
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the charge stabilization and transport in naphthalimide-based semiconductors via a fused-ring and core-engineering strategy†
Raúl
González-Núñez‡
a,
Matías J.
Alonso-Navarro‡
 bc,
Fátima
Suárez-Blas‡
bc,
Elena
Gala
bc,
M. Mar
Ramos
c,
José L.
Segura
*b and
Rocío
Ponce Ortiz
*a
bc,
Fátima
Suárez-Blas‡
bc,
Elena
Gala
bc,
M. Mar
Ramos
c,
José L.
Segura
*b and
Rocío
Ponce Ortiz
*a
aDepartment of Physical Chemistry, University of Málaga, Málaga, 29071, Spain. E-mail: rocioponce@uma.es
bDepartment of Organic Chemistry, Complutense University of Madrid, Faculty of Chemistry, Madrid 28040, Spain. E-mail: segura@ucm.es
cChemical and Environmental Technology, Department. Univ. Rey Juan Carlos, Móstoles, 28933, Spain
First published on 11th March 2024
Abstract
The synthesis and characterization of a family of rationally designed compounds based on naphthalimide units attached, through conjugated nitrogenated linkers (i.e. pyrazine and imidazoline units), to fused thiophene-based moieties is shown. This combination of different donor–acceptor moieties allows fine tuning of the HOMO and LUMO energy levels, and thus the modulation of their electronic properties. A comprehensive physical chemistry study is carried out, in which the nature of the neutral and charged species are analyzed and their electrical performance is understood in terms of molecular and supramolecular characteristics.
Introduction
The tunable electronic and structural properties of organic semiconductors together with their flexibility, low molecular weight, high solution processability and low cost in comparison with inorganic semiconductors are some of the key points for the current interest in the development of π-conjugated small molecules and polymers for a wide variety of applications. Among them, their use as active materials stands out in organic light-emitting diodes (OLEDs),1,2 organic phototransistors (OPTs),3–5 organic solar cells (OSCs),6–9 organic field-effect transistors (OFETs)10–13 and, more recently, in wearable electronics14,15 or memory arrays.16,17 However, despite the significant advances in organic electronics over the last few decades, the performances of the organic devices are often below that exhibited by the inorganic counterparts, mainly attributed to lower charge carrier mobilities.18 For this reason, it is mandatory to design and synthesize novel organic semiconductors with enhanced processability and tunable properties to overcome current limitations.19,20In this regard, different molecular building blocks have been used for the synthesis of π functional materials with tunable properties and high performances in organic electronic devices.7,21–23 The design and synthesis of ambipolar materials are receiving a great deal of attention with the aim of obtaining materials with good charge transport abilities and tunable HOMO/LUMO energy levels.24,25 On the one hand, imide units, and more specifically naphthalimide derivatives, are often used as strong electron-acceptor moieties to obtain processable organic materials with low-lying LUMO energy levels and good optical, electrochemical and electrical properties.26–30 On the other hand, the covalent linkage of naphthalimide units with strong donor scaffolds such as oligothiophenes or triarylamines has shown to be a promising approach to obtain ambipolar materials.31,32 Thus, in our research groups we have widely demonstrated that the combination of donor and acceptor units through different rigid and conjugated linkers can pave the way to functional materials with tunable and, sometimes unexpected, optical and electrochemical properties and good n-type or ambipolar characteristics in OFETs or OSCs.33–40
In the last 5 years, the fused-ring electron acceptor strategy (FREA) has become efficient in the modulation of the donor ability of organic materials without introducing strong donor moieties. This is the case of ITIC/IDIC or Y6 fused ring molecules with strong electron-acceptors in the terminal position in which it is possible to fine tune their donor abilities by controlling the length of the donor units.41,42
In this article, we have designed, synthesized and characterized a novel family of compounds based on the combination of electron-withdrawing naphthalimide units with strong thiophene-based donor moieties through different rigid and conjugated nitrogenated linkers. In order to fine-tune the HOMO energy levels, we have extended the length of the thiophenic unit and tested different isomers. As for the LUMO, the introduction of imidazole or pyrazine connectors covalently linked to the naphthalimide unit has made it possible to precisely modulate its energy levels. In addition to this, we have demonstrated that the use of a fused-ring strategy in the donor moiety can drastically change the charge transport properties of the organic semiconductors, obtaining electrical behavior as p-type materials in OFETs in contrast with the previously reported n-type performance of the non-fused oligothiophene–naphthalimide assembly.38
Results and discussion
As mentioned above, there is currently a great deal of interest in the development of organic materials by adopting the fused-ring electron acceptor strategy (FREA). In this regard, we envisaged the potential of using this strategy in combination with electron-acceptor naphthalimides connected through different planar and conjugated linkers. Thus, in this article we describe the synthesis and an exhaustive optical, electrochemical and electrical characterization of a novel family of six organic semiconductors based on naphthalimides with multiple fused rings (Scheme 1(a)). With this aim, we developed a novel one-pot procedure to obtain in situ the pyrazine or the imidazole conjugated linkers between the two electroactive units, naphthalimide 1,2-diketone (NID)/naphthalimide anhydride (NIA) and the thiophene-based benzo[c][1,2,5]thiadiazole (BTD) derivatives.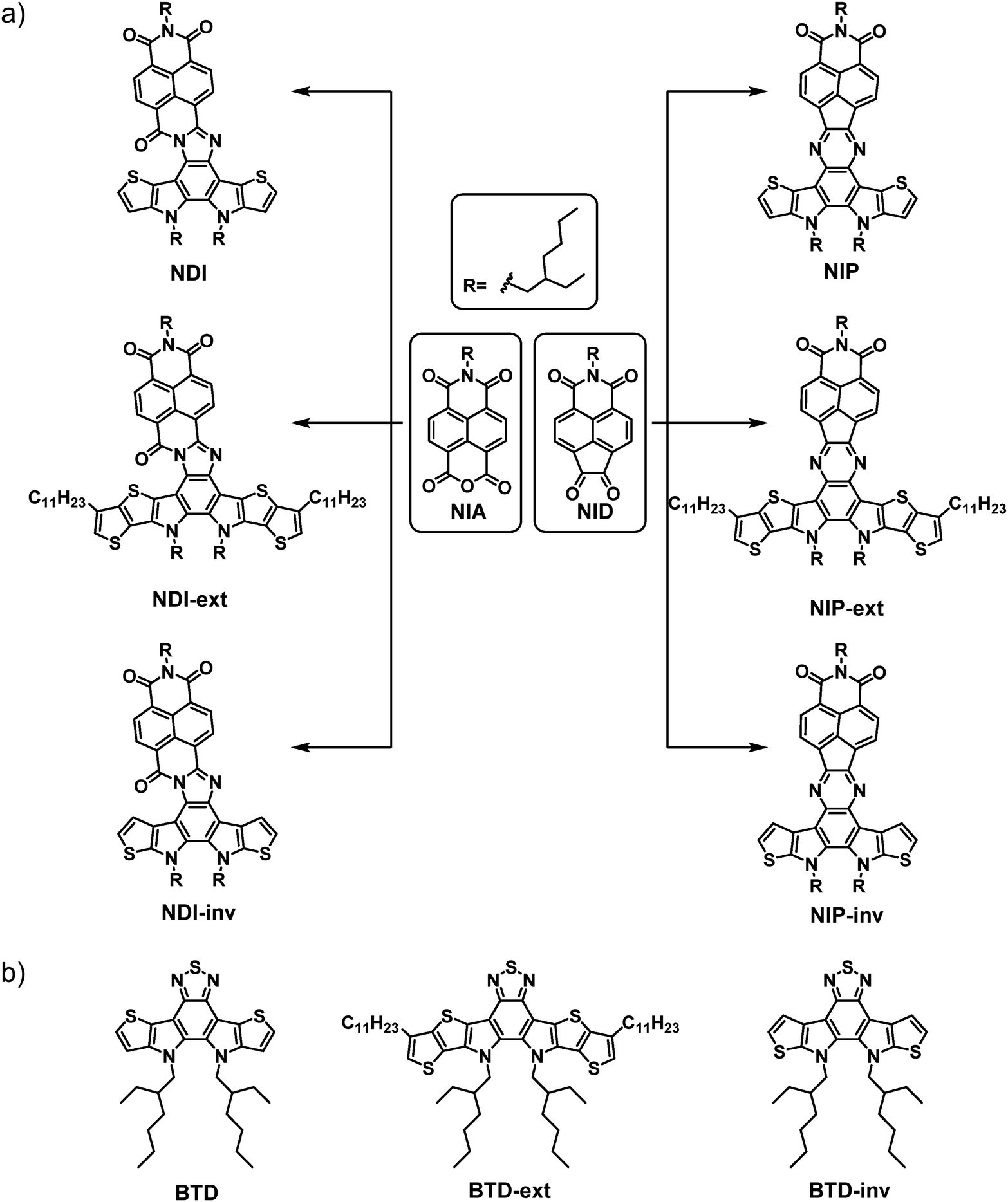 | ||
| Scheme 1 (a) Schematic representation of the naphthalimide-based semiconductors described in this work, and (b) benzothiadiazole-based reference materials described in the literature. | ||
The synthetic route starts by obtaining the functionalized naphthalimide derivatives NID and NIA, previously published by our research group.38,43 In addition, we synthesized the corresponding benzothiadiazole (BTD)-based assemblies BTD, BTD-ext and BTD-inv following the synthetic procedures previously reported by Nakamura et al.,44 He et al.45 and Zou et al.,46 respectively (Scheme 1(b)). Then, a one-pot methodology not previously reported for this type of compounds was developed for the synthesis of the target compounds. Thus, the thiadiazol rings of the BTD-based reference materials were opened in acid media to form the corresponding unstable diamine derivatives which subsequently condensate with both NID and NIA moieties to obtain the donor–acceptor π-conjugated semiconductors endowed with pyrazine (NIP, NIP-inv and NIP-ext) or imidazole (NDI, NDI-inv and NDI-ext) linkers (Scheme 1(a)).
