
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrongly fluorescent indolizine-based coumarin analogs†
Jaqueline S. A.
Badaro
 a,
Antoni
Wrzosek
b,
Olaf
Morawski
c,
Adam
Szewczyk
*b,
Irena
Deperasińska
*c and
Daniel T.
Gryko
*a
a,
Antoni
Wrzosek
b,
Olaf
Morawski
c,
Adam
Szewczyk
*b,
Irena
Deperasińska
*c and
Daniel T.
Gryko
*a
aInstitute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: dtgryko@icho.edu.pl
bNencki Institute of Experimental Biology, Polish Academy of Sciences, Pasteur 3, 02-093 Warsaw, Poland. E-mail: a.szewczyk@nencki.gov.pl
cInstitute of Physics, Polish Academy of Sciences, Al. Lotników 32/46, 02-668 Warsaw, Poland. E-mail: deper@ifpan.edu.pl
First published on 2nd October 2024
Abstract
It is now possible to prepare 2-oxo-2H-pyrano[2,3-b]indolizine-3-carboxylates with an ordered arrangement of various substituents directly from pyridinium salts and diethyl 2-(ethoxymethylene)malonate, allowing for refined control of their photophysical properties. Facile entry into some previously unknown derivatives is disclosed to demonstrate the potential of this method. The use of substituted picolinium salts, as well as further functionalization of the pyrrole ring, permitted easy introduction of new moieties upon the dye, which enabled fine-tuning of the photophysical properties. The obtained dyes possess absorption and emission spectrum in the blue–green region and fluorescence quantum yields reaching 92%. The parent 2-oxo-pyrano[2,3-b]indolizine-3-carboxylate turned out to be an electron-deficient system with a low-lying LUMO, an electronic transition energy of 2.7 eV and possessing a large oscillator strength. Almost complete overlap of the HOMO and LUMO in the 2-oxo-pyrano[2,3-b]indolizine core is responsible for the large fluorescence quantum yields for almost all prepared derivatives. The reason for maintaining the large emission intensity in polar solvents is that the increase in the dipole moment is accompanied with a significant change in its orientation in space. Fluorescence imaging studies have proven that 2-oxo-pyrano[2,3-b]indolizines penetrate the membrane of living cells. A positively charged analog was synthesized and used to stain intracellular organelles in the H9c2 cell line. This compound did not penetrate the cell membrane, however after permeabilization, it specifically stained the nucleus.
Introduction
Pyrano[2,3-b]indolizin-2-ones are a unique class of heterocycles that, contrary to their analogues pyrano[3,2-b]indol-2-ones1–12 and pyrano[2,3-b]indol-2-ones,13–22 have been greatly ignored after they were synthesized by Kakehi et al. in the 80s23–25 (together with their isomers pyrano[3,2-a]indolizin-2-ones)25,26 (Fig. 1). However, their distinct structure, composed of a fused tricyclic system, in which a pyran ring is fused to an indolizine moiety, offers a wide range of opportunities for the design and development of novel compounds, exhibiting immense potential for applications in various fields, ranging from medicinal chemistry to materials science. Structurally, pyrano[2,3-b]indolizin-2-ones can be considered analogues of coumarins27–34 and benzocoumarins.35–42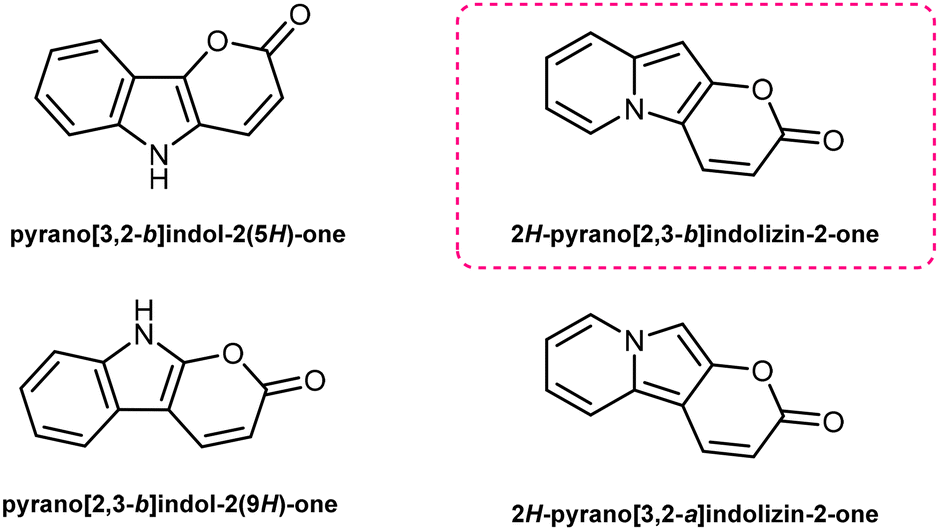 | ||
| Fig. 1 Pyrano-indol-2-ones and pyrano-indolizin-2-ones. | ||
The synthesis of pyrano[2,3-b]indolizines was investigated by Kakehi and co-workers mainly from 1980 to 1998, driven by the need to establish an efficient and reliable methodology. Numerous synthetic routes have been explored, involving the use of pyridinium salts as starting materials and the formation of various intermediates, e.g. 3-bis(methylthio)methylene-2,3-dihydroindolizin-2-ones,23 2,3-dihydroindolizin-2-ones,24 3-vinylindolizines25 as well as the hydrolysis of pyrano[2,3-b]indolizine-2-imines.43 These synthetic efforts have resulted in the successful preparation of a diverse array of pyrano[2,3-b]indolizine derivatives, expanding the chemical space available for further exploration and application.
In the context of search for novel fluorescent platforms we resolved to explore 2-oxo-pyrano[2,3-b]indolizine-3-carboxylates because of the easily available starting materials and the strong fluorescence emission claimed by the authors in their paper, but not sufficiently explored.24 The Kakehi's synthesis (Scheme 1) consisted of four steps: (1) quaternization of 2,6-dimethylpyridine with ethyl 2-bromoacetate; (2) Dieckmann condensation to form the 5-methylindolizin-2-ol intermediate; (3) Friedel–Crafts reaction of the intermediate with diethyl 2-(ethoxymethylene)malonate; (4) final intramolecular cyclization via transesterification with the formation of the lactone ring and additional ethanol elimination. It is noteworthy that the last three steps occur in one pot. However, the drawback of this methodology is that a long reaction time (7 days) is required, and the yield is relatively poor (38%). By solving the synthetic drawbacks and shedding light on the properties of this class of compounds, we aimed to provide a comprehensive understanding of this intriguing heterocyclic system, thus paving the way for further research and development of novel compounds with enhanced properties and biological application.
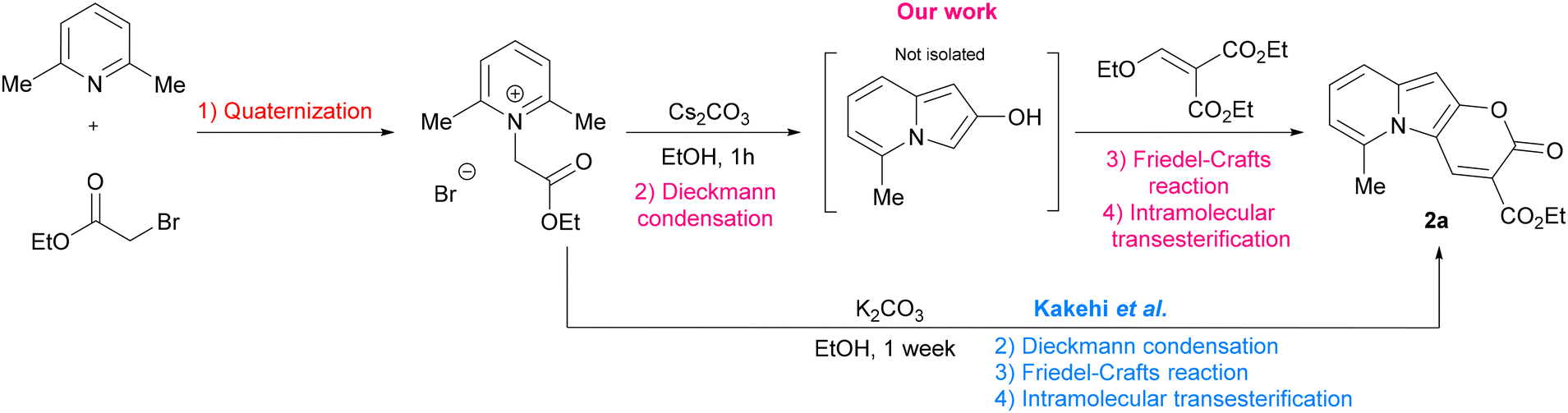 | ||
| Scheme 1 Synthesis of ethyl 6-methyl-2-oxo-pyrano[2,3-b]indolizine-3-carboxylate. | ||
Results and discussion
Synthesis
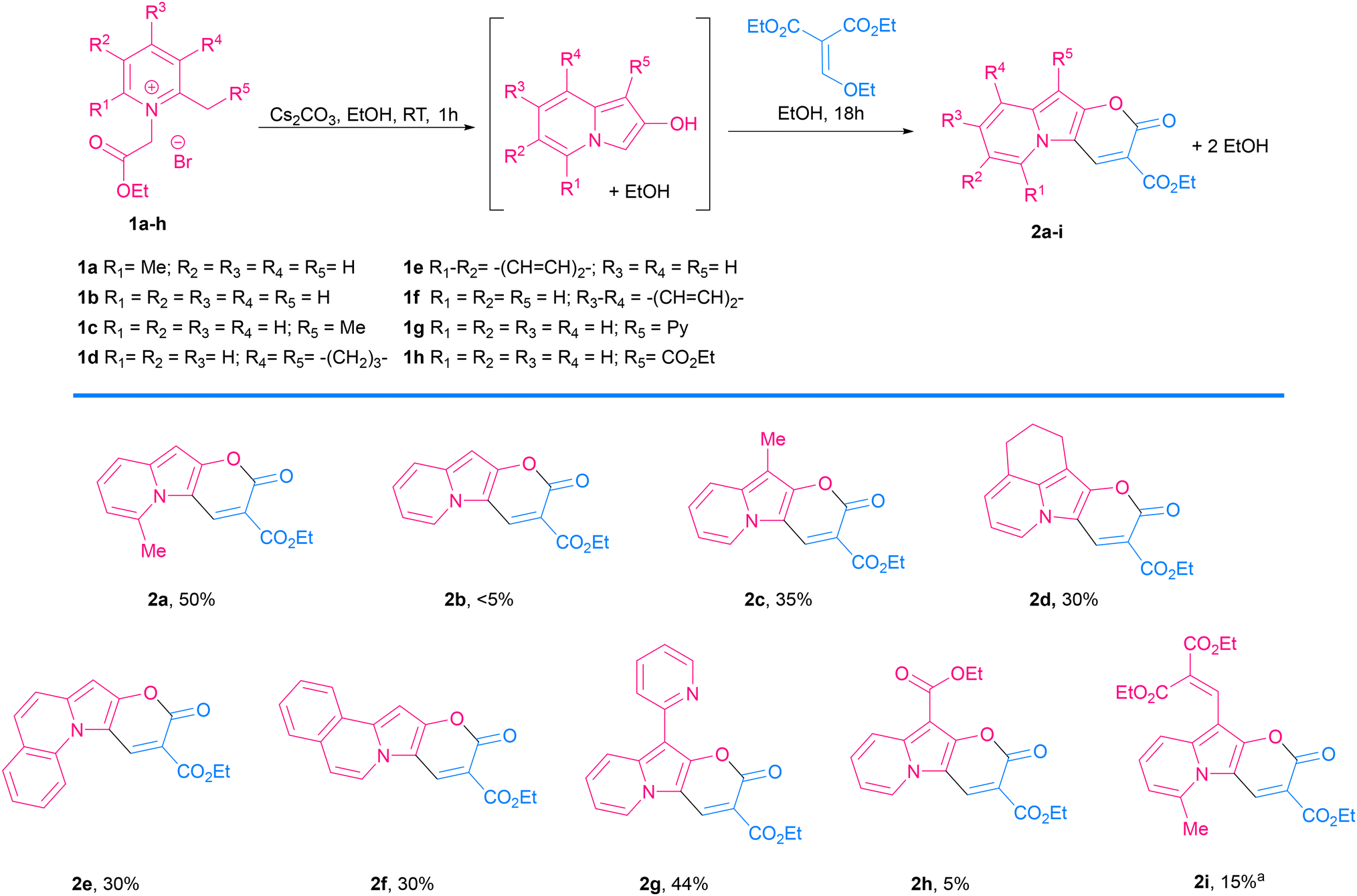 | ||
Scheme 2 Scope of 2-oxo-pyrano[2,3-b]indolizine-3-carboxylate. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Obtained by simultaneous addition of malonate. Obtained by simultaneous addition of malonate. | ||
Consistent with what was observed by Kakehi and co-workers in 1986, the reaction with 2,3-cyclopentenopyridinium salts did not lead to the indolizine product,52 while the reaction with a 5,6,7,8-tetrahydroquinolinium salt led to the expected product 2d in 30% yield. 2-Methylquinolinium and 1-methylisoquinolinium salts also reacted nicely and gave π-expanded indolizine products with a free R5 position (2e, 2f) in 30 and 28% yield, respectively, while 3-methylisoquinolin-2-ium salt did not react as expected.
