
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient catalytic activity of NiO and CeO2 films in benzoic acid removal using ozone†
Daynahi Franco Peláeza,
Julia Liliana Rodríguez S. *a,
Tatyana Poznyaka,
Hugo Martínez Gutiérrezb,
J. Alberto Andraca Adamec,
Luis Lartundo Rojasb and
Claudia Jazmín Ramos Torresb
*a,
Tatyana Poznyaka,
Hugo Martínez Gutiérrezb,
J. Alberto Andraca Adamec,
Luis Lartundo Rojasb and
Claudia Jazmín Ramos Torresb
aLaboratorio de Investigación en Ingeniería Química Ambiental, ESIQIE-Instituto Politécnico Nacional, Zacatenco, Ciudad de México 07738, Mexico. E-mail: jlrodriguezs@ipn.mx; ozliliana@yahoo.com.mx
bCentro de Nanociencias y Micro y Nanotecnologías, Instituto Politécnico Nacional, Zacatenco, Ciudad de México, 07738, Mexico
cDepartamento Ciencias Básicas, UPIIH—Instituto Politécnico Nacional, México City, 42050, Mexico
First published on 26th January 2024
Abstract
This research focuses on the synthesis of NiO and CeO2 thin films using spray pyrolysis for the removal of benzoic acid using ozone as an oxidant. The results indicate that the addition of CeO2 films significantly enhances the mineralization of benzoic acid, achieving a rate of over 80% as the CeO2 films react with ozone to produce strong oxidant species, such as hydroxyl radicals, superoxide radicals, and singlet oxygen as demonstrated by the presence of quenchers in the reaction system. The difference in catalytic activity between NiO and CeO2 films was analyzed via XPS technique; specifically, hydroxyl oxygen groups in the CeO2 film were greater in number than those in the NiO film, thus benefitting catalytic oxidation as these species are considered active oxidation sites. The effects of nozzle-substrate distances and deposition time during the synthesis of the films on benzoic acid removal efficiency were also explored. Based on XRD characterization, it was established that the NiO and CeO2 films were polycrystalline with a cubic structure. NiO spherical nanoparticles were well-distributed on the substrate surface, while some pin holes and overgrown clusters were observed in the CeO2 films according to the SEM results. The stability of the CeO2 films after five consecutive cycles confirms their reusability. The retrieval of films is easy because it does not require additional separation methods, unlike the catalyst in powder form. The obtained results indicate that the CeO2 films have potential application in pollutant removal from water through catalytic ozonation.
1. Introduction
The search for new materials is a topic of broad and current interest owing to the lack of highly efficient catalysts for environmental applications. Advanced oxidation processes (AOPs), such as the Fenton reaction, ozonation, persulfate, and photodegradation, have been used to enhance the degradation of diverse organic pollutants.1–5 Most of the AOPs require the presence of a catalyst to initiate or increase the degradation of toxic organic compounds in water, and their efficiency depends on the catalyst properties.6,7 For example, heterogeneous catalytic ozonation is an excellent remediation technology for the elimination of recalcitrant toxic compounds from water.4,7 It is commonly used when the catalyst dispersed into the water is a solid powder, which favors ozone decomposition to produce reactive oxygen species, such as free radicals (superoxide radical (O2˙−), hydroxyl radicals (˙OH)), and non-radicals (singlet oxygen (1O2)).4,8–10 Complex post-treatment is required to recover catalysts that exist in the form of powder; in addition, loss of material is inevitable during several cycles of reuse.Thin films have attracted considerable attention in the last few years owing to their ease of fabrication and low cost. Numerous methods have been developed to deposit thin films, such as sol–gel,11 electrochemical deposition, sputtering,12 and spray pyrolysis techniques.13,14 Compared to these methods, spray pyrolysis has some advantages, including simple operation, higher stability of the obtained coatings, adaptability (for preparing single and multi-layered films), and an open-atmosphere process.15,16 Recently, the ultrasonic spray pyrolysis method has been used to create a wide variety of thin films, and their properties depend on a set of controllable parameters, such as substrate temperature,17 solution concentration,18 carrier gas ratio,19 solvent type,20 nozzle-substrate distance,21 and deposition time.22 These parameters can be modulated to generate various nanostructures, crystal orientations, and single and multi-layered films.
Although several attempts have been made to produce catalysts in the form of films, there is not enough research on evaluating their catalytic activity in organic pollutant removal from aqueous solutions with ozone as the oxidizing agent. C. M. Aguilar-Melo et al.23 demonstrated that nickel oxide (NiO) achieved higher catalytic performance in naproxen removal in comparison with conventional ozonation because of the improvement in reactive oxygen species (ROS) generation. Similar results were obtained by I. C. Guzmán et al.24 in the elimination of 4-chlorophenol and 4-phenolsulfonic acid using a combination of ozone and a cerium oxide (CeO2) film as the catalyst, which exhibited remarkable catalytic performance due to electron transfer of the redox pair (Ce3+/Ce4+).
Among transition metal oxides, NiO is cheaper than other noble metals and has demonstrated high catalytic activity towards some oxidation reactions that involve ozone.25 Another transition metal oxide widely used as a catalyst in heterogeneous catalytic ozonation is rare-earth CeO2 because of its tunable nanoscale morphologies, high Ce4+/Ce3+ redox activity, excellent stability, low toxicity, and high abundance. Furthermore, it possesses abundant oxygen vacancies (OV), which often favor the formation of ROS. L. Wu et al.26 showed that the formation of two neighboring Ce3+ is possible due to the absence of an oxygen atom from the CeO2 lattice, generating Frenkel-type OV (movement of the oxygen atom from the lattice site to an interstitial site), and this was the critical descriptor of ozone decomposition. Therefore, it is expected that NiO and CeO2 films can provide excellent catalytic activity towards pollutant removal in the presence of ozone. To the best of our knowledge, the effects of the nozzle-to-substrate distance (NSD) during synthesis on the structural properties of both these catalysts in ozonation have not been studied.
Benzoic Acid (BA) is commonly found in the effluents of several industries, mainly the manufacturing units of dyes, food, cosmetics, and pharmaceuticals.27,28 Furthermore, it is the main precursor used in the production of phenols and certain polymers; hence BA is a major pollutant in wastewater of these industries,29 and its removal from water is a longstanding issue. BA could be completely decomposed by UVC-persulfate oxidation within 1 h, and it was mineralized up to 96%, resulting in the generation of sulfate radicals (SO4˙−) and ˙OH from S2O82− ions under a UVC light.27 A. Sennaoui et al.30 studied the oxidation of BA by the electro-Fenton process using boron-doped diamond/carbon-felt cells, achieving complete elimination by around 2 h, and complete mineralization was achieved in 5 h. Both methods were effective in BA removal; however, the employment of UVC light and electrodes is expensive, increasing the cost of the treatment system. In this sense, catalytic ozonation might be a better alternative to obtain a higher mineralization degree in the system.
The main objective of this study is to investigate BA removal by ozone in the presence of NiO and CeO2 films synthesized by ultrasonic spray pyrolysis. In addition, the effects of NSD and deposition time on the characteristics and catalytic activity of the thin film were analyzed. Then, the most efficient film was selected to study the influence of the number of glass plates containing the film on target organic compound degradation, as well as its stability. Finally, the involvement of specific ROS was confirmed by the addition of scavengers, such as tert-butanol, sodium azide and p-benzoquinone, to the reaction medium.
2. Materials and methods
2.1. Experimental conditions of thin film synthesis
The glass substrates (“Lauka”, 2.5 × 7.5 cm2) were cleaned by rinsing in acetone, ethyl alcohol, and deionized water using an ultrasonic bath for 20 min each. After these treatments, the substrates were covered and stored to avoid impurities. The thin films were deposited by using ultrasonic spray pyrolysis equipment with a 1.5 cm diameter spray nozzle. A solution of 0.1 M cerium nitrate hexahydrate (Ce(NO3)3·6H2O (99%)) or nickel nitrate hexahydrate (Ni(NO3)2·6H2O (97%)) in deionized water was used as the precursor.The NSD was varied (1 and 3.7 cm), while the other preparative parameters, including substrate temperature (T = 450 °C), concentration of the precursors (0.1 M), carrier gas pressure (20 psi), deposition time (15 or 30 min), and quantity of the spraying solution, were kept constant. The obtained thin films of CeO2 and NiO were labeled as MeHxTy, where Me = Ni, Ce; H = substrate-to-nozzle distance (x = 1 for 1 cm, 2 for 3.7 cm); T = deposition time (y = 1 (15 min), 2 (30 min)). The two values of NSD were chosen according to the operational characteristics of the equipment.
