
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of (±)-auranthine confirmed its refined structure†
Marie Reinkera,
Svetlana A. Kalinina *b and
Dmitrii V. Kalinin*a
*b and
Dmitrii V. Kalinin*a
aInstitute of Pharmaceutical and Medicinal Chemistry, University of Münster, Corrensstraße 48, Münster 48149, Germany. E-mail: dmitrii.kalinin@uni-muenster.de
bInstitute of Food Chemistry, University of Münster, Corrensstraße 45, Münster 48149, Germany. E-mail: s_kali03@uni-muenster.de
First published on 4th January 2024
Abstract
Auranthine, isolated in 1986 from Penicillium aurantiogriseum, is a fungal benzodiazepine. Through the successful total synthesis of (±)-auranthine, we confirmed the refined structure of natural (−)-auranthine. We established that natural (−)-auranthine is a fused quinazolino benzodiazepine dione 1 featuring an acyclic aliphatic nitrile moiety, thereby disproving the proposed structure 2.
Introduction
Auranthine (1) is a fungal benzodiazepine first isolated in 1986 from a plant pathogen Penicillium aurantiogriseum.1 The analysis of the 1H-NMR, 13C-NMR, and mass spectra led to the proposal of the initial structure of auranthine as structure 2 (Fig. 1).1 No absolute configuration of auranthine 2 has been reported at this point. It was suggested that auranthine 2 is naturally produced by the fungus in a condensation reaction between two molecules of anthranilic acid and a molecule of glutamine.2 Since benzodiazepines are known to have a diverse spectrum of biological activities and are often used as therapeutic agents,3 auranthine, a newly isolated benzodiazepine with a rather unusual structure 2, has attracted the attention of synthetic chemists.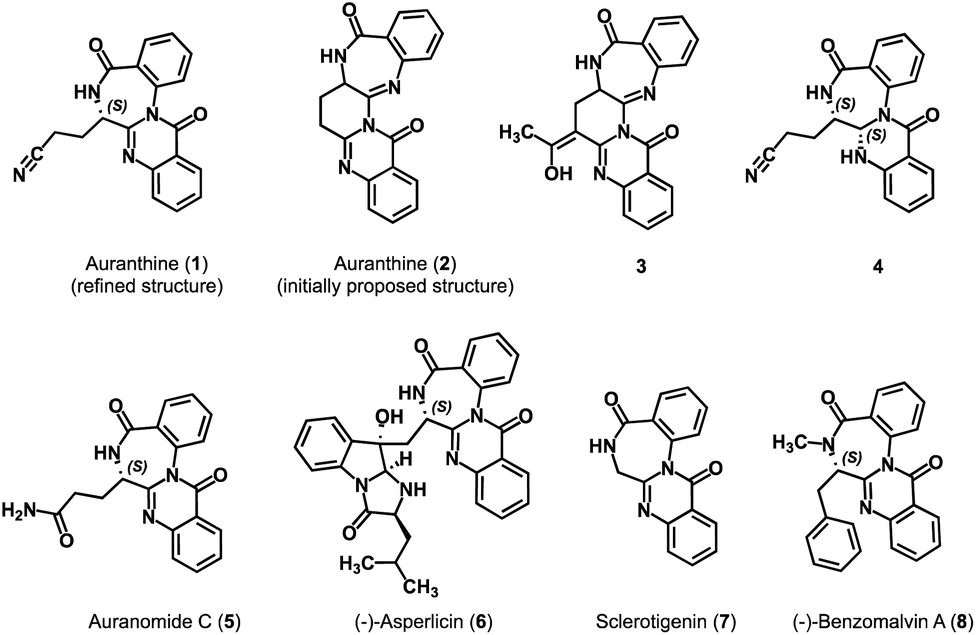 | ||
| Fig. 1 Refined structure of auranthine (1), initially proposed structure of auranthine (2), (semi)synthetic derivatives of auranthine 3 and 4 as well as structurally related natural products 5–8. | ||
In 2002, the research group of Bergman attempted the synthesis of auranthine 2.4 Despite not achieving the desired auranthine 2, the team successfully synthesized compound 3, a structurally relevant C-acetyl derivative of auranthine 2.4 Later, in 2010, a group led by Argade, starting from Cbz-protected glutamic anhydride and Boc-protected o-aminobenzyl amine, successfully accomplished the first total synthesis of proposed racemic auranthine 2 over seven synthetic steps.5 The authors achieved explicit confirmation of the structure of the synthesized racemic auranthine 2 through a combination of NMR analysis and X-ray diffraction. Nevertheless, the 1H-NMR spectrum of the synthesized compound failed to match the spectral profile of the natural auranthine isolated back in 1986.5 This observation raised significant doubts regarding the accuracy of the initially proposed structure of auranthine (2).