To have an insight into their chemical structure, these novel materials have been characterized by 1H and 13C NMR spectroscopy, FT-IR spectroscopy and mass spectrometry (MALDI-HRMS) (a complete characterization is reported in the ESI,† Fig. S1–S36).
In this regard, 1H-NMR analyses of these novel assemblies show slight differences in the chemical shifts and multiplicities of the signals. For example, as depicted in Fig. S7 and S15 (ESI†), the comparison between NIP and NIP-inv shows that the position of the sulfur atoms plays a crucial role in the chemical shift of the β-hydrogen of the thienopyrrole unit, being up-shielded up to 0.6 ppm for the inverted one. In addition, in the case of NIP-inv and NDI-inv, the planarity of the central core (detailed below in the molecular structures and packing section) and the space proximity of the β-hydrogen to the electrowithdrawing units (both naphthalimide and thiadiazole) lead to more deshielded chemical shifts for hydrogen atoms, phenomena which agrees with both experimental and theoretical information. Moreover, a comparison of the chemical shifts of the α-hydrogen atoms of both NIP and NIP-ext shows a slight downshift of the larger one due to the greater distance between that hydrogen atom and the electron-withdrawing pyrazine. When the imidazole-based semiconductors NDI-X were studied, we observed that the number of signals were doubled in comparison with the NIP analogues. This may be related to the loss of the plane of symmetry in the NIP derivatives due to the presence of the unsymmetric imidazoline linker, as previously observed in other works.38 In all cases the signals related to the naphthalimide hydrogen atoms in the pyrazine or imidazole-based materials remain practically unaltered within each family. However, by comparing the chemical shifts of the hydrogen atoms in the two different families, we noticed that the presence of the extra carbonyl group in the NDI series deshields one of the signals up to 9.3 ppm (Fig. S9, S13 and S17, ESI†).
Molecular structures and packing
The structure of the studied compounds was optimized by density-functional theory (DFT) using the B3LYP functional and the 6-31G** basis set as implemented in the Gaussian 16 program. The calculations show a moderate distortion of around 5–10 degrees in the benzothiadiazole fragment in all the molecules under study. In addition, while comparing the different naphthalimide derivatives, we found that all imidazole derivatives (NDI) have a higher torsion angle in the connecting unit (of about 16°) compared to the pyrazine derivatives (NIP), regardless of the length of the π-conjugation chain or the isomer studied. In contrast, NIP derivatives show practically completely planar structures, as shown in Fig. 1 and Fig. S37 (ESI†).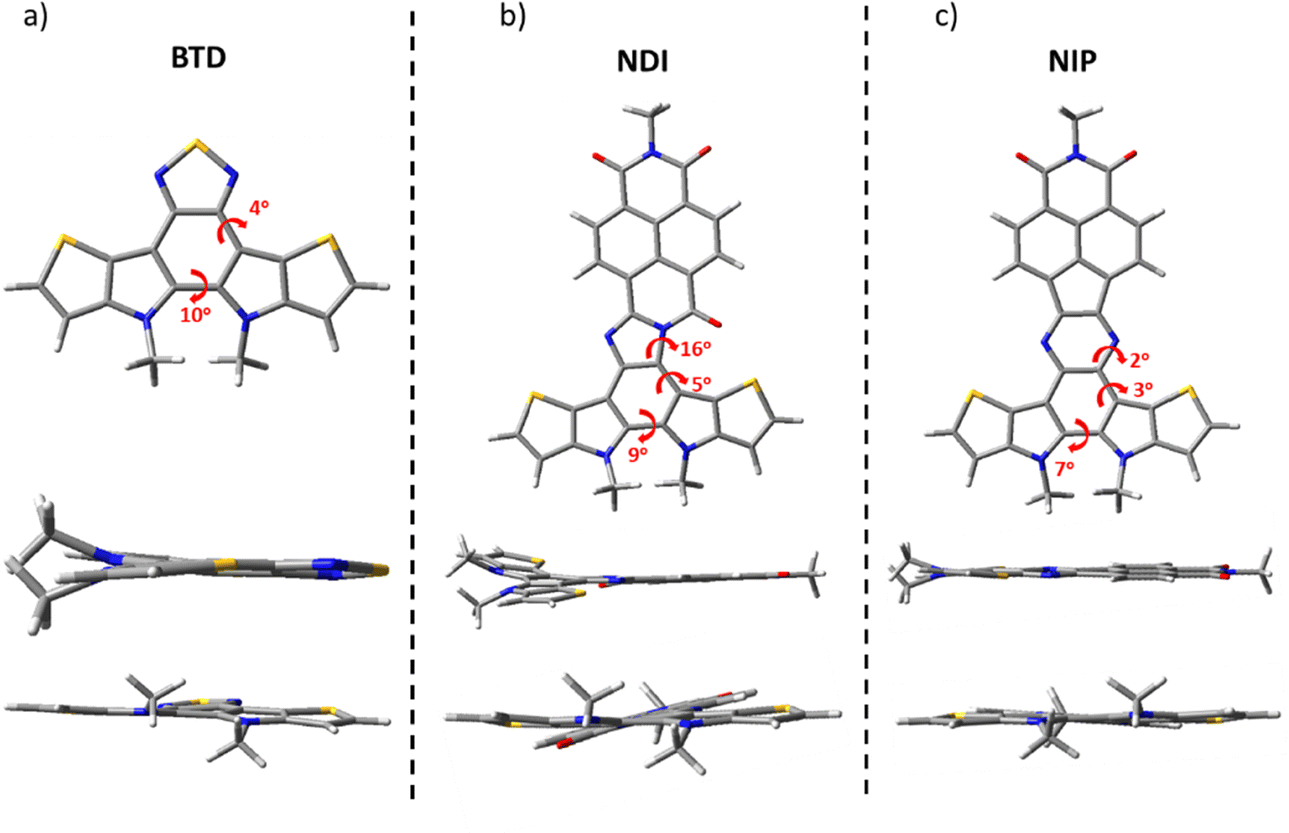 | ||
| Fig. 1 DFT-computed global minimum structures for (a) BTD, (b) NDI and (c) NIP at the B3LYP/6-31G** level of theory. | ||
Table S1 (ESI†) shows the calculated MO energy values along with the calculated reorganization energies for charge transport (p-type and n-type) for all these π-extended semiconductors. As can be seen, the introduction of the naphthalimide unit remarkably stabilizes the LUMO energy level, although this stabilization is significantly lower for the pyrazine derivatives with respect to the imidazole derivatives, and these phenomena are also observed in other oligothiophene–naphthalimide assemblies, mainly because of the presence of an electron-withdrawing carbonyl group in the amidine moiety, as we previously demonstrated.34,47 Moreover, this energy is very similar for the extended and inverted compounds, indicating that the LUMO energy is basically dictated by the naphthalimide unit. On the other hand, the introduction of the aryleneimide fragment does not have a remarkable effect on the HOMO energy levels, which, in contrast, are slightly stabilized when the number of fused thiophenes decreases (i.e.NDI-extvs.NDI or NIP-extvs.NIP) or for isomers with the inversion of the more external thiophene rings (BTD/NDI/NIPvs.BTD-inv/NDI-inv/NIP-inv), as compared in Tables S1–S10 (ESI†). Thus, substantially lower energy gaps are found for the NDI derivatives, showing the extended derivative, NDI-ext, the lowest value of 1.67 eV. Note that all the studied systems show HOMO energy levels close to −5 eV, being thus appropriate for hole injection from gold electrodes. In contrast, only LUMO energy levels suitable for electron injection, i.e. deeper than −3 eV, are registered in the NDI derivatives.
The internal reorganization energies for electron and hole transport, calculated for the investigated semiconductors, are also summarized in Table S1 (ESI†). The reorganization energy (λ) is a parameter that takes into account the structural reorganization required to accommodate the charge defect during charge transport, small reorganization energies being a prerequisite for efficient charge transport. In our case, all systems have reorganization energy values for p-type transport lower than for n-type transport. This indicates that positive charges accommodation is favored versus negative charges, thus facilitating hole transport. Focusing on the NIP and NDI families, the calculated values indicate comparable reorganization energies for electron transport within the same family, independently of the length of the thienopyrrole core or the isomeric structure. This indicates that the electron defect is basically stabilized through the naphthalimide unit. In contrast, lower reorganization energies for hole transport are registered upon extension of the thienopyrrole unit, which agrees with the easier accommodation of positive charges through a longer conjugated core.
Optical and electrochemical properties
The introduction of linear and branched alkyl chains at the imide, pyrrole and thiophene sites in these systems allows solubility enhancement with values above 30 mg mL−1. Because of that, their optical and electrochemical properties can be studied by UV-Vis spectroscopy in solution and solid states (thin-films) and by cyclic voltammetry analyses, respectively. Therefore, in Tables 1 and 2, the most relevant optical and electrochemical parameters obtained from the experimental measurements are summarized.| UV-Visa | ||||||
|---|---|---|---|---|---|---|
| [C] (μM) | λ max (nm) | ε λ max (nm) | λ ICT (nm) | λ onset (nm) |
E
optg
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (eV)
(eV) |
|
| a UV-Vis absorption in CHCl3 solution. b Absorption maxima in solution. c Molar extinction coefficient to the referred wavelength. d Onset wavelength for the absorption band. e Energy band gap derived from the low-energy absorption edge using the equation Eoptg = 1240/λonset. | ||||||
| BTD | 5 | 316 | 26930 |
441 | 495 | 2,51 |
| NIP | 5 | 395 | 42360 |
583 | 690 | 1,79 |
| NDI | 5 | 374 | 35300 |
670 | 834 | 1,49 |
| BTD-ext | 5 | 369 | 133217 |
470 | 546 | 2.27 |
| NIP-ext | 5 | 372 | 197484 |
618 | 740 | 1.67 |
| NDI-ext | 5 | 360 | 73825 |
674 | 867 | 1.43 |
| BTD-inv | 5 | 368 | 14015 |
426 | 486 | 2.55 |
| NIP-inv | 5 | 398 | 22928 |
551 | 664 | 1.87 |
| NDI-inv | 5 | 363 | 21869 |
641 | 785 | 1.57 |
| Cyclic voltammetrya | ||||||
|---|---|---|---|---|---|---|
|
E
1/2RedI (V) |
E
1/2RedII (V) |
E
1/2OxI (V) |
E HOMO (eV) | E LUMO (eV) |
E
elecg
(eV) |
|
|
a Cyclic voltammetry recorded in DCM/TBAPF6 (0.1 M) at a scan rate of 0.10 V s−1 using Pt as working and the counter electrode, and Fc/Fc+ as reference.
