DOI:
10.1039/D3RA04008A
(Paper)
RSC Adv., 2024,
14, 2354-2368
Dispersed MnO2 nanoparticles/sugarcane bagasse-derived carbon composite as an anode material for lithium-ion batteries†
Received
14th June 2023
, Accepted 5th December 2023
First published on 11th January 2024
Abstract
Bagasse-derived carbon electrodes were developed by doping with nitrogen functional groups and compositing with high-capacity MnO2 nanoparticles (MnO2/NBGC). The bagasse-derived biochar was N-doped by refluxing in urea, followed by the deposition of MnO2 nanoparticles onto its porous surface via the hydrothermal reduction of KMnO4. Different initial KMnO4 loading concentrations (i.e. 5, 10, 40, and 100 mM) were applied to optimize the composite morphology and the corresponding electrochemical performance. Material characterization confirmed that the carbon composite has a mesoporous structure along with the dispersion of MnO2 nano-particles on the N-containing carbon surface. It was found that the 5-MnO2/NBGC sample exhibited the highest electrochemical performance with a reversible capacity of 760 mA h g−1 at a current density of 186 mA g−1. It delivered reversible capacities of 488 and 390 mA h g−1 in cycle tests at 372 and 744 mA g−1, respectively, for 150 cycles and presented good reversibility with nearly 100% coulombic efficiency. In addition, it could exert high capacities up to 388 and 301 mA h g−1 even under high current densities of 1860 and 3720 mA g−1, respectively. Moreover, most of the prepared composite products showed high rate capability with great reversibility up to more than 90% after testing at a high current density of 3720 mA g−1. The great electrochemical performance of the MnO2/NBGC nanocomposite electrode can be attributed to the synergistic impact of the hierarchical architecture of the MnO2 nanocrystals deposited on porous carbon and the capacitive effect of the N-containing defects within the carbon material. The nanostructure of the MnO2 particles deposited on porous carbon limits its large volume change during cycling and promotes good adhesion of MnO2 nanoparticles with the substrate. Meanwhile, the capacitive effect of the exposed N-functional groups enables fast ionic conduction and reduces interfacial resistance at the electrode interface.
Introduction
Currently, climate change, which is mainly caused by extensive carbon dioxide emissions from fossil fuel combustion, drives the demand for sustainable electrical energy generators and storage devices. Lithium-ion batteries (LIBs) have drawn much attention as effective energy storage and conversion devices for various electrical equipment ranging from small portable devices and electricity-driven automotives to electrical power plants owing to their high energy density, excellent cyclic stability, long lifetime, and low self-discharge features.1 The anode material is considered as one of the key components that determine the performance of LIBs. Generally, most commercial LIBs use graphite as the active anode material because of its feasibility for mass production. However, the production cost of high-quality graphite is quite high; it requires a long reaction process and consumes a high amount of fossil fuel-based energy. Moreover, its theoretical capacity is only about 370 mA h g−1 due to the limitation of the stoichiometric intercalation of lithium ions within the nominal graphitic layer.2 In the past decades, hard carbon (HC) has emerged as an alternative carbonaceous anode material for LIBs to enhance the ionic storage capacity of typical crystalline graphite. This non-graphitic carbon is derived from small domains of graphene-like stacking layers (with dilated d-spacing) and embedded microporous regions (a “house of cards” structure).3 This unique microstructure allows both ionic intercalation between the carbon layers and ionic adsorption on the surface defect sites, as well as ionic filling of the micropores.4 HC is usually derived from the pyrolysis of biomass and polymeric carbon precursors, such as pitch, phenolic and epoxy resins. The resulting electrochemical ionic storage capability is generally influenced by the carbon precursor type, material morphology, carbonization temperature, and heteroatom doping.5–7
Recently, the sustainable concept of converting agricultural waste into porous carbon electrode materials is rapidly developing.8,9 One of the promising biomass-based carbon precursors is sugarcane bagasse, which naturally has a high density of xylem and phloem that can facilitate the formation of porous carbon materials with abundant transport channels.3,10 So far, research on the development of sugarcane-bagasse-derived carbon materials as potential anodes for LIBs has mostly been devoted to optimizing the preparation approach or employing elaborate methods to formulate highly porous structures.10–12 Many studies have focused on doping with heteroatoms, such as nitrogen, sulphur, and phosphorus-containing functional groups, to promote charge storage capability and ionic conduction, which ultimately increase the cycling stability of the carbon electrode materials. The capacitive effect arising from their electron-donating characteristic leads to a reduction in interfacial resistance between the electrode and the aqueous electrolyte.13,14
Meanwhile, transition metal oxides (MO) have also emerged as attractive electrode materials for LIBs. In particular, manganese oxide, as MnO2, has been regarded as a promising electrode material owing to its high theoretical capacity of 1230 mA h g−1, abundance, low cost, and environment-friendly nature. Moreover, it is the main composing material in primary alkaline batteries and can be recycled from disposed batteries. However, as an anode material, MnO2 typically shows fast capacity fading due to the large volume change during the lithium insertion-desertion process. One of the potential methods to improve the mechanical stability of MnO2-based electrodes is to deposit it onto a supporting material or form a porous structure.15,16
Therefore, this research was aimed at developing a modified bagasse-derived carbon anode material for LIBs. The electrochemical performance of the bagasse-based carbon material was enhanced by doping it with nitrogen functional groups using urea as the precursor, followed by the deposition of different amounts of MnO2 nanoparticles on the carbon substrate via the reduction of KMnO4 at various initial concentrations. The doped N-containing functional groups in conjunction with the MnO2 nanocomposite are expected to promote ionic conduction and lithium storage capability. Meanwhile, the MnO2 nanoparticles deposited onto the bagasse-based porous carbon substrate would improve the cycling stability of the MnO2 compound from a large volume expansion during the conversion process. The carbon composite products prepared with different KMnO4 loadings were analyzed to investigate the impact of KMnO4 loading on composite morphology and corresponding electrochemical performance.
Experimental details
Sodium hydroxide (NaOH), calcium chloride (CaCl2) and urea (CH4N2O) were obtained from Kemaus. Hydrochloric acid (HCl, 37%) and potassium permanganate (KMnO4) were obtained from QRëC. All chemicals were used as received without purification.
Preparation of N-containing sugarcane-bagasse-derived carbon material (NBGC)
7.5 g of sugarcane bagasse was soaked in 10% w/v NaOH for 30 minutes, washed with DI water until a pH of around 6.5–7 was reached and dried at 80 °C overnight. Dried sugarcane bagasse was then dispersed in 0.5 M sulfuric acid inside a Teflon-lined autoclave and heated at 180 °C for 24 hours. Subsequently, the solid residue was filtered, washed with DI water, and dried at 80 °C overnight to obtain the biochar product. Later, the obtained biochar was mixed with CaCl2 and urea at a carbon material![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CaCl2:urea weight ratio of 1:2:2 in 50 mL DI water and refluxed at 80 °C for 4 hours. After cooling to room temperature, the solid product was filtered and pyrolyzed at 800 °C for 2 hours under a nitrogen atmosphere at a heating rate of 5 °C min−1. After the pyrolysis process, the N-doped carbon material was soaked in 2 M HCl under stirring for 1 hour and washed with DI water until the pH reached around 6–7. After drying in a vacuum oven at 105 °C for 12 hours, the N-doped sugarcane-bagasse-derived carbon material (NBGC) was achieved as a fine black powder.
:CaCl2:urea weight ratio of 1:2:2 in 50 mL DI water and refluxed at 80 °C for 4 hours. After cooling to room temperature, the solid product was filtered and pyrolyzed at 800 °C for 2 hours under a nitrogen atmosphere at a heating rate of 5 °C min−1. After the pyrolysis process, the N-doped carbon material was soaked in 2 M HCl under stirring for 1 hour and washed with DI water until the pH reached around 6–7. After drying in a vacuum oven at 105 °C for 12 hours, the N-doped sugarcane-bagasse-derived carbon material (NBGC) was achieved as a fine black powder.
Synthesis of MnO2 nanoparticle-dispersed N-doped sugarcane-bagasse-derived porous carbon composite (MnO2/NBGC)
0.3 g of the obtained NBGC was dispersed in 80 mL of aqueous potassium permanganate (KMnO4) solutions with different initial concentrations of 5, 10, 40 and 100 mM, respectively. Each mixture was well mixed by sonication for 1 hour. After transferring to a Teflon-lined autoclave, the mixture was heated up at 140 °C for 2 hours and then cooled to room temperature. The solid product was filtered, washed with DI water, and dried at 80 °C overnight. The achieved MnO2/NBGC carbon composite products were named based on the different initial KMnO4 concentrations (i.e., 5, 10, 40, and 100 mM) as 5-MnO2/NBGC, 10-MnO2/NBGC, 40-MnO2/NBGC, and 100-MnO2/NBGC, respectively. As a reference, pure MnO2 was solely synthesized by employing the same procedure without NBGC addition. In detail, 180 mL of a 50 mM KMnO4 solution was sonicated for an hour and then transferred to the Teflon-lined autoclave. The solution was heated at 140 °C for 24 hours. The obtained solid was filtered, washed, and dried at 80 °C overnight.
Material characterization
The crystallographic information of the products was obtained using X-ray diffraction (XRD, Bruker D8 Advance) with Cu-Kα radiation λ = 1.54060 Å and 2θ = 10° to 80°. The morphologies of the carbon composites with porous MnO2 coatings were characterized by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) with a high tension of 120 kV (HITACHI-HT7700). The surface elemental analysis of the samples was carried out using energy-dispersive X-ray spectroscopy (FEI, QUANTA 450), and the bulk C, H, and N contents were analyzed by an Elemental Analyzer (LECO, CHN628 Series). X-ray photoelectron spectroscopy (XPS) was used to analyse the surface functional groups on the carbon composite products. N2 adsorption–desorption measurement (Quantachrome ASIC-2) was employed to obtain the specific surface area based on the Brunauer–Emmett–Teller theory (BET).
Electrochemical performance evaluation
The electrochemical performance of the MnO2/NBGC carbon composite products was assessed by assembling coin cells (CR2032). The anode material slurry was composed of 80 wt% of the prepared MnO2/NBGC carbon composite, 10 wt% of Super P conductive carbon, and 10 wt% of polyvinylidene fluoride (PVDF) in N-methylpyrrolidone, which was used as a binder. After vigorous mixing for 3 hours, the mixture was cast on a copper foil, followed by drying overnight at 70 °C in a vacuum oven. Then, the cast anode was cut into circle-shaped specimens of radius 14 mm, which approximately contained 2 mg of the dried material. Subsequently, the prepared anode electrode was assembled with a lithium metal counter electrode, Celgard2320 (PP/PE/PP) separator, and 1 M LiPF6 in the electrolyte mixture of 1:1:1 v/v EC:EMC:DMC. The testing cells were charged and discharged between 0.01 and 3.0 V relative to Li/Li+ at various current densities by using an electrochemical charge–discharge tester (Gelon company Neware BTS-4000). Cyclic voltammetry (CV) measurements were performed in the potential window of −0.1 to 3 V relative to Li/Li+ at a scan rate of 1–2.2 mV s−1 to investigate the electrode reaction mechanism by using a potentiostat/galvanostat (Autolab PGSTAT101). Finally, electrochemical impedance spectroscopy (EIS) (VersaSTAT3 Potentiostat Galvanostat, Prinston Applied Research, USA) was used to measure the electrochemical impedance of the electrode in the frequency range of 0.1–100000 Hz and at the voltage of 10 mV.