Additionally, α-picoline derivatives (1g and 1h) bearing EWG groups (2-Py and CO2Et) attached to the methylene group (R5) furnished the target product (2g, 2h) in 44% and 5% yield respectively. It is noteworthy that in those cases, due to the highly acidic character of the proton at the α-position, the quaternization of the pyridine with ethyl 2-bromoacetate led to a 3-substituted indolizin-2-ol directly, even in the absence of base. In such cases the base was added later, only to promote the last two steps: nucleophilic substitution and transesterification.
Meanwhile, the reaction from a 4-bromopyridinium salt and diethyl 2-(ethoxymethylene)malonate did not produce the expected product, probably due to a less reactive indolizin-2-ol intermediate. Similarly, the reaction with a 4-dimethylaminopyridinium salt did not give the target product, in this case probably due to the difficulty in the formation of the indolizine intermediate caused by the effect of the electron-donating group, which turned the proton in the α-position less acidic, thus the reagent is less prone to cyclization.
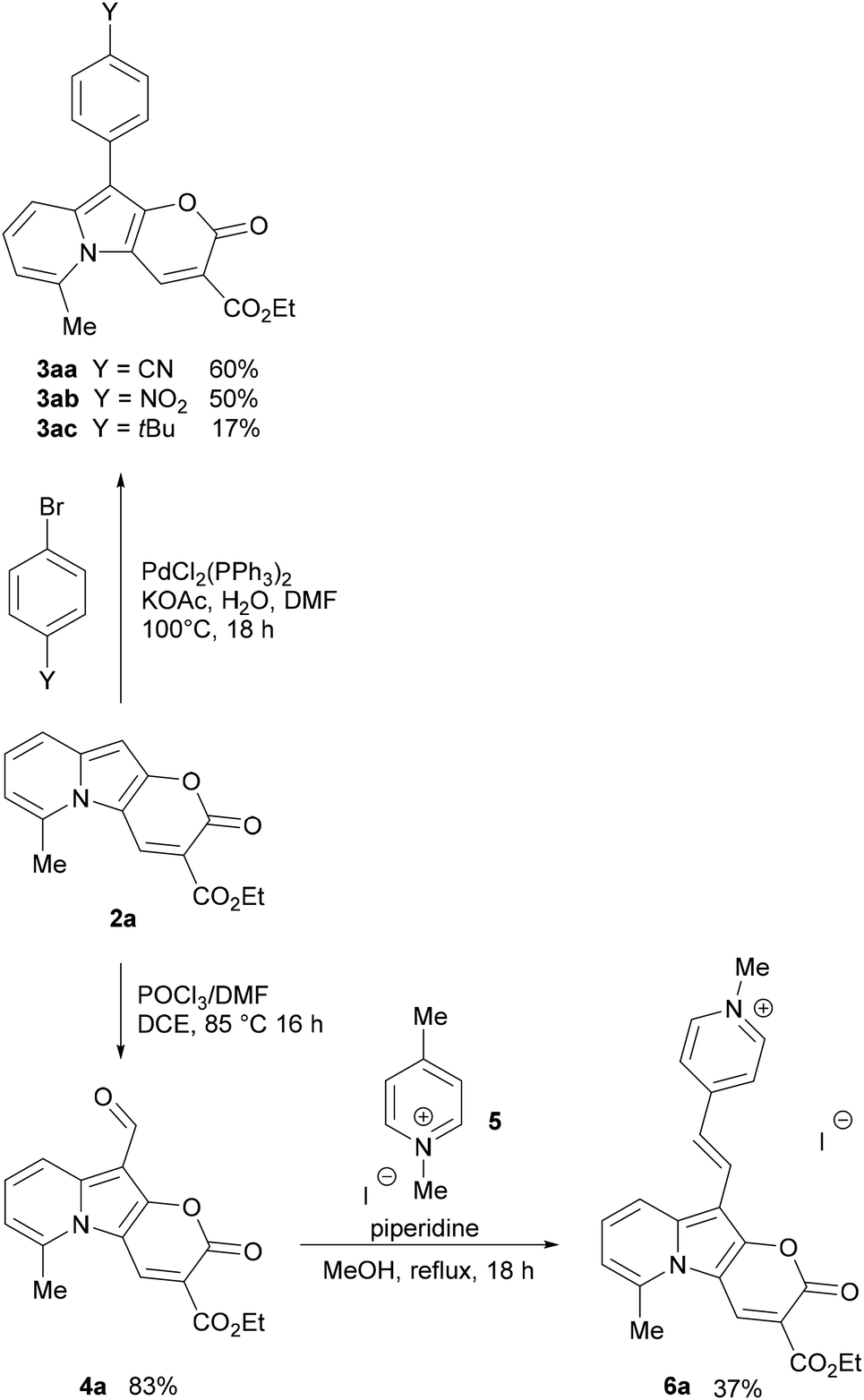 | ||
| Scheme 3 Synthesis of 3aa, 3ab, 3ac and 4a from 2a, and synthesis of 6a from 4a. | ||
The Vilsmeier–Haack formylation also successfully occurred at the electron-rich position, resulting in the formation of 4a in 83% yield.
To probe the potential of this class of dyes, Knoevenagel condensation was carried out between 4a and 1,4-dimethylpyridin-1-ium iodide (5) (Scheme 3) forming a new styryl53–55 dye 6a, which was obtained in 37% yield and later evaluated for cell staining (vide infra).
Considering the principles of green chemistry,56 the reactions leading to compounds 2a–2i and 4a are considered to have moderate atom economy (AE) efficiency, in the range of 40–60%. Meanwhile, direct arylation products (3aa–3ac) and Knoevenagel condensation were obtained with AE efficiency above 80%, which is considered optimal according to the above principles. For all reactions, the reaction mass efficiency is very low, less than 10%, mainly due to the significant impact of the solvent in the calculation.
Photophysical properties
UV-Vis absorption and emission spectra were recorded in nonpolar toluene (dielectric constant ε = 2.38), moderately polar DCM (ε = 8.93), and polar DMSO (ε = 46.45) for all dyes, except 6a, which was measured only in DCM and DMSO and will be discussed separately and correlated to its precursor 4a.A comparison between the photophysical parameters for 2-oxo-pyrano[2,3-b]indolizine-3-carboxylates and those available for similar structures (Fig. 2) such as 11-benzyl-7-hydroxyisochromeno[4,3-b]indol-5-one9 (Φfl = 42% in DCM, with λabs = 382 and λem 496) or methyl 2-oxo-4-(pyrrolidin-1-yl)-2,5-dihydropyrano[3,2-b]indole-3-carboxylate57 (λabs = 335 in EtOH) reveals a significant bathochromic shift for absorption, while emission and fluorescence quantum yield (Φfl) remain in the same range.
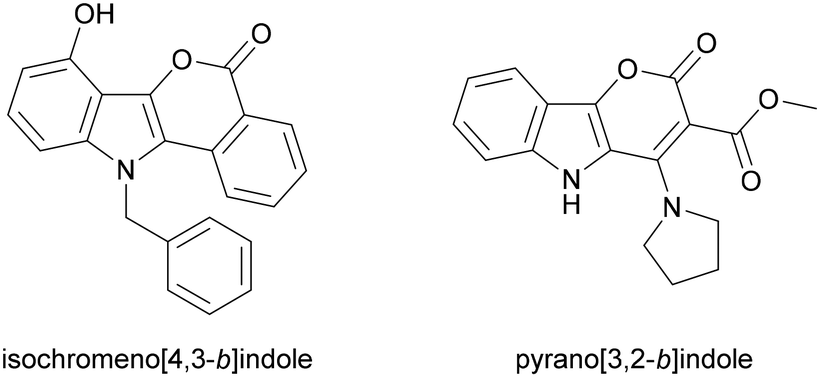 | ||
| Fig. 2 Indoles structures used as reference to discuss photophysical properties of synthesized dyes. | ||
The collected data of compounds in DCM are presented in Table 2 and the spectra of selected dyes are compared in Fig. 4, while the remaining data for the other solvents are available in Table S5 in ESI.† Absorption maxima (λabs) of the dyes obtained are located within 420–470 nm in all solvents, while the emission maxima (λem) falls between 440 and 520 nm for almost all of them and is more bathochromically shifted in polar solvents (solvatochromism). It is worth noting that compound 3ab represents an exception to this trend in both DCM and DMSO, showing two emission bands overlapping, one at the ‘expected’ wavelength (491 nm) and another one around 642 nm. Excitation spectra were recorded to prove that both signals come from the same molecule and are not due to impurities in the sample. This property can be explained by hypothesizing that this structure, constituting of a strong donor, 2-oxo-pirano[2,3-b]indolizine, and a strong acceptor, nitrophenyl, can undergo internal charge transfer (ICT) in polar solvents and generate two possible excited states according to the geometry and consequently allow for two possible emitting paths.
