the iodine atom were also refined, restricting the thermal factors of the I1 and I2 atoms to the same value. It should be noted that, given the limitation of experimental data acquired on high pressure samples, we cannot precisely determine the occupation factor of the iodine atom, however the increase in the occupation of the I2 site on sample compression is clear.
2.3. Density functional theory calculations
In this work simulations were performed with the well-known plane wave pseudopotential method in the framework of density-functional theory (DFT, ref. 15) implemented in the Vienna Ab initio Simulation Package, VASP.16,17 We used the projector augmented wave (PAW) pseudopotentials (ref. 18 and 19) provided in the VASP pseudopotentials database, and the exchange correlation term was described using the generalized gradient approximation (GGA), using the AM0520,21 functional. Brillouin zone k-point sampling was performed using a dense Monkhorst-Pack22 with a grid of 6 × 6 × 6. A plane-wave basis set with an energy cutoff of 600 eV was used to ensure accurate and highly converged results. For the different analyzed structures, all degrees of freedom, lattice parameters, and internal atomic parameters were fully relaxed with self-consistent convergence criteria of 0.003 eV Å−1, 10−6 eV and below 0.1 GPa for the atomic forces, the self-consistent total energy convergence, and the diagonal terms of the stress tensor, respectively. From our simulations we obtain a set of volume, energy, and pressure data that can be fitted using an equation of state, EOS.The major, intermediate and minor principal compression axes (κ2, κ3 and κ1) respectively, were determined using the PASCaL web tool for principal axis strain calculations,23 with magnitudes of 10.1(1), 5.88(5) and 2.50(7) × 10−3 GPa−1. The compressibility is highly anisotropic, with κ2 being four times more compressible than κ1, for example. The anisotropic compressibility can be explained in terms of the crystal structure. The respective directions of the major, intermediate and minor compression axes are: [010], [409] and [301]. Therefore, the major compression axis, κ2, is exactly parallel to the crystallographic b axis, which also corresponds to the Ba–O bond direction. It is also perpendicular to the chains of BaO11 units which go through the crystal structure (Fig. S3†). Therefore, the BaO11 chains are pushed into the space occupied by the H2O molecule, rather than into each other. The intermediate compression axis, κ3, is in the direction between the c axis and the ac face diagonal, therefore it points roughly along the BaO11 chains. The minor compression axis, κ1, is roughly parallel to the a axis, and therefore pushes the edge sharing BaO11 units (which link the chains together), pushing one BaO11 chain into the adjacent chain.
3. Results and discussion
3.1. Ambient pressure crystal structure of Ba(IO3)2·H2O
The crystal structure of barium di-iodate monohydrate, Ba(IO3)2·H2O, as determined by room-temperature single-crystal X-ray diffraction (SCXRD), was first reported in ref. 24, where it was found to have a monoclinic lattice (space group I2/c, Z = 4) isostructural to the di-bromate and di-chlorate analogs Ba(BrO3)2·H2O and Ba(ClO3)2·H2O.25–27 Subsequent room-temperature and low-temperature single-crystal neutron diffraction studies, combined with Raman and infrared vibrational studies, confirmed an earlier hypothesis that the H2O molecule in Ba(IO3)2·H2O exhibits dynamical disorder at ambient temperature.28,29 The water H atom positions exhibit less disorder than the water O atoms to which they are bonded.The ambient pressure structure of Ba(IO3)2·H2O as determined by SCXRD in this work is