Results and discussion
The morphologies of the bagasse-derived carbon material products and hydrothermally synthesized MnO2 nanoparticles were observed by using FESEM (Fig. 1a–g) and TEM (Fig. 2a–f). The bagasse-derived carbon materials (BGC) showed honeycomb-like porous structures that depict their natural xylem and phloem transporting channels with macropores of diameter about 5–8 μm and relatively thick walls. After refluxing bagasse with urea to insert N-containing functional defects, the obtained N-doped bagasse-based carbon (NBGC) material exhibited a slightly smoother surface compared to pristine bagasse. The collapse of some connecting walls within the parent bagasse carbon during the reflux process resulted in merged pores, leading to a larger pore size in the N-containing carbon compound, as listed in Table 1. Composites with dispersed manganese dioxide (MnO2) nanoparticles at various loading contents on N-containing bagasse-derived porous carbon (MnO2/NBGCs) were prepared by using different concentrations of the potassium permanganate (KMnO4) precursor (e.g. 5, 10, 40, and 100 mM). Likewise, the MnO2/NBGC composites obtained with different KMnO4 loading concentrations retained the honeycomb-like porous structure of the parent bagasse-based carbon substrate. The porous structure of the MnO2/NBGC composite can accommodate the large volume expansion of the MnO2 compound during the Li+ insertion/desertion process. It was found that the increase in the applied KMnO4 precursor led to more agglomeration of the MnO2 nanoparticles, ultimately resulting in thicker MnO2 coatings on the porous surface. This thick coating layer can block the pore channels and cause a reduction in the specific surface area of the electrode material. As the reference, pure MnO2 nanoparticles were also synthesized by the hydrothermal reaction in the absence of the bagasse-based carbon substrate. As seen in Fig. 1g, without the carbon substrate, the synthesized MnO2 nanocrystals exhibited radial growth and became spherical coral-like particles with an average diameter of around 1 μm. Similarly, the TEM images shown in Fig. 2c–f also reveal a coating layer of MnO2 on the bagasse-derived carbon surface. Meanwhile, only clusters of carbon material without any coating layer or spike-like coating were observed in the cases of BGC and NBGC. At a low concentration of KMnO4, as in samples 5-MnO2/NBGC and 10-MnO2/NBGC, only a thin MnO2 coating layer was formed on the surface of porous carbon. At higher loading concentrations of the KMnO4 precursor, as in the samples 40-MnO2/NBGC and 100-MnO2/NBGC, the rapid growth of the MnO2 clusters led to thicker coating layers on the carbon surface, forming numerous tiny nanoflakes. This obtained caterpillar-like nanostructure was similar to the porous MnO2 nanocomposite reported by Xia et al.16 who synthesized MnO2 coating layers on carbon nanotubes via a similar procedure. These MnO2 nanoflakes were interconnected and uniformly distributed on the carbon surface. By controlling the KMnO4 loading concentration, the thickness of the MnO2 layer could be practically varied. The mechanism of MnO2 nanocrystal growth on the carbon material can be explained by the reduction reaction of KMnO4.17 Briefly, the N-doped carbon material (NBGC) was initially dispersed well in a KMnO4 solution, which was subjected to a hydrothermal reaction to produce the final carbon composite product. During mixing within the KMnO4 solution at room temperature, nanocrystalline MnO2 seeds are generated on the surface of the porous carbon substrate via a slow redox process, according to eqn (1).16,18 Once the solution further undergoes the hydrothermal reaction, the MnO2 nanoparticles grow from the preformed nanocrystalline seeds due to the decomposition of KMnO4 in water, according to eqn (2).16 The flaky morphology is formed due to the preferred growth of the layered birnessite-type MnO2 along the ab plane.19 Here, the layer of nano-structured MnO2 particles coated the surfaces of the porous carbon substrate uniformly to form nanocomposite products.| | |
4MnO4− + 3C + 2H2O → 4MnO2 + CO32− + 2HCO3−
| (1) |
| | |
4MnO4− + 2H2O → 4MnO2 + 4OH− + 3O2
| (2) |
 |
| | Fig. 1 SEM images of the prepared bagasse-based carbon materials: (a) bagasse-derived carbon material (BGC), (b) N-doped bagasse-derived carbon material (NBGC), and MnO2/N-doped bagasse-derived carbon composites with different loading concentrations (MnO2/NBGC): (c) 5-MnO2/NBGC, (d) 10-MnO2/NBGC, (e) 40-MnO2/NBGC, (f) 100-MnO2/NBGC, and (g) hydrothermally synthesized pure MnO2. | |
 |
| | Fig. 2 TEM images of the prepared bagasse-based carbon material: (a) bagasse-derived carbon material (BGC), (b) N-doped bagasse-derived carbon material (NBGC), MnO2/N-doped bagasse-derived carbon composite at different loading concentrations (MnO2/NBGC): (c) 5-MnO2/NBGC, (d) 10-MnO2/NBGC, (e) 40-MnO2/NBGC, and (f) 100-MnO2/NBGC. | |
Table 1 Surface morphology and contents of the crystalline bagasse-derived carbon composites with different MnO2 loading contents, as analyzed by Raman spectroscopy
| Carbon composite |
Specific surface area (m2 g−1) |
Pore volume (cm3 g−1) |
Avg. pore size (nm) |
Id/Ig |
| BGC |
271.80 |
0.34 |
5.01 |
1.296 |
| NBGC |
210.63 |
0.29 |
5.59 |
1.251 |
| 5-MnO2/NBGC |
183.11 |
0.23 |
4.97 |
1.233 |
| 10-MnO2/NBGC |
178.93 |
0.23 |
5.82 |
1.187 |
| 40-MnO2/NBGC |
144.51 |
0.17 |
4.76 |
1.231 |
| 100-MnO2/NBGC |
105.61 |
0.11 |
4.01 |
1.245 |
Surface elemental analysis using Energy-Dispersive Spectroscopy (EDS) and the C, H and N content analysis of the bagasse-derived carbon material and the MnO2-doped carbon composite products with different degrees of doping are summarized in Table S1.† Based on surface EDS mapping, as a reference, the bagasse precursor was subjected to the hydrothermal reaction and carbonized at 800 °C but refluxed without urea addition. It transformed into a carbon material (BGC) that contained a high C content of about 92 wt%. However, oxygen-containing functional groups, such as oxy, hydroxyl, and carbonyl groups, which are typically found for bio-based carbon, were retained. In the NBGC compound, refluxing in the urea solution resulted in a nitrogen functional group content of approximately 5 wt%. Subsequently, dispersing NBGC in KMnO4 under a hydrothermal condition could deposit manganese oxide onto the carbon substrate, as indicated by the emergence of Mn content in the composite products. Increasing the loading concentration of the KMnO4 precursor caused the generation of more manganese oxide, as greater Mn wt% was observed. This was also confirmed by the results from the elemental analyzer, which demonstrated that C and N decreased as they were consumed during the formation of MnO2. However, the differences in the amounts of C and N determined from EDS mapping and the elemental analyzer are attributed to the different collecting techniques. For instance, EDS generally maps the elements only on the surface and over some selected areas, whereas the C H N elemental analyzer provides bulk elemental analysis. The lower amount of C and N observed in the bulk analysis may result from the normalization of these elements with O and Mn from MnO2 and other related O-containing organic functional groups. Results from both techniques agree well with the observed SEM and TEM images, which revealed a thicker coating layer on the porous substrate for samples with higher KMnO4 loading contents. Notably, the N-containing defects are expected to support ionic conductivity and promote lithium-ion capacity. On the one hand, MnO2 accommodates high Li+ storage capability because of its high theoretical capacity. However, the formation of a thick MnO2 coating may not only block the exposed nitrogen functional groups and retard ionic transport but also readily inhibit the growth of its good crystalline structure.
Nitrogen adsorption/desorption analysis was conducted to investigate the pore morphology of BGC, NBGC, and the MnO2/NBGC carbon composites. The sorption isotherms illustrated in Fig. S1† present type IV isotherms in the medium to high relative pressure range of about 0.5–1.0 P/P0, indicating mesoporous material characteristics. H3-type hysteresis loops at high pressures and nearly parallel adsorption and desorption branches at relative pressures between 0.05 and 0.5 were observed for all samples, revealing the presence of slit-shaped pores that enable the aggregation of plate-like particles. As summarized in Table 1, pristine bagasse-based porous carbon (BGC) showed a Brunauer–Emmett–Teller (BET) specific surface area of approximately 271.80 m2 g−1 with a specific pore volume of 0.34 cm3 g−1 and a pore size of 5.01 nm. Doping with N-containing compounds resulted in a reduction in the BET specific surface area to 210.63 m2 g−1 with a larger average pore size and a smaller specific pore volume. This is consistent with its corresponding SEM image (Fig. 1b), which reveals merged pores formed due to the collapse of the connecting walls within BGC. Further loading MnO2 on the obtained N-doped carbon tended to decrease the BET specific surface area of the carbon composite product. Increasing the MnO2 loading concentration led to smaller pore volumes and average pore sizes, which can be explained by the formation of thicker MnO2 coating layers that clog the pores of the bagasse-based parent carbon substrate. On the one hand, high content of MnO2 can promote electrode capacity because of its high theoretical capacity of 1230 mA h g−1. However, coating a very thick MnO2 layer can cause a reduction in the active specific surface area and weak adhesion of the MnO2 layers on the porous carbon substrate, as well as shield the capacitive effect of the doped N-containing defects within the carbon material.
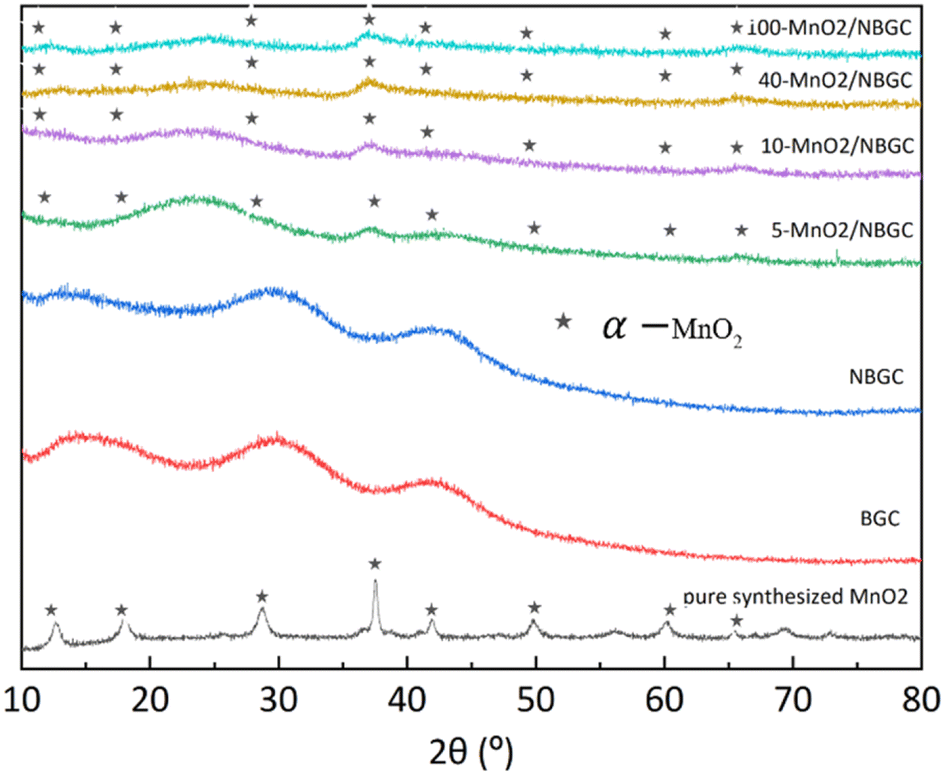
The X-ray diffraction patterns of BGC, NBGC, the carbon composite of porous NBGC with dispersed MnO2 particles at various loading concentrations (MnO2/NBGC), and pure manganese dioxide (MnO2) are shown in Fig. 3. Bagasse-derived carbon (BGC) and its corresponding N-doped compound (NBGC) exhibited broad diffraction peaks at 2θ of approximately 17°, 32° and 44°, indicating the characteristic (002) and (100) planes of the amorphous carbon structure with a low graphitization fraction.20 Pure MnO2 synthesized from the hydrothermal reaction had XRD diffraction peaks at 2θ of 12.8, 18.1, 28.9, 37.5, 42, 49.9, 60.3 and 65.4°, representing the (110), (200), (310), (211), (301), (411), (600) and (002) planes of hollandite-type α-MnO2, respectively (JCPDS 44-0141).21–23 Compared to BGC and NBGC, the MnO2/NBGC composite products at all MnO2 loading concentrations showed common XRD peaks at 2θ of approximately 38° and 67°, consistent with the main characteristic crystalline planes of α-MnO2. All the diffraction peaks obtained from the deposited MnO2 nanoparticles were quite weak and broad, denoting their nanocrystalline nature. Interestingly, in the XRD patterns of all the MnO2/NBGC carbon composite samples, the XRD peak at 2θ = 32°, which represents the amorphous structure of the bagasse-derived parent carbon, was shifted to a lower diffraction angle of around 2θ = 25°. This implies that the inserted MnO2 nanoparticles caused the expansion of the carbon plane structure such that the d-spacing of the carbon plane increased from about 1.5 Å to 1.9 Å, as calculated based on Bragg's law. This wider d-spacing can ultimately facilitate better accessibility of Li+ or larger ions, such as Na+ and K+. As the concentration of MnO2 doping increased, the diffraction peaks from MnO2 became stronger, whereas the diffraction peaks from the carbon parent became almost invisible. This indicates the growth of thicker MnO2 layers covering the porous carbon substrate.