| Cmp | Solv | λ abs [nm] | λ em [nm] | Δ
|
ε [M−1 cm−1] | Φ fl |
|---|---|---|---|---|---|---|
| Fluorescence quantum yields measured with Coumarin 153 in EtOH as reference (Φfl = 0.55).a Fluorescence quantum yield measured with Rhodamine 6G in EtOH as reference (Φfl = 0.95). | ||||||
| 2a | DCM | 446 | 464 | 900 | 30500 |
0.71 |
| 2b | DCM | 449 | 470 | 1000 | 16300 |
0.77 |
| 2c | DCM | 468 | 494 | 1100 | 22900 |
0.61 |
| 2d | DCM | 467 | 499 | 1400 | 24600 |
0.64 |
| 2e | DCM | 456 | 470 | 700 | 39700 |
0.73 |
| 2f | DCM | 436 | 458 | 1100 | 39700 |
0.33 |
| 2g | DCM | 466 | 500 | 1500 | 17600 |
0.92 |
| 2h | DCM | 433 | 452 | 1000 | 29200 |
0.81 |
| 2i | DCM | 446 | 485 | 1800 | 21000 |
0.25 |
| 3aa | DCM | 453 | 490 | 1700 | 22928 |
0.64 |
| 3ab | DCM | 452 | 491/642 | 6500 | 27700 |
0.03 |
| 3ac | DCM | 457 | 514 | 2400 | 19600 |
0.65 |
| 4a | DCM | 426 | 448 | 1200 | 31800 |
0.77 |
| 6a | DCM | 509 | 575 | 2300 | 24400 |
0.44 |
The fluorescence quantum yields (Φfl) are in most cases on the same level regardless of the solvent and they vary among the studied 2-oxopyrano[3,2-b]indolizines from 0.92 for dye 2g in DCM to being barely detectable for 3ab in DCM and DMSO due to the preferred non-emissive decay (Fig. 3). Exceptions to this trend are 2c, 2d, 2g that show lower fluorescence in toluene; and 3ab, which shows a reverse trend by having the highest Φfl in the less polar solvent. The comparison of the photophysical properties of 2a, 2b and 2c (Fig. 4A), which vary their position of a methyl group (or is absent), reveals that this alkyl group at position R5 shifts both absorption and emission bathochromically (2cvs.2b) by about 20 nm, while the same group at position R1 slightly shifts both signals hypsochromically (2avs.2b).
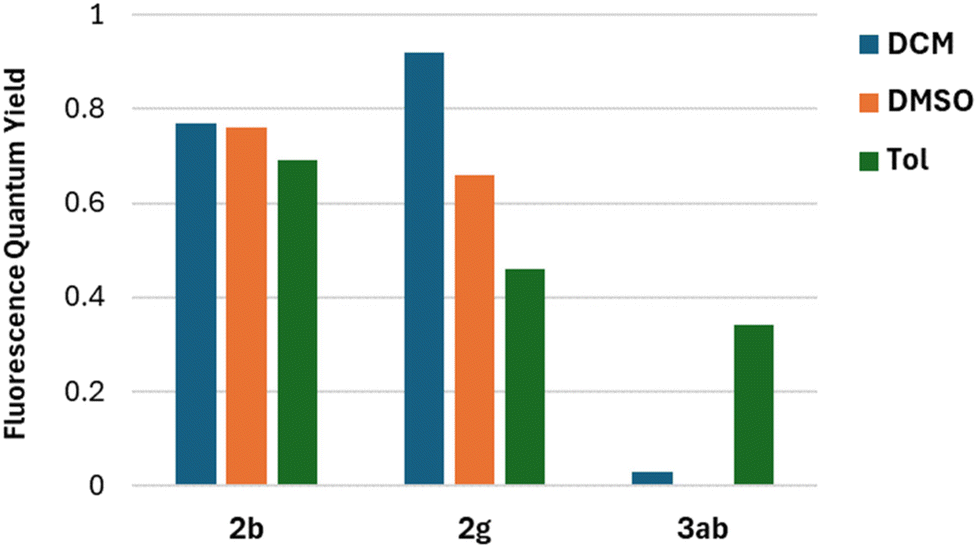 | ||
| Fig. 3 Comparison of Fluorescence quantum yield in different solvents for representative compounds 2b, 2g and 3ab. | ||
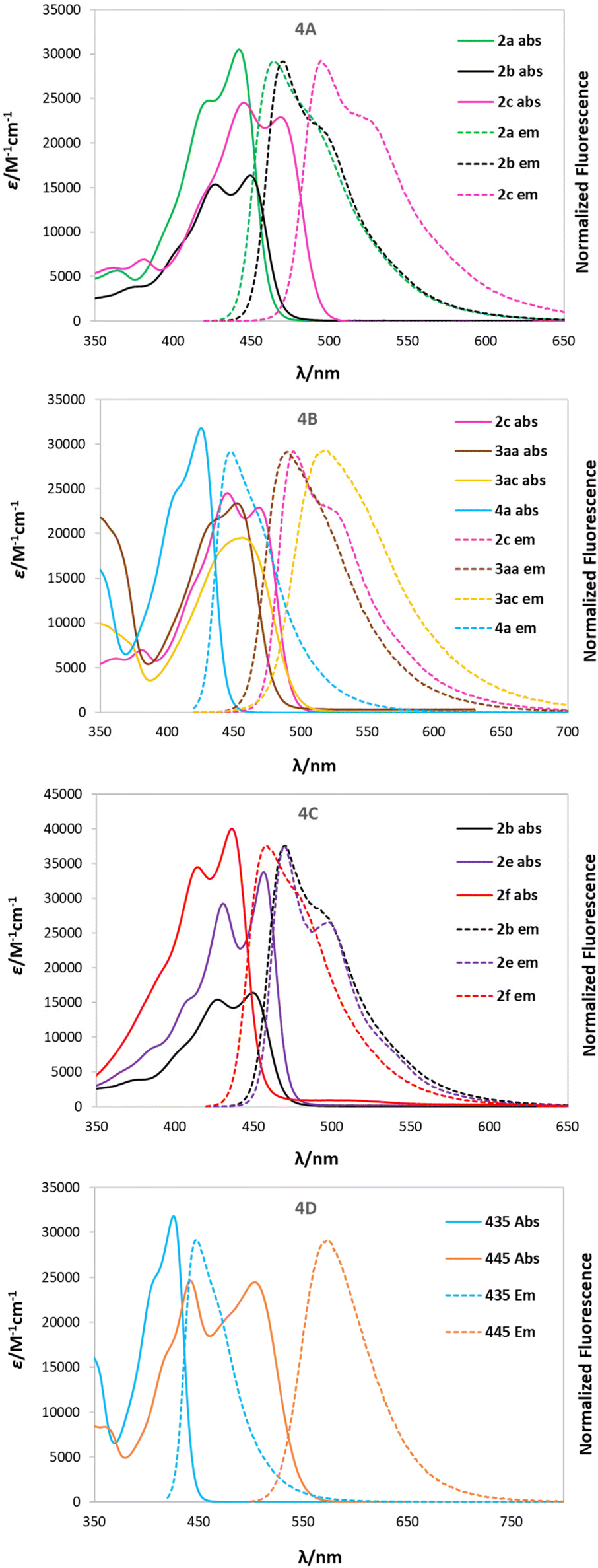 | ||
| Fig. 4 Absorption and emission spectra of some representative 2-oxo-pyrano[2,3-b]indolizine-3-carboxylate derivatives in DCM. (A) Normalized absorption (solid line) and normalized emission (dashed line) of 2a, 2b, 2c (green, black, pink) (B) normalized absorption (solid line) and normalized emission (dashed line) of 2c, 3aa, 3ac and 4a (pink, brown, yellow and light blue) (C) normalized absorption (solid line) and normalized emission (dashed line) of 2b, 2e and 2f (black, purple, red) (D) normalized absorption (solid line) and normalized emission (dashed line) of 4a and 6a (light blue, orange). | ||
Moreover, different electronic character of the substituent in R5 (Fig. 4B) influences both λabs and λem. If CHO is present (4a), λabs = 426 nm and λem = 448 nm, while if Me is present (2c), λabs = 468 nm and λem = 494. The presence of a π-extended system, as in 3aa and 3ac, slightly blue shifts the absorption. Additionally, having a tBu group in the para position (3ac) red shifts the emission, consequently increasing the Stokes shift (2400 cm−1). Interestingly, the effect of the π-expansion of the heterocyclic core strongly depends on the position of the fused benzene ring. In the case of dye 2f a blue shift is observed accompanied with a decrease of the Φfl (0.33 vs. 0.77) compared to 2b (Fig. 4C).
The addition of a methylpyridinium conjugated through an alkene to the 2-oxo-pirano[2,3-b]indolizine system (6a) allowed for an increased dipole moment that translates to a bathochromic shift of 83 nm for λabs and almost 130 nm for λem when compared to 4a (Fig. 4D). Even though a decrease in the fluorescence quantum yield (0.44 vs. 0.77) was observed comparing 6a and 4a, the emission is still high suggesting that, in comparison to 3ab, the decay through a radiative channel is favoured.
Fluorescence kinetic studies were performed for selected compounds: 2a representing “typical” 2-oxo-pyrano[2,3-b]indolizine, 3ab exhibiting dual fluorescence and salt 6a. In all solvents fluorescence of dyes 2a and 6a decays exponentially (Fig. S11†) with decay times, τF, about 3.5 and 2.0 ns respectively (Table S6†). For both compounds τF changes slightly with solvent's polarity (represented by dielectric constant, ε) and the change is parallel to that of Φfl, – for 2a both τF and Φfl grow with increasing ε whereas for 6a they decrease (Table S6†). Consequently, the radiative rate constant kr = Φfl/τF of the S1 → S0 transition remains constant and amounts to 1.3 × 108 s−1 and 2.1 × 108 s−1 respectively (Table S6†). Such high value is typical for the fully allowed ππ* transitions. For dye 3ab kinetic traces are complex. In non-polar toluene fluorescence decays exponentially (Fig. S12a†) with characteristic time 2.62 ns (Table S7†). Radiative rate is 8.8 × 107 s−1, much less than values obtained for dyes 2a and 6a. For polar solvents where dual fluorescence from LE and CT states is observed emission decays are non-exponential and depend on wavelength of observation. For DCM the high energy band of the LE state with maximum at 487 nm two components are clearly visible in the decay trace (Fig. S12b†), and deconvolution yields 20 and 356 ps decay times. Intensity of emission originating from low energy CT band with maximum at 700 nm grows at early times and reaches maximum much later than the LE profile (Fig. S12b†). This points to precursor (LE) – successor (CT) scheme and suggests that that initial population of the CT state is null. Indeed, deconvolution reveals grow time of 55 ps and decay time of 356 ps (Table S7†). Negative amplitude of the grow time is comparable to the (positive) amplitude of the decay time what confirms that CT state is not directly excited and is populated from the LE state. This provides experimental proof for the TICT concept hypothesised below. Long component of the LE profile has same decay time as decay time of CT trace, pointing to thermalization of both states. However, grow time of CT is a bit longer than the short decay time of LE so one may expect presence of a transition state or a slow relaxation to emissive CT structure, so the simple two states model used commonly for TICT can be not adequate. In more polar DMSO kinetic profiles are faster (Fig. S12c†) and the LE decay time decreases down to 15 ps (Table S7†) pointing to the increase of the intramolecular CT rate constant. Formation of TICT enhances also non-radiative relaxation, hence the Φfl decreases by two orders of magnitude.
