
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling the beauty of cyclopropane formation: a comprehensive survey of enantioselective Michael initiated ring closure (MIRC) reactions
Ramkumar
Moorthy
,
Waidath
Bio-Sawe
,
Sagar S.
Thorat
and
Mukund P.
Sibi
 *
*
Department of Chemistry and Biochemistry, North Dakota State University, Fargo, North Dakota 58108-6050, USA. E-mail: Mukund.Sibi@ndsu.edu
First published on 31st May 2024
Abstract
This review presents a comprehensive overview of the synthesis of cyclopropanes using enantioselective Michael Initiated Ring Closure (MIRC) reactions. Cyclopropane-containing compounds possess unique structural and chemical properties that render them valuable in various fields, such as medicine, agrochemistry, and materials science. The MIRC approach has emerged as a versatile and efficient method for generating cyclopropane rings with excellent enantioselectivity. The review details fundamental concepts, reaction mechanisms, and synthetic strategies employed in MIRC reactions. A short introduction is provided to highlight the use of chiral substrates and nucleophiles to synthesize enantioenriched cyclopropanes. Additionally, recent advancements, challenges, and future prospects in the field are also discussed. This review serves as a valuable resource for researchers and practitioners interested in harnessing the potential of MIRC reactions for the synthesis of chiral cyclopropane derivatives, offering a comprehensive and up-to-date overview of the subject matter.
 Ramkumar Moorthy | Dr Ramkumar Moorthy received his MS in chemistry from the University of South Dakota in 2010. In 2010, he joined Prof. Mukund Sibi's group in North Dakota State University to do his PhD, and his research was focused on developing novel asymmetric approaches for the installation of chiral quaternary and tertiary centers by 1,3-dipolar cycloaddition and MIRC reactions. In 2016, he joined Prof. Daniel A. Harki at the University of Minnesota for his postdoctoral studies, where his research is focused on design and synthesis of small molecules as anticancer or antiviral agents. |
 Waidath Bio-Sawe | Waidath Bio-Sawe obtained her bachelor's degree in Biochemistry at St Cloud State University in 2015. She pursued her graduate studies in organic chemistry at North Dakota State University (NDSU) under the mentorship of Dr Mukund Sibi. She worked in the area of cooperative catalysis, enantioselective stereodivergent catalysis, and obtained her Ph.D. in 2023. Her interests include method development, enantioselective catalysis, photocatalysis, and environmental remediation. |
 Sagar S. Thorat | Sagar Sudam Thorat was born in 1992 in the village Chinchani (Ambak), Maharashtra, India. In 2014, he completed his post-graduation in Organic Chemistry from the School of Chemistry at Punyashlok Ahilyadevi Holkar University, Solapur, India. Under the guidance of Dr Ravindar Kontham, he successfully defended his PhD thesis at CSIR-National Chemical Laboratory in Pune, India in 2021. Presently, Sagar holds the position of a postdoctoral research associate in the research group led by Prof. Mukund Sibi at North Dakota State University. His area of scientific interests includes the total synthesis of biologically active natural products, transition metal salt-catalysed domino reactions, and the synthesis of biobased monomers for applications in polymer chemistry. |
 Mukund P Sibi | Prof. Mukund P Sibi earned his PhD from the City University of New York. After postdoctoral studies, he began his independent career at North Dakota State University where he is currently a University Distinguished Professor. His research focus is on radical chemistry, development of enantioselective reactions, total synthesis, and green chemistry. Prof. Sibi has received the prestigious Arthur C. Cope Scholar Award from the American Chemical Society in 2008 in recognition of his contributions to the field of radical chemistry. In 2021 he was elected as a Fellow of AAAS for his contribution to radical chemistry as well as building biomedical research infrastructure. |
1. Introduction
Ever since August Freund first synthesized the cyclopropane ring in 1882,1 it has held a crucial and flexible position in chemical synthesis and pharmacological research. The strained three-membered ring possesses a unique reactivity and serves as an important building block for the synthesis of complex compounds. The incorporation of cyclopropane ring into pharmacologically active drug molecules has enhanced metabolic stability and target binding through the modulation of molecular conformation.2 The success of this approach is evident through numerous examples of structurally diverse cyclopropanes found in both developmental drug candidates and commercialized products.3,4The ability to access enantioenriched cyclopropanes is of importance as it enables the synthesis of chiral molecules with a wide array of biological and functional properties. To address the specific synthetic challenges posed by the strained ring, various methods for constructing cyclopropanes have been developed. Among these methods, there are four major transformations described in the literature, with a focus on disconnections involving a formal [2 + 1] cycloaddition (Fig. 1).
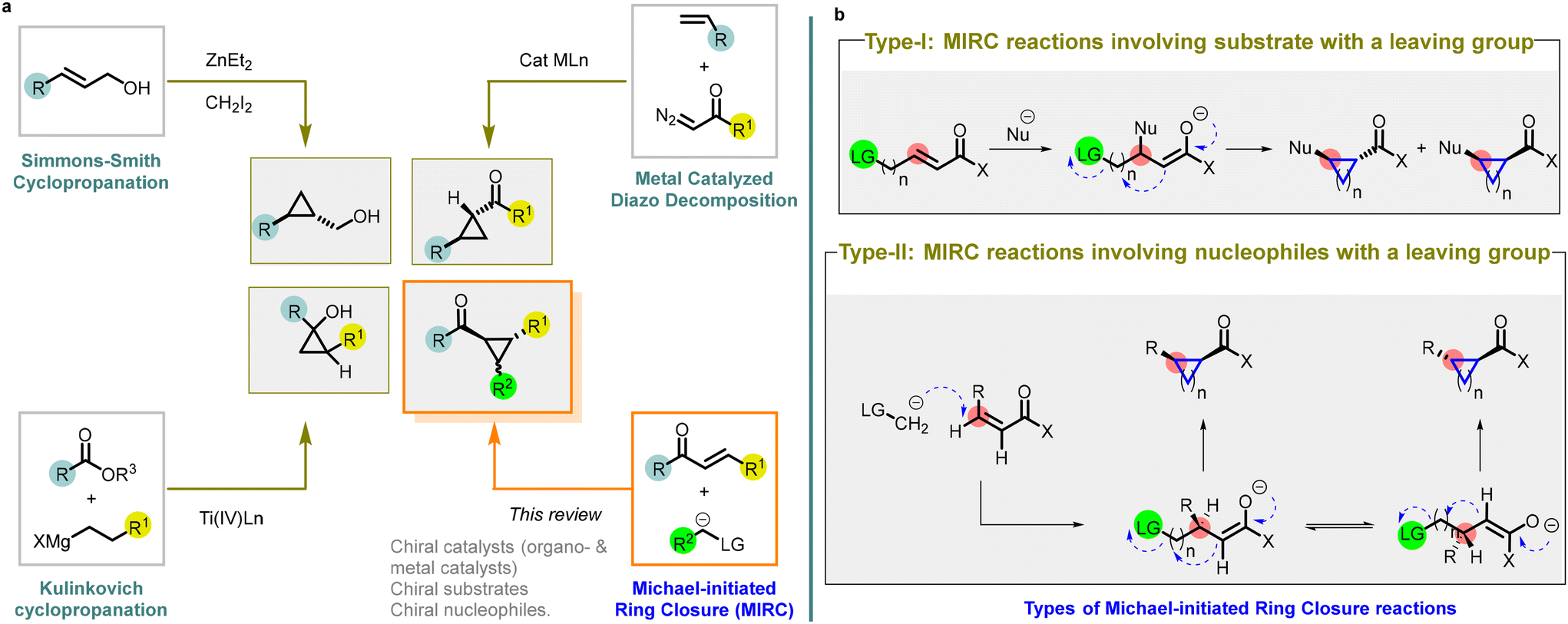 | ||
| Fig. 1 (a) Different synthetic strategies utilized for the preparation of cyclopropanes; (b) different types of MIRC reactions. | ||
The Simmons–Smith cyclopropanation is a widely used method for incorporating cyclopropane into compounds through the reaction of an organozinc carbenoid with an alkene or an alkyne5,6 Achieving high reactivity and stereoselectivity often requires the presence of an oxygen or nitrogen-directing group. Asymmetric variations employing zinc- or boron-centered catalysts have expanded the options for stereoselective synthesis, leading to the desired stereochemical outcomes in cyclopropane synthesis (entry a, Fig. 1).
Diazo compound decomposition with transition metal catalysts is another popular method for cyclopropane synthesis.7–10 It involves reacting a diazoalkane with a late transition metal catalyst, generating a nucleophilic metal carbenoid that can transfer to an electrophilic alkene, such as α,β-unsaturated carbonyl compounds. This versatile transformation allows for various substituents on both the diazo compound and the alkene substrate. Extensive research on asymmetric variations, resembling the Simmons–Smith cyclopropanation, has resulted in highly selective processes using readily accessible bisoxazoline and pyridine bisoxazoline ligands. These advances in asymmetric catalysis significantly expand the synthetic possibilities for accessing chiral cyclopropanes (entry a, Fig. 1).
The Kulinkovich cyclopropanation method is unique for cyclopropane synthesis.11,12 It employs a titanium catalyst to combine an ester or an amide with a Grignard reagent containing an α-hydrogen, resulting in oxygen- or nitrogen-substituted cyclopropanes. Additionally, asymmetric variants enable the synthesis of enantioenriched cyclopropanes (entry a, Fig. 1).
Among these strategies, the utilization of Michael Initiated Ring Closure (MIRC) reaction (or Michael/alkylation cascade reaction) has emerged as a powerful approach to access chiral cyclopropanes with high stereoselectivity.13–15 MIRC reaction involves Michael addition followed by a subsequent intramolecular cyclization, resulting in the formation of the cyclopropane ring. Little and Dawson were the pioneers who reported a MIRC reaction by combining a haloenolate with nitrogen and sulfur nucleophiles.16 Their ground-breaking work involved the synthesis of disubstituted derivatives of cyclopropane, cyclopentane, cyclohexane, and cycloheptane through this reaction.
There are two types of Michael Initiated Ring Closure (MIRC) reactions (entry b, Fig. 1).
Type-I: MIRC reactions involving substrate with a leaving group
In type-I reaction, the formation of cyclopropanes is achieved through nucleophilic addition to electrophilic substrates that contain a leaving group.
A broad range of nucleophiles can be employed in these reactions, including alkoxides, thiolates, cyanides, enolates, Grignard reagents, hydrides, phosphites, and phosphonites.
Type-II: MIRC reactions involving nucleophile with a leaving group
In type-II reaction, the leaving group is located on the nucleophile itself.
The nucleophiles used in these reactions includes α-halo carbanions, but the heteroatom-derived ylides are the most effective reagents for transferring methylene to electron-deficient olefins. Ylides derived from sulfur, phosphorus, arsenic, and tellurium have all been successfully employed as precursors for cyclopropane formation.
Asymmetric variants of MIRC reactions have gained significant interest for their ability to generate enantiomerically enriched cyclopropane products. These reactions typically employ chiral catalysts or auxiliaries to control the stereochemistry during the Michael addition and subsequent ring closure steps. The development of efficient and selective enantioselective MIRC reactions has opened new avenues for synthesizing complex chiral cyclopropane derivatives.
Although previous reviews have covered cyclopropanation reactions,17–38 there is currently no comprehensive account solely focused on Michael Initiated Ring Closure (MIRC) cyclopropanation. Therefore, we present a detailed account on enantioselective cyclopropanation, serving as a valuable resource for synthetic organic and medicinal chemists. In this review, we categorize MIRC cyclopropanation into three groups based on the chirality inducer employed: 1. chiral substrates, 2. chiral nucleophiles, and 3. chiral catalysts (including organocatalysts and metal catalysts). To facilitate readers understanding, we provide a concise description of substrate scope, putative reaction mechanisms, and any additional steps required to achieve the synthesis of the target natural product or its intermediate, if applicable. While we have made efforts to conduct an extensive literature search, we apologize in advance for any inadvertent omission of essential references.
2. Diastereoselective MIRC reactions leading to enantioenriched cyclopropanes
Stereoselectivity in Michael-initiated ring closing cyclopropanation reactions can be achieved by employing with chiral substrates (Michael acceptors) and chiral nucleophiles (Michael Donors). The presence of inherent structural asymmetry in chiral substrates which possess a chiral auxiliary (or a chiral center) is crucial for controlling the stereochemistry of the resulting cyclopropane product. On the other hand, chiral nucleophiles, which possess a chiral auxiliary or coordinating group, play a pivotal role in determining the stereochemistry of the cyclopropane product (Fig. 2). Previous reviews extensively discuss strategies involving chiral substrates and chiral nucleophiles. In this overview, we provide a concise summary of these types of cyclopropanations. | ||
| Fig. 2 General scheme for diastereoselective MIRC cyclopropanation using chiral substrate (Michael acceptors) and chiral nucleophile (Michael donors). | ||
2.1. Chiral substrates (Michael acceptors)
Synthetic chemists employ the strategic addition of reagents to chiral substrates to achieve stereoselective reactions, and for this review, MIRC cyclopropanation reactions being extensively explored for a wide range of asymmetric Michael acceptors. These reactions involve the use of various auxiliaries (such as Evans's oxazolidinones,39 Seebach's oxazolidine,40,41 chiral lactams,42–49 diphenyltetrahydrooxazinone,50,51 pinanone-derived reagent, α,β-didehydroamino acid derivatives,52 dehydroalanine53) and sulfoxides54–61 attached to the substrate, which play a critical role in controlling the stereochemistry of the resulting cyclopropane product (entry a, Fig. 3). Careful selection of the appropriate chiral auxiliary or sulfoxide enables high diastereoselectivity, leading to the formation of enantioenriched cyclopropanes. Factors like steric hindrance, electronic effects, and non-covalent interactions guide the choice of auxiliary or sulfoxide. By strategically utilizing these chiral components, chemists can access a diverse array of enantioenriched cyclopropanes, enabling the synthesis of complex molecules with precise stereochemical control. Various types of cyclopropanes have been synthesized using these substrates (entry b, Fig. 3). | ||
| Fig. 3 Diastereoselective synthesis of cyclopropanes: (a) types of auxiliaries used in Michael acceptors, (b) different cyclopropanes synthesized using chiral auxiliaries. | ||
Enantioenriched cyclopropanes have also been successfully synthesized using a variety of chiral building blocks containing acyclic enones,62–69 cyclic enones,70–77 acyclic enoates,78–92 unsaturated lactones,93–95 and amides as electron-withdrawing groups, in addition to employing chiral auxiliaries. This versatile synthetic method serves as a crucial step in the synthesis of numerous biologically important compounds and natural products. Notable examples include the synthesis of potent taxol analogues,62 glutamate analogues,78L-DCG-IV,63L-CCGI (selective activators of specific isotypes of metabotropic glutamate receptors),89 24,25-ethanovitamin D3 lactones (novel vitamin D receptor antagonists),68 eicosanoids,69 17S,20S-methanofusidic acid (a fusidic acid antibiotic),93 and majusculoic acid (an antifungal agent),94 among many others (Fig. 4). This synthetic approach enables the efficient construction of diverse molecules with precise stereochemistry, facilitating the exploration of their biological activities and potential therapeutic applications.
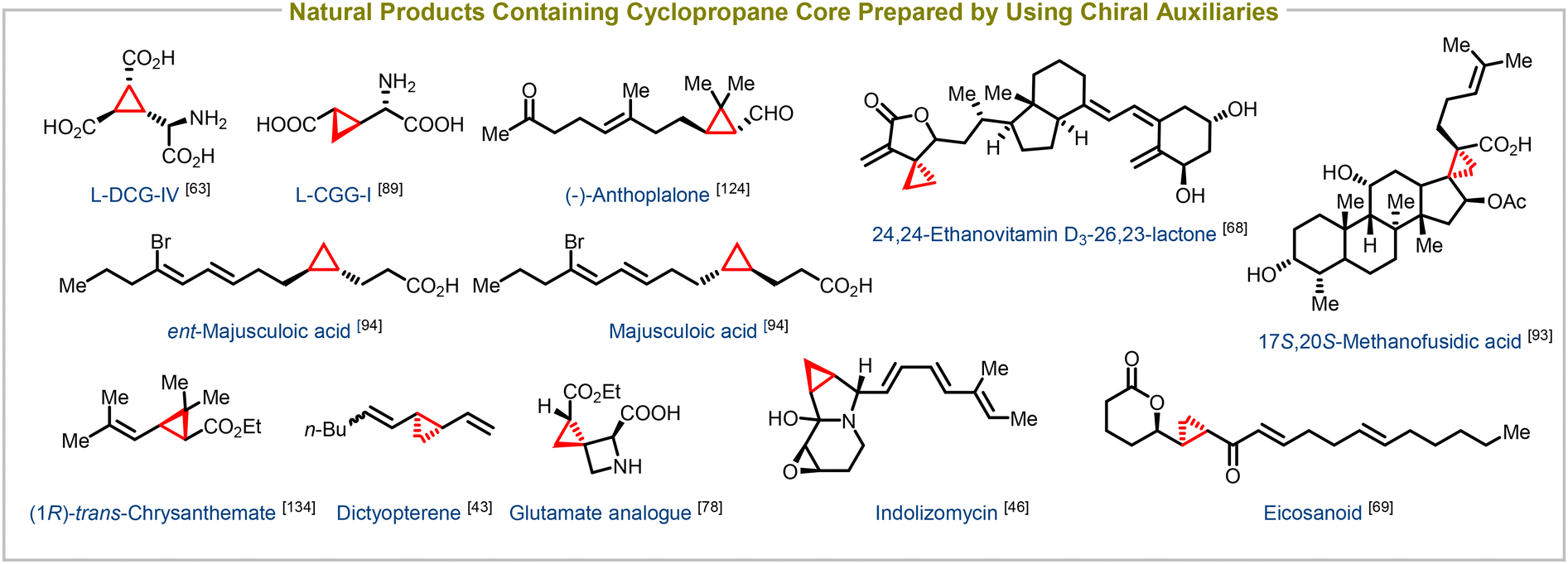 | ||
| Fig. 4 Natural products containing cyclopropane core structure synthesized using chiral substrates and chiral nucleophiles. | ||
2.2. Chiral nucleophiles
Chiral nucleophiles serve as a key reagent in an alternate strategy to obtain enantiomerically enriched cyclopropanes.96 This approach involves the addition of nucleophiles that contain chiral auxiliaries to an achiral substrate. Initial studies involved chiral aminosulfoxonium ylides to access chiral cyclopropanes.97–109 The nucleophiles possess chiral sulfur,110–117 phosphorus,118–124 nitrogen,125–129 and tellurium ylides,130 as well as chiral sulfoxides131–134 and oxazolidinones135,136 (entry a, Fig. 5). They possess a remarkable ability to undergo nucleophilic addition to electron-deficient Michael acceptors, resulting in the formation of chiral cyclopropane rings. | ||
| Fig. 5 Diastereoselective synthesis of cyclopropanes: (a) types of auxiliaries used in Michael acceptors, (b) different cyclopropanes synthesized using chiral auxiliaries. | ||
The literature showcases the synthesis of various types of cyclopropanes, some of which are illustrated in Fig. 5 (entry b). The chirality inherent in these nucleophiles allows for stereochemical control over the reaction, enabling selective formation of specific cyclopropane diastereomers. By utilizing chiral nucleophiles in Michael-initiated cyclopropanation reactions, chemists can access structurally diverse and stereochemically complex cyclopropane derivatives. These derivatives find applications in various fields, including medicinal chemistry and natural product synthesis.
3. Chiral catalysts
3.1 Organocatalysts
In the past two decades a large number of scientists including the recent Nobel laureates List and MacMillan have made significant contributions that has revitalized the field of organocatalysis, establishing it as a crucial component of asymmetric catalysis alongside organometallic and enzymatic catalysis.137,138 The utilization of organocatalytic methods has proven to be highly effective in achieving efficient synthesis. These methods are characterized by their ability to accurately predict stereochemistry, accommodate diverse functional groups, and promote metal-free processes.Organocatalysts play a crucial role in facilitating enantioselective Michael Initiated Ring Closure (MIRC) reaction.139 These catalysts interact with the reactants to direct the synthesis of highly enantioselective cyclopropane derivatives through processes such as hydrogen bonding and Lewis acid/base interactions. An advantage of MIRC reaction is its capability to operate under mild reaction conditions without the need for metal catalysts. This characteristic significantly expands the possibilities for synthesizing complex chiral cyclopropanes.
In 1999, Arai, Shioiri, and co-workers reported the first asymmetric cyclopropanation reaction using phase-transfer catalysis (PTC) (Scheme 1). They utilized α-bromocycloalkenones (1) and nitriles (2) in a Michael addition, proton transfer, and intramolecular alkylation sequence employing catalyst 3 (cinchona alkaloid derived quaternary ammonium salt) and potassium carbonate as base. The reaction proceeded through intermediate 4, which yielded highly functionalized bicyclic compounds (5) with moderate to good enantioselectivities (45–83% ee).140

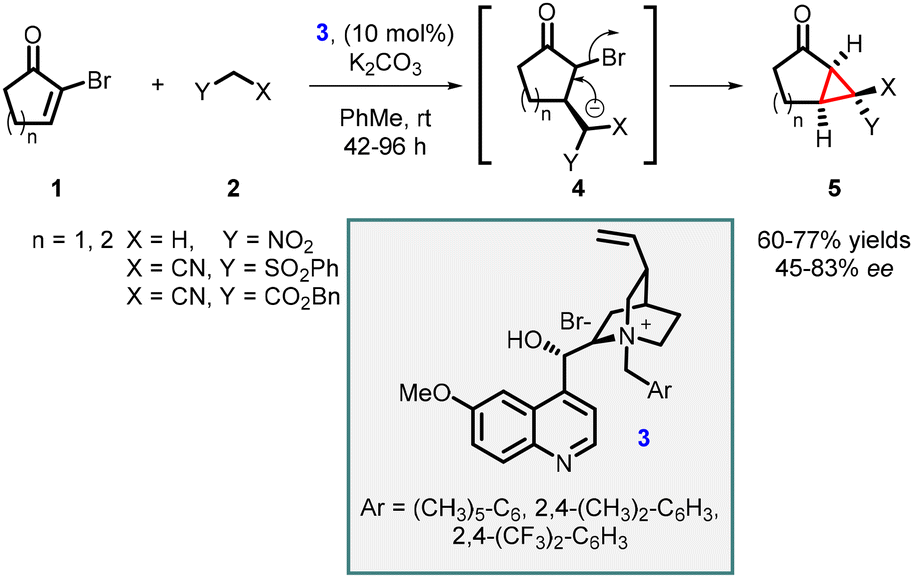 | ||
| Scheme 1 PTC catalyzed asymmetric cyclopropanation of α-bromocycloalkenones. | ||
In 2003, Gaunt and co-workers reported enantioselective cyclopropanation utilizing a cinchona alkaloid derived quaternary ammonium salt. They achieved this by employing a Michael addition of a chiral ammonium ylide (11), derived from cinchona alkaloids (8, 9), to an α,β-unsaturated carbonyl compound (7). This step was crucial in generating the cyclopropane structure with high stereoselectivity (Scheme 2).141
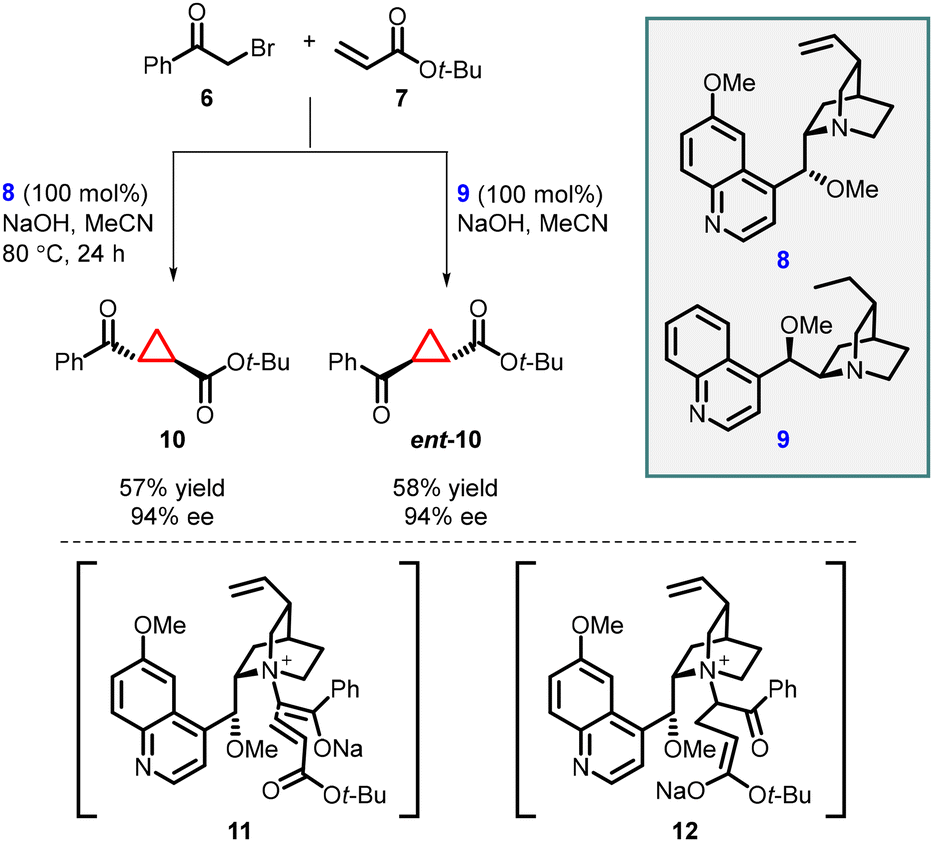 | ||
| Scheme 2 Chiral ammonium ylide-mediated asymmetric cyclopropanation of tert-butyl acrylate. | ||
To accomplish asymmetric variation of MIRC reaction, they first formed a quaternary ammonium salt in situ using phenacyl bromine 6 and a stoichiometric amount of chiral cinchonine 8 through a nucleophilic substitution process. Subsequently, deprotonation with sodium hydroxide yielded the ammonium ylide. The chiral ammonium ylide was then reacted with tert-butyl acrylate 7 resulting in the formation of chiral cyclopropane (10) in 57% yield and good stereoselectivity (94% ee). Interestingly, when cinchonine 9 was used instead of cinchonine 8 under the same reaction conditions, the opposite enantiomer, ent-10, was obtained with a 58% yield and the same stereochemistry (94% ee) (Scheme 2).
The reaction mechanism can be explained by a Michael addition of the ammonium ylide (11) to tert-butyl acrylate, followed by the release of a chiral tertiary amine during the 3-exo-tet-cyclization process (12) to yield cyclopropane 10.
Following their initial report on stoichiometric asymmetric cyclopropanation, the Gaunt group went on to develop a catalytic enantioselective version using a series of cinchona alkaloid catalysts derived from quinine or quinidine. Through a thorough screening of bases, they discovered that using a larger metal cation (transitioning from Na to Cs) of the base resulted in improved yields of the cyclopropane product. This breakthrough allowed for the efficient utilization of a variety of easily accessible or commercially available cinchona alkaloid derivatives (8, 16, 17, 18) in the asymmetric cyclopropanation reactions. These reactions involved α–bromo esters or amides (13) and vinyl ketones or ester (14), yielding the corresponding cyclopropanes (15) with good diastereo- and enantioselectivities (Scheme 3).142
 | ||
| Scheme 3 Chiral ammonium ylide-mediated asymmetric cyclopropanation of α,β-unsaturated carbonyl compounds. | ||
Gaunt and coworkers expanded MIRC reaction to enantioselective intramolecular cyclopropanation by utilizing substrate 19. For the intramolecular variant, catalysts 8 and 9, derived from cinchona alkaloids, were used for the reaction resulting in moderate yield and enantioselectivity 67% and 64% ee respectively. However, when NaBr was introduced to aid in the formation of the quaternary salt of the catalyst (Scheme 4, entry a) the reaction gave excellent enantioselectivity (94% ee).143
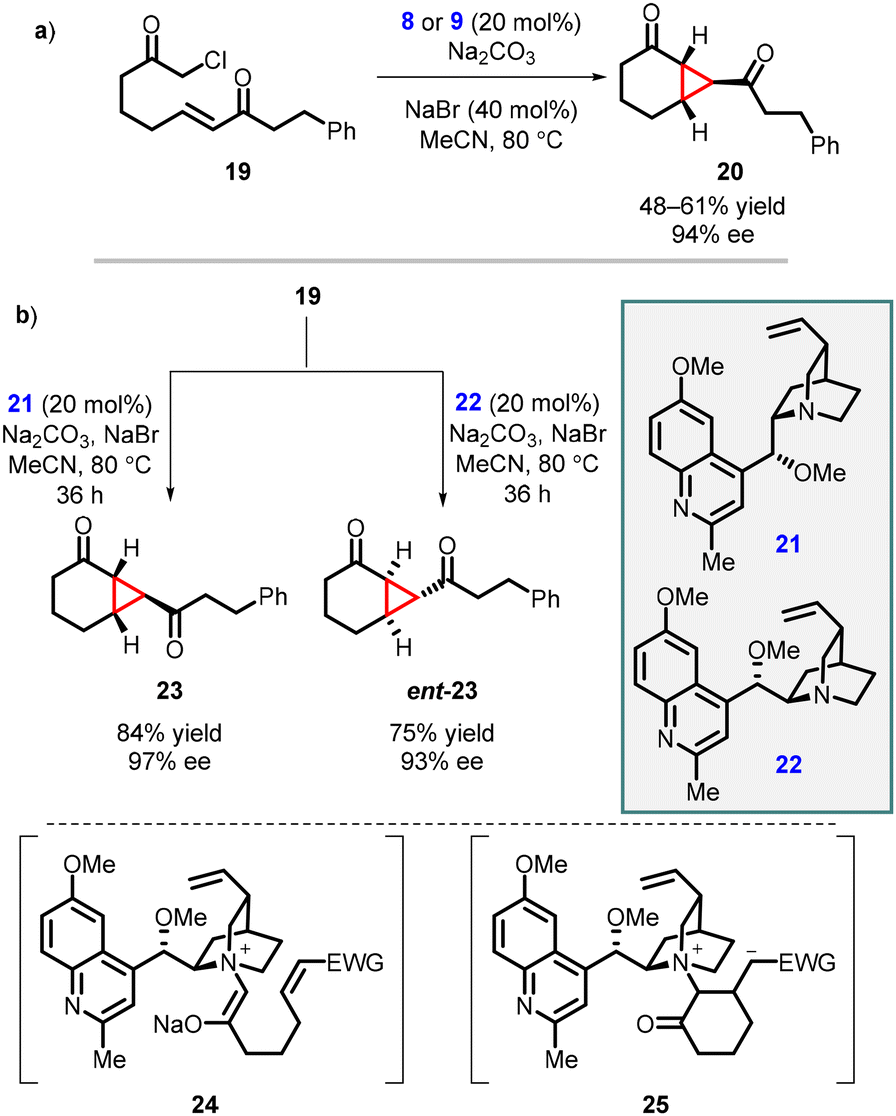 | ||
| Scheme 4 Intramolecular asymmetric cyclopropanation using modified cinchona alkaloid. | ||
Two years after their initial report on intramolecular MIRC reaction, Gaunt and coworkers used novel cinchona alkaloid derivatives 21 or 22 to improve the yield and enantioselectivity for substrate 19. The reaction involves two steps: an intramolecular Michael addition (24) followed by an intramolecular cyclopropanation via nucleophilic substitution (25), taking place within a chiral ammonium ylide, resulting in the formation of [4.1.0]-bicycloalkane 23 (Scheme 4 entry b). The substrate 19 was treated with the modified cinchona catalyst 21 (20 mol%), in the presence of Na2CO3 and NaBr in MeCN at 80 °C for 36 h, resulting in the formation of cyclopropane 23 with an 84% yield and 97% enantiomeric excess. By using catalyst 22 under the same reaction conditions, enantiomer ent-23 was obtained with a 75% yield and 93% ee (Scheme 4, entry b). The modified cinchona alkaloid derivatives 21 and 22 prevented side reactions caused by N-alkylation of the quinoline ring nitrogen atom thereby improving the yields of both 23 and ent-23.144
In 2006, Kojima and coworkers presented an enantioselective synthesis of tetra-substituted cyclopropanes by utilizing chiral cinchonidine catalyst 29 in MIRC reaction involving chloromethyl ketones (26) and β-substituted methylidenemalononitriles (27) (Scheme 5).145 This reaction yielded trans-cyclopropanes (28) with good enantioselectivities up to 82% ee. Notably, the Cinchonidine catalyst functioned as a chiral Brønsted base catalyst, and the presence of hydrogen bonding played a crucial role in achieving high enantioselectivity. This innovative approach highlighted the versatility and effectiveness of cinchonidine in promoting asymmetric transformations.
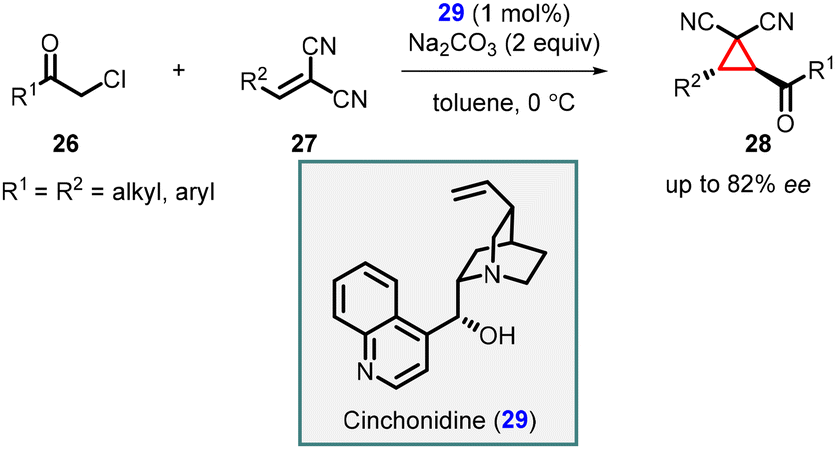 | ||
| Scheme 5 Enantioselective cyclopropanation of β-substituted methylidenemalononitriles. | ||
Total synthesis of an eicosanoid was developed by Kumaraswamy and coworkers who adopted Gaunt's method of Michael-initiated ring closure reaction for the cyclopropanation step using the (DHQD)Pyr (18) as catalyst. They applied the protocol by functionalizing the vinyl ketone 31, derived from pentane-1,5-diol (30), and tert-butyl bromoacetate, resulting in the formation of the key cyclopropyl moiety 34. To access the opposite enantiomer 36, they employed the cinchona alkaloid derivative 33 as catalyst. The cyclopropane intermediate 34 served as a pivotal building block for the synthesis of the natural product eicosanoid 35. This was accomplished by subjecting compound 34 to catalytic asymmetric transfer hydrogenation, following Noyori's protocol, followed by subsequent manipulations of the functional groups (Scheme 6).146
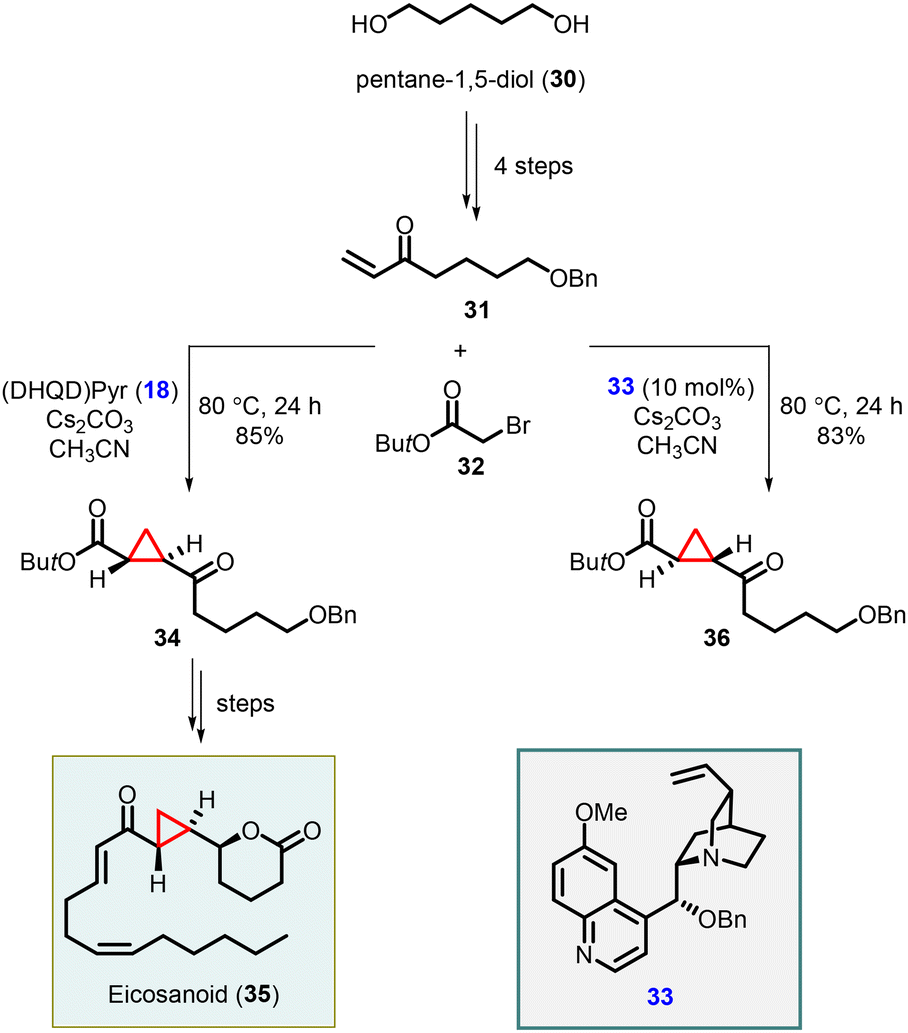 | ||
| Scheme 6 Enantioselective cyclopropanation in total synthesis of eicosanoid. | ||
Kumaraswamy and coworkers have also reported the total synthesis of solandelactone A and B which involves the synthesis of cyclopropyl δ-lactonealdehyde 40 as a key intermediate. The cyclopropyl moiety of the intermediate 40 was synthesized using asymmetric MIRC reaction which involves reaction of 2-bromo-N,O-dimethylacetamide (37) and enone (38), utilizing organocatalyst 18 and 33 (Scheme 7).147
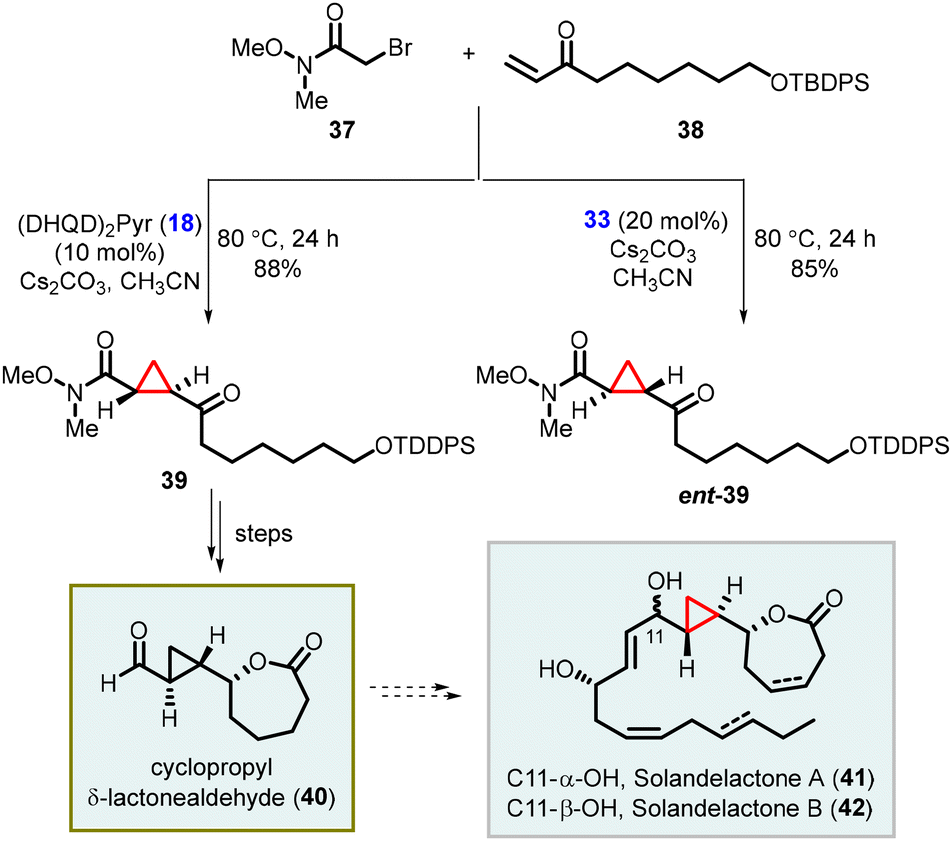 | ||
| Scheme 7 Synthesis of cyclopropyl δ-lactonealdehydes through enantioselective cyclopropanation. | ||
In 2009, Yan and coworkers showcased an enantioselective conjugate addition of dimethyl bromomalonate 44 to nitroalkenes (43) using the 6′-demethyl quinine (46) catalyst. The reaction was followed by an intramolecular cyclopropanation employing 1,4-diazabicyclo[2.2.2]octane (DABCO), resulting in the formation of nitrocyclopropanes 45 with 47–78% yields and high diastereoselectivity (>99% de) and enantioselectivity (>99% ee) (Scheme 8).148
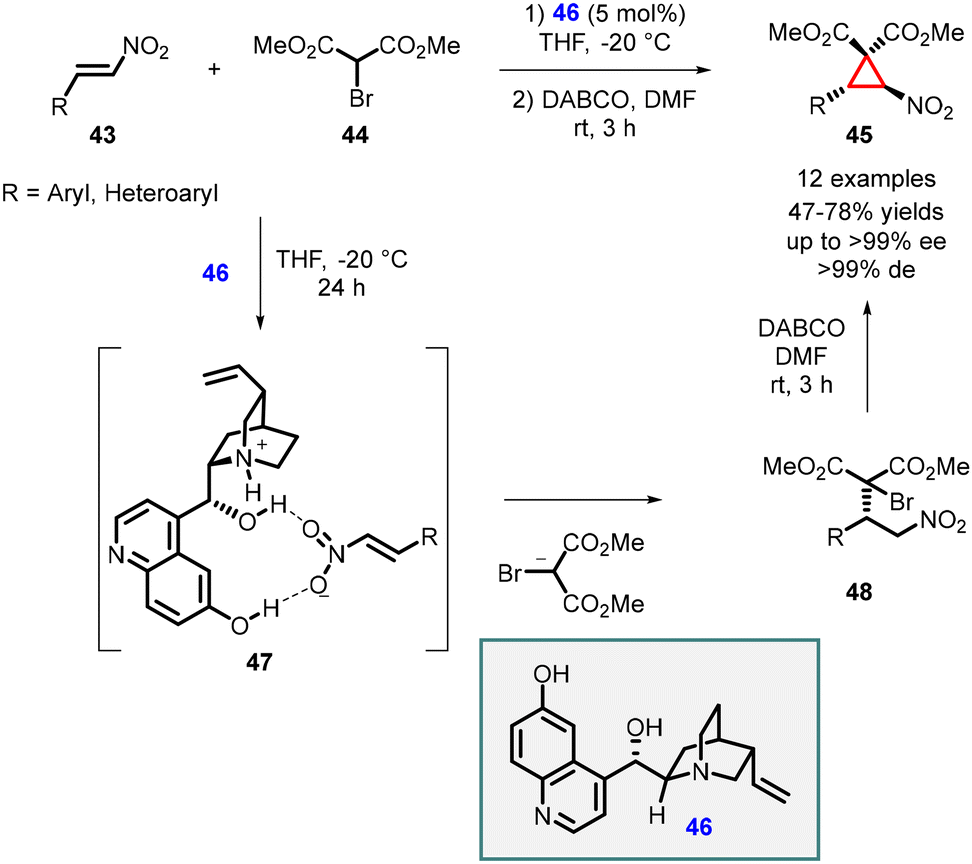 | ||
| Scheme 8 6′-Demethyl quinine catalyzed enantioselective synthesis of nitrocyclopropanes. | ||
The cinchona alkaloid 46 played a crucial role as a bifunctional catalyst In the MIRC reaction. The nitrogen atom of the catalyst facilitated the abstraction of the α-proton from dimethyl bromomalonate 44, while the hydroxyl groups of catalyst (46) formed hydrogen bonds with the nitro group of the β-nitroalkene 43 which is depicted in the transition state 47. This interaction activated the β-nitrostyrene 43, enabling an efficient asymmetric Michael addition. Subsequent treatment of the Michael-adduct (48) with DABCO in DMF at room temperature for 3 hours yielded trans-cyclopropane (45). Notably, no cis-cyclopropane was observed based on NMR analysis.
