
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganoboron–thiophene-based polymer electrodes for high-performance lithium-ion batteries†
Yunfei Baic,
Ting Liua,
Huayu Pengb,
Han Zhaob,
Qingchen Fanb,
Xiaobo Pan *bc,
Lian Zhou*b and
Hao Zhao*a
*bc,
Lian Zhou*b and
Hao Zhao*a
aSchool of Physics and Electronic Information, Yantai University, Yantai 264005, People's Republic of China. E-mail: zhaohaolc@126.com
bNew Energy (Photovoltaic) Industry Research Center, Qinghai University, Xining 810006, People's Republic of China. E-mail: zhoulianminai@126.com
cState Key Laboratory of Applied Organic Chemistry, Key Laboratory of Nonferrous Metal Chemistry and Resources Utilization of Gansu Province, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, P. R. China. E-mail: boxb@lzu.edu.cn
First published on 28th February 2024
Abstract
Polymer electrodes are drawing widespread attention to the future generation of lithium-ion battery materials. However, weak electrochemical performance of organic anode materials still exists, such as low capacity, low rate performance, and low cyclability. Herein, we successfully constructed a donor–acceptor thiophene-based polymer (PBT-1) by introducing an organoboron unit. The charge delocalization and lower LUMO energy level due to the unique structure enabled good performance in electrochemical tests with a reversible capacity of 405 mA h g−1 at 0.5 A g−1 and over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at 1 A g−1. Moreover, electron paramagnetic resonance (EPR) spectra revealed that the unique stable spin system in the PBT-1 backbone during cycling provides a fundamental explanation for the highly stable electrochemical performance. This work offers a reliable reference for the design of organic anode materials and expands the potential application directions of organoboron chemistry.
000 cycles at 1 A g−1. Moreover, electron paramagnetic resonance (EPR) spectra revealed that the unique stable spin system in the PBT-1 backbone during cycling provides a fundamental explanation for the highly stable electrochemical performance. This work offers a reliable reference for the design of organic anode materials and expands the potential application directions of organoboron chemistry.
Introduction
The quest for next-generation sustainable lithium-ion battery (LIB) electrode materials has become a hot topic to overcome the mineral resource constraints and environmental pollution problems currently faced by conventional batteries.1 Organic materials can be seen as prospective Li-ion electrodes because of their lightweight, tunable molecular structure, environmental friendliness, and flexibility.2,3 In comparison with extensive research into organic cathode materials, organic anode materials have not yet achieved rapid research development, and conventional anode materials such as graphite cannot match well with organic cathodes in rate performance and electrochemical stability.4–6 Developing efficient organic anode materials is equally important in the future improvement of organic lithium-ion battery performance.7Polythiophene and its derivatives are prospective anode materials for n-type conjugated polymers; however, low redox activity, slow ion mobility, and poor cycling stability at low potentials limit their development for rechargeable batteries.8–12 In contrast, donor–acceptor (D–A) conjugated polymers usually have narrow band gaps, high conductivity, and low electrode potentials, which can alleviate the disadvantages of thiophene-derived polymer materials as anode materials.13–15 The redox activity is further enhanced by optimizing the electronic structure via deliberately incorporating electron acceptor groups into the thiophene polymer architecture, facilitating efficient electron migration along the polymer backbone during charge and discharge processes.16,17 Organoboron molecules, as classic electron-deficient groups, could form strong p–π* conjugations with π-conjugated systems. Thus, boron-based polymers typically exhibit lower lowest unoccupied molecular orbital (LUMO) energy levels, higher electrical conductivity, and distinctive photophysical and electronic properties.18–21 However, the effect of the integration of organoboron units in polymers on the properties of lithium batteries has rarely been studied. The ongoing development of potential application directions is important for organoboron chemistry. In 2019, Susumu Oda et al. synthesized tetracoordinate boron-fused double helices as LIB cathode materials with a capacity of 63 mA h g−1 and a cycling stability of only 20 cycles.22 In our recent studies, the boron-containing radical material PTMA–PSBpin and the quinone-based boron-containing material B–AQ have been prepared, and both have shown good electrochemical stability.23,24 The incorporation of organoboron units effectively adjusts electron energy levels, enhances electron transport characteristics, and improves electrode redox activity. This motivates us to further explore the performance of novel boron-containing electrode materials for LIBs.
In our previous study, we have synthesized a linear boron-containing polymer, PBT-1, using thiophene as the core electron donor unit and two dimesitylborane units as the electron acceptors for electronic structure modification, as shown in Scheme 1, which exhibited excellent photocatalytic properties.25 This excellent photocatalytic property implies the outstanding electron migration property of the material, and for this, we tested its lithium anode material performance, which was also outstanding. In this work, the introduction of boron units compensates for the above-mentioned shortcomings of thiophene derivatives, thus enhancing the π-electron delocalization among molecular skeletons, which significantly increases the intermolecular forces and promotes charge transfer. The PBT-1 battery exhibits fast charging/discharging capability, which, upon activation, can reach up to 405 mA h g−1 at a high current density of 0.5 A g−1, and a good cycling stability beyond 10000 cycles at 1 A g−1. In addition, the varied electron paramagnetic resonance (EPR) spectra reflected an enrichment of the spin system with the introduction of Li+ in the PBT-1 skeleton, and the unchanged g factor indicated the stabilization of free electrons in the D–A structure of the polymer, which supports the high performance of organoboron–thiophene-based LIBs.