2.2. Characterization of the thin films
The film thickness was measured with a Uvisel Ellipsometer (model LT M200AGMS HORIBA) with a xenon lamp (75 W), and the thickness values were obtained via simulation by employing a Tauc Lorentz dispersion formula. Crystallite structure information was obtained by Grazing Incidence X-Ray Diffraction (GIXRD) using an XPERT-Pro diffractometer with Cu Kα radiation (40 mA and 45 kV). The grazing incidence angle of the X-ray beam was fixed at 0.5°, and the 2-theta scanning angle was varied from 10° to 90°. Morphological analysis was performed with a Scanning Electron Microscope (SEM) model Jeol-JSM 7800 F. Finally, the surface roughness was determined by using an Atomic Force Microscope (AFM) MultiMode V equipped with a minicontroller NanoEscope V (BRUKER, USA). The chemical compositional analysis of the samples was carried out by X-ray Photoelectron Spectroscopy (XPS) using a Thermo Scientific (Waltham, MA, USA) K-Alpha instrument equipped with an Al Kα X-ray source (1486.6 eV) running at a power of 150 W. XPS narrow scans using an X-ray spot size of 400 μm2 were collected using pass energies of 200 and 20 eV for the survey and high-resolution spectra, respectively. XPS core level spectra were analyzed using XPSpeak 4.1 software and fitted using a Gaussian–Lorentzian mix function.2.3. Ozonation experiments
Ozonation experiments were conducted in two systems; the first system involved BA (115 mg L−1) removal by conventional ozonation, and in the second system, the catalytic ozonation of BA was carried out in the presence of six glass plates with thin films of CeO2 or NiO. All the tests used the same cylindrical reactors with a reaction volume of 400 mL. Ozone gas was continuously generated from HTU500G (Azco Industries Limited, Canada) using a pure oxygen source and distributed via a porous glass plate at the bottom of the reactor. The gas flow and the concentration of ozone were maintained at 0.5 L min−1 and 30 ± 0.2 mg L−1, respectively, to avoid the phenomenon of mass transfer. Residual ozone in the gas phase was measured using BMT 946 BT (BMT Messtechnik, Berlín). Scavenger tests were performed to demonstrate the influence of ROS on catalytic ozonation; tert-butanol (TBA), sodium azide (NaN3) and p-benzoquinone (p-BQ) at 50 mg L−1, 50 mM and 0.2 M were used as scavengers of ˙OH, O2˙−, and 1O2, respectively.2.4. Analytical methods
The concentration of BA was analyzed by High-Performance Liquid Chromatography (HPLC, PerkinElmer Flexar) using a C-18 YMC column at a flow rate of 0.5 mL min−1 and 230 nm of wavelength. The mobile phase was 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 (v/v) acetonitrile:water adjusted to 2.5 pH with phosphoric acid. The oxalic acid concentration was also examined by HPLC under the analytical condition reported by C. M. Aguilar-Melo et al.23 The total organic carbon (TOC) was determined by using a TOC analyzer (Shimadzu TOC-500 A).
:50 (v/v) acetonitrile:water adjusted to 2.5 pH with phosphoric acid. The oxalic acid concentration was also examined by HPLC under the analytical condition reported by C. M. Aguilar-Melo et al.23 The total organic carbon (TOC) was determined by using a TOC analyzer (Shimadzu TOC-500 A).
3. Results and discussion
3.1. Characterization of the thin films
The film thicknesses were determined by ellipsometry and are presented in Table 1. It can be observed that at a shorter spray distance (1 cm), films with greater thickness were obtained than those formed at the nozzle-substrate distance of 3.7 cm. For the NiO sample, the thickness was 33.78 nm and 15.37 nm, and for the CeO2 sample, it was 15.37 nm and 6.8 nm at NSDs of 1 cm and 3.7 cm, respectively (Table 1). However, the crystal structure is amorphous of the samples at a distance from the NSD of 1 cm (Fig. 1). At a greater NSD, the crystalline structure was improved, and with the deposition time increasing from 15 to 30 minutes, the thickness of the NiO film increased from 15.37 nm to 29.06 nm and that of the CeO2 film changed from 6.80 nm to 59.89 nm, respectively.| Sample | 2θ (°) | FWHM(c) | D (nm) | Thickness |
|---|---|---|---|---|
| NiH1T1 | — | — | — | 33.78 |
| NiH2T1 | 37.22 | 0.639 | 28.6 | 15.37 |
| NiH2T2 | 37.22 | 0.325 | 94.2 | 29.06 |
| CeH1T1 | — | — | — | 15.37 |
| CeH2T1 | 28.59 | 1.100 | 12.1 | 6.80 |
| CeH2T2 | 28.59 | 0.922 | 15.6 | 59.89 |
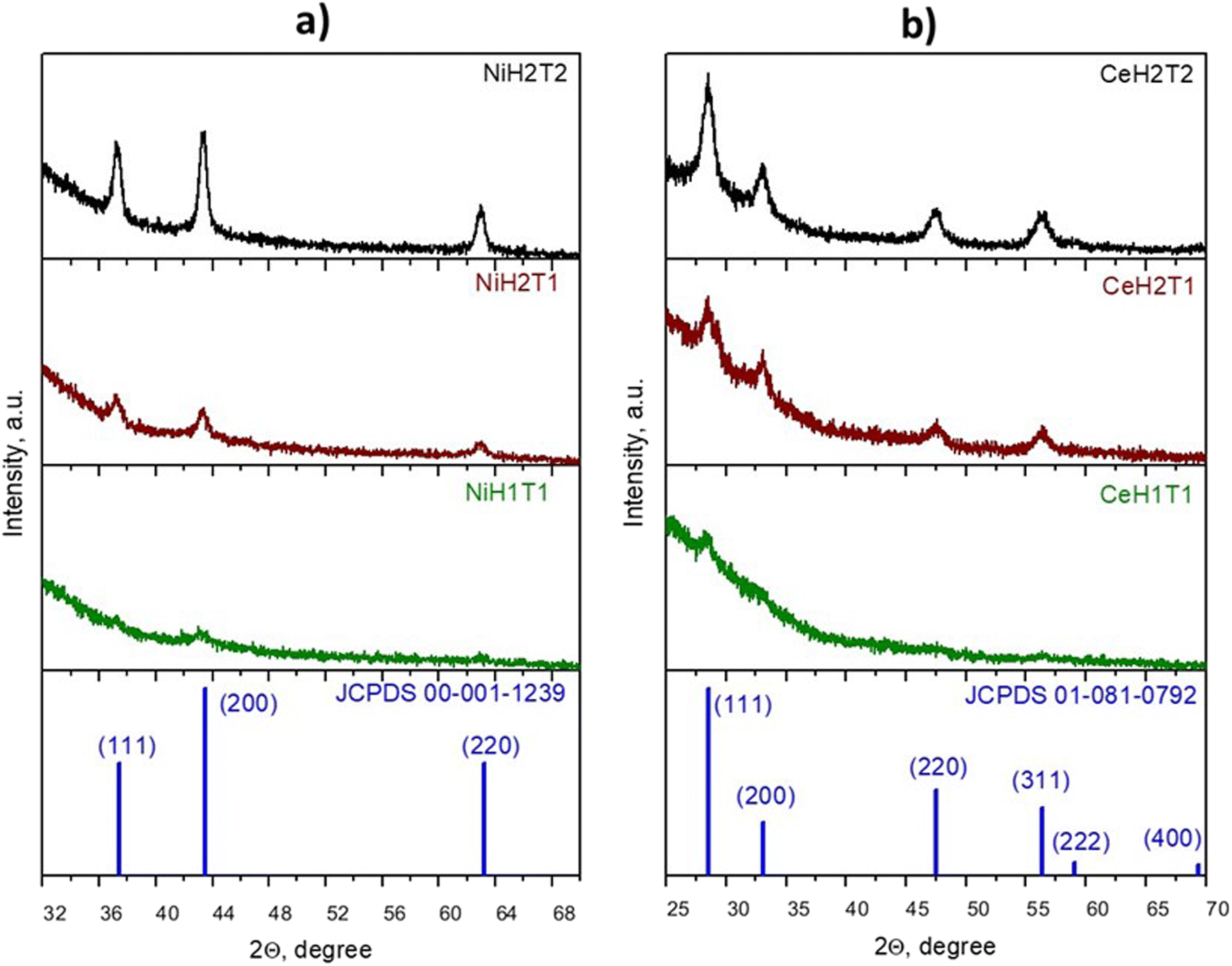 | ||
| Fig. 1 The XRD patterns of the as-prepared (a) NiO and (b) CeO2 thin films on glass substrates at 1 cm (H1) and 3.7 cm (H2) substrate-to-nozzle distance and at 15 min (T1) and 30 min (T2) deposition time. | ||
The X-ray diffraction patterns (XRDP) of the as-deposited NiO and CeO2 thin films are shown in Fig. 1a and b, respectively, which reveal that all the samples were polycrystalline with a cubic structure. The NiO thin films displayed three main peaks at 2θ = 37.33°, 43.34° and 62.97°, representing the (111), (200) and (220) plane directions, respectively, and no impurity phases were detected (JCPDS 00-001-1239). Fig. 1b shows the main characteristic orientations of CeO2 along the (111), (200), (220), and (311) directions, which are in accordance with JCPDS 01-081-0792.