b Estimated from ELUMO = −5.1 eV − E1/2RedI.
c Estimated from EHOMO = −5.1 eV − E1/2OxI.
d Estimated from Egapelec = EHOMO–ELUMO.
e Estimated from Egapopt = EHOMO–ELUMO.
|
||||||
| BTD | — | — | 0.42 | −5.52 | −3.01e | — |
| NIP | −1.37 | −1.74 | 0.37 | −5.47 | −3.73 | 1.73 |
| NDI | −1.13 | −1.43 | 0.37 | −5.47 | −3.97 | 1.50 |
| BTD-ext | — | — | 0.26 | −5.36 | −3.08e | — |
| NIP-ext | −1.36 | −1.65 | 0.27 | −5.37 | −3.74 | 1.63 |
| NDI-ext | −1.12 | −1.49 | 0.26 | −5.36 | −3.98 | 1.38 |
| BTD-inv | — | — | 0.56 | −5.66 | −3.11e | — |
| NIP-inv | −1.37 | −1.77 | 0.49 | −5.59 | −3.73 | 1.86 |
| NDI-inv | −1.14 | −1.49 | 0.46 | −5.56 | −3.96 | 1.60 |
As shown in Fig. 2, the substitution of the benzothiadiazole core for naphthalimide units through planar and conjugated linkers not only promote a red shift in the lowest-energy absorption band of the UV-Vis spectra, but also enhances the molar absorption coefficient (ε) of these derivatives. This phenomenon is especially remarkable in all pyrazine-based semiconductors, with ε values up to two times higher in comparison with those observed for the BTD analogues, probably because of the π-extended structure and enhanced planarity,48 as is depicted in Fig. 1.
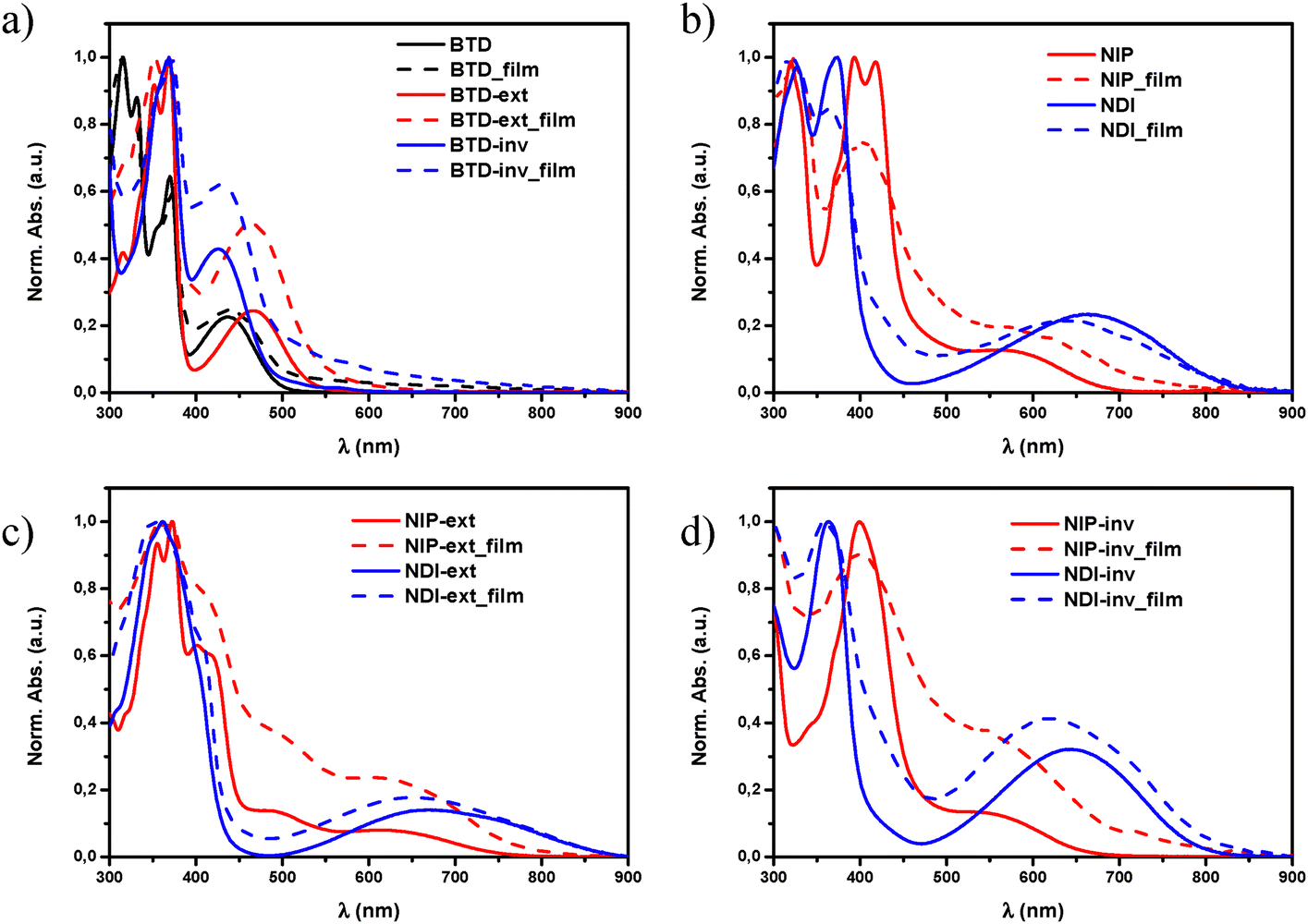 | ||
| Fig. 2 UV-Vis absorption spectra of (a) BTD (black), BTD-ext (red) and BTD-inv (blue) in chloroform solution (solid) and thin-film (dashed), (b) NIP (red) and NDI (blue) in chloroform solution (solid) and thin-film (dashed), (c), NIP-ext (red) and NDI-ext (blue) in chloroform solution (solid) and thin-film (dashed) and (d) NIP-inv (red) and NDI-inv (blue) in chloroform solution (solid) and thin-film (dashed). | ||
Regarding the highest-energy band (λmax) in solution, it is observed that all pyrazine assemblies present a red-shifted absorption maxima in comparison with their imidazole-based analogues, especially in the smaller systems (395 vs. 374 nm for NIP/NDI and 398 vs. 363 nm for NIP-inv/NDI-inv), which is in good agreement with the different UV-Vis absorption parameters observed for similar oligothiophene–naphthalimide assemblies.34,47 The effect of the thiophene extension in the π-conjugated systems was also studied, observing that both NIP-ext and NDI-ext derivatives present similar λmax values in solution in comparison with the unfunctionalized BTD-ext analogue. However, as far as the lowest-energy absorption band is concerned, the extension in the donor fragment increases this absorption up to the NIR region, being more pronounced in the NDI-ext derivative than in the pyrazine-based NIP-ext, and also in comparison with the smaller semiconductors NDI and NIP (740 and 867 nm for NIP-ext/NDI-extvs. 690 vs. 834 nm in NIP/NDI), phenomena observed in Fig. 2(b)–(d) for the NIP and NDI, and their corresponding extended and inverted derivatives.
On the other hand, when thiophene inversion was evaluated (NIP-inv and NDI-inv) we observed a blue-shifted effect of λmáx in NDI-inv compared to BTD-inv but, when a pyrazine unit is introduced in NIP-inv, a red-shifted behavior occurs. In addition to this, the inversion of the fused thiophene unit leads to (i) an intense decrease of the absorption molar coefficient (ε) in these new materials, probably because of the lack of linearity of the donor fragment and (ii) blue-shifted effect in the ICT band in terms of λonset values with respect to non-inverted systems. This trend found in the electronic absorption spectra are endorsed by TD-DFT calculations, as shown in the ESI† (Tables S2–S10).
The analysis of the UV-Vis absorption spectra of these systems as thin films and in solution does not show significant differences (Fig. 2(b)–(d)). This coincidence seems to indicate that, despite the large π-conjugated T-shape of these systems, the introduction of long branched alkyl chains in their structure promotes not only good processability but also avoids the formation of big supramolecular aggregates in the films.30,35
DFT theoretical calculations were carried out in order to understand the nature of the electronic absorption bands. The lower energy absorption bands in all the studied systems are theoretically ascribed to the HOMO–LUMO transition. As indicated by the frontier molecular orbital topologies shown in Fig. 3 and Fig. S38 (ESI†), this electronic transition entails a clear intramolecular charge transfer character, from the more electron-rich part of the semiconductor, the thienopyrrole part, to the more electron deficient unit, the naphthalimide group. This electronic absorption band is more accentuated in imidazole derivatives compared to pyrazine derivatives, both for extended or inverted derivatives.