Wang and coworkers successfully synthesized enantiopure functionalized nitrocyclopropanes by utilizing a quinine-derived amine, specifically 9-amino-9-deoxyepiquinine (53), as an effective catalyst (Scheme 9).149 The conjugate addition of bromonitromethane 51 to α,β-unsaturated enone systems was efficiently catalyzed by the salt derived from the combination of cinchona alkaloid 53 with mandelic acid. The reaction tolerated cyclic as well as aromatic enones (49 and 50) as substrates, resulting in high yields of nitrocyclopropane products (52 and 55) with excellent stereoselectivities (>98 and >99% ee). This approach demonstrated the effectiveness of the 9-amino-9-deoxyepiquinine as an organocatalyst in facilitating the synthesis of enantioenriched nitrocyclopropanes through MIRC reaction.
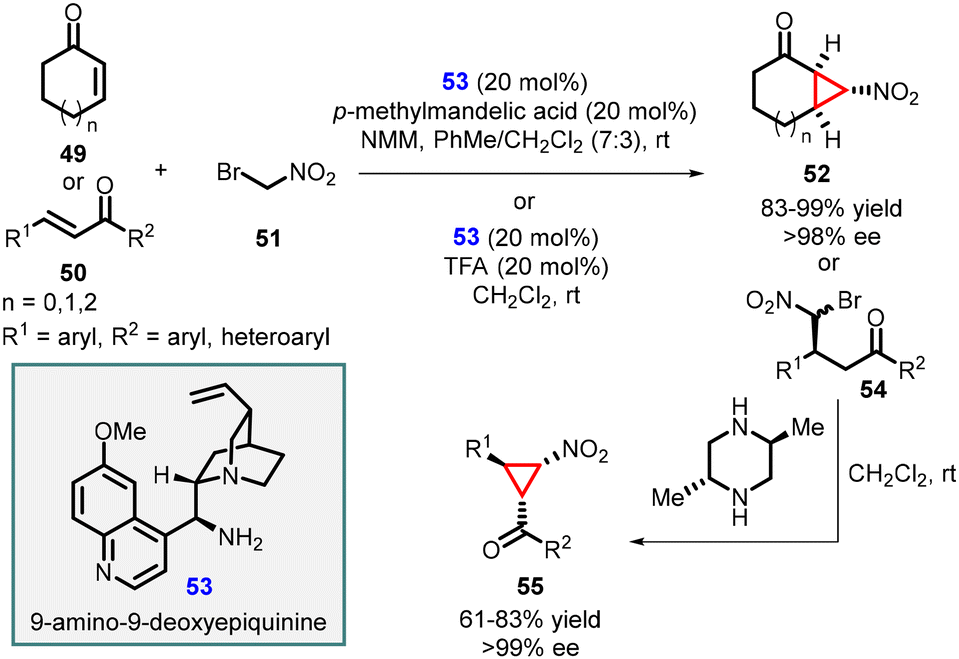 | ||
| Scheme 9 9-Amino-9-deoxyepiquinine catalyzed enantioselective synthesis of functionalized nitrocyclopropanes. | ||
Adamo and coworkers have reported an efficient enantioselective synthesis of highly substituted cyclopropanes through MIRC reaction. The reaction involves 4-nitro-5-styrylisoxazoles (56), a class of cinnamate synthetic equivalent, with 2-bromomalonate esters (44) using a cinchona alkaloid derived phase-transfer catalyst (57) (Scheme 10).150 Remarkably, this method yielded trans-cyclopropylisoxazoles (58) as a single diastereomer with excellent yields (up to 99%) and good to excellent enantiomeric excess (84–96% ee). The presence of a free hydroxy group of the catalyst 57 plays a vital role in achieving high enantioselectivity. This is attributed to the ability of the hydroxy group to form a hydrogen bond with the bromomalonate substrate (44).
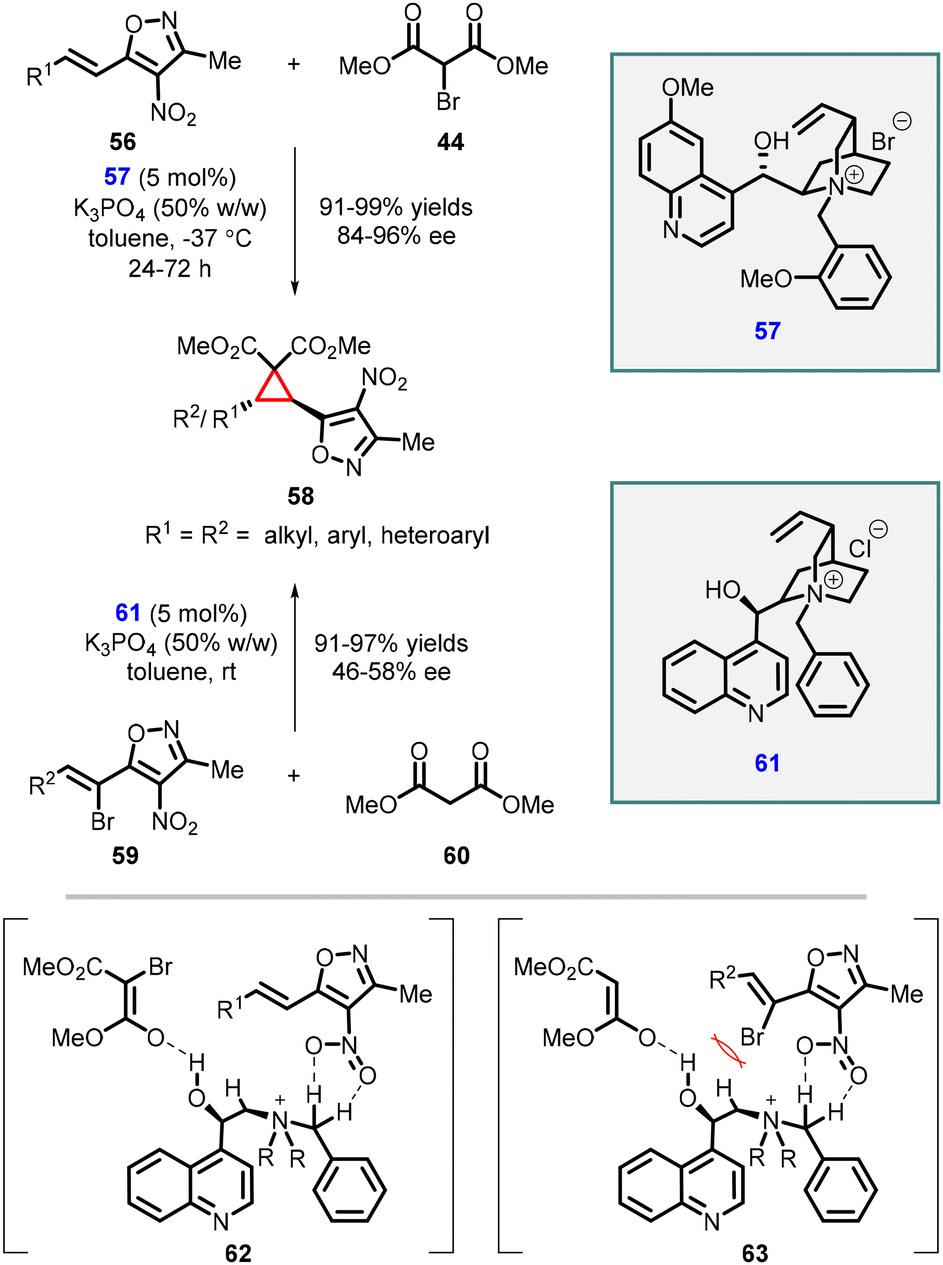 | ||
| Scheme 10 Enantioselective cyclopropanation of 4-nitro-5-styrylisoxazoles. | ||
In 2015, the study was extended to malonate esters 60 as a nucleophile resulting in the formation of the same cyclopropanes 58 with excellent yields (91–97%) and poor enantioselectivities (46–58% ee) (Scheme 10).151
The observed difference in enantioselectivity between these two approaches can be attributed to a comparison of transition states 62 and 63. In compound 59, the presence of a bromine atom shields the nitro group (NO2), limiting its interaction with the phase-transfer catalyst (PTC). This interaction, which is absent in compounds 56, is responsible for higher enantioselectivity. Therefore, the lower enantioselectivity observed can be correlated to the limited interaction between the nitro group and the PTC due to the presence of the bromine atom.
In a significant breakthrough, Waser and coworkers introduced an asymmetric MIRC reaction between bromomalonate (64) and chalcones (65). This transformation was achieved using Cinchona alkaloids (67) as chiral catalyst under phase transfer catalysis conditions, resulting in the synthesis of highly substituted cyclopropanes (66) (Scheme 11).152 Catalyst 67, derived from quinidine and 9-(chloromethyl)-anthracene proved to be highly effective in facilitating the reaction, leading to high yields (up to 98%) and enantioselectivity (up to 91![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 er). The successful outcome of the reaction relied on the utilization of a cinchona alkaloid ammonium salt catalyst containing a free OH group, along with the optimized liquid/liquid reaction conditions. Notably, the process demonstrated excellent performance with both electron-donating and electron-deficient chalcones.
:9 er). The successful outcome of the reaction relied on the utilization of a cinchona alkaloid ammonium salt catalyst containing a free OH group, along with the optimized liquid/liquid reaction conditions. Notably, the process demonstrated excellent performance with both electron-donating and electron-deficient chalcones.
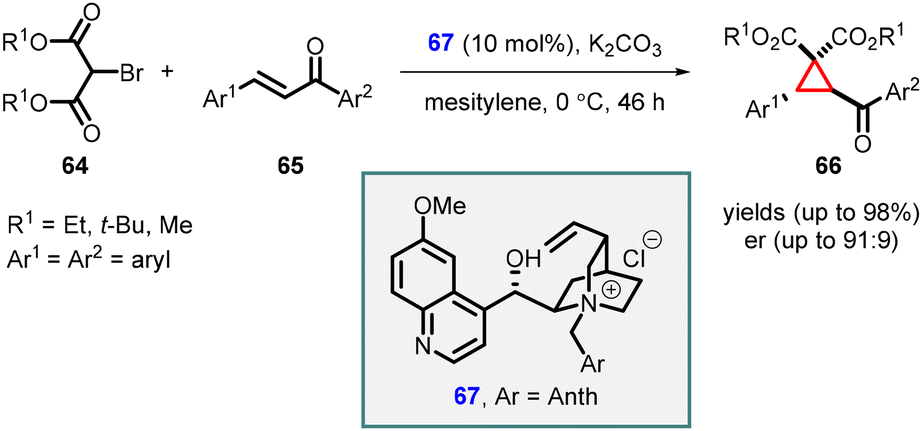 | ||
| Scheme 11 Enantioselective cyclopropanation of chalcones. | ||
Cobb and coworkers have recently disclosed a remarkable finding regarding the enantioselective cyclopropanation of conjugated cyanosulfones (68). This transformation resulted in the formation of a single diastereomer with remarkably high enantioselectivity, reaching up to 96% enantiomeric excess (ee). The high stereoselectivity for the MIRC reaction was attributed to the development of a novel bifunctional catalyst based on the cinchona alkaloid framework, specifically the cupreine organocatalyst (70). The reaction proceeds through the simultaneous activation of the pronucleophile bromomalonate (44) and the electrophilic cyanosulfone (68), facilitated by coordination to the cupreine-derived catalyst which is shown in transition state (71) (Scheme 12).153
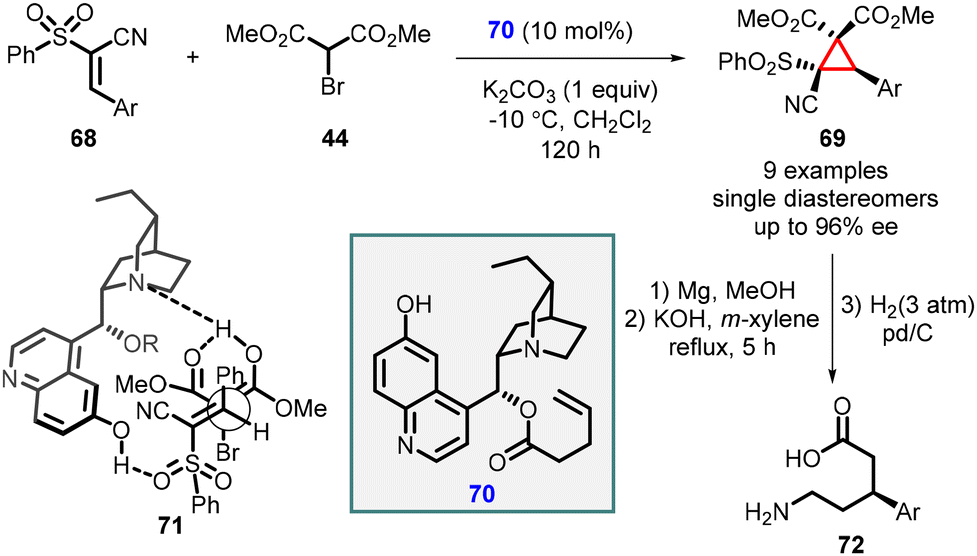 | ||
| Scheme 12 Enantioselective cyclopropanation of conjugated cyanosulfones. | ||
The resulting cyclopropanes (69) can be further transformed into the corresponding δ-amino acids (72) through a magnesium-initiated radical desulfonylation-ring opening process.
Adamo and coworkers reported highly enantioselective MIRC reaction using (Z)-3-substituted-2-(4-pyridyl)-acrylonitrile (73), a reactive class of Michael acceptor, and 2-bromomalonate esters (64). The reaction was catalyzed by a Cinchona derived phase-transfer catalyst (75) which resulted in the rapid synthesis of highly functionalized cyclopropanes 74 with two quaternary centers (Scheme 13).154 The transformation produced cis-cyclopropanes as single diastereoisomers, exhibiting excellent yields (68–95%) and good enantioselectivity (up to 83% ee). The reaction demonstrated broad substrate scope and was successfully scaled up to gram level without compromising yield or stereochemical outcome. Additionally, a one-pot multicomponent procedure proved to be effective in this context.
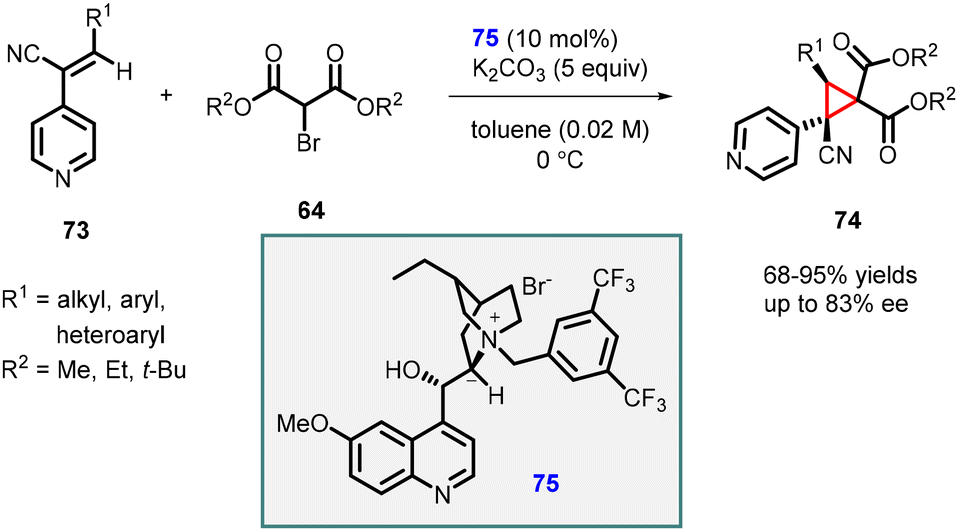 | ||
| Scheme 13 Enantioselective cyclopropanation of (Z)-3-substituted-2-(4-pyridyl)-acrylonitrile. | ||
Synthesis of cyclopropane-based purine analogues was reported by Xie and Guo using C2-symmetric hydroquinidine(anthraquinone-1,4-diyl)diether (DHQN)2AQN (79) as an organocatalyst. The system was efficient for enantioselective cyclopropanation of α-purine acrylate 76 and tert-butyl-2-bromoacetate 77, leading to the formation of cyclopropyl purine analogs 78. The reaction exhibited high yields (up to 98%), and excellent stereoselectivity (>20:1 dr, 97% ee) (Scheme 14).155
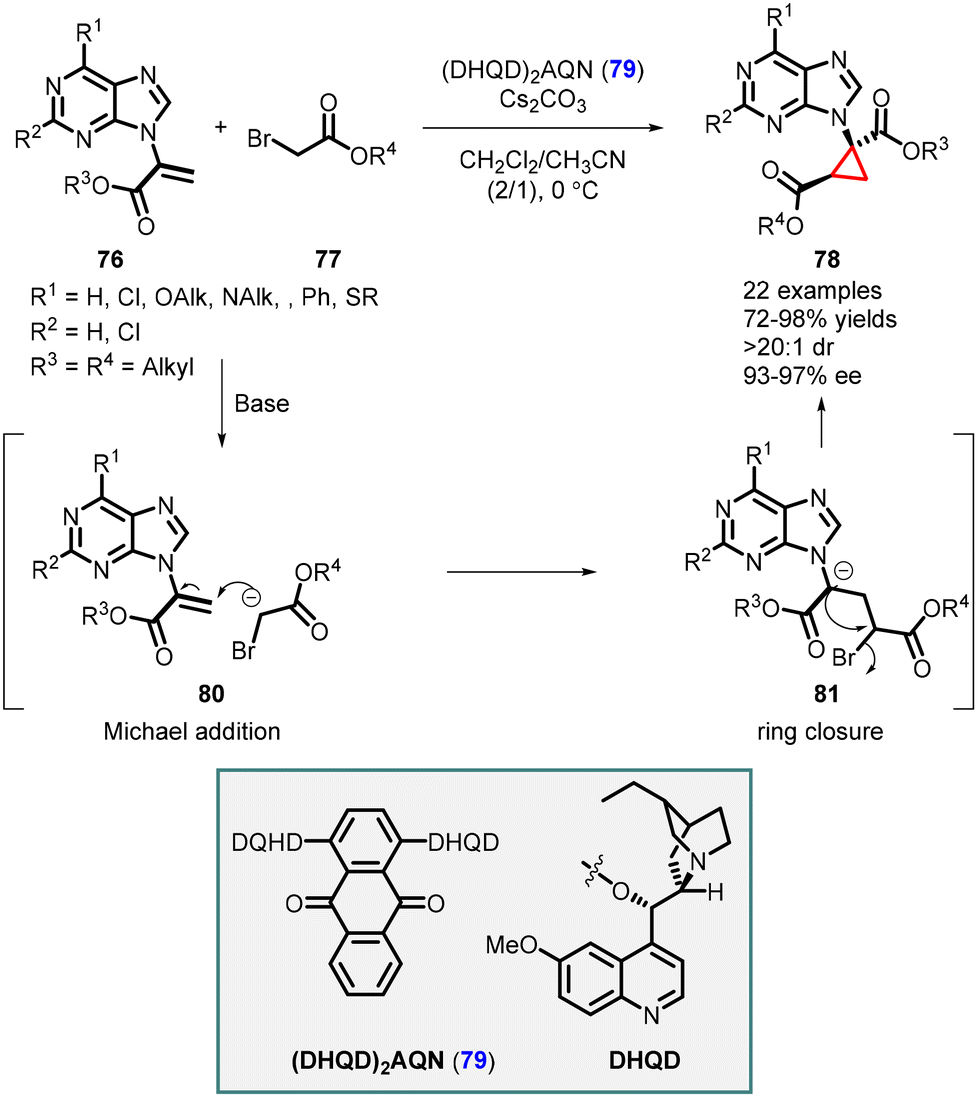 | ||
| Scheme 14 Enantioselective cyclopropanation of α-purine acrylate. | ||
The mechanism involves the formation of a hydrogen bond between the enol form of tert-butyl-2-bromoacetate 77 and the catalyst 79. This interaction facilitates a Michael addition (intermediate 80) to the purine acrylate 76, followed by intramolecular alkylation (intermediate 81). The release of a bromine atom results in the formation of cyclopropyl purine analogue 78, with two stereogenic centers, one of which is a quaternary center.
In an interesting development, Xie and Guo were able to extend the studies to cyclopropane-based pyrimidine analogues (84) containing a quaternary center. The asymmetric version of the MIRC reaction was achieved using (DHQD)2AQN (79) as an organocatalyst and the reaction involves the coupling of α-pyrimidine substituted acrylates (82) with bromo-carboxylic esters (83) (Scheme 15).156
 | ||
| Scheme 15 Enantioselective cyclopropanation of α-pyrimidine substituted acrylates. | ||
Notably, the cyclopropyl pyrimidine analogues synthesized exhibited axis chirality, which was observed for the first time in this context. This axis chirality arises from the rotationally restricted N–C bond in the N–COR3 moiety of the molecules.
Roiser and Waser reported the synthesis of highly enantioenriched spirocyclopropanes (chiral spiro[2.5]octa-4,7-dien-6-ones) (87). The reaction involved the addition of cinchona alkaloid-based chiral ammonium ylides 86 to para-quinone methides 85 in the presence of Cs2CO3 (6 equiv.) as base in CH2Cl2 at reflux condition for 96 h (Scheme 16).157 The reaction yielded spirocyclopropanes 87 with a high diastereomeric ratio of >95:5 and an enantiomeric ratio of >99.9:0.1, making it a valuable method for the synthesis of chiral spirocyclopropanes.
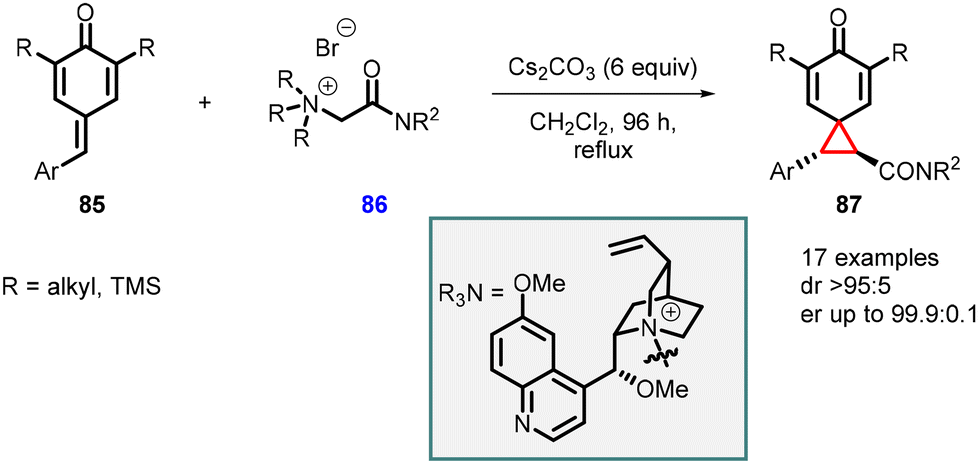 | ||
| Scheme 16 Enantioselective cyclopropanation of para-quinone methides. | ||
Ji, Peng, and Zeng reported the synthesis of highly functionalized cyclopropa[c]coumarin derivatives (90) through a MIRC reaction of 3-substituted coumarins (88) and 2-bromomalonate (89), (Scheme 17).158 The reaction utilized chiral phase-transfer catalyst (PTC) (91), which resulted in the desired cyclopropanation transformation in average yield (75%) and poor enantioselectivity (41% ee). It is important to note that the asymmetric version of the reaction was performed for only one substrate.
 | ||
| Scheme 17 Enantioselective cyclopropanation of 3-substituted coumarins. | ||
Sun and coworkers subsequently employed the catalyst (DHQ)2PYR 94 for the highly enantioselective synthesis of cyclopropa[c]coumarins (93). In this method, the catalyst (DHQ)2PYR 94 reacts with tert-butyl 2-bromoacetate 13, leading to the formation of an ylide 96 with the assistance of the base Cs2CO3. The intermediate 96 then undergoes a Michael addition to the coumarin (92), followed by nucleophilic substitution, resulting in the formation of the corresponding cyclopropa[c]coumarins (93) with good to excellent yields of up to 97% and enantioselectivities of up to 97% ee (Scheme 18).159
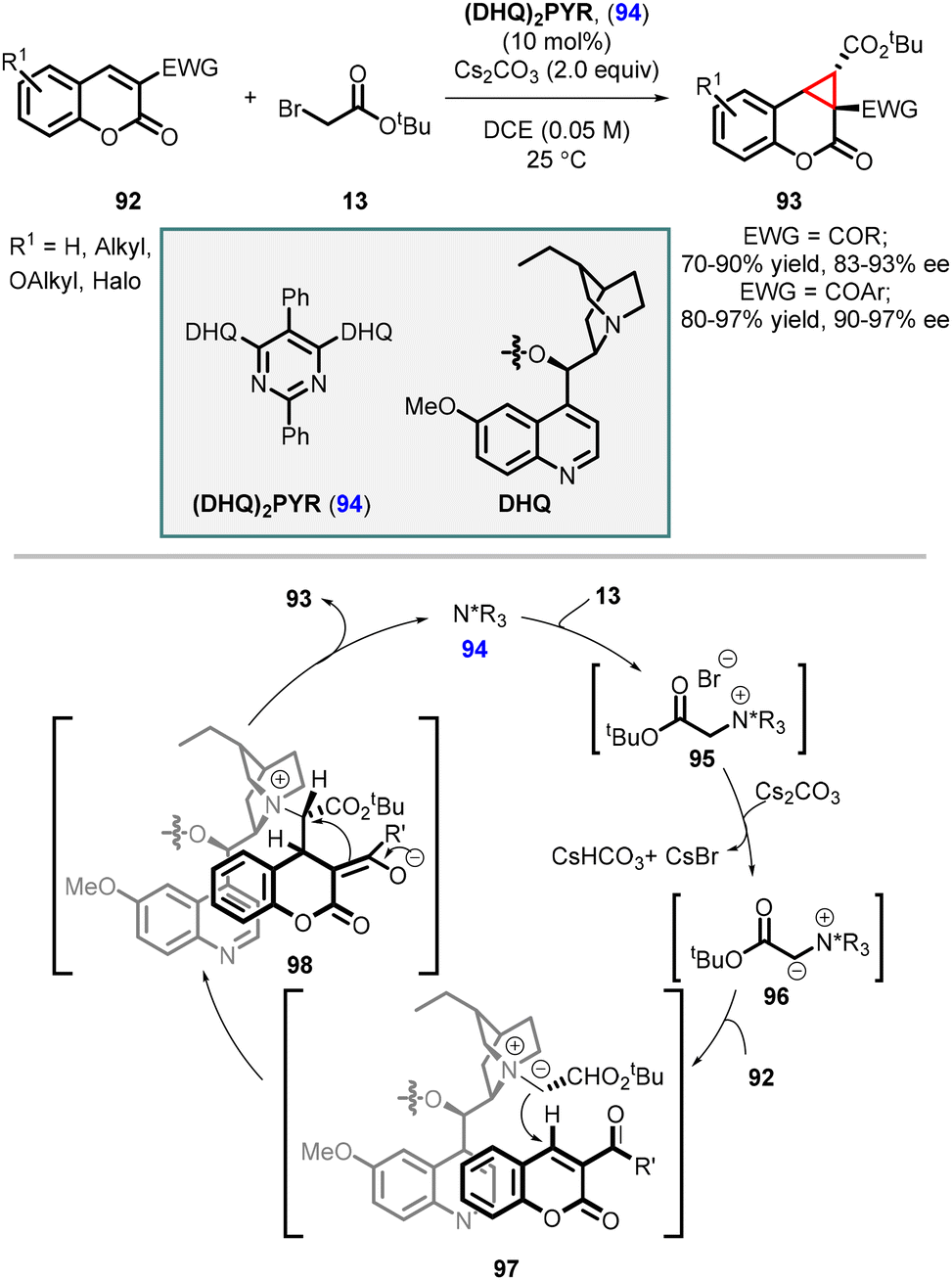 | ||
| Scheme 18 (DHQ)2PYR catalyzed enantioselective MIRC cyclopropanation of coumarins. | ||
Recently, Blay, Vila, and coworkers have made significant progress in developing an asymmetric MIRC reaction. They successfully demonstrated the reaction between arylidenepyrazolones (99) and dialkyl 2-bromomalonates (64) (Scheme 19).160 The catalyst employed in this process was a commercially available organocatalyst, (DHQ)2AQN (79). The reaction conditions were mild, leading to the formation of chiral spirocyclopropyl pyrazolones (21 examples) with yields ranging from 30–81%. The diastereoselectivity for the reaction varied from 60:40 to >95:5, and the enantioselectivity obtained was modest to high ranging from 26–93% ee.
 | ||
| Scheme 19 Enantioselective MIRC cyclopropanation of arylidenepyrazolones. | ||
Singh and coworkers have recently introduced an efficient and practical method for the synthesis of highly enantioenriched spiro-cyclopropyl oxindole derivatives (103). This approach involves a cinchonidine-derived bifunctional urea catalyst 104 and employs an asymmetric vinylogous Michael addition of 4-nitroisoxazole derivatives (102) to N-Boc isatylidene malonates (101), followed by intramolecular alkylation.
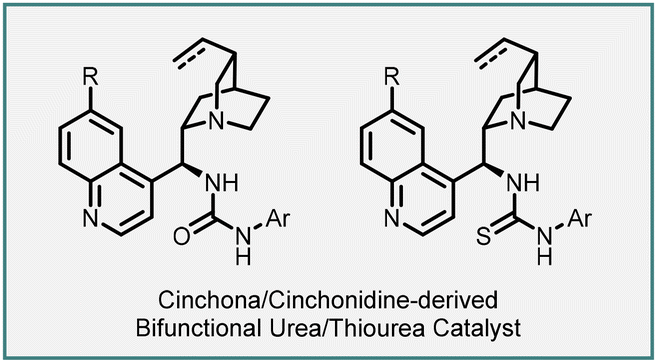
The novel organocatalyst yielded highly substituted spiro-3,3′-cyclopropyl oxindoles (103) with tert-spiro quaternary stereogenic centers. The reaction exhibited excellent diastereoselectivity (>20:1, dr) and enantioselectivity (up to >99%). The desired products were obtained in high yields (up to 93%) (Scheme 20).161
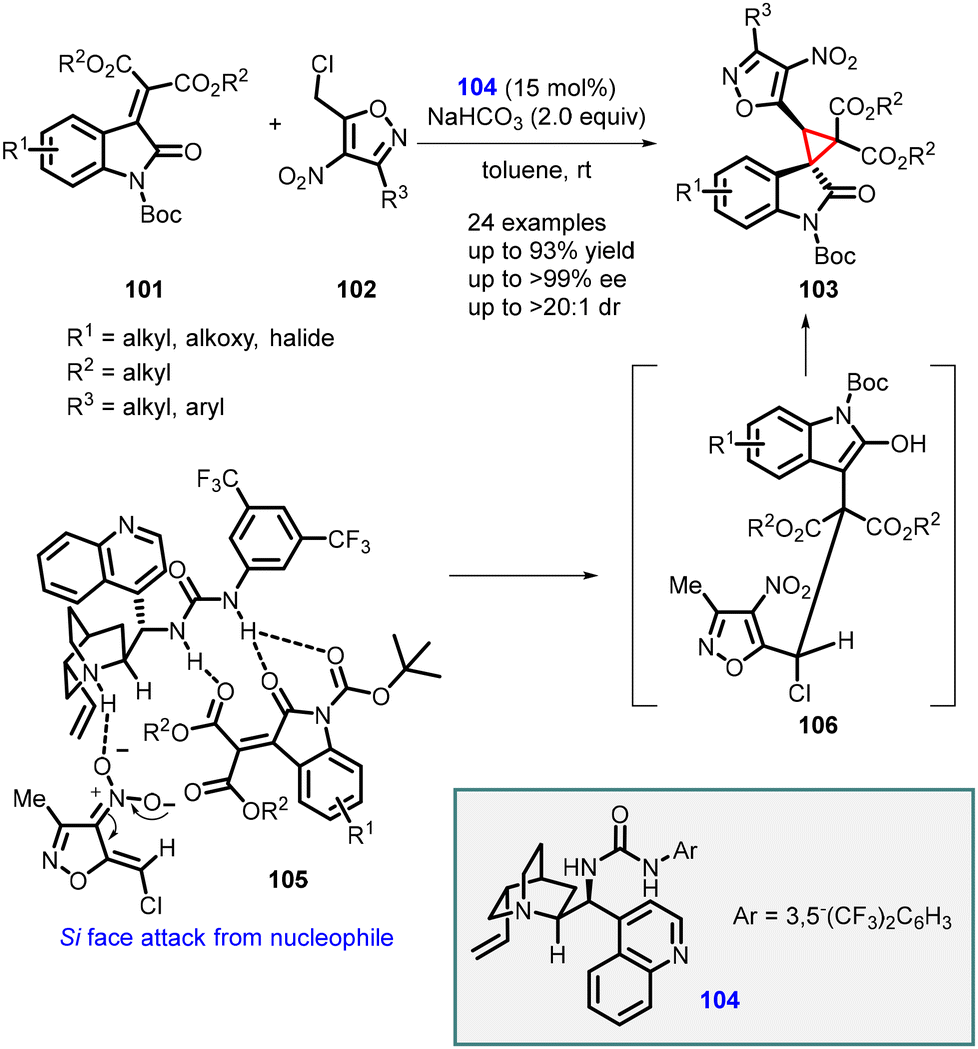 | ||
| Scheme 20 Asymmetric synthesis of spiro-3,3′-cyclopropyl oxindoles using cinchonidine-derived bifunctional urea catalyst. | ||
The observed stereochemical outcome of the vinylogous MIRC reaction was rationalized based on a transition state 105. This transition state involves the simultaneous activation of the reaction partners through H-bonding interactions facilitated by the bifunctional urea catalyst.