When the spraying solution emerges from an atomizer, the droplets (made up of solvent and solute) travel different NSDs and hence experience diverse temperature zones (temperature gradient in the vapor phase). This is due to some forces, such as gravitational and friction (droplet-air) forces, playing an important role in the reaction kinetics of the droplets.21
At a short NSD (1 cm, NiH1T1), the droplets in the liquid phase hit the substrate surface and evaporate after a certain time. When the distance is increased, that is, at 3.7 cm (NiH2T1), the droplets have more time to absorb the thermal energy and evaporate the solvent before the droplets reach the substrate; therefore, the crystallinity improves upon increasing the NSD from 1 to 3.7 cm. Similar results were obtained by R. J. Deokate et al.21 for the deposition of CdIn2O4 films; their results also demonstrated that the density of oxygen vacancies improves with increasing NSD, and this effect improves catalytic activity in the ozonation process as the oxygen vacancies serve as active sites.31
From the patterns obtained, the crystallite size was calculated for each sample except for NiH1T1 and CeH1T1 because they produced no peaks in XRD. The crystallite size was obtained using the Debye–Scherrer eqn (1).
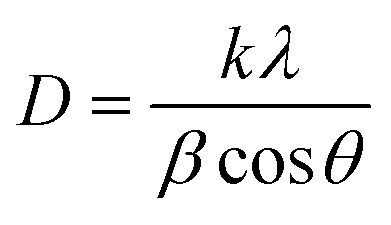 | (1) |
Notably, the intensity of the peaks of the NiO and CeO2 thin films enhanced with an increase in the NSD and deposition time. When the deposition time was increased from 15 min (NiH2T1) to 30 min (NiH2T2), an increase in crystal size from 28.6 nm to 94.2 nm was observed for the samples NiH2T1 and NiH2T2 (see Table 1) due to the presence of a higher quantity of metallic oxides on the substrate. Similar results were obtained for CeO2, which indicates that a longer deposition time is favorable for crystal formation (T2 = 30 min), consistent with previous publications.32,33
The morphology of the NiO thin films formed at different NSDs and deposition times was analyzed using SEM, as displayed in Fig. 2.
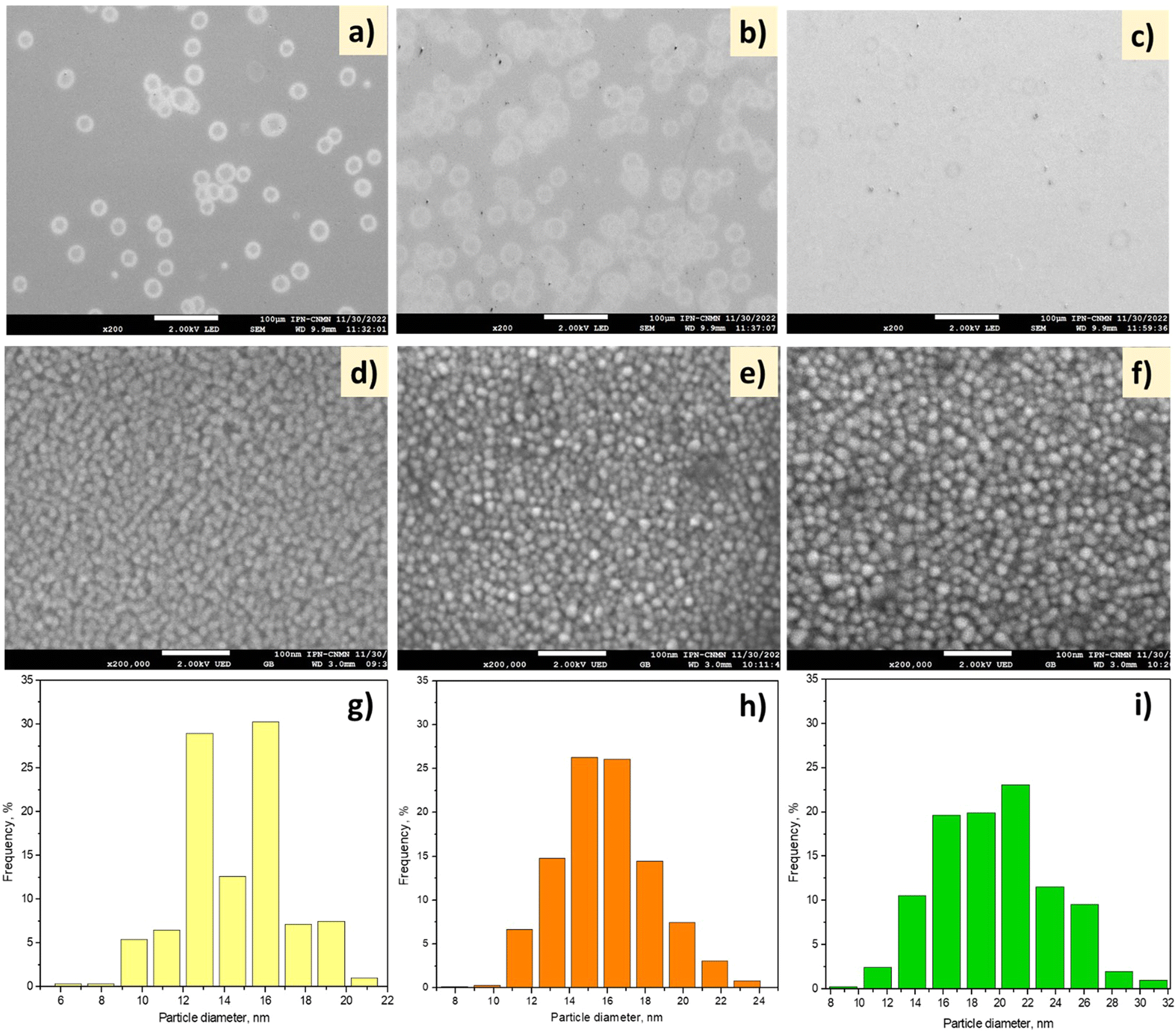 | ||
| Fig. 2 (a–f) SEM images and (g–i) particle size distributions of (a, d, and g) NiH1T1, (b, e, and h) NiH2T1 and (c, f, and i) NiH2T2. Scale bars: (a–c) 100 μm and (d–f) 100 nm. | ||
At both 1 and 3.7 cm spraying distances (Fig. 2a and b), the formation of hollow spherical particles with an average diameter of 20 μm was observed. These particles are derived from the coagulation of the incoming (and deposited) wet aerosol droplets. Upon increasing the spray time to 30 min, slight deposition of the hollow spherical particles was observed in the image (Fig. 2c), and they were of smaller size than those observed at 15 min. Fig. 2d–f show the micrographs of the NiO thin films at the scale of 100 nm. At 1 cm spray distance (NiH1T1), the deposition of NiO nanoparticles with spherical morphology and sizes approx. 6–21 nm was observed. The shape of the grains was maintained when the NSD and deposition time were changed (Fig. 2d–f). Fig. 2g–i show the size distributions of the nickel oxide nanoparticles; in the case of NiH1T1, the average particle size was ∼14.5 nm (Fig. 2g).
The average particle size increased with the NSD from 14.5 nm to 15.5 nm (Fig. 2g and h). When the deposition time was doubled, the average particle size increased from 15.5 to 19 nm. Furthermore, the particles tended to agglomerate faster when the deposition time was increased (Fig. 2i).