shown in Fig. 1a. The starting atomic positions and lattice parameters used in the refinement were all taken from ref. 28. In the present work, the unit cell parameters of Ba(IO3)2·H2O at ambient conditions were determined to be: a = 9.0494(2) Å, b = 7.9814(2) Å, c = 9.9187(3) Å and β = 92.147(2)°. The optimized unit cell parameters determined from the density functional theory calculations at ambient pressure are in excellent agreement: a = 8.96826 Å, b = 8.11657 Å, c = 10.00231 Å and β = 90.865°. CIF files from the present work are available free of charge from the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers 2341132–2341134.† SCXRD refinement details for selected pressure are included in Table S1 in the ESI† and example two-dimensional SCXRD precession images are available in Fig. S4.†
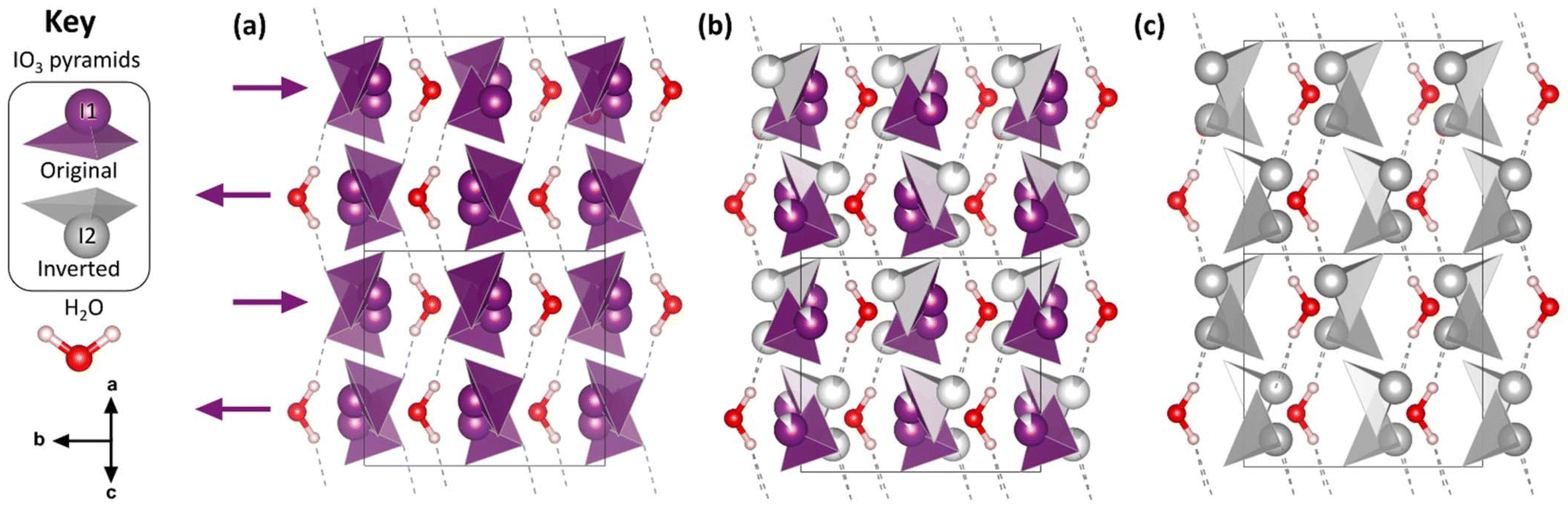 | ||
| Fig. 1 The crystal structure of Ba(IO3)2·H2O. The Ba atoms have been omitted for clarity. See Fig. S2† for a version of Fig. 1 which includes the Ba atoms. (a) The structure at ambient pressure. (b) The structure at high pressure (9.59(5) GPa) where the occupation of the I(2) site is 12.5%. (c) The hypothetical fully-inverted structure. The crystal structure is viewed along the ac body diagonal, perpendicular to the b axis: uvw projection vector (101), hkl upwards vector (100), space group I2/a (no. 15, setting 3). Hydrogen bonds are shown as dashed lines. The key shows the essential molecular units of the structure. The black arrows below the key indicate the crystal axes. | ||
The essential building blocks of the Ba(IO3)2·H2O crystal structure (IO3 and H2O) are shown in the key in Fig. 1. In the crystal structure there is one crystallographically unique iodine atom I1 and it occupies the 8e Wyckoff position. The iodine atom is coordinated by three oxygen atoms (O1, O2, and O3) each forming single I–O covalent bonds, thereby forming the previously described IO3 pyramids. It is important to note for later discussion that all of the IO3 pyramids in the crystal structure are identical in terms of their chemical environment. In total, the structure contains four crystallographically distinct oxygen atoms, with the fourth one (O4) belonging to the H2O molecule. The water molecule has an ideal trigonal planar coordination, as shown in Fig. 2a. It forms two equivalent H1⋯O2 hydrogen bonds, each to the O2 vertex of an IO3 pyramid, and a Ba1–O4 metal–oxygen bond which points exactly parallel to the crystallographic b-axis. This description of the trigonal coordination of the water molecule (Fig. 2a) constitutes the chemical formula, Ba(IO3)2·H2O, and crystal motif which shall be used later to discuss the high-pressure evolution of the crystal structure. The ‘V-shaped’ IO3⋯HOH⋯O3I motifs are related by a 21 screw axis in the a direction. Consequently, they are arranged into layers along the (101) body diagonal, pointing in alternating directions along the b-axis according to the layer which they are in, as shown by the purple arrows in Fig. 1a. The crystallographically unique Ba atom (Ba1) is located within the layers and it is coordinated by oxygen atoms forming octadecahedral BaO11 units (see Fig. S2a†). Of these 11 oxygen atoms, six come from corner-sharing with six IO3 pyramids, four come from edge-sharing with two IO3 pyramids, and one comes from H2O, which forms the shortest Ba–O distance. The BaO11 units share edges to form two parallel chains that run along the crystallographic c-axis (see Fig. S2b and S2c†).
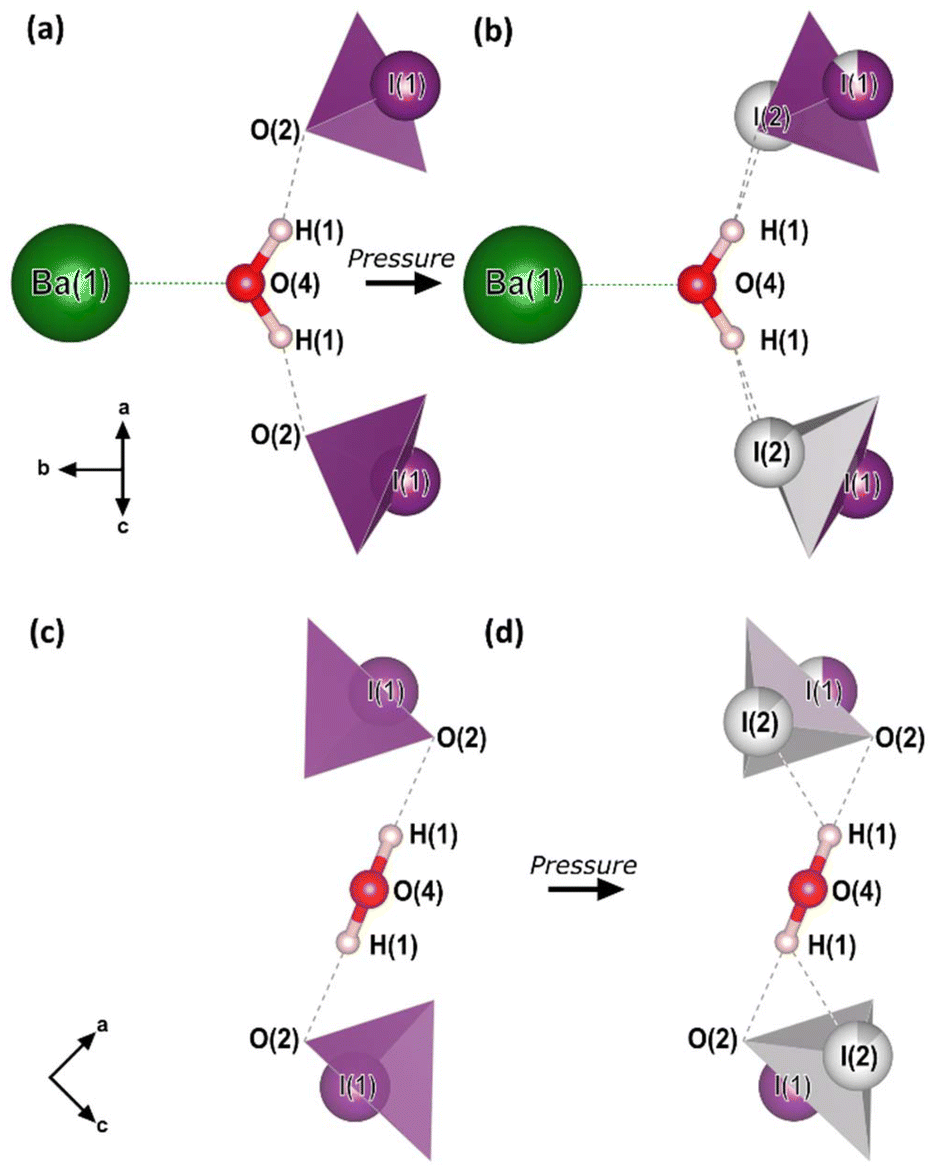 | ||