Singh and coworkers extended the idea in a subsequent study, involving the enantioselective synthesis of novel spirocyclopropyl oxindole (108) from N-boc-isopropylidene oxindole (107) and 5-(chloromethyl)-3-methyl-4-nitroisoxazole (102) (Scheme 21).162 The reaction employed the same catalyst 104 and exhibited a similar stereochemical outcome. The catalyst deprotonated compound 102, generating an anionic nucleophile, while activating tert-butyl 2-oxo-3-(propan-2-ylidene) indoline-1-carboxylate (107) through H-bonding. The nucleophile then attacked substrate 107, resulting in the formation of an enolate. The facilitation of the chiral catalyst 104 in the substitution step, and its anion binding activity promoted SN2-type reactions, ultimately leading to the formation of the major stereoisomer of compound 108 (Scheme 21. See 109 and 110).
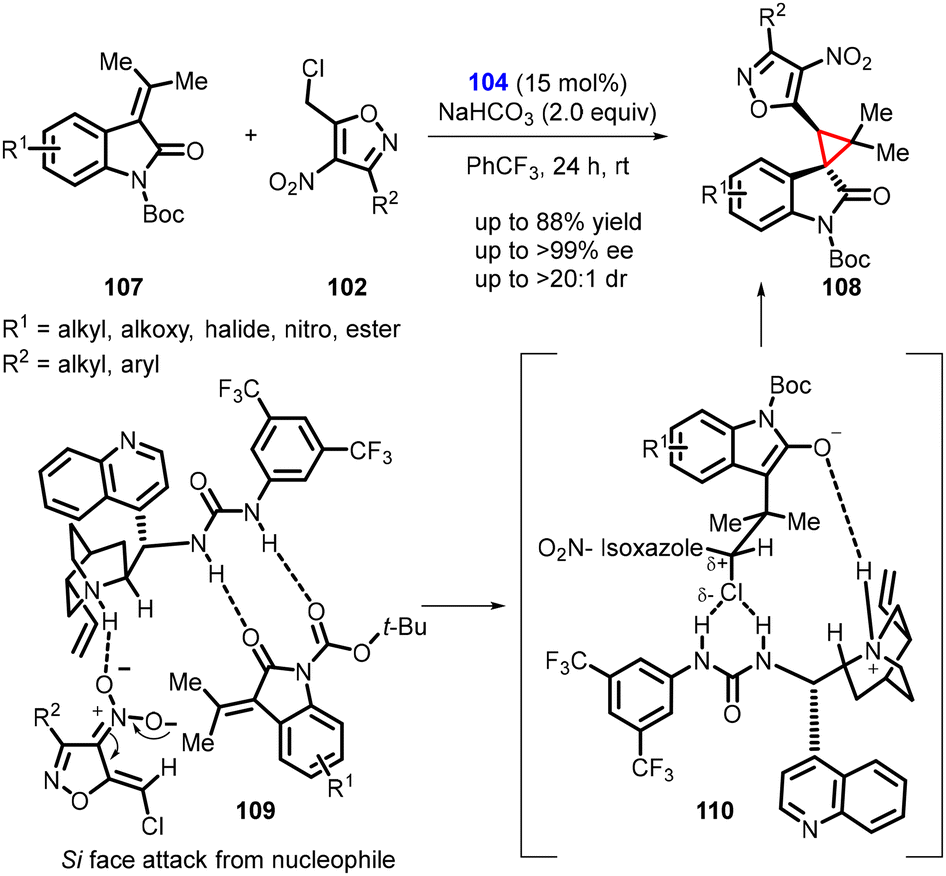 | ||
| Scheme 21 Asymmetric cyclopropanation of N-boc-isopropylidene oxindole and 5-(chloromethyl)-3-methyl-4-nitroisoxazole. | ||
In 2006, Connon developed an elegant and convenient MIRC reaction involving β-nitrostyrenes (111) and 2-chloromalonates (112). Chiral thiourea (114) served as a catalyst, and base was required for the final cyclization step. The reaction gave cyclopropanes (113) in good yields with excellent diastereoselectivities (>98% dr). Mechanistically, the reaction proceeded through the addition of chloromalonate to nitroalkenes, followed by base catalyzed intramolecular alkylation. However, the observed enantioselectivity for different substrates were modest, with values ranging from moderate to low, reaching a maximum of 47% ee (Scheme 22).163
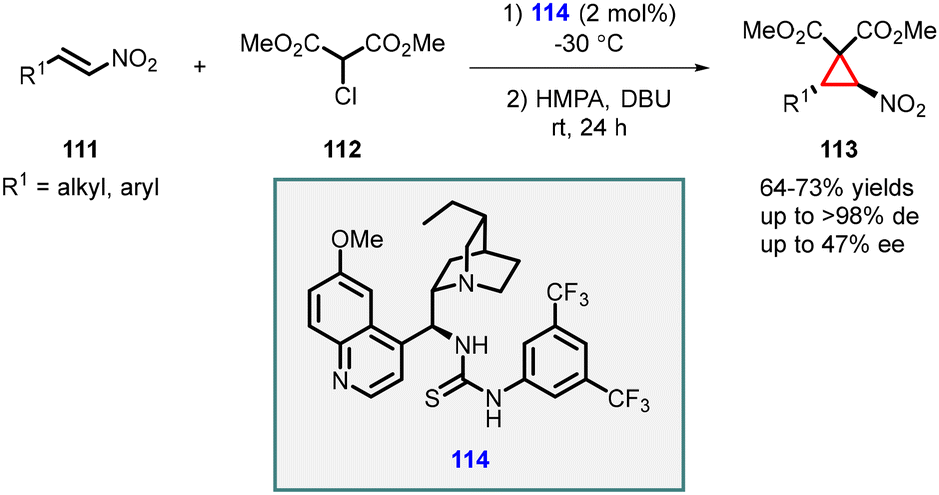 | ||
| Scheme 22 Chiral thiourea catalyzed stereoselective synthesis of heavily functionalized nitrocyclopropanes. | ||
Marini and coworkers reported the synthesis of enantioenriched cyclopropanes containing vicinal tertiary and quaternary stereocenters using a MIRC reaction. The reaction involves the conjugate addition of α-substituted cyanoacetates (116) to readily accessible, less reactive β-substituted vinyl selenones (115) using a bifunctional thiourea catalyst (114). The reaction proceeds through formation of the Michael adduct (118), which undergoes intramolecular alkylation induced by a de-ethoxycarbonylation process.
Two distinct conditions were utilized for the de-ethoxycarbonylation step: (1) use of LiCl in HMPA (a Krapcho-type protocol), and (2) a more environmentally friendly strategy employing EtONa in EtOH (Scheme 23).164 As a result, the Z-cyclopropanes were obtained as exclusive isomers with yields ranging from moderate to high, accompanied by moderate to good enantioselectivity (54–74% ee).
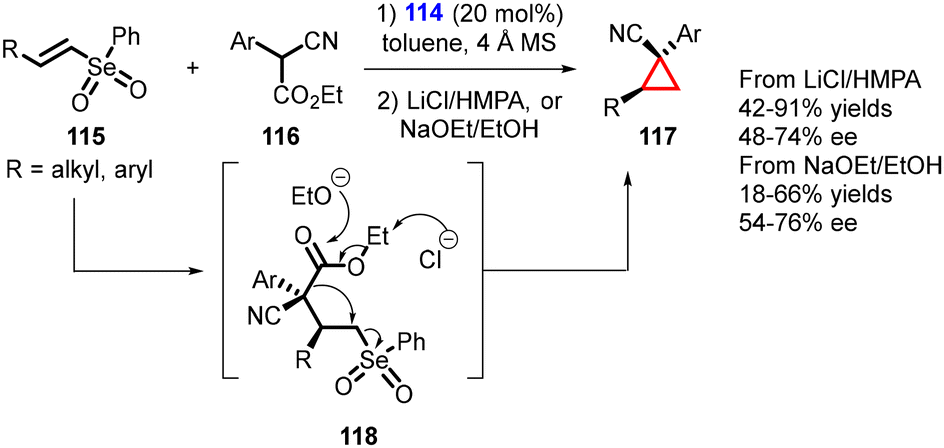 | ||
| Scheme 23 Organocatalyzed MIRC cyclopropanation reaction of β-substituted vinyl selenones. | ||
In 2011, Bencivenni and Bartoli described a new variant for the asymmetric synthesis of spirocyclopropyl oxindoles utilizing a MIRC strategy. The reaction involved treatment of 3-alkenyl oxindoles (119) with bromonitromethane (120) in presence of cinchona alkaloid-derived bifunctional thiourea catalyst (114). The reaction resulted in the formation of spiro nitrocyclopropane oxindoles (spiro 3,3′-cyclopropyl oxindoles) (121) with moderate to high diastereoselectivity (>5:1) and excellent enantioselectivity (>99% ee) (Scheme 24).165
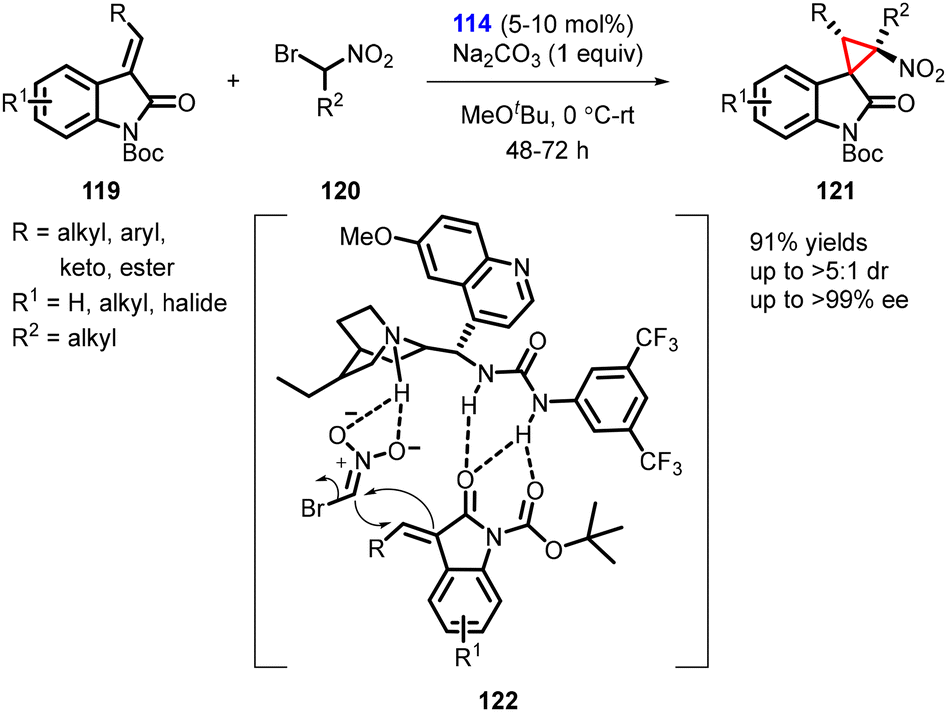 | ||
| Scheme 24 MIRC cyclopropanation of 3-alkenyl oxindoles with bromonitromethane. | ||
Catalyst 114 played a dual role by activating both the electrophilic oxindole and the Michael donor bromonitromethane. The activation occurred through hydrogen-bond interactions controlled by the thiourea moiety of the catalyst, which facilitated the formation of cyclopropane from the Si-face of the double bond.
Lattanzi and coworkers have developed a direct and efficient method to synthesize activated cyclopropanes containing homologated carbonyl groups. The method involves K2CO3-promoted tandem MIRC reaction between γ-hydroxyenone derived diphenylphosphinates (123) and 1,3-indandione (124). In preliminary studies, it was discovered that the cinchona alkaloids derived thiourea 114 served as a suitable catalyst for the development of an asymmetric version of this process (Scheme 25).166
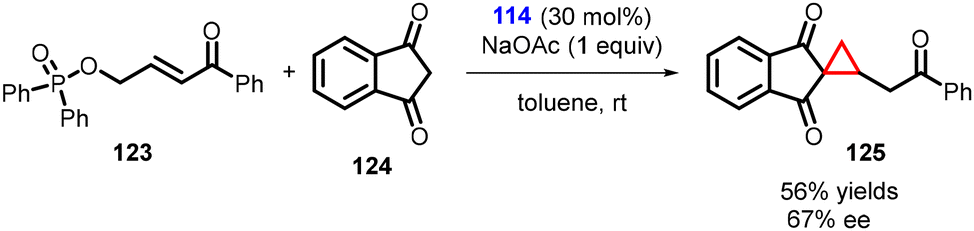 | ||
| Scheme 25 Cinchona alkaloids derived thiourea catalyzed MIRC cyclopropanation containing homologated carbonyl groups. | ||
Malkov and coworkers utilized Michael addition strategy for the synthesis of spirocyclopropanes containing two adjacent quaternary carbon centers. They employed α-chloro-β-dicarbonyl derivatives (127) in combination with alkylideneoxindoles (126) to generate the desired spirocyclopropanes (128). These products featured valuable ketone or ester groups and exhibited excellent diastereomeric ratios (up to 99:1) and enantioselectivity (80–98% ee) (Scheme 26).167
 | ||
| Scheme 26 Michael addition of α-chloro-β-dicarbonyl derivatives to alkylideneoxindoles. | ||
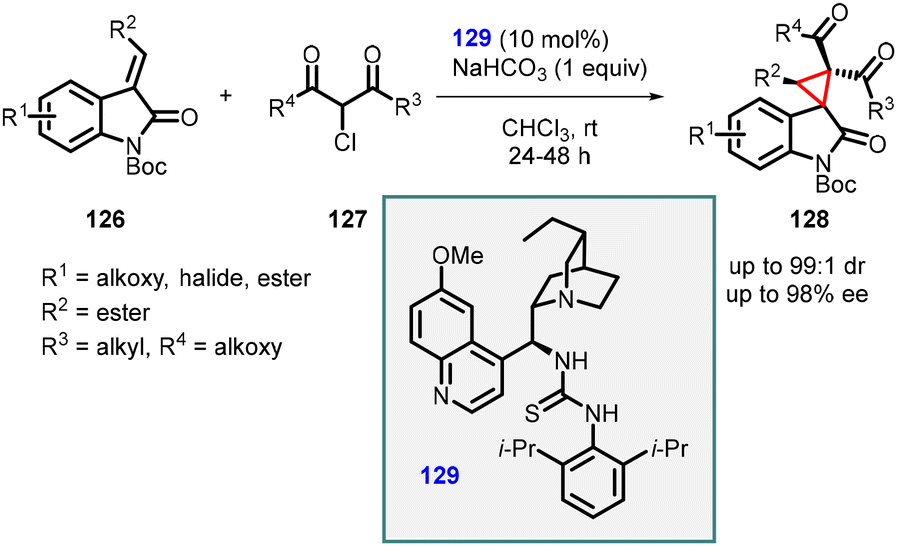
Malkov group utilized 129 as a catalyst for the MIRC reaction, which is similar to the one employed by Bencivenni and Bartoli, thereby facilitating the formation of cyclopropane from Si-facial. The presence of 2′,5′-diisopropyl substitution on the aromatic ring of chiral catalyst 129 proved beneficial in restricting the rotation of the C–N bond, thereby enhancing the reaction's diastereoselectivity.
Lu and coworkers developed a MIRC strategy for the asymmetric synthesis of spirocyclopropyl oxindoles. The approach involves direct coupling of dinucleophilic oxindoles with a suitable dielectrophile, such as α-halogenated nitroolefins (Scheme 27).168 The N-Boc oxindole 130 (serving as the dinucleophile C1 synthon) was reacted with bromonitroolefins 131 (acting as the dielectrophilic C2 synthon) in the presence of a novel thiourea catalyst 134 (10 mol%). Ammonium carbonate was used as an HBr scavenger to mitigate its generation during the reaction. This reaction resulted in the formation of two diastereoisomers, 133 and 132, in good yields, moderate to excellent diastereoselectivity, and excellent enantioselectivity (90–97% ee).
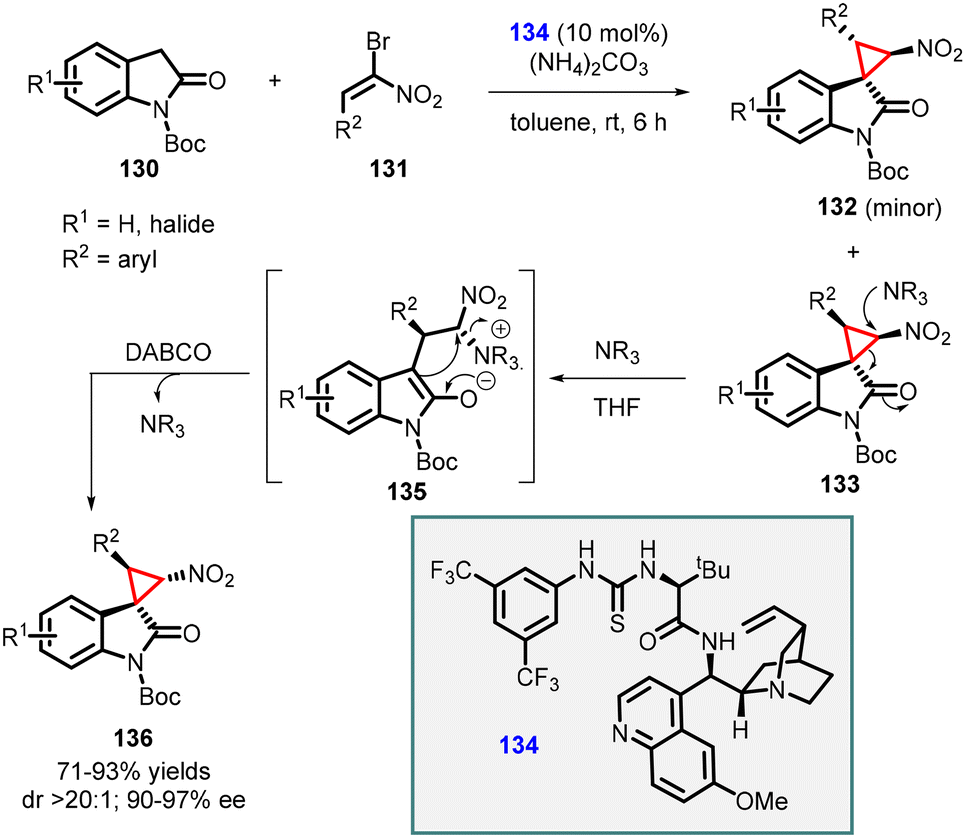 | ||
| Scheme 27 Chiral amino acid-derived multifunctional thiourea catalyzed cyclopropanation of oxindoles with nitroolefins. | ||
In addition, a diastereodivergent approach was developed by subjecting compound 133 to DABCO which acts as a nucleophile. Upon the addition of DABCO, a conventional cyclopropane ring-opening and closing mechanism was observed. Interestingly, the presence of the nucleophile did not affect the other diastereomer 132, thereby influencing the diastereoselectivity of the reaction.
Lin and coworkers reported a cascade reaction which involves Michael addition and intramolecular alkylation steps between 2-arylidene-1,3-indandiones (137) and 1-bromo nitroalkanes (120). The initial step of the cascade reaction involves Michael addition of nitro alkyl to the indandiones (137), followed by alkylation with the bromo group to the enolate resulting in spironitrocyclopropanes (138) formation (Scheme 28).169 The reaction was efficiently catalyzed by a bifunctional tertiary amine/thiourea catalyst (139) derived from cinchona alkaloids. The reaction yielded good yields (63–88%) and with excellent stereoselectivities (up to 19:1 dr and 98% ee).
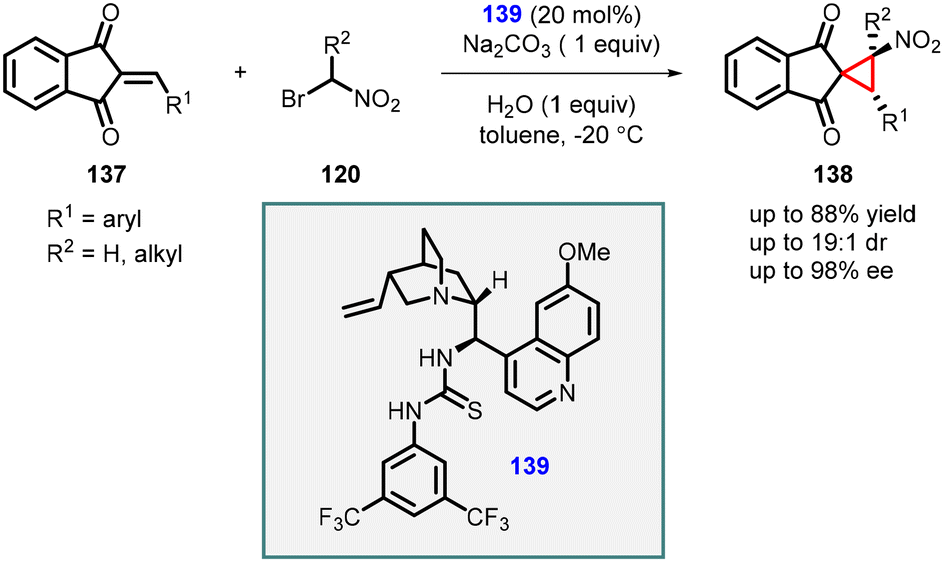 | ||
| Scheme 28 MIRC cyclopropanation of 2-arylidene-1,3-indandiones with 1-bromo nitroalkanes. | ||
Kanger and coworkers reported the synthesis of spirocyclopropyl oxindoles using MIRC reaction. In this method, N-Boc-3-chlorooxindole 140, unsaturated aromatic 1,4-diketone 141 and chiral thiourea catalyst 139 was used in the synthesis of spirocyclopropanes (142) (Scheme 29).170 The chlorooxindole (140) underwent conjugate addition with the unsaturated 1,4-diketone (141), followed by an intramolecular nucleophilic chloride displacement to yield cyclopropane (142). The mechanism is similar to the one proposed by Malkov and coworkers.
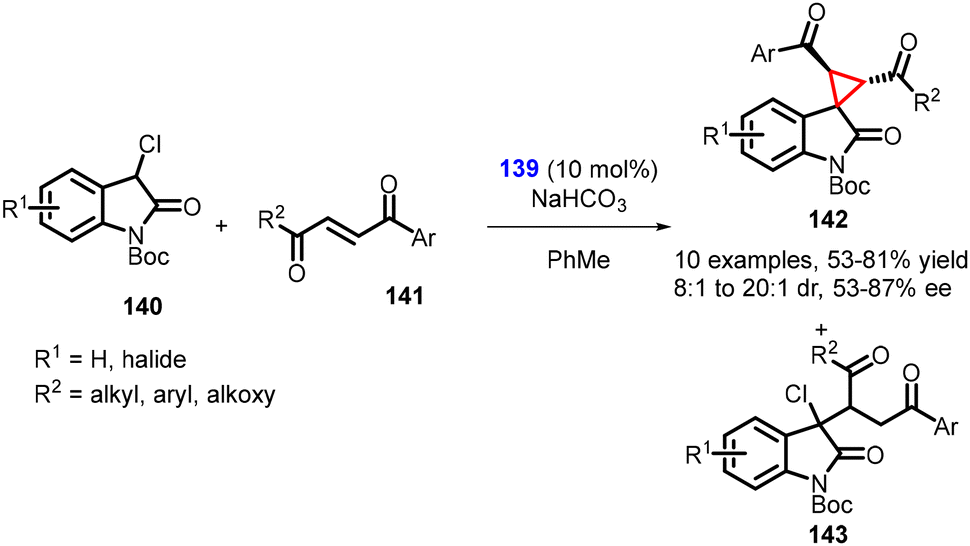 | ||
| Scheme 29 MIRC cyclopropanation of N-Boc-3-chlorooxindole with unsaturated aromatic 1,4-diketone. | ||
It is worth noting that the nucleophilic substitution reaction of chlorooxindole (140) to unsymmetrical, unsaturated 1,4-diketones (two reactive electrophilic centers) proceeded with regioselectivity. However, a small amount of uncyclized Michael adduct (143) was produced in all reactions. The reaction resulted in the desired cyclopropanes (142) in good yields (58–81%), diastereoselectivity (dr = 8:1 to 20:1), and enantioselectivity (87% ee).
In 2022 by Ouyang and Zheng reported stereoselective spirooxindoles synthesis by utilizing α-cyano chalcones (145) as the Michael acceptors, 3-chlorooxindole 144 as the nucleophile, and (147) derived from multifunctional quinine-based aminoindanol-thiourea as catalyst (Scheme 30).171 The reaction yielded the desired product (146) with high yields (up to 92%), diastereoselectivity (up to 12:1), and enantioselectivity (93% ee). Density functional theory (DFT) calculations provided insights into the reaction mechanism, revealing the significance of potential intramolecular hydrogen bonds (148) within the chiral catalyst for effective stereocontrol.
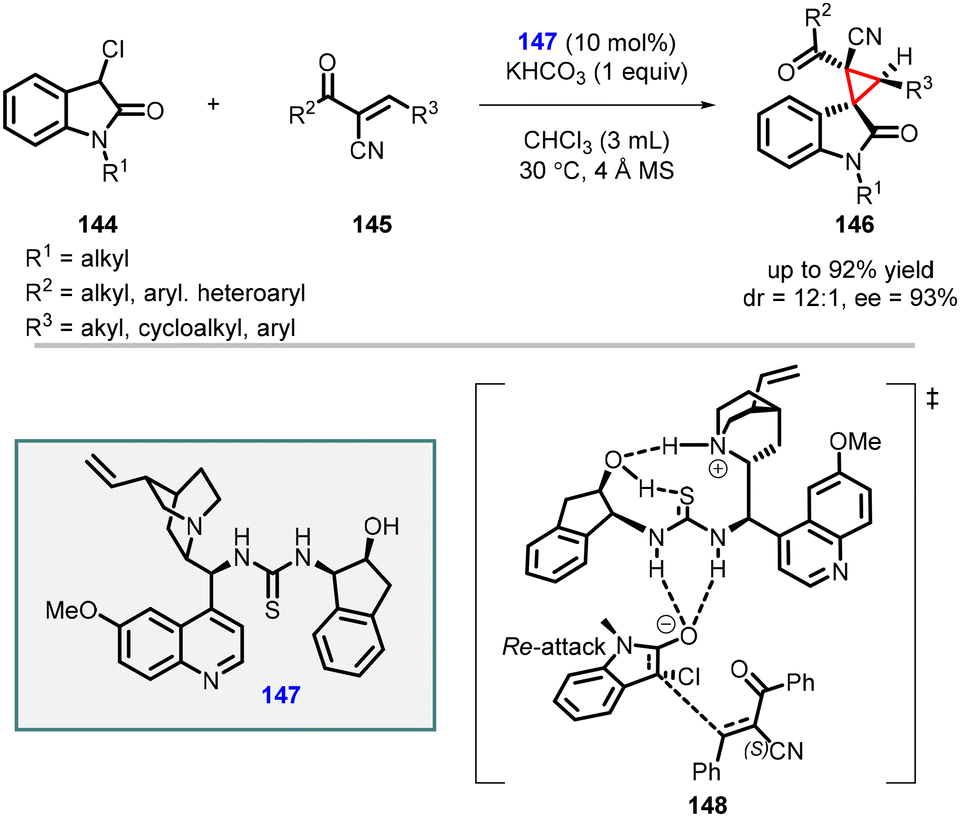 | ||
| Scheme 30 MIRC cyclopropanation of chlorooxindole with α-cyano chalcones. | ||
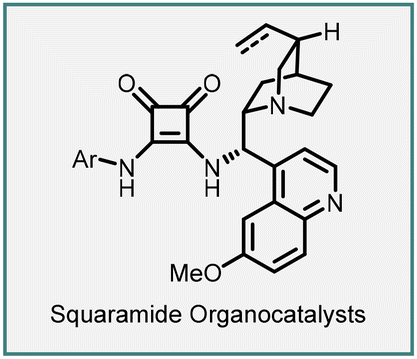
In 2005, Du and coworkers reported the synthesis of spiro-pyrazolone–cyclopropane oxindoles (151) through a tandem Michael addition/alkylation cascade reaction (Scheme 31).172 The reaction involves 3-chlorooxindoles (150) and arylidenepyrazolones (149) in the presence of DIPEA to yield the product (151) in excellent yield (75–99%) and diastereoselectivity (5:1 to >25:1).
 | ||
| Scheme 31 Michael/alkylation cascade cyclopropanation of chlorooxindole with arylidenepyrazolones. | ||
The proposed reaction mechanism involves the initial formation of an enolate by deprotonation of the halo oxindole derivative using DIPEA. The enolate then underwent a Michael addition on the β-carbon of the Michael acceptor (arylidenepyrazolone) through a Re-face nucleophilic approach. Subsequently, an intramolecular nucleophilic displacement of the chlorine atom occurred in the presence of the base, resulting in the formation of the spiro-pyrazolone–cyclopropane-oxindole (151) product.
An enantioselective variant was demonstrated by employing a bifunctional squaramide catalyst (152). The reaction involves treating 149 and 150 with 5 mol% of the catalyst 152 and 100 mol% K2CO3, resulting in the cyclopropanes (151) bearing three contiguous stereocenters, including two vicinal quaternary centers. The reaction gave modest diastereoselectivity (87:13) and enantioselectivity (up to 74% ee), but the yields obtained were excellent ranging from 91% to 99%.
In a related study Du and coworkers, an asymmetric [2 + 1] cycloaddition reaction between 3-chlorooxindoles (150) and 5-alkenylthiazolones (155) was investigated (Scheme 32).173 This reaction was catalyzed by hydroquinine squaramide 157, with Na2CO3 as the base in acetonitrile at −10 °C. These optimized reaction conditions resulted in higher yields of the desired products, reaching up to 99%. Moreover, the reaction exhibited excellent diastereoselectivity (>99:1) and the enantioselectivity was also significantly improved, with enantiomeric excess (ee) values reaching up to 99%.
 | ||
| Scheme 32 Michael/alkylation cascade cyclopropanation of chlorooxindole with 5-alkenylthiazolones. | ||
Du and coworkers extended the MIRC reaction for the asymmetric synthesis of spironitro-chromanonecyclopropanes (159) using bromonitroalkane 51 as nucleophile. The reaction proceeded through a Michael addition/alkylation cascade cyclization reaction, which involves unsaturated 4-chromanones (158), bromonitroalkane 51, and chiral squaramide (160) as catalyst (Scheme 33).174 The reaction resulted in the formation of highly substituted cyclopropanes with three contiguous stereocenters, with moderate to good yields (54–90%), excellent diastereoselectivity (98:2), and exceptional enantioselectivity, with an enantiomeric excess (ee) exceeding 99%.
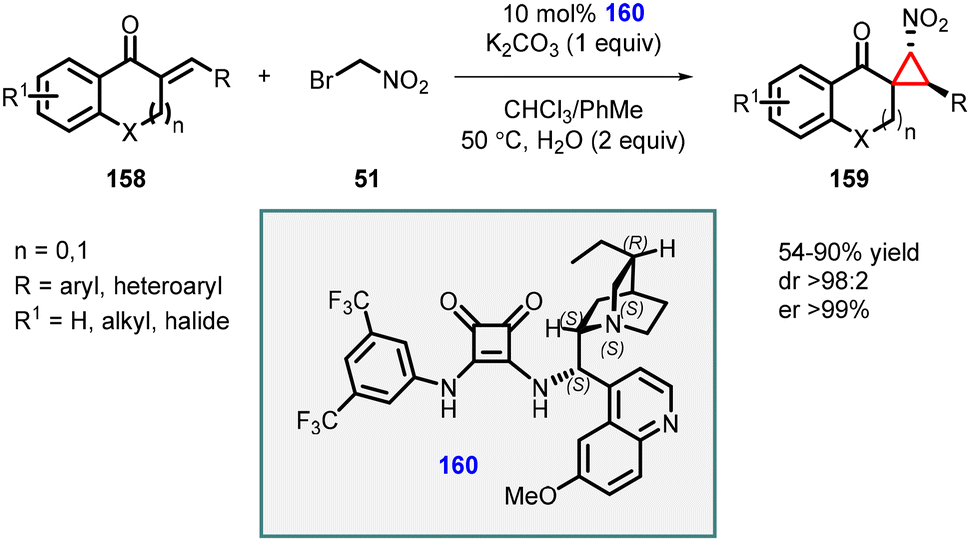 | ||
| Scheme 33 Chiral squaramide catalyzed MIRC cyclopropanation cascade of unsaturated 4-chromanones with bromonitroalkane. | ||
Yuan and coworkers reported an asymmetric synthesis of spirooxindole cyclopropa[c]coumarin compounds (163) achieved using a bifunctional squaramide catalyst (157) through a [2 + 1] Michael/intramolecular cyclization cascade reaction (Scheme 34).175 Notably, to confirm the formation of spirocyclic compounds, the authors conducted dynamic high-resolution mass spectrometry studies by treating 3-acylcoumarins (161) and 3-halooxindoles (162) with catalyst 157.
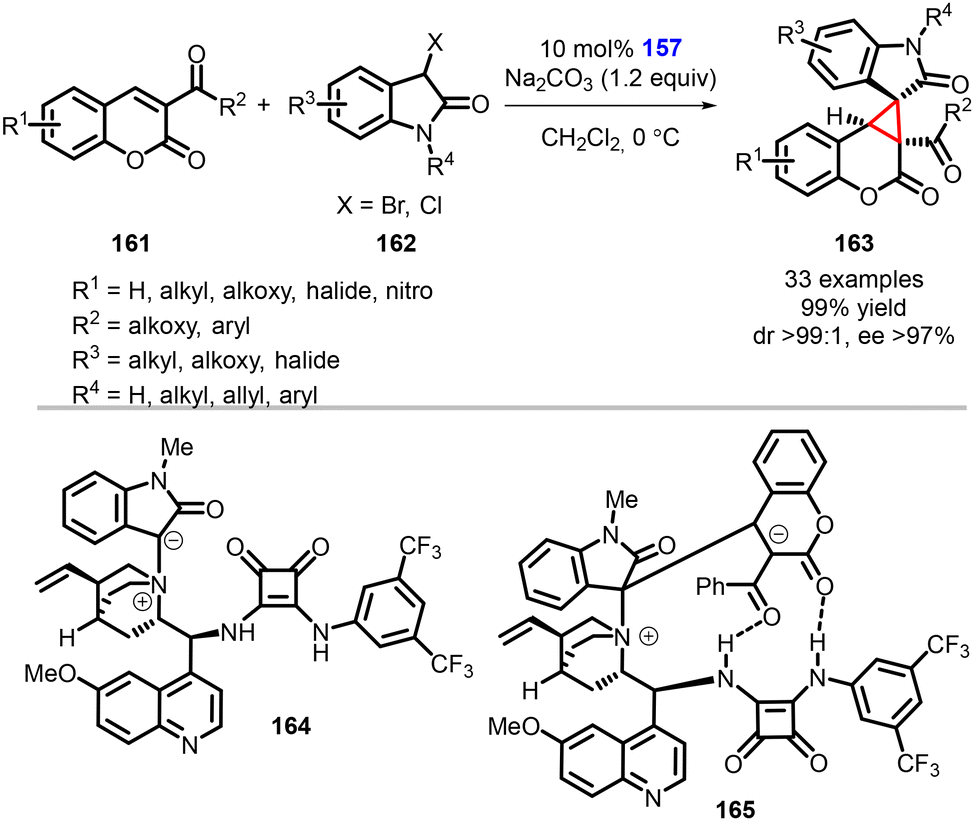 | ||
| Scheme 34 Chiral squaramide catalyzed MIRC cyclopropanation of 3-acylcoumarins with 3-halooxindoles. | ||
During the model reaction under standard conditions, it was observed that the reaction proceeded smoothly over a duration of 30 minutes. The dynamic high-resolution mass spectrometry analysis detected the presence of the ammonium ylide or ammonium enolate 164, as well as the zwitterionic intermediate 165. These observations provided further evidence and mechanistic insight for the formation of the desired spirocyclic compounds.
Lei and coworkers reported a similar approach involving the [2 + 1] annulation reaction with 2-arylidene-1,3-indanediones (167), 3-bromooxindoles (166), and a bifunctional squaramide catalyst (169). This reaction resulted in the formation of a series of novel bi-spiro[indanedione–oxindole–cyclopropane] (168) compounds through a tandem Michael addition/alkylation cascade reaction (Scheme 35).176 Like the protocol described by Yuan et al., high-resolution mass spectrometry (HRMS) studies were conducted to elucidate the mechanism of this tandem [2 + 1] annulation reaction, revealing the involvement of an in situ generated ammonium ylide followed by an intramolecular cyclization process.
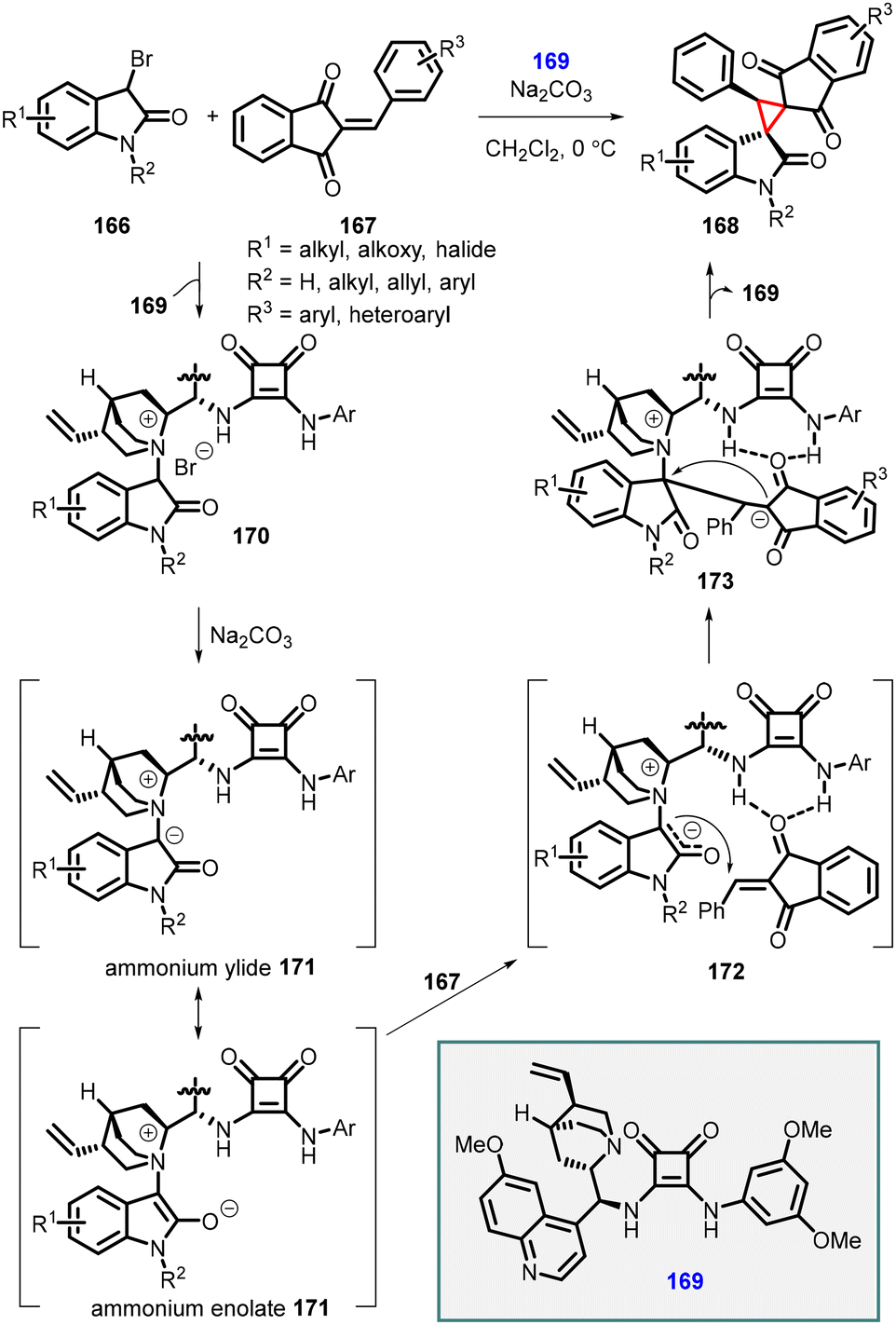 | ||
| Scheme 35 Chiral squaramide catalyzed MIRC cyclopropanation of 2-arylidene-1,3-indanediones with 3-bromooxindoles. | ||
The proposed mechanism for [2 + 1] annulation is as follows: step 1: substrate 166 reacts with catalyst (169), leading to the formation of ammonium salt 170. Step 2: Na2CO3 deprotonates ammonium salt 170, generating a nucleophilic ammonium ylide or enolate intermediate 171. In 172, the double hydrogen-bonding interaction involving the N–H in catalyst 164 facilitates a Re-face Michael addition of the 2-phenylidene-1,3-indanedione 167, resulting in the formation of the zwitterionic intermediate 173. Step 3: the intramolecular alkylation process occurs, with the Re-face attack at the C3-position of the oxindole moiety, leading to the formation of the desired chiral product 168 and concomitant release of catalyst 169.