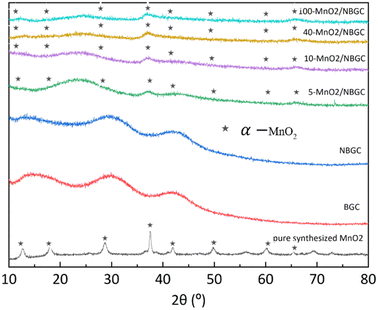 |
| | Fig. 3 The XRD spectra of BCG, NGGC, 5-MnO2/NBGC, 10-MnO2/NBGC, 40-MnO2/NBGC, 100-MnO2/NBGC, and pure MnO2. | |
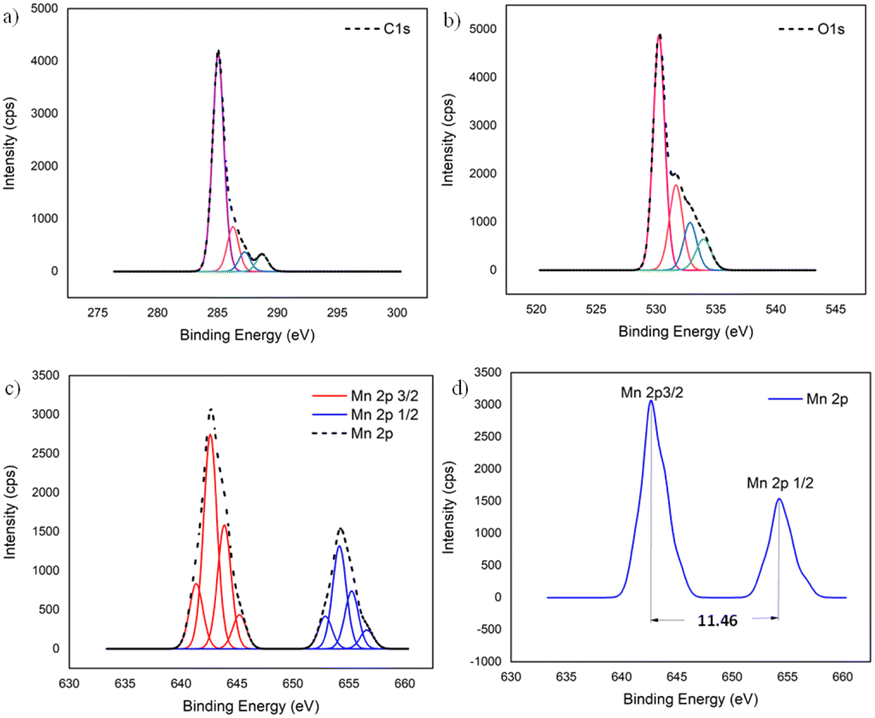
To further confirm the XRD results, the oxidation state and the composition of manganese oxide in the MnO2/NBGC nanocomposite products, as well as those of the bagasse-derived carbon material, were examined by X-ray photoelectron spectroscopy (XPS). As shown in Fig. S2a and b,† the wide survey XPS spectrum of BGC and NBGC comprised binding energy peaks at 285 and 533 eV, representing C 1s and O 1s. Meanwhile, as seen in Fig. S2c,† the XPS survey spectra of the MnO2/NBGC composites with different MnO2 loading concentrations showed a predominant signal of Mn 2p at the binding energy of around 642 eV in addition to the C 1s and O 1s peaks at 285 and 531 eV, respectively (Table S2†). Notably, signals of the N-containing defects in the carbon material were not clearly observed in all samples. This may be because of the very low nitrogen content (∼5 wt% detected by EDS elemental mapping) in NBGC. According to the lower penetration depth of the XPS technique compared with that of the EDS technique, the MnO2 coating layer may shield the signal of the N functional groups underneath in the case of MnO2/NBGC composites. The deconvoluted XPS spectra of the MnO2/NBGC composites are reported in Fig. 4. Fig. 4a shows the deconvoluted C 1s spectrum with four binding energy peaks at 284.9, 286.2, 287.3 and 288.7 eV, which represent graphitic carbon (C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), C–O, CO, and CO–O, respectively.13,24 The deconvoluted O 1s XPS spectrum, shown in Fig. 4b, indicates the bonds between manganese and lattice oxygen, as well as surface-adsorbed oxygen, including Mn–O–Mn, Mn–O–H and C–O/CO, which correspond with the binding energy peaks at 530.2, 531.7 and 532.9 eV, respectively.15,24 In Fig. 4c and d, the Mn 2p spectra exhibit well-defined valence states at binding energies approximately equal to 643.3 and 654.8 eV, which represent the Mn 2p3/2 and Mn 2p1/2 states, respectively. The difference between the two binding energy signals of Mn 2p was about 11.5 eV, which is the typical characteristic of Mn4+ in MnO2.25
C), C–O, CO, and CO–O, respectively.13,24 The deconvoluted O 1s XPS spectrum, shown in Fig. 4b, indicates the bonds between manganese and lattice oxygen, as well as surface-adsorbed oxygen, including Mn–O–Mn, Mn–O–H and C–O/CO, which correspond with the binding energy peaks at 530.2, 531.7 and 532.9 eV, respectively.15,24 In Fig. 4c and d, the Mn 2p spectra exhibit well-defined valence states at binding energies approximately equal to 643.3 and 654.8 eV, which represent the Mn 2p3/2 and Mn 2p1/2 states, respectively. The difference between the two binding energy signals of Mn 2p was about 11.5 eV, which is the typical characteristic of Mn4+ in MnO2.25
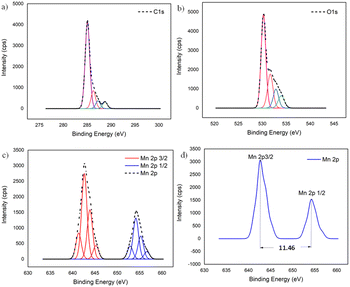 |
| | Fig. 4 The deconvoluted XPS spectra of MnO2/NBGC: (a) C 1s, (b) O 1s, and (c) Mn 2p; (d) binding energy difference between Mn 2p3/2 and Mn 2p1/2. | |
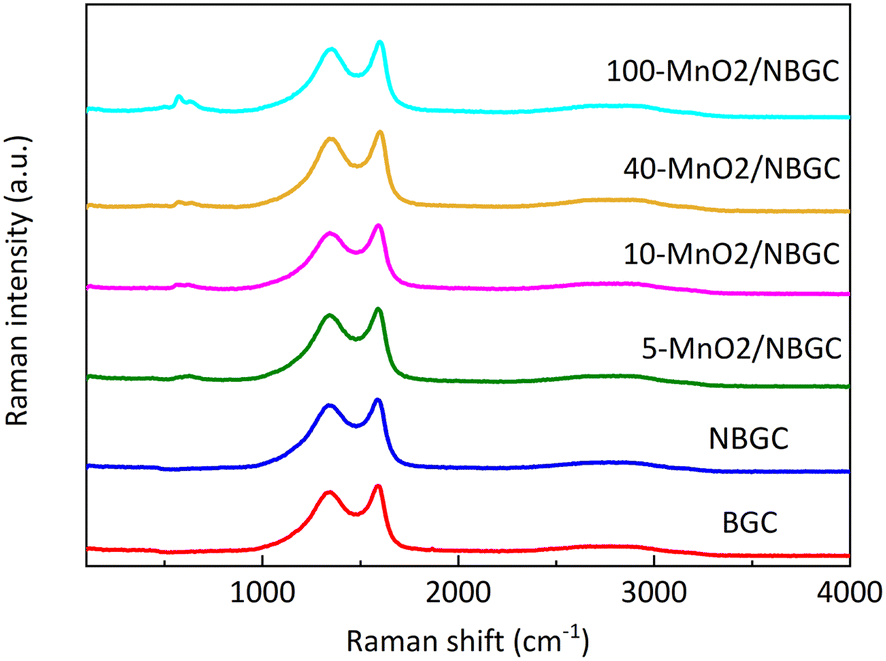
Raman spectroscopy was further employed to analyze the ordered structures of the MnO2/NBGC composite products (Fig. 5). The G band and D band of the carbon structure were observed around 1586–1604 cm−1 and 1336–1354 cm−1, respectively. The weak bands at 630 cm−1 were related to the symmetric stretching vibration of M–O in the MnO6 groups.26 The findings reveal that somehow doping carbon with N-containing functional groups and loading it with MnO2 nanoparticles via thermal reduction of KMnO4 result in a carbon structure with better crystallinity, as reflected by the lower values of Id/Ig for NBGC and all MnO2/NBGC samples (Table 1). Perhaps, either refluxing under basic conditions or the hydrothermal reduction of KMnO4 might catalyze the conversion of the bio-based amorphous carbon material into a carbon framework with better crystallinity. However, a continuous increase in MnO2 loading by enhancing KMnO4 concentration, such as the cases of 40-MnO2/NBGC, and 100-MnO2/NBGC, tended to drive the formation of more disordered carbon structures, as observed by their higher Id/Ig ratios.