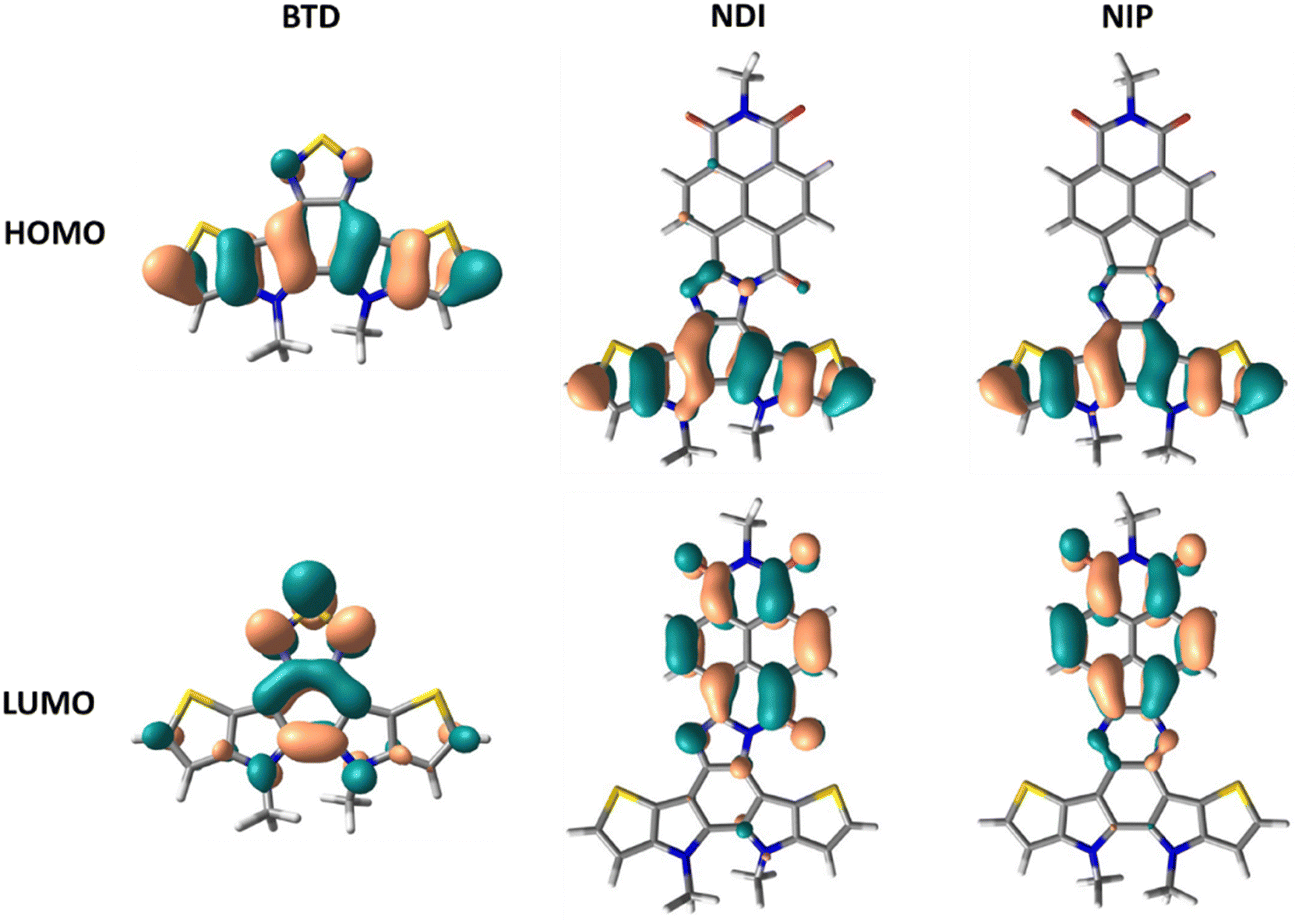 | ||
| Fig. 3 DFT/B3LYP/6-31G** molecular orbital topologies of BTD, NDI and NIP. | ||
To study the electrochemical properties of these novel assemblies, cyclic voltammetry experiments were carried out under argon atmosphere in dry dichloromethane solutions and using TBAF 0.1 M as the supporting electrolyte (experimental details can be found in the ESI†). As depicted in Table 2 and Fig. 4, ring fusion strategy yields redox amphoteric organic semiconductors with lower oxidation potentials than those corresponding to non-fused oligothiophene–naphthalimide assemblies previously published, where the oligothiophene fragment is a non-fused terthiophene (E1/2Ox (NDI3T): +0.59 V and E1/2Ox (NIP3T): +0.50 V).38,47 In addition, it is also possible to finely tune the potential values of the oxidation processes by extending the length of the thienopyrrole unit (extended systems) or, in the other hand, by changing the position of the sulfur atom of the thiophene moiety (inverted systems). The experimental values of the oxidation processes summarized in Table 2 show that, despite their similar chemical structures, slight modifications in the position of the heterocyclic sulfur atoms easily modify the oxidation processes. Therefore, NIP and NDI have lower oxidation potentials in comparison with those observed for the isomeric inverted analogues NIP-inv and NDI-inv. Furthermore, the oxidation ability of these semiconductors can be significantly improved by extending the effective conjugation in the thienopyrrole core. Thus, the oxidation potentials of the larger NIP-ext and NDI-ext semiconductors, show a shift of ca. 0.15 V to less positive potentials in comparison with that of the parent smaller derivatives NIP and NDI.
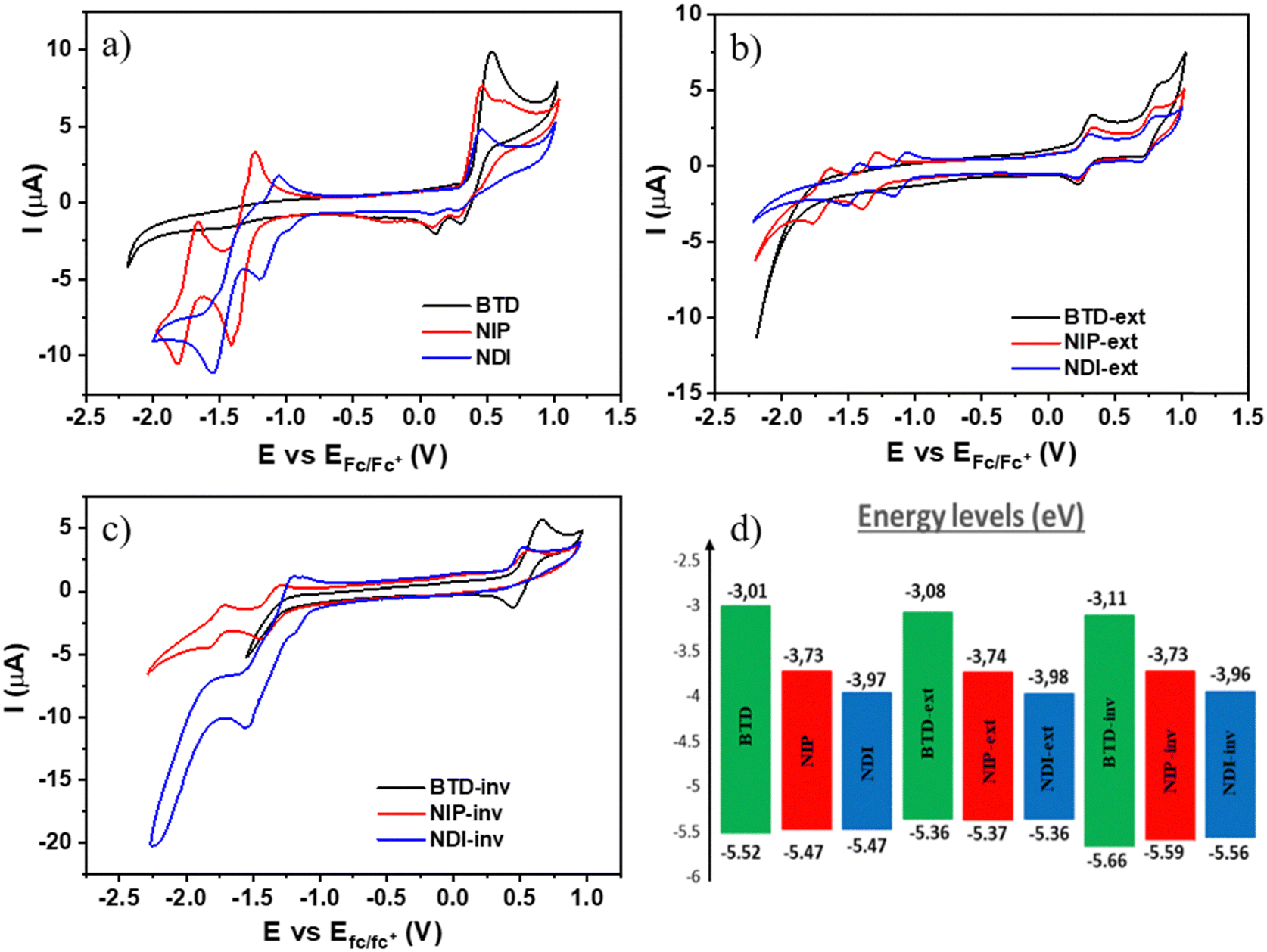 | ||
| Fig. 4 Benzothiadiazole and naphthalimide based semiconductors comparison for (a) BTD (black), NIP (red) and NDI (blue) (b) BTD-ext (black), NIP-ext (red) and NDI-ext (blue) (c) BTD-inv (black), NIP-inv (red) and NDI-inv (blue) and (d) the corresponding energy levels diagram estimated from the experimental electrochemical values. | ||
Regarding the electron-acceptor abilities of these organic semiconductors, it becomes clear that the electron-withdrawing character of these materials can be remarkably modified by introducing naphthalimide units in the core to replace the benzo[c][1,2,5]thiadiazole unit. As shown in Table 2, this chemical modification paved the way for obtaining new π-conjugated assemblies with better electron-accepting abilities. Thus, the naphthalimide-based derivatives show reduction potential values as good as −1.12 V in comparison with −2.2 V determined for some benzothiadiazole-based semiconductors, as can be noticed in Fig. 4(a)–(c).44,49 Note however that, in the benzothiadiazole-based systems studied here (BTD, BTD-ext and BTD-inv), no reduction processes were recorded in the electrochemical window of the working electrode in dichloromethane solvent. Furthermore, we have also finely tuned the electron accepting abilities of these naphthalimide-based semiconductors using the two different rigid and conjugated connectors, imidazole and pyrazine. An improvement in the reduction ability of around 0.2 V is observed for the imidazole-based assemblies (NDI, NDI-ext and NDI-inv) in comparison with the parent pyrazine-based analogues (NIP, NIP-ext and NIP-inv).