 | ||
| Scheme 1 Synthetic route of the conjugated polymer PBT-1. | ||
Results and discussion
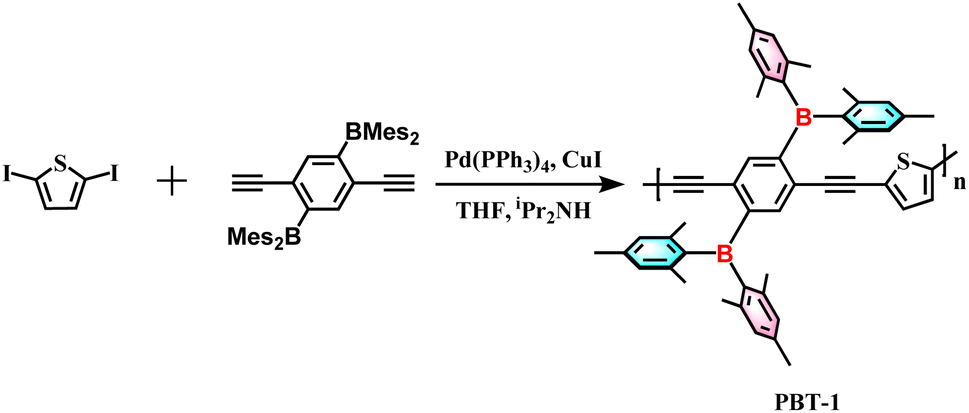
The polymer PBT-1 was synthesized via the Sonogashira–Hagihara coupling polymerization method following standard procedures of (2,5-diethynyl-1,4-phenylene)bis(dimesitylborane) with 2,5-diiodothiophene under reflux in a tetrahydrofuran (THF) and diisopropylamine (iPr2NH) mixture (Scheme 1, detailed in ESI†). After repeated dissolution and precipitation by methanol and hexane, the crude products were further purified by centrifugation to form a yellow powdery solid, PBT-1, in 74% yield. The corresponding structural data and photophysical properties when PBT-1 was used as a photocatalyst have been characterized and analyzed in detail and those are not be repeated herein.25 The chemical structure of PBT-1 was determined by 1H and 11B nuclear magnetic resonance (NMR) (Fig. S1 and S2†) and energy-dispersive spectroscopy (EDS) analyses (Fig. S3†). The signals of Mes2B units can be identified at the chemical shift range of 50–70 ppm in 11B NMR, and EDS analysis illustrates the homogeneous dispersion of C, S, and B. Moreover, the microscopic morphology of the materials is a major factor in lithium storage behavior.26,27 Scanning electron microscopy (SEM) imaging illustrated that the stable skeleton design of PBT-1 exhibits a porous structure similar to that of foam materials, with skeleton diameters ranging from 50 to 200 nm and pore diameters ranging from 200 to 600 nm (Fig. S4†). Nitrogen adsorption isotherm depicting the porosity of PBT-1 was tested at 77 K (Fig. S5†), revealing characteristic H4 hysteresis loops and type II isotherms.28 Using the Brunauer–Emmett–Teller (BET) model, the surface area of the materials was determined to be 134.8 m2 g−1, which is not a high specific surface area for porous material but the electrolyte pores provided are more important.7,9,26 Adsorption grows rapidly under low relative pressures as the curve becomes convex due to the strong interaction of the adsorbate on the surface. Moreover, it progressively forms multilayer adsorption as the relative pressure increases, which is a typical mesoporous physical adsorption process. In addition, the hysteresis loops do not show a saturation adsorption plateau, indicating the presence of more than one pore size, indicating that the material is a mixture of microporous and mesoporous materials.29 The inherent high porosity and appropriate surface area of PBT-1 provide a large contact area with electrolytes, reducing the ionic diffusion distance.30 However, the large pore size distribution cannot lead to effective adsorption capacity contribution, and the adsorption charge is not stabilized for storage.31–33First, the polymer's electronic structure was evaluated using density functional theory (DFT) calculations at the B3LYP/6-31G level (ORCA 5.0.1) to identify the redox activity of the PBT-1 structure (Fig. 1a). Because the redox action derives from the n-type doping/de-doping process, electronic structure analysis can preliminarily identify the redox performance of the materials.16 The construction of the D–A polymer combined with the electron-deficient organoboron unit results in an increase in the concentration of the highest occupied molecular orbital (HOMO) on the linear backbone and a greater distribution of LUMO orbitals on the organoboron unit. As shown in the HOMO distribution, the high level of charge dispersion on the polymer skeleton implies that the material may have better electrical conductivity. Moreover, PBT-1 has an elevated electron affinity and electrical conductivity due to the lower LUMO levels and a narrower bandgap resulting from the intense p–π* cross interaction of that boron group.34,35 These properties are probably caused by the combination of the electron-deficient organoboron moieties and the electron-rich thiophene moieties to form the D–A structure, which, in turn, enhances the battery performance. Molecular electrostatic potential (MESP) analysis calculated by Multiwfn 3.8 (dev)36 shows that PBT-1 has a clear polarity distribution (Fig. 1b), which leads to the creation of a strong dipolar structure (0.378 debye), facilitating rapid charge transfer.37
 | ||
| Fig. 1 (a) Calculated HOMO and LUMO energy levels and (b) MESP map of the polymer fragments. | ||
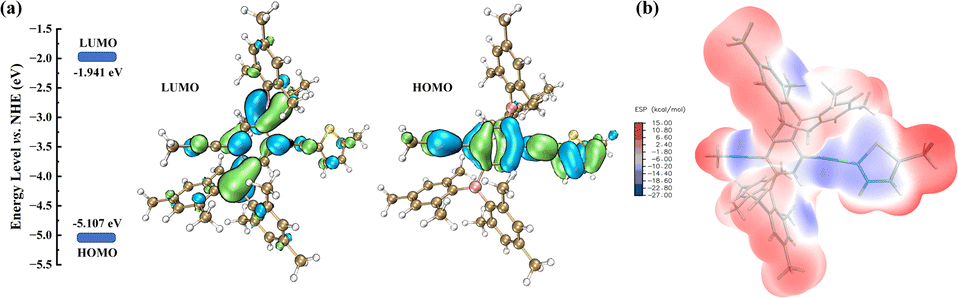