Theoretical analysis
The DFT and TDDFT O3LYP/6-31G(d,p) calculations were performed to corroborate the experimental data. The choice of the O3LYP functional allows for the best reconstruction of the spectral data in comparison to our attempts using other functionals (Table S1†). Calculations included the optimization of molecular structures in their ground (S0) and lowest electronic excited (S1) states along with the determination of the corresponding absorption and fluorescence energies. Corresponding data for the molecules in the three solvents, toluene, DCM and DMSO, are collected in Table S2.† The calculated dipole moments for the S0 and S1 states are summarized in Table S3.† The two highest occupied (HOMO's) and two lowest unoccupied molecular orbital (LUMO's) energy levels are shown in the diagram in Fig. S1,† while the values of HOMO and LUMO energies are given and compared in Table S4.† More detailed results of calculations are in the ESI.†Dye 2b lacking any substituents can be considered a model chromophore for this family of heterocycles (Scheme 2). This is a molecule with planar geometry in both S0, and S1, states. The electronic transition between them has a π–π* character and essentially it is described by a single HOMO/LUMO electronic configuration. A diagram of the calculated electronic states of heterocycle 2b with a short analysis is presented in ESI, Fig. S2.†
According to the computational results, the geometry of 2b in the excited state undergoes relatively small changes. This result is consistent with the small Stokes shift observed experimentally. Small changes in geometry are manifested by small Franck–Condon factors (Fig. S3†) which well reproduce the experimental spectrum (Fig. 1). The small values of the calculated spin–orbit coupling elements (Hso) between the excited S1 state and the energetically close triplet states (Fig. S2†) together with the small Franck–Condon factors are indicative of relatively small rate constants of non-radiative processes,58,59 which in the experiment (Table 2) is visible as significant values of Φfl achieved for dye 2b.
The calculation results show also, that 2b is a molecule with a large dipole moment, increasing in the excited state (9.6 D for S0 state and 12 D in S1 – Table S3†). In this light, slight red-shifts of its electronic spectra (both observed, Table 2 and Table S5† and calculated, Table S2†) as the solvent polarity increases may seem surprisingly small. This phenomenon, has also been observed in the case of various other molecules,60,61 and can be explained with the use of classical theory of the solvent effect,60,61 and is related to the fact that dipole moments are vector quantities (and not scalars). When the increase in the dipole moment in the S1 state (relative to the S0 state) is accompanied by a change of orientation in space, the solvent effect is smaller than expected based on values alone (see Fig. S4†).
Fig. S2† shows the shapes of the HOMO and LUMO of molecule 2b, providing high oscillator strength for the transition between the S0 and S1 states. They will constitute a reference orbital in the presentation of results for other 2-oxopyrano[3,2-b]indolizines.
The substitution of molecule 2b causes changes in the energy of the molecular orbitals (Fig. S1†), including changes in the spacing between them (Table S4†). This is reflected by the shifts of the electronic spectra, since the electronic transition has an initial and final state, the magnitude of this shift depends on the energy changes in both states.
In Fig. 5, correlation between the absorption S0 → S1 energy and the energy difference of the HOMO and LUMO for the studied 2-oxopyrano[2,3-b]indolizines is presented. This correlation demonstrates that transitions between the S0 and S1 states for all molecules, as in parent dye 2b, are described by a single HOMO/LUMO electronic configuration. At the same time, two groups of molecules can be distinguished, the first with moderate changes in the HOMO/LUMO energy gaps and excitation energies (in relation to 2b), and the other group of two molecules with large changes in these quantities.
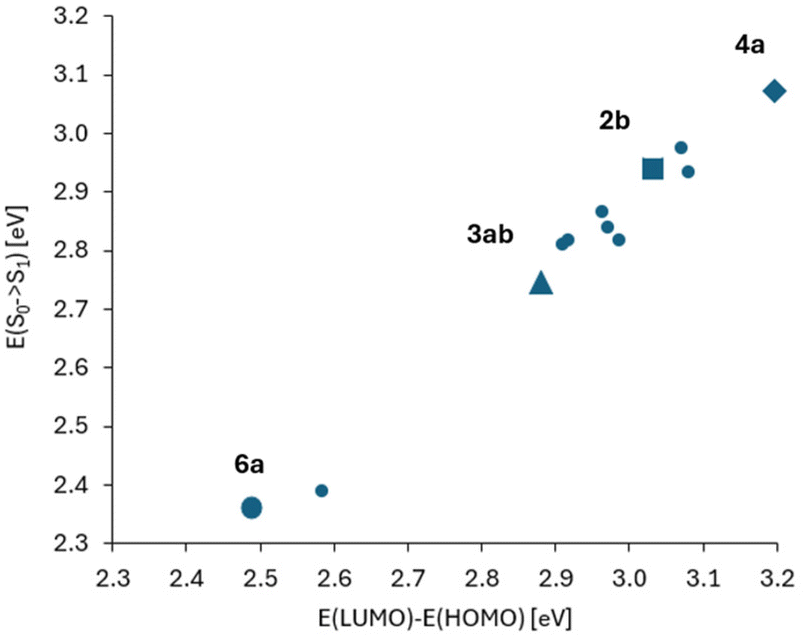 | ||
| Fig. 5 Correlation between the absorption energy and the energy difference of HOMO and LUMO orbitals (numerical data in Tables S2 and S4†). The point corresponding to molecule 2b is marked with a square. Two distant points (for lower energies, i.e. with red shift in comparison to 2b) correspond to molecules 3ab and 6a. These two molecules also differ from the others in large values and changes of the dipole moment between S0 and S1 states (Table S3†). | ||
Fig. 6 (part of the diagram in Fig. S1†) illustrates the variation of the E(HOMO)–E(LUMO) gap and shows the shapes of these orbitals for four representatives of the studied family of 2-oxopyrano[2,3-b]indolizines: 2b – as a model chromophore, 3ac – a dye with observed red-shift in the absorption and fluorescence spectra (relative to the spectra of 2b), 4a – with a blue-shift of the spectra and dye 6a – as a molecule with large red-shift of electronic spectra (see also Fig. 4).
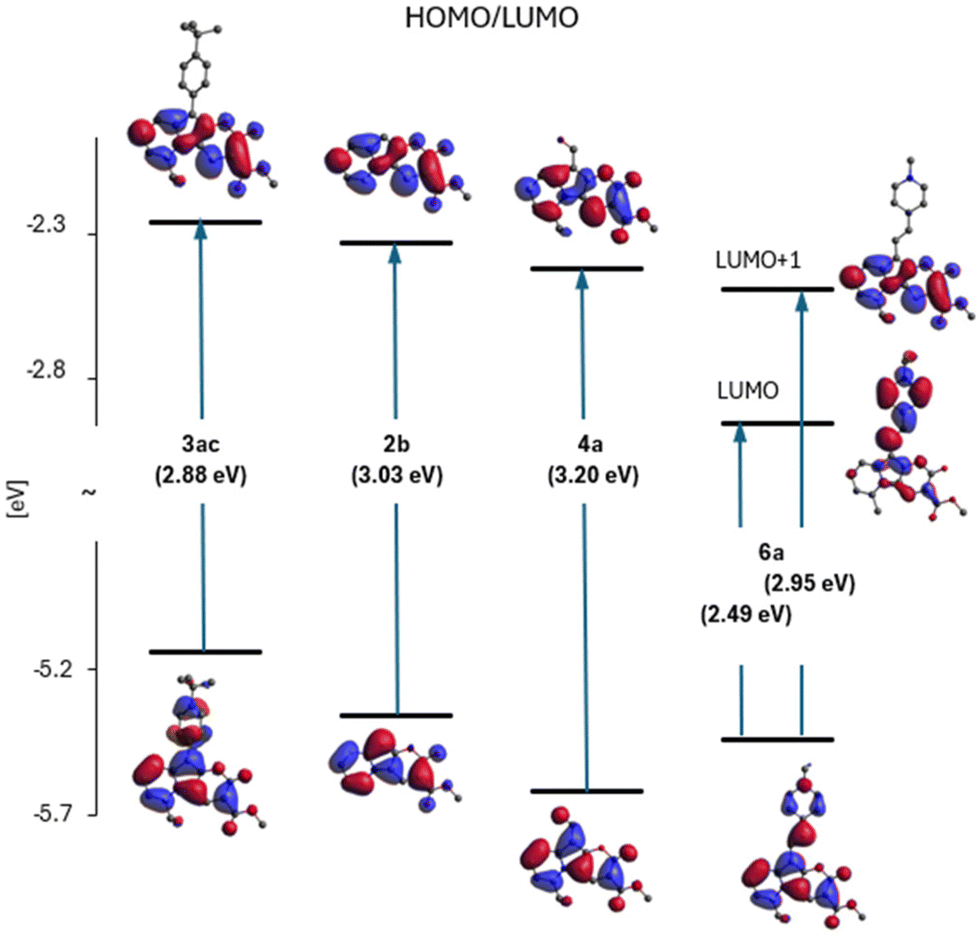 | ||
| Fig. 6 Diagram of orbital energies: 2b as the model heterocycle and its three derivatives, 3ac, 4a and 6a in DMSO. The numbers in brackets indicate the size of the HOMO/LUMO energy gaps. In the case of 6a two LUMOs are present in the considered energy region and in contrary to previous cases lowest of them is localised on substituent. | ||
Based on Fig. 6, it can be concluded that the effect of substitution of molecule 2b produces two qualitatively different effects depending on the electron affinity of the substituent. In the case of compounds 3ac and 4a, this is a modification effect, i.e. their HOMO and LUMO are the modified (to a larger or smaller extent) HOMO and LUMO of molecule 2b, but this is no longer the case of the LUMO of molecule 6a. Although the HOMO of 6a is still a modified HOMO of molecule 2b, the LUMO is the orbital located on the substituent (while the orbital located on core 2b lies above it, as shown in Fig. 6).
In addition to the diagram shown on Fig. 6, two other diagrams are shown in the ESI (Fig. S5 and S6†): HOMO/LUMO energy changes from methylation of the dye 2b in different positions (2avs. 2c) and with π-extension (2evs. 2f) of 2b. Methylation of 2b at the R5 position causes the largest change in HOMO energy leading to a red shift of the 2c spectra.
In contrast to the substitution with a single bond, the π-expansion of 2b becomes a problem not only at the level of the HOMOs, but also at the level of the (so far untouched) LUMO of molecule 2b.
Therefore, the reason for the large red shift of the 6a (and 3ab) spectra is a qualitative change of LUMO from localized on 2b to localized on the substituent. The S0 → S1 transition becomes, instead of the modified 2b excitation, intramolecular charge-transfer between the core and the substituent.
Both orbitals are extended into the attached ring. The energy effect depends on the geometry in which the expansion is carried out (ELUMO(2e) < ELUMO(2f)).