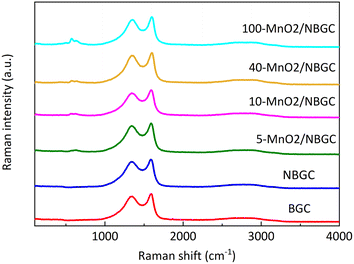 |
| | Fig. 5 The Raman spectra of BGC, NBGC and MnO2/NBGC with different MnO2 loadings. | |
Electrochemical performance evaluation
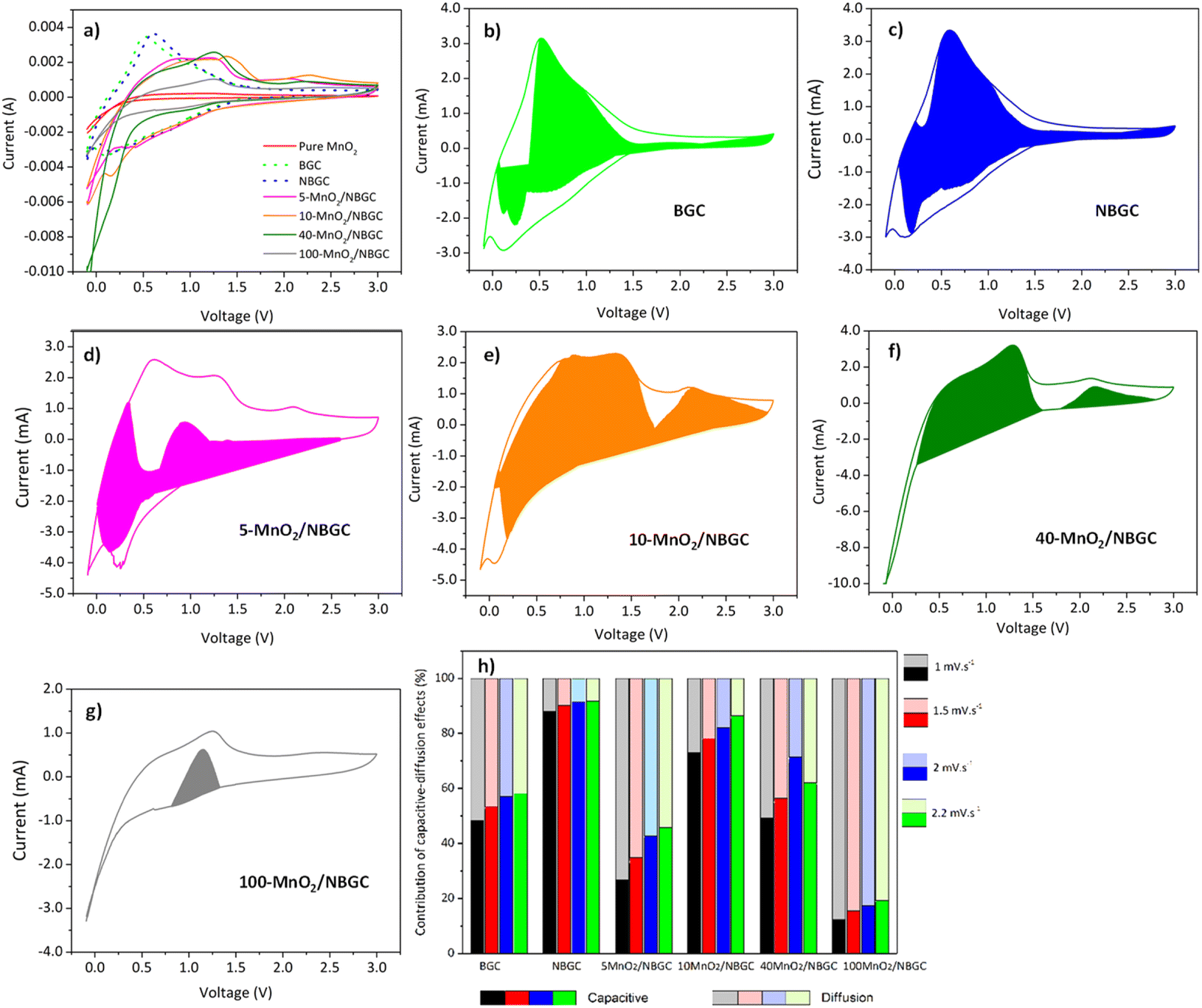
Cyclic voltammetry (CV) was used to investigate the lithium storage phenomena of the bagasse-based carbon materials (BGC and NBGC) and the MnO2/NBGC composites when used as anode materials in the potential window of −0.01–3.0 V vs. Li/Li+ at a scan rate of 2 mV s−1. As shown in Fig. 6a, the 1st CV scans of BGC and NBGC presented broad cathodic peaks at around 0.3 and 0.4 V, respectively, corresponding to the decomposition of the electrolyte into a solid coating interface (SEI). Meanwhile, the obvious anodic peak at approximately 0.6–0.7 V corresponds to the de-intercalation of Li+ from the carbon structure. The sloped broad anodic peak in the range of 0.7–1.3 V arises from the extraction of Li+ from the pores.27,28 In general, the CV signal for the de-insertion of Li+ from the carbon material with a large surface area in the 1000–2000 m2 g−1 range will appear as a broad anodic peak.29–32 The obvious anodic peaks of our carbon material that does not hold a very high specific surface area (∼271.80 m2 g−1) may imply the greater influence of Li+ de-intercalation from the MnO2 compound over the de-insertion process typically found in high-surface-area biomass-based porous carbon materials. In the CV scans of the MnO2/NBGC composites, the oxidation peaks observed at 1.2 V and 2.3 V represent the 2-step oxidation reaction of Mn0 to Mn2+ and Mn2+ to Mn4+, respectively.33 The more distinct anodic peaks of the composites with high MnO2 loading, such as 40-MnO2/NBGC and 100-MnO2/NBGC, may have resulted from the greater content of MnO2 in these composites. The cathodic peaks appearing at ∼1 V and 0.4 V correspond to the reduction of manganese oxide to metallic manganese (Mn4+ to Mn0) in conjunction with the formation of Li2O34 and SEI formation, respectively. The reaction mechanism can be described as shown in eqn (3) and (4); during discharge, MnO2 is reduced to Mn, and Li+ is converted to Li2O. This lithiation typically leads to the formation of nanometer-scale metal clusters embedded in a Li2O matrix accompanied by a large volume expansion. The formation of Mn/Li2O leads to a theoretical capacity of 1230 mA h g−1 for MnO2. During charging, Mn is oxidized to form MnO2, while Li2O is decomposed into Li+.35–37| | |
MnO2 + 4Li+ + 4e− ↔ 2Li2O + Mn
| (3) |
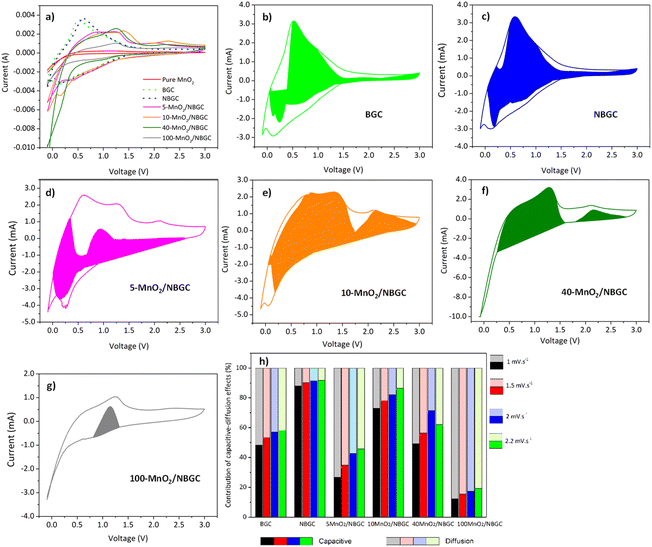 |
| | Fig. 6 (a) Cyclic voltammograms (CVs) of BGC, NBGC and MnO2/NBGC composite at various MnO2 loading contents at 2 mV s−1, CVs at scan rates of 2 mV s−1 and the corresponding capacitive currents (colored region) of (b) BGC, (c) NBGC, (d) 5-MnO2/NBGC, (e) 10-MnO2/NBGC, (f) 40-MnO2/NBGC, (g) 100-MnO2/NBGC, and (h) capacity contribution ratios at various scan rates of BGC, NBGC and MnO2/NBGC composite at various MnO2 loading contents. | |
Furthermore, the lithium-ion storage mechanisms of BGC, NBGC, and the MnO2/NBGC composites with different doping content were investigated by using cyclic voltammetry to observe their electrochemical behavior at varied scanning rates. The relationship between the current and the scanning rate is depicted by the following eqn (5),38–41 and the charge storage process is also described by eqn (5).
where the measured current
i follows the power law relationship with the scan rate
ν. Both
a and
b are fitting parameters obtained from the intercept and slope of the plot log(
i)
vs. log(
ν), respectively. There are two defined values for
b,
i.e. 0.5 and 1.0. At
b = 0.5, the current is proportional to the square root of the sweep rate
ν, and the current response is purely controlled by ion diffusion in the electroactive material, reflecting that the current and capacity result from the faradaic intercalation/extraction processes. Meanwhile, at
b = 1.0, the current is linearly proportional to the sweep rate
ν, which is considered capacitive response; here, the current and capacity originate from the charge-transfer process involving the surface atoms and the non-faradaic double layer effect or surface adsorption process. Therefore, for a system governed by the synergistic effect between ionic diffusion and the surface capacitive effect, the relationship between the current and the scan rate at a fixed potential can be described as follows:
| | |
I(V)/ν1/2 = k1ν1/2 + k2
| (7) |
where both
k1 and
k2 are constants at each fixed potential, while
k1ν and
k2ν1/2 correspond to the current contributions of the surface capacitive effects and diffusion-controlled intercalation/extraction processes, respectively. To quantitatively estimate the current contributions of the surface capacitive effects and diffusion-controlled insertion/extraction processes, a series of
k1 and
k2 for each potential was solved by plotting
I(
V)/
ν1/2 vs. ν1/2, in which the slope and
y-axis intercept of the plot correspond to
k1 and
k2, respectively. The ratio of capacitive contribution to the total capacity was determined from the integrated area of the plot (
k1ν vs. potential) and the total integrated area of the CV curve. Fig. S3a–f
† show the CVs of BGC, NBGC and MnO
2/NBGC doped with different loadings of MnO
2 at various sweep rates between 0.1 and 2.2 mV s
−1, respectively. It can be observed that all anodic peaks had shifted to higher potentials, and the cathodic peaks moved to lower potentials with the increase in scanning rate. This reflects high polarization at high sweep rates.
Fig. 6b–g present the ratios of capacitive contribution to the total capacity obtained by integrating the area of capacitive current (
k1ν)
vs. potential curve in comparison with the total area of the CV curves at the selected scan rates of 2 mV s
−1. While, the capacity contribution ratios between capacitive effect to the diffusion controlled process are summarized in
Fig. 6h. The values of
k1 and
k2 at various potentials were calculated by employing
eqn (7). Regarding the behavior, the samples were classified into two groups, namely (i) BGC and NBGC and (ii) the MnO
2/NBGC composites with various MnO
2 contents, to analyze the kinetics of the ionic storage mechanism. For BGC and NBGC, the capacities mainly depended on the surface capacitive effect, as indicated by the high capacitive contribution, that is, roughly more than half, including 48–53% for BGC and 88–92% for NBGC. The high capacitive contributions may be attributed to the high surface area of the materials. Especially, the significantly high capacitive contribution (more than 80%) in the case of NBGC may result from its abundant exposed electron rich-N functional groups that readily attract the lithium ions. Compared with their parent,
i.e. NBGC, the MnO
2/NBGC composites showed less influence of the capacitive effect on their capacities. With the addition of MnO
2, it was clearly seen that the ionic storage mechanism was increasingly controlled by the synergistic impact of the surface capacitive effect and diffusion-controlled insertion/extraction processes. The capacitive contributions of the MnO
2/NBGC composites at varying scan rates ranging from 1–2.2 mV s
−1 were 27–46%, 73–86%, 49–71%, and 12–19%, for 5-MnO
2/NBGC, 10-MnO
2/NBGC, 40-MnO
2/NBGC, and 100-MnO
2/NBGC, respectively. Interestingly, with the increase in MnO
2 doping, the ionic capacity seemed to basically rely on the capacitive effect or adsorption at the surface of the active MnO
2/NBGC electrode composites up to an optimum condition. With a further increase in MnO
2 doping, the ionic storage process predominantly depended on ionic diffusion-controlled intercalation/extraction to the crystalline structure of the active material. Notably, 5-MnO
2/NBGC, which had a comparable specific surface area and N content but a relatively lower amount of MnO
2 than 10-MnO
2/NBGC (see
Tables 1 and S1
†), showed quite a low capacitive contribution. The reason for the greater role of the diffusion-controlled process in the case of the 5-MnO
2/NBGC sample may be the better crystallinity of its structure as it was grown with a more diluted KMnO
4 precursor. A high initial KMnO
4 concentration may cause very rapid growth of MnO
2 crystals on the carbon surface, leading to the formation of MnO
2 nanocrystals with denser or lower crystallinity. Hence, 5-MnO
2/NBGC, which has a better crystalline structure of the MnO
2 active material, would better support the process of Li
+ intercalation/extraction. This can promote its ionic capacity thereby high storage capability of the MnO
2 compound. The 10-MnO
2/NBGC sample, which possesses lower MnO
2 crystallinity while holding a comparable surface area to 5-MnO
2/NBGC, would then prefer ionic storage
via surface adsorption processes. Further increasing MnO
2 doping on the NBGC, as the cases of 40-MnO
2/NBGC and 100-MnO
2/NBGC, did not only reduce the specific surface area due to blockage of the porous structure, but the thick MnO
2 coating would also shield the exposed N-functional groups on NBGC. In addition, their MnO
2 nanocrystallites may exhibit low crystallinity under a dense crystalline growth. This may result in a lower capacitive contribution to the ionic capacity and also low capacity due to limited ionic intercalation in the poor crystalline structures. In particular, 100-MnO
2/NBGC, which contained a substantially high MnO
2 content while possessing the lowest surface area, acted more like bulk MnO
2, thereby favoring an ionic storage mechanism
via the intercalation/extraction processes. Moreover, the fraction of capacitive contribution to the total capacity of all the MnO
2/NBGC composites mostly increased with the scanning rate and current density (
Fig. 6h), indicating that at high charge/discharge rates, they are likely to rely on the surface capacitive effect or surface adsorption process to sustain their ionic capacity. The high contribution of pseudocapacitance would be beneficial to the electrochemical performance of electroactive materials at high rate capacities.