The electrochemical test of PBT-1 was performed in the 2032 half-cell with 1 M LiPF6 in ethylene carbonate/ethyl methyl carbonate/dimethyl carbonate (EC/EMC/DMC, 1:1:1 volume ratio) as the electrolyte. PBT-1, acetylene black, and sodium alginate were mixed in a mass ratio of 5:4:1 to form the working electrodes. Fig. 2a shows the cyclic voltammetry (CV) curves of PBT-1-based cells with a potential window of 0.01–3.0 V at a scan rate of 0.2 mV s−1. A strong cathodic peak was discovered in the first cycle, which disappeared in other cycles due to the irreversible decomposition of electrolytes and the formation of solid electrolyte interphase (SEI) films.38 PBT-1 displays relatively good cycling stability in subsequent CV cycles. Galvanostatic charge–discharge was measured at 45 mA g−1 with the voltage window of 0.01–3.0 V (vs. Li+/Li) (Fig. 2b and c). The discharge curve in the first cycle shows relatively poor initial coulombic efficiency (Fig. S6†), which can be ascribed to the SEI film formation and irreversible reaction with solvated lithium ions, which results in an irrecoverable capacity loss in the first cycle.39–42 Apart from the first discharge curve, the capacity remained stable at 150 mA h g−1 until the fiftieth cycle, highlighting the great cycling stability and reversibility of PBT-1 as a high-performance organic anode. Based on the n-type doping behavior of conjugated polymers, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in the thiophene ring and C
C in the thiophene ring and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C at the linkage between the benzene ring and the thiophene should be the most likely locations for lithium ion embedding.9,10,43–45 Moreover, PBT-1 exhibits a lower voltage plateau (∼0.25 V) compared to the reported organic anode materials, which is beneficial for the expansion of the full cell voltage window and avoids problems such as fast capacity decay. This is a critical feature that facilitates the potential application of this material. Fig. 2d shows the rate property of PBT-1 at different current densities in the range from 0.5 to 10 A g−1. In subsequent long-cycle tests, the capacity showed an increasing trend; therefore, the rate performance test was performed near the 5000 cycles, where the capacity had stabilized and could avoid the objective impact on the rate performance test caused by the activation process of increasing capacity. PBT-1 shows 242 mA h g−1 at 0.5 A g−1. Due to the incomplete charge/discharge behavior at large current densities, the capacity drops significantly with the increase in current density. The capacity is still 121 mA h g−1 when the current density reaches 2 A g−1 as required for conventional power-type batteries. When the current density was measured under 20× magnification, the PBT-1 cell still released 34 mA h g−1. With the current density restored to 0.5 A g−1, PBT-1's discharge capacity immediately returned to 215 mA h g−1 and restored gradually to the initial level at 0.5 A g−1. These results indicated the excellent long-term cycle life and good rate property of the as-synthesized organoboron–thiophene-based electrode.
C at the linkage between the benzene ring and the thiophene should be the most likely locations for lithium ion embedding.9,10,43–45 Moreover, PBT-1 exhibits a lower voltage plateau (∼0.25 V) compared to the reported organic anode materials, which is beneficial for the expansion of the full cell voltage window and avoids problems such as fast capacity decay. This is a critical feature that facilitates the potential application of this material. Fig. 2d shows the rate property of PBT-1 at different current densities in the range from 0.5 to 10 A g−1. In subsequent long-cycle tests, the capacity showed an increasing trend; therefore, the rate performance test was performed near the 5000 cycles, where the capacity had stabilized and could avoid the objective impact on the rate performance test caused by the activation process of increasing capacity. PBT-1 shows 242 mA h g−1 at 0.5 A g−1. Due to the incomplete charge/discharge behavior at large current densities, the capacity drops significantly with the increase in current density. The capacity is still 121 mA h g−1 when the current density reaches 2 A g−1 as required for conventional power-type batteries. When the current density was measured under 20× magnification, the PBT-1 cell still released 34 mA h g−1. With the current density restored to 0.5 A g−1, PBT-1's discharge capacity immediately returned to 215 mA h g−1 and restored gradually to the initial level at 0.5 A g−1. These results indicated the excellent long-term cycle life and good rate property of the as-synthesized organoboron–thiophene-based electrode.