| Fig. 2 The crystal motif and chemical formula, Ba(IO3)2·H2O. (a and c) The crystal motif of the low-pressure (0–5 GPa) crystal structure, before pyramidal inversion is observed. (b and d) The crystal motif of the high-pressure (5–15 GPa) crystal structure, showing the partial occupation of both I1 and I2 sites. All of the atomic positions shown were determined from SCXRD in this work with the exception of the H atom positions which are taken from the ambient-pressure single-crystal neutron diffraction study of ref. 28. The H atom positions from our optimized DFT calculations agree with those of ref. 28. The black arrows indicate the crystal axes. | ||
3.2. High-pressure evolution of the crystal structure of Ba(IO3)2·H2O
To explore the mechanism driving the inversion of the IO3 pyramids, here we examine the geometric evolution of the IO3 pyramids based on Rietveld refinement of SCXRD measurements. The numerical quantities given in this section are based on the ESRF data only, although the data from all experiments agree and are shown in the figures. The description of the pressure evolution of the crystal structure begins at ambient pressure and follows the compression of the crystal. Below, the description is separated into two subsections: (1) before pyramidal inversion is observed (0 to 5.10(5) GPa), and, (2) pressure-induced pyramidal inversion (5.10(5) to 14.84(5) GPa).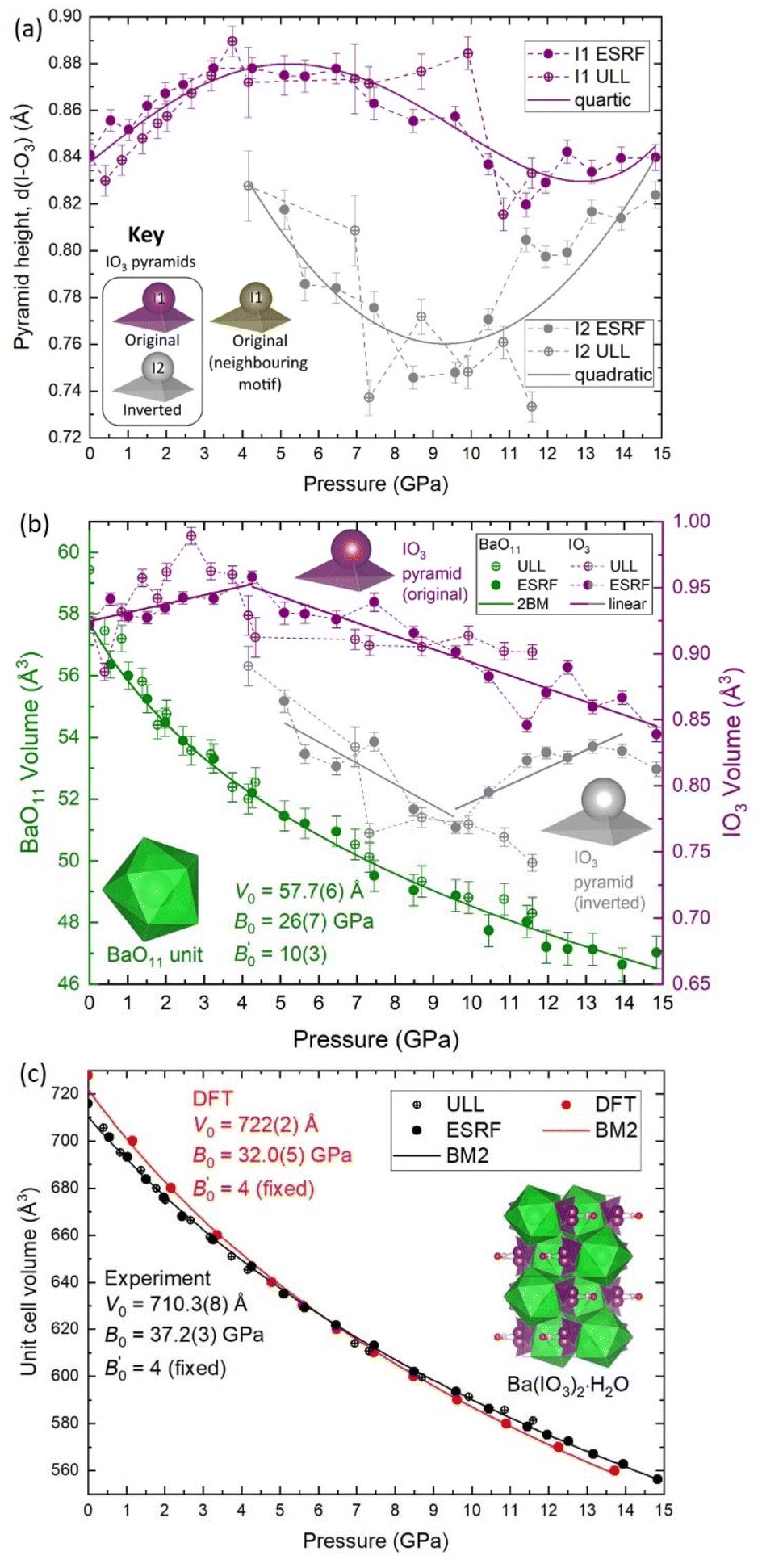 | ||
| Fig. 3 The pressure evolution of the polyhedral geometry and volume determined from SCXRD. (a) The IO3 pyramid height, d(I–O3). (b) The volume of the original and inverted IO3 pyramid (right y-axis), and the volume of the BaO11 unit (left y-axis). (c) The volume-pressure equation of state (EOS) of Ba(IO3)2·H2O according to SCXRD (black) and DFT calculations (red). The experimental values determined from SCXRD at the ULL and the ESRF for the BaO11 unit in part (b) and for the unit cell in part (c) were fitted with single Birch–Murnaghan equations of state. In (c) the error in unit cell volume is smaller than the symbols. The insets show schematic representations of the IO3 pyramids to assist the reader with the description of the IO3 evolution. The error on all experimental pressure measurements is ±0.05 GPa. | ||
One possible explanation for the increase in height of IO3 pyramids below 5 GPa is the growing electrostatic interaction between the electron-dense I1 iodine atom lone electron pairs (LEPs) and the electrophilic (δ+) hydrogen atom of the water molecule. To better visualize this interaction, the reader is referred to Fig. 4a, which shows the repeating pattern of the crystal motif which was shown in detail in Fig. 2a. As previously described, the H2O molecule forms two equivalent hydrogen bonds, each to the O2 vertex of an IO3 pyramid, shown by the solid red lines. Although the H2O molecule is hydrogen bonded to these two IO3 pyramids, it is actually coordinated by four IO3 pyramids in total, where the two extra IO3 pyramids come from neighboring motifs, as shown in Fig. 4b and more detail in Fig. 4c and d. The two IO3 pyramids of the original motif (shown in purple) are related by a 180° rotation about the Ba1–O4 bond (parallel to the b-axis). Therefore, their orientations relative to the H2O molecule are identical. In that orientation, the LEP of the I1 atom points away from the H2O molecule. In other words, the water molecule can “see” only the triangular bases of the IO3 pyramids to which it is hydrogen bonded (see Fig. 2a and c). In contrast, the IO3 pyramids which come from the neighboring motifs (shown in yellow) are oriented so that their LEPs point toward the H2O molecule of the original motif (see Fig. 4d). It should be reiterated here that the purple and yellow IO3− pyramids are crystallographically identical, therefore, if the ‘yellow’ pyramids get taller, so do the ‘purple’ pyramids. Apart from the H atoms, the nearest atom to the O4 atom of the H2O molecule is the O2 vertex of the IO3 pyramid to which it is hydrogen bonded. The d(O4–O2) distance varies very little over the entire pressure range studied, varying between 2.75 and 2.65 Å, as shown by the red data in Fig. 5. (The data in Fig. 5 are color-coded to match the interatomic distances of interest shown in Fig. 4.) The next nearest atom to the O4 atom of the H2O molecule is the iodine I1 atom from the neighboring motifs. The d(O4–I1) distance decreases from 3.5 Å to 3.3 Å as pressure is increased from 0 to 5.10(5) GPa, as shown by the yellow data in Fig. 5. In other words, as pressure is increased, the I1 atom approaches LEP-first towards the water molecule (see Fig. 4d), thereby increasing interaction between the I1 LEP and the Hδ+ of the water molecule and increasing the IO3 pyramid height. Following the convention of Jeffrey et al.31,32 a hydrogen bond in the range 3.5 to 3.3 Å is compatible with ‘weak’ hydrogen bonding, whereas a hydrogen bond in the range 2.6 to 2.8 Å is compatible with ‘moderate’ bonding. Therefore, the hypothesis is that below 5.10(5) GPa there is a competition between the O2 vertex of IO3, and the LEP of the I1 atom from the neighboring motif, to form hydrogen bonds with the water molecule. The hydrogen bond to the O2 vertex of the IO3 pyramid is dominant since it is shorter (2.7 Å vs. 3.3 Å), however between 0 and 5.10(5) GPa the I1 approaches the water molecule more quickly, indicating that the ILEP becomes more competitive with increasing pressure. Looking at Fig. 4c and d, it is clear that a slight rotation of the H2O molecule about the Ba–O4 bond (crystallographic b-axis) would favor the ILEP–Hδ+ electrostatic attraction because the position of the I(1) atoms fulfills the strict directionality requirements of hydrogen bonding. At 5.10(5) GPa, the angle subtended by the O2 vertex and Ba1 atom about the water molecule is φ(O2–O4–Ba1) = 109.6(4)° (see Fig. 4d). The angle subtended by the I1 and Ba atom about the water molecule, φ(I1–O4–Ba1) = 123.3(2)°, which is closer to the angle defined by the hydrogen atoms according to ref. 28, φ(H–O4–Ba1) = 122.0(4)°, which would favor the hydrogen bond interaction with the I1 atoms. Since the H atom positions cannot be determined from SCXRD, the H positions shown throughout this article are taken from the neutron diffraction study of ref. 28, although it is plausible that the H2O molecule may rotate to a new equilibrium position, forming bifurcated H-bonds with both the O2 vertex and the I1 LEP. From these results it is clear is that between 0–5.10(5) GPa it appears to be energetically favourable for the IO3 pyramids to increase in height and volume, possibly due to the strengthening electrostatic interaction between the I1 LEP and the Hδ+ atoms of the H2O molecule. However, the trend of increasing IO3 pyramid volume with increasing pressure cannot physically continue indefinitely and, as described in the next section, the trend is broken at higher pressures.
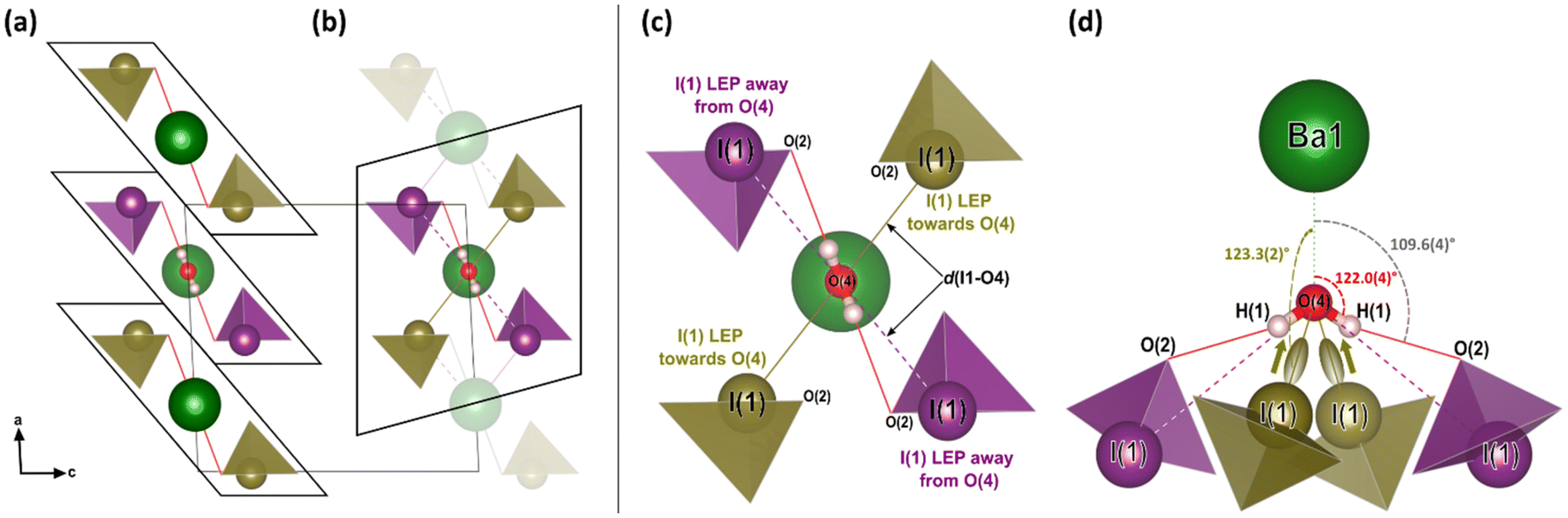 | ||
| Fig. 4 The low-pressure crystal structure of Ba(IO3)2·H2O. (a) The repeating motif corresponding to the chemical formula of barium di-iodate monohydrate including the hydrogen bonds (solid red lines) to the O2 vertex of the IO3 pyramids. (b) An alternative motif that emphasizes the importance of the neighbouring IO3 pyramids (yellow) whose I atoms are closer to the water O4 atom. (c) A detailed expanded view of part (b) which shows atom labels and the important interatomic distances (which are plotted as a function of pressure in Fig. 3b). (d) A rotated view of part (c) to clarify the interaction of the I1 electron pair with the water molecule. The black arrows indicate the crystal axes. The Ba1–O4–H1 angle of 122.0° (red) is taken from the ambient pressure neutron diffraction measurements of ref. 28. | ||