On the other hand, the SEM surface images of the CeO2 films deposited on glass substrates at two different NSDs and deposition times are shown in Fig. 3. At a 1 cm spray distance, the formation of plate-type hollow particles with a diameter of around 2–25 μm was observed (Fig. 3a); increasing the spray distance to 3.7 cm (Fig. 3b) led to the deposition of smaller particles than those obtained by spraying from a 1 cm distance. Unlike the short NSD, at 3.7 cm (longer spray distance), the particles were fully dried before they reached the substrate, causing some agglomeration, as observed in Fig. 3. Furthermore, at 30 min deposition time, the form of the droplets had changed slightly, which could be due to the amount of deposited material (Fig. 3c). At the scale of 100 nm, some pin holes and overgrown clusters were observed on the CeO2 film surface, as seen in Fig. 3d. Some cracks appeared in the deposited film, which may be caused by the strain of the thickened deposits at 3.7 cm NSD and 30 min deposition time (Fig. 3e and f).
 | ||
| Fig. 3 SEM images of (a and d) CeH1T1, (b and e) CeH2T1, and (c and f) CeH2T2. Scale bars: (a–c) 100 μm and (d–f) 100 nm. | ||
Fig. 4 displays the AFM results of the as-prepared films. With an increase in NSD, for both materials (Ni and Ce), the surface roughness decreased from 17.83 nm to 11.63 nm for NiH1T1 and NiH2T1, respectively, and from 24.70 nm to 19.36 nm for CeH1T1 and CeH2T1, respectively. At a 1 cm spray distance, the coated area is smaller, and the deposited nickel and cerium oxide films presented rougher surfaces because the sprayed solvent would not evaporate immediately from the substrate surface and generate aggregation. In summary, at a shorter NSD, a greater amount of solvent emerges from the nozzle and strikes the substrate, generating surfaces with high roughness. At 3.7 cm (NiH2T1 and CeH2T1), the droplets have more time to absorb thermal energy, and evaporation happens when they hit the substrate surface; as a result, the roughness surface is reduced. On increasing the spray time to 30 min, the roughness surface increased, reaching 43.63 nm and 30.42 nm in NiH2T2 and CeH2T2, respectively, due to an increase in the number of droplets, which generate more deposited material. These results are evidenced by the thickness of NiO and CeO2 on the substrate (Table 1).
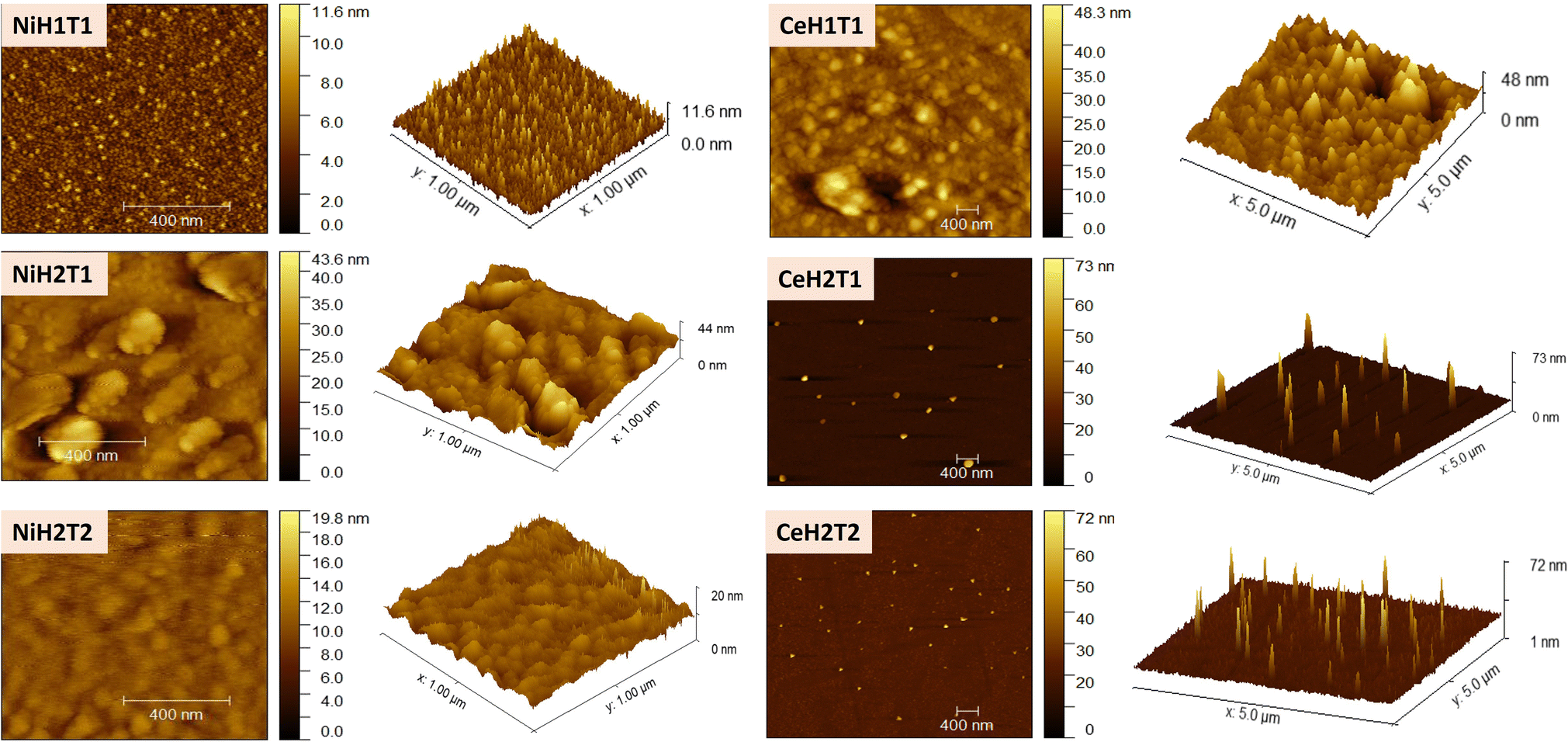 | ||
| Fig. 4 AFM images of the as-prepared NiO and CeO2 films. | ||
3.2. Thin film activity
The activity of the NiO and CeO2 thin films was examined by BA ozonation experiments conducted in aqueous solutions, and complete elimination of BA was obtained in all the studied systems at around 30 min (Fig. S1†). Almost no significant difference in BA degradation was observed among the six catalysts analyzed. Nevertheless, an important parameter that affects the catalytic activity is the mass of metallic oxide deposited on the glass plates. Thus, the thickness of the films was determined by an ellipsometer, and the deposited mass of NiO and CeO2 was calculated considering that all surfaces were homogeneous and well-covered with the metal oxides. Fig. 5a and c show the data of the eliminated BA mass normalized, respectively to the mass of NiO and CeO2 as a function of the ozonation time.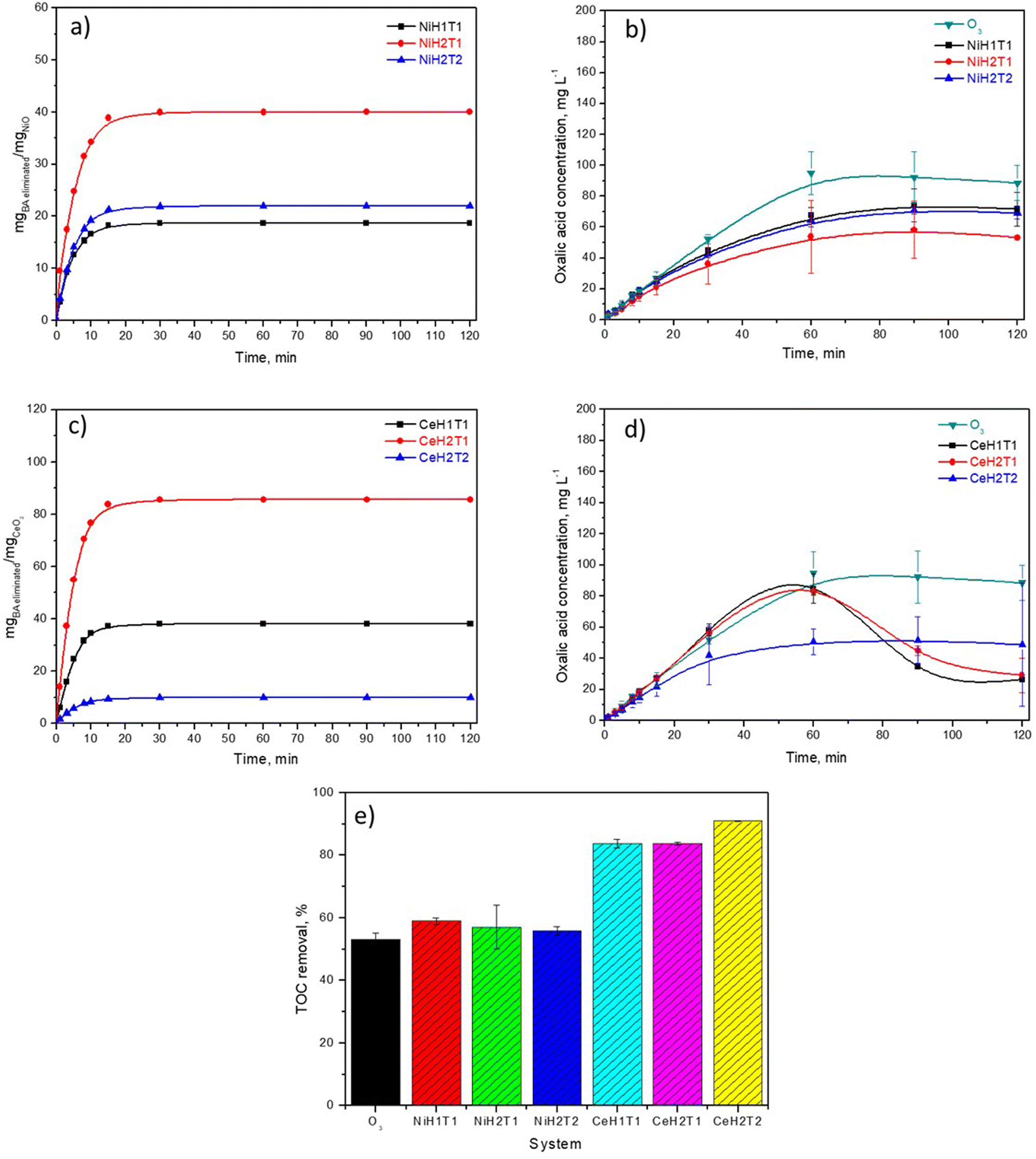 | ||
| Fig. 5 (a and c) The normalized curves of the amount of eliminated BA per mg of the catalyst and (b and d) the OA concentration profiles in the presence of (a and b) the NiO and (c and d) CeO2 thin films; (e) TOC removal by conventional and catalytic ozonation. | ||
The following trend was obtained for the elimination of BA per mg catalyst: CeH2T1 > CeH1T1 ∼ NiH2T1 > NiH2T2 > NiH1T1 > CeH2T2, which is in concordance with increasing film thickness (Table 1). Interestingly, the highest BA removal was obtained with the catalyst with minimal thickness; this could be due to ozone contact at a superficial level.