 | ||
| Fig. 2 (a) CV curves of the PBT-1 electrode measured at a scan rate of 0.2 mV s−1. (b and c) Charge and discharge curves at 45 mA g−1. (d) Rate capability of PBT-1 at different current densities. | ||
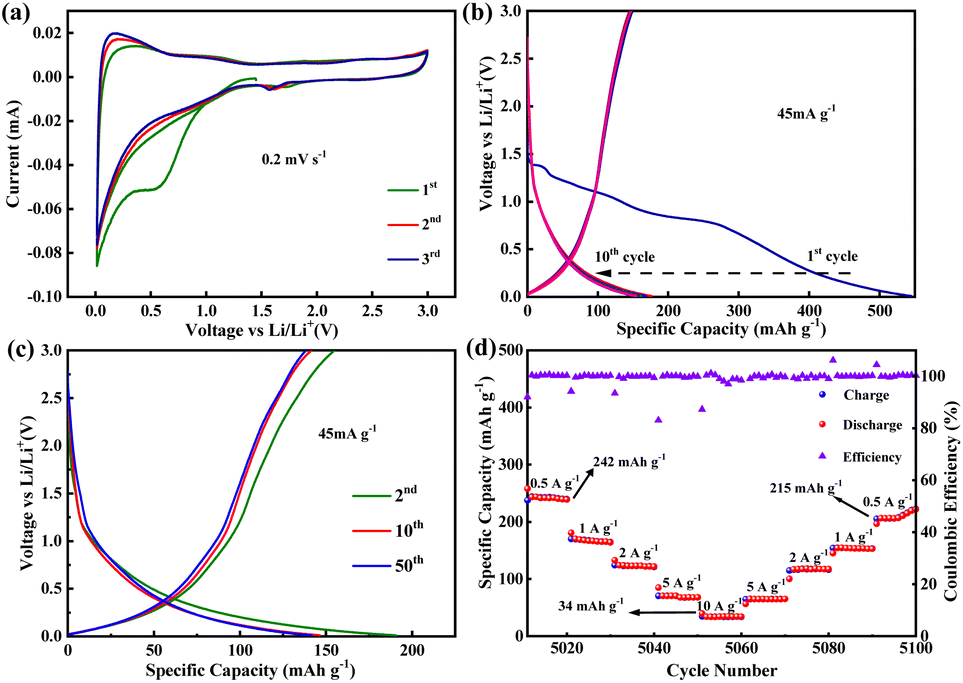
In the cycling stability test for PBT-1, a unique electrochemical performance was demonstrated. Initially, when PBT-1 was cycled at a low current density of 45 mA g−1, besides the low coulombic efficiency in the first five cycles caused by the SEI formation and electrochemical activation of electrodes, the coulombic efficiency remained near 98% for the next 145 cycles, and the capacity did not decay or even increase, from 152 mA h g−1 at cycle 5 to 190 mA h g−1 at cycle 150, an increase of a quarter (Fig. 3a). Such a growing trend aroused our interest in further exploring the changing trend thereafter. By increasing the multiplier to reduce the single-turn charge/discharge time, a 5000-turn cycle test was achieved at 500 mA g−1, as shown in Fig. 3b. The capacity showed a substantial increase, from an initial stabilization of 110 mA h g−1 to 405 mA h g−1 after 1060 cycles (about 1050 minutes), a nearly 4-fold increase. While the capacity was growing rapidly, the coulombic efficiency did not show any abnormal changes and remained at 99%, indicating that there were no other side reactions during this capacity growth, and the capacity growth might be related to the progressive activation of the alkyne bond and benzene ring.46–49 The impedance semicircle of activated PBT-1 showed a significant increase including the increase in solid electrolyte interface resistance and charge transfer resistance (Fig. S7†), which should be ascribed to the electrolyte decomposition at the electrode/electrolyte interface.40 After reaching the peak capacity, a smooth decreasing stage is entered, and the capacity remains 185 mA h g−1 after 5000 turns, averaging 0.13‰ decrease per turn vs. peak capacity, which is still a 68% increase compared to the initial capacity. A comparison of the Nyquist curves of the electrodes before and after cycling is shown in Fig. S7.† Both electrodes showed good charge transfer impedance (RCT, 50 Ω approx.) and ion transport properties. A small increase in RCT was observed, consistent with the change in the electrode/electrolyte interface resistance caused by surface morphology remodeling of the electrode after long cycling. A decrease in bulk-phase diffusion impedance indicates that the ion transport channels are opened after activation.50 Although the relevant changes in this process can be initially inferred from the impedance changes, a more direct study of this process using novel in situ or ex situ characterization tools should be a major issue for further research in subsequent studies of similar long-cycle phenomena. By further increasing the charge/discharge multiplier, PBT-1 completed a cycle test of 10000 turns at 1 A g−1 current density (Fig. 3c). The peak capacity occurred at 2100 cycles (about 730 minutes), indicating that the slower charge/discharge rate