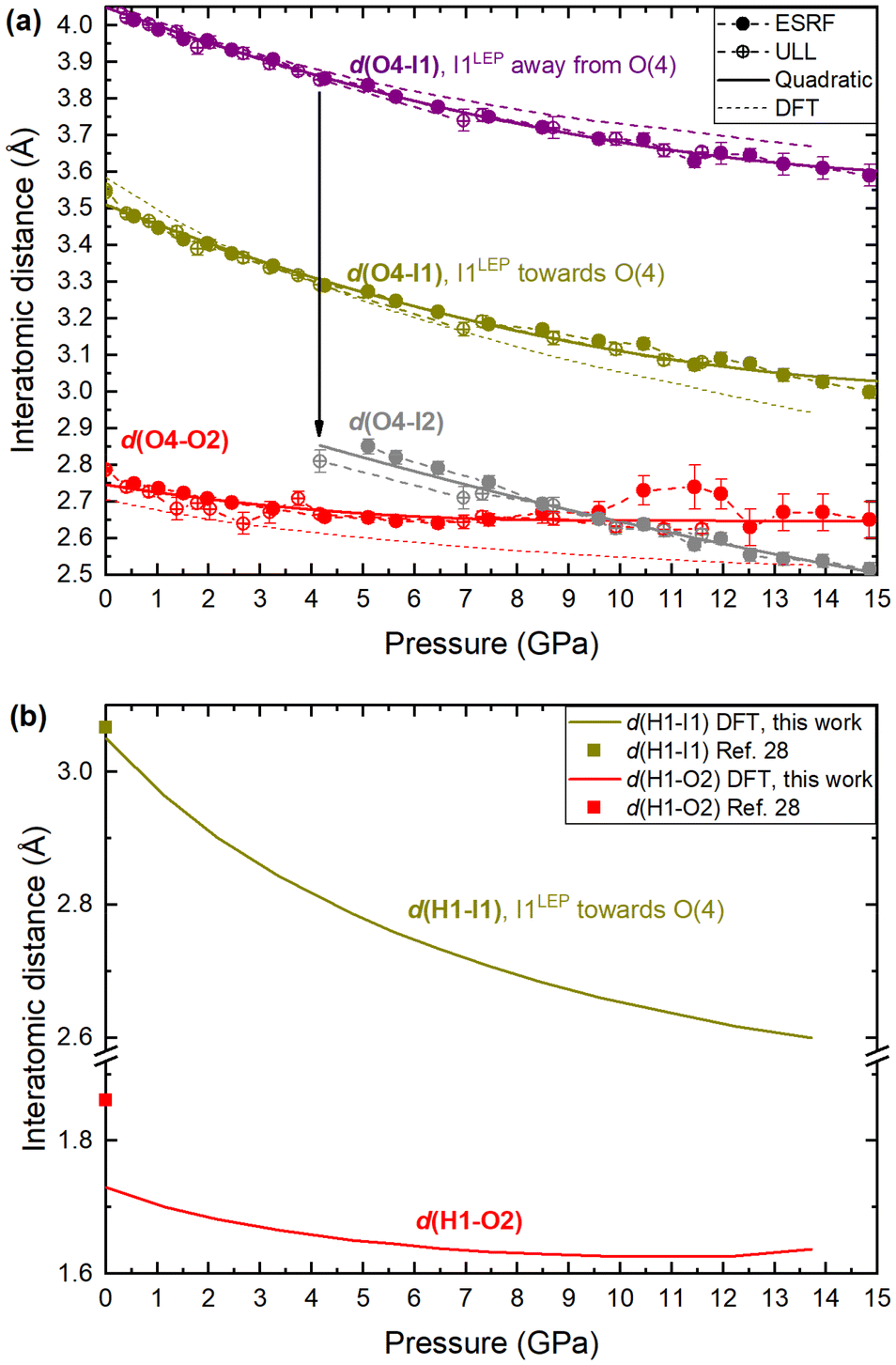 | ||
| Fig. 5 Interatomic distances in Ba(IO3)2·H2O with increasing pressure according to SCXRD and DFT calculations. (a) The interatomic distance between the iodine atom and the O4 oxygen atom of the H2O molecule, d(I–O4). d(O4–O2), which corresponds to the hydrogen bond between the O4 vertex of the IO3 pyramid and the oxygen atom of the H2O molecule at ambient pressure, is included for comparison (red). (b) The hydrogen bond distances, d(H1–I1) and d(H1–O2), according to DFT calculations. The symbols at ambient pressure are taken from the neutron diffraction study of ref. 28. The error on all experimental pressure measurements is ±0.05 GPa. | ||
Firstly, regarding the I1/I2 site occupation, at 5.10(5) GPa, the lowest pressure at which the I2 atom was observed, the occupation of the I2 site is only 2.6%, meaning that most of the iodine atoms remain in the original I1 position. The occupation of the I2 site increases with pressure up to 17.5% at 14.84(5) GPa (see Fig. S9†). Therefore, within the pressure range 5.10(5) to 14.84(5) GPa, the distribution of original I1 pyramids and inverted I2 pyramids shifts towards the inverted I2 pyramid with increasing pressure, although the equilibrium is always biased towards the original I1 pyramid. This suggests that the two configurations must be increasingly energetically similar with increasing pressure. This hypothesis is supported by DFT calculations discussed later in this section. The Ba(IO3)2·H2O structure with a distribution of original/inverted IO3 pyramids is shown with an I2 site occupancy of 12.5% in Fig. 1b.
Secondly, regarding the IO3 pyramid height and volume, strictly speaking pyramidal inversion, as defined in ref. 1–3, refers to conversion between two equivalent isomers, however in this case the two pyramids are not equal. The inverted I2 pyramid is always shorter than the original I1 pyramid (see Fig. 3a) and it always has a lower volume (see Fig. 3b). For example, at 5.10(5) GPa the original I1 pyramid has a volume of 0.9318 Å3, and the inverted I2 pyramidal volume of 0.8641 Å3, which constitutes a volume decrease upon inversion of approximately 7%, thereby making the original I1 pyramid more favourable than the inverted I2 pyramid from a purely thermodynamic perspective since the total internal energy (U) can be referred to U = Q − PV under the assumption that there is no heat (Q) transfer during the pyramidal inversion. That agrees with the fact that the majority remains in the original I1 configuration as observed in SCXRD. The appearance of the I2 pyramid also coincides with a change in the pressure evolution of the I1 pyramid height and volume as observed by the change in gradients in Fig. 2a and b. Between 5–10 GPa, both the I1 and I2 pyramids decrease in height and volume with increasing pressure, which is the expected result for a crystal under compression. Above 10 GPa the behaviour changes again, with I1 volume continuing to decrease under compression, albeit with a relatively constant pyramid height remaining with pressure, and the I2 height and volume beginning to increase again with increasing pressure. This observation is consistent with the explanation offered in the previous section for the increasing height of the I1 pyramid below 5 GPa. Specifically, as the I2 atom approaches the H2O molecule more closely than the water O2 atom, the ILEP–Hδ+ interaction becomes stronger, causing the height of the pyramid to increase.