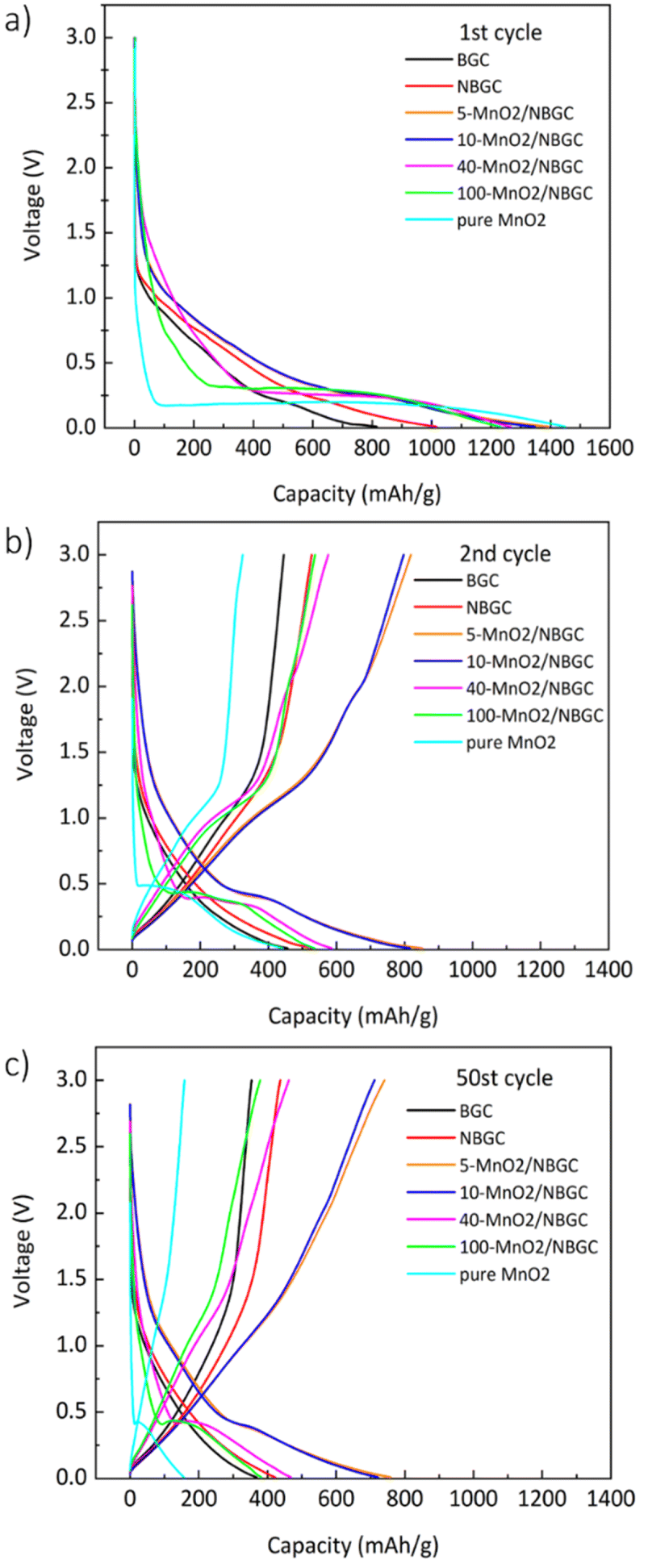
The voltage profiles of BGC, NBGC and the MnO2/NBGC composites at a current density of 186 mA g−1 (approximately 0.5C based on the theoretical capacity of graphite, ∼370 mA h g−1 as the reference) in the 1st, 2nd and 50th cycles are presented in Fig. 7. For BGC and NBGC, only a sloped discharge potential, as generally found in carbon-based electrodes, could be observed. In the case of the MnO2/NBGC composites, an obvious plateau was observed at around 0.45 V in the discharge process, corresponding to the reduction of Mn4+ to Mn0. In the charging process, the sloped potentials at around 1.51 V and 2.25 V were ascribed to the two steps of Mn oxidation, in agreement with the CV scan result. It was found that the composites with greater MnO2 loadings (such as 40-MnO2/NBGC and 100-MnO2/NBGC) exhibited similar characteristics to pure MnO2, where the long plateau potential at 0.45 V was obviously observed. The 1st cycle discharge capacities of most MnO2/NBGC composites were higher than the theoretical capacity of MnO2 (1230 mA h g−1), reflecting the mixed processes of ionic storage. The extreme decay of the specific capacity in the 2nd cycle resulted from the formation of the SEI due to the decomposition of the electrolyte at the electrode interface during the 1st discharge process. The formation of the coating layer limits ionic accessibility to the electrode in the subsequent cycles.36 This irreversible capacity from the first cycle is highly dependent on the mobility of Li+ and e− during the Li extraction process. Li+ transport can be enhanced by the formation of a nanoporous SEI structure at the end of the first discharge cycle that can facilitate the penetration of the electrolyte into the nanopores of the electrode. However, electron transport in the electrode may not be affected since the electrolyte cannot supply free electrons to the electrode. Therefore, electron mobility is highly dependent on the original particle size. In the following cycles, charging and discharging were quite reversible due to the stability of the SEI microstructure after several cycles. After 50 cycles, only the MnO2/NBGC composites with medium to low MnO2 loadings, namely 5-MnO2/NBGC and 10-MnO2/NBGC, exhibited high capacity retention of more than 90% relative to the capacity obtained in the 2nd cycle.
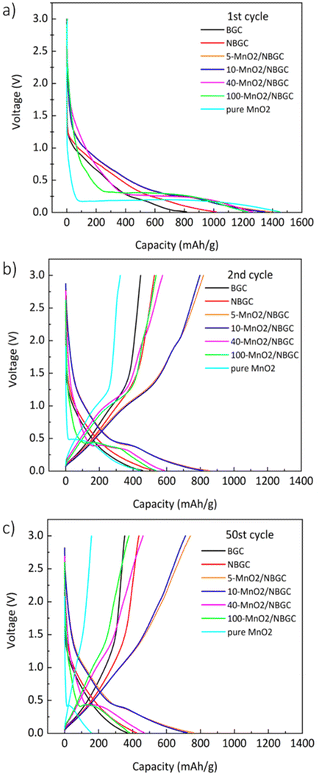 |
| | Fig. 7 The voltage profiles of bagasse-derived carbon material (BGC), N-doping bagasse-derived carbon (NBGC), and MnO2/NBGC composites with various MnO2 loadings at the (a) 1st cycle, (b) 2nd cycle, and (c) 50th cycle. | |
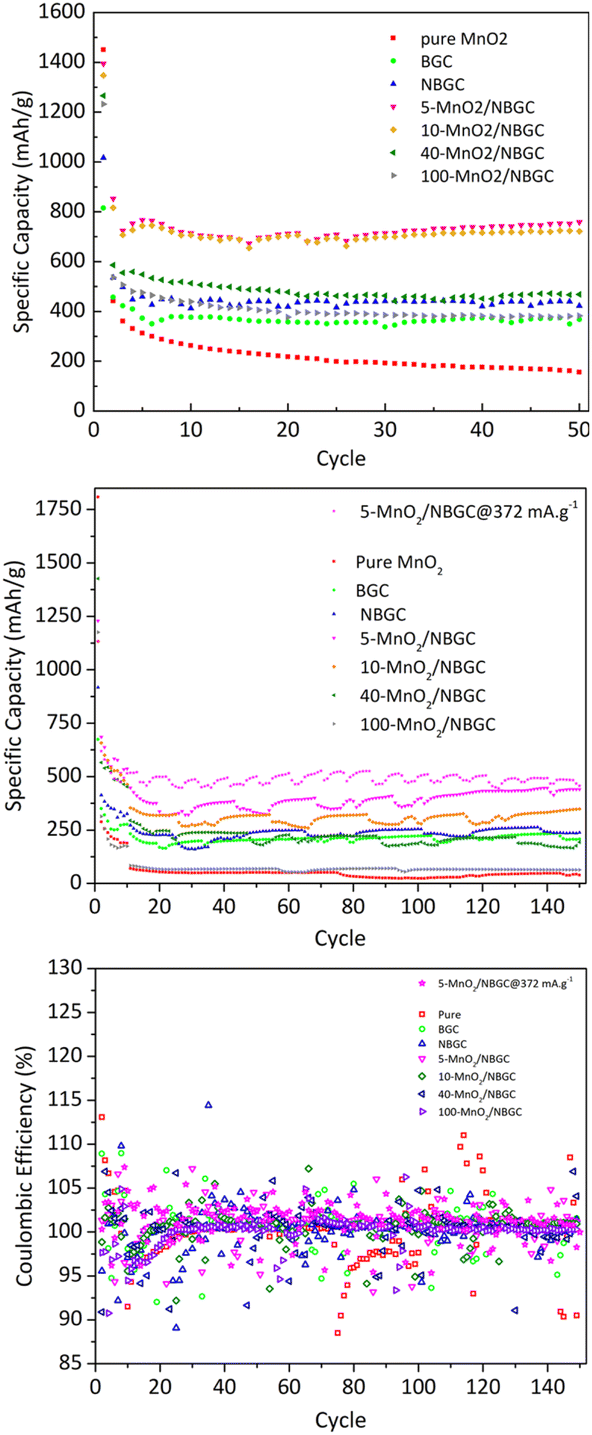
As shown in Fig. 8, the cycling performances of the bagasse-derived carbon anode material and its composites with various MnO2 loadings were compared with that of pure MnO2 nanoparticles to explore the benefit of the doped N-functional groups and the MnO2 nanoparticles dispersed on the carbon porous substrate, as well as the impact of the MnO2 coating layer on the corresponding electrochemical performance. As shown in Fig. 8a, the half-cell tests were cycled between 0 and 3 V at a current density of 186 mA g−1 for 50 cycles. The initial capacity of the parent carbon materials, namely BGC and NBGC, the doped MnO2/NBGC composites, including 5-MnO2/NBGC, 10-MnO2/NBGC, 40-MnO2/NBGC, and 100-MnO2/NBGC, and the pure synthesized MnO2 were 815, 1016, 1394, 1348, 1265, 1232 and 1450 mA h g−1, respectively. These obtained capacities were higher than the theoretical capacities of graphite and the MnO2 crystalline material, reflecting the combination of the ionic storage mechanisms. In the 2nd cycle, all samples had substantial capacity decay reaching 457, 535, 853, 816, 586, 536, and 442 mA h g−1, respectively. This capacity decay typically results from the influence of SEI formation. However, all samples showed great coulombic efficiencies of almost 100% during the 50-cycle tests. Regarding the reduction in carbon content with increasing MnO2 doping amount (see Table S1†), the obtained capacities of the MnO2/NBGC composites were quite dependent on the MnO2 compound. After 50 cycles, all MnO2/NBGC composites with different MnO2 loading concentrations and even the un-doped bagasse-based carbon materials exhibited much better cycling performance than that of the pure MnO2 electrode. After 50 cycles, 5-MnO2/NBGC, 10-MnO2/NBGC, 40-MnO2/NBGC, and 100-MnO2/NBGC could retain specific capacities of 759, 722, 468, and 382 mA h g−1, which correspond to about 54%, 54%, 37%, and 31% capacity retention, respectively. Meanwhile, the parent porous carbon substrates NBGC and BGC exhibited specific capacities of 422 and 368 mA h g−1, demonstrating capacity retention of 45% and 42%, respectively. Although the pure MnO2 electrode expressed a very high initial capacity (∼1450 mA h g−1), its 2nd cycle capacity considerably dropped and continually decreased through the end of the 50th cycle, finally holding a specific capacity of only 156 mA h g−1, corresponding to 11% retention. This clearly reveals the dramatic instability of the bulk pure MnO2 compound. Compared with their parent substrates (i.e. NBGC and BGC) and pure MnO2, the superior capacity of the MnO2/NBGC composites reflects the influence of the high-capacity MnO2 active compound in addition to the capacitive effect of the N-functional groups and the large surface area of the parent carbon substrate. However, the composite samples that contained a high density of deposited MnO2, such as 40-MnO2/NBGC and 100-MnO2/NBGC, behaved more like the bulk MnO2, hence displaying significantly lower capacity than the samples with less MnO2 doping, namely 5-MnO2/NBGC and 10-MnO2/NBGC. Relative to the capacity observed in the 2nd cycle, where the formed SEI layer was quite stable, the samples BGC, NBGC, 5-MnO2/NBGC, 10-MnO2/NBGC, 40-MnO2/NBGC, 100-MnO2/NBGC, and pure MnO2 demonstrated 81%, 79%, 89%, 88%, 80%, 71%, and 35% capacity retention, respectively. All the bagasse-based electrodes exhibited high capacity retention above ∼80%, except for pure MnO2, which still showed low retention of about 35%. The poor cycling stability of the pure MnO2 electrode is attributed to its typically large volume expansion, which causes weaker adhesion with the current collector after several cycling tests.16 Considering the impact of the MnO2 coating amount on the electrochemical performance of the composite electrodes, increasing the MnO2 loading amount either led to a reduction in the specific capacity of the composites or resulted in MnO2 structures with poor crystallinity due to the dense crystalline growth. With thin MnO2 coatings, the 5-MnO2/NBGC and 10-MnO2/NBGC samples exhibited high performance because of the synergistic effect of the high-capacity MnO2 active material and the supported capacitive effect arising from the exposed N-containing defects. In addition, the thin coating allowed better adhesion of MnO2 with the carbon surface and could better tolerate the high stress from its large expansion. In contrast, thick MnO2 coatings, as found in the 40-MnO2/NBGC and 100-MnO2/NBGC samples, would not only block the pore channels of the parent carbon material, leading to a reduction in the specific surface area but also would shield the capacitive effect of the N-containing functional groups on the carbon substrate from the contacting electrolyte. In addition, the low crystalline quality resulting from rapid crystalline growth at high concentrations of MnO2 would limit the diffusion-controlled intercalation/extraction process of Li+ through the MnO2 compound. Large MnO2 agglomerates would also result in weak adhesion with the substrate and may generate non-uniform expansion during cycling, causing instability or detachment of some MnO2 coatings after several cycles. To evaluate the long-term cyclic performance of the MnO2/NBGC electrode materials, their as-assembled LIBs were cycled at 774 mA g−1 (∼2C based on the theoretical capacity of graphite with ∼370 mA h g−1 as the reference) for 150 cycles, as depicted in Fig. 8b. The cells assembled from 5-MnO2/NBGC, 10-MnO2/NBGC, 40-MnO2/NBGC, 100-MnO2/NBGC, NBGC, BGC, and pure synthesized MnO2 exhibited average reversible discharge capacities of 390.03, 306.94, 211.63, 66.29, 233.32, 208.11 and 43.05 mA h g−1, respectively. All samples showed great reversibility with the coulombic efficiency reaching nearly 100%, as shown in Fig. 8c. The observed long-term capacity behaviour at high rates also confirms the synergistic influence of the high-capacity MnO2 compound and the pseudocapacitive effect arising from the N-containing defects and the large surface area, as previously mentioned. For instance, the composite comprising a thin MnO2 coating exhibited high storage capability compared to the samples that were coated with thick MnO2 layers. It was found that even at a relatively high current density, 5-MnO2/NBGC exhibited a specific capacity of 390 mA h g−1 (487.6 mA h g−1 at 372 mA g−1), which is higher than the capacity of typical graphite (∼370 mA h g−1). Thicker MnO2 coating layers at elevated MnO2 doping levels resulted in declined ionic capacity. Interestingly, the 100-MnO2/NBGC sample with a very thick MnO2 coating exhibited a substantial drop in capacity, even lower than that of BGC, which is the carbon material without N-doping. Moreover, it behaved more like the bulk pure MnO2 instead of the MnO2/NBGC carbon composite, delivering a comparatively low capacity as the unstable MnO2. The high pseudocapacitive effect in NBGC, which does not hold a MnO2, could promote high rate capability, leading to a higher specific capacity than that of the 40-MnO2/NBGC sample.