From the first reduction and oxidation potentials obtained by CV experimental measurements (Fig. 4(a)–(c) and Fig. S48–S56, ESI†) in dry dichloromethane (Table 2) it is possible to estimate the highest occupied molecular orbital (HOMO) levels and the lowest unoccupied molecular orbital (LUMO) levels of all these fused π-extended assemblies. As depicted in Fig. 4(d), the replacement of the thiadiazole core by a naphthalimide unit greatly stabilizes the LUMO energy level, providing low-lying LUMO energy values of around −4.0 eV. The stabilization is less significant for the pyrazine-containing derivatives than for the imidazole-containing analogues; the phenomena are also consistent with the DFT experiments and with other previous results.34 This is related to the fact that the presence of three carbonyl groups in the naphthalimide structure enhances the electron-withdrawing ability of this kind of compound with respect to pyrazine analogues. On the other hand, the introduction of these electroactive moieties does not have remarkable impact on the HOMO energy levels, being only slightly destabilized when the number of fused thiophenes increases (thienopyrrole vs. thienothiophenepyrrole). In addition, the HOMO values are moderately stabilized for the inverted thienopyrrole derivatives in comparison with the thienopyrrole analogues.
Thus, the great impact found in the LUMO energy level on the introduction of the aryleneimide moieties in conjunction with the subtle modification of the HOMO energy level produces in all cases a reduction in the electrochemical bandgap of the new assemblies. Furthermore, there is a good agreement between the optical (Eoptgap) and the electrochemical band gaps (Eelecgap) determined for all these donor–acceptor semiconductors.
Spectroelectrochemical studies
In order to analyze the charged species, Fig. S58–S63 (ESI†) show the evolution of the UV/Vis/NIR spectra obtained by progressive spectroelectrochemical oxidation and reduction of a solution with a low concentration of all studied compounds in the presence of a large excess of tetrabutylammonium hexafluorophosphate supporting electrolyte (TBAHFP). BTD is considered in this study as the main building block common to all the systems investigated. It presents two positively charged species (at 930 mV and 1200 mV) ascribed to radical cation and dication oxidized species, respectively (Fig. S58, ESI†). During reduction, a negatively charged species is formed (at −1490 mV), which can be assigned to the radical anion species (Fig. S59, ESI†). Introduction of a naphthalimide unit into the benzothiadiazole core (NDI and NIP) significantly diminishes the potential needed for the stabilization of the two positively charged species in the oxidation process, being 300 mV and 580 mV for NDI and 680 mV and 1150 mV for NIP (Fig. 5(a)). This potential decrease is remarkable considering that the HOMO is basically located on the BTD fragment, whose energy level undergoes minimal changes with the naphthalimide introduction. However, when analyzing the charge distribution upon one-electron and two-electron oxidation processes (Fig. S64–S66, ESI†) it becomes clear that while the BTD fragment bears the majority of the positive charge (approx. 92%) the naphthalimide fragments also help in the stabilization of the charged species.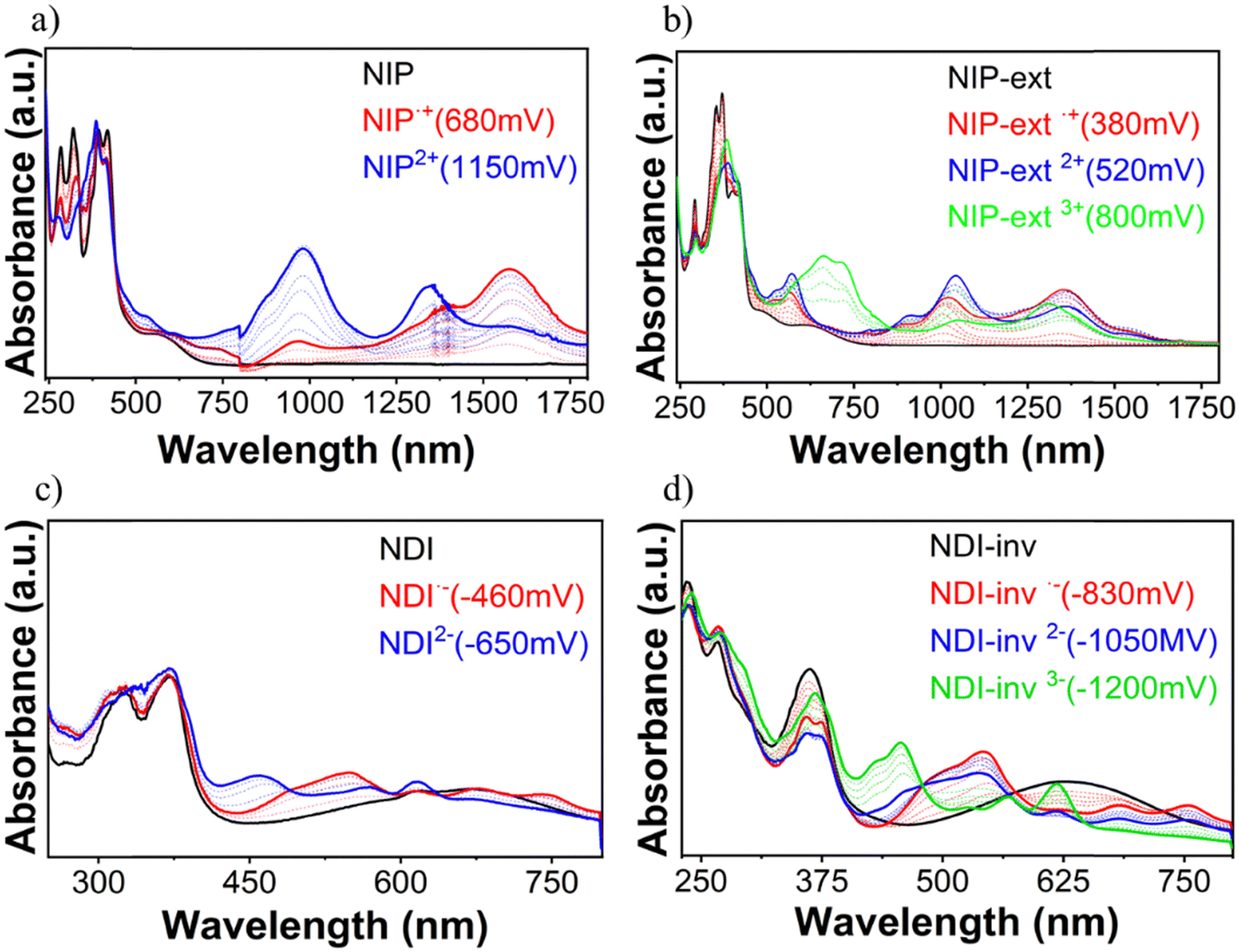 | ||
| Fig. 5 UV/Vis/NIR spectra changes at room temperature upon oxidation (top) and reduction (bottom). (a) for NIP (b) for NIP-ext, (c) for NDI and (d) for NDI-inv within an OTTLE cell in dichloromethane in the presence of 0.1 M (n-Bu)4NPF6 as the supporting electrolyte. | ||
In addition, in NDI and NIP semiconductors two negatively charged species are stabilized upon reduction, attributed to radical anion (−460 mV for NDI and −480 mV for NIP) (Fig. S59, ESI†) and dianion (−650 mV for NDI and −1000 mV for NIP), view Fig. 5(c), whereas only one reduced species is stabilized in BTD. The stabilization of the dianion species together with the decrease of the reduction potentials agrees with the remarkable tuning of the LUMO energy level found in Fig. 4(d). To support this fact, DFT calculations predict that most of the negative injected charge (approx. 95%) is delocalized over the naphthalimide groups (Fig. S67 and S68, ESI†).
Furthermore, upon extension of the π-conjugated BTD core in these derivatives, differences can be observed between NDI-ext and NIP-ext semiconductors. While no further oxidation species are found in NDI-ext with respect to NDI (Fig. S60, ESI†), in the case of NIP-ext three different oxidized species are stabilized, at 380 mV, 520 mV and 800 mV, which are ascribed to radical cation, dication and trication species (Fig. 5(b)), only the first two were found in the NIP molecule (Fig. 5(a)). This is explained considering that the extension of the thienopyrrole chain favors the stabilization of the positive charge through a longer conjugated path, as evidenced by the HOMO topology in Fig. S38 (ESI†), especially in NIP-ext due to its enhanced planarity. Besides, DFT calculations show that in NIP-ext (Fig. S75 and S76, ESI†) the naphthalimide units help to stabilize the injected positive charge to a further extent compared to NDI-ext. In contrast, no remarkable differences in the stabilization of reduced species are found upon extension, stabilizing only radical anion and dianion species, as in NIP and NDI.
On the other hand, π-conjugation in the thienopyrrole chain, as expected, is somewhat hindered in the inverted derivatives which also has an impact on the charge stabilization. In this sense, reduction processes are facilitated for NDI-inv, with the recording of three new spectral profiles attributed to the radical anion (−830 mV), dianion (−1050 mV) and trianion (−1200 mV) species (Fig. 5(d)), compared to the stabilization of only the radical anion and dianion species in NDI (Fig. 5(c)). In contrast, only one oxidized species is formed (Fig. S62, ESI†). Thus, inversion in NDI strongly favors reductive over oxidative processes.
In contrast, no such effect is found for NIP-inv, showing spectral changes similar to those of NIP, with stabilization of only two reduced and two oxidized species (Fig. S62 and S63, ESI†). This points out to the role of the pyrazine connector in promoting an improved communication between the napthalimide and benzothiadiazole units, due to the more planar skeleton found in DFT calculations.