Thirdly, regarding the distance between the I atom and the O4 atom of the H2O molecule, at 5.1 GPa the I–O distance in the original I1 pyramid is d(I1–O4) = 3.30 Å, whereas the I–O distance in inverted I2 pyramid the distance is d(I2–O4) = 2.80 Å (see yellow vs. grey data in Fig. 5a), making the I2 atom much more competitive than the I1 atom with the hydrogen-bonded O2 vertex in terms of distance from the H2O molecule (see red vs. grey data in Fig. 5a). The I2 atom even becomes the closest atom to the H2O molecule above 10 GPa. The shortening of the I–O4 distance upon pyramidal inversion also is shown in Fig. 2 by the appearance of the grey I2 atoms. This decrease in the I–OH2 distance constitutes a change from ‘weak’ to ‘moderate’ hydrogen bonding according to the aforementioned convention of Jeffrey et al.31,32 The H atom may form bifurcated hydrogen bonds with the I2 atom and the O4 atom, as shown in Fig. 2b and d. Therefore, the inverted I2 pyramid is competitive with the original I1 pyramid due to two main factors: the formation of shorter hydrogen bonds due to its proximity to the H2O molecule; and the decrease in pyramidal volume. The hydrogen bond distances (H–O and H–I) based on the geometrically optimised structures from DFT calculations are shown in Fig. 5b, supporting the hypothesis that the H–I bond distance (yellow) becomes increasingly competitive with the H–O distance (red) with increasing pressure. The interatomic distances determined from DFT shown in Fig. 5a (dashed lines) show excellent agreement with the distances determined via SCXRD. From these results it is clear is that between 5.10(5)–14.84(5) GPa the SCXRD measurements unambiguously reveal pressure-induced pyramidal inversion in the IO3 pyramids.
It is worth mentioning that one potential contribution to the kinetic barreier between the original and inverted iodine site is the I1–I2 distance. For example, in the low-pressure crystal structure (Fig. 1a), the shortest I–I distance is d(I1–I1) = 3.580(2) Å. In a hypothetical fully inverted structure (Fig. 1c) the shortest I–I distance is d(I2–I2) = 3.11(4) Å. However, in the real high-pressure crystal structure (Fig. 1b), which contains a distribution of I1 and I2 pyramids, the shortest I–I distance is d(I1–I2) = 2.692 Å, which is similar to the I–I covalent bond length in gaseous I2 molecules (2.665 A, ref. 33) and shorter than the I–I distance in solid crystalline iodine (2.728(1) Å, ref. 34). The proximity of neighboring I1 and I2 atoms may contribute to the potential barrier to pyramidal inversion.
In order to quantify the competition between the I1 and I2 sites we performed additional DFT calculations where the occupation of the I1 and I2 sites was approximated in the following way. The experimentally determined Ba(IO3)2·H2O structures, which were refined against the SCXRD patterns, had partial I1 and I2 site occupations, where the total occupation (I1 + I2) is equal to 1. DFT calculations cannot be performed for partially occupied atomic sites. Therefore, to approximate, for example, an I2 site with a partial occupation of 0.25, one quarter of the I1 atoms in the crystal structure were replaced with their analogue I2 atoms. The Ba(IO3)2·H2O until cell contains eight I atoms, therefore in the example case of 25% IO3 inversion, there would be six original I1 atoms and two inverted I2 atoms. In order to treat the atoms individually in this way it was necessary to remove the monoclinic crystal symmetry (C2/c) and treat the structure as triclinic (P1), except in the cases of 0% and 100% I2, in which cases it was treated as monoclinic. This approach constitutes an approximation because the real site occupation, derived from SCXRD, is the time-average site occupation, i.e. at any random instant the I2 site may or may not be occupied with a statistical probability proportional to the observed occupation. In the DFT calculations, the I2 site is either always or never occupied. Therefore, the DFT calculations do not account for any kinetic effects.
Fig. 6a shows the calculated enthalpy for the Ba(IO3)2·H2O structure as a function of the percentage of inverted I2 pyramids, from 0% to 100%, in increments of 12.5%. All nine structures were calculated using the DFT optimized lattice parameters from the case of 0% I2 at 12.2 GPa: a = 12.2910 Å, b = 7.1088 Å, c = 8.7465 Å and β = 131.77°. According to the SCXRD data, at 12.2 GPa the occupation of the I2 site is approximately 17%. According to the DFT data in Fig. 6a, there is a local minimum in the enthalpy between 37.5 and 50% I2. The energy difference between 0% and 50% configurations is ∼30 meV at ambient pressure. For comparison, the thermal energy at room temperature is kBT = 25 meV. Therefore, the 0% and 50% states they are almost equally probable according to thermodynamics. This implies that, experimentally, a 25% pyramidal inversion is to be the expected observation at 12.2 GPa barring any kinetic barriers. In order to approximate the kinetic barrier we can use the enthalpy data of Fig. 6a. To move from the initial (0%) to the final state (50%), the system must transition through a higher enthalpy intermediate state (25%), therefore the transition rate is influenced by an activation energy, which is the magnitude of the potential barrier (commonly referred to as the kinetic barrier) separating the enthalpy minima. In Fig. 6a the greatest difference between the local enthalpy maximum (25%) and the two enthalpy minima (0% and 50%) gives an activation energy of Ea = 54 meV. The minimum proportion of pyramidal inversion is therefore given by the exponential function e−Ea/KBT, where at 300 K, KBT = 25 meV. This leads to e−55/25 = 0.11, which is which is equivalent to 11% inversion. It is therefore reasonable to posit that the 17.5% pyramidal inversion which we observe experimentally is a plausible outcome, since it is situated between the minimum (11%) and maximum (25%) values predicted by our model.