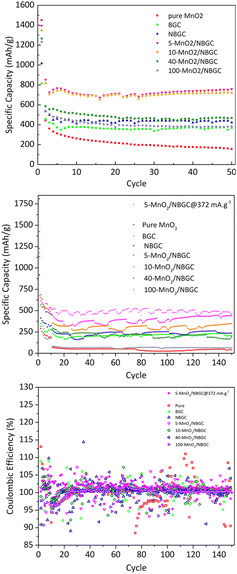 |
| | Fig. 8 Cycling performance of BGC, NBGC, MnO2/NBGC at various MnO2 loadings and pure synthesized MnO2: (a) cycling at a current density of 186 mA g−1, (b) cycling at a current density of 774 mA g−1 and (c) their corresponding coulombic efficiency at a current density of 774 mA g−1. | |
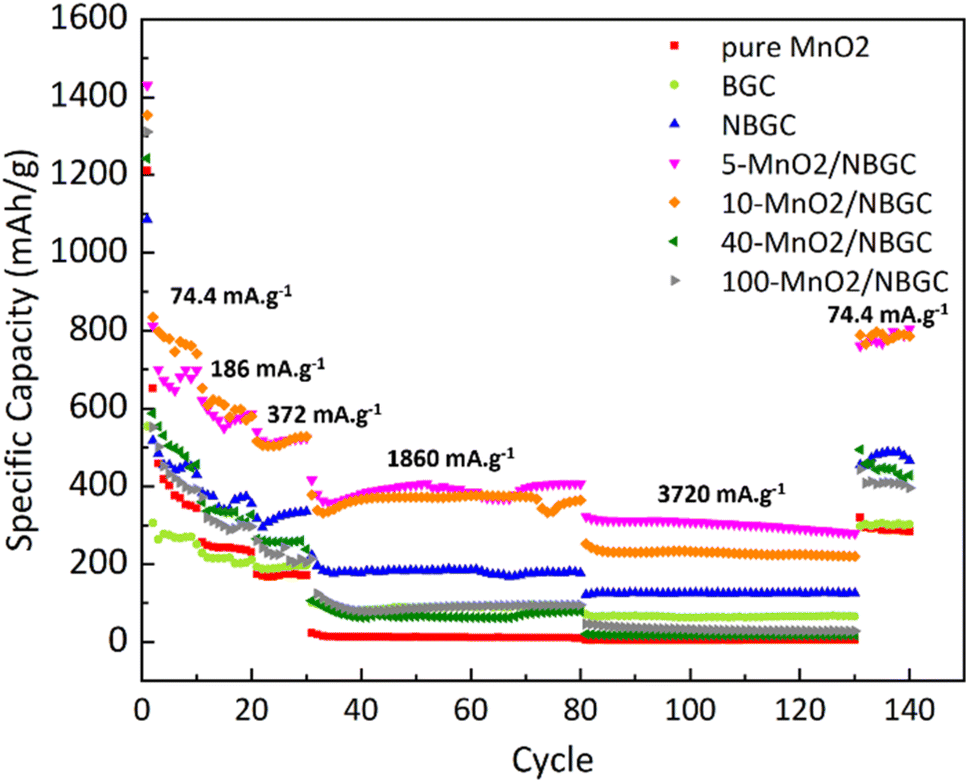
As shown in Fig. 9, different current densities were applied to investigate the rate performance of the MnO2/NBGC samples with various MnO2 loadings and compared with their parent bagasse-derived porous carbons (NBGC and BGC), as well as, the synthesized pure MnO2. Different current densities, including 74.4 mA g−1, 186 mA g−1, 372 mA g−1, 1860 mA g−1, 3720 mA g−1, which are approximately equivalent to about 0.2C, 0.5C, 1C, 5C, and 10C, respectively, (based on the 1C graphite theoretical capacity of about 372 mA g−1) were consecutively applied for 10 cycles, 10 cycles, 10 cycles, 50 cycles, and 50 cycles, respectively, and eventually restored back to 74.4 mA g−1 for 10 cycles. As summarized in Table 2, all samples exhibited good rate performance, achieving great capacity retention of more than 70% even under high applied current densities of 1860 mA g−1 and 3720 mA g−1 over almost 100 cycles. Compared with the MnO2 composite samples, pure MnO2 exhibited poor reversibility with quite a low capacity retention of ∼59%. Similar to the trend achieved in the cycling performance assessment, regarding the synergistic impact of the inserted high-capacity MnO2 compound and N-doping, the MnO2/NBGC composite samples with low to medium MnO2 loadings, namely the 5-MnO2/NBGC and 10-MnO2/NBGC composites, displayed distinctively great rate performances. Interestingly, 5-MnO2/NBGC could deliver a high specific capacity of up to 388 and 301 mA h g−1 at high current densities of 1860 and 3720 mA g−1, or about 5C and 10C, respectively. At low to relatively high current densities (74.4–1860 mA g−1), though exhibited comparable performance 10-MnO2/NBGC was found to exert a slightly higher capacity than 5-MnO2/NBGC. This may imply its favorable thermodynamics-controlled ionic storage at low current density due to higher MnO2 doping. However, when tested at a very high current density of 3720 mA g−1, 5-MnO2/NBGC showed distinctively higher capacity than 10-MnO2/NBGC. Their comparable performance at low to medium current densities may be attributed to their similar specific surface area and comparable amounts of N-containing groups, which support the accessibility of the electrolyte to the electrode material and promote the pseudo capacitive effect. However, higher amounts of loaded MnO2 may form thicker coating layers that shield the exposed N-functional groups on the carbon substrate and drive poor MnO2 crystalline growth. In addition, depositing MnO2 at higher KMnO4 concentrations may increase the dispersion of the MnO2 clusters, leading to lower dimensional stability because of their non-uniform and large volume expansion under high current densities, such as 3720 mA g−1. Both 5-MnO2/NBGC and 10-MnO2/NBGC showed great reversibility with high capacity retention of more than 90% and even full recovery in the case of 5-MnO2/NBGC, when restored back to the low rate of 74.4 mA g−1. Notably, such high-capacity compound MnO2 seems to play a greater role than the surface capacitive effect when operated under low current densities, such as 74.4 mA g−1. However, the higher specific capacity of NBGC than those of the high MnO2-loaded composites, such as 40-MnO2/NBGC and 100-MnO2/NBGC, at high current densities suggests a major impact of the capacitive effect arising from the doped N-containing groups, as well as, the large surface area on the ionic conduction and stability of the MnO2 coating layer to sustain the capacity at high rates. In agreement with the results from the cycling test, thick MnO2 coating layers may not be able to tolerate large dimensional changes under fast rates and cause weak adhesion, leading to a dramatic capacity drop possibly due to the detachment of MnO2 from the carbon substrate.