Field-effect transistor characterization
Field-effect transistors were fabricated to evaluate the charge transport properties of the studied semiconductors. We fabricated thin-film transistors in a top-contact/bottom-gate structure evaporation of the semiconductor onto SAM-treated substrates (both octadecyltrichlorosilane and hexamethyldisilazane were used for the surface treatment of the SiO2 dielectric layer). The deposited thin films were characterized using XRD (Fig. 6(c)) and AFM techniques (Fig. 6(a) and (b)).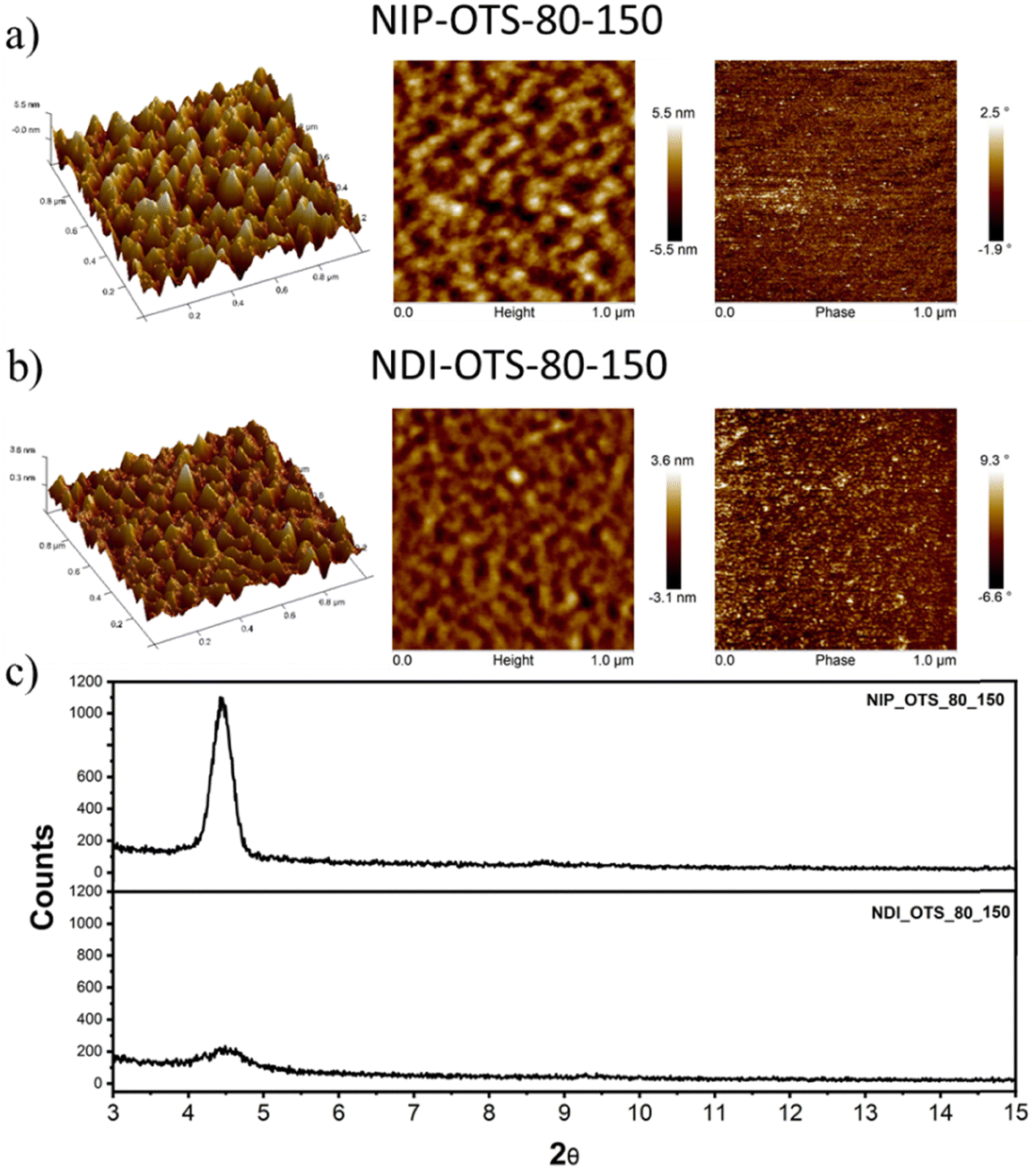 | ||
| Fig. 6 (a) and (b) AFM images of thin films of the NIP and NDI semiconductors deposited onto OTS-modified substrates. (c) XRD plots for indicated semiconductors onto OTS-modified substrates. | ||
After the deposition of the semiconductors, gold electrodes were patterned using shadow masks with different predefined channel lengths and widths. The charged carrier mobilities (μ), threshold voltage (VT) and current on/off ratio (ION/IOFF) were extracted from the saturated region in the device transfer curves and the device parameters are summarized in Table 3 (complete device characterization is shown in the ESI†). The field effect mobilities for the naphthalimide derivatives are modest, with a maximum hole mobility of 10−2 cm2 V−1 s−1 for NIP (see output and transfer plots in Fig. 7) for films deposited on preheated substrates at 80 °C and after thermal annealing at 150 °C for 3 hours. However, the introduction of imidazole as a spacer ring (NDI), provokes a decrease in the field effect mobility of two orders of magnitude (10−4 cm2 V−1 s−1) under the same deposition conditions. If we analyze the effect of the extension of the π-conjugated system in these derivatives, we find that the field-effect mobilities sharply decrease in the case of the NIP-ext and NDI-ext with values of 10−6 cm2 V−1 s−1 for both semiconductors (view Tables S13 and S14, ESI†). On the other hand, the inverse derivatives (NDI-inv and NIP-inv) are not active. XRD measurements (Fig. 6) show higher crystallinity for NIP derivatives compared to NDI derivatives, and hence the higher field effect mobility. Furthermore, XRD measurements show completely amorphous films for NIP-ext and NDI-ext (Fig. S83 and S84, ESI†) and NIP-inv and NDI-inv (Fig. S85 and S86, ESI†), reason that could be behind their poor electronic properties. A similar information can be inferred by AFM images, showing a slightly more homogeneous texture for NIP samples compared to NDI ones (Fig. 6(a) and (b)). In addition, upon lengthening of the thienopyrrole chain, in NIP-ext and NDI-ext, AFM images show irregular surfaces (Fig. S80, ESI†). These results are in acccordance with XRD measurements and indicate a slightly improved morphology for shorter molecular systems, especially in the case of NIP derivatives. These results are in agreement with NIP derivatives having improved electrical performance (two orders of magnitude higher) than NDI derivatives, while the extension of the π-conjugated chain worsens the field-effect mobility values for both derivatives (NIP-ext and NDI-ext).
| Compound | Deposition conditions | Annealing | μ h (cm2 V−1 s−1) | V T (V) | I ON/IOFF |
|---|---|---|---|---|---|
| NDI | OTS, 80 °C | 150 °C, 3 h | 2 × 10−4 | −6 | 2 × 104 |
| NIP | OTS, 80 °C | 150 °C, 3 h | 1 × 10−2 | −20 | 2 × 104 |
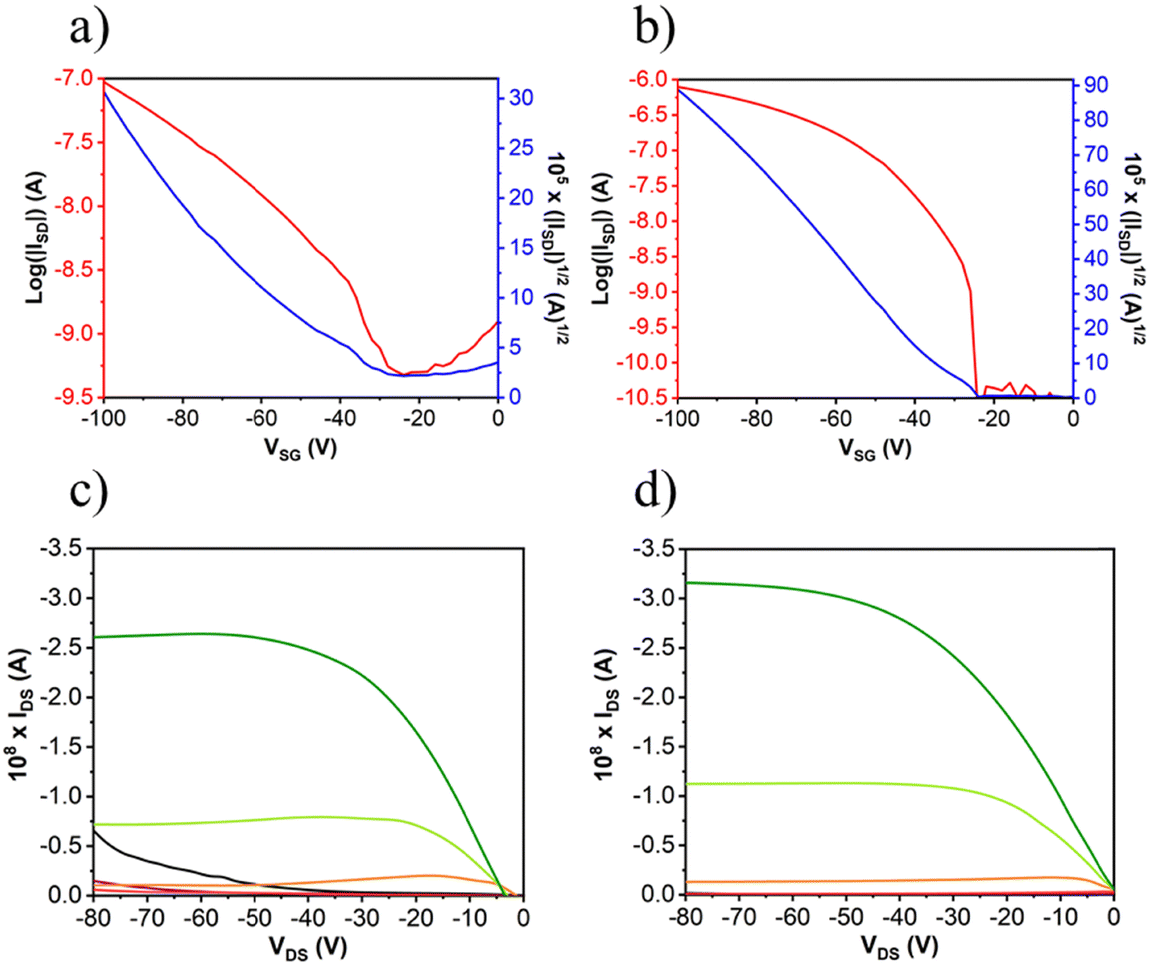 | ||
| Fig. 7 OFET transfer and output characteristic of (a) and (c) for NDI and (b) and (d) for NIP. | ||
Conclusions
In this contribution we have designed, synthetized and characterized a novel family of compounds based on the combination of electron-withdrawing naphthalimide units with strong donor moieties based on benzothiadiazole, BTD, through two different rigid and conjugated nitrogenated linkers (i.e. pyrazine and imidazoline linkers). Different synthetic modifications have been carried out, in which either the BTD unit was extended, or different isomers were employed. The effect of these synthetic approaches was analyzed by a complete physico-chemical analysis in which the stabilization of charged species has been primarily analyzed. In general, we observed remarkable differences in the LUMO energy levels upon introduction of imidazole or pyrazine connectors covalently linked to the naphthalimide unit. In contrast, only moderate changes are found in the HOMO energy levels over the whole family of compounds.Spectroelectrochemical studies demonstrated the formation of both reduced and oxidized, charged species in all the systems under study. However, modifications on the chemical structures have an impact on the stabilization of such charged species. In particular, the results highlight the role of the pyrazine connector in promoting an improved communication between the napthalimide and benzothiadiazole units, due to the more planar conjugated skeleton. Thus, the system stabilizing the higher number of charge species is NIP-ext, showing two reduced (radical anion and dianion) and up to three oxidized species (up to the trication). Thus, the use of the pyrazine connector is demonstrated to be more efficient for charge stabilization.