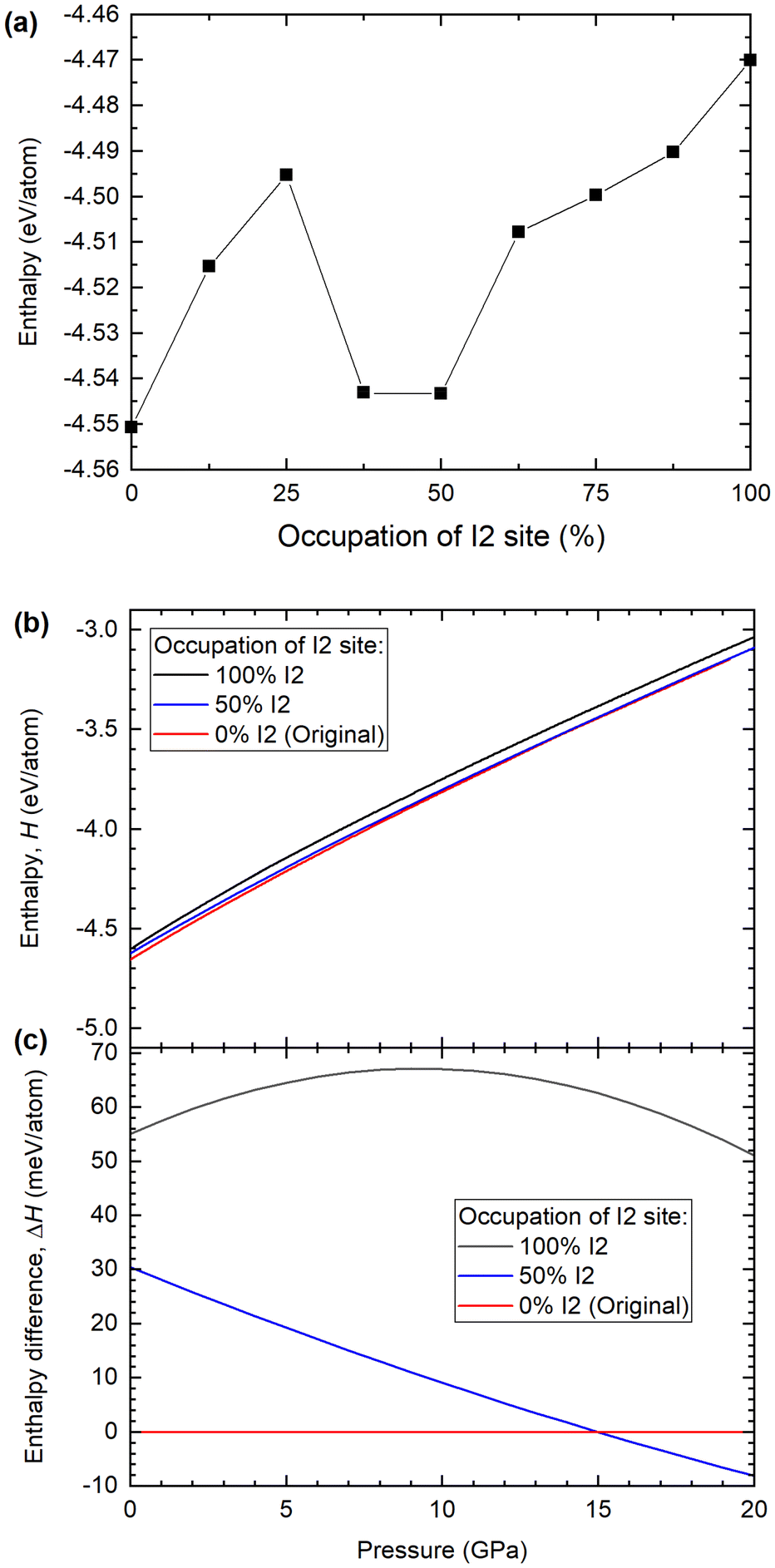 | ||
| Fig. 6 Enthalpy of competing Ba(IO3)2·H2O configurations according to density functional theory calculations. (a) Calculated enthalpy for the Ba(IO3)2·H2O structure at 12.2 GPa with 0, 12.5, 25, 37.5, 50, 62.5, 75, 87.5 and 100% IO3 inversion. (b) The enthalpy and (c) enthalpy of the 50 and 100% configurations relative to the 0% configuration. | ||
According to the SCXRD experiments, the percentage of I2 inversion increases with pressure. Therefore, the enthalpies of the 0, 50% and 100% I2 configurations were calculated as a function of pressure, as shown in Fig. 6b and c. The enthalpy difference between the 0% and 50% I2 configurations decreases monotonically with increasing pressure, which is consistent with the experimental observation that the I2 occupation increases monotonically with increasing pressure. At pressures higher than 15 GPa, the 50% I2 configuration becomes the most energetically favorable (Fig. 6c). Therefore, according to DFT calculations and experimental SCXRD measurements, increasing pressure shifts the distribution of I1/I2 iodine atoms towards the I2 site.
4. Conclusion
In this work, inversion of the pyramidal IO3 molecule has been observed in the solid state in crystals of barium di-iodate mono-hydrate, Ba(IO3)2·H2O, under hydrostatic compression via single-crystal synchrotron X-ray diffraction up to 15 GPa. The percentage of inverted pyramids increases with pressure, from 2.5% at 5.10(5) GPa to 17.5% at 14.84(5) GPa. The geometric evolution of the IO3 pyramids has been investigated based on the atomic positions determined from the SCXRD data, finding that the distance between the O4 atom of the water molecule and the iodine atom may play a key role in the stabilization of the inverted IO3 pyramids. The energetic competition between the original and inverted IO3 pyramids as a function of pressure was investigated by density functional theory calculations, finding that the original and inverted IO3 pyramids are energetically competitive.This work opens clear directions for future work. For example, does pyramidal inversion occur in the anhydrous version of barium di-iodate? α-Ba(IO3)2 exhibits a similar monoclinic structure (C2/c) wherein the Ba2+ and IO3− ions have the same site symmetry as they do in the hydrated version studied here.35 A similar high-pressure study of α-Ba(IO3)2 would unequivocally answer the question of whether the presence of the water molecule plays a role in the pyramidal inversion observed in this work. Similarly, does pyramidal inversion occur in the isostructural di-bromate and di-chlorate analogs Ba(BrO3)2·H2O and Ba(ClO3)2·H2O?25–27 Can the IO3 pyramidal inversion be encouraged by heating, rather than by hydrostatic compression? According to the DFT calculation results this is theoretically plausible, but it has not been tested experimentally. Finally, there remains the unanswered question about what happens at pressures above 15 GPa. According to the SCXRD results the volume of the inverted IO3 pyramid may be increasing with compression, similar to what occurs with the original IO3 pyramid at lower pressures. The trend of increasing volume/atom cannot continue indefinitely and may lead to a phase transition.
Author contributions
The manuscript was written through the contribution of all authors. All author have given approval to the final version of the manuscript.Data availability
Crystallographic data for Ba(IO3)2·H2O at ambient pressure, 11.59 GPa and 14.84 GPa have been deposited at the CCDC under deposition numbers 2341132–2341134 and can be obtained from https://www.ccdc.cam.ac.uk.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Spanish Research Agency (AEI) and Spanish Ministry of Science, Innovation, and Universities (MCIN) under grant PID2022-138076NB-C41/C44, and MALTA Consolider Team Network (RED2022-134388-T), co-financed by EU FEDER funds (https://doi.org/10.13039/501100011033). This work was also supported by the Generalitat Valenciana under Grant No. PROMETEO CIPROM/2021/075-GREENMAT and MFA/2022/007. R.T. acknowledges funding from the Generalitat Valenciana for the postdoctoral Fellowship No. CIAPOS/2021/20. J.S.-M. acknowledges the Spanish Ministry of Science, Innovation and Universities for the PRE2020-092198 fellowship. The authors are grateful to Samuel Gallego-Parra for assistance in performing measurements at the ESRF. This study forms part of the Advanced Materials program and is supported by MCIU with funding from European Union Next Generation EU (PRTR-C17.I1) and by the Generalitat Valenciana.References
- L. Ayarde-Henriquez, C. Guerra, M. Duque-Norena and E. Chamorro, Revisiting the bonding evolution theory: a fresh perspective on the ammonia pyramidal inversion and bond dissociations in ethane and borazane, Phys. Chem. Chem. Phys., 2023, 25, 27394–27408 RSC.