A byproduct generated during BA degradation was oxalic acid (OA), which is considered highly recalcitrant in conventional ozonation. Therefore, the concentration profiles of OA for as-prepared catalysts were obtained by HPLC (Fig. 5b and d). The following trend was obtained for OA removal at 120 min: CeH1T1 ∼ CeH2T1 (20 mg L−1) < CeH2T2 ∼ NiH2T1 (50 mg L−1) < NiH2T2 ∼ NiH1T1 (70 mg L−1) < O3 (90 mg L−1). The increased OA removal efficiencies (recalcitrant byproduct generated by the ozonation process) of the catalytic systems are attributed to the probable generation of ROS, such as ˙OH.
TOC removal in the catalytic ozonation system with CeO2 thin films reached approximately 90% and 84% for CeH2T2 and CeH1T1 ∼ CeH1T2, respectively (Fig. 5e). The NiO thin films exhibited ∼30% lower TOC than catalytic ozonation with CeO2 films. This demonstrates that the mineralization of BA was enhanced in the presence of the CeO2 thin films. According to the literature, the presence of large amounts of oxygen vacancies and Ce3+ favors the production of ROS, which attack several byproducts like oxalic acid and get highest mineralization level.34 Conventional ozonation of BA resulted in 53% TOC removal in 120 min, which is similar to the value reported by Z. Wang et al.35 X. Huang et al.36 demonstrated that some hydroxylated intermediates of BA improved its removal by conventional ozonation. These hydroxylated derivatives are more active in attacking ozone, resulting in an enhancement in ozone decomposition and yield of ROS.35
In general, the presence of NiO thin films did not cause a significant difference in BA removal compared with conventional ozonation, which is probably due to the similarity in the amount and type of ROS generated. The CeO2 thin films presented higher catalytic activity toward BA removal and its intermediates than the NiO films; therefore, CeH2T1 was chosen for subsequent experiments.
To understand the BA removal ability of the catalysts better, XPS characterization was applied to determine the chemical composition of fresh NiO and CeO2 films. Fig. 6 shows the high-resolution Ni 2p, Ce 3d and O 1s XPS regions calibrated to C 1s (284.5 eV). The Ni 2p high-resolution spectrum was deconvoluted into several peaks, as shown in Fig. 6a, and peak assignments are provided in Table S1.† The XPS fitting analysis of the Ni 2p region confirmed that the surface of the NiO film contained NiO, NiOOH and Ni(OH)2. The presence of these species of nickel is supported by the respective satellite signals. The O 1s regions of the NiO and CeO2 films could be fitted to three peaks, as shown in Fig. 6b and d. The first peak (OI) was assigned to lattice oxygen (O2−), the second oxygen (OII) peak corresponded with surface-adsorbed oxygen, and the third (OIII) was attributed to oxygen-containing groups, such as hydroxyl species. It was observed that the hydroxyl oxygen groups in the CeO2 film were greater in number than those in the NiO film, thus benefitting catalytic oxidation as these species are considered active oxidation sites. Probably, the presence of these groups is responsible for the higher activity of CeO2. The high-resolution Ce 3d spectrum of CeO2 was assigned to different peaks; the letters v and u are used to represent the Ce 3d3/2 and Ce 3d5/2 spin–orbit components, respectively (Fig. 6c). The spin–orbit peaks of v0, v′, v′′′ and u′ were assigned to Ce3+, whereas the rest of the components corresponded to Ce4+.37 This result indicates the coexistence of Ce3+ and Ce4+ in the CeO2 films; the energy positions of the peaks are summarized in Table S2.† The generation of ˙OH is possible due to the oxidation of Ce3+ by ozone (eqn (2) and (3)), and this species participates actively in the removal of BA and its by-products, eventually achieving mineralization (eqn (4)).
| Ce3+ + O3 + H+ → Ce4+ + HO3˙ | (2) |
| HO3˙ → ˙OH + O2 | (3) |
| ˙OH + organic compounds → CO2 + H2O | (4) |
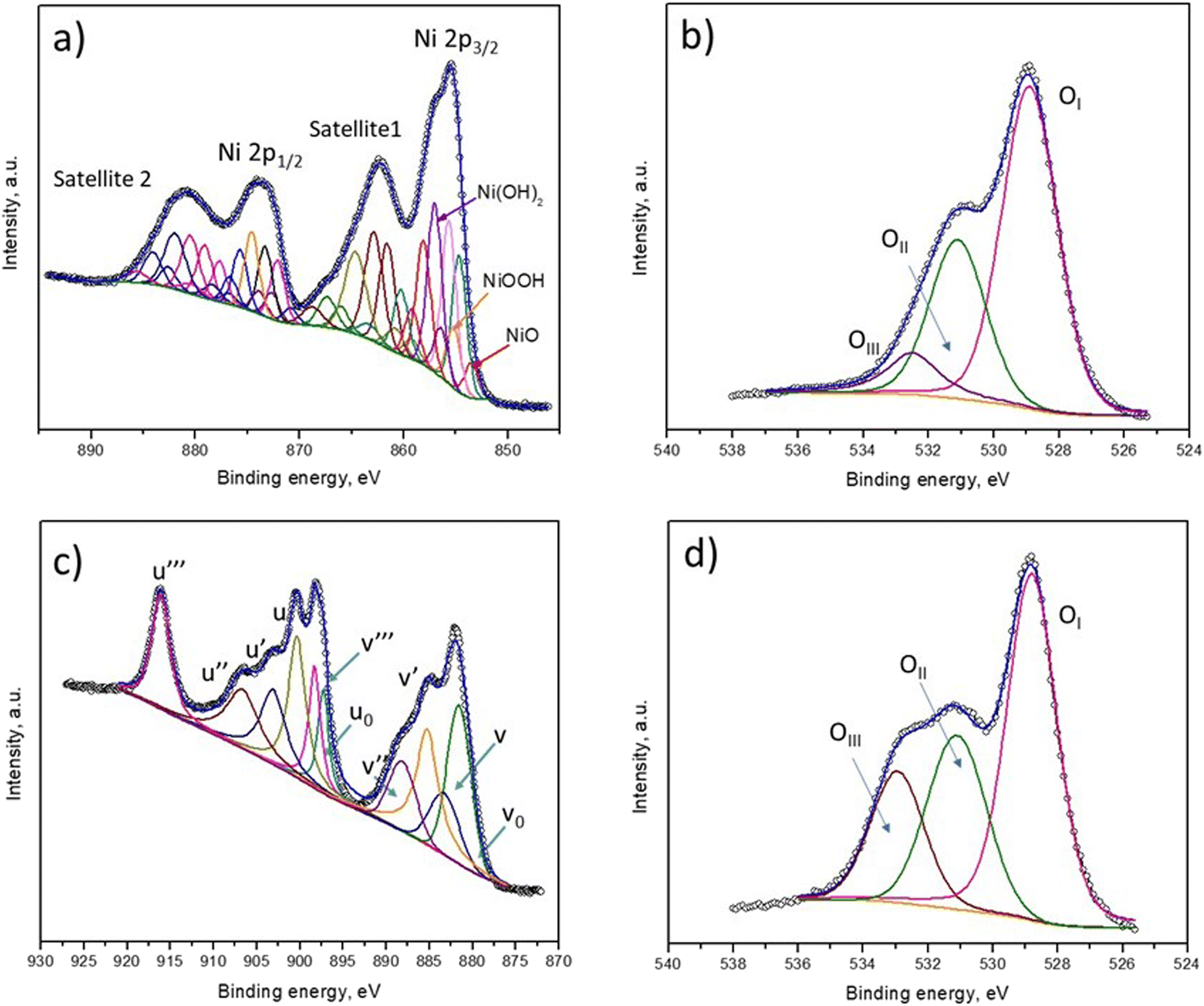 | ||
| Fig. 6 Deconvolution of the (a) Ni 2p, (b and d) O 1s and (c) Ce 3d XPS spectra of the (a and b) fresh NiO and (c and d) CeO2 thin films. | ||
3.3. Effect of the number of glass plates with CeH2T1 thin films on BA removal