Field-effect transistors were fabricated, finding that the use of this fused-ring strategy in the donor moiety can drastically change the charge transport properties of the organic semiconductors, obtaining electrical behavior as p-type semiconductors. The electrical performances of the systems studied are modest, with a maximum hole mobility of 10−2 cm2 V−1 s−1 for NIP. Furthermore, the introduction of imidazole as a spacer ring (NDI), provokes a decrease in the field effect mobility of two orders of magnitude (10−4 cm2 V−1 s−1) under the same deposition conditions. Other derivatives, either by extending the conjugation or by rendering different isomers, showed decreased or completely extinguished electrical performances, despite the improved stabilization of charged species in the extended systems. XRD and AFM images showed rather amorphous films for all the studied systems except for NIP, which could be the reason supporting the higher electrical performance.
Author contributions
Raúl González-Núñez: investigation, data curation, visualization, writing – original draft, writing – review & editing. Matías J. Alonso-Navarro: investigation, data curation, visualization, writing – original draft, writing – review & editing. Fátima Suárez-Blas: investigation, data curation, visualization. Elena Gala: investigation, data curation, visualization. M. Mar Ramos: visualization, writing – review & editing. José L. Segura: conceptualization, investigation, visualization, supervision, writing – review & editing, funding acquisition, project administration. Rocío Ponce Ortiz: conceptualization, investigation, visualization, supervision, writing – review & editing, funding acquisition, project administration.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by MICINN (PID2022-138908NB-C33, PID2022-139548NB-I00 and TED2021-129886B-C43) and the UCM (INV.GR.00.1819.10759). R GN thanks the MICINN for a FPI predoctoral fellowship (PRE2020-092327). FSB and MJAN gratefully acknowledge Universidad Rey Juan Carlos for their predoctoral and postdoctoral contracts. Computer resources, technical expertise and assistance was provided by the SCBI (Supercomputing and Bioinformatics) center of the University of Málaga and are gratefully acknowledged. The Vibrational spectroscopy (EVI) lab of the Research Central Services (SCAI) of the University of Málaga is also gratefully acknowledged.References
- S. Li, K. Liu, X.-C. Feng, Z.-X. Li, Z.-Y. Zhang, B. Wang, M. Li, Y.-L. Bai, L. Cui and C. Li, Synthesis and macrocyclization-induced emission enhancement of benzothiadiazole-based macrocycle, Nat. Commun., 2022, 13, 2850 CrossRef CAS PubMed.
- T. Tsukada, Y. Shoji, K. Takenouchi, H. Taka and T. Fukushima, A carbon-functionality-appended diborylacetylene available for a component of organic synthesis and OLEDs, Chem. Commun., 2022, 58, 4973–4976 RSC.
- Y.-S. Guan, J. Qiao, Y. Liang, H. K. Bisoyi, C. Wang, W. Xu, D. Zhu and Q. Li, A high mobility air-stable n-type organic small molecule semiconductor with high UV-visible-to-NIR photoresponse, Light: Sci. Appl., 2022, 11, 236 CrossRef CAS PubMed.
- Y.-C. Lin, W.-C. Yang, Y.-C. Chiang and W.-C. Chen, Recent Advances in Organic Phototransistors: Nonvolatile Memory, Artificial Synapses, and Photodetectors, Small Sci., 2022, 2, 2100109 CrossRef CAS.
- A. Tavaslı, B. Gurunlu, D. Güntürkün and R. Isci, A Review on Solution-Processed Organic Phototransistors and Their Recent Developments, Electronics, 2022, 11, 316 CrossRef.
- W. Gao, F. Qi, Z. Peng, F. R. Lin, K. Jiang, C. Zhong, W. Kaminsky, Z. Guan, C.-S. Lee, T. J. Marks, H. Ade and A. K. Y. Jen, Achieving 19% Power Conversion Efficiency in Planar-Mixed Heterojunction Organic Solar Cells Using a Pseudosymmetric Electron Acceptor, Adv. Mater., 2022, 34, 2202089 CrossRef CAS.
- D. Luo, W. Jang, D. D. Babu, M. S. Kim, D. H. Wang and A. K. K. Kyaw, Recent progress in organic solar cells based on non-fullerene acceptors: materials to devices, J. Mater. Chem. A, 2022, 10, 3255–3295 RSC.
- R. Ma, C. Yan, J. Yu, T. Liu, H. Liu, Y. Li, J. Chen, Z. Luo, B. Tang, X. Lu, G. Li and H. Yan, High-Efficiency Ternary Organic Solar Cells with a Good Figure-of-Merit Enabled by Two Low-Cost Donor Polymers, ACS Energy Lett., 2022, 7, 2547–2556 CrossRef CAS.
- L. Xue, X. Liu, Q. Wang, M. Yang, S. Du, C. Yang, J. Tong, Y. Xia and J. Li, Improved Performance of Organic Solar Cells by Utilizing Green Non-Halogen Additive to Modulate Active-Layer Morphology, Energy Technol., 2022, 2200504 CrossRef CAS.
- J. Chen, J. Yang, Y. Guo and Y. Liu, Acceptor Modulation Strategies for Improving the Electron Transport in High-Performance Organic Field-Effect Transistors, Adv. Mater., 2022, 34, 2104325 CrossRef CAS.
- K. Liu, B. Ouyang, X. Guo, Y. Guo and Y. Liu, Advances in flexible organic field-effect transistors and their applications for flexible electronics, npj Flexible Electron., 2022, 6, 1 CrossRef CAS.
- B. Peng, Z. He, M. Chen and P. K. L. Chan, Ultrahigh On-Current Density of Organic Field-Effect Transistors Facilitated by Molecular Monolayer Crystals, Adv. Funct. Mater., 2022, 2202632 CrossRef CAS.
- X. Ren, Z. Lu, X. Zhang, S. Grigorian, W. Deng and J. Jie, Low-Voltage Organic Field-Effect Transistors: Challenges, Progress, and Prospects, ACS Mater. Lett., 2022, 4, 1531–1546 CrossRef CAS.
- D. Lv, Q. Jiang, Y. Shang and D. Liu, Highly efficient fiber-shaped organic solar cells toward wearable flexible electronics, npj Flexible Electron., 2022, 6, 38 CrossRef CAS.
- J. S. Heo, J. Eom, Y.-H. Kim and S. K. Park, Recent Progress of Textile-Based Wearable Electronics: A Comprehensive Review of Materials, Devices, and Applications, Small, 2018, 14, 1703034 CrossRef PubMed.
- Z. Chen, S. Duan, X. Zhang, B. Geng, Y. Xiao, J. Jie, H. Dong, L. Li and W. Hu, Organic Semiconductor Crystal Engineering for High-Resolution Layer-Controlled 2D Crystal Arrays, Adv. Mater., 2022, 34, 2104166 CrossRef CAS PubMed.
- Y. Wei, Y. Geng, K. Wang, H. Gao, Y. Wu and L. Jiang, Organic ultrathin nanostructure arrays: materials, methods and applications, Nanoscale Adv., 2022, 4, 2399–2411 RSC.
- G. Schweicher, G. Garbay, R. Jouclas, F. Vibert, F. Devaux and Y. H. Geerts, Molecular Semiconductors for Logic Operations: Dead-End or Bright Future?, Adv. Mater., 2020, 32, 1905909 CrossRef CAS PubMed.
- S. Wang, L. Peng, H. Sun and W. Huang, The future of solution processing toward organic semiconductor devices: a substrate and integration perspective, J. Mater. Chem. C, 2022, 10, 12468–12486 RSC.
- H. Bronstein, C. B. Nielsen, B. C. Schroeder and I. McCulloch, The role of chemical design in the performance of organic semiconductors, Nat. Rev. Chem., 2020, 4, 66–77 CrossRef CAS PubMed.
- J. Panidi, D. G. Georgiadou, T. Schoetz and T. Prodromakis, Advances in Organic and Perovskite Photovoltaics Enabling a Greener Internet of Things, Adv. Funct. Mater., 2022, 32, 2200694 CrossRef CAS.
- C. Xu, Z. Zhao, K. Yang, L. Niu, X. Ma, Z. Zhou, X. Zhang and F. Zhang, Recent progress in all-small-molecule organic photovoltaics, J. Mater. Chem. A, 2022, 10, 6291–6329 RSC.