The calculation results for excited states indicate that most of the considered molecules retain features characteristic of parent dye 2b. The fluorescence energies of the molecules optimized in the excited state are not very far from the absorption energies, and these transitions are described by high oscillator strengths (Table S1†). Although these results are in agreement with the experiments, (small Stokes shift, large emission efficiencies) these facts may seem surprising at first. This is because in these dyes there is a convenient relaxation path, which is a rotation around a single bond connecting the substituents with the heterocyclic core 2b. We can expect two effects to play a role: a difference between the dihedral angles corresponding to the energy minimum in the ground and excited states, as well as the formation of rotational conformers. Differences between geometries optimized in the ground and excited states do indeed exist. For example, in systems containing a phenyl substituent at position R5, such as 3aa, in the ground state, the aromatic ring makes an angle of 41.3° with the 2b plane, while in the optimized excited state it makes an angle of 30.7°; a similar rotation occurs in 3ac, from 35.9° to 29.5°. However, they do not have a major impact on the spectroscopic properties because the LUMO is not extended to the substituent (Fig. 6). The exceptions are two molecules: 3ab and 6a, because their LUMOs are located on the substituent (Fig. 6, S7 and S8†). In both cases, the dark rotational conformers were optimised in the excited state (Table S1†). These conformers are characterised by zero oscillator strength for S1 → S0 transition at the perpendicular arrangement of the planes of the heterocyclic core and the substituent ring. The difference between 6a and 3ab is that the dark form of 3ab is the only form optimized in the excited state in polar DMSO. This result is consistent with experimental: while 6a in this solvent emits with significant efficiency, the emission yield of 3ab is extremely low. This means that the relaxation of 6a to the dark state is slower than the decay via the radiative channel or inhibited by a barrier due to a more complex link between substituent and the heterocyclic core.
In turn, the bright form of 3ab is experimentally observed and optimised in calculations for the S1 state, in a non-polar medium only. Fig. S8† shows a diagram of electronic states of isolated molecule 3ab with both optimised forms of the excited state. This is a diagram typical of TICT systems.62
Overall, it is worth to acknowledge the consistency between experimental and theoretical results. The ratio of radiative rate constant of 2a and 6a, 1.3/2.1 = 0.62 is very similar to the ratio oscillator strength calculated for them, 0.64:1.06 (Tables S1 and S7†). Also, the radiative rate obtained experimentally for 3ab is two/three times lower than for 2a/6a, exactly like the oscillator strength calculated for 3ab and 2a/6a (Tables S1 and S7†). For 3ab the theoretically predicted TICT mechanism is fully confirmed by fluorescence kinetic study. These as well as other correspondence between theory and experiment supports confidence for the HOMO and LUMO overlap statements.
Imaging studies
To prove the potential applicability of 2-oxo-pyrano[2,3-b]indolizines as a fluorescent probes, incubation of rat embryonic cardiomyoblast-derived cells H9c2 with selected dyes 2g, 2c, 4a and 6a was performed. The results showed that, neutral dyes from this family (i.e.2g, 2c and 4a) penetrate the living cell membrane and, as they are uncharged, accumulate nonspecifically in a wide range of cellular structures (Fig. S13†). In the case of dye 6a which is a quaternary pyridinium salt, imaging experiments did not lead to accumulation of the dye intracellularly (Fig. 7A). To promote the access to the cytosol, H9c2 were permeabilized with digitonin (100 μg ml−1). Addition of digitonin to the incubation medium resulted in permeabilization of the cell membrane and the successful penetration of the dye into the cytosol and cell organelles (Fig. 7B). It turned out that under these conditions, compound 6a, as seen in the presented image (Fig. 7B), the dye was accumulated in the structure of the nucleus and the nuclear membrane. In order to compare the dye 6a with the classical cell nuclear stain, co-staining with Hoechst 33342 was performed (Fig. 7C). It appears that the fluorescence of these two dyes overlaps except for some intranuclear structures, which are more strongly stained by pyranoindolizine 6a (Fig. 7C and D). Additional intranuclear structures more strongly stained by the dye 6a may prove useful in distinguishing specific cell nuclear structures.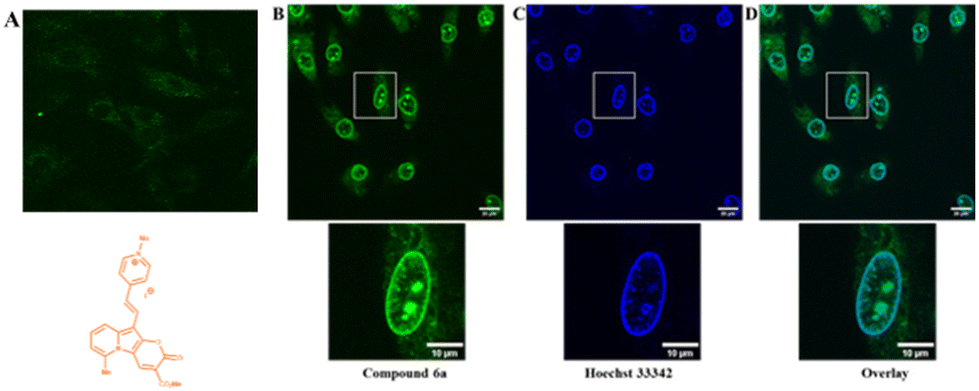 | ||
| Fig. 7 Intracellular localization of compound 6a in H9c2 cells detected using confocal fluorescence microscopy. (A) Image of the cells incubated with dye 6a in a non-permeabilized cell membrane. Lower diagram structure of the dye. (B) Fluorescence of cells with digitonin (100 μg ml−1) permeabilized membrane. (C) Fluorescence of permeabilized cell co-incubated with 0.5 μM Hoechst 33342 recorded with a 405 nm excitation wavelength and emission ranges from 420–520 nm. (D) Overlay of B and C. Lower pictures represents zoom upper square marked cells for B, C and D. The fluorescence of 6a was recorded with a 473 nm excitation wavelength and emission ranges from 510–610 nm. | ||
Conclusions
In conclusion, the synthesis of pyrano[2,3-b]indolizin-2-ones from picolinium salts and diethyl 2-(ethoxymethylene) malonate under basic conditions could be optimized to reach 50% yields for the one-pot procedure. The presence of an electron-rich pyrrolic position can be harnessed to open the avenue towards various novel derivatives. The presence of different substituents on the starting picoline as well as post-functionalization allowed for detailed studies on their impact on the photophysical properties in polar and non-polar solvents. It was found that the fluorescence intensity is large for all studied pyrano[2,3-b]indolizin-2-ones regardless of the substitution pattern which originates from the almost complete overlap of the HOMO and LUMO. Combined experimental and computational study enabled to conclude that pyrano[2,3-b]indolizin-2-one core can be considered as an electron-acceptor and in most of the derivatives examined in this work, the consequence of π-expansion was the extension of the HOMO and the modification of the electronic transition energy in the range of 0.2 eV. We discovered that a simultaneous increase in the dipole moment and change of its orientation in space is the reason for maintaining the large emission intensity as well as the small Stokes shift and solvatochromism, in polar solvents. Studies on the staining of the H9c2 cell line with exemplary 2-oxo-pyrano[2,3-b]indolizines has proven that uncharged dyes easily penetrate the living cells’ membrane. On the other hand styryl derivative 6a does not, it specifically stains structures associated with the cell nucleus of permeabilized cells. All reported findings may serve as the guide enabling modulation of this class of molecules towards the design of improved dyes for cell imaging applications.Experimental section
General synthetic information
All reagents and solvents were purchased from commercial sources and used as received. Reactions with moisture and oxygen sensitive compounds were performed under an argon atmosphere. The progress of reactions was monitored by thin-layer chromatography (TLC), performed on aluminum foil plates, covered with silica gel 60 F254 (Merck). Pure products were achieved by means of column chromatography with silica gel 60 (230–400 mesh). The identity and purity of final products were established by 1H and 13C NMR spectrometry as well as by MS-spectrometry (APCI-MS or ESI-MS). All reported 1H NMR and 13C NMR spectra were recorded on Varian AM 500 MHz, or Varian AM 600 MHz spectrometers. Chemical shifts (ppm) were determined with TMS as internal reference; J values are given in Hz. All melting points were measured with EZ-Melt apparatus.Synthesis of precursors for pyrano[2,3-b]indolizin-2-ones
Pyridinium salts 1a,481b,491c,501d,481e,511f,50 and bis(2-pyridyl)methane 0g63 were synthesized according to previously published procedures, and spectroscopic data are consistent.
General procedure for the synthesis of 2-oxo-2H-pyrano[2,3-b]indolizine-3-carboxylates (condition A)
In a 20 mL Schlenk flask, pyridinium salt (6 mmol, 1 equiv.) and Cs2CO3 (6 mmol, 1 equiv.) were added and dissolved in 5 mL of ethanol. After 1–2 hours a color change in the mixture was observed and fluorescence, typical for indoles, was visible when irradiated with UV lamp. Diethyl 2-(ethoxymethylene) malonate (6 mmol, 1 equiv.) was added, and the reaction mixture was stirred overnight at room temperature. After completion, the mixture was extracted with DCM, and the combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by column chromatography (silica, AcOEt/hexane 1:1).
:1); m.p. 189.0 °C. 1H NMR (500 MHz, CDCl3) δ 8.97 (s, 1H), 7.46 (d, J = 8.8 Hz, 1H), 7.25 (dd, J = 8.8, 7.0 Hz, 1H), 6.77 (d, J = 7.0 Hz, 1H), 6.43 (s, 1H), 4.41 (q, J = 7.1 Hz, 2H), 2.89 (s, 3H), 1.41 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 165.8, 158.3, 153.7, 142.1, 138.4, 138.1, 126.9, 117.0, 114.5, 112.7, 100.8, 89.0, 61.2, 22.0, 14.5 ppm. HRMS (ESI): m/z calcd for C15H13NO4Na, 294.0742 [M + Na]+; found, 294.0741.
:1); m.p. 199.5 °C. 1H NMR (500 MHz, CDCl3) δ 8.81 (s, 1H), 8.27 (d, J = 6.9 Hz, 1H), 7.50 (d, J = 9.0 Hz, 1H), 7.26–7.20 (m, 1H), 6.90 (t, J = 6.9 Hz, 1H), 6.33 (s, 1H), 4.41 (q, J = 7.1 Hz, 2H), 1.41 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 165.1, 158.8, 152.9, 140.5, 133.7, 126.3, 124.6, 119.4, 113.0, 111.2, 101.9, 88.2, 61.3, 14.5. HRMS (ESI): m/z calcd for C14H11NO4Na, 280.0586 [M + Na]+; found, 280.0583.
:1); m.p. 215.8 °C. 1H NMR (500 MHz, CDCl3) δ 8.76 (s, 1H), 8.23 (dt, J = 6.9, 1.1 Hz, 1H), 7.45 (dt, J = 9.0, 1.2 Hz, 1H), 7.21 (ddd, J = 9.0, 6.8, 1.1 Hz, 1H), 6.86 (td, J = 6.9, 1.2 Hz, 1H), 4.40 (q, J = 7.1 Hz, 2H), 2.30 (s, 3H), 1.41 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 165.3, 159.2, 150.7, 139.7, 133.1, 125.6, 124.5, 117.5, 112.6, 110.8, 100.8, 96.6, 61.1, 14.5, 6.2 ppm. HRMS (ESI): m/z calcd for C15H13NO4Na, 294.0742 [M + Na]+; found, 294.0739.