The number of glass plates with thin films used in the catalytic system is an important parameter to evaluate the influence of catalyst concentration (thin film) on BA degradation because an increment in the number of active sites can favor the catalytic activity. According to the results, almost no discernible decrease in BA degradation was observed when the number of glass plates with CeO2 thin films in the system was increased (Fig. 7a). However, OA decomposition was favored in the presence of CeO2 thin films. Even the OA concentration profile changed; in the presence of one glass plate with CeO2 thin film, the profile showed accumulation of OA, and when the number of plates with thin films was greater than one, the OA profile indicated accumulation-decomposition at 120 min (Fig. 7b). This was evidenced by the TOC removal efficiency of six glass plates of CeO2 thin films reaching nearly 83%, which is 20%, 11% and 23% greater than those of conventional ozonation, catalytic ozonation in the presence of one and four plates with CeO2 thin films, respectively (Fig. 7c). This demonstrates that the mineralization degree of BA is enhanced by six glass plates with thin films of CeO2. | ||
| Fig. 7 Effect of the number of glass plates with CeO2 thin films (CeH2T1) on (a) BA removal, (b) OA concentration profiles, and (c) TOC removal; (d) the effect of various quenchers on the BA degradation efficiency of CeH2T1. | ||
To verify the contribution of ROS (O2˙−, ˙OH and 1O2) to BA degradation, quenchers, such as TBA, p-BQ and NaN3, were employed, as displayed in Fig. 7d. According to the literature, ˙OH, O2˙−, and 1O2 react with TBA, p-BQ and NaN3, respectively, generating inert intermediates.38,39 The presence of TBA during BA removal tended to drastically diminish (68%) the mineralization degree of the catalytic ozonation system. Therefore, it is proposed that ˙OH has a meaningful role in BA degradation. When p-BQ was added to the reaction, there was an inhibition of catalytic ozonation (61%), demonstrating that O2˙− also participated in BA removal. Importantly, p-BQ can react with ˙OH and O3 considering that its kO2˙− = 3.5–7.8 × 108 M−1 s−1, k˙OH = 1.2 × 109 M−1 s−1 and kO3 = 2.5 × 103 M−1 s−1. Hence, a high concentration of p-BQ (50 mM) was used in the system to exclude the influence of ˙OH and O3. In this sense, NaN3 has been commonly used to quench 1O2.40 Fig. 7d shows that in the presence of NaN3, TOC removal was significantly suppressed from 84.2% to 1.1% in the presence of CeH2T1. Therefore, the scavenger studies demonstrate that ˙OH, O2˙−, and 1O2 participate actively in BA removal.
3.4. Stability of the CeH2T1 thin films
The stability and catalytic activity of CeH2T1 during BA degradation are the other main parameters to be considered and were evaluated after 5 continuous runs. As shown in Fig. 8a, no significant increase or decrease in BA degradation efficiency was found during the five successive cycles. However, TOC removal diminished by approximately 20% after the fifth cycle in comparison with the first cycle (Fig. 8b). | ||
| Fig. 8 (a) BA degradation and (b) TOC removal by CeH2T1 at different cycles. | ||
Based on the results of these five successive cycles, the activity of the CeH2T1 thin film in the ozonation process diminished until the fourth cycle and reached a plateau. The reduction in the catalytic activity of the CeH2T1 thin film in this study can be associated with the adsorption of the intermediates generated during BA degradation or the leaching of metal ions after each cycle. Importantly, the leaching of metal ions might be much lower than that in the case of a powder catalyst because the amount of catalyst on the film surface is much less than the surface of the powder catalyst. To demonstrate the stability of the material, after the 5th BA degradation cycle, the CeH2T1 film was analyzed by XDR and SEM, as shown in Fig. 9(a–c). According to the XRD results (Fig. 9a), the crystal structure of the CeH2T1 film remained unchanged before and after the 5th cycle, and no extra diffraction peaks were detected due to the recycling of the thin film. Fig. 9(b and c) show that the thin film maintained its morphology after the BA degradation reaction, and the recycled film surface was exclusively smoother than the fresh film surface.
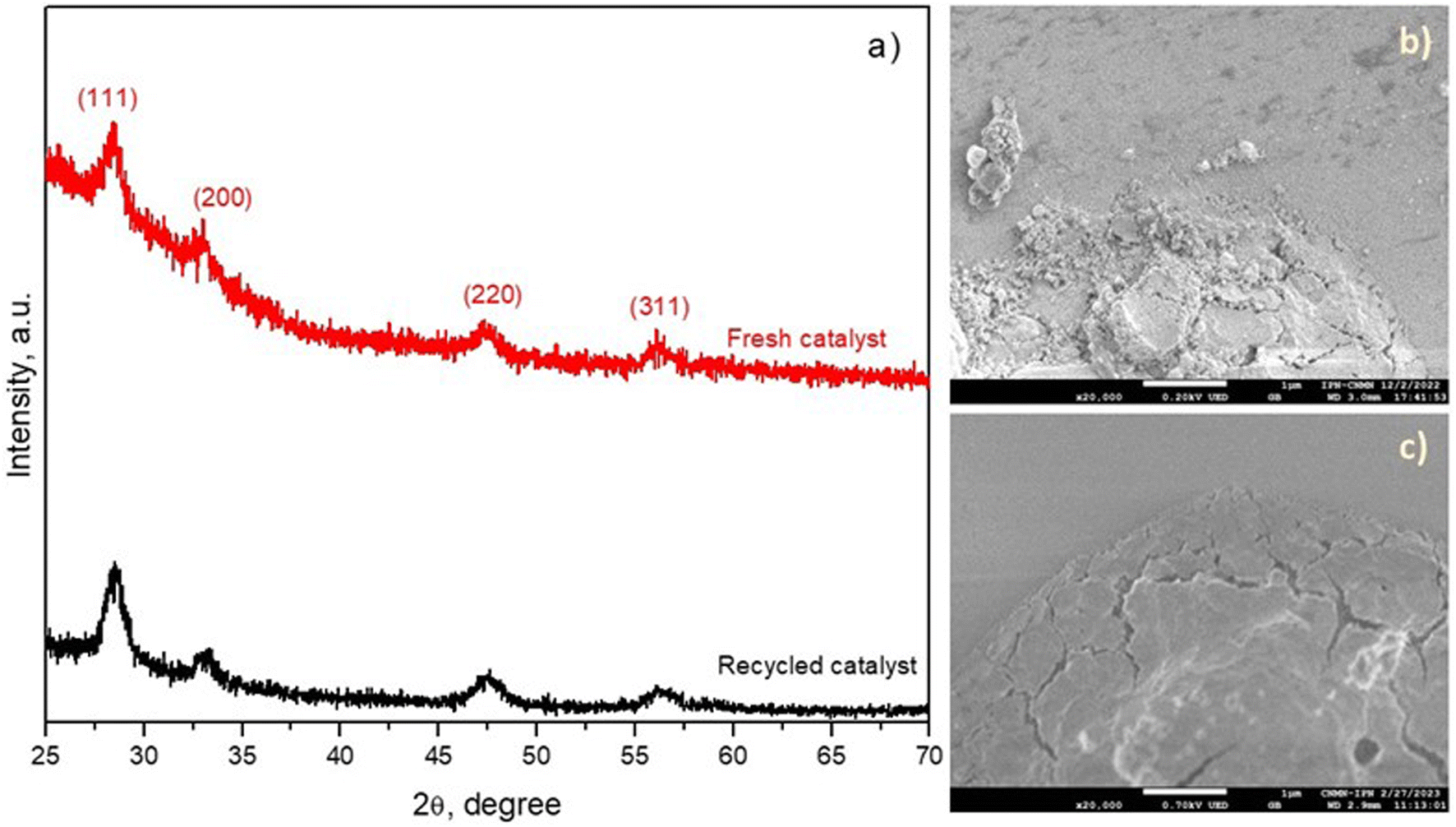 | ||
| Fig. 9 (a) XRD patterns and SEM images of the (b) fresh and (c) recycled CeH2T1 thin films (after the 5th run). | ||
Our results were compared with studies that have reported BA removal using several advanced oxidation processes. As shown in Table 2, the catalytic performance of the CeH2T1 thin film in this investigation is similar or even superior to those of other reported materials. Furthermore, it is worth highlighting that most of the research studies have used catalysts in the powder form; therefore, the present study provides guidance for the synthesis of thin films for applications in environmental remediation, specifically in water, using lower amounts of metal oxide than catalysts in the powder form. Furthermore, the recovery of films did not require additional separation methods, unlike catalysts in the powder form.