3.1.4.1 Reactions of unsaturated aldehydes.
(a) The addition of α-halocarbonyl compounds to conjugated aldehydes (enals). General reaction mechanism: the halocarbonyl compounds, specifically Cl/Br-malonates, exhibit intriguing ambiphilic characteristics in their reactivity. They can act as both nucleophiles in Michael additions and electrophiles in alkylations (Scheme 36). This ambiphilicity is fundamental to understanding their mode of reactivity in MIRC reactions. The prolinol 175 activates the enal 174, by generating the iminium ion 176. The bulky TMS-protected diphenyl ether unit effectively shields the Si face of the iminium ion. As a result, the halomalonate attacks the Re face of the iminium ion, leading to the formation of an enamine intermediate. This enamine then undergoes intramolecular alkylation 178, resulting in formation of the desired cyclopropane product 179. Finally, the catalyst is regenerated through hydrolysis.
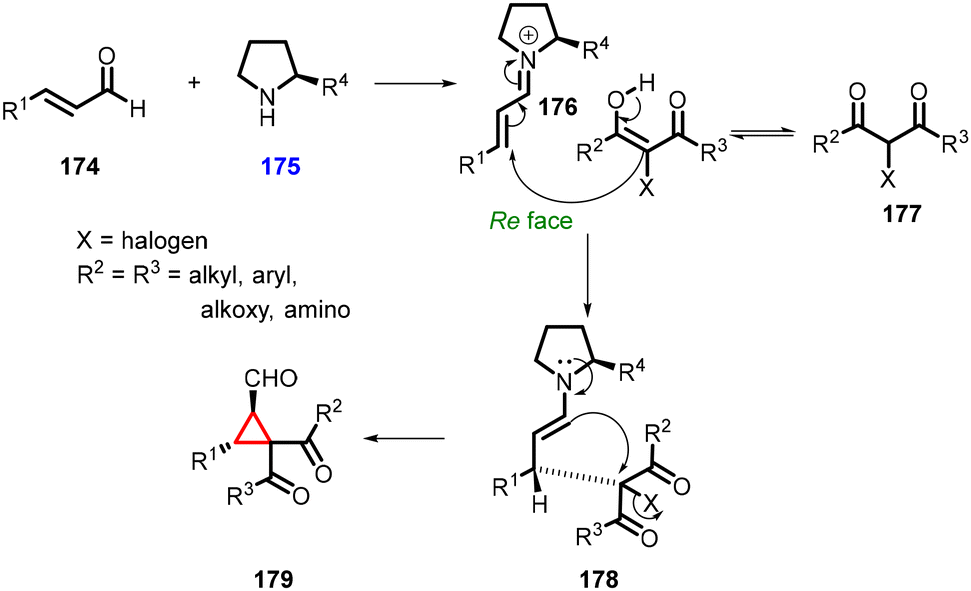 | ||
| Scheme 36 Proposed mechanism for the cyclopropanation of enals with halocarbonyl compounds. | ||
Córdova and coworkers made a significant discovery involving the TMS-protected prolinol (181 and 182) catalyzed MIRC reaction for synthesizing chemo- and enantioenriched 2-formylcyclopropanes (180) (Scheme 37).177,178 The organocatalytic cyclopropanation of α,β-unsaturated aldehyde (174) was performed with bromomalonate (64) in CHCl3 at room temperature, resulting in good yields (60–80%), excellent diastereoselectivity (ranging from 9:1 to >25:1), and enantioselectivity (93–99% ee). The use of triethylamine was crucial for achieving optimal reactivity in this process.
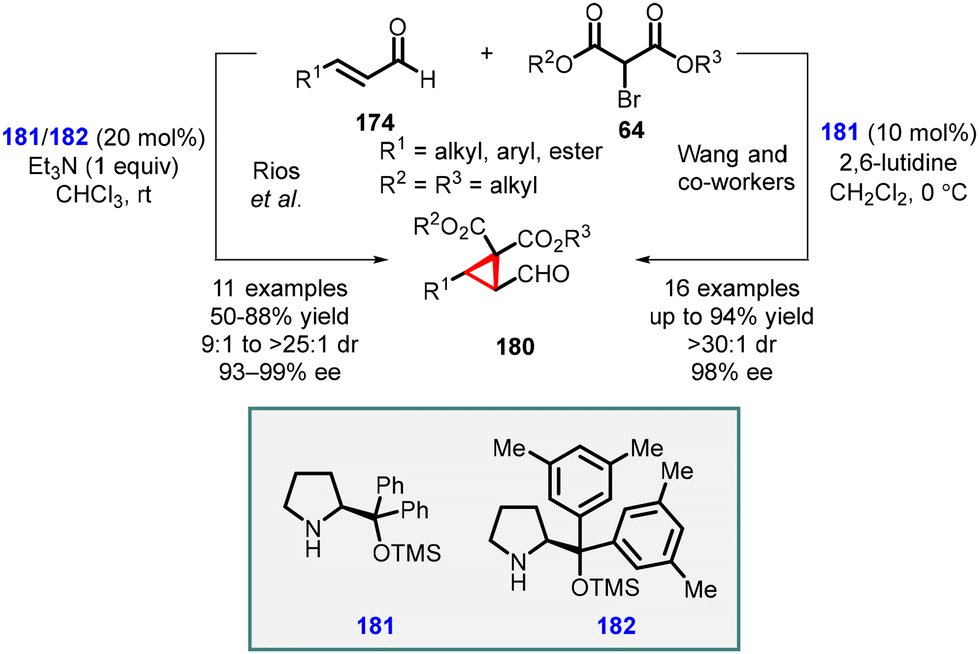 | ||
| Scheme 37 Enantioselective MIRC cyclopropanation of enals with bromomalonates. | ||
Wang and coworkers also reported MIRC reaction for synthesizing cyclopropanes, utilizing bromomalonates and enals. By employing catalyst (181) and 2,6-lutidine as a base, they successfully obtained highly functionalized chiral cyclopropanes (180) with good enantioselectivities (up to 98% ee).179
Rois and coworkers successfully utilized 2-bromo-3-keto esters (183) as nucleophiles for cyclopropanation (184). The reaction yielded excellent levels of chemo-, diastereo-, and enantioselectivity by using α,β-unsaturated aldehydes as substrates (174) (Scheme 38).180 The cyclopropanes were obtained in high yields, excellent diastereoselectivity (>25:1) and enantioselectivity (99% ee). Interestingly, the studies revealed a predominant formation of the diastereomer where the ester group is positioned cis to the formyl group. This observation provides valuable insights into the stereochemical outcome of the cyclopropanation reaction.
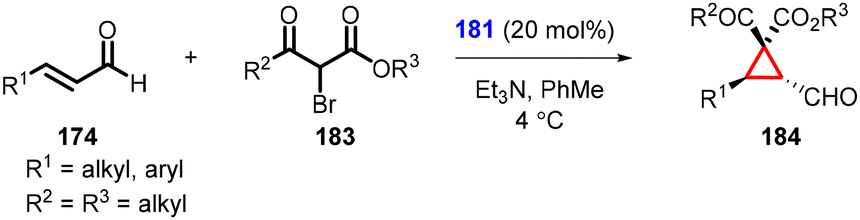 | ||
| Scheme 38 Enantioselective MIRC cyclopropanation of enals with 2-bromo-3-keto esters. | ||
Terrasson and coworkers expanded the MIRC cyclopropanation reaction for more challenging α-substituted α,β-unsaturated aldehydes (185) as substrates with bromomalonate (89) using catalyst 181 (Scheme 39).181 The protocol enables the synthesis of cyclopropanes (186) with a quaternary carbon stereocenter, in high yields and excellent enantioselectivity (up to 97% ee). However, it is worth noting that this method is limited to β-unsubstituted unsaturated systems, as the reactivity significantly decreases with β-substitution.
 | ||
| Scheme 39 Enantioselective MIRC cyclopropanation of α-substituted α,β-unsaturated aldehydes. | ||
Kim and coworkers devised a complex one-pot domino strategy involving Michael addition/alkylation and sequential aza-cyclization for the synthesis of chiral cyclopropane-fused tetrahydroquinolines (189). The transformation utilizes aldehydes (187) and α-bromomalonic esters (64). The reaction proceeds in several steps: 1. an asymmetric domino Michael/alkylation reaction occurs between N-protected ortho-aminophenyl α,β-unsaturated aldehydes (187), utilizing triethylamine and chiral catalyst 181. 2. Cyclization of dialkyl bromomalonates (64) resulting in the formation of intermediate (188). 3. The final product, cyclopropane-fused tetrahydroquinolines (189), was obtained through a DBU-induced intramolecular attack of the carbamate nitrogen onto the formyl group (Scheme 40).182
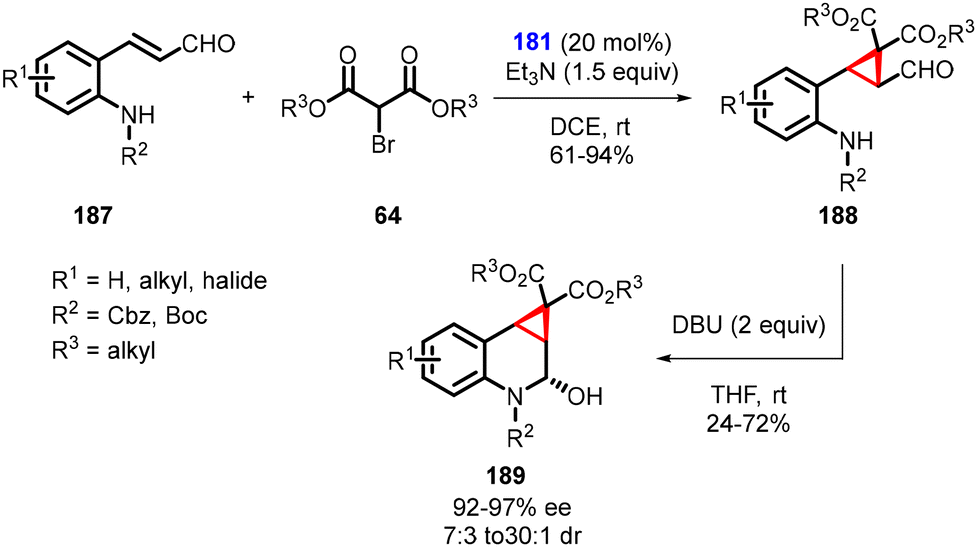 | ||
| Scheme 40 MIRC reaction in the synthesis of cyclopropane-fused tetrahydroquinolines. | ||
In 2015, Nishii and coworkers achieved a significant milestone by successfully completing the first enantioselective total synthesis of (+)-podophyllic aldehydes A (192), B (193), and C (194) belonging to the dihydronaphthalene-type lignans in overall yields of 30% (16 steps), 26% (16 steps), and 43% (8 steps) respectively. The starting materials used were α,β-unsaturated aldehyde (190) and α-bromomalonate (44).
The cyclopropane synthesis by MIRC reaction was the key step in the total synthesis of (+)-podophyllic aldehydes A, B, and C. The reaction involved treating aldehyde (190) with α-bromomalonate 44 in the presence of Jørgensen-Hayashi amine catalyst 181, 2,6-lutidine, and dichloromethane to yield cyclopropane intermediate (191) in high yield (91%) and excellent enantioselectivity (95% ee) (Scheme 41).183
 | ||
| Scheme 41 MIRC cyclopropanation reaction in total synthesis of podophyllic aldehydes. | ||
Yang and coworkers made a recent breakthrough by introducing a small modification to the aldehyde used by Nishii et al. By using the aldehyde (E)-3-((S)-1-[(R)-1-phenylethyl)aziridin-2-yl]acrylaldehyde (195), they synthesized enantioenriched cyclopropylaziridine (196) using (181) as organocatalyst and diethyl bromomalonate 89 as nucleophile in good yield (68%) and excellent diastereoselectivity (>30:1) (Scheme 42).184 This interesting design represents a significant advancement in the field of stereoselective synthesis.
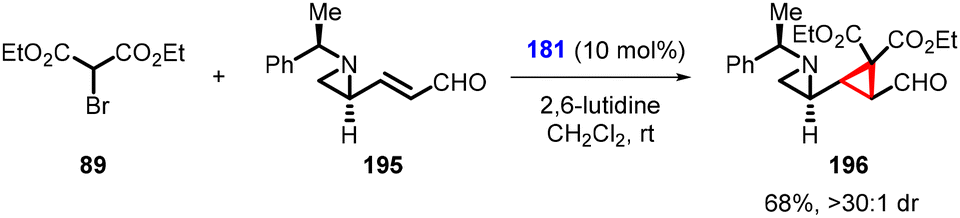 | ||
| Scheme 42 Stereoselective synthesis of cyclopropylaziridines. | ||
In 2013, Martínez and coworkers reported a reaction involving a novel catalyst derived from chiral diarylprolinols (199). This new family of organocatalysts exhibited enhanced capabilities in catalyzing the enantioselective MIRC cyclopropanation of α,β-unsaturated aldehydes (197) with diethyl bromomalonate (89) in water.
The catalyst was specifically designed with the intention of incorporating a tertiary amino group at the 4-position of the pyrrolidine scaffold. This feature provides a basic site that can effectively deprotonate the pronucleophile. Additionally, a bulky diaryltrialkylsilyloxymethyl group was strategically placed at the 2-position to control the geometry of the iminium ion and introduce the necessary steric bias for stereoinduction.
Using catalyst 199, Martínez et al., successfully synthesized cyclopropanes (198) with remarkable levels of diastereoselectivity (>19:1) and enantioselectivity (>99% ee) (Scheme 43).185
 | ||
| Scheme 43 Asymmetric MIRC cyclopropanation using novel organocatalyst which acts as an external base. | ||
Pericàs and coworkers reported an organocatalytic enantioselective continuous-flow cyclopropanation reaction using α,β-unsaturated aldehydes. They introduced a solid-supported diarylprolinol catalyst (200) for this purpose. A series of cyclopropanes (198) were successfully synthesized with moderate yields, exceptional diastereoselectivity (>95:5), and enantioselectivity of up to 96% (Scheme 44).186
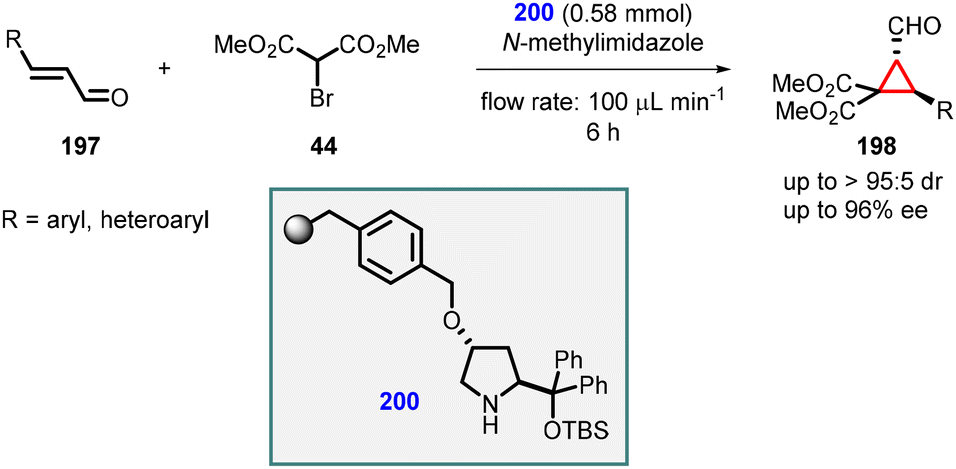 | ||
| Scheme 44 Enantioselective MIRC cyclopropanation using flow chemistry. | ||
Phillips and Barros developed a noteworthy domino Michael addition/intramolecular alkylation reaction using α,β-unsaturated aldehyde (202) and α-bromophosphonoacetates (201) (Scheme 45).187 This reaction enables the synthesis of α-cyclopropylphosphonates (203, 204) using organocatalyst 181 in high yields, good diastereoselectivity (dr = 68:32 to 83:17) and high enantioselectivity (>99% ee). However, the reaction was limited to a narrow class of substrates.
 | ||
| Scheme 45 MIRC cyclopropanation of α,β-unsaturated aldehydes and α-bromophosphonoacetates. | ||
In 2021, Ye and coworkers employed chloroacetophenone as a nucleophile in the enantioselective cyclopropanation reaction with α,β-unsaturated aldehydes using prolinol organocatalyst 181 (Scheme 46).188 The cyclopropanes (206) were successfully synthesized with excellent yields (up to 85%), exceptional diastereoselectivity (>30:1), and enantioselectivity (96% ee).
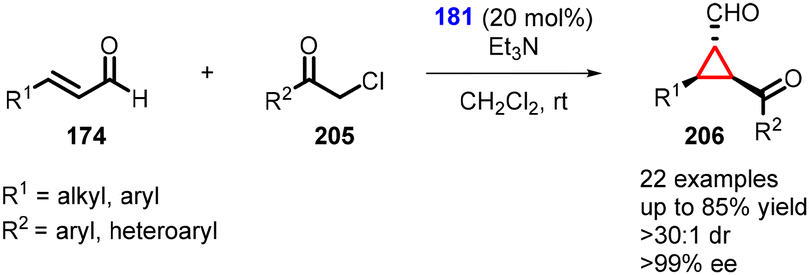 | ||
| Scheme 46 MIRC cyclopropanation of α,β-unsaturated aldehydes and chloroacetophenone. | ||
(b) The addition of α-halonitro compounds to conjugated aldehydes. In 2008, Cordova and coworkers introduced a novel method for the nitrocyclopropanation of α,β-unsaturated aldehydes (197) using bromonitromethane (51) as a nucleophile and Jørgensen-Hayashi amine (181) as catalyst. The reaction furnished cyclopropanes (207) and (208) with moderate yields (29–63%), low diastereoselectivity (3
:1) and excellent enantioselectivity (91–99% ee). (Scheme 47).189
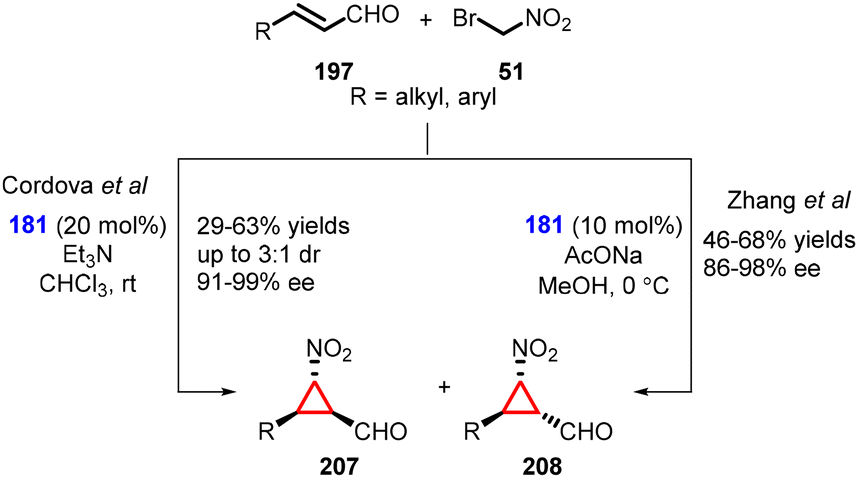 | ||
| Scheme 47 MIRC cyclopropanation of α,β-unsaturated aldehydes with bromonitromethane. | ||
Zhang and coworkers reported a similar transformation for the construction of cyclopropanes using MIRC reaction. The only difference was reaction conditions, AcONa was used as base and MeOH as solvent (Scheme 47).190
In 2015, Risi and coworkers reported an unprecedented one-pot four-step organocatalytic process for the synthesis of bicyclic nitrocyclopropane 6-nitro-3-thiabicyclo[3,1,0]hexane-1-carbaldehyde (211). The reaction involved the use of bromonitromethane as nucleophile. The reaction began with the unsaturated aldehyde (197) reacting with 1,4-dithiane-2,5-diol 209 in the presence of catalyst (181) (20 mol%) and benzoic acid (10 mol%) as additive in CH2Cl2 at 40 °C for 2 h, under an inert atmosphere. This step involved a domino sulfa-Michael-addition/aldol condensation reaction. Subsequently, a mixture of bromonitromethane and triethylamine was added, leading to a domino Michael-addition/α-alkylation reaction which yielded the bicyclic nitrocyclopropane (211) in moderate yields (of up to 45%) and high stereoselectivity (100:0 dr, 95:5 er) (Scheme 48).191
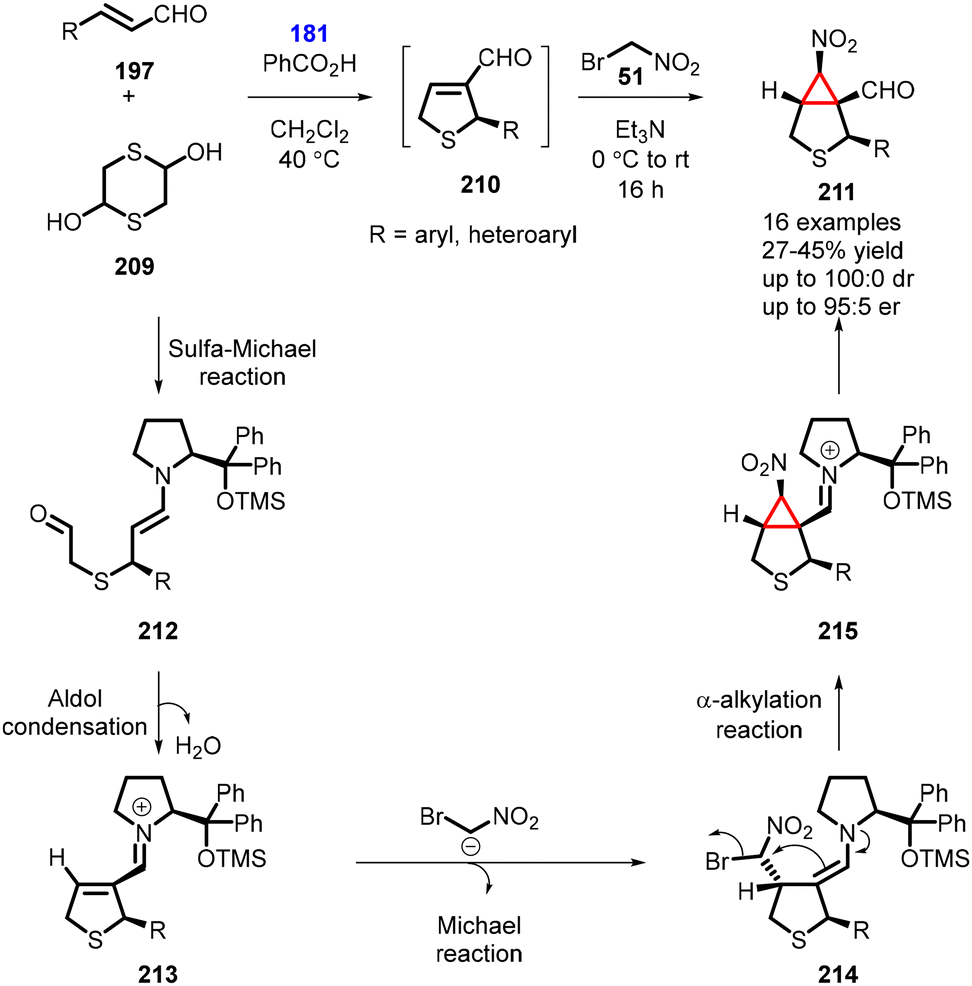 | ||
| Scheme 48 One pot four-step MIRC cyclopropanation. | ||
The plausible reaction mechanism involves the initial sulfa-Michael reaction between aldehyde (197) and in situ generated mercaptoacetaldehyde, forming the enamine intermediate (212). This intermediate then undergoes an intramolecular aldol reaction and subsequent dehydration, resulting in the formation of the iminium-ion intermediate (213). Bromonitromethane then adds to this intermediate, leading to the formation of 214. The bromine atom is subsequently displaced via an intramolecular nucleophilic substitution, generating the second cycle. The chiral organocatalyst is regenerated, ultimately leading to the formation of nitrocyclopropane 211 (Scheme 48).
(c) The addition of α-halooxindoles to conjugated aldehydes. Malkov and Kanger developed two distinct syntheses of spirocyclopropyl oxindole derivatives using 3-chlorooxoindoles (150) through a domino Michael-addition/substitution reaction (Scheme 49).192 When the reaction was applied to enals 216, the enamine catalyst 220 was utilized, resulting in the formation of desired spirocyclopropyl oxindole derivatives (217) with good to high yields (44–76%) and high stereoselectivities (>19
:1 dr; >99% ee). Conversely, when the reaction involved Michael acceptors 218, the hydroquinine-derived squaramide 160 emerged as the most effective catalyst, delivering the desired spirocyclopropyl oxindole derivatives 219 with excellent yields (71–99%) and high stereoselectivities (>30:1 dr; up to 99% ee).
 | ||
| Scheme 49 MIRC cyclopropanation reaction with chlorooxoindoles. | ||
Melchiorre and coworkers reported a fascinating reaction for the asymmetric synthesis of cyclopropane spirooxindoles (222) from 3-chlorooxindoles (150), dienals (221), and catalyst 181 through vinylogous organocatalytic cascade MIRC reaction. This reaction represents a rare example of highly regioselective and asymmetric 1,6-addition to 2,4-dienals. The presence of a bulky R2 group at the β-site of the dienals is crucial for achieving δ-regioselective 1,6-addition, which is key to the successful outcome of the reaction. Various cyclopropanes (222) were synthesized with good to excellent yields (52–91%), along with excellent stereoselectivity (6:1 to 15:1 dr and 92–99% ee) (Scheme 50).193
 | ||
| Scheme 50 MIRC cyclopropanation of 2,4-dienals and oxindoles. | ||
(d) The addition of benzyl and heterobenzyl halides to conjugated aldehydes. Rios and co-workers accomplished efficient synthesis of chiral trisubstituted diarylcyclopropane carbaldehydes (224, 225, and 226) using substituted benzyl chlorides as an ambiphilic (nucleophilic and electrophilic) reagents (223) and α,β-unsaturated aldehydes (197) as Michael acceptor (Scheme 51).194 The transformation was carried out using Jørgensen-Hayashi amine 181 as catalyst.
 | ||
| Scheme 51 MIRC cyclopropanation of benzyl chlorides and α,β-unsaturated aldehydes. | ||
Mechanistic investigations revealed that the reaction proceeded through an initial activation of the α,β-unsaturated aldehyde (197) by catalyst 181, forming the iminium species 227. The Michael addition of the benzyl anion (derived from 223 using DIPEA as base) to 227 generated the enamine intermediate 228, which underwent chloride displacement to yield the cyclopropane intermediate 229. Finally, hydrolysis led to the formation of the desired products 224, 225, and 226, while the catalyst was recovered. It is worth noting that the nucleophilic substitution could occur from both directions due to the presence of a chloride group at the achiral β position, resulting in relatively poor diastereoselectivity.
In the same year, Li and Wang reported a similar transformation by substituting the organocatalyst with chiral diphenylprolinol TBDMS ether (234) and using triethylamine as a base under mild reaction conditions (Scheme 52).195
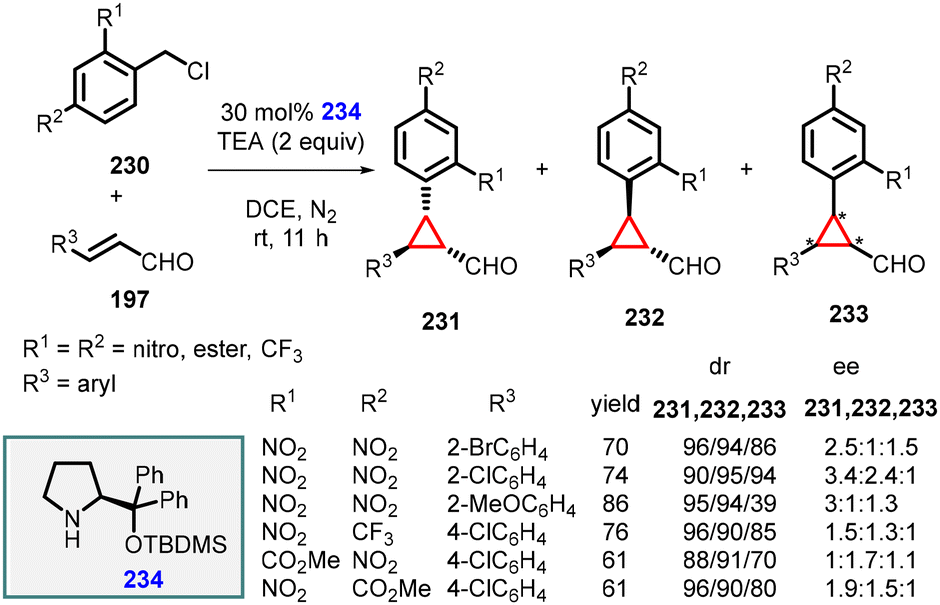 | ||
| Scheme 52 MIRC cyclopropanation of benzyl chlorides and α,β-unsaturated aldehydes using catalyst 234. | ||
Veselý and coworkers applied a similar approach for the enantioselective synthesis of 1,2,3-trisubstituted cyclopropanes using heteroaryl chloride (102) as an ambiphilic reagent. In this method, the MIRC reaction was performed between 5-(chloromethyl)-3-methyl-4-nitroisoxazole (102) and α,β-unsaturated aldehydes (235), using catalyst 239 resulting in the formation of the desired cyclopropanes (236, 237, 238) in high yields (up to 98%), moderate diastereoselectivity and excellent enantioselectivity (99% ee), and (Scheme 53).196
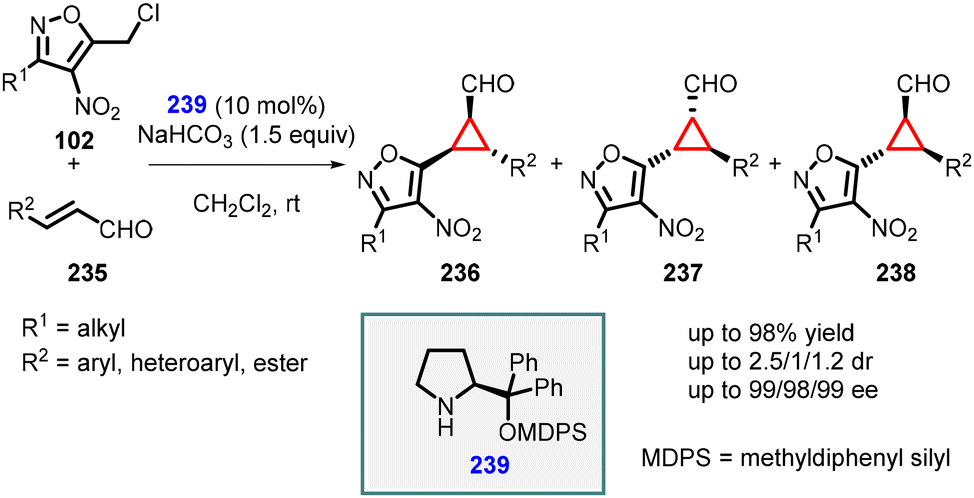 | ||
| Scheme 53 MIRC cyclopropanation of 5-(chloromethyl)-3-methyl-4-nitroisoxazole and α,β-unsaturated aldehydes. | ||
Two years later, Veselý and coworkers published another noteworthy method for synthesizing cyclopropane carbaldehydes (241) derived from BODIPY (Scheme 54).197 The approach involved a cascade reaction between readily available BODIPY derivatives (240), α,β-unsaturated aldehydes (235), and commercially available Jørgensen catalyst (242). The resulting BODIPY-derived cyclopropanes were obtained in high yields (66–98%), diastereomeric ratios ranging from 3/2 to >20/1, and high enantiomeric excess (92–99%) for the major diastereomer. The stereochemistry of the final product was investigated in detail through density functional theory (DFT) calculations of the intermediates involved in the reaction. These findings provide valuable insights into the mechanism and stereochemistry of the cascade reaction for the synthesis of BODIPY-derived cyclopropane carbaldehydes.
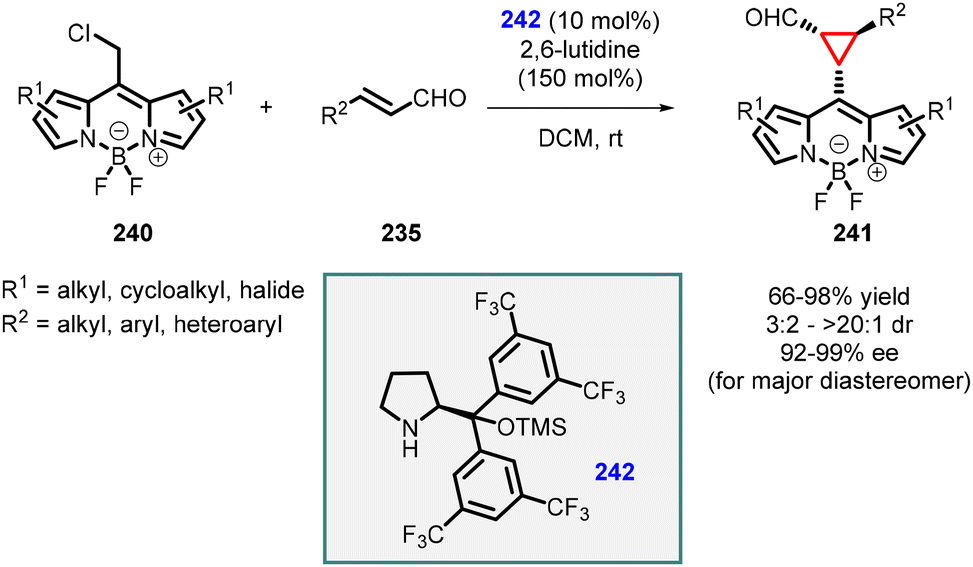 | ||
| Scheme 54 MIRC cyclization to synthesize cyclopropane carbaldehydes derived from BODIPY. | ||
(e) The addition of ylides to conjugated aldehydes. Fochi and Bernardi recently introduced a novel approach for synthesizing the 1,1a,2,7b-tetrahydrocyclopropa[c]chromene scaffold (245) through enantioselective aminocatalytic Corey–Chaykovsky-type cyclopropanation reactions with unsaturated aldehydes (243), utilizing sulfoxonium ylides (244). This method represents a convenient utilization of sulfoxonium ylides in asymmetric amino-catalysis (181). The reaction yielded several ring-fused derivatives (specifically, cyclopropane-fused chromanes) (245). The chromanol derivative (245) was subjected to derivatization through Wittig olefination, resulting in the formation of 246 in moderate yields (up to 79%), excellent diastereoselectivity (E/Z > 9
:1), and excellent enantioselectivities (up to 97% ee) (Scheme 55).198 The product obtained predominantly existed as the E-isomer, which allowed for easy isolation and accurate determination of the enantiomeric excess.
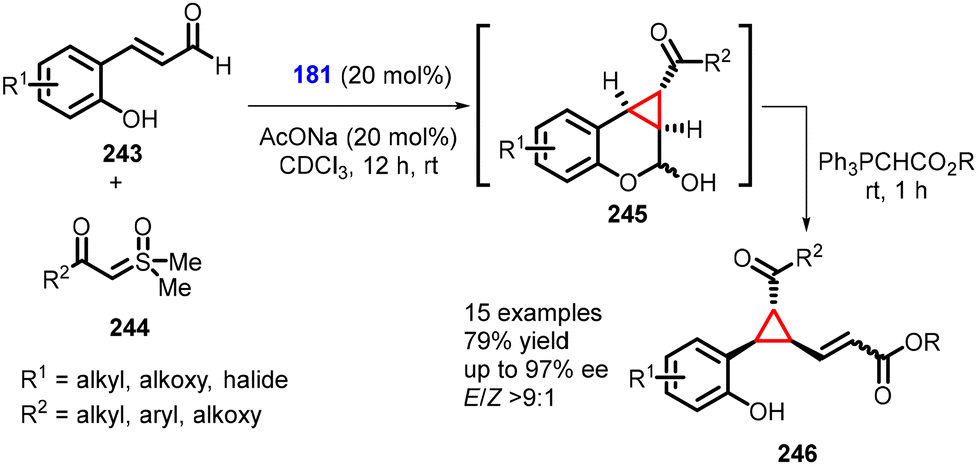 | ||
| Scheme 55 MIRC cyclopropanation of unsaturated aldehydes and stabilized sulfoxonium ylides. | ||
Zhao and Cao reported the synthesis of chiral cyclopropanes (248) utilizing α,β-unsaturated aldehydes, arsonium ylide (247) and diphenylprolinol silylether (181) as a catalyst (Scheme 56).199 The configuration of these compounds was determined by the intermediate (E)-iminium 249 and the steric approach control, which are both influenced by the bulky diarylcarbinol silyl ether moiety in the catalyst.
 | ||
| Scheme 56 MIRC cyclopropanation using arsonium ylide. | ||
Apart from enals, other unsaturated carbonyl compounds such as ketones, nitro compounds, and amides are also valuable substrates in the MIRC reaction when prolinol organocatalysts are employed.