:1); m.p. 244.1 °C. 1H NMR (600 MHz, CDCl3) δ 8.76 (s, 1H), 8.05 (dd, J = 6.9, 0.8 Hz, 1H), 6.90 (dd, J = 6.9, 0.8 Hz, 1H), 6.79 (t, J = 6.8 Hz, 1H), 4.40 (q, J = 7.1 Hz, 2H), 2.93–2.86 (m, 4H), 2.03 (p, J = 6.1 Hz, 2H), 1.41 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 165.4, 159.3, 148.0, 139.5, 133.6, 130.8, 121.8, 121.5, 113.4, 111.5, 99.9, 98.3, 61.1, 27.4, 22.0, 19.8, 14.5 ppm. HRMS (ESI): m/z calcd for C17H15NO4Na, 320.0899 [M + Na]+; found, 320.0902.
:1); m.p. not measured, decomposed around 280 °C. 1H NMR (500 MHz, CDCl3) δ 9.36 (s, 1H), 8.32 (d, J = 8.5 Hz, 1H), 7.82 (dd, J = 7.9, 1.3 Hz, 1H), 7.77–7.72 (m, 1H), 7.54–7.48 (m, 2H), 7.41 (d, J = 9.2 Hz, 1H), 6.48 (s, 1H), 4.46 (q, J = 7.1 Hz, 2H), 1.45 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 165.5, 157.9, 154.3, 140.0, 138.0, 134.6, 130.6, 130.1, 127.8, 125.1, 124.3, 117.8, 115.6, 114.2, 103.5, 91.7, 61.6, 14.5 ppm. HRMS (APCI): m/z calcd for C18H13NO4, 307.0845 [M−]; found, 307.0847.
:1); m.p. 261.2 °C. 1H NMR (600 MHz, CDCl3) δ 8.79 (d, J = 0.8 Hz, 1H), 8.11–8.08 (m, 1H), 8.01 (d, J = 7.1 Hz, 1H), 7.74–7.69 (m, 1H), 7.65–7.58 (m, 2H), 7.11 (d, J = 7.2 Hz, 1H), 6.84 (s, 1H), 4.41 (q, J = 7.1 Hz, 2H), 1.41 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 165.0, 158.8, 152.9, 138.6, 134.0, 129.6, 129.2, 128.7, 127.6, 124.3, 121.0, 113.2, 112.5, 103.3, 88.8, 61.4, 14.4 ppm. HRMS (ESI): m/z calcd for C18H13NO4Na, 330.0742 [M + Na]+; found, 330.0746.
:1); m.p. 252.6 °C. 1H NMR (500 MHz, CDCl3) δ 9.02 (d, J = 9.1 Hz, 1H), 8.83 (s, 1H), 8.67–8.64 (m, 1H), 8.33 (d, J = 6.8 Hz, 1H), 8.23 (d, J = 8.1 Hz, 1H), 7.78 (td, J = 7.8, 1.9 Hz, 1H), 7.41 (ddd, J = 9.1, 6.9, 1.2 Hz, 1H), 7.16 (ddd, J = 7.5, 4.8, 1.1 Hz, 1H), 7.03 (td, J = 6.9, 1.3 Hz, 1H), 4.42 (q, J = 7.1 Hz, 2H), 1.43 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 164.8, 158.4, 151.4, 150.4, 148.9, 139.5, 136.7, 133.4, 127.9, 124.3, 122.6, 122.1, 120.9, 114.3, 111.1, 101.7, 101.2, 61.3, 14.5. HRMS (ESI): m/z calcd for C19H15N2O4, 335.1032 [M + H]+; found, 335.1031.
:1); m.p. 227.9 °C. 1H NMR (500 MHz, CDCl3) δ 8.82 (s, 1H), 8.43 (d, J = 8.8 Hz, 1H), 8.37 (d, J = 6.8 Hz, 1H), 7.50 (t, J = 8.8 Hz, 1H), 7.13 (t, J = 6.8 Hz, 1H), 4.42 (q, J = 24.5 Hz, 2H), 4.40 (q, J = 24.5 Hz, 2H), 1.43 (t, J = 21.3, Hz, 3H), 1.40 (t, J = 21.3, Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 164.5, 162.4, 157.6, 152.3, 140.8, 134.0, 129.1, 124.6, 120.4, 115.1, 110.8, 104.2, 93.8, 61.5, 60.5, 14.5, 14.4 ppm. HRMS (ESI): m/z calcd for C17H15NO6Na, 352.0797 [M + Na]+; found, 352.0798.
General procedure for the synthesis of 2-oxo-2H-pyrano[2,3-b]indolizine-3-carboxylates (condition B)
In a 20 mL Schlenk flask, pyridinium salt (6 mmol, 1 equiv.), potassium carbonate (6 mmol, 1 equiv.) and diethyl 2-(ethoxymethylene)malonate (6 mmol, 1 equiv.) were added and dissolved in 10 mL of DMF. The reaction mixture changed color and was stirred for 24 hours at room temperature. After completion, the mixture was extracted with DCM, and the combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by column chromatography (silica, AcOEt/hexane 1:1).
:1); m.p. 210.5 °C. 1H NMR (600 MHz, CDCl3) δ 8.92 (s, 1H), 7.77 (s, 1H), 7.65 (d, J = 8.6 Hz, 1H), 7.36 (dd, J = 8.9, 7.1 Hz, 1H), 6.85 (d, J = 7.1 Hz, 1H), 4.53 (q, J = 7.1 Hz, 2H), 4.35 (q, J = 7.1 Hz, 2H), 4.25 (q, J = 7.2 Hz, 2H), 2.86 (s, 3H), 1.34 (t, J = 7.2 Hz, 3H), 1.32 (t, J = 7.2 Hz, 3H), 1.28 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 166.7, 165.1, 165.1, 156.5, 151.5, 141.6, 138.9, 138.1, 128.9, 128.7, 124.7, 116.4, 115.1, 113.3, 102.9, 96.9, 62.4, 61.6, 61.5, 22.1, 14.4, 14.2, 14.0 ppm. HRMS (APCI): m/z calcd for C23H22NO8, 442.1345 [M − H]−; found, 440.1350.
General procedure for direct arylation
A Schlenk flask flushed with argon, was charged with ethyl 6-methyl-2-oxo-2H-pyrano[2,3-b]indolizine-3-carboxylate (0.55 mmol, 1 equiv.), aryl bromide (0.66 mmol, 1.2 equiv.), potassium acetate (1.11 mmol, 2 equiv.), NMP (3 mL) and PdCl2(PPh3)2 (0.027 mmol, 0.05 equiv.). The solution was stirred at 100 °C for 10 min. Afterwards, water (1.11 mmol, 2 equiv.) was added, and the heating was continued at the same temperature overnight. Then, the mixture was cooled to room temperature and extracted with DCM. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by column chromatography (silica, AcOEt/hexane 1:1).
:1); m.p. 306.1 °C. 1H NMR (600 MHz, CDCl3) δ 9.06 (s, 1H), 7.84–7.81 (m, 2H), 7.80–7.77 (m, 3H), 7.38 (dd, J = 8.9, 7.0 Hz, 1H), 6.89 (d, J = 7.0 Hz, 1H), 4.42 (q, J = 7.1 Hz, 2H), 2.96 (s, 3H), 1.42 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 165.3, 157.4, 150.5, 139.4, 138.9, 138.3, 135.1, 132.7, 129.0, 128.0, 118.9, 115.6, 115.4, 112.5, 110.4, 102.3, 101.4, 61.5, 22.3, 14.4 ppm. HRMS (APCI): m/z calcd for C22H15N2O4, 371.1032 [M − H]−; found, 371.1036.
:1); m.p. 284.3 °C. 1H NMR (500 MHz, CDCl3) δ 9.07 (s, 1H), 8.36 (d, J = 8.8 Hz, 2H), 7.89 (d, J = 8.8 Hz, 2H), 7.83 (d, J = 8.9 Hz, 1H), 7.40 (dd, J = 9.0, 7.0 Hz, 1H), 6.91 (d, J = 7.1 Hz, 1H), 4.43 (q, J = 7.1 Hz, 2H), 2.97 (s, 3H), 1.42 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 165.3, 157.3, 150.7, 146.3, 139.4, 139.0, 138.3, 137.2, 129.0, 128.2, 124.3, 115.8, 115.5, 112.5, 102.5, 101.1, 61.5, 22.3, 14.4 ppm. HRMS (APCI): m/z calcd for C21H15N2O6, 391.0930 [M − H]−; found, 391.0932.
:1); m.p. 241.6 °C. 1H NMR (600 MHz, CD2Cl2) δ 8.93 (s, 1H), 7.71 (d, J = 8.7 Hz, 1H), 7.57–7.51 (m, 2H), 7.51–7.46 (m, 2H), 7.24 (dd, J = 8.9, 6.9 Hz, 1H), 6.76 (d, J = 7.0 Hz, 1H), 4.27 (q, J = 7.1 Hz, 2H), 2.84 (s, 3H), 1.32–1.29 (m, 12H). 13C NMR (126 MHz, CDCl3) δ 165.7, 158.1, 150.3, 140.1, 138.5, 137.9, 128.9, 128.5, 127.0, 126.8, 125.9, 116.1, 114.9, 112.5, 103.3, 101.1, 61.2, 34.7, 31.3, 22.2, 14.5 ppm. HRMS (APCI): m/z calcd for C25H26NO4, 404.1862 [M + H]+; found, 404.1861.
Procedure for the synthesis of styryl dye
:1) affording 4a (230 mg, 83%) as yellow crystals. Rf = 0.40 (SiO2, AcOEt/hexane, 3:1); m.p. 241.4 °C. 1H NMR (500 MHz, CDCl3) δ 10.29 (s, 1H), 9.03 (s, 1H), 8.54 (d, J = 8.8 Hz, 1H), 7.60 (dd, J = 8.8, 7.2 Hz, 1H), 7.05 (d, J = 7.2 Hz, 1H), 4.43 (q, J = 7.1 Hz, 2H), 2.99 (s, 3H), 1.43 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 181.8, 164.8, 156.6, 155.8, 140.6, 139.1, 139.0, 131.5, 118.5, 118.0, 112.2, 103.7, 102.3, 61.7, 22.1, 14.4. ppm. HRMS (APCI): m/z calcd for C16H12NO5, 298.0714 [M − H]−; found, 298.0715.