| Advanced oxidation process | Experimental conditions | Treatment efficiency | Ref. |
|---|---|---|---|
| a [BA] = initial concentration of BA; [catalyst] = catalyst concentration: [O3] ozone concentration; WE = working electrode; CE = counter electrode. | |||
| Catalytic ozonation | [BA] = 115 mg L−1 | TOC removal: CeH2T2 (90%) > CeH1T1 ∼ CeH2T1 (84%) > NiH1T1 (59%) > NiH2T1 (57%) > NiH2T2 (56%) at 120 min | This study |
| mNiO = 2.5 mg | |||
| mCeO2 = 1.2 mg | |||
| [O3] = 30 ± 0.2 mg L−1 | |||
| pH = 3.5 ± 0.1 | |||
| Fenton reaction | [BA] = 40.0 μM | BA removal 25.3% with the Fenton process at 20 min. In the presence of reducing agents, BA removal was 44%, 58% and 72% with the addition of cysteine, ascorbic acid, and hydroxylamine in the reaction medium, respectively | 48 |
| [FeSO4·7H2O] = 10.0 μM | |||
| [H2O2] = 0.4 mM | |||
| pH = 3.0 | |||
| [Hydroxylamine] = [cystine] = [ascorbic acid] = 0.1 mM | |||
| Electrochemical oxidation | [BA] = 10 mM | BA removal 40% at 90 min | 49 |
| [Na2SO4] = 0.4 M | |||
| [NaH2PO4] = [Na2HPO4] = 0.1 M | |||
| pH = 6 | |||
| Photocatalytic process | [BA] = 100 mg L−1 | BA elimination at 135 min: MgFe2O4/ZnO/GO (45%) > MgFe2O4/ZnO (43%) > ZnO (39%) > MgFe2O4 (36%) | 50 |
| [Catalysts] = 0.05 g L−1 | |||
| Sunlight | |||
| Catalytic ozonation | [BA] = 1 mM | 60% and 90% of TOC removal with O3 and O3-catalyst, respectively, at 90 min | 35 |
| [O3] = 38 mg L−1 | |||
| [Fe-shaving] = 50 g L−1 | |||
| pH = 6.8 | |||
| Ozonation | [BA] = 0.2, 5 and 25 μM | BA removal: 20% (0.2 μM) < 85% (5 μM) at 10 min with [O3] = 1 mg L−1 | 36 |
| [O3] = 1 and 5 mg L−1 | TOC removal: 10% using [O3] = 5 mg L−1 and [BPA] = 25 μM | ||
| pH = 2.3 ± 0.1 | |||
| Photocatalysis and photo-Fenton-like oxidation (PO/PFO) | [BA] = 50 mg L−1 | 95% of BA removal at 80 min | 51 |
| [Fe–TiO2/AC] = 0.5 g L−1 | |||
| [H2O2] = 2 mM | |||
| pH = 4 | |||
| UV light (254 nm) = 6 W | |||
| Photocatalysis (Ph) and photoelectrocatalysis (PE) | [BA] = 1 mM | BA removal: PE (65.7%) > Ph (18.3) at 400 min | 52 |
| WE [glass/FTO/ZnO] = 64 cm2 and CE = Pt | COD removal: 59.3 to 17.6 mg L−1 | ||
| Voltage = 1.5 V | |||
| [HClO4] = 10 mM | |||
| UV light | |||
During heterogeneous catalytic ozonation, ozone decomposition is believed to occur through its interactions with a catalyst to generate free radicals.9,41–43 According to experiments performed in the presence of scavengers in the reaction medium, the CeH2T1 films promote the formation of radicals, such as ˙OH and O2˙−, due to ozone decomposition. Considering the formation of oxidant species (O3, ˙OH, O2˙−, and 1O2) and the byproducts formed (identified by HPLC; Fig. S2†) during the catalytic ozonation process, a probable pathway of BA removal is proposed, as illustrated in Fig. S3.† BA removal is initiated by its rapid decarboxylation to form phenol. Subsequently, phenol oxidation occurs through the hydroxylation of the aromatic ring to generate catechol and hydroquinone,44 which decompose into maleic and fumaric acids via benzene ring cleavage. Further, their elimination generates oxalic and acetic acids.45,46 Finally, these are oxidized to CO2 and H2O. The proposed reaction pathway of benzoic acid removal coincides with that reported by Y. Xu et al.47 using ozone in the presence of vanadium oxide deposited on zeolite as the catalyst.
4. Conclusion
The ultrasonic spray pyrolysis technique was used to successfully produce NiO and CeO2 thin films. The XRD results show that the crystal structure of NiO and CeO2 was cubic without additional phases. The surface morphology of the NiO films reveals well-distributed nanoparticles on glass, while CeO2 films reveal irregular particle aggregation and some pin holes. The presence of a catalyst in the reaction medium did not change the concentration profile of BA removal in comparison with conventional ozonation. Therefore, in both processes, the complete elimination of BA was achieved at around 30 min. However, the combination of CeO2 films with ozone increased TOC removal to 84–90% by 120 min in comparison with conventional ozonation (53%) and ozonation catalyzed by NiO films (around 57%). This is probably due to the difference in the amount of ROS generated, such as ˙OH, O2˙−, and 1O2, and the formation of the hydroxyl oxygen groups on the CeO2 film. Therefore, the CeO2 film can be a promising catalyst for the elimination of recalcitrant pollutants from water using ozone as the oxidant. It also has the advantage of not requiring additional techniques for separation from the reaction medium.Author contributions
Daynahi Franco Peláez: conceptualization, methodology, formal analysis, investigation, data curation, visualization. Julia Liliana Rodríguez S.: conceptualization, methodology, formal analysis, supervision, resources, funding acquisition, project administration, visualization, writing – review & editing. Tatyana Poznyak: methodology, formal analysis, supervision. Hugo Martínez Gutiérrez: SEM analysis and investigation. J. Alberto Andraca Adame: DRX analysis and writing – review & editing. Luis Lartundo Rojas: XPS analysis and revision. Claudia Jazmín Ramos Torres: determination of thickness.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors acknowledge to Consejo Nacional de Ciencia y Tecnología, al proyecto aprobado por el Fondo Sectorial de Investigación para la educación (Project No. A1-S-30591), and Instituto Politécnico Nacional (Projects No. 20230946 and Innovation 20231138).References
- S. Wu, Z. Yang, Z. Zhou, X. Li, Y. Lin, J. J. Cheng and C. Yang, J. Hazard. Mater., 2023, 459, 132133 CrossRef CAS PubMed.
- S. Wu, L. Shen, Y. Lin, K. Yin and C. Yang, Chem. Eng. J., 2021, 414, 128872 CrossRef CAS.
- J. Xie, C. Yang, X. Li, S. Wu and Y. Lin, Chemosphere, 2023, 339, 139659 CrossRef CAS PubMed.
- C. V. Rekhate and J. K. Srivastava, Chem. Eng. J. Adv., 2020, 3, 100031 CrossRef CAS.
- T. D. Kusuma, M. S. V. Naga Jyothi, C. P. Rao and S. M. Maliyekkal, in Nanomaterials and Nanocomposites for Environmental Remediation, ed. S. P. Singh, K. Rathinam, T. Gupta and A. K. Agarwal, Springer Singapore, Singapore, 2021, pp. 275–305 Search PubMed.
- R. Andreozzi, V. Caprio, A. Insola and R. Marotta, Catal. Today, 1999, 53, 51–59 CrossRef CAS.
- W. Chen, H. He, J. Liang, X. Wei, X. Li, J. Wang and L. Li, J. Hazard. Mater., 2023, 443, 130302 CrossRef CAS PubMed.
- S. P. Ghuge and A. K. Saroha, J. Environ. Manage., 2018, 211, 83–102 CrossRef CAS PubMed.
- E. Issaka, J. N.-O. Amu-Darko, S. Yakubu, F. O. Fapohunda, N. Ali and M. Bilal, Chemosphere, 2022, 289, 133208 CrossRef CAS PubMed.