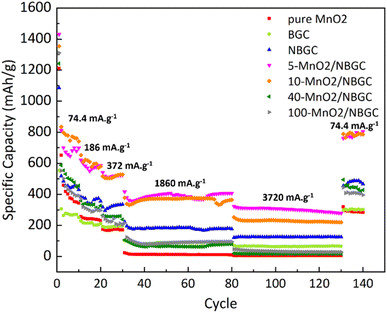 |
| | Fig. 9 Rate performance of bagasse-derived carbon material (BGC), N-doping bagasse-derived carbon (NBGC), MnO2/NBGC carbon composite with various MnO2 loading concentrations and the pure hydrothermally synthesized MnO2. | |
Table 2 The specific capacity of bagasse-derived carbon material and its composites with different MnO2 loadings under varying current density
| Carbon composite |
Average specific capacity at varied current densities (mA h g−1) |
Capacity retention (%) |
| 74.4 (mA g−1) |
186 (mA g−1) |
372 (mA g−1) |
1860 (mA g−1) |
3720 (mA g−1) |
74.4 (mA g−1) |
| Pure MnO2 |
493 |
242 |
171 |
12 |
4 |
292 |
59 |
| BGC |
300 |
212 |
192 |
89 |
65 |
301 |
100 |
| NBGC |
521 |
363 |
321 |
183 |
125 |
475 |
91 |
| 5-MnO2/NBGC |
767 |
580 |
519 |
388 |
301 |
780 |
101 |
| 10-MnO2/NBGC |
833 |
603 |
514 |
365 |
228 |
784 |
94 |
| 40-MnO2/NBGC |
578 |
332 |
256 |
77 |
17 |
447 |
77 |
| 100-MnO2/NBGC |
526 |
308 |
225 |
93 |
32 |
410 |
77 |
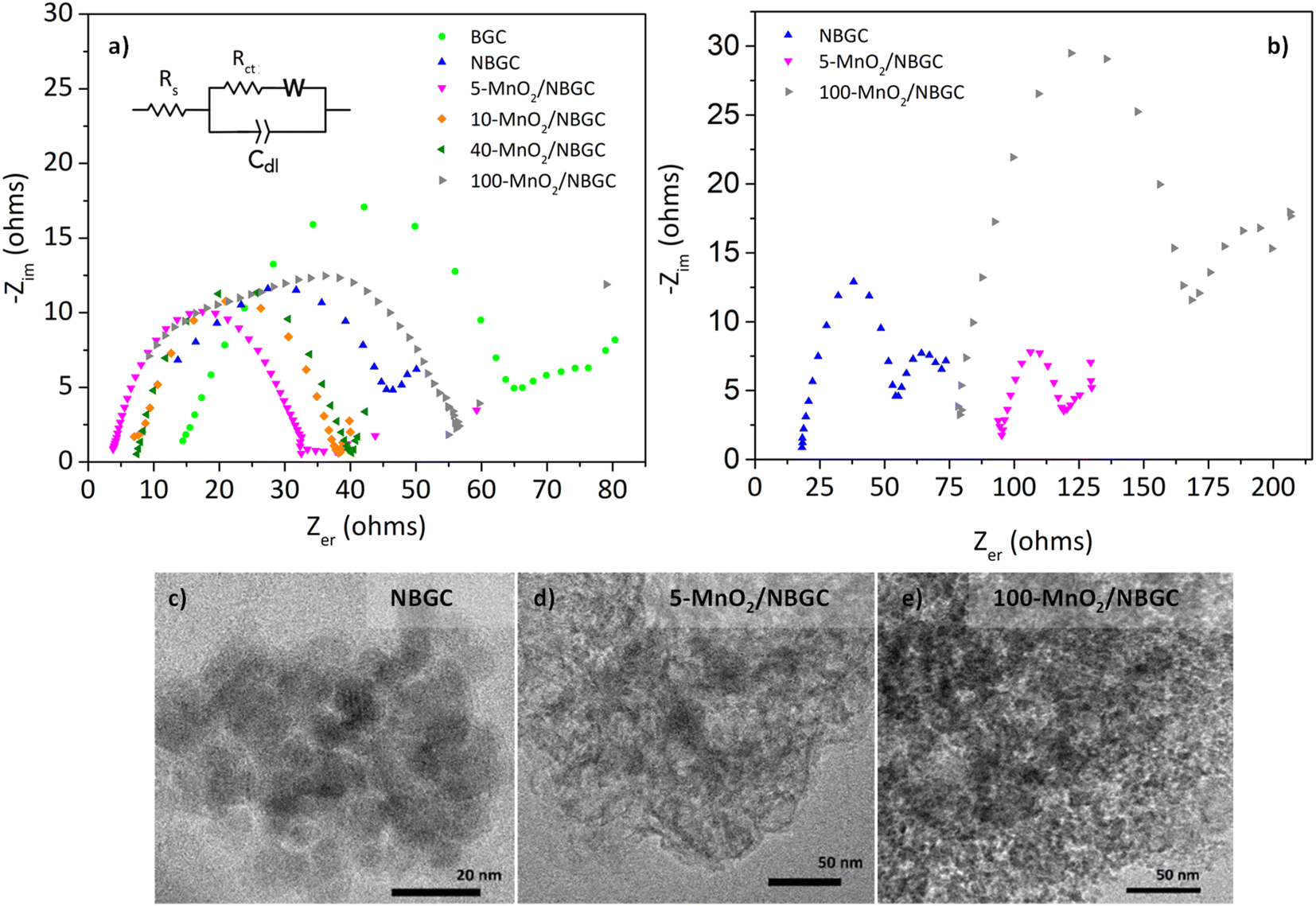
The electrochemical impedances of BGC, NBGC, 5-MnO2/NBGC, 10-MnO2/NBGC, 40-MnO2/NBGC and 100-MnO2/NBGC were analyzed by using electrochemical impedance spectroscopy (EIS) in the frequency range of 0.1–100000 Hz and a voltage of 10 mV, as illustrated in Fig. 10a. Based on the electrical equivalent circuit (EEC) shown as the inset, all the Nyquist plots composed of a single semicircle in the high-to-medium frequency range and a diagonal inclined line in the middle-to-low frequency range, corresponding to the charge transfer resistance (Rct) and lithium ionic diffusion ability, respectively. The electrochemical charge transfer resistance (Rct) of each sample was obtained from the extrapolated intercept of the semicircle plot in the medium-frequency range on the X-axis. The electrochemical impedance generally reflects the ion transport ability within the bulk electrolyte through the electrode material. In particular, the charge transfer resistance denotes the transport of ions through the interface between the electrode material and electrolyte solution. BGC, NBGC, 5-MnO2/NBGC, 10-MnO2/NBGC, 40-MnO2/NBGC, and 100-MnO2/NBGC revealed Rct values of 65.2, 46.1, 33.2, 38.5, 41.0 and 57.5 ohms, respectively. These can be ranked as Rct (5-MnO2/NBGC) < Rct (10-MnO2/NBGC) < Rct (40-MnO2/NBGC) < Rct (NBGC) < Rct (100-MnO2/NBGC) < Rct (BGC). This ionic conduction trend is quite consistent with that of their specific capacities. Moreover, it is most likely influenced by the crystallinity of the doped MnO2 nanoclusters and the specific surface area. For instance, a better crystalline structure when combined with a large specific surface area tends to promote higher ionic conduction. Furthermore, the reduction in ionic conductivity of the MnO2/NBGC electrode after cycling at 774 mA g−1 for 150 cycles was explored by TEM and the EIS of representative carbon composite electrode materials, namely NBGC, 5-MnO2/NBGC, and 100-MnO2/NBGC, as illustrated in Fig. 10c–e. The Nyquist plots of the used electrode material seemed to depict an additional second semicircle at medium frequencies, reflecting the formation of a secondary phase or additional resistance generated after the cycling tests. They also showed higher charge transfer resistances compared with the fresh electrodes and could be arranged as Rct (NBGC) < Rct (5-MnO2/NBGC) < Rct (100-MnO2/NBGC). A comparison of the TEM images of the spent electrodes relative to the fresh ones depicted in Fig. 2 revealed that the NBGC and 5-MnO2/NBGC electrode materials did not clearly show morphology changes, whereas, the obvious flaky structure of the MnO2 coating layer for the 100-MnO2/NBGC sample disappeared after long cycling. This may imply the degradation of the unstable thick MnO2 coating layer due to non-uniform expansion during cycling, eventually leading to its detachment from the porous carbon substrate and causing large resistance due to the collapse of the crystalline structure.
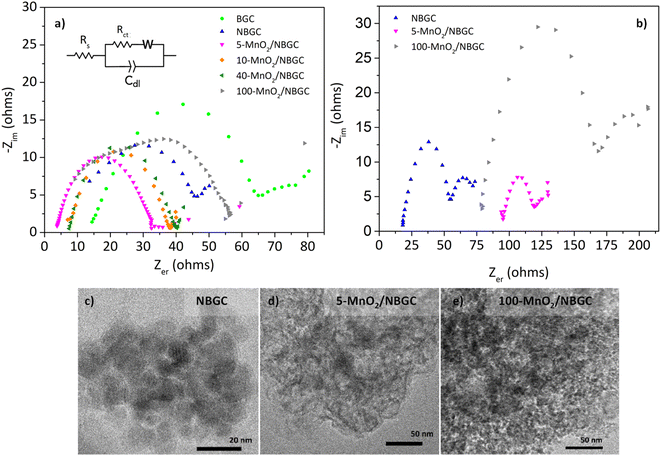 |
| | Fig. 10 Electrochemical impedance of (a) the fresh carbon composite anode electrodes and (b) carbon composite anode electrodes after cycling at 774 mA g−1 for 150 cycles; the TEM images of some spent carbon composite electrodes: (c) NBGC, (d) 5-MnO2/NBGC, and (e) 100-MnO2/NBGC. | |
Table 3 demonstrates the comparison of previously reported electrochemical performances of manganese oxide compound/carbon composites in various fabricated structures. Most of the developed manganese oxide/carbon composite electrodes exhibited a capacity of more than 400 mA h g−1, which is higher than the theoretical capacity of the typical graphite electrode. This may be attributed to the utilization of carbon nanotubes or graphene sheets as the highly conductive carbon substrate with enhanced specific surface for the nanocrystalline growth of high-capacity manganese oxide compounds. However, though porous graphene or carbon nanotubes deliver high ionic capacity at high current densities, the materials are expensive and require elaborate fabricating methods. In comparison with the manganese oxide/carbon composite electrodes from previous studies, our best sample 5-MnO2/NBGC delivers quite a high reversible capacity (∼722 mA h g−1 at a moderate current density of 186 mA g−1, ∼0.5C based on theoretical graphite capacity). Moreover, it shows relatively high capacity and good stability at high current densities: 488 at 372 mA g−1, and 390 at 744 mA g−1. This great storage capability may arise from the synergistic effect of the stable MnO2 nanocrystalline structures deposited onto the porous carbon substrate with enhanced surface area containing high-capacitive N-functional groups. In addition, the appropriate density of the MnO2 nanocrystalline particles on the porous substrate promotes the formation of good crystalline and optimizes the obtained specific surface area of the composite products. The proposed utilization of agricultural wasted sugarcane bagasse, might promote the concept of using sustainable raw materials to produce a higher-value electrode material for electrochemical storage devices that can sustain comparable performance to expensive electrode materials.
Table 3 Comparison of the electrochemical performance of various composites of manganese oxide compounds and carbon materials
| Sample |
Material |
Specific capacity (mA h g−1) |
Potential range (V) |
References |
| MnO2/CNT |
Composite of coated MnO2 nanoflakes on connected carbon nanotubes |
620 at 200 mA g−1 after 50 cycles |
0.01–3 |
16 |
| α-MnO2/GNS |
Composite of α-MnO2 nanosheets distributed on graphene surface |
693.7 at 0.05C and 387.7 at 0.2C after 30 cycles |
0.01–3 |
34 |
| MnO2/CNTs |
Composite of hybrid coaxial structured MnO2 and carbon nanotubes |
500 at 50 mA g−1 after 15 cycles |
0.02–3.2 |
36 |
| C/MnO-L |
Composite of commercial MnO coated with sugar-derived carbon material |
400 at 400 mA g−1 |
0.01–3 |
42 |
| N-MnO/GNS |
Composite of N-doped MnO distributed on graphene nanosheets |
772 at 100 mA g−1 after 90 cycles |
0–3 |
43 |
| MnO@C core–shell nanoplates |
Composite of core–shell MnO nanoplates coated with carbon material |
770 at 200 mA g−1 after 30 cycles |
0.001–3 |
44 |
| MnO2/CNHs |
Composite of MnO2 nanoflakes coated on carbon nanohorns (CNHs) |
565 at 100 mA g−1 after 60 cycles |
0.05–3 |
45 |
| MnO2 NFs@GF |
Interconnected MnO2 nanoflakes (NFs) assembled on CVD-grown graphene (GF) foam |
1200 mA h g−1 at 500 mA g−1 after 300 cycles and 500 mA h g−1 at 5 A g−1 |
0.01–3.00 |
46 |
| GMG |
Composite of graphene-coated MnO2 deposited on graphene nanoribbons (GNRs) |
612 mA h g−1 at 400 mA g−1 after 250 cycles |
0.01–3.00 |
47 |
| MnO2 nanotube/graphene |
Thin film composites of layered-by-layered graphene and MnO2 nanotubes |
495 mA h g−1 at 100 mA g−1 after 40 cycles |
0.01–3.00 |
48 |
| 5-MnO2/NBGC |
Composite of dispersed MnO2 nanoparticles on the N-doped bagasse-derived porous carbon material |
722 at 186 mA g−1 after 50 cycles, 488 at 372 mA g−1 after 150 cycles, and 390 at 744 mA g−1 after 150 cycles |
0.01–3 |
This work |
To further align the academic and industrial practices for the potential utilization of our proposed 5-MnO2/NBGC anode material, the energy density of the full-cell fabricated using 5-MnO2/NBGC coupled with an LFP cathode was calculated based on the method presented by Son et al.49 The calculated full-cell energy density was then compared with the case where the common graphite anode material was employed. The calculated results for both cases are illustrated in Fig. S4.† Basically, the energy density of the full cell mainly relies on the practical capacity of the cathode material and the nominal voltage between the cathode and the anode. The nominal voltage of the full cell may be roughly approximated from the potential difference between the plateau voltages derived from separate half-cell GCD tests of each cathode and anode material vs. Li/Li+. With the same cathode material, the anode material which provides lower voltage may gather a higher output of nominal voltage when coupled in the full cell. Based on the energy density calculation shown in ESI,† with the same electrode mass loading, the calculated full-cell energy densities for the LFP-5MnO2/NBGC and LFP–graphite electrode couples were 137.42 and 151.16 A h kg−1, respectively. The lower full-cell energy density of the LFP-5MnO2/NBGC couple compared to that of the LFP–graphite pair results from its higher half-cell testing nominal voltage (0.45 V) relative to 0.15 V for typical graphite. Though it is predicted to deliver slightly lower full cell energy density compared with the common graphite anode, 5-MnO2/NBGC showed quite an impressive rate performance (i.e. 388 mA h g−1 at a current density of 1860 mA g−1, approximately 5C) and good cycling stability. This promising rate performance and cycling stability of 5-MnO2/NBGC may promote its potential usage in batteries for fast-rate applications and ensure a longer lifetime.