3.1.4.2 Reactions of unsaturated ketones. Lattanzi and coworkers conducted a study in which they employed 2-arylidene-1,3-indandiones (137) as Michael acceptors, along with α-bromo-methylmalonate (44), in the presence of diarylprolinol derivative (251) as the catalyst. This innovative approach led to the synthesis of spirocyclopropanes (250) with high yields (74–96%) and moderate to good enantioselectivity (60–85%) (Scheme 57).200
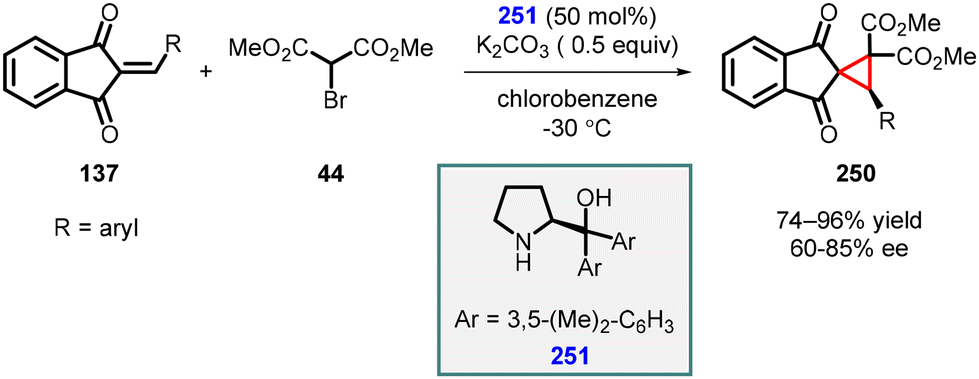 | ||
| Scheme 57 MIRC cyclopropanation of 2-arylidene-1,3-indandiones and α-bromo-methylmalonate. | ||
3.1.4.3 Reactions of unsaturated nitro compounds. Lattanzi and coworkers demonstrated MIRC reaction utilizing nitroalkenes (43) as Michael acceptor and α-bromo-methylmalonate (44) as nucleophile for the synthesis of chiral nitrocyclopropanes (45). In this study, α,α-(2-naphthyl) prolinol silyl ether 252 was used as catalyst at 30 mol% loading. The reaction exhibited complete trans-diastereoselectivity, although the enantioselectivity was relatively low (≤49% ee) (Scheme 58).201
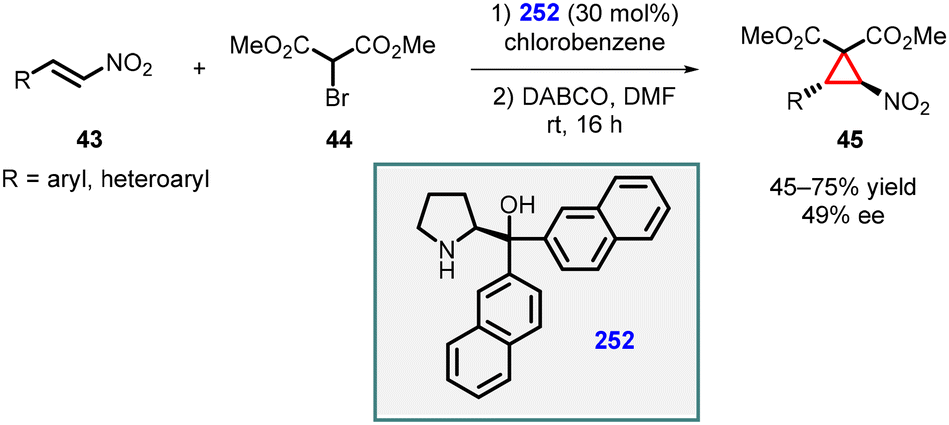 | ||
| Scheme 58 MIRC cyclopropanation of nitroalkenes using α,α-(2-naphthyl) prolinol silyl ether catalyst. | ||
Alemán and coworkers presented an interesting one-pot transformation utilizing bromonitroalkenes (253) as Michael acceptor. This transformation resulted in the formation of cyclopropanes (255), with aldehyde (254), which exhibited good yields and excellent diastereoselectivity (up to 98:2) and enantioselectivity (up to 97% ee) (Scheme 59).202 The one-pot reaction involves a two-step process: 1. an aminocatalytic reaction between aldehydes and bromonitroalkenes; and 2. an intramolecular bromo substitution catalyzed by DABCO as a base.
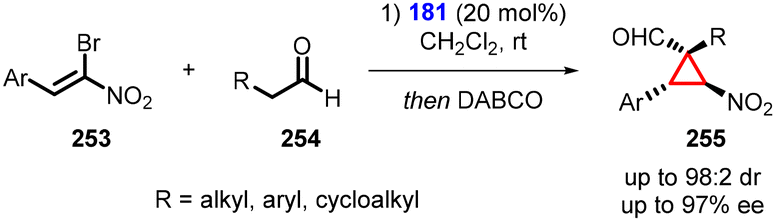 | ||
| Scheme 59 One pot MIRC cyclopropanation of nitroalkenes. | ||
3.1.4.4 Reactions of unsaturated amides. Noole and co-workers developed three straightforward and effective approaches for accessing the spiro-cyclopropane framework. Among these strategies, one involves a MIRC reaction between unsaturated aldehydes (257) and 3-chlorooxindoles (256) utilizing prolinol (220) as the preferred catalyst. This method exhibits remarkable activity, favorable selectivity, and smooth conversion within a time frame of 4–24 hours (Scheme 60). The desired spiro-cyclopropane compounds are synthesized by employing iminium catalysis with catalyst 220. These reactions proceed in high yields while demonstrating excellent diastereoselectivity (>20
:1 dr) and enantioselectivity (>99% ee).203
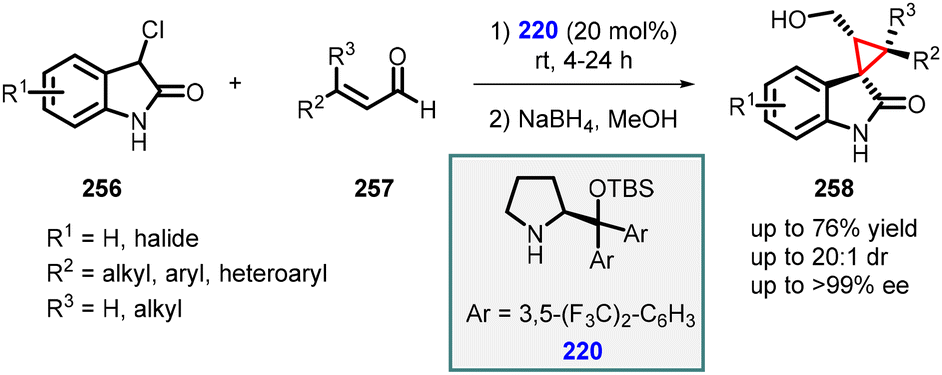 | ||
| Scheme 60 Strategies utilized in the synthesis of spiro-cyclopropane scaffold. | ||
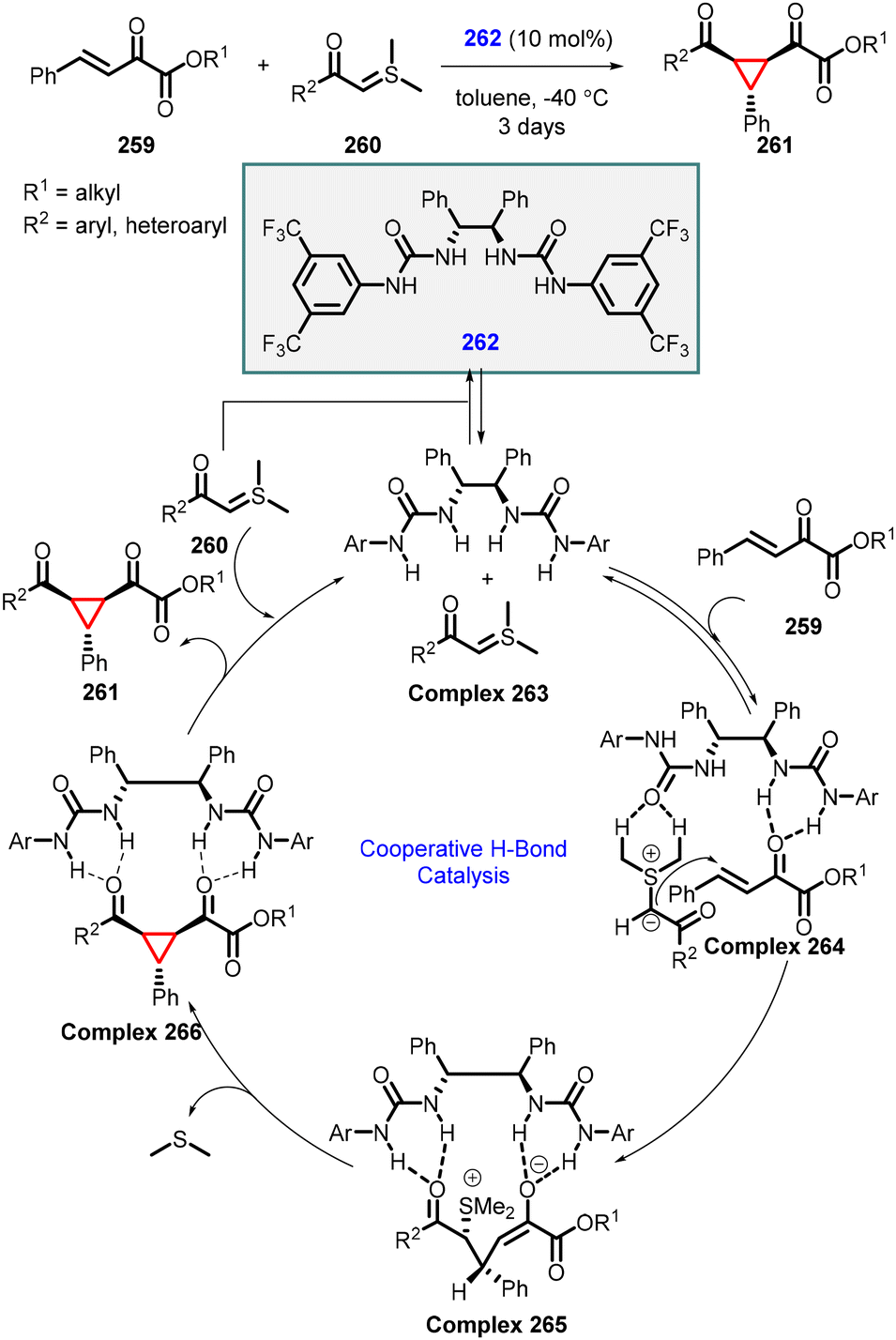
In 2011, Xiao and coworkers developed an enantioselective MIRC cyclopropanation reaction of β,γ-unsaturated α-ketoesters with sulfur ylides using a hydrogen-bonding urea catalyst (262) (Scheme 61).204 The reaction mechanism involves the formation of hydrogen bond between catalyst 262 and ylide (260), resulting in the formation of complex 263. Complex 263 then reacts with unsaturated ketoester (259) to form a ternary complex 264, where the hydrogen bonds guide the reactant into the appropriate position and induce the observed face selectivity in intermediate 265. Finally, intermediate 265 undergoes intramolecular SN2 displacement to yield the desired cyclopropane (261) while regenerating urea catalyst 262 for the subsequent catalytic cycle. A diverse array of cyclopropanes can be synthesized with moderate to good yields, and enantioselectivity (up to 80% ee).
 | ||
| Scheme 61 Urea catalyzed asymmetric MIRC cyclopropanation of β,γ-unsaturated α-ketoesters and sulfur ylides. | ||
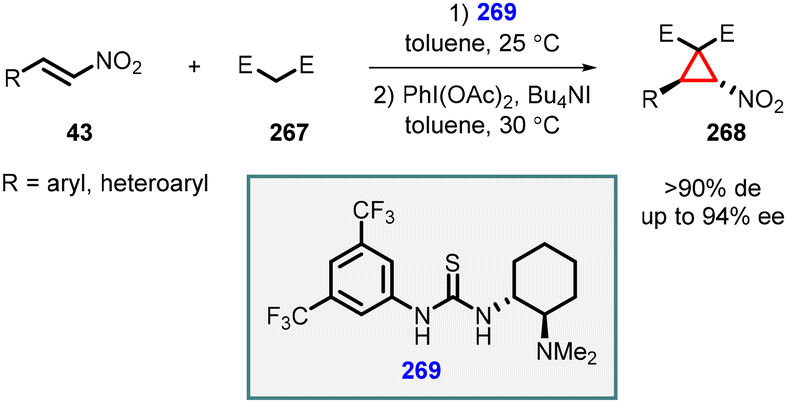
Fan and coworkers demonstrated organocatalyzed one-pot asymmetric synthesis of a range of functionalized cyclopropanes (268) through an efficient oxidative cyclopropanation of Michael adducts of nitroalkenes (43) with activated methylene compounds (267). This transformation was achieved by employing a combination of iodobenzene diacetate and tetrabutylammonium iodide. The highly efficient thiourea organocatalyst 269 was utilized for this reaction, leading to the synthesis of functionalized nitrocyclopropanes (268) in moderate to good yields, along with excellent diastereoselectivity (>90%de) and enantioselectivity (up to 94%ee) (Scheme 62).205
 | ||
| Scheme 62 Oxidative MIRC cyclopropanation of the Michael adduct of nitroalkenes with activated methylene compounds catalyzed by thiourea. | ||
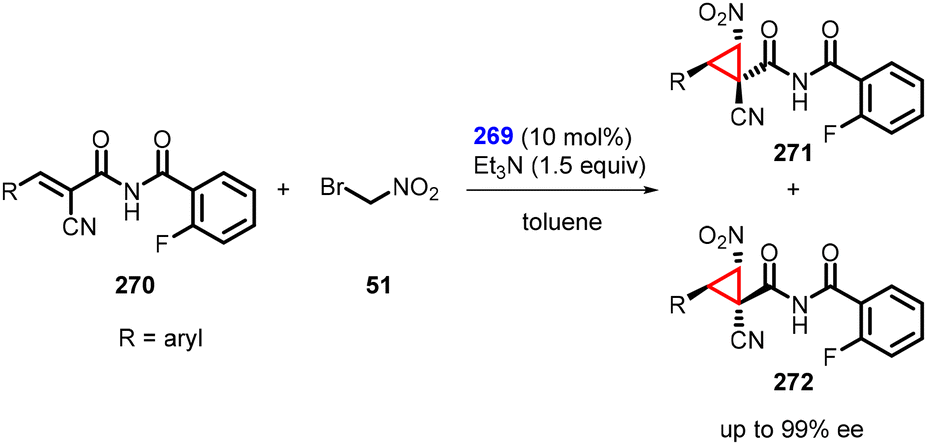
In a separate study, Inokuma and coworkers reported asymmetric MIRC reaction using bifunctional thiourea catalyst (269) to synthesize a wide range of cyclopropanes (271 and 272) from α-cyano-α,β-unsaturated imides (270) and bromonitromethane (51). The reaction gave outstanding enantioselectivity (up to 99%) (Scheme 63).206 Notably, the catalyst also demonstrated remarkable efficacy in facilitating the preparation of these α-cyanoimides through Knoevenagel condensation.
 | ||
| Scheme 63 MIRC cyclopropanation of α-cyano-α,β-unsaturated imides and bromonitromethane catalyzed by thiourea. | ||
An intriguing study was conducted by Yan and coworkers utilizing chiral bifunctional thiourea–primary amine (275) catalyst to address the issue of poor diastereoselectivity in the synthesis of asymmetric nitro cyclopropanes from cyclic enones (273) (Scheme 64).207 The reaction demonstrated a limited scope; however, it achieved excellent enantioselectivity (88–99% ee). Notably, the presence of both acid (PhCO2H) and base (NMM) additives was found to be crucial for the success of the reaction. The inclusion of PhCO2H enhances both enantioselectivity and yield, while NMM is essential for product 274 formation.
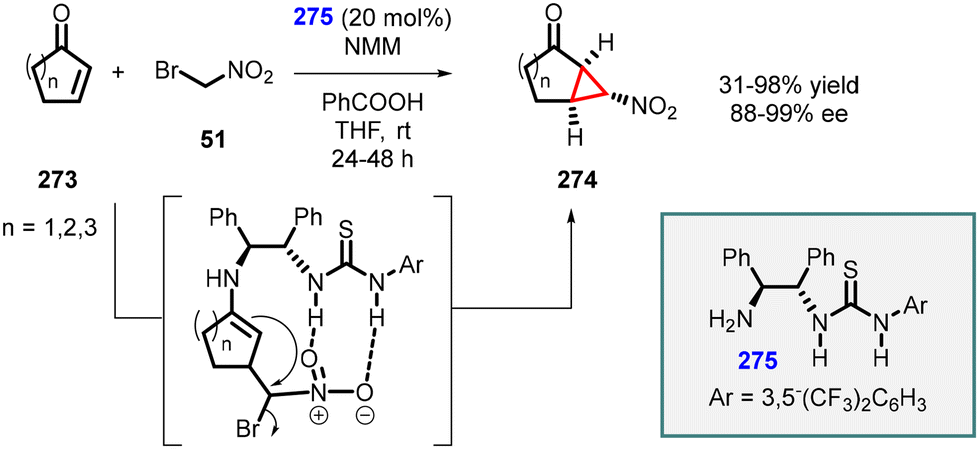 | ||
| Scheme 64 MIRC cyclopropanation of cyclic enones and bromonitromethane catalyzed by chiral bifunctional thiourea–primary amine catalyst. | ||

Bakó and co-workers attempted enantioselective MIRC cyclopropanation reactions using innovative chiral phase-transfer catalysts, including carbohydrate-based crown ethers (284–288). The MIRC cyclopropanation reaction was studied between diethyl bromomalonate (89) and different types of Michael acceptors, such as chalcones (276), 2-arylidenemalononitriles (278), 2-benzylidene-1,3-diphenyl-1,3-propanediones (280), 2-arylidene-1,3-indandiones (137), and cyanosulfones (282) under solid–liquid phase transfer catalytic conditions (Scheme 65).208–211
 | ||
| Scheme 65 MIRC cyclopropanation using chiral α-D-glucopyranoside-based crown ether. | ||
The reactions were carried out in a solvent mixture of THF and diethyl ether (in a 4:1 ratio) or DCM. Dry Na2CO3 served as the base, added in twofold excess, while 10/15 mol% of the crown ether catalyst was employed at room temperature. The results showed that enantioenriched cyclopropanes were obtained with moderate to good yields with varying levels of enantioselectivity. This research shed light on the potential of these novel chiral phase-transfer catalysts for controlling the stereoselectivity of MIRC cyclopropanation reactions.
In 2011, Ryu reported the novel use of chiral oxazaborolidinium 294 as an organocatalyst for MIRC cyclopropanation reaction between α,β-unsaturated aldehydes (289) with aryl- and alkyl diazoacetates (290) as ylides (Scheme 66).212 The catalyst coordinates with the oxygen atom of aldehyde (289), shielding the re face and enabling the attack of diazoacetate (290) from the si face, leading to intermediate (293). Subsequent cyclization, accompanied via nitrogen loss, yields formyl cyclopropanes 291 with moderate to excellent yields (up to 93%), excellent diastereoselectivity (up to 98%) and enantioselectivity (up to 95% ee).
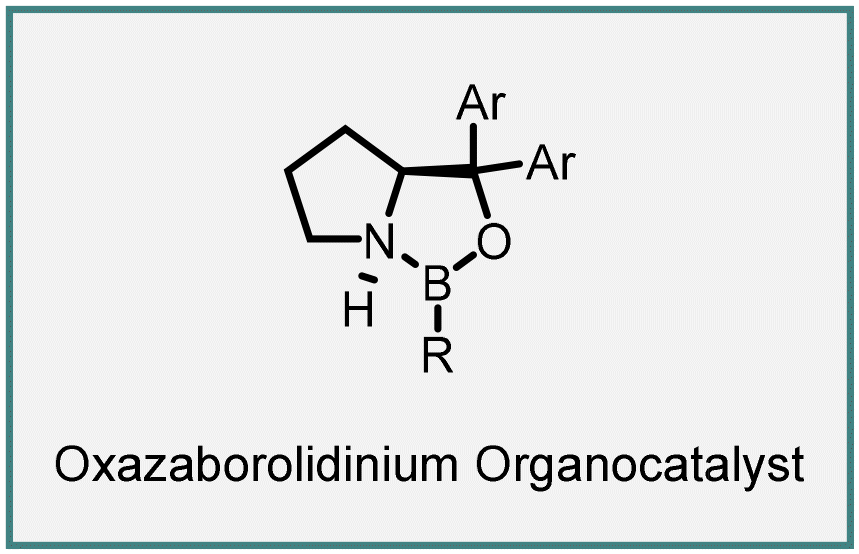
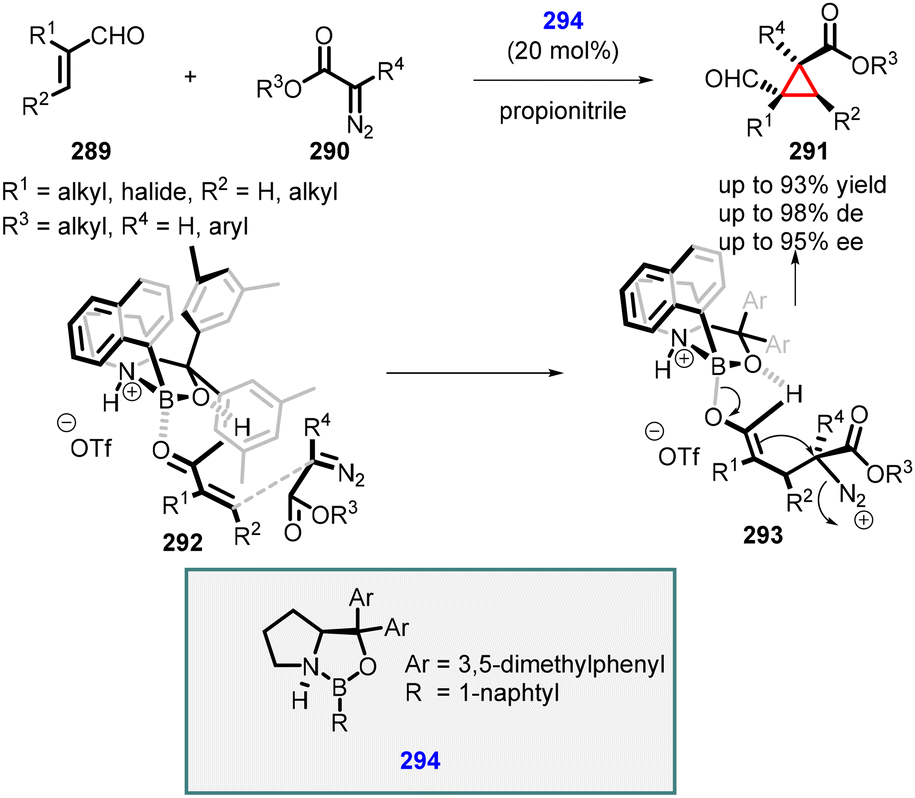 | ||
| Scheme 66 MIRC cyclopropanation of α,β-unsaturated aldehydes and alkyl-and aryl diazoacetates catalyzed by chiral oxazaborolidinium. | ||
Building upon the successful outcomes achieved with α-aryl diazo compounds, Ryu and coworkers expanded the asymmetric MIRC cyclopropanation reaction to α-alkyl diazo compounds (295). The reaction resulted in a series of highly functionalized cyclopropanes (297) with high trans-diastereoselectivity and exceptional enantioselectivity (up to >99% ee) when employing 296 as catalyst (Scheme 67).213 Notably, the methodology was successfully applied in the total synthesis of (+)-hamavellone B, marking an important milestone in the field.
 | ||
| Scheme 67 MIRC cyclopropanation of alkyl diazoacetates catalyzed by chiral oxazaborolidinium catalyst and application in total synthesis. | ||
:1) and enantioselectivity (89–96% ee).
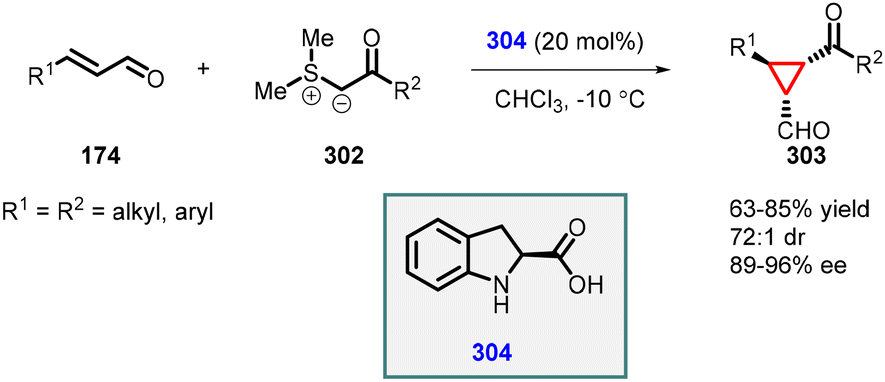 | ||
| Scheme 68 Johnson–Corey–Chaykovsky cyclopropanation using chiral secondary amine catalyst (2-carboxylic acid dihydroindole). | ||
Ley and coworkers have reported an enantioselective MIRC cyclopropanation reaction involving cyclohexenone and bromonitromethane employing chiral 5-(pyrrolidin-2-yl)-1H-tetrazole (307) as organocatalyst. Chiral nitrocyclopropanes (306) were synthesized in high yield (80%) and good enantioselectivity (77% ee). The efficiency of the reaction was further aided by the addition of morpholine as an additive (entry a, Scheme 69).215
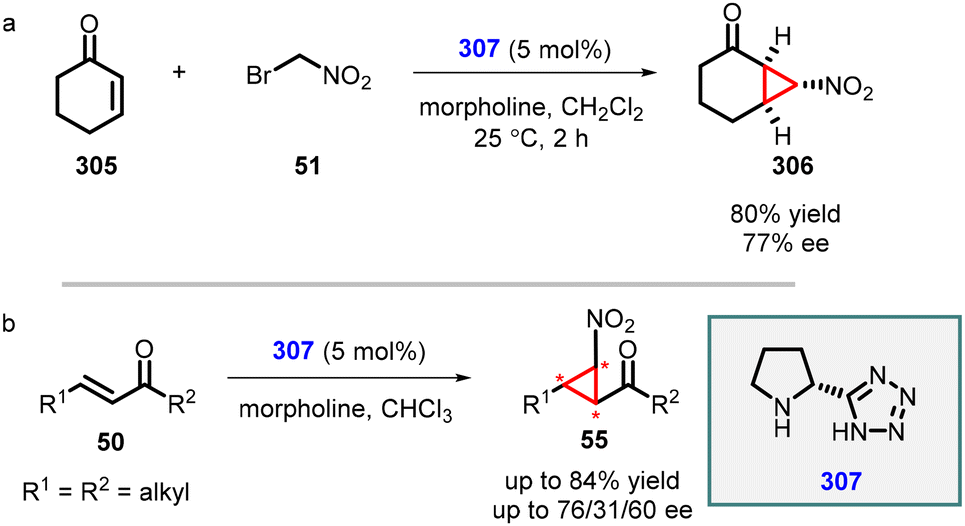 | ||
| Scheme 69 MIRC cyclopropanation of cyclohexenones and bromonitromethane using proline tetrazole catalyst. | ||
Subsequently, they explored MIRC reaction by expanding the scope of Michael acceptor to various cyclic and acyclic enones. However, the reaction resulted in poor to average diastereo- and enantioselectivity (entry b Scheme 69).216
In 2007, Arvidsson and coworkers made significant advancement in the enantioselective Corey–Chaykovsky cyclopropanation process by modifying the organocatalyst 2-carboxylic acid dihydroindole (304). They replaced the carboxylic acid with a tetrazole group, leading to the development of novel organocatalyst (310). The MIRC reaction performed with the new catalyst significantly improved the yields (up to 93%), diastereoselectivity (up to 98% dr), and enantioselectivity (up to 99% ee). The larger size of the tetrazole group, in comparison to the carboxylic acid, created steric hindrance during the nucleophilic attack step by the sulfonium ylide, consequently increasing the enantioselectivity of the reaction (Scheme 70).217
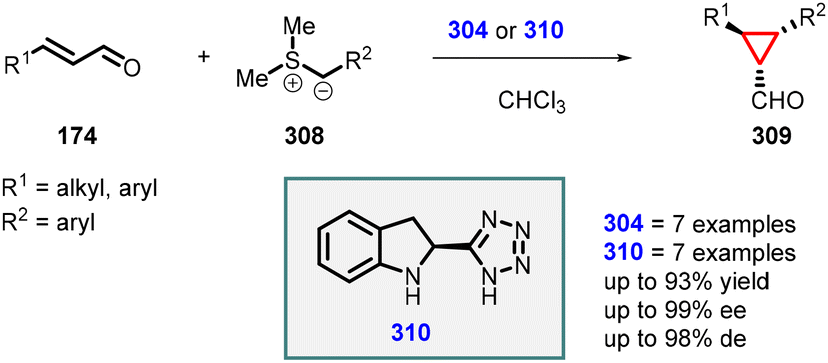 | ||
| Scheme 70 Tetrazole-organocatalyst catalyzed MIRC cyclopropanation of unsaturated aldehydes and sulfur ylide. | ||
In the same year, Arvidsson and coworkers further modified the organocatalyst by incorporating a sulfonamide group resulting in a new organocatalyst. A comparison of their findings with previously reported work showed newly developed organocatalysts (311 and 312) gave excellent diastereoselectivity (up to 98% dr) and enantioselectivity (up to 99% ee) for the cyclopropanes (303). However, the yields achieved with these modified catalysts were relatively low (58%) (Scheme 71).218
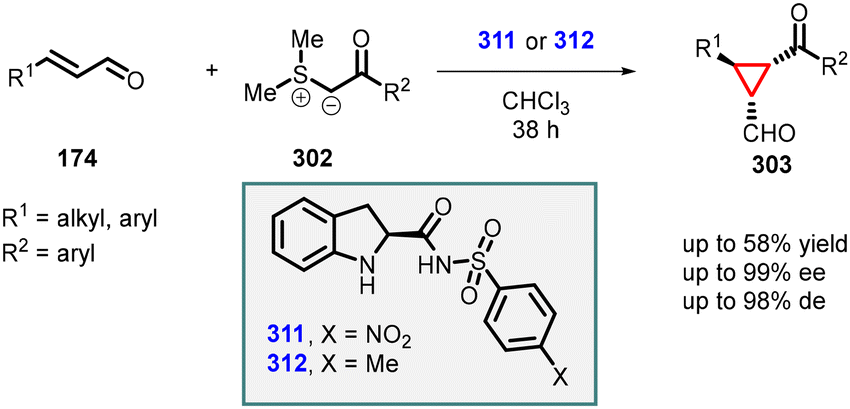 | ||
| Scheme 71 Sulfonamide-organocatalyst catalyzed MIRC cyclopropanation. | ||
In an important contribution by Kudo and coworkers, it was demonstrated that resin-supported N-terminal Pro-containing peptide (315) as catalyst in the asymmetric cyclopropanation reaction of α,β-unsaturated aldehydes (313) with sulfur ylides (302) under aqueous conditions resulted in good yield (>83%) and excellent enantioselectivity (>98% ee) (Scheme 72).219
 | ||
| Scheme 72 Peptide-catalyzed MIRC cyclopropanation of α,β-unsaturated aldehydes with sulfur ylides. | ||
Feng and coworkers successfully developed an asymmetric cyclopropanation method for α,β-unsaturated ketones (316) using stabilized ylides (302) as nucleophiles. The reaction was carried out in the presence of 20 mol% diamine catalyst (318) and benzoic acid as additive. The MIRC reaction yielded chiral cyclopropanation adducts (317) in moderate yields (up to 68%) and excellent enantioselectivity (up to 93% ee) (Scheme 73).220
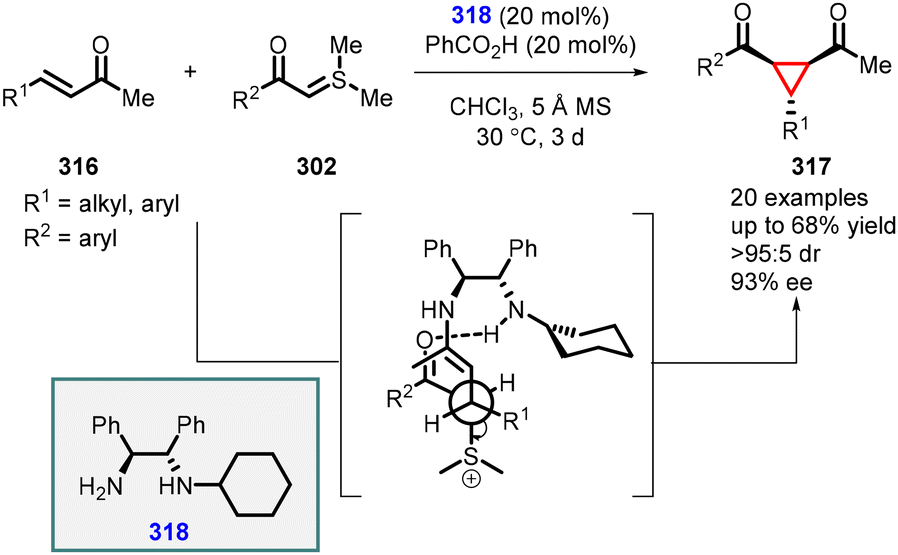 | ||
| Scheme 73 Chiral diamine-catalyzed MIRC cyclopropanation of α,β-unsaturated ketones. | ||
To understand the stereoinduction of the MIRC reaction, the authors proposed a transition state where the primary amine of the catalyst formed an enamine with the ketone, while the secondary amine guided the ylide through hydrogen bonding. This cooperative interaction between the catalyst and reactants played a significant role in achieving excellent stereo- and enantioselectivity in the reaction.
Chein and coworkers introduced a series of novel chiral tetrahydroselenophenes (321) as organocatalysts derived from (S)-diphenyl(tetrahydroselenophen-2-yl)methanol. The authors demonstrated their efficacy as chiral organocatalysts in the first organoselenium-catalyzed asymmetric cyclopropanation reactions. These selenium-containing catalyst generate selenonium ylide intermediates by reacting with benzyl bromide, which further reacts with (E)-chalcones (319) to generate a diverse array of cyclopropanes (320) (27 examples), exhibiting exceptional enantioselectivity (99% ee) (Scheme 74).221
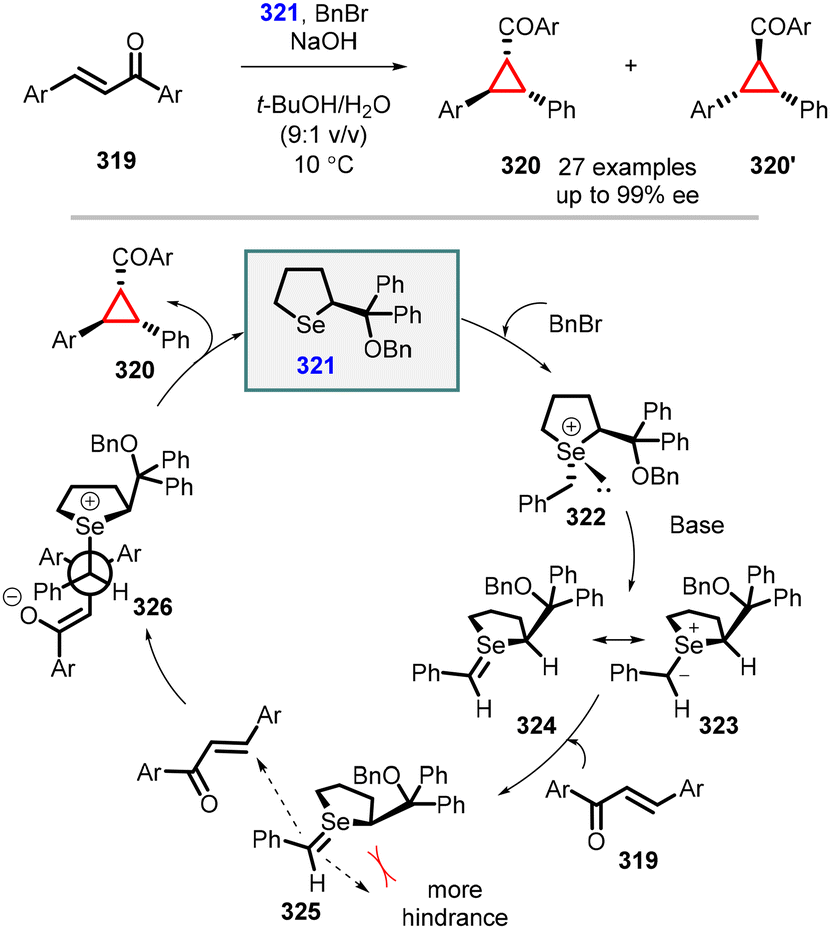 | ||
| Scheme 74 Organoselenium-catalyzed MIRC cyclopropanation. | ||
The mechanism of this reaction involves the initial conversion of the selenide catalyst (321) into the selenonium salt 322 by reacting with benzyl bromide under basic conditions. Salt 322 then forms the corresponding ylide 323/324. The bulky side chain in 324 is strategically positioned to shield the si face of the ylide carbon, directing the approach of the Michael acceptor from the re face. This leads to the formation of intermediate 326, adopting the favorable anti–anti conformation. Subsequent intramolecular cyclization of the Michael adduct produces the major diastereomer, yielding the (R,R)-cyclopropane product.
Chein and coworkers subsequently expanded their MIRC reaction by developing an innovative and environmentally friendly approach for the synthesis of enantioselective cyclopropane scaffolds using (S)-(thiolan-2-yl)diphenylmethanol benzyl ether (330) as an organocatalyst. This method utilizes a solvent system comprising t-BuOH/H2O, a low-cost base (NaOH), and a reusable catalyst, making it a greener alternative.