- P. International Union of and C. Applied, 2006, 10.1351/goldbook.P04956.
- A. Rauk, L. C. Allen and K. Mislow, Pyramidal Inversion, Angew. Chem., Int. Ed. Engl., 1970, 9, 400–414 CrossRef CAS.
- A. Rauk, L. C. Allen and E. Clementi, Electronic Structure and Inversion Barrier of Ammonia, J. Chem. Phys., 1970, 52, 4133–4144 CrossRef CAS.
- G. W. Koeppl, D. S. Sagatys, G. S. Krishnamurthy and S. I. Miller, Inversion barriers of pyramidal (XY3) and related planar (
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) XY) species, J. Am. Chem. Soc., 1967, 89, 3396–3405 CrossRef CAS.
XY) species, J. Am. Chem. Soc., 1967, 89, 3396–3405 CrossRef CAS. - Y. Kikukawa, K. Seto, S. Uchida, S. Kuwajima and Y. Hayashi, Solid-State Umbrella-type Inversion of a VO5 Square-Pyramidal Unit in a Bowl-type Dodecavanadate Induced by Insertion and Elimination of a Guest Molecule, Angew. Chem., Int. Ed., 2018, 57, 16051–16055 CrossRef CAS PubMed.
- A. Liang, R. Turnbull and D. Errandonea, A review on the advancements in the characterization of the high-pressure properties of iodates, Prog. Mater. Sci., 2023, 136, 101092 CrossRef CAS.
- V. R. Kalinin, V. V. llyukhin and N. V. Belov, Crystal structure of triclinic modification of potassium iodate, Dokl. Akad. Nauk SSSR, 1978, 239(3), 590–593 CAS.
- A. Liang, L.-T. Shi, R. Turnbull, F. J. Manjón, J. Ibáñez, C. Popescu, M. Jasmin, J. Singh, K. Venkatakrishnan, G. Vaitheeswaran and D. Errandonea, Pressure-induced band-gap energy increase in a metal iodate, Phys. Rev. B, 2022, 106, 235203 CrossRef CAS.
- M. D. Baer, V.-T. Pham, J. L. Fulton, G. K. Schenter, M. Balasubramanian and C. J. Mundy, Is Iodate a Strongly Hydrated Cation?, J. Phys. Chem. Lett., 2011, 2, 2650–2654 CrossRef CAS.
- G. E. Ericksen, M. E. Mrose and J. W. Marinenko, J. Res. U.S. Geol. Surv., 1974, 471–478 CAS.
- S. T. Shen, Y. T. Yao and T.-Y. Wu, Depolarization of Raman Lines and Structure of Chlorate, Bromate and Iodate Ions, Phys. Rev., 1937, 51, 235–238 CrossRef CAS.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. Sheldrick, SHELXT - Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- W. Kohn and L. J. Sham, Self-Consistent Equations Including Exchange and Correlation Effects, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- J. Hafner, Ab initio simulations of materials using VASP: Density-functional theory and beyond, J. Comput. Chem., 2008, 29, 2044–2078 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- A. E. Mattsson, R. Armiento, J. Paier, G. Kresse, J. M. Wills and T. R. Mattsson, The AM05 density functional applied to solids, J. Chem. Phys., 2008, 128, 084714 CrossRef PubMed.
- R. Armiento and A. E. Mattsson, Functional designed to include surface effects in self-consistent density functional theory, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 085108 CrossRef.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- M. Lertkiattrakul, M. L. Evans and M. J. Cliffe, PASCal Python: A Principal Axis Strain Calculator, J. Open Source Softw., 2023, 8, 5556 CrossRef.
- H. D. Lutz, E. Alici and W. Buchmeier, Kristallstrukturen des Sr(BrO3)2·H2O, Ba(BrO3)2·H2O, Ba(IO3)2·H2O, Pb(CIO3)2·H2O und Pb(BrO3)2·H2O, Z. Anorg. Allg. Chem., 1986, 535, 31–38 CrossRef CAS.
- S. K. Sikka, S. N. Momin, H. Rajagopal and R. Chidambaram, Neutron–Diffraction Refinement of the Crystal Structure of Barium Chlorate Monohydrate Ba(ClO3)2·H2O, J. Chem. Phys., 1968, 48, 1883–1889 CrossRef CAS.
- G. Kartha, Structure of halogenates of the type A (BO3)2·H2O, Proc. Natl. Acad. Sci., India, Sect. A, 1953, 38, 1–12 CrossRef.
- G. Kartha, Structure of barium chlorate monohydrate Ba(ClO3)2·H2O, Acta Crystallogr., 1952, 5, 845–846 CrossRef CAS.
- T. Kellersohn, H. D. Lutz, T. Vogt, R. G. Delaplane and I. Olovsson, Structural instabilities of the trigonally coordinated water molecules in Ba(IO3)2·H2O and Pb(ClO3)2·H2O studied by X-ray and neutron diffraction at 25 and 295 K, Acta Crystallogr., Sect. B: Struct. Sci., 1992, 48, 166–172 CrossRef.
- H. D. Lutz and N. Lange, Dynamic behaviour of water molecules with trigonal coordination in solid hydrates, J. Mol. Liq., 1990, 46, 255–261 CrossRef CAS.
- R. J. Angel, M. Alvaro and J. Gonzalez-Platas, EosFit7c and a Fortran module (library) for equation of state calculations, Z. Kristallogr. - Cryst. Mater., 2014, 229, 405–419 CrossRef CAS.
- T. Steiner, The Hydrogen Bond in the Solid State, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- G. A. Jeffrey, An introduction to Hydrogen Bonding, Oxford University Press, Oxford, 1997 Search PubMed.
- L. V. Gurvich, I. V. Veyts and C. B. Alcock, Thermodynamic Properties of Individual Substances, Hemisphere Pub. Co., New York, 1989 Search PubMed.
- F. Bertolotti, A. V. Shishkina, A. Forni, G. Gervasio, A. I. Stash and V. G. Tsirelson, Intermolecular Bonding Features in Solid Iodine, Cryst. Growth Des., 2014, 14, 3587–3595 CrossRef CAS.
- V. Petricek, K. Maly, B. Kratochvil, J. Podlahova and J. Loub, Barium diiodate, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 2130–2132 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2341132–2341134. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qi01021c |
| This journal is © the Partner Organisations 2024 |