Photophysical measurements
HPLC grade solvents were purchased from Sigma-Aldrich and used as obtained. For optical studies, solutions of molecules at low concentrations were used to avoid dimerization or reabsorption effects. All absorption and fluorescence spectra were taken at room temperature (21 °C). A Shimadzu UV/VIS-NIR spectrophotometer, model UV-3600i Plus, was used for absorption spectra measurement. Fluorescence spectra were recorded with an FS5 spectrofluorometer from Edinburgh Instruments. Fluorescence quantum yields (Φfl) of molecules in solvents at 21 °C were determined using Coumarin 153 in EtOH (Φfl = 0.38) and Rhodamine 6 g in EtOH (Φfl = 0.95) as references. Solutions of low absorbance (A < 0.1) were used to avoid reabsorption or concentration quenching. Refractive index correction for solvents have been performed in the calculations of quantum yields. Molar absorption coefficient, ε, was calculated from the absorbance, A, of a solution of the given molar concentration, c, in a cuvette of length, l, according to Lambert Beer formula:| A = c ε l |
Fluorescence kinetics studies were performed using the time correlated single photon counting technique. A mode-locked Coherent Mira-HP picosecond laser pumped by a Verdi 18 laser was used for excitation. The fundamental pulses of the Mira laser were up-converted to 387.5 nm. The temporal width of the excitation pulses was ∼280 fs and the instrument response function (IRF) about 40 ps. Fluorescence was dispersed with a 0.25 m Jarrell-Ash monochromator and detected with a HMP-100-07 hybrid detector coupled to an SPC-150 PC module, (Becker&Hickl GmbH). Fluorescence decays were analyzed with deconvolution software using a nonlinear least squares procedure with the Marquardt algorithm. A standard χ2 test and Durbin–Watson (DW) parameter along with residual and autocorrelation function plots were used to assess the quality of a fit. The estimated accuracy for the determination of decay time was below 10 ps.
Computational details
The calculations were performed using the Gaussian 16 program.65 The solvent effect was introduced in PCM procedure. The SOC elements were also calculated using the Orca program.66Biological studies
The rat embryonic cardiomyoblast-derived H9c2 cells were loaded with the fluorophore at the final concentration of 500 nM in DMEM medium supplemented with 10% foetal bovine serum, 2 mM glutamine, 100 U ml−1 penicillin, and 100 μg ml−1 streptomycin, and incubated at 37 °C in a humidified atmosphere containing 5% CO2 for 15 to 60 minutes. After that, the incubation medium was replaced with FluoroBrite™ DMEM. The measurements were performed with the use of an Olympus IX83 confocal microscope with the silicone objective 60× UPLSAPO 60XS. Registered data were transferred to the ImageJ and analysed for presentation.Author contributions
Conceptualization: D. T. G. and J. S. A. B.; investigation: J. S. A. B., A. W., I. D.; supervision: D. T. G., A. S.; visualization: J. S. A. B., A. W., I. D.; writing – original draft: J. S. A. B., A. W., I. D.; writing – review and editing: D. T. G., J. S. A. B.All the authors discussed the results and commented on the manuscript.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Polish National Science Center, Poland (grants OPUS 2020/37/B/ST4/00017 and HARMONIA 2018/30/M/ST5/00460). This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowka-Curie grant agreement no 101007804. The work was financially supported by the Foundation for Polish Science (TEAM POIR.04.04.00-00-3CF4/16-00). Calculations were performed at the Interdisciplinary Center for Mathematical and Computational Modeling (ICM) University of Warsaw under computational allocation G95-1734. We thank Joseph Milton for amending the manuscript.References
- K. Sun, S. Jin, S. Fang, R. Ma, X. Zhang, M. Gao, W. Zhang, T. Lu and D. Du, N-Heterocyclic carbene-catalyzed formal [3 + 3] annulation of alkynoic acid esters with indolin-3-ones: access to functionalized pyrano[3,2-b]indol-2-ones, Org. Chem. Front., 2019, 6, 2291–2295 RSC.
- X.-Y. Meng, M.-Y. Sun, F.-J. Zhao, Y.-J. Dang, B. Jiang and S.-J. Tu, Domino Reaction of 2,2-Dihydroxyindene-1,3-dione with Aromatic Amines: Efficient Synthesis of Isochromeno[4,3-b]indol-5(11H)-one Derivatives, Synthesis, 2014, 3207–3212 CAS.
- N. S. Masterova, S. Yu. Ryabova, L. M. Alekseeva, M. I. Evstratova, S. S. Kiselev and V. G. Granik, Novel pyrano[3,2-b]indole derivatives: synthesis and some properties, Russ. Chem. Bull., 2010, 59, 637–641 CrossRef CAS.
- N. Monakhova, J. Korduláková, A. Vocat, A. Egorova, A. Lepioshkin, E. G. Salina, J. Nosek, E. Repková, J. Zemanová, H. Jurdáková, R. Górová, J. Roh, G. Degiacomi, J. C. Sammartino, M. R. Pasca, S. T. Cole, K. Mikušová and V. Makarov, Design and Synthesis of Pyrano[3,2-b]indolones Showing Antimycobacterial Activity, ACS Infect. Dis., 2021, 7, 88–100 CrossRef CAS PubMed.
- A. P. Gaywood and H. McNab, Methylene Meldrum's Acid Derivatives of Indoxyl and Their Cyclization Reactions under Flash Vacuum Pyrolysis Conditions, Synthesis, 2010, 1361–1364 CAS.
- P. C. Unangst and R. E. Brown, Indole esters as heterocyclic synthons. II. Preparation of 1,3-oxazino[5,6-b]indoles and 3-substituted-pyrano[3,2-b]indoles, J. Heterocycl. Chem., 1984, 21, 283–288 CrossRef CAS.
- H. K. Kadam and S. G. Tilve, Unique synthesis of isochromenoindolone via reductive-oxidative cyclisation approach, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2019, 58, 104–108 Search PubMed.
- X. Zhang, W. Hou, D. Zhang-Negrerie, K. Zhao and Y. Du, Hypervalent Iodine-Mediated Intramolecular trans-Aminocarboxylation and Oxoaminocarboxylation of Alkynes: Divergent Cascade Annulations of Isocoumarins under Metal-Free Conditions, Org. Lett., 2015, 17, 5252–5255 CrossRef CAS PubMed.
- S. Pathak, D. Das, A. Kundu, S. Maity, N. Guchhait and A. Pramanik, Synthesis of 4-hydroxyindole fused isocoumarin derivatives and their fluorescence “Turn-off” sensing of Cu(II) and Fe(III) ions, RSC Adv., 2015, 5, 17308–17318 RSC.
- J. L. Bullington and J. H. Dodd, Synthesis of tetrahydroindeno[1,2-b]indol-10-ones and their rearrangement to [2]benzopyrano[4,3-b]indol-5-ones, J. Org. Chem., 1993, 58, 4833–4836 CrossRef CAS.
- A. Sugimoto, K. Sakamoto, Y. Fujino, Y. Takashima and M. Ishikawa, Synthesis and Inhibitory Effect on Platelet Aggregation of 2-Phenyl-1 (2H)-phthalazinone Derivatives, Chem. Pharm. Bull., 1985, 33, 2809–2820 CrossRef CAS PubMed.
- A. J. Frew, G. R. Proctor and J. V. Silverton, Reactions of the sodium salts of some heterocyclic β-ketoesters with dimethyl acetylenedicarboxylate, J. Chem. Soc., Perkin Trans. 1, 1980, 1251–1256 RSC.
- A. Arcadi, S. Cacchi, F. Marinelli and P. Pace, 5-Alkyl-5-[2-(o-iodophenylcarbamoyl)vinyl] Derivatives of Meldrum's Acid as Substrates for the Intramolecular Heck Reaction: Application to the Synthesis of Carbazoles, Synlett, 1993, 743–744 CrossRef CAS.
- N. L. Nam and I. I. Grandberg, Condensation of oxindole with acetoacetic ester and acetylacetone, Chem. Heterocycl. Compd., 2006, 42, 1010–1011 CrossRef CAS.
- W.-Z. Zhang, M.-W. Yang and X.-B. Lu, Carboxylative cyclization of substituted propenyl ketones using CO2: transition-metal-free synthesis of α-pyrones, Green Chem., 2016, 18, 4181–4184 RSC.
- G. H. Ko, C. Maeng, H. Jeong, S. H. Han, G. U. Han, K. Lee, H. C. Noh and P. H. Lee, Rhodium(III)-Catalyzed Sequential C–H Activation and Cyclization from N-Methoxyarylamides and 3-Diazooxindoles for the Synthesis of Isochromenoindolones, Chem. – Asian J., 2021, 16, 3179–3187 CrossRef CAS PubMed.
- S. Park, J. Lee, K. J. Shin, E. Oh and J. H. Seo, Consecutive One-Pot versus Domino Multicomponent Approaches to 3-(Diarylmethylene)oxindoles, Molecules, 2017, 22, 503 CrossRef PubMed.
- Q. Chen, Y. Teng and F. Xu, Lanthanide Silylamide-Catalyzed Synthesis of Pyrano[2,3-b]indol-2-ones, Org. Lett., 2021, 23, 4785–4790 CrossRef CAS PubMed.
- A. S. Kumar and R. Nagarajan, Cyclization Routes for the Synthesis of Functionalized Pyrano[2,3-b]indolones, Pyrazolo[3,4-b]indoles, and Furo[2,3-b]indoles, Synthesis, 2013, 1235–1246 CAS.
- M. Abdel-Megid, A versatile molecule for the synthesis of some new polynuclear bioactive heterocyclic systems, Int. J. Chem., 2006, 16, 149–157 CAS.
- R. Frutos-Pedreño and J.-A. García-López, 2-Arylacetamides as Versatile Precursors for 3-Aminoisocoumarin and Homophthalimide Derivatives: Palladium-Catalyzed Cascade Double Carbonylation Reactions, Adv. Synth. Catal., 2016, 358, 2692–2700 CrossRef.
- X. Peng, Y. Huang, W. Wang, S. Li, G. Hao, S. Ren and Y. R. Chi, N-Heterocyclic Carbene-Catalyzed Remote Enantioselective C–C Bond Formation via 1,6-Addition with Formyl Enynes, ACS Catal., 2024, 14, 2127–2133 CrossRef CAS.
- A. Kakehi, S. Ito, K. Nakanishi, K. Watanabe and M. Kitagawa, Preparation of New Nitrogen-bridged Heterocycles. A Facile Synthetic Method of Pyrano[2,3-b]indolizinone Derivatives, Bull. Chem. Soc. Jpn., 1980, 53, 1115–1120 CrossRef CAS.
- A. Kakehi, S. Ito, K. Watanabe, M. Kitagawa, S. Takeuchi and T. Hashimoto, Preparation of new nitrogen-bridged heterocycles. Synthesis and some reactions of 2,3-dihydroindolizin-2-one derivatives, J. Org. Chem., 1980, 45, 5100–5104 CrossRef CAS.
- A. Kakehi, S. Ito, S. Murakami and H. Sano, Preparation of New Nitrogen-bridged Heterocydes. 8. Syntheses of Some Fused Indolizine Derivatives via the Acid-catalyzed Cyclizations of Functionalized 1- and 3-Vinylindolizines, Bull. Chem. Soc. Jpn., 1984, 57, 548–552 CrossRef CAS.
- A. Kakehi, S. Ito and K. Matsubara, Preparation of New Nitrogen-Bridged Heterocycles. 39. One-Pot Synthesis of 2H-Pyrano[3,2-a]indolizin-2-one Derivatives, Bull. Chem. Soc. Jpn., 1995, 68, 2409–2415 CrossRef CAS.