is more favorable to the activation of PBT-1. Although the cycle test of PBT-1 is accompanied by the growth and reduction in capacity, the growth of capacity is inevitably a more popular feature in practice, while the decay behavior all appears after nearly 1000 turns with a 0.065‰ decrease per turn. After rational activation, PBT-1 has a considerable reversible capacity contribution at high currents and is capable of thousands of stable cycles, which is an important feature when it is used to match high-power cathode materials in real-world applications. In addition, the PBT-1 electrode as an anode material in LIBs only shows a conventional level of capacity, but exhibited better high-power long-cycle characteristics and practical applications in lithium-ion batteries than in the reported polymer anode materials, as shown in Table S1.†
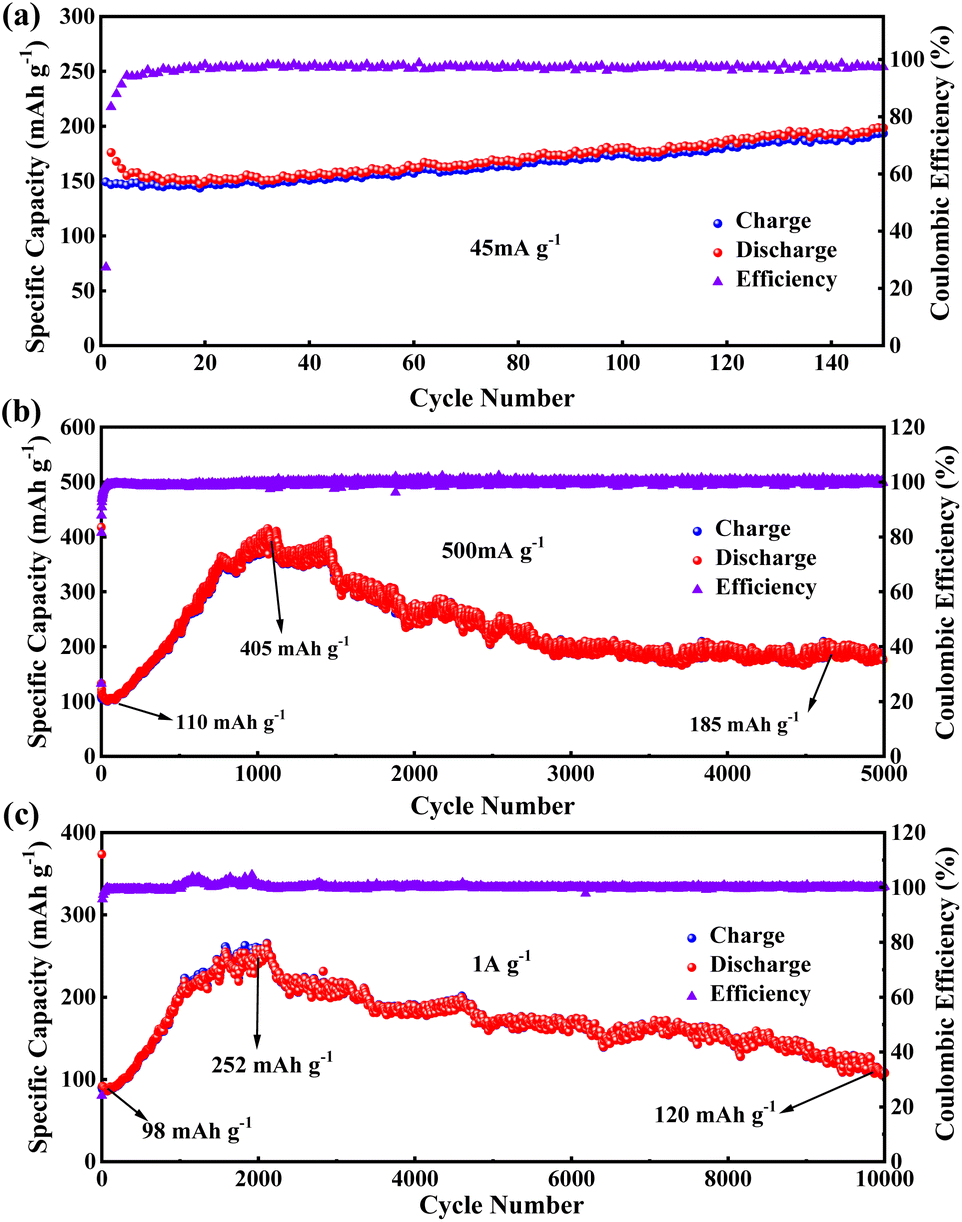 | ||
| Fig. 3 Cycling stability of PBT-1 at (a) 45, (b) 500, and (c) 1000 mA g−1. | ||
To deeply understand the charging and discharging process of PBT-1, EPR spectroscopy was performed at different potentials (Fig. 4). For the ex situ test, the coin cells were fully discharged/charged (0.01 and 3.0 V) at a current density of 45 mA g−1, disassembled and then washed in a glove box for EPR measurement. The observed first derivative and absorption spectra are shown in Fig. 4b–d. From the original EPR data, the EPR intensity is almost at the same magnitude at the pristine, fully lithiated, and delighted stages (orange zone in Fig. 4b). Thus, in order to analyze the variation in the EPR spectra, as depicted in Fig. 4c, normalization was performed. It was clear that all three spectra were centered at 3362 G with a g factor of 2.0043. In the full discharge/charge spectra, the sharp peak was due to the deposition of lithium dendrite, whereas the broad peak corresponded to the free electrons that were stabilized with delocalization in the D–A structure of the polymers. In the open circuit voltage (OCV) state, the electrolyte was decomposed by the highly active alkali metal (lithium); thus, trace lithium may react with the electrolyte and be deposited onto the bulk lithium surface, resulting in a dendrite signal in the OCV EPR spectra. In Fig. 4c, the linewidth of the EPR spectra changed upon lithiation/delithiation. In the fully discharged state, with Li+ insertion into the PBT-1 skeleton and dendrite accumulation on the PBT-1 electrode surface, the EPR linewidth was broadened compared to that in the OCV state. The line broadening reflected an increment of the spin system due to the introduction of Li+ into the PBT-1 skeleton.51,52 Accordingly, the EPR line narrowed when the cell was fully charged. Although the EPR linewidth was changed, the g factor of the EPR signal was maintained during cycling. This suggests that the spin system is preserved in the D–A structure on the charging/discharging intermediates. The EPR analysis suggested that the higher rate and cycle performance were presumably because of the structural stability of the PBT-1 framework.