To verify this, in 2018, we chemically modified natural auranthine to obtain its reduced derivative 4 (Fig. 1).6 The chemical modification of auranthine was required to obtain a derivative capable of producing X-ray-diffractable crystals as natural auranthine failed to give suitable crystals. The X-ray crystallography data of semi-synthetic derivative 4 revealed that its structure significantly differs from the originally proposed structure of auranthine 2. Based on the confirmed structure of 4 and subsequently performed detailed analysis of natural auranthine using NMR, mass, and CD spectroscopy as well as DFT calculations, we refined the original structure of auranthine 2 and suggested that auranthine is a fused quinazolino benzodiazepinedione 1 bearing an acyclic aliphatic nitrile moiety (Fig. 1).6 The refined auranthine (1) shares structural similarity with other known fungal secondary metabolites such as auranomide C (5),7 asperlicin (6),8,9 sclerotigenin (7),10 and benzomalvin A (8)11 (Fig. 1), total synthesis of which has also been reported.12–15
Although we have presented substantial evidence proving the proposed structure of auranthine (1), it is important to acknowledge that these findings, despite their strength, do not provide absolute certainty. Of all the spectroscopic data at our disposal, the X-ray crystal structure of derivative 4 emerged as the most convincing and definitive piece of evidence supporting the refined structure of auranthine (1). Nevertheless, compound 4 was obtained by the treatment of natural auranthine with a reducing agent NaBH3CN comprising a nitrile moiety. Thus, there is a small possibility that the nitrile group in derivative 4 may have been derived from the reducing agent rather than intrinsic to the original structure of auranthine (1). To unambiguously establish the accuracy of the refined structure of auranthine (1), it was necessary to perform its total synthesis. We herein report the total synthesis of racemic auranthine (1).
Results and discussion
To confirm the refined structure of auranthine (1), its total synthesis should be performed. Since auranthine (1) and another fungal metabolite auranomide C (5) differ only in the structure of the aliphatic side chain (amide moiety vs. nitrile moiety, Fig. 1), we decided that auranthine (1) could be synthetically accessed via a dehydration of the amide moiety of auranomide C (5). For this purpose, at first, auranomide C (5) is to be synthesized following one of the suggested routes A or B, both starting from a common precursor – isatoic anhydride (9) (Scheme 1).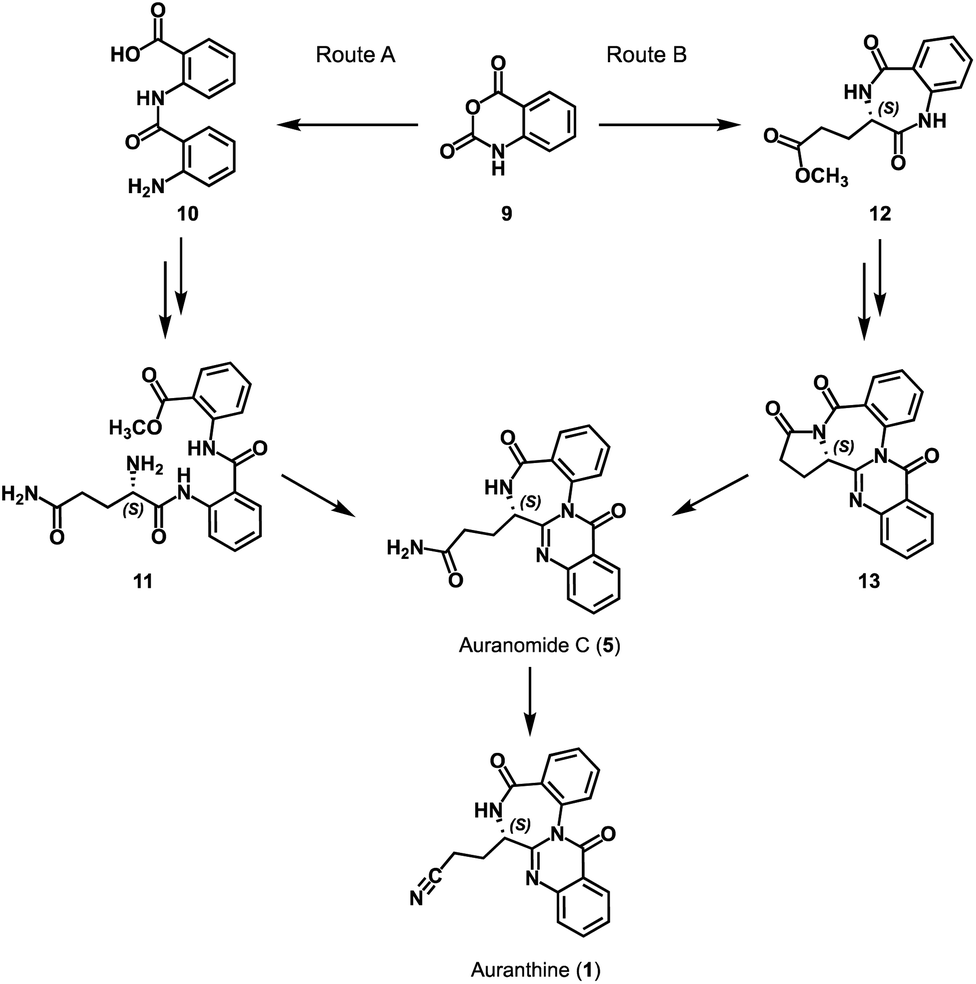 | ||
| Scheme 1 Planned synthetic routes (A and B) towards auranthine (1). | ||
Rote A is inspired by the reported synthesis of structurally related quinazolino[3,2-a]benzodiazepinediones including natural products sclerotigenin (7) and asperlicin C but not auranomide C (5).13 In this synthetic route, isatoic anhydride (9) is converted into amide 10 consisting of two anthranilic acid moieties, which then is transformed into the anthranilate-containing tripeptide 11, subsequent double dehydrocyclization of which should yield auranomide C (5) (Scheme 1).13 Alternatively, auranomide C (5) can be accessed following synthetic route B. Here, key intermediate benzodiazepine 12 over several steps is converted into quinazolinobenzodiazepine 13, followed by its lactam ring opening to afford auranomide C (5) as previously reported.14