- G. Jones, W. R. Jackson and A. M. Halpern, Medium effects on fluorescence quantum yields and lifetimes for coumarin laser dyes, Chem. Phys. Lett., 1980, 72, 391–395 CrossRef CAS.
- J. A. Van Gompel and G. B. Schuster, Photophysical behavior of ester-substituted aminocoumarins: a new twist, J. Phys. Chem., 1989, 93, 1292–1295 CrossRef CAS.
- B. B. Raju and S. M. B. Costa, Photophysical properties of 7-diethylaminocoumarin dyes in dioxane–water mixtures: hydrogen bonding, dielectric enrichment and polarity effects, Phys. Chem. Chem. Phys., 1999, 1, 3539–3547 RSC.
- M. Sulpizi, P. Carloni, J. Hutter and U. Rothlisberger, A hybrid TDDFT/MM investigation of the optical properties of aminocoumarins in water and acetonitrile solution, Phys. Chem. Chem. Phys., 2003, 5, 4798–4805 RSC.
- K. Azuma, S. Suzuki, S. Uchiyama, T. Kajiro, T. Santa and K. Imai, A study of the relationship between the chemical structures and the fluorescence quantum yields of coumarins, quinoxalinones and benzoxazinones for the development of sensitive fluorescent derivatization reagents, Photochem. Photobiol. Sci., 2003, 2, 443–449 CrossRef CAS PubMed.
- M. El-Kemary and W. Rettig, Multiple emission in coumarins with heterocyclic substituents, Phys. Chem. Chem. Phys., 2003, 5, 5221–5228 RSC.
- X. Liu, Z. Xu and J. M. Cole, Molecular Design of UV–vis Absorption and Emission Properties in Organic Fluorophores: Toward Larger Bathochromic Shifts, Enhanced Molar Extinction Coefficients, and Greater Stokes Shifts, J. Phys. Chem. C, 2013, 117, 16584–16595 CrossRef CAS.
- M. Cavazzini, S. Quici, S. Orlandi, C. Sissa, F. Terenziani and A. Painelli, Intimately bound coumarin and bis(alkylaminostyryl)benzene fragments: synthesis and energy transfer, Tetrahedron, 2013, 69, 2827–2833 CrossRef CAS.
- D. Kim, S. Singha, T. Wang, E. Seo, J. H. Lee, S.-J. Lee, K. H. Kim and K. H. Ahn, In vivo two-photon fluorescent imaging of fluoride with a desilylation-based reactive probe, Chem. Commun., 2012, 48, 10243–10245 RSC.
- I. Kim, D. Kim, S. Sambasivan and K. H. Ahn, Synthesis of π-Extended Coumarins and Evaluation of Their Precursors as Reactive Fluorescent Probes for Mercury Ions, Asian J. Org. Chem., 2012, 1, 60–64 CrossRef CAS.
- D. Kim, Q. P. Xuan, H. Moon, Y. W. Jun and K. H. Ahn, Synthesis of Benzocoumarins and Characterization of Their Photophysical Properties, Asian J. Org. Chem., 2014, 3, 1089–1096 CrossRef CAS.
- Y. W. Jun, H. R. Kim, Y. J. Reo, M. Dai and K. H. Ahn, Addressing the autofluorescence issue in deep tissue imaging by two-photon microscopy: the significance of far-red emitting dyes, Chem. Sci., 2017, 8, 7696–7704 RSC.
- Y. J. Reo, Y. W. Jun, S. W. Cho, J. Jeon, H. Roh, S. Singha, M. Dai, S. Sarkar, H. R. Kim, S. Kim, Y. Jin, Y. L. Jung, Y. J. Yang, C. Ban, J. Joo and K. H. Ahn, A systematic study on the discrepancy of fluorescence properties between in solutions and in cells: super-bright, environment-insensitive benzocoumarin dyes, Chem. Commun., 2020, 56, 10556–10559 RSC.
- Y. Jung, N. K. Park, S. Kang, Y. Huh, J. Jung, J. K. Hur and D. Kim, Latent turn-on fluorescent probe for the detection of toxic malononitrile in water and its practical applications, Anal. Chim. Acta, 2020, 1095, 154–161 CrossRef CAS PubMed.
- A. Mukhopadhyay, T. Hossen, I. Ghosh, A. L. Koner, W. M. Nau, K. Sahu and J. N. Moorthy, Helicity-Dependent Regiodifferentiation in the Excited-State Quenching and Chiroptical Properties of Inward/Outward Helical Coumarins, Chem. – Eur. J., 2017, 23, 14797–14805 CrossRef CAS PubMed.
- A. Mukhopadhyay, V. K. Maka and J. N. Moorthy, Remarkable influence of ‘phane effect’ on the excited-state properties of cofacially oriented coumarins, Phys. Chem. Chem. Phys., 2017, 19, 4758–4767 RSC.
- A. Kakehi, S. Ito, H. Suga, S. Takano and K. Hirata, Preparation of New Nitrogen-Bridged Heterocycles. 45. Smooth Hydrolysis of 6-Membered Heterocyclic 2-Imines with Partial Aromaticity, Chem. Pharm. Bull., 1998, 46, 1632–1634 CrossRef CAS.
- C. Galli, “CESIUM ION EFFECT” AND MACROCYCLIZATION. A CRITICAL REVIEW, Org. Prep. Proced. Int., 1992, 24, 285–307 CrossRef CAS.
- S. S. Bhalodiya, M. P. Parmar, D. B. Upadhyay, C. D. Patel, D. P. Vala, D. Rajani and H. M. Patel, Cs2CO3-promoted one-pot synthesis of novel tetrahydrobenzofuran-4(2H)-ones: In vitro antimicrobial, antimalarial activity and in silico docking study, Results Chem., 2024, 7, 101304 CrossRef CAS.
- P. A. Khardina, E. M. Buev, V. S. Moshkin and V. Y. Sosnovskikh, Novel synthesis of 2,3-dihydrobenzofuran-3-amines from salicylic aldehydes: Trimethylsilyl as traceless activating group, Tetrahedron Lett., 2024, 139, 154992 CrossRef CAS.
- P. Li, Y. Ji, W. Chen, X. Zhang and L. Wang, The facile synthesis of 2-bromoindoles via Cs2CO3-promoted intramolecular cyclization of 2-(gem-dibromovinyl)anilines under transition-metal-free conditions, RSC Adv., 2012, 3, 73–78 RSC.
- J. Ezquerra and J. Alvarez-Builla, 2-Methylpyridinium salts as 1,4-dinucleophiles. II. Westphal condensation with substituted pyridinium substrates, J. Heterocycl. Chem., 1986, 23, 1151–1157 CrossRef CAS.
- Z. Chen, S. Zhang, X. Qi, S. Liu, Q. Zhang and Y. Deng, Fluorescent quinolizinium ionic liquids (salts) with unexpectedly high quantum yields up to >99%, J. Mater. Chem., 2011, 21, 8979–8982 RSC.
- T. S. Chinta Rao, S. Saha, G. B. Raolji, B. Patro, P. Risbood, M. J. Difilippantonio, J. E. Tomaszewski and S. V. Malhotra, Microwave assisted Westphal condensation and its application to synthesis of sempervirine and related compounds, Tetrahedron Lett., 2013, 54, 487–490 CrossRef CAS.
- J. V. Greenhill, H. Loghmani-Khouzani and D. J. Maitland, Tautomerism in ketomethylquinolines. Part 2. Further results on 2-ketomethylquinolines, J. Chem. Soc., Perkin Trans. 1, 1991, 2831–2840 RSC.
- A. Kakehi, S. Ito and T. Yotsuya, Preparation of New Nitrogen-Bridged Heterocycles. XIII.: Syntheses of Some Tricyclic and Tetracyclic Indolizine Derivatives with Antiallergic Activity, Chem. Pharm. Bull., 1986, 34, 2435–2442 CrossRef CAS PubMed.
- S. Wu, Q. Cao, X. Wang, K. Cheng and Z. Cheng, Design, synthesis and biological evaluation of mitochondria targeting theranostic agents, Chem. Commun., 2014, 50, 8919–8922 RSC.
- A. S. Brown, L.-M. Bernal, T. L. Micotto, E. L. Smith and J. N. Wilson, Fluorescent neuroactive probes based on stilbazolium dyes, Org. Biomol. Chem., 2011, 9, 2142–2148 RSC.
- Y. Liu, B. Wang, J.-T. Hou, P. Xie, W. Li and S. Wang, Molecular engineering and bioimaging applications of C2-alkenyl indole dyes with tunable emission wavelengths covering visible to NIR light, Bioorg. Chem., 2023, 141, 106905 CrossRef CAS PubMed.
- 12 Principles of Green Chemistry, https://www.acs.org/greenchemistry/principles/12-principles-of-green-chemistry.html, (accessed 31 July 2024).
- G. Kobayashi, Y. Matsuda, R. Natsuki, Y. Tominaga, T. Okamura and A. Itamura, Studies on Indole Derivatives. XVII. The Synthesis of Indoxyl Derivatives. (3) The Reactions of 3-Hydroxy-2-(1-methylthiovinyl)-indole Derivatives with Amines, Yakugaku Zasshi, 1973, 93, 964–970 CrossRef CAS PubMed.
- R. R. Valiev, V. N. Cherepanov, G. V. Baryshnikov and D. Sundholm, First-principles method for calculating the rate constants of internal-conversion and intersystem-crossing transitions, Phys. Chem. Chem. Phys., 2018, 20, 6121–6133 RSC.
- A. Manian, R. A. Shaw, I. Lyskov, W. Wong and S. P. Russo, Modeling radiative and non-radiative pathways at both the Franck–Condon and Herzberg–Teller approximation level, J. Chem. Phys., 2021, 155, 054108 CrossRef CAS PubMed.
- A. Kawski, On the Estimation of Excited-State Dipole Moments from Solvatochromic Shifts of Absorption and Fluorescence Spectra, Z. Naturforsch., A: Phys. Sci., 2002, 57, 255–262 CrossRef CAS.
- Ł. Kielesiński, I. Deperasińska, O. Morawski, K. V. Vygranenko, E. T. Ouellette and D. T. Gryko, Polarized, V-Shaped, and Conjoined Biscoumarins: From Lack of Dipole Moment Alignment to High Brightness, J. Org. Chem., 2022, 87, 5961–5975 CrossRef PubMed.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Structural Changes Accompanying Intramolecular Electron Transfer: Focus on Twisted Intramolecular Charge-Transfer States and Structures, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed.
- G. Dyker and O. Muth, Synthesis of Methylene- and Methine-Bridged Oligopyridines, Eur. J. Org. Chem., 2004, 4319–4322 CrossRef CAS.
- K. Senthil, S. Kalainathan and A. R. Kumar, Effect of additives on the large-size growth of 4-N,N-dimethylamino-4-N-methyl stilbazolium naphthalene-2-sulfonate (DSNS) single crystal: an efficient stilbazolium derivative NLO crystal with potential terahertz wave properties, CrystEngComm, 2014, 16, 9847–9856 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta Jr., F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 (Revision A.03), Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- F. Neese, Software update: the ORCA program system, version 4.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, UV/vis, fluorescence spectra and computational details. See DOI: https://doi.org/10.1039/d4qo01216j |
| This journal is © the Partner Organisations 2024 |