In this process, a sulfur ylide, generated in situ, reacts with (E)-chalcones via a Johnson–Corey–Chaykovsky reaction. This transformation affords a range of cyclopropanes with excellent yields and enantioselectivity (up to 95% ee) (Scheme 75).222
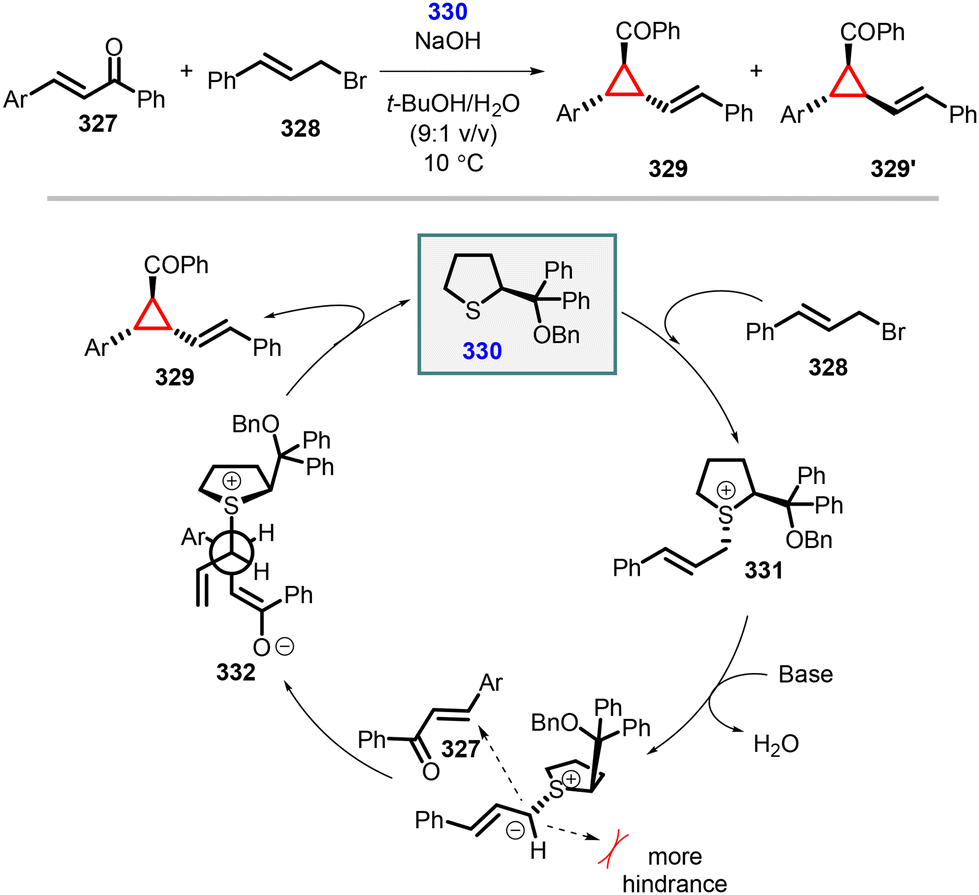 | ||
| Scheme 75 (Thiolan-2-yl)diphenylmethanol benzyl ether-catalyzed MIRC cyclopropanation of chalcones. | ||
Li and coworkers developed a novel methodology for enantioselective cyclopropanation utilizing chiral sulfide (336) as an organocatalyst (Scheme 76).223 In this process, the electron-deficient diene 333 is treated with bromides 334 in the presence of a chiral catalyst, namely (1R,4R,5R)-4,7,7-trimethyl-6-thiabicyclo[3.2.1]octane 336, and NaHCO3 in acetonitrile. The reaction yielded the desired product, vinylcyclopropanes (335) with a high yield (99%) and excellent stereoselectivity (>19:1 dr, 98:2 er).
 | ||
| Scheme 76 (1R,4R,5R)-4,7,7-Trimethyl-6-thiabicyclo[3.2.1]octane catalyzed cyclopropanation. | ||
The reaction proceeds via the formation of a chiral sulfur ylide through the combination of 2-bromoalkyl ethanone (334) with the sulfide catalyst (336). Subsequently, the ylide undergoes vinylogous addition to diene (333), followed by an intramolecular nucleophilic substitution, leading to the release of catalyst (336) and formation of vinylcyclopropane moiety (335).
Studer and coworkers have developed an asymmetric cyclopropanation of α,β-unsaturated aldehydes (197) using stabilized sulfonium ylides (337) (Scheme 77).224 This method employs a chiral NHC catalyst precursor (339), DABCO as base, and benzoquinone 340 as the oxidant. The reaction enables the synthesis of a diverse range of highly functionalized chiral cyclopropanes (338) with moderate yields (47–74%) and excellent diastereoselectivity (10:1 dr) and enantioselectivity (>99% ee).
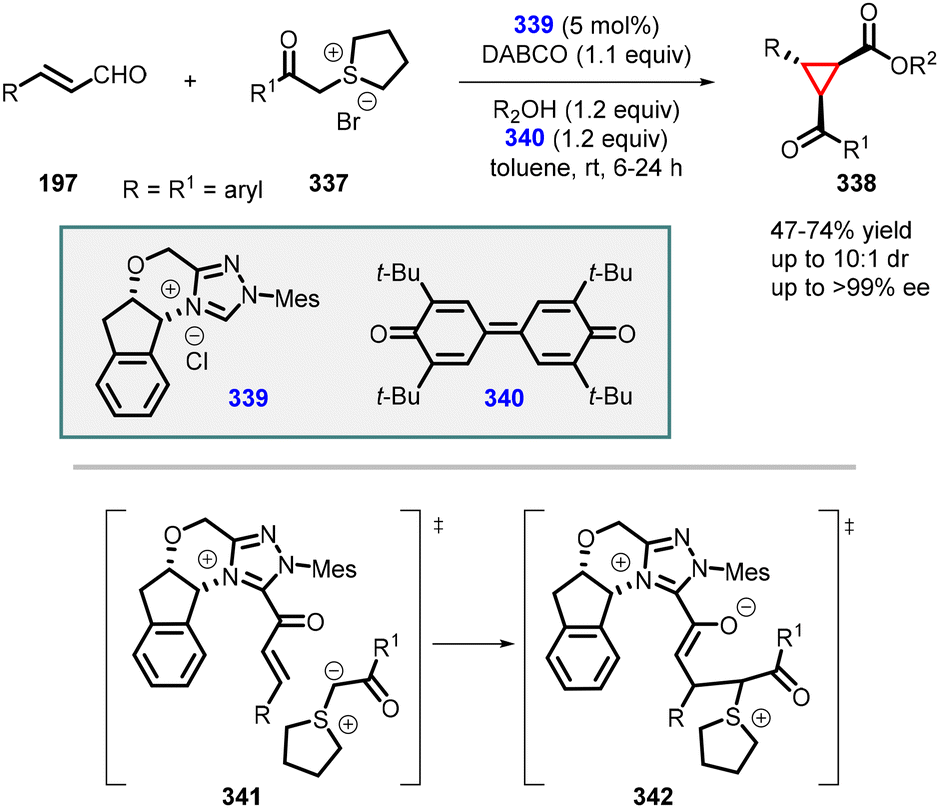 | ||
| Scheme 77 NHC catalyzed enantioselective MIRC cyclopropanation. | ||
The reaction mechanism begins with the formation of an acylazolium ion intermediate (341) by the reaction between carbene catalyst (339) and enal (197) in the presence of a mild oxidant (340). This intermediate, 341, then undergoes a stereoselective Michael addition using ylide 337, followed by an intramolecular alkylation reaction leading to the formation of intermediate (342). Finally, after the removal of NHC with isopropanol, the desired cyclopropane product (338) is obtained.
3.2 Metal catalysts
Metal catalysts play a crucial role in Michael-initiated ring-closing cyclopropanation, a key process in organic chemistry. These catalysts, including metals like ruthenium, magnesium, copper, lanthanum, nickel, indium, palladium, rhodium, and others in the form of complexes, are essential for accelerating the formation of the cyclopropane ring. By utilizing these catalysts, chemists can not only increase the efficiency of the reaction but also exert control over its yield, stereochemistry, and regioselectivity. This ability to activate and interact with the reactants enables the generation of highly valuable molecules containing cyclopropane unit. The discovery of metal catalysts in Michael-initiated ring-closing cyclopropanation has significantly expanded the synthetic toolkit available to chemists, providing them with a diverse range of methods for creating complex and valuable compounds.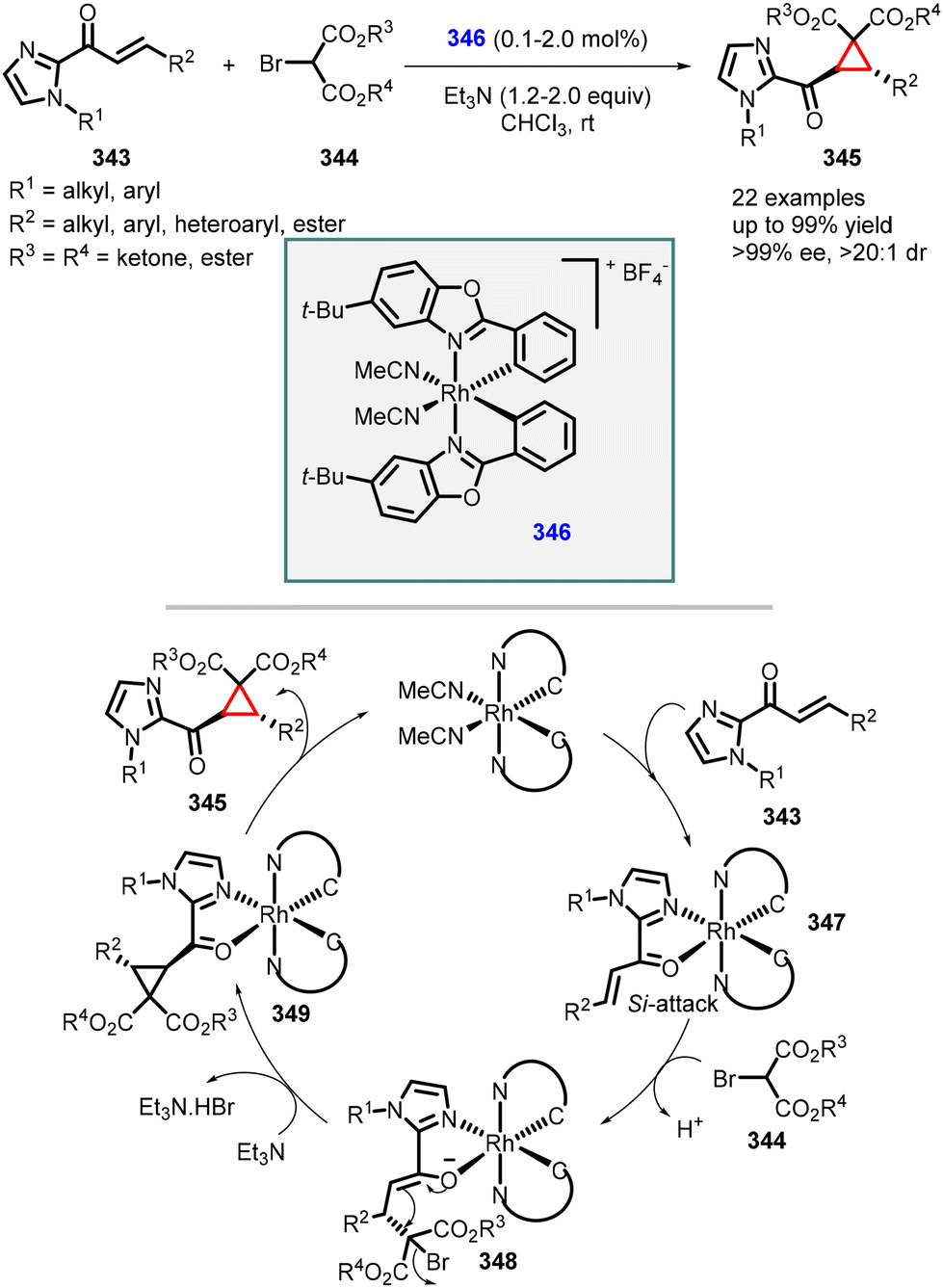 | ||
| Scheme 78 Chiral Rh(III) complex-catalyzed cascade MIRC reactions for enantioselective synthesis of cyclopropanes. | ||
The reaction proceeds through several key steps. First, substrate (343) was activated by the rhodium catalyst, forming an intermediate 347 through bidentate N,O-coordination. The bulky t-Bu group on the ligand shields the Re face of 347, allowing nucleophilic attack of diethyl α-bromomalonate 344 to occur from the Si face. This leads to the formation of an enolate anion, intermediate 348. Next, intermediate 348 undergoes cyclization, resulting in the formation of a coordinated intermediate 349 with a cyclopropane skeleton. The desired cyclopropane adduct (345) was released from the coordinated intermediate 349 through ligand exchange with substrate (343), initiating a new catalytic cycle.
Gong and coworkers pursued a similar approach by employing sulfoxonium ylides (302) as nucleophiles in conjunction with α,β-unsaturated 2-acyl imidazoles (350). The reaction was catalyzed by a chiral rhodium complex (352) and the reaction gave favorable outcomes, as demonstrated in Scheme 79,226 with a mechanism similar to the previously mentioned by Kang et al.
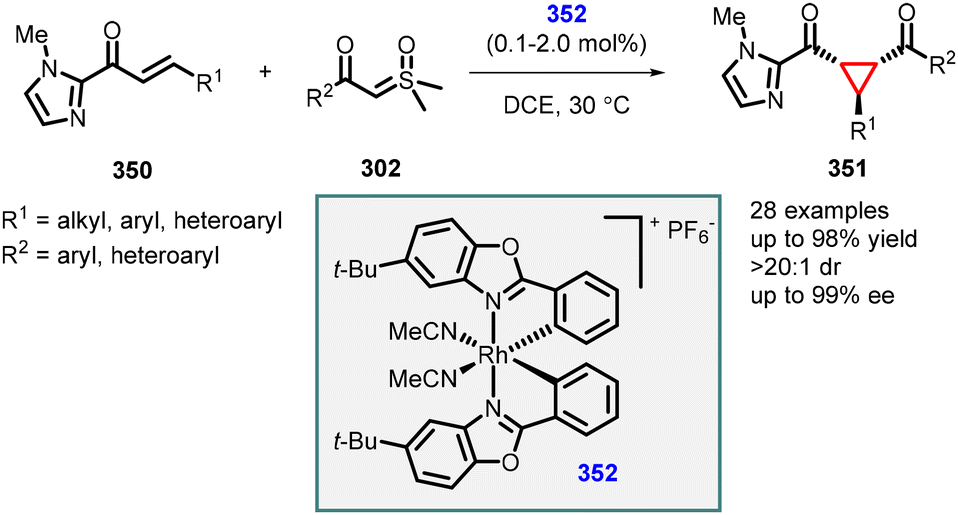 | ||
| Scheme 79 Chiral Rh(III) complex-catalyzed cascade MIRC cyclopropanation of α,β-unsaturated 2-acyl imidazoles with sulfoxonium ylides. | ||
Li and coworkers reported an enantioselective MIRC reaction between sulfoxonium ylides (302) and β,γ-unsaturated ketoesters (353) using the same catalytic system (352) (Scheme 80).227 The reaction furnished 1,2,3-trisubstituted cyclopropanes (354) in moderate to high yields (48–89%) and excellent diastereo- and enantioselectivity (dr >20:1 and up to 99% ee).
 | ||
| Scheme 80 Chiral Rh(III) complex-catalyzed cascade MIRC cyclopropanation of sulfoxonium ylides and β,γ-unsaturated ketoesters. | ||
Mechanistic studies demonstrated the key role of a weak coordination mode between the chiral rhodium catalyst (352) and the β,γ-unsaturated carbonyl esters (353) in achieving high levels of diastereoselectivity and enantioselectivity for the product formation as shown in the Scheme 80.
In the synthesis of chiral functionalized cyclopropanes (360), Cramer and coworkers employed chiral cyclopentadienyl Rh(III) complexes 361 as catalysts. The substrates used were olefins with N-enoxyphthalimides (358) and electron-deficient olefins (359). The MIRC cyclopropanation yielded chiral cyclopropanes (360) in high yields (up to 90%), excellent diastereoselectivity (>20:1), and excellent enantiomeric excesses of 95% (Scheme 81).228 The reaction involves enantioselective alkenyl C–H bond functionalization, providing an avenue for accessing chiral cyclopropanes under mild, open-flask reaction conditions. To demonstrate the synthetic applicability of this method, the authors successfully utilized cyclopropanation as a crucial step in synthesizing both the oxylipin family of natural products and the kynurenine 3-monooxygenase inhibitor UPF-648.
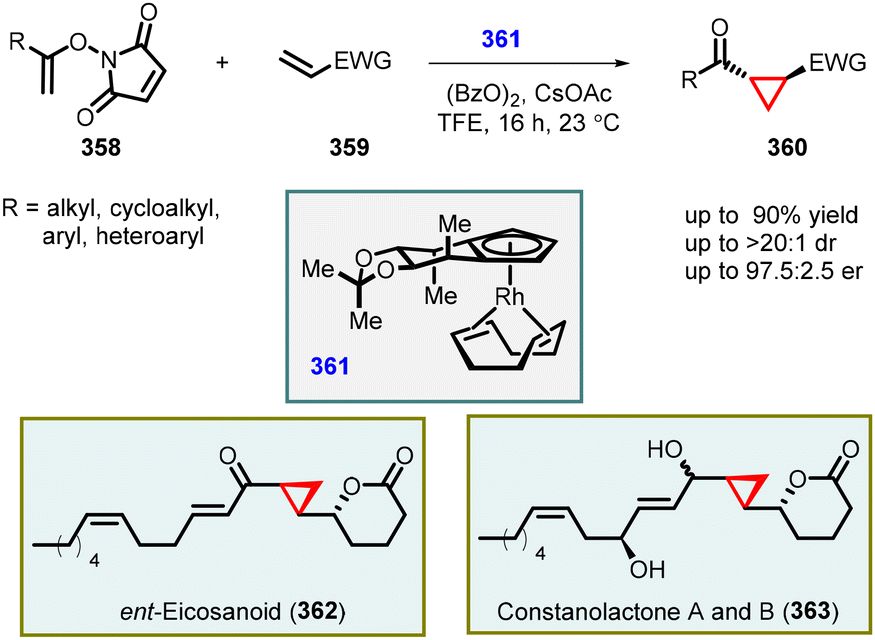 | ||
| Scheme 81 Chiral Rh(III) complex-catalyzed cyclopropanation of N-enoxyphthalimides and electron-deficient olefins. | ||
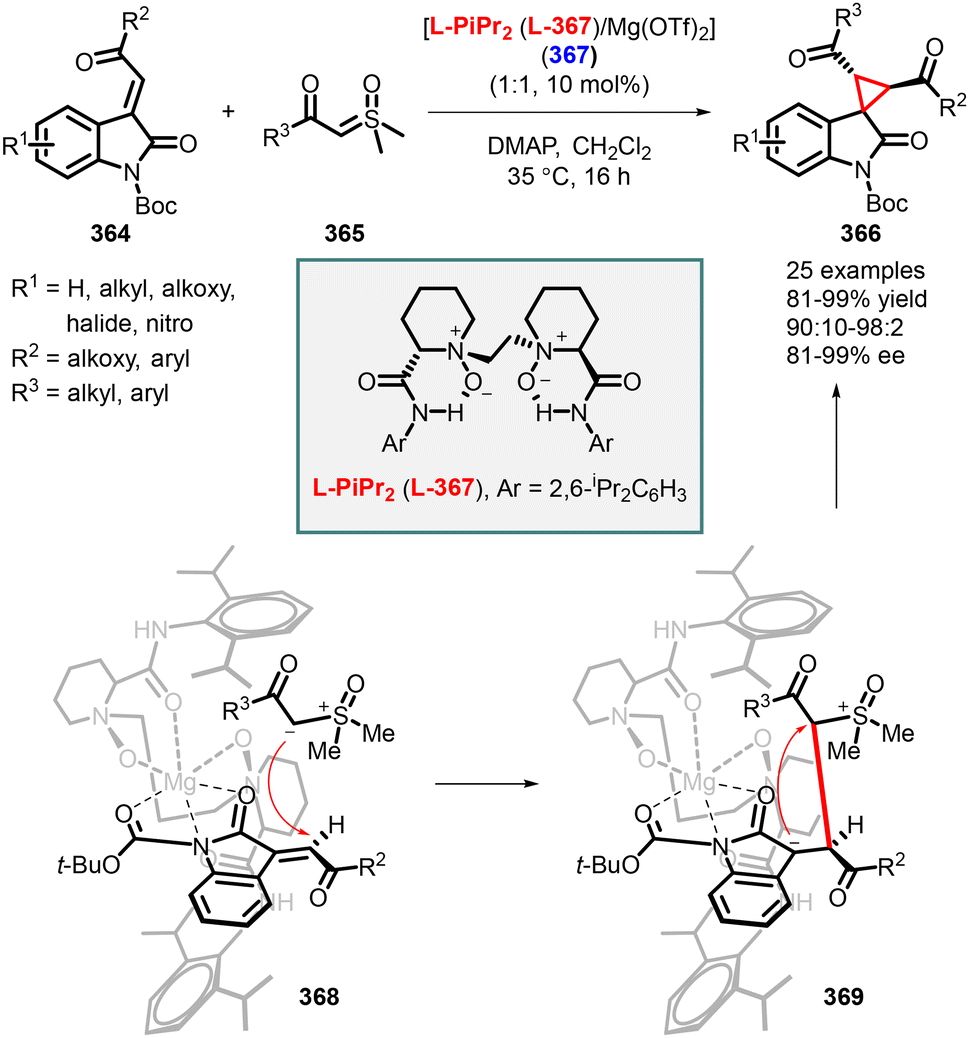 | ||
| Scheme 82 N,N′-Dioxide (L-PiPr2)/Mg(OTf)2 complex catalyzed cyclopropanation of 3-alkenyl-oxindoles and sulfoxonium ylides. | ||
Feng and coworkers expanded their study to a chiral sulfur ring-opening/cyclopropanation reaction involving 1-alkyl-3-oxotetrahydro-1H-thiophen-1-ium salts (cyclic sulfur ylides) 370 and (E)-3-(oxyethylidene)-2-oxoindolines 364. Notably, this reaction was catalyzed by a chiral N,N′-dioxide (L2-PiPr3) (L-372) complex, featuring a slightly different aromatic substituent, in combination with Mg(OTf)2. The reaction resulted in the formation of sulfur-containing syn, anti spirocyclopropyloxindoles (371) that possesses three contiguous chiral centers (Scheme 83).230
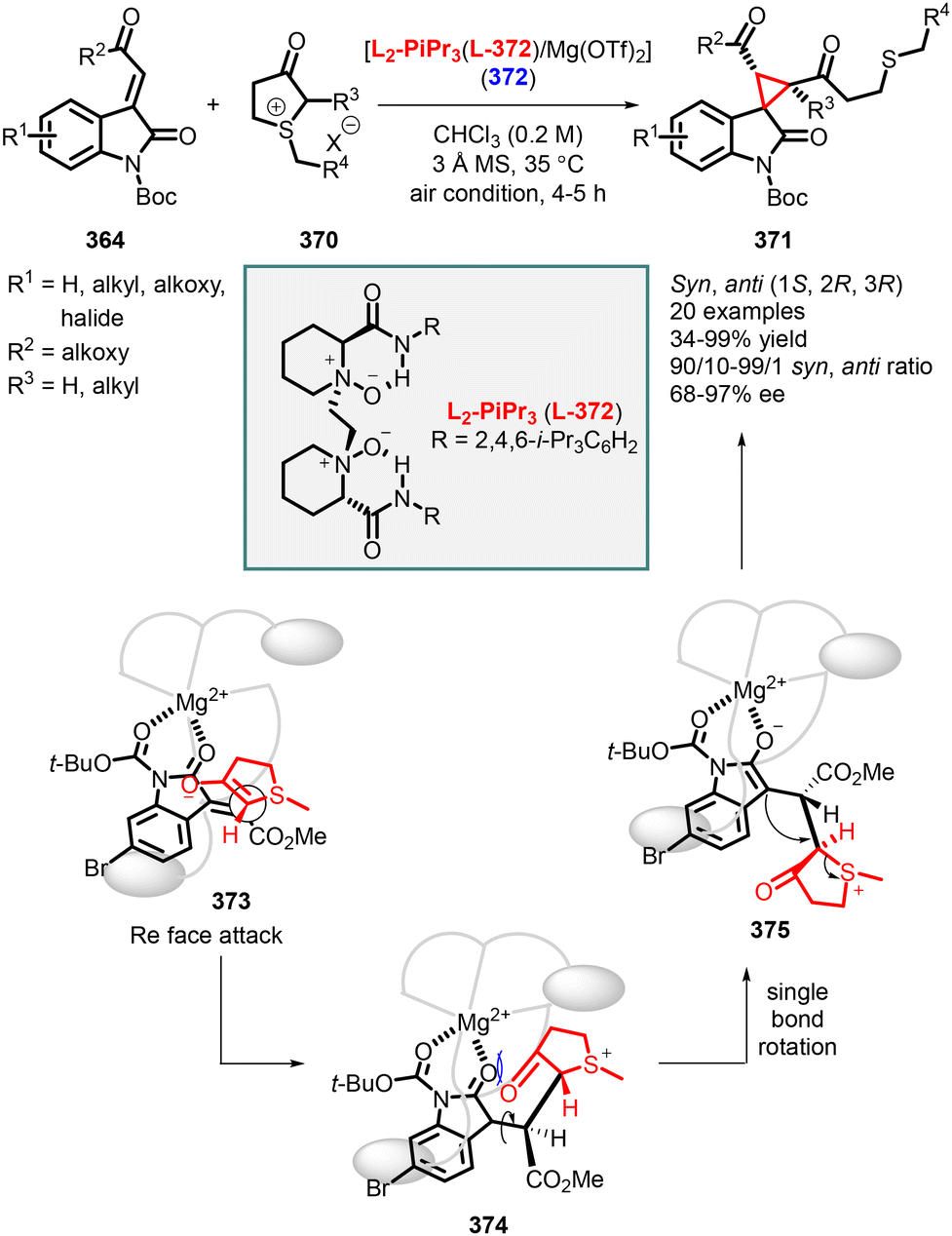 | ||
| Scheme 83 Ring opening cyclopropanation of cyclic sulfur ylides using Mg metal mediated catalyst. | ||
The authors proposed transition-state models to explain the origin of stereoselectivity. In this process, the chiral N,N′-dioxide ligand (L2-PiPr3) coordinates with Mg(II), adopting an octahedral geometry with four oxygens. Following this, the two oxygens of (E)-3-(oxyethylidene)-2-oxoindolines 364 coordinates with Mg(II) in a bidentate manner. The neighboring amide group of the ligand provides shielding to the Si face of compound (364). Consequently, the Re face of the nucleophilic ylide (370) approaches the Re face of compound (364).
During the ring-closing step, which proceeds through an SN2 reaction, the enolate anion must attack from the opposite position as the releasing sulfur salt. If the enolate approaches the thianone (370) with its Si face (intermediate 374), the repulsive effect intensifies. This prompts a rotation of the C–C single bond connected to the enolate, allowing the enolate anion's Re face to attack the thianone (370). The resulting process is an SN2 reaction that yields the (1S,2R,3R)-product (371).
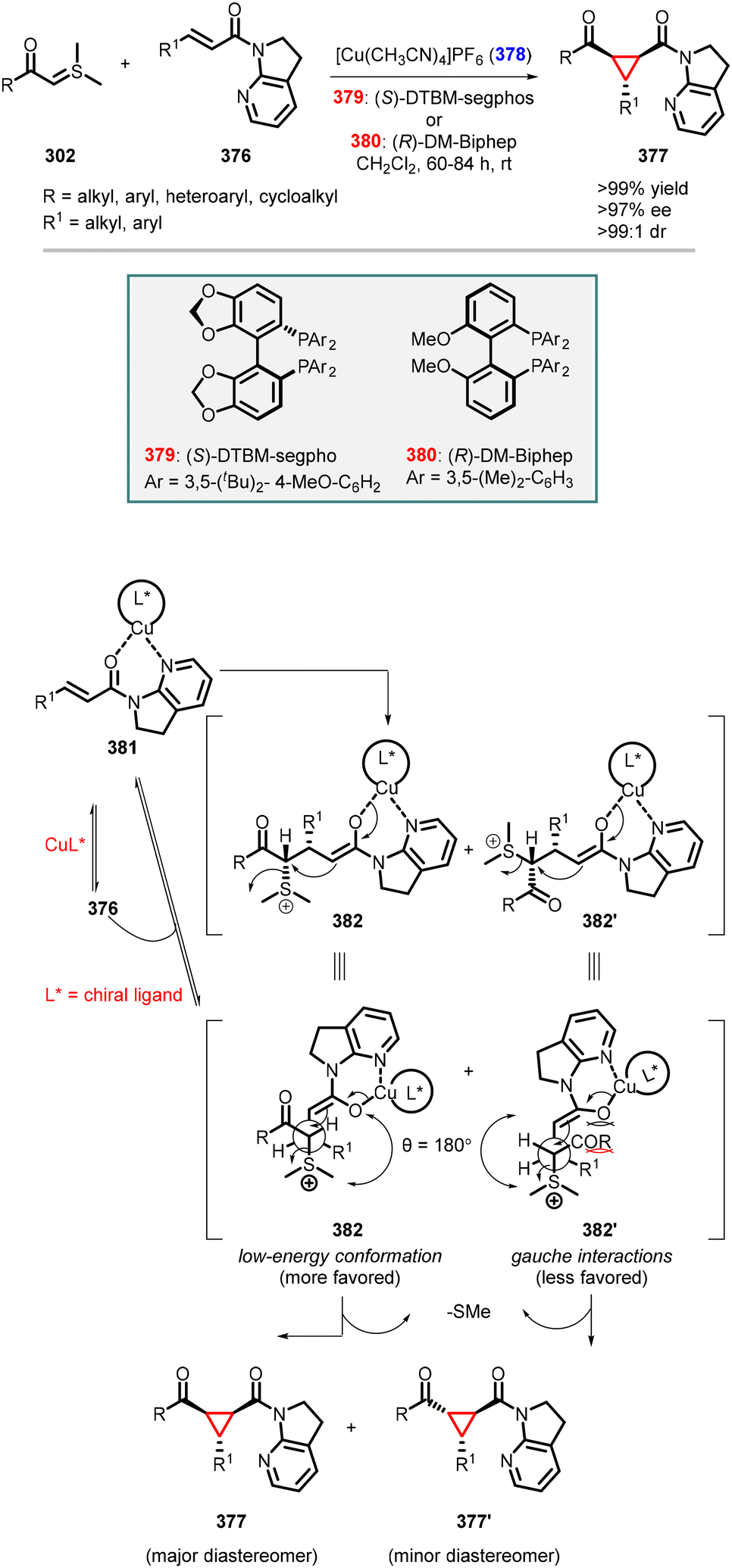 | ||
| Scheme 84 Cu(I) complex catalyzed MIRC cyclopropanation of 7-azaindoline amides, with stabilized sulfur ylides. | ||
Michael acceptor (376) reacts with the Cu(I)/L* complex, forming a Z-configured complex 381. This complex enhances the electrophilicity of (376) for the nucleophilic attack by the ylide (302) in a chiral environment. Through an irreversible process, the initial asymmetric C–C bond formation occurs at the β-position. The resulting major Cu enolate intermediate 382 undergoes intramolecular SN2 displacement, leading to the formation of the major diastereomer (377). In contrast, the minor diastereomer (377′) is formed from the minor Cu-enolate intermediate 382′. The formation of the major diastereomer (377) is favored because it involves a more stable intermediate 382, whereas intermediate 382′ experiences significant steric interactions, resulting in the formation of the minor diastereomer (377′) through a higher-energy transition state.
For β-alkyl and β-aryl substrates, two different ligand systems were used. These systems successfully produced the desired products in high yields (>99%) and selectivity (>99:1, >97% ee), with 1–2 mol% of catalyst loading.
In 2010, Feringa and coworkers reported an enantioselective copper-catalyzed domino reaction for synthesizing trans-1-alkyl-2-substituted cyclopropanes (385) (Scheme 85).232 The reaction involves a combination of Grignard reagents and 4-chloro-α,β-unsaturated esters/thioesters/ketones (383) in the presence of the chiral ligand (R)-Tol-BINAP and CuI as the pre-catalyst. The presence of the internal chloro as a leaving group enabled a tandem conjugate addition-enolate trapping process, resulting in the formation of trans-1-alkyl-2-substituted cyclopropanes (385) with moderate to high yields (50–95%) and good to excellent enantiomeric excess values (up to 98%). This versatile reaction demonstrated its utility by producing key intermediates for the formal syntheses of cascarillic acid (386) and grenadamide (387).
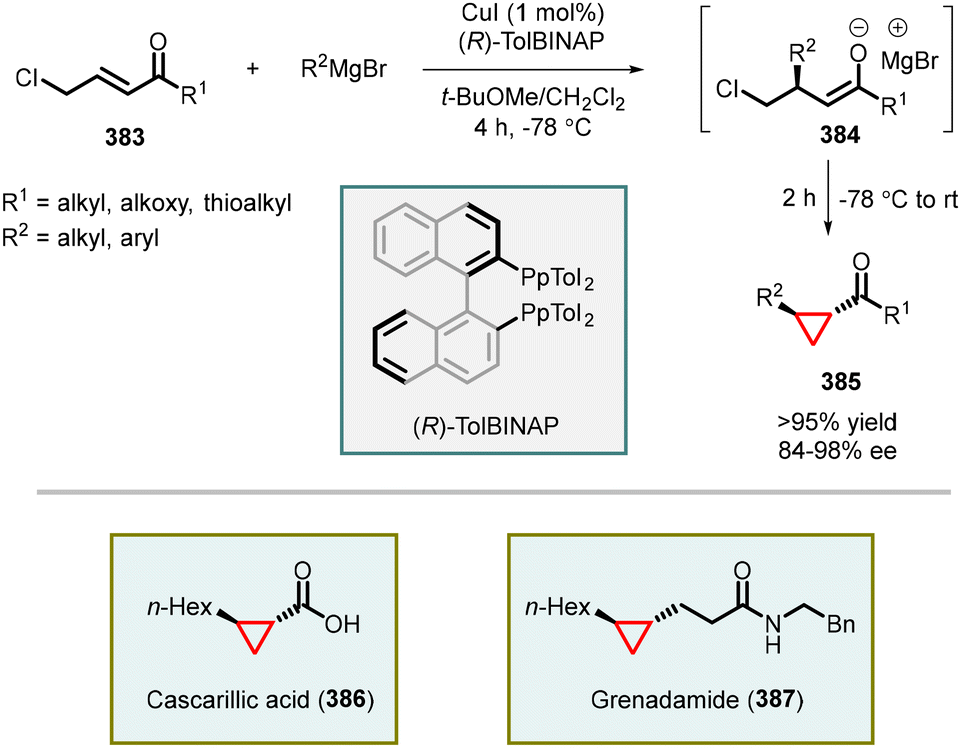 | ||
| Scheme 85 MIRC cyclopropanation of 4-chloro-α,β-unsaturated esters/thioesters/ketones with Grignard reagents. | ||
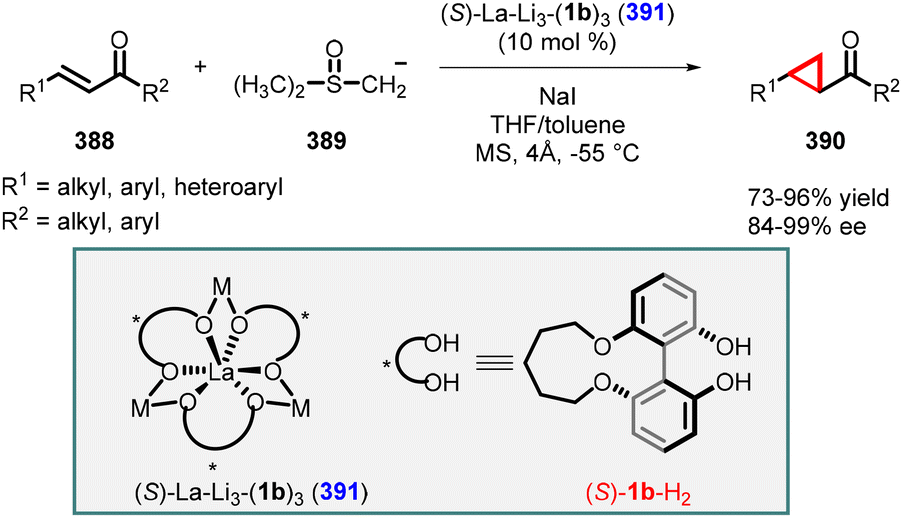 | ||
| Scheme 86 La-Li3-(biphenyldiolate)3/NaI complex catalyzed MIRC cyclopropanation of enones with dimethyloxosulfonium methylide. | ||
Feng and coworkers reported a significant advancement in the synthesis of highly functionalized cyclopropanes through a La-catalyzed enantioselective MIRC reaction. In this process, 2-cyano-3-arylacrylates (392) was reacted with 2-bromomalonates 89, utilizing a N,N′-dioxide L2-RaEt2–lanthanum(III) complex (394) as a Lewis acid catalyst. The desired highly functionalized chiral cyclopropanes (393) were obtained in high yields (up to 93%), excellent diastereomeric ratios (>95:5 dr) and high enantiomeric excess (up to 91% ee) (Scheme 87).234
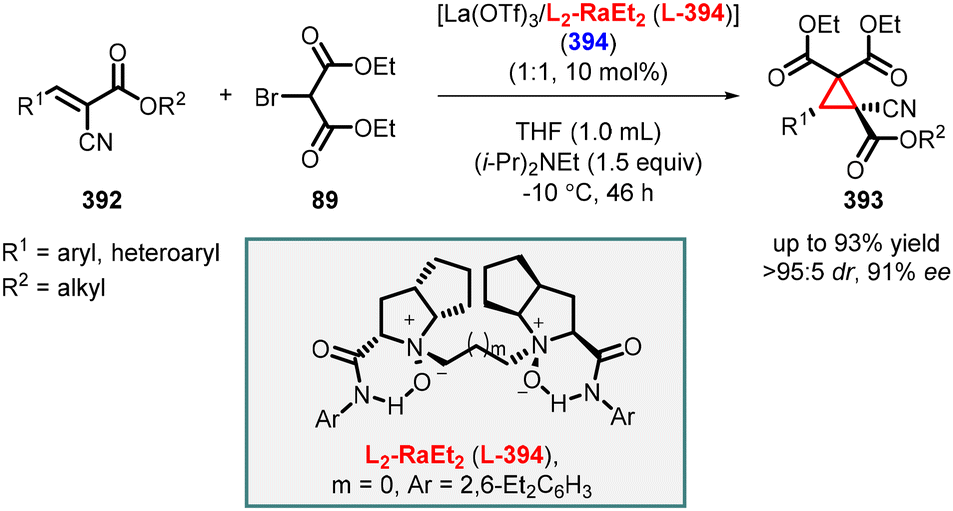 | ||
| Scheme 87 N,N′-Dioxide–lanthanum(III) complex catalyzed enantioselective MIRC cyclopropanation. | ||
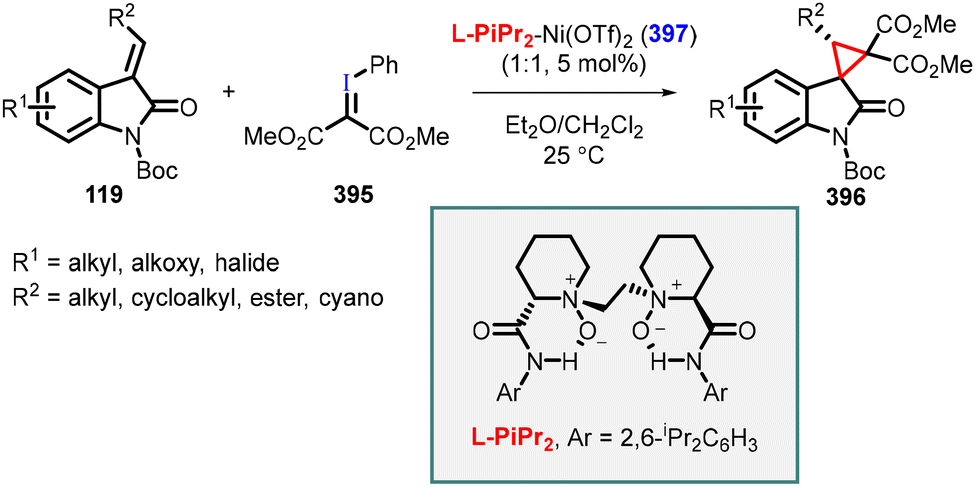 | ||
| Scheme 88 N,N′-Dioxide–Ni(II) complex catalyzed enantioselective MIRC cyclopropanation. | ||
A domino Michael addition/cyclization reaction was reported by Luo and co-workers for the enantioselective synthesis of functionalized chiral cyclopropanes from β,γ-unsaturated α-keto esters (353) and diazo esters (398). This transformation was achieved using a binary Lewis acid catalyst consisting of 5 mol% InBr3 and an equivalent quantity of chiral calcium phosphate (400). The reaction was carried out in dichloroethane (DCE) at room temperature. The resulting cyclopropanes (399) were obtained in moderate to good yields (50–77%) and as a single diastereomer and with excellent enantiomeric excess (>99% ee). In this process, the bidentate coordination of the β,γ-unsaturated α-keto esters with the cationic indium complex creates a rigid system for chiral identification. It was proposed that weak π-interactions between the chiral phosphoric acid and the diazo ylide facilitates a facial attack, followed by backside attack for the cyclization with the loss of nitrogen (Scheme 89).236
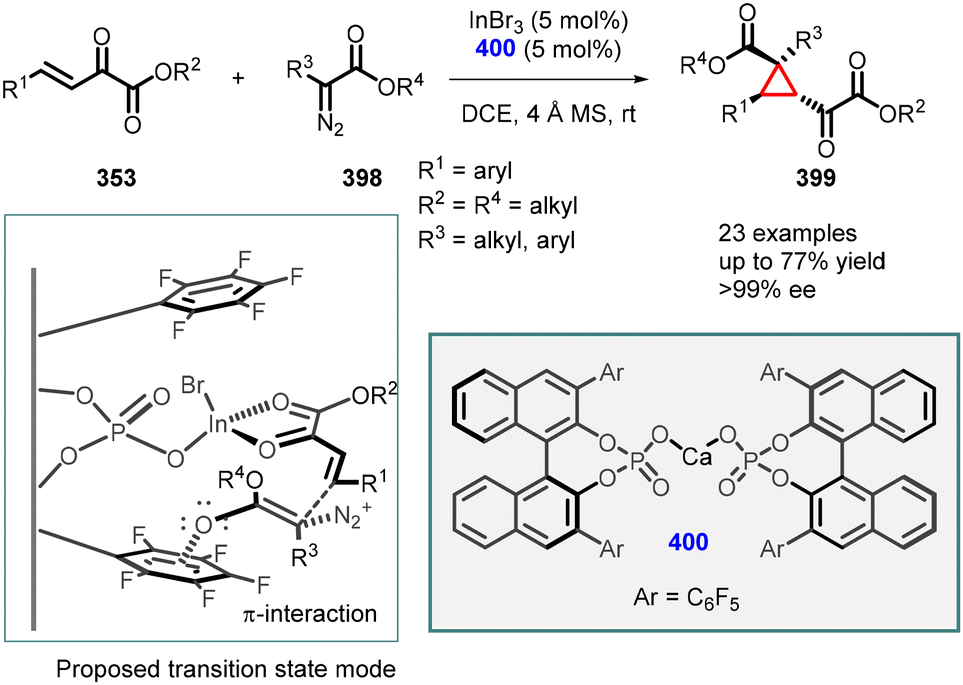 | ||
| Scheme 89 Binary Lewis acid (InBr3 and chiral calcium phosphate) catalyzed enantioselective MIRC cyclopropanation. | ||
in 2018, Feng, Liu and coworkers developed a diastereodivergent asymmetric Michael addition-alkylation reaction using 3-Cl oxindoles (150) and β,γ-unsaturated α-ketoesters (401) (Scheme 90).237 They achieved high yields (50–99%) and excellent enantioselectivity (72–99% ee) of rel-(1R,2S,3R) spiro cyclopropane oxindoles by modifying metal catalysts, ligands, and temperature. Different metal catalysts resulted in the formation of distinct diastereoisomers: rel-(1S,2S,3R) via intramolecular trapping and (iso)-402 through a direct substitution pathway. These findings shed light on the reaction mechanism and provide valuable insights for future applications.