Conclusions
Composites of the N-doped bagasse-derived porous carbon with dispersed MnO2 nanoparticles (MnO2/NBGC) were fabricated via a practical method. Controlled loading of MnO2 nanoparticles on the porous carbon substrate could be achieved by varying the KMnO4 precursor concentration in the reduction reaction. Increasing the MnO2 loading content caused the formation of thick flake-like MnO2 coating layers on the porous carbon surface, leading to a reduction in the specific surface area and shielding of the capacitive effect originating from the N-containing functional groups. In addition, high concentrations of the KMnO4 precursor caused the formation of MnO2 with low crystallinity. The superior electrochemical performance of the MnO2/NBGC nanocomposite electrode was achieved from the synergistic impact of the hierarchical architecture according to appropriate MnO2 loading on the porous carbon and the capacitive effect from the N-containing defects on the carbon material. These combined effects could facilitate great adhesion of the MnO2 nanoparticles on the substrate, limiting its large volume expansion and enabling fast ion and electron transport. The optimized carbon composite, namely 5-MnO2/NBGC, exhibited a high reversible capacity of about 760 mA h g−1 at a current density of 186 mA g−1 after 50 cycles, 488 mA g−1 at 372 mA g−1 and up to 390 mA g−1 at 774 mA g−1 after 150 cycles. It also showed good reversibility, with the coulombic efficiency reaching nearly 100%. Moreover, it delivered good rate capability with high specific capacity up to 388 and 301 mA h g−1 even when tested under high current densities of 1860 and 3720 mA g−1, respectively, for at least 50 cycles. Compared to similar MnO2/carbon composite anode materials reported in previous studies, our proposed MnO2/NBGC anode material exhibits quite impressive performance and utilizes sustainable agricultural wasted, sugarcane bagasse as the base material. The approximated energy density calculation of the LFP-coupled 5-MnO2/NBGC full cell expectedly revealed a lower energy density compared to that of the LFP–graphite anode couple, regarding its higher nominal half-cell testing voltage. However, the good rate and cycling performance of the proposed 5-MnO2/NBGC anode may benefit potential usage in batteries for fast-rate applications with a longer lifetime. Here, the proposed MnO2/NBGC composites show potential for further development not only as promising electrode materials for rechargeable batteries (Li+, Na+, and K+ batteries) and supercapacitors but also as active materials for other applications, such as catalysis and sensing.
Author contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Krittaporn Pongpanyanate, Supacharee Roddecha, and Chanita Piyanirund. The first draft of the manuscript was written by Krittaporn Pongpanyanate, and Supacharee Roddecha. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to thank the Kasetsart University Research and Development Institute (KURDI) and Faculty of Engineering, Kasetsart University for the financial support during this project. We are also grateful for the equipment and academic facility by the department of Chemical Engineering, Kasetsart University. In addition, we would also like to thank Dirayanti for gathering final revised electrochemical performance assessment.
Notes and references
- K. Kim, G. Daniel, V. G. Kessler, G. A. Seisenbaeva and V. G. Pol, Nanomaterials, 2018, 8, 608 CrossRef PubMed
 .
. - Y. Li, Y. Lu, P. Adelhelm, M. M. Titirici and Y. S. Hu, Chem. Soc. Rev., 2019, 48(17), 4655 RSC .
- P. C. Rath, J. Patra, H. T. Huang, D. Bresser, T. Y. Wu and J. K. Chang, ChemSusChem, 2019, 12(10), 2302 CrossRef CAS PubMed .
- K. Chayambuka, G. Mulder, D. L. Danilov and P. H. L. Notten, Adv. Energy Mater., 2018, 8(16), 1800079 CrossRef .
- G. Wang, M. Yu and X. Feng, Chem. Soc. Rev., 2021, 50(4), 2388 RSC .
- S. Lin, L. Mo, F. Wang and Z. Shao, J. Alloys Compd., 2021, 873, 159705 CrossRef CAS .
- L. Xie, C. Tang, Z. Bi, M. Song, Y. Fan, C. Yan, X. Li, F. Su, Q. Zhang and C. Chen, Adv. Energy Mater., 2021, 11(38), 2101650 CrossRef CAS .
- G. Zhong, H. Xie, Z. Xu, S. Xu, S. Xu, Z. Cai, X. Fu, W. Liao and R. Miao, ChemistrySelect, 2019, 4(12), 3432 CrossRef CAS .
- K. Le Van and T. T. L. Thi, Prog. Nat. Sci.: Mater. Int., 2014, 24(3), 191 CrossRef CAS .
- S. Agarkar, P. Yadav, R. Fernandes, D. Kothari, A. Suryawanshi and S. Ogale, Electrochim. Acta, 2016, 212, 535 CrossRef CAS .
- R. Luan, D. Xu, H. Pan, C. Zhu, D. Wang, X. Meng, Y. Li, M. Imtiaz, S. Zhu and J. Ma, J. Energy Storage, 2019, 22, 60 CrossRef .
- M. Chen, D. Yan, X. Zhang, Z. Yu, G. Zhu, Y. Zhao, S. Lu, G. Chen, H. Xu and A. Yu, Mater. Lett., 2017, 196, 276 CrossRef CAS .
- H. Wan and X. Hu, Solid State Ionics, 2019, 341, 115030 CrossRef CAS .
- D. B. Babu and K. Ramesha, Carbon, 2019, 144, 582 CrossRef CAS .
- P. Wu, M. Gao, S. Yu, M. Feng, S. Liu and J. Fu, Electrochim. Acta, 2020, 354, 136681 CrossRef CAS .
- H. Xia, M. Lai and L. Lu, J. Mater. Chem., 2010, 20(33), 6896 RSC .
- D. Yan, P. Yan, S. Cheng, J. Chen, R. Zhuo, J. Feng and G. Zhang, Cryst. Growth Des., 2009, 9(1), 218 CrossRef CAS .
- X. Jin, W. Zhou, S. Zhang and G. Z. Chen, Small, 2007, 3(9), 1513 CrossRef CAS PubMed .
- H. T. Zhu, J. Luo, H. X. Yang, J. K. Liang, G. H. Rao, J. B. Li and Z. M. Du, J. Phys. Chem. C, 2008, 112, 17089 CrossRef CAS .
- J. Ou, Y. Zhang, L. Chen, Q. Zhao, Y. Meng, Y. Guo and D. Xiao, J. Mater. Chem. A, 2015, 3(12), 6534 RSC .
- J. Jang, T. Kim, Y. Choi, I. Park, D. Lim, S. E. Shim and S. H. Baeck, J. Nanosci. Nanotechnol., 2016, 16(5), 5195 CrossRef CAS .
- X. Wang, X. Wang, W. Huang, P. J. Sebastian and S. Gamboa, J. Power Sources, 2005, 140(1), 211 CrossRef CAS .
- T.-H. Ting, J. Chin. Chem. Soc., 2009, 56, 1225 CrossRef CAS .
- F. Gao, S. H. Qin, Y. H. Zang, J. F. Gu and J. Y. Qu, N. Carbon Mater., 2020, 35(2), 121 CrossRef CAS .
- Z. Fan, J. Yan, T. Wei, L. Zhi, G. Ning, T. Li and F. Wei, Adv. Funct. Mater., 2011, 21(12), 2366 CrossRef CAS .
- M. Jayashree, M. Parthibavarman, R. BoopathiRaja, S. Prabhu and R. Ramesh, J. Mater. Sci.: Mater. Electron., 2020, 31(9), 6910 CrossRef CAS .
- R. Muruganantham, F. M. Wang, R. A. Yuwono, M. Sabugaa and W. R. Liu, Energy Fuels, 2021, 35(13), 10878 CrossRef CAS .
- A. P. Nowak, A. L. Oleksiak, B. Wicikowska and M. Gazda, J. Solid State Electrochem., 2017, 21(8), 2251 CrossRef CAS .
- L. Qie, W. M. Chen, Z. H. Wang, Q. G. Shao, X. Li, L. X. Yuan, X. L. Hu, W. X. Zhang and Y. H. Huang, Adv. Mater., 2012, 24(15), 2047 CrossRef PubMed .
- J. Huang, Y. Lin, M. Ji, G. Cong, H. Liu, J. Yu, B. Yang, C. Li, C. Zhu and J. Xu, Appl. Surf. Sci., 2020, 504, 144398 CrossRef CAS .
- Z. Li, Z. Xu, X. Tan, H. Wang, C. M. B. Holt, T. Stephenson, B. C. Olsenab and D. Mitlin, Energy Environ. Sci., 2013, 6, 871 RSC .
- W. Shi, Y. Zhang, Z. Q. Tian, Z. Pan, J. Key and P. K. Shen, J. Power Sources, 2018, 398, 149 CrossRef CAS .
- J. G. Lee, Y. Kwon, J. Y. Ju, S. Choi, Y. Kang, W. R. Yu and D. W. Kim, J. Appl. Electrochem., 2017, 47(8), 865 CrossRef CAS .
- L. Xing, C. Cui, C. Ma and X. Xue, Mater. Lett., 2011, 65(14), 2104 CrossRef CAS .
- M. S. Wu, P. C. J. Chiang, J. T. Lee and J. C. Lin, J. Phys. Chem. B, 2005, 109(49), 23279 CrossRef CAS PubMed .
- A. L. M. Reddy, M. M. Shaijumon, S. R. Gowda and P. M. Ajayan, Nano Lett., 2009, 9(3), 1002 CrossRef CAS PubMed .
- L. Feng, Z. Xuan, H. Zhao, Y. Ba, J. Guo, C. W. Su and X. Chen, Nanoscale Res. Lett., 2014, 9(1), 290 CrossRef PubMed .
- H. Lindström, S. Södergren, A. Solbrand, H. Rensmo, J. Hjelm, A. Hagfeldt and S. E. Lindquist, J. Phys. Chem. B, 1997, 101, 7717 CrossRef .
- Q. Zhu, M. Wang, B. Nan, H. Shi, X. Zhang, Y. Deng, L. Wang, Q. Chen and Z. Lu, Core/shell nanostructured Na3V2(PO4)3/C/TiO2 composite nanofibers as a stable anode for sodium-ion batteries, J. Power Sources, 2017, 362, 147 CrossRef CAS .
- Y. H. Du, X. Y. Liu, X. Y. Wang, J. C. Sun, Q. Q. Lu, J. Z. Wang, A. Omar and D. Mikhailova, Freestanding strontium vanadate/carbon nanotube films for long-life aqueous zinc-ion batteries, Rare Met., 2022, 41(2), 415 CrossRef CAS .
- C. Liu, Q. Lu, M. V. Gorbunov, A. Omar, I. G. G. Martinez, P. Zhao, M. Hantusch, A. D. C. Permana, H. He, N. Gaponik and D. Mikhailova, Ultrasmall CoS nanoparticles embedded in heteroatom-doped carbon for sodium-ion batteries and mechanism explorations via synchrotron X-ray techniques, J. Energy Chem., 2023, 79, 373 CrossRef CAS .
- K. Zhong, X. Xia, B. Zhang, H. Li, Z. Wang and L. Chen, J. Power Sources, 2010, 195(10), 3300 CrossRef CAS .
- K. Zhang, P. Han, L. Gu, L. Zhang, Z. Liu, Q. Kong, C. Zhang, S. Dong, Z. Zhang, J. Yao, H. Xu, G. Cui and L. Chen, ACS Appl. Mater. Interfaces, 2012, 4(2), 658 CrossRef CAS PubMed .
- X. Zhang, Z. Xing, L. Wang, Y. Zhu, Q. Li, J. Liang, Y. Yu, T. Huang, K. Tang, Y. Qian and X. Shenc, J. Mater. Chem., 2012, 22(34), 17864 RSC .
- H. Lai, J. Li, Z. Chen and Z. Huang, ACS Appl. Mater. Interfaces, 2012, 4(5), 2325 CrossRef CAS PubMed .
- J. Deng, L. Chen, Y. Sun, M. Ma and L. Fu, Carbon, 2015, 92, 177–184 CrossRef CAS .
- L. Li, A. R. O. Raji and J. M. Tour, Adv. Mater., 2013, 25(43), 6298 CrossRef CAS PubMed .
- A. Yu, H. W. Park, A. Davies, D. C. Higgins, Z. Chen and X. Xiao, J. Phys. Chem. Lett., 2011, 2, 1855 CrossRef CAS .
- Y. Son, H. Cha, C. Jo, A. S. Groombridge, T. Lee, A. Boies, J. Cho and M. De Volder, Mater. Today Energy, 2021, 21, 100838 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ac,
Chanita Piyanirunda,
Thanya Phraewphiphatd and
Panitat Hasinbc
*ac,
Chanita Piyanirunda,
Thanya Phraewphiphatd and
Panitat Hasinbc