- D. Corzo, D. Rosas-Villalva, A. C, G. Tostado-Blázquez, E. B. Alexandre, L. H. Hernandez, J. Han, H. Xu, M. Babics, S. De Wolf and D. Baran, High-performing organic electronics using terpene green solvents from renewable feedstocks, Nat. Energy, 2023, 8, 62–73 CrossRef.
- H. Lee, D. Lee, D. H. Sin, S. W. Kim, M. S. Jeong and K. Cho, Effect of donor–acceptor molecular orientation on charge photogeneration in organic solar cells, NPG Asia Mater., 2018, 10, 469–481 CrossRef CAS.
- J. Zhao, C. Yao, M. U. Ali, J. Miao and H. Meng, Recent advances in high-performance organic solar cells enabled by acceptor–donor–acceptor–donor–acceptor (A–DA′D–A) type acceptors, Mater. Chem. Front., 2020, 4, 3487–3504 RSC.
- X. Li, J. Guo, L. Yang, M. Chao, L. Zheng, Z. Ma, Y. Hu, Y. Zhao, H. Chen and Y. Liu, Low Bandgap Donor–Acceptor π-Conjugated Polymers From Diarylcyclopentadienone-Fused Naphthalimides, Front. Chem., 2019, 7, 362 CrossRef CAS PubMed.
- I. Torres-Moya, J. R. Carrillo, M. V. Gómez, A. H. Velders, B. Donoso, A. M. Rodríguez, Á. Díaz-Ortiz, J. T. López Navarrete, R. P. Ortiz and P. Prieto, Synthesis of D–π–A high-emissive 6-arylalkynyl-1,8-naphthalimides for application in Organic Field-Effect Transistors and optical waveguides, Dyes Pigm., 2021, 191, 109358 CrossRef CAS.
- L. Wang, Y. Chen, W. Tao, K. Wang, Z. Peng, X. Zheng, C. Xiang, J. Zhang, M. Huang and B. Zhao, Polymerized Naphthalimide Derivatives as Remarkable Electron-Transport Layers for Inverted Organic Solar Cells, Macromol. Rapid Commun., 2022, 2200119 CrossRef CAS PubMed.
- Z. Wang, J. Zhao, H. Dong, G. Qiu, Q. Zhang and W. Hu, An asymmetric naphthalimide derivative for n-channel organic field-effect transistors, Phys. Chem. Chem. Phys., 2015, 17, 26519–26524 RSC.
- M. J. Alonso-Navarro, A. Harbuzaru, M. Martínez-Fernández, P. Pérez Camero, J. T. López Navarrete, M. M. Ramos, R. Ponce Ortiz and J. L. Segura, Synthesis and electronic properties of nitrogen-doped π-extended polycyclic aromatic dicarboximides with multiple redox processes, J. Mater. Chem. C, 2021, 9, 7936–7949 RSC.
- J. Zhang, X. Zhang, H. Xiao, G. Li, Y. Liu, C. Li, H. Huang, X. Chen and Z. Bo, 1,8-Naphthalimide-Based Planar Small Molecular Acceptor for Organic Solar Cells, ACS Appl. Mater. Interfaces, 2016, 8, 5475–5483 CrossRef CAS PubMed.
- S. Gámez-Valenzuela, I. Torres-Moya, A. Sánchez, B. Donoso, J. T. López Navarrete, M. C. Ruiz Delgado, P. Prieto and R. Ponce Ortiz, Extended π-Conjugation and Structural Planarity Effects of Symmetrical D–π–A–π–D Naphthalene and Perylene Diimide Semiconductors on n-type Electrical Properties, Chem. – Eur. J., 2023, e202301639 CrossRef PubMed.
- R. Adel, E. Gala, M. J. Alonso-Navarro, E. Gutierrez-Fernandez, J. Martín, M. Stella, E. Martinez-Ferrero, A. de la Peña, A. Harbuzaru, M. M. Ramos, R. P. Ortiz, J. L. Segura and M. Campoy-Quiles, Comparing the microstructure and photovoltaic performance of 3 perylene imide acceptors with similar energy levels but different packing tendencies, J. Mater. Chem. C, 2022, 10, 1698–1710 RSC.
- M. J. Alonso-Navarro, E. Gala, M. M. Ramos, R. Ponce Ortiz and J. L. Segura, Oligothiophene-Naphthalimide Hybrids Connected through Rigid and Conjugated Linkers in Organic Electronics: An Overview, Electron. Mater., 2021, 2, 222–252 CrossRef.
- M. J. Alonso-Navarro, A. Harbuzaru, P. de Echegaray, I. Arrechea-Marcos, A. Harillo-Baños, A. de la Peña, M. M. Ramos, J. T. López Navarrete, M. Campoy-Quiles, R. Ponce Ortiz and J. L. Segura, Effective interplay of donor and acceptor groups for tuning optoelectronic properties in oligothiophene–naphthalimide assemblies, J. Mater. Chem. C, 2020, 8, 15277–15289 RSC.
- P. de Echegaray, M. J. Mancheño, I. Arrechea-Marcos, R. Juárez, G. López-Espejo, J. T. López Navarrete, M. M. Ramos, C. Seoane, R. P. Ortiz and J. L. Segura, Synthesis of Perylene Imide Diones as Platforms for the Development of Pyrazine Based Organic Semiconductors, J. Org. Chem., 2016, 81, 11256–11267 CrossRef CAS PubMed.
- A. de la Peña, I. Arrechea-Marcos, M. J. Mancheño, M. C. Ruiz Delgado, J. T. López Navarrete, J. L. Segura and R. Ponce Ortiz, Tuning of the Electronic Levels of Oligothiophene–Naphthalimide Assemblies by Chemical Modification, Chem. – Eur. J., 2016, 22, 13643–13652 CrossRef PubMed.
- R. P. Ortiz, H. Herrera, R. Blanco, H. Huang, A. Facchetti, T. J. Marks, Y. Zheng and J. L. Segura, Organic n-Channel Field-Effect Transistors Based on Arylenediimide-Thiophene Derivatives, J. Am. Chem. Soc., 2010, 132, 8440–8452 CrossRef CAS PubMed.
- R. P. Ortiz, H. Herrera, C. Seoane, J. L. Segura, A. Facchetti and T. J. Marks, Rational Design of Ambipolar Organic Semiconductors: Is Core Planarity Central to Ambipolarity in Thiophene–Naphthalene Semiconductors?, Chem. – Eur. J., 2012, 18, 532–543 CrossRef CAS.
- R. Ponce Ortiz, H. Herrera, M. J. Mancheño, C. Seoane, J. L. Segura, P. Mayorga Burrezo, J. Casado, J. T. López Navarrete, A. Facchetti and T. J. Marks, Molecular and Electronic-Structure Basis of the Ambipolar Behavior of Naphthalimide–Terthiophene Derivatives: Implementation in Organic Field-Effect Transistors, Chem. – Eur. J., 2013, 19, 12458–12467 CrossRef CAS PubMed.
- B. Jia, J. Wang, Y. Wu, M. Zhang, Y. Jiang, Z. Tang, T. P. Russell and X. Zhan, Enhancing the Performance of a Fused-Ring Electron Acceptor by Unidirectional Extension, J. Am. Chem. Soc., 2019, 141, 19023–19031 CrossRef CAS PubMed.
- Z. Liu, X. Zhang, P. Li and X. Gao, Recent development of efficient A–D–A type fused-ring electron acceptors for organic solar, Sol. Energy, 2018, 174, 171–188 CrossRef CAS.
- H. Herrera, P. de Echegaray, M. Urdanpilleta, M. J. Mancheño, E. Mena-Osteritz, P. Bäuerle and J. L. Segura, Linear and star-shaped naphthalimide-fused pyrazinacenes, Chem. Commun., 2013, 49, 713–715 RSC.
- S.-i Kato, T. Furuya, A. Kobayashi, M. Nitani, Y. Ie, Y. Aso, T. Yoshihara, S. Tobita and Y. Nakamura, π-Extended Thiadiazoles Fused with Thienopyrrole or Indole Moieties: Synthesis, Structures, and Properties, J. Org. Chem., 2012, 77, 7595–7606 CrossRef CAS PubMed.
- D. Mo, H. Chen, J. Zhou, N. Tang, L. Han, Y. Zhu, P. Chao, H. Lai, Z. Xie and F. He, Alkyl chain engineering of chlorinated acceptors for elevated solar conversion, J. Mater. Chem. A, 2020, 8, 8903–8912 RSC.
- J. Yuan, Y. Zhang, L. Zhou, C. Zhang, T.-K. Lau, G. Zhang, X. Lu, H.-L. Yip, S. K. So, S. Beaupré, M. Mainville, P. A. Johnson, M. Leclerc, H. Chen, H. Peng, Y. Li and Y. Zou, Fused Benzothiadiazole: A Building Block for n-Type Organic Acceptor to Achieve High-Performance Organic Solar Cells, Adv. Mater., 2019, 31, 1807577 CrossRef PubMed.
- M. J. Alonso-Navarro, A. Harbuzaru, R. González-Núñez, M. Mar Ramos, J. L. Segura and R. Ponce Ortiz, Tunable electroactive oligothiophene-naphthalimide semiconductors via end-capped engineering: Cumulative effects beyond the linker, J. Mater. Chem. C, 2023, 11, 10852–10863 RSC.
- M. S. Vezie, S. Few, I. Meager, G. Pieridou, B. Dörling, R. S. Ashraf, A. R. Goñi, H. Bronstein, I. McCulloch, S. C. Hayes, M. Campoy-Quiles and J. Nelson, Exploring the origin of high optical absorption in conjugated polymers, Nat. Mater., 2016, 15, 746–753 CrossRef CAS PubMed.
- S.-i Kato, T. Furuya, M. Nitani, N. Hasebe, Y. Ie, Y. Aso, T. Yoshihara, S. Tobita and Y. Nakamura, A Series of π-Extended Thiadiazoles Fused with Electron-Donating Heteroaromatic Moieties: Synthesis, Properties, and Polymorphic Crystals, Chem. – Eur. J., 2015, 21, 3115–3128 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qm00029c |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2024 |