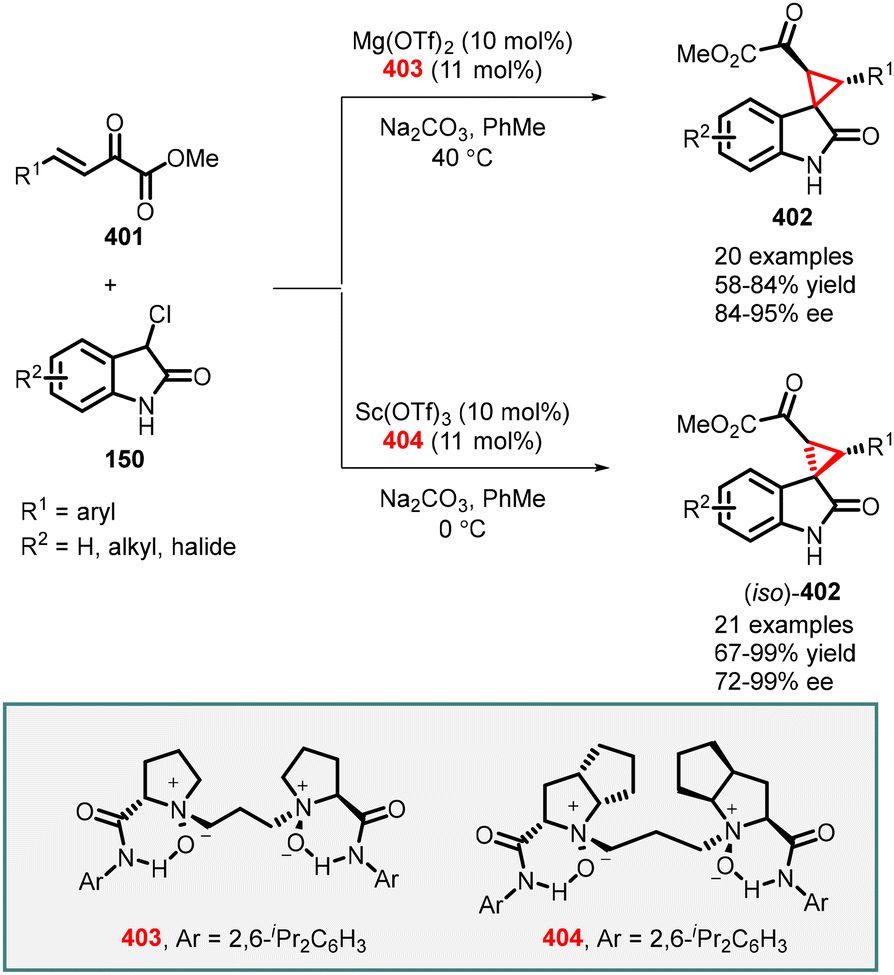 | ||
| Scheme 90 Catalytic enantioselective MIRC cyclopropanation of β,γ-unsaturated α-ketoesters. | ||
In a subsequent publication, the same research group employed a scandium complex derived from a chiral N,N′-dioxide ligand 409 as a catalyst in the enantioselective Michael-initiated addition between α-substituted vinyl ketones 406 and α-substituted α-diazoesters 405. The reactions were conducted at 0 °C, and compounds 406 and 405 underwent a MIRC reaction, leading to the formation of chiral tetrasubstituted cyclopropanes 407 as exclusive diastereomers.238 The reactions exhibited moderate to good yields (42–65%) and consistently excellent enantioselectivities (94–99% ee) (Scheme 91). In addition to the cycloadducts, a competing C–H insertion pathway resulted in the production of chiral E-enone derivatives 408. These compounds were also obtained with high enantioenrichment (72–95% ee) and modest yields (24–57%).
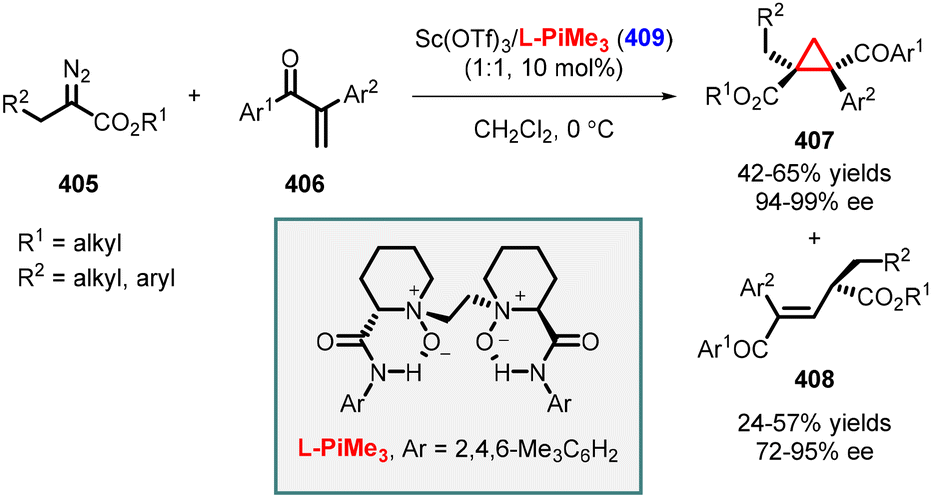 | ||
| Scheme 91 N,N′-Dioxide–Sc(III) complex catalyzed enantioselective MIRC cyclopropanation. | ||
In 2007, Maruoka and coworkers demonstrated the effectiveness of acid-catalyzed Michael-initiated cyclopropanation reactions with aryldiazoacetates as reactive nucleophiles and α-substituted acroleins (Scheme 92). The aim was to prepare sterically congested cyclopropanes with high diastereoselectivity. The authors investigated the viability of an asymmetric variation utilizing (S)-BINOL-Ti(Oi-Pr)4 (2:1 molar ratio) as the catalyst. The cyclopropanation reactions between tert-butyl phenyldiazoacetate (410) and α-benzoyloxyacrolein (411) yielded a cyclopropane product (412) with good diastereoselectivity (12:1 dr) but poor enantioselectivity (57% ee for the major isomer).239
 | ||
| Scheme 92 Titanium/(S)-BINOLate catalyzed enantioselective MIRC cyclopropanation. | ||
3.3 Guideline for catalyst selection for the enantioselective construction of cyclopropane rings
In this review, we have summarized catalytic MIRC reactions for the stereoselective construction of cyclopropane rings. Catalytic methodologies ranging from organocatalysis to metal based chiral Lewis-acid catalysis have been discussed in this review. Given the extensive amount of literature covered, it is appropriate to inform the readers which methodology would be appropriate while planning a synthesis. The chirality in these catalytic MIRC reactions is achieved through chiral electrophiles (acceptors) or through chiral Michael nucleophiles (donors). For chiral electrophiles, especially α,β-unsaturated aldehydes or ketones, organocatalysis based on chiral secondary amines (prolinol or imidazolidinone derived) (see section 3.1.4) could be the method of choice. The electrophiles can also be activated through chiral Lewis acid catalysis (see section 3.2). In this case, an auxiliary group capable of chelating to the metal center is often a requirement. For reactions with chiral nucleophiles, organocatalysis based on cinchona alkaloid derived catalysts (for the generation of ammonium ylide as a Michael initiator) (see section 3.1.1) could be the method of choice.This review aims to guide researchers in selecting the most effective method for their specific cyclopropane targets. The analysis of enantioselectivity, substrate scope, synthetic efficiency, and reproducibility serves as a valuable resource for the selection.
4. Conclusions
This comprehensive review is focused on catalytic enantioselective Michael initiated ring closure (MIRC) cyclopropanation reaction. The review highlights the relevance of MIRC reactions in achieving high enantioselectivity for the product cyclopropanes and provides insights into the various methods, catalysts, substrates, and nucleophiles utilized in this transformation. Furthermore, the analysis delves into the examination of factors that influence reactivity and selectivity, including catalyst selection, reaction conditions, and functional groups. Despite notable advancements in cyclopropane synthesis, there are persistent obstacles that need to be addressed. These include gaining a better understanding of the reaction mechanism, exploring alternative systems to enhance selectivity, and reducing catalyst loading to promote environmentally friendly practices. Ongoing efforts are crucial to tackle these challenges and drive advancements in the field of asymmetric cyclopropanation, which holds significant importance in the synthesis of pharmaceuticals, and advanced materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank NSF-2102605 and North Dakota State University for financial support. The authors also thank Prof. Hari Subramanian for critical reading and help in the preparation of the manuscript.References
- A. Freund, Ueber Trimethylen, J. Prakt. Chem., 1882, 26, 367 CrossRef.
- T. T. Talele, The “Cyclopropyl Fragment” is a Versatile Player that Frequently Appears in Preclinical/Clinical Drug Molecules, J. Med. Chem., 2016, 59, 8712 CrossRef CAS PubMed.
- M. M. Heravi, S. Asadi, N. Nazari and B. Malekzadeh Lashkariani, Developments of Corey-Fuchs Reaction in Organic and Total Synthesis of Natural Products, Curr. Org. Chem., 2015, 19, 2196 CrossRef CAS.
- Z. Časar, Synthetic Approaches to Contemporary Drugs that Contain the Cyclopropyl Moiety, Synthesis, 2020, 52, 1315 CrossRef.
- A. B. Charette and A. Beauchemin, in Organic Reactions, 2004, p. 1 Search PubMed.
- H. Y. Kim and P. J. Walsh, Efficient Approaches to the Stereoselective Synthesis of Cyclopropyl Alcohols, Acc. Chem. Res., 2012, 45, 1533 CrossRef CAS PubMed.
- M. P. Doyle and D. C. Forbes, Recent Advances in Asymmetric Catalytic Metal Carbene Transformations, Chem. Rev., 1998, 98, 911 CrossRef CAS PubMed.
- H. M. L. Davies and E. G. Antoulinakis, in Organic Reactions, 2004, p. 1 Search PubMed.
- V. K. Singh, A. DattaGupta and G. Sekar, Catalytic Enantioselective Cyclopropanation of Olefins Using Carbenoid Chemistry, Synthesis, 1997, 137 CrossRef CAS.
- B.-W. Pan, Y. Shi, S.-Z. Dong, J.-X. He, B.-S. Mu, W.-B. Wu, Y. Zhou, F. Zhou and J. Zhou, Highly stereoselective synthesis of spirocyclopropylthiooxindoles and biological evaluation, Org. Chem. Front., 2022, 9, 2640 RSC.
- J. K. Cha and O. G. Kulinkovich, in Organic Reactions, 2012, p. 1 Search PubMed.
- Y. A. Konik and D. G. Kananovich, Asymmetric synthesis with titanacyclopropane reagents: From early results to the recent achievements, Tetrahedron Lett., 2020, 61, 152036 CrossRef CAS.
- H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Stereoselective Cyclopropanation Reactions, Chem. Rev., 2003, 103, 977 CrossRef CAS PubMed.
- H.-U. Reissig and R. Zimmer, Donor−Acceptor-Substituted Cyclopropane Derivatives and Their Application in Organic Synthesis, Chem. Rev., 2003, 103, 1151 CrossRef CAS PubMed.
- O. G. Kulinkovich, The Chemistry of Cyclopropanols, Chem. Rev., 2003, 103, 2597 CrossRef CAS PubMed.
- R. D. Little and J. R. Dawson, MIRC (Michael Initiated Ring Closure) Reactions Formation of Three, Five, Six and Seven Membered Rings, Tetrahedron Lett., 1980, 21, 2609 CrossRef CAS.
- G. L. Beutner and D. T. George, Opportunities for the Application and Advancement of the Corey–Chaykovsky Cyclopropanation, Org. Process Res. Dev., 2023, 27, 10 CrossRef CAS.
- A. L. Gabbey, K. Scotchburn and S. A. L. Rousseaux, Metal-catalysed C–C bond formation at cyclopropanes, Nat. Rev. Chem., 2023, 7, 548 CrossRef CAS PubMed.
- R. Munir, A. F. Zahoor, S. Javed, B. Parveen, A. Mansha, A. Irfan, S. G. Khan, A. Irfan, K. Kotwica-Mojzych and M. Mojzych, Simmons–Smith Cyclopropanation: A Multifaceted Synthetic Protocol toward the Synthesis of Natural Products and Drugs: A Review, Molecules, 2023, 28, 5651 CrossRef CAS PubMed.
- E. Sansinenea and A. Ortiz, The Chemistry of Cyclopropanes and New Insights into Organocatalyzed Asymmetric Cyclopropanation, Eur. J. Org. Chem., 2022, e202200210 CrossRef CAS.
- Z.-L. Chen, Y. Xie and J. Xuan, Visible Light-Mediated Cyclopropanation: Recent Progress, Eur. J. Org. Chem., 2022, e202201066 CrossRef CAS.
- C. Apel and M. Christmann, Cyclopropanes in organocatalytic transformations, Tetrahedron, 2021, 82, 131760 CrossRef CAS.
- L. G. Menchikov, E. V. Shulishov and Y. V. Tomilov, Recent advances in the catalytic cyclopropanation of unsaturated compounds with diazomethane, Russ. Chem. Rev., 2021, 90, 199 CrossRef CAS.
- M. Mato, A. Franchino, C. García-Morales and A. M. Echavarren, Gold-Catalyzed Synthesis of Small Rings, Chem. Rev., 2021, 121, 8613 CrossRef CAS PubMed.
- A. Pons, L. Delion, T. Poisson, A. B. Charette and P. Jubault, Asymmetric Synthesis of Fluoro, Fluoromethyl, Difluoromethyl, and Trifluoromethylcyclopropanes, Acc. Chem. Res., 2021, 54, 2969 CrossRef CAS PubMed.
- A. P. Sakla, P. Kansal and N. Shankaraiah, Syntheses and Applications of Spirocyclopropyl Oxindoles: A Decade Review, Eur. J. Org. Chem., 2021, 757 CrossRef CAS.
- E. M. D. Allouche and A. B. Charette, Cyclopropanation Reactions of Semi-stabilized and Non-stabilized Diazo Compounds, Synthesis, 2019, 51, 3947 CrossRef CAS.
- A. G. Herraiz and M. G. Suero, New Alkene Cyclopropanation Reactions Enabled by Photoredox Catalysis via Radical Carbenoids, Synthesis, 2019, 51, 2821 CrossRef CAS.
- Y. V. Tomilov, L. G. Menchikov, R. A. Novikov, O. A. Ivanova and I. V. Trushkov, Methods for the synthesis of donor-acceptor cyclopropanes, Russ. Chem. Rev., 2018, 87, 201 CrossRef CAS.
- D. Y. K. Chen, R. H. Pouwer and J.-A. Richard, Recent advances in the total synthesis of cyclopropane-containing natural products, Chem. Soc. Rev., 2012, 41, 4631 RSC.
- C. Ebner and E. M. Carreira, Cyclopropanation Strategies in Recent Total Syntheses, Chem. Rev., 2017, 117, 11651 CrossRef CAS PubMed.
- M. Bos, T. Poisson, X. Pannecoucke, A. B. Charette and P. Jubault, Recent Progress Toward the Synthesis of Trifluoromethyl- and Difluoromethyl-Substituted Cyclopropanes, Chem. – Eur. J., 2017, 23, 4950 CrossRef CAS PubMed.
- S. Meninno and A. Lattanzi, Asymmetric organocatalytic journey into the world of three-membered rings, Catal. Today, 2017, 285, 39 CrossRef CAS.
- P. I. Arvidsson, P. P. Bose and T. Naicker, in Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, 2015 Search PubMed.
- G. Bartoli, G. Bencivenni and R. Dalpozzo, Asymmetric Cyclopropanation Reactions, Synthesis, 2014, 46, 979 CrossRef.
- H. Pellissier, Recent developments in asymmetric cyclopropanation, Tetrahedron, 2008, 64, 7041 CrossRef CAS.
- S. Ma, D. Mandalapu, S. Wang and Q. Zhang, Biosynthesis of cyclopropane in natural products, Nat. Prod. Rep., 2022, 39, 926 RSC.
- W. Wu, Z. Lin and H. Jiang, Recent advances in the synthesis of cyclopropanes, Org. Biomol. Chem., 2018, 16, 7315 RSC.
- A. Mamai and J. S. Madalengoitia, Lewis acid mediated diastereoselective and enantioselective cyclopropanation of Michael acceptors with sulfur ylides, Tetrahedron Lett., 2000, 41, 9009 CrossRef CAS.
- R. Chinchilla, C. Nájera, S. García-Granda and A. Menéndez-Velázquez, Synthesis of (R)- and (S)-2,3-methanovaline from (2S)-N-benzoyl-2-tert-butyl-4-methylene-1,3-oxazolidin-5-one, Tetrahedron Lett., 1993, 34, 5799 CrossRef CAS.
- S. G. Pyne, K. Schafer, B. W. Skelton and A. H. White, Asymmetric Synthesis of Protected 2-Substituted Cyclopropane Amino Acids, Aust. J. Chem., 1998, 51, 127 CrossRef CAS.
- D. Romo and A. I. Meyers, Chiral non-racemic bicyclic lactams. Vehicles for the construction of natural and unnatural products containing quaternary carbon centers, Tetrahedron, 1991, 47, 9503 CrossRef CAS.
- D. Romo, J. L. Romine, W. Midura and A. I. Meyers, Diastereoselective cyclopropanations of chiral bicyclic lactams leading to enantiomerically pure cyclopropanes. Application to the total synthesis of CIS-(1S, 3R)-deltamethrinic acid and R-(-)- dictyopterene C, Tetrahedron, 1990, 46, 4951 CrossRef CAS.
- A. I. Meyers, J. L. Romine and S. A. Fleming, A novel asymmetric synthesis of substituted cyclopropanes, J. Am. Chem. Soc., 1988, 110, 7245 CrossRef CAS.
- M. Es-Sayed, P. Devine, L. E. Burgess, A. de Meijere and A. I. Meyers, An asymmetric synthesis of novel aminocyclopropyl carboxylic acids (ACC), J. Chem. Soc., Chem. Commun., 1995, 141, 10.1039/C39950000141.
- M. D. Groaning and A. I. Meyers, An asymmetric synthesis of the key precursor to (−)-indolizomycin, Tetrahedron Lett., 1999, 40, 4639 CrossRef CAS.
- A. I. Meyers and D. Romo, Chiral bicyclic lactams. An asymmetric synthesis of cis-(1s, 3r) deltamethrinic acid, Tetrahedron Lett., 1989, 30, 1745 CrossRef CAS.
- D. Romo and A. I. Meyers, An asymmetric route to enantiomerically pure 1,2,3-trisubstituted cyclopropanes, J. Org. Chem., 1992, 57, 6265 CrossRef CAS.
- M. E. Jung and J. J. Chang, Enantiospecific Formal Total Synthesis of (+)-Fawcettimine, Org. Lett., 2010, 12, 2962 CrossRef CAS PubMed.
- R. M. Williams and G. J. Fegley, Asymmetric syntheses of 1-aminocyclopropane-1-carboxylic acid derivatives, J. Am. Chem. Soc., 1991, 113, 8796 CrossRef CAS.
- R. M. Williams and G. J. Fegley, Asymmetric synthesis of (1S,2R)-(+)-2-phenyl-1-aminocyclopropane-1-carboxylic acid, J. Org. Chem., 1993, 58, 6933 CrossRef CAS.
- M. Calmes, J. Daunis and F. Escale, Synthesis of enantiomerically pure 2-alkyl 1-amino cyclopropane-1-carboxylic acid, Tetrahedron: Asymmetry, 1996, 7, 395 CrossRef CAS.
- E. Buñuel, S. D. Bull, S. G. Davies, A. C. Garner, E. D. Savory, A. D. Smith, R. J. Vickers and D. J. Watkin, Asymmetric synthesis of substituted 1-aminocyclopropane-1-carboxylic acids via diketopiperazine methodology, Org. Biomol. Chem., 2003, 1, 2531 RSC.
- W. H. Midura, J. A. Krysiak and M. Mikołajczyk, The first synthesis of enantiomerically pure cyclopropylphosphonate analogues of nucleotides via asymmetric cyclopropanation of chiral (1-diethoxyphosphoryl)vinyl p-tolyl sulfoxide, Tetrahedron: Asymmetry, 2003, 14, 1245 CrossRef CAS.
- W. H. Midura, Asymmetric Cyclopropanation of Optically Active Vinyl Sulfoxides: A New Synthetic Approach to Biologically Active Compounds, Phosphorus, Sulfur Silicon Relat. Elem., 2005, 180, 1285 CrossRef CAS.
- M. Mikołajczyk, Asymmetric cyclopropanation of chiral (1-phosphoryl)vinyl sulfoxides: A new approach to constrained analogs of biologically active compounds, Pure Appl. Chem., 2005, 77, 2091 CrossRef.
- M. Mikołajczyk, Acyclic and cyclic aminophosphonic acids: asymmetric syntheses mediated by chiral sulfinyl auxiliary, J. Organomet. Chem., 2005, 690, 2488 CrossRef.
- W. H. Midura, J. A. Krysiak, M. Cypryk, M. Mikołajczyk, M. W. Wieczorek and A. D. Filipczak, Asymmetric Cyclopropanation of Optically Active (1-Diethoxyphosphoryl)vinyl p-Tolyl Sulfoxide with Sulfur Ylides: A Rationale for Diastereoselectivity, Eur. J. Org. Chem., 2005, 653 CrossRef CAS.
- J. L. García Ruano, C. Fajardo, M. R. Martín, W. Midura and M. Mikołajczyk, Asymmetric cyclopropanation of (5S,SS)-3-p-tolylsulfinyl-5-ethoxyfuran-2(5H)-one with sulfonium ylides: influence of the sulfur ylide substituents on stereoselectivity, Tetrahedron: Asymmetry, 2004, 15, 2475 CrossRef.
- M. Mikołajczyk, W. H. Midura, E. Michedkina, A. D. Filipczak and M. W. Wieczorek, Stereoselective Cyclopropanation of 2-[(S)-(4-Methylphenyl)sulfinyl]cyclopent-2-en-1-one with Sulfur Ylides and α-Halo Carbanions. Preliminary Communication, Helv. Chim. Acta, 2005, 88, 1769 CrossRef.
- E. F. Altenhofer, M. J. Lawler, P. Kumar, L. A. Joyce, M. Fowler-Watters, T. Pei and Z. Li, Synthesis of a novel cyclopropyl phosphonate nucleotide as a phosphate mimic, Chem. Commun., 2021, 57, 6808 RSC.
- P. A. Wender, T. E. Glass, N. E. Krauss, M. Mühlebach, B. Peschke and D. B. Rawlins, The Pinene Path to Taxanes. 4. Approaches to Taxol and Taxol Analogs through Elaboration of Aromatic C-Ring Precursors, J. Org. Chem., 1996, 61, 7662 CrossRef CAS PubMed.
- D. Ma, Y. Cao, Y. Yang and D. Cheng, Stereochemically Controlled Cyclopropanation of (S)-Glyceraldehyde Acetonide-Derived Olefins. Synthesis of (2S,1′R,2′R,3′R)-2-(2′,3′-Dicarboxycyclopropyl)glycine, Org. Lett., 1999, 1, 285 CrossRef CAS.
- D. Ma and Y. Jiang, Stereocontrolled cyclopropanation of Garner's aldehyde derived enones, Tetrahedron: Asymmetry, 2000, 11, 3727 CrossRef CAS.
- D. Ma, Y. Cao, W. Wu and Y. Jiang, Synthesis of (2S,1′R,2′R,3′R)-2-(2′,3′-Dicarboxycyclopropyl)-glycine via the Stereochemically Controlled Cyclopropanation of (S)-Glyceraldehyde Acetonide-Derived Enones, Tetrahedron, 2000, 56, 7447 CrossRef CAS.
- Y. Jiang and D. Ma, Synthesis of enantiopure substituted dihydrofurans via the reaction of (S)-glyceraldehyde acetonide- or Garner aldehyde acetonide-derived enones with sulfonium ylides, Tetrahedron: Asymmetry, 2002, 13, 1033 CrossRef CAS.
- D. K. Mohapatra, S. R. Chaudhuri, G. Sahoo and M. K. Gurjar, Stereoselective synthesis of the polyketide chain of nagahamide A, Tetrahedron: Asymmetry, 2006, 17, 2609 CrossRef CAS.
- N. Saito, M. Masuda, H. Saito, K. Takenouchi, S. Ishizuka, J.-i. Namekawa, M. Takimoto-Kamimura and A. Kittaka, Synthesis of 24,24-Ethanovitamin D3 Lactones Using Ruthenium-Catalyzed Intermolecular Enyne Metathesis: Potent Vitamin D Receptor Antagonists, Synthesis, 2005, 2533 CAS.
- V. Angamuthu, W.-J. Chang and D.-R. Hou, Anti-addition of Dimethylsulfoxonium Methylide to Acyclic α,β-Unsaturated Ketones and Its Application in Formal Synthesis of an Eicosanoid, ACS Omega, 2017, 2, 4088 CrossRef CAS PubMed.
- W. G. Dauben and T. A. Lewis, Enantioselective Synthesis of a Bicyclo[3.1.0]hexane A-Ring Synthon for 1α, 25-Dihydroxyvitamin D3, Synlett, 1995, 857 CrossRef CAS.
- W. Lee and M. J. Miller, Concise Synthesis of 4-Acylamino Analogues of 2-Aminobicyclo[3.1.0]hexane-2,6-dicarboxylic Acids (LY354740) from an Acylnitroso Diels−Alder Cycloadduct, J. Org. Chem., 2004, 69, 4516 CrossRef CAS PubMed.
- W. Wan, G. Ma, W. Gao, J. Wang, L. Li, S. Rao, C. Zheng, H. Jiang, H. Deng and J. Hao, A facile total synthesis of drospirenone isomers containing 14β-hydrogen configuration, Org. Biomol. Chem., 2013, 11, 6597 RSC.
- M. Leiva de Faria, R. de, A. Magalhães, F. C. Silva, L. G. de O. Matias, M. A. Ceschi, U. Brocksom and T. John Brocksom, Enantiodivergent syntheses of cycloheptenone intermediates for guaiane sesquiterpenes, Tetrahedron: Asymmetry, 2000, 11, 4093 CrossRef CAS.
- U. Sakee, N. Kongkathip and B. Kongkathip, The First Syntheses of 16β-Chloro- and 16β-Bromo-cyproterone Acetate, Synth. Commun., 2003, 33, 1695 CrossRef CAS.
- A. V. Samet, A. M. Shestopalov, D. N. Lutov, L. A. Rodinovskaya, A. A. Shestopalov and V. V. Semenov, Preparation of chiral cyclopropanes with a carbohydrate fragment from levoglucosenone, Tetrahedron: Asymmetry, 2007, 18, 1986 CrossRef CAS.
- E. T. Ledingham, C. J. Merritt, C. J. Sumby, M. K. Taylor and B. W. Greatrex, Stereoselective Cyclopropanation of (–)-Levoglucosenone Derivatives Using Sulfonium and Sulfoxonium Ylides, Synthesis, 2017, 49, 2652 CrossRef CAS.
- H. Xiong, B. Chen, T. F. Durand-Réville, C. Joubran, Y. W. Alelyunas, D. Wu and H. Huynh, Enantioselective Synthesis and Profiling of Two Novel Diazabicyclooctanone β-Lactamase Inhibitors, ACS Med. Chem. Lett., 2014, 5, 1143 CrossRef CAS PubMed.
- B. Bechi, D. Amantini, C. Tintori, M. Botta and R. d. Fabio, Stereocontrolled synthesis of 5-azaspiro[2.3]hexane derivatives as conformationally “frozen” analogues of L-glutamic acid, Beilstein J. Org. Chem., 2014, 10, 1114 CrossRef PubMed.
- Y. Matsumura, M. Inoue, Y. Nakamura, I. L. Talib, T. Maki and O. Onomura, A convenient method for synthesis of optically active 2,3-methanopipecolic acid, Tetrahedron Lett., 2000, 41, 4619 CrossRef CAS.
- M. Pohlman and U. Kazmaier, Efficient Stereoselective Syntheses of Cyclic Amino Acids via Michael-Induced Ring-Closing Reactions, Org. Lett., 2003, 5, 2631 CrossRef CAS PubMed.
- A. N. Kasatkin, A. N. Kulak, R. K. Biktimirov and G. A. Tolstikov, Reactions of organomagnesium and organolithium compounds with diethyl (2,3-epoxybutylidene)malonate. A simple synthesis of cyclopropane carboxylates, Tetrahedron Lett., 1990, 31, 4915 CrossRef CAS.
- S. Tang, X. Xie, X. Huo, Q. Liang, X. She and X. Pan, Synthesis of 1,2,3-trisubstituted cyclopropanes by MIRC reactions of dithianyllithiums with monocarboxylic vinyl epoxide analogues, Tetrahedron Lett., 2006, 47, 205 CrossRef CAS.
- R. C. Dhakal and R. K. Dieter, Regio- and Stereocontrol in the Michael-Initiated Ring-Closure Reactions of γ,δ-Epoxy-α,β-unsaturated Esters, Ketones, Sulfones, and Amides, J. Org. Chem., 2013, 78, 12426 CrossRef CAS PubMed.
- A. Krief, W. Dumont, P. Pasau and P. Lecomte, Stereoselective synthesis of methyl (1R) trans- and (1R) cis-hemicaronaldehydes from natural tartaric acid: Application to the synthesis of s-bioallethrin and deltamethrin insecticides§, Tetrahedron, 1989, 45, 3039 CrossRef CAS.
- G. Stork and M. Kahn, A highly stereoselective osmium tetroxide-catalyzed hydroxylation of γ-hydroxy α,β-usaturated esters, Tetrahedron Lett., 1983, 24, 3951 CrossRef CAS.
- A. Krief and P. Lecomte, Streoselective syntheses of cyclopropane derivatives from γ-alkoxy-α,β-unsaturated carbonyl compounds and isopropylidene transfer reagents, Tetrahedron Lett., 1993, 34, 2695 CrossRef CAS.
- A. Krief, L. Provins and A. Froidbise, Diastereoselective synthesis of dimethyl cyclopropane-1,1-dicarboxylates from a γ-alkoxy-alkylidene malonate and sulfur and phosphorus ylides, Tetrahedron Lett., 1998, 39, 1437 CrossRef CAS.
- A. Krief, L. Provins and A. Froidbise, Asymmetric Synthesis of Cyclopropane-1,1-Dicarboxylates from a γ-Alkoxy-Alkylidenemalonate, Synlett, 1999, 1936 CrossRef CAS.
- D. Ma and Z. Ma, Facile Synthesis of (2S,1′S,2′S)-2-(Carboxycyclopropyl)glycine, an Isotype-Selective Agonist of Metabotropic Glutamate Receptors, Tetrahedron Lett., 1997, 38, 7599 CrossRef CAS.
- A. Krief, W. Dumont and P. Pasau, From tartaric acid to the most biologically active insecticides: Straightforward enantioselective synthesis of pyrethrins, Tetrahedron Lett., 1988, 29, 1079 CrossRef CAS.
- A. Krief and W. Dumont, From tartaric acid to the most biologically active insecticides: Straightforward enantioselective synthesis of deltamethrin, Tetrahedron Lett., 1988, 29, 1083 CrossRef CAS.
- R. E. J. Cebula, M. R. Hanna, C. R. Theberge, C. A. Verbicky and C. K. Zercher, Ylide-mediated bis-cyclopropane formation: A reversal in substrate-mediated facial selectivity, Tetrahedron Lett., 1996, 37, 8341 CrossRef CAS.
- T. Duvold, A. Jørgensen, N. R. Andersen, A. S. Henriksen, M. Dahl Sørensen and F. Björkling, 17S,20S-Methanofusidic acid, a new potent semi-synthetic fusidane antibiotic, Bioorg. Med. Chem. Lett., 2002, 12, 3569 CrossRef CAS PubMed.
- R. Chen, L. Li, N. Lin, R. Zhou, Y. Hua, H. Deng and Y. Zhang, Asymmetric Total Synthesis of (+)-Majusculoic Acid via a Dimerization–Dedimerization Strategy and Absolute Configuration Assignment, Org. Lett., 2018, 20, 1477 CrossRef CAS PubMed.
- S. Canova, R. Lépine, A. Thys, A. Baron and D. Roche, Synthesis and biological properties of macrolactam analogs of the natural product macrolide (−)-A26771B, Bioorg. Med. Chem. Lett., 2011, 21, 4768 CrossRef CAS PubMed.
- L.-Q. Lu, T.-R. Li, Q. Wang and W.-J. Xiao, Beyond sulfide-centric catalysis: recent advances in the catalytic cyclization reactions of sulfur ylides, Chem. Soc. Rev., 2017, 46, 4135 RSC.
- C. R. Johnson, E. R. Janiga and M. Haake, Chemistry of sulfoxides and related compounds. X. Ylides from salts of sulfoximines, J. Am. Chem. Soc., 1968, 90, 3890 CrossRef CAS.
- C. R. Johnson and C. W. Schroeck, Chemistry of sulfoxides and related compounds. XV. Synthesis of optically active cyclopropanes and oxiranes using an optically active oxosulfonium methylide, J. Am. Chem. Soc., 1968, 90, 6852 CrossRef CAS.
- C. R. Johnson, M. Haake and C. W. Schroeck, Chemistry of sulfoxides and related compounds. XXVI. Preparation and synthetic applications of (dimethylamino)phenyloxosulfonium methylide, J. Am. Chem. Soc., 1970, 92, 6594 CrossRef CAS.
- C. R. Johnson and J. P. Lockard, (Dimethylamino)-phenyl-(2-phenylvinyl)-oxosulfonium fluoroborate. A model reagent for ethylene transfer to dibasic nucleophiles, Tetrahedron Lett., 1971, 12, 4589 CrossRef.
- C. R. Johnson and C. W. Schroeck, Chemistry of sulfoxides and related compounds. XXXIV. Mechanism of sulfonium ylide reactions. Synthesis of cyclopropanes and oxiranes of high optical purity, J. Am. Chem. Soc., 1971, 93, 5303 CrossRef CAS.
- C. R. Johnson and E. R. Janiga, Chemistry of sulfoxides and related compounds. XLIV. Nucleophilic alkylidene transfer reagents. Ethylides, isopropylides, and cyclopropylides derived from salts of sulfoximines, J. Am. Chem. Soc., 1973, 95, 7692 CrossRef CAS.
- C. R. Johnson and C. W. Schroeck, Chemistry of sulfoxides and related compounds. XLV. Asymmetric syntheses using optically active oxosulfonium alkylides, J. Am. Chem. Soc., 1973, 95, 7418 CrossRef CAS.
- C. R. Johnson, C. W. Schroeck and J. R. Shanklin, Chemistry of sulfoxides and related compounds. XLVIII. Mechanism of nucleophilic alkylidene transfer by sulfonium and oxosulfonium ylides, J. Am. Chem. Soc., 1973, 95, 7424 CrossRef CAS.