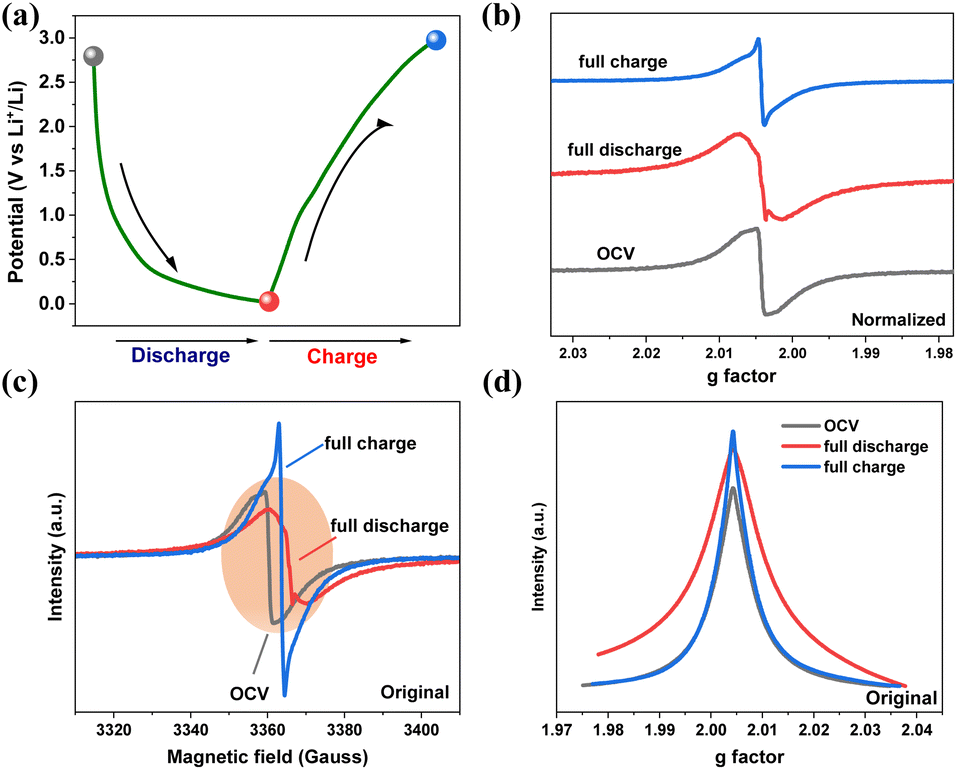 | ||
| Fig. 4 (a) Charge/discharge profile of the PBT-1 anode at the second cycle at 0.2 A g−1, and the select points were denoted in the curve. Normalized (b) and original (c) first-derivative EPR spectra at the selected points indicated in (a). (d) Absorption EPR spectra at the three selected potentials. | ||
Conclusions
In conclusion, a thiophene-based D–A conjugated polymer constructed with organoboron units was used as an anode in LIBs. The boron unit optimized the electrode performance by adjusting the electronic structure of the thiophene skeleton. The charge delocalization and lower LUMO energy level enabled PBT-1 to exhibit impressive rate properties and cycling stability. PBT-1 delivered a maximum reversible charging/discharging capacity of 405 mA h g−1 at a large current density of 0.5 A g−1 upon activation and high cycling stability with more than 10000 cycles at 1 A g−1. Moreover, the EPR signal was almost of the same intensity, and the g factor manifested as the same (g = 2.0043). The spin system of PBT-1 was stable during Li+ intercalation/deintercalation, which accounted for the excellent electrochemical performance. However, there are still significant limitations in the explanation of the energy storage mechanism and activation process in this work, and further research on this issue should be emphasized as a reference for related work. Overall, this study provides an effective method for the novel molecular engineering of high-performance organic LIBs and will also expand the potential application directions of organoboron chemistry.