- Y. Wang, Y. Lin, S. He, S. Wu and C. Yang, J. Hazard. Mater., 2024, 461, 132538 CrossRef CAS PubMed.
- A. B. Khatibani, Indian J. Phys., 2021, 95, 243–252 CrossRef CAS.
- R. Prajesh, V. Goyal, M. Nahid, V. Saini, A. K. Singh, A. K. Sharma, J. Bhargava and A. Agarwal, Sens. Actuators, B, 2020, 318, 128166 CrossRef CAS.
- N. Y. Abu-Thabit and A. S. H. Makhlouf, in Advances in Smart Coatings and Thin Films for Future Industrial and Biomedical Engineering Applications, ed. A. S. H. Makhlouf and N. Y. Abu-Thabit, Elsevier, 2020, pp. 3–35 Search PubMed.
- S. R. Sriram, S. R. Parne, N. Pothukanuri and D. R. Edla, J. Anal. Appl. Pyrolysis, 2022, 164, 105527 CrossRef CAS.
- H. Das, N. Debnath, A. Toda, T. Kawaguchi, N. Sakamoto, S. Manjura Hoque, K. Shinozaki, H. Suzuki and N. Wakiya, Adv. Powder Technol., 2018, 29, 283–288 CrossRef CAS.
- S. Rahemi Ardekani, A. Sabour Rouh Aghdam, M. Nazari, A. Bayat, E. Yazdani and E. Saievar-Iranizad, J. Anal. Appl. Pyrolysis, 2019, 141, 104631 CrossRef CAS.
- A. S. Enigochitra, P. Perumal, C. Sanjeeviraja, D. Deivamani and M. Boomashri, Superlattices Microstruct., 2016, 90, 313–320 CrossRef CAS.
- S. Khot, S. Phalake, S. Mahadik, M. Baragale, S. Jagadale, V. Burungale, Y. Navale, V. Patil, V. Patil, P. Patil and S. Patil, Mater. Today: Proc., 2021, 43, 2694–2697 CAS.
- S. Jongthammanurak, M. Witana, T. Cheawkul and C. Thanachayanont, Mater. Sci. Semicond. Process., 2013, 16, 625–632 CrossRef CAS.
- G. Grincienė, M. Franckevičius, R. Kondrotas, R. Giraitis, R. Juškėnas, G. Niaura, A. Naujokaitis, J. Juodkazytė, L. Tamašauskaitė-Tamašiūnaitė and V. Pakštas, Semicond. Sci. Technol., 2018, 33, 095013 CrossRef.
- R. J. Deokate, A. V. Moholkar, G. L. Agawane, S. M. Pawar, J. H. Kim and K. Y. Rajpure, Appl. Surf. Sci., 2010, 256, 3522–3530 CrossRef CAS.
- R. Sharma, A. D. Acharya, S. B. Shrivastava, T. Shripathi and V. Ganesan, Optik, 2014, 125, 6751–6756 CrossRef CAS.
- C. M. Aguilar-Melo, J. L. Rodríguez, I. Chairez, I. Salgado, J. A. Andraca Adame, J. A. Galaviz-Pérez, J. Vazquez-Arenas and T. Poznyak, Catalysts, 2020, 10, 884 CrossRef CAS.
- I. C. Guzmán, J. L. Rodríguez, T. Poznyak, I. Chairez, I. Hernández and R. T. Hernández, Catal. Commun., 2020, 133, 105827 CrossRef.
- M. Stoyanova, P. Konova, P. Nikolov, A. Naydenov, S. Christoskova and D. Mehandjiev, Chem. Eng. J., 2006, 122, 41–46 CrossRef CAS.
- L. Wu, J. Wang, C. Yang, X. Gao, Y. Fang, X. Wang, W. D. Wu and Z. Wu, Appl. Catal., B, 2023, 323, 122152 CrossRef CAS.
- V. E. Sugihartono, N. N. N. Mahasti, Y.-J. Shih and Y.-H. Huang, Chemosphere, 2022, 296, 133663 CrossRef CAS PubMed.
- Z. He, M. D. Hayat, S. Huang, X. Wang and P. Cao, J. Electroanal. Chem., 2018, 812, 74–81 CrossRef CAS.
- M. P. Rayaroth, C.-S. Lee, U. K. Aravind, C. T. Aravindakumar and Y.-S. Chang, Chem. Eng. J., 2017, 315, 426–436 CrossRef CAS.
- A. Sennaoui, S. Alahiane, F. Sakr, M. Tamimi, E. H. Ait Addi, M. Hamdani and A. Assabbane, J. Environ. Chem. Eng., 2019, 7, 103033 CrossRef CAS.
- L. Xia, W. Liang, G. Chen, W. Li and M. Gao, Catal. Lett., 2022, 152, 1669–1677 CrossRef CAS.
- M. Messaoudi, M. S. Aida, N. Attaf, T. Bezzi, J. Bougdira and G. Medjahdi, Mater. Sci. Semicond. Process., 2014, 17, 38–42 CrossRef CAS.
- G. Kenanakis, N. Katsarakis and E. Koudoumas, Thin Solid Films, 2014, 555, 62–67 CrossRef CAS.
- C. A. Orge, J. J. M. Órfão, M. F. R. Pereira, A. M. Duarte de Farias, R. C. R. Neto and M. A. Fraga, Appl. Catal., B, 2011, 103, 190–199 CrossRef CAS.
- Z. Wang, X. Lin, Y. Huang and L. Ma, J. Hazard. Mater., 2022, 431, 128620 CrossRef CAS PubMed.
- X. Huang, X. Li, B. Pan, H. Li, Y. Zhang and B. Xie, Water Res., 2015, 73, 9–16 CrossRef CAS PubMed.
- J. Wang, X. Quan, S. Chen, H. Yu and G. Liu, J. Hazard. Mater., 2019, 368, 621–629 CrossRef CAS PubMed.
- H. Zhao, Y. Dong, P. Jiang, G. Wang, J. Zhang, K. Li and C. Feng, New J. Chem., 2014, 38, 1743–1750 RSC.
- J. Ma, S. Zhang, X. Duan, Y. Wang, D. Wu, J. Pang, X. Wang and S. Wang, Chemosphere, 2021, 267, 129287 CrossRef CAS PubMed.
- M. Xu, Y. Zhang, H. Yin, J. Wang, A. Li and P. F.-X. Corvini, Appl. Catal., B, 2023, 322, 122085 CrossRef CAS.
- B. Wang, H. Zhang, F. Wang, X. Xiong, K. Tian, Y. Sun and T. Yu, Catalysts, 2019, 9, 241 CrossRef.
- J. Gomes, R. Costa, R. M. Quinta-Ferreira and R. C. Martins, Sci. Total Environ., 2017, 586, 265–283 CrossRef CAS PubMed.
- B. Kasprzyk-Hordern, M. Ziółek and J. Nawrocki, Appl. Catal., B, 2003, 46, 639–669 CrossRef CAS.
- W. Xiong, N. Chen, C. Feng, Y. Liu, N. Ma, J. Deng, L. Xing and Y. Gao, Environ. Sci. Pollut. Res., 2019, 26, 21022–21033 CrossRef CAS PubMed.
- J. A. Zazo, J. A. Casas, A. F. Mohedano, M. A. Gilarranz and J. J. Rodríguez, Environ. Sci. Technol., 2005, 39, 9295–9302 CrossRef CAS PubMed.
- M. K. Ramseier and U. v. Gunten, Ozone: Sci. Eng., 2009, 31, 201–215 CrossRef CAS.
- Y. Xu, Q. Wang, B. A. Yoza, Q. X. Li, Y. Kou, Y. Tang, H. Ye, Y. Li and C. Chen, Front. Chem., 2019, 7, 384 CrossRef CAS PubMed.
- D.-Q. He, Y.-J. Zhang, D.-N. Pei, G.-X. Huang, C. Liu, J. Li and H.-Q. Yu, J. Hazard. Mater., 2020, 382, 121090 CrossRef CAS PubMed.
- A. Arts, K. P. van den Berg, M. T. de Groot and J. van der Schaaf, Curr. Res. Green Sustainable Chem., 2021, 4, 100217 CrossRef CAS.
- J. Arshad, F. M. A. Alzahrani, S. Munir, U. Younis, M. S. Al-Buriahi, Z. A. Alrowaili and M. F. Warsi, Ceram. Int., 2023, 49, 18988–19002 CrossRef CAS.
- G. Tekin, G. Ersöz and S. Atalay, J. Environ. Chem. Eng., 2018, 6, 1745–1759 CrossRef CAS.
- R. D. Suryavanshi, S. V. Mohite, A. A. Bagade, S. K. Shaikh, J. B. Thorat and K. Y. Rajpure, Mater. Res. Bull., 2018, 101, 324–333 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07316e |
| This journal is © The Royal Society of Chemistry 2024 |