- C. R. Johnson and R. W. Herr, Substituent effects in the ring expansion reactions of isopropenylcycloalkanols by tert-butyl hypochlorite, J. Org. Chem., 1973, 38, 3153 CrossRef CAS.
- C. R. Johnson, R. A. Kirchhoff, R. J. Reischer and G. F. Katekar, Chemistry of sulfoxides and related compounds. XLII. Nucleophilic alkylidene transfer reagents. Anions of N-(p-tolylsulfonyl)sulfoximines, J. Am. Chem. Soc., 1973, 95, 4287 CrossRef CAS.
- C. R. Johnson and P. E. Rogers, Preparation and applications of (dialkylamino)methyloxosulfonium methylides. Synthesis of cyclopropanes and oxiranes, J. Org. Chem., 1973, 38, 1793 CrossRef CAS.
- C. R. Johnson, Utilization of sulfoximines and derivatives as reagents for organic synthesis, Acc. Chem. Res., 1973, 6, 341 CrossRef CAS.
- M. R. Barbachyn and C. R. Johnson, in Asymmetric Synthesis, ed. J. D. Morrison and J. W. Scott, Academic Press, 1984, p. 227 Search PubMed.
- A. Solladié-Cavallo, A. Diep-Vohuule and T. Isarno, Two-Step Synthesis of trans-2-Arylcyclopropane Carboxylates with 98–100% ee by the Use of a Phosphazene Base, Angew. Chem., Int. Ed., 1998, 37, 1689 CrossRef.
- V. K. Aggarwal, H. W. Smith, R. V. H. Jones and R. Fieldhouse, Catalytic asymmetric cyclopropanation of electron deficient alkenes mediated by chiral sulfides, Chem. Commun., 1997, 1785 RSC.
- V. K. Aggarwal, H. W. Smith, G. Hynd, R. V. H. Jones, R. Fieldhouse and S. E. Spey, Catalytic cyclopropanation of electron deficient alkenes mediated by chiral and achiral sulfides: scope and limitations in reactions involving phenyldiazomethane and ethyl diazoacetate, J. Chem. Soc., Perkin Trans. 1, 2000, 3267 RSC.
- V. K. Aggarwal, E. Alonso, G. Fang, M. Ferrara, G. Hynd and M. Porcelloni, Application of Chiral Sulfides to Catalytic Asymmetric Aziridination and Cyclopropanation with In Situ Generation of the Diazo Compound, Angew. Chem., Int. Ed., 2001, 40, 1433 CrossRef CAS PubMed.
- V. K. Aggarwal and E. Grange, Asymmetric Sulfonium Ylide Mediated Cyclopropanation: Stereocontrolled Synthesis of (+)-LY354740, Chem. – Eur. J., 2006, 12, 568 CrossRef PubMed.
- S. Ye, Z.-Z. Huang, C.-A. Xia, Y. Tang and L.-X. Dai, A Novel Chiral Sulfonium Yilde: Highly Enantioselective Synthesis of Vinylcyclopropanes, J. Am. Chem. Soc., 2002, 124, 2432 CrossRef CAS PubMed.
- X.-M. Deng, P. Cai, S. Ye, X.-L. Sun, W.-W. Liao, K. Li, Y. Tang, Y.-D. Wu and L.-X. Dai, Enantioselective Synthesis of Vinylcyclopropanes and Vinylepoxides Mediated by Camphor-Derived Sulfur Ylides: Rationale of Enantioselectivity, Scope, and Limitation, J. Am. Chem. Soc., 2006, 128, 9730 CrossRef CAS PubMed.
- K. Huang and Z.-Z. Huang, A Practical and Controllable Enantioselective Synthesis of 2-Phenyl-1-cyclopropanecarboxylates via a Camphor-Derived Sulfonium Ylide, Synlett, 2005, 1621 CrossRef CAS.
- S. Hanessian, D. Andreotti and A. Gomtsyan, Asymmetric synthesis of enantiomerically pure and diversely functionalized cyclopropanes, J. Am. Chem. Soc., 1995, 117, 10393 CrossRef CAS.
- S. Hanessian, L.-D. Cantin, S. Roy, D. Andreotti and A. Gomtsyan, The synthesis of enantiomerically pure, symmetrically substituted cyclopropane phosphonic acids—A constrained analog of the GABA antagonist phaclophen, Tetrahedron Lett., 1997, 38, 1103 CrossRef CAS.
- S. Hanessian, A. Griffin and P. V. Devasthale, Synthesis of conformationally constrained potential inhibitors of mammalian metalloproteinases, Bioorg. Med. Chem. Lett., 1997, 7, 3119 CrossRef CAS.
- S. Goumain, H. Oulyadi, P. Jubault, C. Feasson and J.-C. Quirion, Attempts at asymmetric electrosynthesis of α-fluorinated cyclopropylphosphonamides, J. Chem. Soc., Perkin Trans. 1, 2001, 701 RSC.
- C. Duquenne, S. Goumain, P. Jubault, C. Feasson and J.-C. Quirion, Electrosynthesis of α-Arylated β-Substituted Cyclopropylphosphonates. Synthesis of a Phosphonic Analogue of Minalcipran, Org. Lett., 2000, 2, 453 CrossRef CAS PubMed.
- S. Goumain, P. Jubault, C. Feasson and J.-C. Quirion, First synthesis of α-fluorinated cyclopropylphosphonates using magnesium electrochemical activation, Tetrahedron Lett., 1999, 40, 8099 CrossRef CAS.
- M. Marinozzi and R. Pellicciari, Novel enantioselective synthesis of (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine (DCG-IV), Tetrahedron Lett., 2000, 41, 9125 CrossRef CAS.
- S. Kojima, K. Fujitomo, Y. Shinohara, M. Shimizu and K. Ohkata, Stereoselective formation of activated cyclopropanes with pyridinium ylides bearing a (−)-8-phenylmenthyl group as the chiral auxiliary, Tetrahedron Lett., 2000, 41, 9847 CrossRef CAS.
- S. Kojima, K. Hiroike and K. Ohkata, Stereoselective synthesis of activated cyclopropanes with an α-pyridinium acetamide bearing an 8-phenylmenthyl group as the chiral auxiliary, Tetrahedron Lett., 2004, 45, 3565 CrossRef CAS.
- N. Kanomata, R. Sakaguchi, K. Sekine, S. Yamashita and H. Tanaka, Enantioselective Cyclopropanation Reactions with Planar-Chiral Pyridinium Ylides: A Substituent Effect and a Remote Steric Effect, Adv. Synth. Catal., 2010, 352, 2966 CrossRef CAS.
- S. Yamada, J. Yamamoto and E. Ohta, Enantioselective cyclopropanation reaction using a conformationally fixed pyridinium ylide through a cation–π interaction, Tetrahedron Lett., 2007, 48, 855 CrossRef CAS.
- F. Couty, O. David, B. Larmanjat and J. Marrot, Strained Azetidinium Ylides: New Reagents for Cyclopropanation, J. Org. Chem., 2007, 72, 1058 CrossRef CAS PubMed.
- W.-W. Liao, K. Li and Y. Tang, Controllable Diastereoselective Cyclopropanation. Enantioselective Synthesis of Vinylcyclopropanes via Chiral Telluronium Ylides, J. Am. Chem. Soc., 2003, 125, 13030 CrossRef CAS PubMed.
- A. J. Walker, Asymmetric carbon-carbon bond formation using sulfoxide-stabilised carbanions, Tetrahedron: Asymmetry, 1992, 3, 961 CrossRef CAS.
- S. G. Pyne, Z. Dong, B. W. Skelton and A. H. White, Cyclopropanation Reactions of Enones with Lithiated Sulfoximines: Application to the Asymmetric Synthesis of Chiral Cyclopropanes, J. Org. Chem., 1997, 62, 2337 CrossRef CAS PubMed.
- T. Toru, S. Nakamura, H. Takemoto and Y. Ueno, Stereoselective Conjugate Addition of an α-Sulfinyl Carbanion to α,β-Unsaturated Esters: Asymmetric Synthesis of Cycloalkanecarboxylates, Synlett, 1997, 449 CrossRef CAS.
- S. Nakamura, Y. Watanabe and T. Toru, Efficient synthesis of chrysanthemate precursor from chiral p-tolyl β-(trimethylsilyl)ethyl sulfoxide, J. Chem. Soc., Perkin Trans. 1, 1999, 3403 RSC.
- N. Shinohara, J. Haga, T. Yamazaki, T. Kitazume and S. Nakamura, Stereoselective Synthesis of Trifluoromethylated Compounds with Controlled Adjacent Tertiary Carbons by Michael Addition to (E)-3-(Trifluoromethyl)acrylates, J. Org. Chem., 1995, 60, 4363 CrossRef CAS.
- P. Ivashkin, S. Couve-Bonnaire, P. Jubault and X. Pannecoucke, Asymmetric Synthesis of Cyclopropanes with a Monofluorinated Quaternary Stereocenter, Org. Lett., 2012, 14, 5130 CrossRef CAS PubMed.
- B. List, R. A. Lerner and C. F. Barbas, Proline-Catalyzed Direct Asymmetric Aldol Reactions, J. Am. Chem. Soc., 2000, 122, 2395 CrossRef CAS.
- K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, New Strategies for Organic Catalysis: The First Highly Enantioselective Organocatalytic Diels−Alder Reaction, J. Am. Chem. Soc., 2000, 122, 4243 CrossRef CAS.
- R. Ballini, A. Palmieri and M. Petrini, Catalysts’ evolution in the asymmetric conjugate addition of nitroalkanes to electron-poor alkenes, Org. Chem. Front., 2022, 9, 6077 RSC.
- S. Arai, K. Nakayama, T. Ishida and T. Shioiri, Asymmetric cyclopropanation reaction Under phase-transfer catalyzed conditions, Tetrahedron Lett., 1999, 40, 4215 CrossRef CAS.
- C. D. Papageorgiou, S. V. Ley and M. J. Gaunt, Organic-Catalyst-Mediated Cyclopropanation Reaction, Angew. Chem., Int. Ed., 2003, 42, 828 CrossRef CAS PubMed.
- C. D. Papageorgiou, M. A. Cubillo de Dios, S. V. Ley and M. J. Gaunt, Enantioselective Organocatalytic Cyclopropanation via Ammonium Ylides, Angew. Chem., Int. Ed., 2004, 43, 4641 CrossRef CAS PubMed.
- N. Bremeyer, S. C. Smith, S. V. Ley and M. J. Gaunt, An Intramolecular Organocatalytic Cyclopropanation Reaction, Angew. Chem., Int. Ed., 2004, 43, 2681 CrossRef CAS PubMed.
- C. C. C. Johansson, N. Bremeyer, S. V. Ley, D. R. Owen, S. C. Smith and M. J. Gaunt, Enantioselective Catalytic Intramolecular Cyclopropanation using Modified Cinchona Alkaloid Organocatalysts, Angew. Chem., Int. Ed., 2006, 45, 6024 CrossRef CAS PubMed.
- S. Kojima, M. Suzuki, A. Watanabe and K. Ohkata, Asymmetric synthesis of activated cyclopropanes catalyzed by cinchonidine as a chiral Brønsted base, Tetrahedron Lett., 2006, 47, 9061 CrossRef CAS.
- G. Kumaraswamy and M. Padmaja, Enantioselective Total Synthesis of Eicosanoid and Its Congener, Using Organocatalytic Cyclopropanation, and Catalytic Asymmetric Transfer Hydrogenation Reactions as Key Steps, J. Org. Chem., 2008, 73, 5198 CrossRef CAS PubMed.
- G. Kumaraswamy, G. Ramakrishna and B. Sridhar, Enantioselective synthesis of cyclopropyl δ-lactonealdehydes and dodecyl-5-ene-1-yne-3-ol: advanced intermediates of solandelactone A and B, Tetrahedron Lett., 2011, 52, 1778 CrossRef CAS.
- Y.-n. Xuan, S.-z. Nie, L.-t. Dong, J.-m. Zhang and M. Yan, Highly Enantioselective Synthesis of Nitrocyclopropanes via Organocatalytic Conjugate Addition of Bromomalonate to α,β-Unsaturated Nitroalkenes, Org. Lett., 2009, 11, 1583 CrossRef CAS PubMed.
- J. Lv, J. Zhang, Z. Lin and Y. Wang, Enantioselective Synthesis of Functionalized Nitrocyclopropanes by Organocatalytic Conjugate Addition of Bromonitroalkanes to α,β-Unsaturated Enones, Chem. – Eur. J., 2009, 15, 972 CrossRef CAS PubMed.
- C. D. Fiandra, L. Piras, F. Fini, P. Disetti, M. Moccia and M. F. A. Adamo, Phase transfer catalyzed enantioselective cyclopropanation of 4-nitro-5-styrylisoxazoles, Chem. Commun., 2012, 48, 3863 RSC.
- L. Piras, M. Moccia, M. Cortigiani and M. F. A. Adamo, Catalysts, 2015, 5, 595 CrossRef CAS.
- R. Herchl and M. Waser, Asymmetric cyclopropanation of chalcones using chiral phase-transfer catalysts, Tetrahedron Lett., 2013, 54, 2472 CrossRef CAS PubMed.
- L. S. Aitken, L. E. Hammond, R. Sundaram, K. Shankland, G. D. Brown and A. J. A. Cobb, Asymmetric cyclopropanation of conjugated cyanosulfones using a novel cupreine organocatalyst: rapid access to δ3-amino acids, Chem. Commun., 2015, 51, 13558 RSC.
- C. Del Fiandra, M. Moccia and M. F. A. Adamo, Enantioselective cyclopropanation of (Z)-3-substituted-2-(4-pyridyl)-acrylonitriles catalyzed by Cinchona ammonium salts, Org. Biomol. Chem., 2016, 14, 3105 RSC.
- J.-P. Li, G.-F. Zhao, H.-X. Wang, M.-S. Xie, G.-R. Qu and H.-M. Guo, Highly Enantioselective Synthesis of Chiral Cyclopropyl Nucleosides via Catalytic Asymmetric Intermolecular Cyclopropanation, Org. Lett., 2017, 19, 6494 CrossRef CAS PubMed.
- H.-X. Wang, W.-P. Li, C. Xia, M.-S. Xie, G.-R. Qu and H.-M. Guo, Enantioselective synthesis of chiral carbocyclic pyrimidine nucleosides via asymmetric cyclopropanation, Tetrahedron Lett., 2019, 60, 151183 CrossRef CAS.
- L. Roiser and M. Waser, Enantioselective Spirocyclopropanation of para-Quinone Methides Using Ammonium Ylides, Org. Lett., 2017, 19, 2338 CrossRef CAS PubMed.
- J.-C. Sun, J.-L. Li, C.-B. Ji, Y.-Y. Peng and X.-P. Zeng, Construction of Cyclopropa[c]coumarins via cascade Michael-alkylation process of 3-cyanocoumarin with 2-bromomalonate, Tetrahedron, 2020, 76, 130852 CrossRef CAS.
- J.-C. Sun, X.-H. Wang, C.-B. Ji, Y.-Y. Peng and X.-P. Zeng, Enantioselective Construction of Chiral Cyclopropa[c]coumarins via Lewis Base-Catalyzed Cyclopropanation, J. Org. Chem., 2020, 85, 14963 CrossRef CAS PubMed.
- L. Carceller-Ferrer, G. Blay, M. C. Muñoz, J. R. Pedro and C. Vila, Diastereo- and Enantioselective Organocatalytic Synthesis of Spirocyclopropyl Pyrazolones, Adv. Synth. Catal., 2024, 366, 698 CrossRef CAS.
- A. Manna, H. Joshi and V. K. Singh, Asymmetric Synthesis of Spiro-3,3′-cyclopropyl Oxindoles via Vinylogous Michael Initiated Ring Closure Reaction, J. Org. Chem., 2022, 87, 16755 CrossRef CAS PubMed.
- A. Manna, H. Joshi and V. K. Singh, Organocatalytic Asymmetric Direct Vinylogous Michael Initiated Ring Closure Reaction of 4-Nitroisoxazole Derivatives to 3-Isopropylidene Oxindoles, J. Org. Chem., 2023, 88, 15937 CrossRef CAS PubMed.
- S. H. McCooey, T. McCabe and S. J. Connon, Stereoselective Synthesis of Highly Functionalized Nitrocyclopropanes via Organocatalyic Conjugate Addition to Nitroalkenes, J. Org. Chem., 2006, 71, 7494 CrossRef CAS PubMed.
- F. Marini, S. Sternativo, F. Del Verme, L. Testaferri and M. Tiecco, A New Stereoselective Synthesis of Cyclopropanes Containing Quaternary Stereocentres via Organocatalytic Michael Addition to Vinyl Selenones, Adv. Synth. Catal., 2009, 351, 1801 CrossRef CAS.
- F. Pesciaioli, P. Righi, A. Mazzanti, G. Bartoli and G. Bencivenni, Organocatalytic Michael–Alkylation Cascade: The Enantioselective Nitrocyclopropanation of Oxindoles, Chem. – Eur. J., 2011, 17, 2842 CrossRef CAS PubMed.
- A. Russo and A. Lattanzi, Base promoted synthesis of activated cyclopropanes bearing homologated carbonyl groups via tandem Michael addition–intramolecular enolate trapping, Org. Biomol. Chem., 2011, 9, 7993 RSC.
- A. Noole, N. S. Sucman, M. A. Kabeshov, T. Kanger, F. Z. Macaev and A. V. Malkov, Highly Enantio- and Diastereoselective Generation of Two Quaternary Centers in Spirocyclopropanation of Oxindole Derivatives, Chem. – Eur. J., 2012, 18, 14929 CrossRef CAS PubMed.
- X. Dou and Y. Lu, Diastereodivergent Synthesis of 3-Spirocyclopropyl-2-oxindoles through Direct Enantioselective Cyclopropanation of Oxindoles, Chem. – Eur. J., 2012, 18, 8315 CrossRef CAS PubMed.
- U. Das, Y.-L. Tsai and W. Lin, An efficient organocatalytic enantioselective synthesis of spironitrocyclopropanes, Org. Biomol. Chem., 2013, 11, 44 RSC.
- M. Ošeka, A. Noole, S. Žari, M. Öeren, I. Järving, M. Lopp and T. Kanger, Asymmetric Diastereoselective Synthesis of Spirocyclopropane Derivatives of Oxindole, Eur. J. Org. Chem., 2014, 3599 CrossRef.
- N. Wang, X. Yan, Z.-T. Hu, Y. Feng, L. Zhu, Z.-H. Chen, H. Wang, Q.-L. Wang, Q. Ouyang and P.-F. Zheng, Intramolecular H-Bonds in an Organocatalyst Enabled an Asymmetric Michael/Alkylation Cascade Reaction to Construct Spirooxindoles Incorporating a Densely Substituted Cyclopropane Motif, Org. Lett., 2022, 24, 8553 CrossRef CAS PubMed.
- J.-H. Li, T.-F. Feng and D.-M. Du, Construction of Spirocyclopropane-Linked Heterocycles Containing Both Pyrazolones and Oxindoles through Michael/Alkylation Cascade Reactions, J. Org. Chem., 2015, 80, 11369 CrossRef CAS PubMed.
- Y.-X. Song and D.-M. Du, Asymmetric synthesis of spirooxindole-fused spirothiazolones via squaramide-catalysed reaction of 3-chlorooxindoles with 5-alkenyl thiazolones, Org. Biomol. Chem., 2019, 17, 5375 RSC.
- B.-L. Zhao and D.-M. Du, Chiral Squaramide-Catalyzed Michael/-Alkylation Cascade Reaction for the Asymmetric Synthesis of Nitro-Spirocyclopropanes, Eur. J. Org. Chem., 2015, 5350 CrossRef CAS.
- W.-C. Yuan, C.-W. Lei, J.-Q. Zhao, Z.-H. Wang and Y. You, Organocatalytic Asymmetric Cyclopropanation of 3-Acylcoumarins with 3-Halooxindoles: Access to Spirooxindole-cyclopropa[c]coumarin Compounds, J. Org. Chem., 2021, 86, 2534 CrossRef CAS PubMed.
- Z.-F. Hao, S.-J. Zhu, Y.-J. Hao, W.-H. Zhang, Y. Zhou, Y.-P. Tian and C.-W. Lei, Enantioselective Synthesis of Bispiro[indanedione-oxindole-cyclopropane]s through Organocatalytic [2 + 1] Cycloaddition, J. Org. Chem., 2023, 88, 7641 CrossRef CAS PubMed.
- R. Rios, H. Sundén, J. Vesely, G.-L. Zhao, P. Dziedzic and A. Córdova, A Simple Organocatalytic Enantioselective Cyclopropanation of α,β-Unsaturated Aldehydes, Adv. Synth. Catal., 2007, 349, 1028 CrossRef CAS.
- I. Ibrahem, G.-L. Zhao, R. Rios, J. Vesely, H. Sundén, P. Dziedzic and A. Córdova, One-Pot Organocatalytic Domino Michael/α-Alkylation Reactions: Direct Catalytic Enantioselective Cyclopropanation and Cyclopentanation Reactions, Chem. – Eur. J., 2008, 14, 7867 CrossRef CAS PubMed.
- H. Xie, L. Zu, H. Li, J. Wang and W. Wang, Organocatalytic Enantioselective Cascade Michael-Alkylation Reactions: Synthesis of Chiral Cyclopropanes and Investigation of Unexpected Organocatalyzed Stereoselective Ring Opening of Cyclopropanes, J. Am. Chem. Soc., 2007, 129, 10886 CrossRef CAS PubMed.
- X. Companyó, A.-N. Alba, F. Cárdenas, A. Moyano and R. Rios, Asymmetric Organocatalytic Cyclopropanation – Highly Stereocontrolled Synthesis of Chiral Cyclopropanes with Quaternary Stereocenters, Eur. J. Org. Chem., 2009, 3075 CrossRef.
- V. Terrasson, A. van der Lee, R. Marcia de Figueiredo and J. M. Campagne, Organocatalyzed Cyclopropanation of α-Substituted α,β-Unsaturated Aldehydes: Enantioselective Synthesis of Cyclopropanes Bearing a Chiral Quaternary Center, Chem. – Eur. J., 2010, 16, 7875 CrossRef CAS PubMed.
- C. Kim and S.-G. Kim, Construction of chiral cyclopropane-fused tetrahydroquinolines: enantioselective organocatalytic Michael/alkylation domino reaction and one-pot aza-cyclization, Tetrahedron: Asymmetry, 2014, 25, 1376 CrossRef CAS.
- J. Ito, D. Sakuma and Y. Nishii, First Total Synthesis of (+)-Podophyllic Aldehydes, Chem. Lett., 2015, 44, 297 CrossRef CAS.
- J. Han, H.-J. Ha and J. W. Yang, Stereoselective Synthesis of Contiguous Cyclopropylaziridines: Enabling the Construction of Versatile Chiral Molecules Including γ-Lactams via Ring-Opening Reactions, Adv. Synth. Catal., 2023, 365, 1970 CrossRef CAS.
- J. I. Martínez, E. Reyes, U. Uria, L. Carrillo and J. L. Vicario, Optimizing the Structure of 4-Dialkylamino-α,α-diarylprolinol Ethers as Catalysts for the Enantioselective Cyclopropanation of α,β-Unsaturated Aldehydes in Water, ChemCatChem, 2013, 5, 2240 CrossRef.
- P. Llanes, C. Rodríguez-Escrich, S. Sayalero and M. A. Pericàs, Organocatalytic Enantioselective Continuous-Flow Cyclopropanation, Org. Lett., 2016, 18, 6292 CrossRef CAS PubMed.
- A. M. Faísca Phillips and M. T. Barros, Enantioselective Organocatalytic Synthesis of α-Cyclopropylphosphonates through a Domino Michael Addition/Intramolecular Alkylation Reaction, Eur. J. Org. Chem., 2014, 152 CrossRef.
- W. Li, X. Li, T. Ye, W. Wu, X. Liang and J. Ye, Asymmetric organocatalytic Michael/α-alkylation reaction of α,β-unsaturated aldehyde with chloroacetophenone, Tetrahedron Lett., 2011, 52, 2715 CrossRef CAS.
- J. Vesely, G.-L. Zhao, A. Bartoszewicz and A. Córdova, Organocatalytic asymmetric nitrocyclopropanation of α,β-unsaturated aldehydes, Tetrahedron Lett., 2008, 49, 4209 CrossRef CAS.
- J.-m. Zhang, Z.-p. Hu, L.-t. Dong, Y.-n. Xuan, C.-L. Lou and M. Yan, Enantioselective conjugate addition of 1-bromonitroalkanes to α,β-unsaturated aldehydes catalyzed by chiral secondary amines, Tetrahedron: Asymmetry, 2009, 20, 355 CrossRef CAS.
- A. Zaghi, T. Bernardi, V. Bertolasi, O. Bortolini, A. Massi and C. De Risi, One-Pot, Four-Step Organocatalytic Asymmetric Synthesis of Functionalized Nitrocyclopropanes, J. Org. Chem., 2015, 80, 9176 CrossRef CAS PubMed.
- A. Noole, M. Ošeka, T. Pehk, M. Öeren, I. Järving, M. R. J. Elsegood, A. V. Malkov, M. Lopp and T. Kanger, 3-Chlorooxindoles: Versatile Starting Materials for Asymmetric Organocatalytic Synthesis of Spirooxindoles, Adv. Synth. Catal., 2013, 355, 829 CrossRef CAS.
- R. César da Silva, I. Chatterjee, E. Escudero-Adán, M. Weber Paixão and P. Melchiorre, Synthesis of Cyclopropane Spirooxindoles by means of a Vinylogous Organocatalytic Cascade, Asian J. Org. Chem., 2014, 3, 466 CrossRef.
- M. Meazza, M. Ashe, H. Y. Shin, H. S. Yang, A. Mazzanti, J. W. Yang and R. Rios, Enantioselective Organocatalytic Cyclopropanation of Enals Using Benzyl Chlorides, J. Org. Chem., 2016, 81, 3488 CrossRef CAS PubMed.
- X. Chen, Y. Yu, Z. Liao, H. Li and W. Wang, Enantioselective synthesis of diarylcyclopropanecarboaldehydes by organocatalysis, Tetrahedron Lett., 2016, 57, 5742 CrossRef CAS.
- V. Dočekal, S. Petrželová, I. Císařová and J. Veselý, Enantioselective Cyclopropanation of 4-Nitroisoxazole Derivatives, Adv. Synth. Catal., 2020, 362, 2597 CrossRef.
- V. Dočekal, T. Koberová, J. Hrabovský, A. Vopálenská, R. Gyepes, I. Císařová, R. Rios and J. Veselý, Stereoselective Cyclopropanation of Boron Dipyrromethene (BODIPY) Derivatives by an Organocascade Reaction, Adv. Synth. Catal., 2022, 364, 930 CrossRef.
- G. D. Bisag, P. Pecchini, M. Mancinelli, M. Fochi and L. Bernardi, Sulfoxonium Ylides in Aminocatalysis: An Enantioselective Entry to Cyclopropane-Fused Chromanol Structures, Org. Lett., 2022, 24, 5468 CrossRef CAS PubMed.
- Y.-H. Zhao, G. Zhao and W.-G. Cao, Organocatalytic asymmetric cyclopropanation of α,β-unsaturated aldehydes with arsonium ylides, Tetrahedron: Asymmetry, 2007, 18, 2462 CrossRef CAS.
- A. Russo, S. Meninno, C. Tedesco and A. Lattanzi, Synthesis of Activated Cyclopropanes by an MIRC Strategy: An Enantioselective Organocatalytic Approach to Spirocyclopropanes, Eur. J. Org. Chem., 2011, 5096 CrossRef CAS.
- A. Russo and A. Lattanzi, Stereoselective synthesis of functionalised cyclopropanes from nitroalkenes via an organocatalysed Michael-initiated ring-closure approach, Tetrahedron: Asymmetry, 2010, 21, 1155 CrossRef CAS.
- J. Luis-Barrera, R. Mas-Ballesté and J. Alemán, One-Pot Asymmetric Synthesis of Cyclopropanes with Quaternary Centers Starting From Bromonitroalkenes under Aminocatalytic Conditions, ChemPlusChem, 2015, 80, 1595 CrossRef CAS PubMed.
- A. Noole, A. V. Malkov and T. Kanger, Asymmetric Organocatalytic Synthesis of Spiro-cyclopropaneoxindoles, Synthesis, 2013, 45, 2520 CrossRef CAS.
- Y. Cheng, J. An, L.-Q. Lu, L. Luo, Z.-Y. Wang, J.-R. Chen and W.-J. Xiao, Asymmetric Cyclopropanation of β,γ-Unsaturated α-Ketoesters with Stabilized Sulfur Ylides Catalyzed by C2-Symmetric Ureas, J. Org. Chem., 2011, 76, 281 CrossRef CAS PubMed.
- R. Fan, Y. Ye, W. Li and L. Wang, Efficient Stereoselective Synthesis of Nitrocyclopropanes by the Oxidative Cyclization of Michael Adducts of Nitroolefins with Activated Methylene Compounds, Adv. Synth. Catal., 2008, 350, 2488 CrossRef CAS.
- T. Inokuma, S. Sakamoto and Y. Takemoto, Enantioselective Nitrocyclopropanation of α,β-Unsaturated α-Cyanoimides Catalyzed by Bifunctional Thiourea, Synlett, 2009, 1627 CAS.
- L.-t. Dong, Q.-s. Du, C.-l. Lou, J.-m. Zhang, R.-j. Lu and M. Yan, Asymmetric Synthesis of Nitrocyclopropanes Catalyzed by Chiral Primary Amines, Synlett, 2010, 266 CAS.
- P. Bakó, Z. Rapi, A. Grün, T. Nemcsok, L. Hegedűs and G. Keglevich, Asymmetric Michael Addition of Malonates to Enones Catalyzed by an α-d-Glucopyranoside-Based Crown Ether, Synlett, 2015, 26, 1847 CrossRef.
- Z. Rapi, T. Nemcsok, Á. Pálvölgyi, G. Keglevich, A. Grün and P. Bakó, Synthesis of l-threitol-based crown ethers and their application as enantioselective phase transfer catalyst in Michael additions, Chirality, 2017, 29, 257 CrossRef CAS PubMed.
- Z. Rapi, T. Nemcsok, A. Grün, Á. Pálvölgyi, G. Samu, D. Hessz, M. Kubinyi, M. Kállay, G. Keglevich and P. Bakó, Asymmetric cyclopropanation reactions catalyzed by carbohydrate-based crown ethers, Tetrahedron, 2018, 74, 3512 CrossRef CAS.
- T. Nemcsok, Z. Rapi, P. Bagi, Y. H. Guan, I. Orbán, G. Keglevich and P. Bakó, Enantioselective cyclopropanation of conjugated cyanosulfones using carbohydrate-based crown ether catalysts, Tetrahedron, 2020, 76, 130965 CrossRef CAS.
- L. Gao, G.-S. Hwang and D. H. Ryu, Oxazaborolidinium Ion-Catalyzed Cyclopropanation of α-Substituted Acroleins: Enantioselective Synthesis of Cyclopropanes Bearing Two Chiral Quaternary Centers, J. Am. Chem. Soc., 2011, 133, 20708 CrossRef CAS PubMed.
- S. Y. Shim, J. Y. Kim, M. Nam, G.-S. Hwang and D. H. Ryu, Enantioselective Cyclopropanation with α-Alkyl-α-diazoesters Catalyzed by Chiral Oxazaborolidinium Ion: Total Synthesis of (+)-Hamavellone B, Org. Lett., 2016, 18, 160 CrossRef CAS PubMed.
- R. K. Kunz and D. W. C. MacMillan, Enantioselective Organocatalytic Cyclopropanations. The Identification of a New Class of Iminium Catalyst Based upon Directed Electrostatic Activation, J. Am. Chem. Soc., 2005, 127, 3240 CrossRef CAS PubMed.
- H. M. Hansen, D. A. Longbottom and S. V. Ley, A new asymmetric organocatalytic nitrocyclopropanation reaction, Chem. Commun., 2006, 4838 RSC.
- V. Wascholowski, H. M. Hansen, D. A. Longbottom and S. V. Ley, A General Organocatalytic Enantioselective Nitrocyclopropanation Reaction, Synthesis, 2008, 1269 CAS.
- A. Hartikka and P. I. Arvidsson, Tetrazolic Acid Functionalized Dihydroindol: Rational Design of a Highly Selective Cyclopropanation Organocatalyst, J. Org. Chem., 2007, 72, 5874 CrossRef CAS PubMed.
- A. Hartikka, A. T. Ślósarczyk and P. I. Arvidsson, Application of novel sulfonamides in enantioselective organocatalyzed cyclopropanation, Tetrahedron: Asymmetry, 2007, 18, 1403 CrossRef CAS.
- K. Akagawa, S. Takigawa, I. S. Nagamine, R. Umezawa and K. Kudo, Peptide-Catalyzed Diastereo- and Enantioselective Cyclopropanation of Aromatic α,β-Unsaturated Aldehydes, Org. Lett., 2013, 15, 4964 CrossRef CAS PubMed.
- J. Wang, X. Liu, S. Dong, L. Lin and X. Feng, Asymmetric Organocatalytic Cyclopropanation of Cinnamone Derivatives with Stabilized Sulfonium Ylides, J. Org. Chem., 2013, 78, 6322 CrossRef CAS PubMed.
- P.-T. Cheng, Y.-H. Tseng and R.-J. Chein, Organoselenium-Catalyzed Asymmetric Cyclopropanations of (E)-Chalcones, Org. Lett., 2021, 23, 8104 CrossRef CAS PubMed.
- Y.-C. Guo, K.-W. Chiu and R.-J. Chein, (Thiolan-2-yl)diphenylmethanol Benzyl Ether-Catalyzed Asymmetric Cyclopropanation of Chalcones, J. Org. Chem., 2023, 88, 559 CrossRef CAS PubMed.
- Q.-Z. Li, X. Zhang, R. Zeng, Q.-S. Dai, Y. Liu, X.-D. Shen, H.-J. Leng, K.-C. Yang and J.-L. Li, Direct Sulfide-Catalyzed Enantioselective Cyclopropanations of Electron-Deficient Dienes and Bromides, Org. Lett., 2018, 20, 3700 CrossRef CAS PubMed.
- A. Biswas, S. De Sarkar, L. Tebben and A. Studer, Enantioselective cyclopropanation of enals by oxidative N-heterocyclic carbene catalysis, Chem. Commun., 2012, 48, 5190 RSC.
- G.-J. Sun, J. Gong and Q. Kang, Chiral Rhodium(III) Complex-Catalyzed Cascade Michael-Alkylation Reactions: Enantioselective Synthesis of Cyclopropanes, J. Org. Chem., 2017, 82, 796 CrossRef CAS PubMed.
- S. Ming, J. Yang, S. Wu, G. Yao, H. Xiong, Y. Du and J. Gong, Catalytic asymmetric cyclopropanation of sulfoxonium ylides catalyzed by a chiral-at-metal rhodium complex, Org. Chem. Front., 2022, 9, 5147 RSC.
- J. Pian, Q. Chen, Y. Luo, Z. Zhao, J. Liu, L. He and S.-W. Li, Asymmetric Synthesis of Chiral Cyclopropanes from Sulfoxonium Ylides Catalyzed by a Chiral-at-Metal Rh(III) Complex, Org. Lett., 2022, 24, 5641 CrossRef CAS PubMed.
- C. Duchemin and N. Cramer, Chiral cyclopentadienyl RhIII-catalyzed enantioselective cyclopropanation of electron-deficient olefins enable rapid access to UPF-648 and oxylipin natural products, Chem. Sci., 2019, 10, 2773 RSC.
- L. Wang, W. Cao, H. Mei, L. Hu and X. Feng, Catalytic Asymmetric Synthesis of Chiral Spiro-cyclopropyl Oxindoles from 3-Alkenyl-oxindoles and Sulfoxonium Ylides, Adv. Synth. Catal., 2018, 360, 4089 CrossRef CAS.
- H. Mei, G. Pan, X. Zhang, L. Lin, X. Liu and X. Feng, Catalytic Asymmetric Ring-Opening/Cyclopropanation of Cyclic Sulfur Ylides: Construction of Sulfur-Containing Spirocyclopropyloxindoles with Three Vicinal Stereocenters, Org. Lett., 2018, 20, 7794 CrossRef CAS PubMed.
- S. K. Pagire, N. Kumagai and M. Shibasaki, Highly Enantio- and Diastereoselective Synthesis of 1,2,3-Trisubstituted Cyclopropanes from α,β-Unsaturated Amides and Stabilized Sulfur Ylides Catalyzed by a Chiral Copper(I) Complex, ACS Catal., 2021, 11, 11597 CrossRef.
- T. den Hartog, A. Rudolph, B. Maciá, A. J. Minnaard and B. L. Feringa, Copper-Catalyzed Enantioselective Synthesis of trans-1-Alkyl-2-substituted Cyclopropanes via Tandem Conjugate Addition−Intramolecular Enolate Trapping, J. Am. Chem. Soc., 2010, 132, 14349 CrossRef CAS PubMed.
- H. Kakei, T. Sone, Y. Sohtome, S. Matsunaga and M. Shibasaki, Catalytic Asymmetric Cyclopropanation of Enones with Dimethyloxosulfonium Methylide Promoted by a La−Li3−(Biphenyldiolate)3 + NaI Complex, J. Am. Chem. Soc., 2007, 129, 13410 CrossRef CAS PubMed.
- Y. Zhang, L. Lin, Y. Chen, X. Liu and X. Feng, N,N′-Dioxide–Lanthanum(III)-Catalyzed Asymmetric Cyclopropanation of 2-Cyano-3-arylacrylates with 2-Bromomalonates, Adv. Synth. Catal., 2017, 359, 1831 CrossRef CAS.
- J. Guo, Y. Liu, X. Li, X. Liu, L. Lin and X. Feng, Nickel(II)-catalyzed enantioselective cyclopropanation of 3-alkenyl-oxindoles with phenyliodonium ylide via free carbene, Chem. Sci., 2016, 7, 2717 RSC.
- X. Zhong, J. Lv and S. Luo, Enantio- and Diastereoselective Cyclopropanation of β,γ-Unsaturated α-Ketoester by a Chiral Phosphate/Indium(III) Complex, Org. Lett., 2017, 19, 3331 CrossRef CAS PubMed.
- Y. Kuang, B. Shen, L. Dai, Q. Yao, X. Liu, L. Lin and X. Feng, Diastereodivergent asymmetric Michael-alkylation reactions using chiral N,N′-dioxide/metal complexes, Chem. Sci., 2018, 9, 688 RSC.
- P. Zhao, S. Wu, C. Ke, X. Liu and X. Feng, Chiral Lewis acid-catalyzed enantioselective cyclopropanation and C–H insertion reactions of vinyl ketones with α-diazoesters, Chem. Commun., 2018, 54, 9837 RSC.
- T. Hashimoto, Y. Naganawa, T. Kano and K. Maruoka, Construction of stereodefined 1,1,2,2-tetrasubstituted cyclopropanes by acid catalyzed reaction of aryldiazoacetates and α-substituted acroleins, Chem. Commun., 2007, 5143 RSC.
| This journal is © the Partner Organisations 2024 |