Author contributions
Yunfei Bai: conceptualization, methodology, experiment, formal analysis, and writing of the original draft. Ting Liu: electronic structure calculations. Huayu Peng: investigation and experiment. Han Zhao: conformational analysis. Qingchen Fan: resources. Xiaobo Pan: conceptualization, methodology, writing of the review and editing, and supervision. Lian Zhou: conceptualization and supervision. Hao Zhao: writing the review and editing, and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the Basic Research Project of Qinghai Province in 2021 (2021-ZJ-721). The authors also thank the Supercomputing Center of Lanzhou University for the DFT calculations.References
- Y. Lu and J. Chen, Nat. Rev. Chem, 2020, 4, 127–142 CrossRef CAS PubMed.
- Z. Song and H. Zhou, Energy Environ. Sci., 2013, 6, 2280–2301 RSC.
- R. Shi, S. Jiao, Q. Yue, G. Gu, K. Zhang and Y. Zhao, Exploration, 2022, 2, 20220066 CrossRef PubMed.
- R. Kaur, V. A. Chhabra, V. Chaudhary, K. Vikrant, S. K. Tripathi, Y. Su, P. Kumar, K.-H. Kim and A. Deep, Int. J. Energy Res., 2022, 46, 13178–13204 CrossRef CAS.
- A. V. Desai, R. E. Morris and A. R. Armstrong, ChemSusChem, 2020, 13, 4866–4884 CrossRef CAS PubMed.
- A. A. Pavlovskii, K. Pushnitsa, A. Kosenko, P. Novikov and A. A. Popovich, Materials, 2023, 16, 117 Search PubMed.
- H. Cheng, J. G. Shapter, Y. Li and G. Gao, J. Energy Chem., 2021, 57, 451–468 CrossRef CAS.
- L. Zhan, Z. Song, J. Zhang, J. Tang, H. Zhan, Y. Zhou and C. Zhan, Electrochim. Acta, 2008, 53, 8319–8323 CrossRef CAS.
- C. Zhang, Y. He, P. Mu, X. Wang, Q. He, Y. Chen, J. Zeng, F. Wang, Y. Xu and J.-X. Jiang, Adv. Funct. Mater., 2018, 28, 1705432 CrossRef.
- L. Zhu, Y. Niu, Y. Cao, A. Lei, X. Ai and H. Yang, Electrochim. Acta, 2012, 78, 27–31 CrossRef CAS.
- K.-S. Lee, Y.-K. Sun, J. Noh, K. S. Song and D.-W. Kim, Electrochem. Commun., 2009, 11, 1900–1903 CrossRef CAS.
- Q. T. Wang, R. R. Li, X. Z. Zhou, J. Li and Z. Q. Lei, J. Solid State Chem., 2016, 20, 1331–1336 CAS.
- C. Deibel, T. Strobel and V. Dyakonov, Adv. Mater., 2010, 22, 4097–4111 CrossRef CAS.
- S.-M. Xu, X. Liang, X. Liu, W.-L. Bai, Y.-S. Liu, Z.-P. Cai, Q. Zhang, C. Zhao, K.-X. Wang and J.-S. Chen, Energy Storage Mater., 2020, 25, 52–61 CrossRef.
- W. Ma, L.-W. Luo, P. Dong, P. Zheng, X. Huang, C. Zhang, J.-X. Jiang and Y. Cao, Adv. Funct. Mater., 2021, 31, 2105027 CrossRef CAS.
- C. Zhang, Y. Qiao, P. Xiong, W. Ma, P. Bai, X. Wang, Q. Li, J. Zhao, Y. Xu, Y. Chen, J. H. Zeng, F. Wang, Y. Xu and J.-X. Jiang, ACS Nano, 2019, 13, 745–754 CrossRef CAS PubMed.
- W. Dai, L. Jiang, J. Wang, Y. Pu, Y. Zhu, Y. Wang and B. Xiao, Chem. Eng. J., 2020, 397, 125476 CrossRef CAS.
- H. Helten, Chem.–Asian J., 2019, 14, 919–935 CrossRef CAS.
- C. Ru, Q. Wei, W. Chen, Q. Guan, Q. Zhang, Y. Ling, C. Tao, D. Qin, J. Wu and X. Pan, ACS Energy Lett., 2020, 5, 669–675 CrossRef CAS.
- C. Ru, P. Chen, X. Wu, C. Chen, J. Zhang, H. Zhao, J. Wu and X. Pan, Adv. Sci., 2022, 9, 2204055 CrossRef CAS.
- P. Chen, C. Ru, L. Hu, X. Yang, X. Wu, M. Zhang, H. Zhao, J. Wu and X. Pan, Macromolecules, 2023, 56, 858–866 CrossRef CAS.
- S. Oda, T. Shimizu, T. Katayama, H. Yoshikawa and T. Hatakeyama, Org. Lett., 2019, 21, 1770–1773 CrossRef CAS PubMed.
- W. Chen, X. Wu, Y. Bai, P. Sun, C. Gong, C. Huang, X. Pan, J. Wu and X. Pan, ACS Appl. Energy Mater., 2020, 3, 9296–9304 CrossRef CAS.
- J. Zhou, Y. Bai, Q. Qiu, S. Liu, H. Qiu, X. Zhang and H. Zhao, Chem. Eng. J., 2022, 452, 139576 CrossRef.
- Q. Wei, X. Yao, Q. Zhang, P. Yan, C. Ru, C. Li, C. Tao, W. Wang, D. Han, D. Han, L. Niu, D. Qin and X. Pan, Small, 2021, 17, 2100132 CrossRef CAS PubMed.
- A. A. AbdelHamid, A. Mendoza-Garcia and J. Y. Ying, Nano Energy, 2022, 93, 106860 CrossRef CAS.
- N. P. Shetti, S. Dias and K. R. Reddy, Mater. Sci. Semicond. Process., 2019, 104, 104684 CrossRef CAS.
- B. Sun, Y. Yuan, H. Li, X. Li, C. Zhang, F. Guo, X. Liu, K. Wang and X. S. Zhao, Chem. Eng. J., 2019, 371, 55–63 CrossRef CAS.
- J. Zou, C. Fan, Y. Jiang, X. Liu, W. Zhou, H. Xu and L. Huang, Microporous Mesoporous Mater., 2021, 327, 111411 CrossRef CAS.
- Y. Liu, G. Sun, X. Cai, F. Yang, C. Ma, M. Xue and X. Tao, J. Energy Chem., 2021, 54, 179–193 CrossRef CAS.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597–1614 RSC.
- Q. Zhu, M. Wang, B. Nan, H. Shi, X. Zhang, Y. Deng, L. Wang, Q. Chen and Z. Lu, J. Power Sources, 2017, 362, 147–159 CrossRef CAS.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- Z. Ding, X. Long, C. Dou, J. Liu and L. Wang, Chem. Sci., 2016, 7, 6197–6202 RSC.
- X. Wang, J. Zhou and W. Tang, Energy Storage Mater., 2021, 36, 1–9 CrossRef.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- T. C. Leung, C. L. Kao, W. S. Su, Y. J. Feng and C. T. Chan, Phys. Rev. B, 2003, 68, 195408 CrossRef.
- Y. Bai, W. Fu, W. Chen, Z. Chen, X. Pan, X. Lv, J. Wu and X. Pan, J. Mater. Chem. A, 2019, 7, 24454–24461 RSC.
- P. Verma, P. Maire and P. Novák, Electrochim. Acta, 2010, 55, 6332–6341 CrossRef CAS.
- Y. Wang, Y. Deng, Q. Qu, X. Zheng, J. Zhang, G. Liu, V. S. Battaglia and H. Zheng, ACS Energy Lett., 2017, 2, 2140–2148 CrossRef CAS.
- C. Wang, Y. Xu, Y. Fang, M. Zhou, L. Liang, S. Singh, H. Zhao, A. Schober and Y. Lei, J. Am. Chem. Soc., 2015, 137, 3124–3130 CrossRef CAS PubMed.
- S. Wang, L. Wang, K. Zhang, Z. Zhu, Z. Tao and J. Chen, Nano Lett., 2013, 13, 4404–4409 CrossRef CAS.
- Q. Yuan, C. Li, X. Guo, J. Zhao, Y. Zhang, B. Wang, Y. Dong and L. Liu, Energy Rep., 2020, 6, 2094–2105 CrossRef.
- L. M. Zhu, A. W. Lei, Y. L. Cao, X. P. Ai and H. X. Yang, Chem. Commun., 2013, 49, 567–569 RSC.
- Y. Yang, C. Wang, B. Yue, S. Gambhir, C. O. Too and G. G. Wallace, Adv. Energy Mater., 2012, 2, 266–272 CrossRef CAS.
- Z. Lei, Q. Yang, Y. Xu, S. Guo, W. Sun, H. Liu, L. P. Lv, Y. Zhang and Y. Wang, Nat. Commun., 2018, 9, 576 CrossRef PubMed.
- S. Wang, Q. Wang, P. Shao, Y. Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2017, 139, 4258–4261 CrossRef CAS PubMed.
- Z. Zhang, Y. Zhou, P. Chen, S. Zeng, W. Nie and Y. Xu, ACS Appl. Energy Mater., 2021, 4, 12882–12891 CrossRef CAS.
- M. Wu, Y. Zhao, H. Zhang, J. Zhu, Y. Ma, C. Li, Y. Zhang and Y. Chen, Nano Res., 2021, 15, 9779–9784 CrossRef.
- H. Zhao, X. Ma, J. Bai, Z. Yang, G. Sun, Z. Zhang, X. Pan, W. Lan, J. Y. Zhou and E. Xie, Nanoscale, 2017, 9, 8192–8199 RSC.
- C. P. Poole and H. A. Farach, Bull. Magn. Reson., 1979, 1, 162–194 Search PubMed.
- M. Kalapsazova, R. Kukeva, S. Harizanova, P. Markov, D. Nihtianova, E. Zhecheva and R. Stoyanova, Batteries, 2023, 9, 144 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra06060h |
| This journal is © The Royal Society of Chemistry 2024 |