At first, we attempted to obtain auranthine (1) via auranomide C (5) following synthetic rout A (Scheme 2). For this, isatoic anhydride (9) was refluxed with anthranilic acid in water to afford bis(anthranilic acid) 10, which was then esterified yielding corresponding methyl ester 14.13 Subsequently performed amide coupling between 14 and N-Cbz-L-glutamine afforded anthranilate-containing tripeptide 15 in 62% yield. The hydrogenolytic Cbz-group removal from 15 allowed for almost quantitative formation of the key intermediate 11 possessing the primary amino group (Scheme 2). It was anticipated that by applying the literature-described procedure,13 intermediate 11 should undergo two intramolecular cyclocondensation reactions forming the 4-quinazolinone scaffold fused with the 1,4-benzodiazepine moiety to afford auranomide C (5). This microwave-assisted reaction was shown to be promoted by metal triflates, which being Lewis acids presumably increase the electrophilicity of the carbonyl C-atoms involved in the corresponding cyclization steps.13 Nevertheless, despite our numerous efforts, under conditions identical to those previously described (microwave irrad. at 140 °C in DMF with Sn(OTf)2),13 compound 11 failed to produce auranomide C (5). Additional efforts to optimize the reaction, such as changing the reaction time, temperature, solvent and Lewis acid, also proved to be fruitless. Presumably, the reaction was incompatible with substrate 11 comprising an additional amide moiety, which in the presence of Lewis acids underwent side intramolecular reactions.
 | ||
Scheme 2 Attempted synthesis of auranthine (1): (a) H2O, 2 h, reflux, 76%; (b) CH3OH, H2SO4, 96 h, reflux, 69%; (c) DCM, EDCI, DMAP, r.t., 18 h, 62%; (d) THF/CH3OH (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), Pd(OH)2/C, H2, r.t., 3 h, 95%; (e) DMF, Sn(OTf)2, μ-wave (30 W), 140 °C, 15 min. :1), Pd(OH)2/C, H2, r.t., 3 h, 95%; (e) DMF, Sn(OTf)2, μ-wave (30 W), 140 °C, 15 min. | ||
Next, we attempted the synthesis of auranomide C (5) following previously reported route B (Scheme 3).14 For this, isatoic anhydride (9) was reacted with dimethyl L-glutamate to undergo two sequential addition–elimination reactions affording the cyclization product 12 exhibiting a 1,4-benzodiazepine-2,5-dione scaffold. It has been reported that derivatives of compound 12 exhibiting a flexible side chain are prone to racemization under reaction conditions used for the 1,4-benzodiazepine-2,5-dione scaffold annulation, making them problematic substrates for enantioselective synthesis. In contrast, under otherwise similar reaction conditions, tricyclic benzodiazepines like compound 16 are reported to retain their chiral information.14,16 Therefore, the synthesis was continued in the direction of the tricyclic intermediate 16 reported earlier,14 which in our hands, however, was obtained as a racemic mixture.
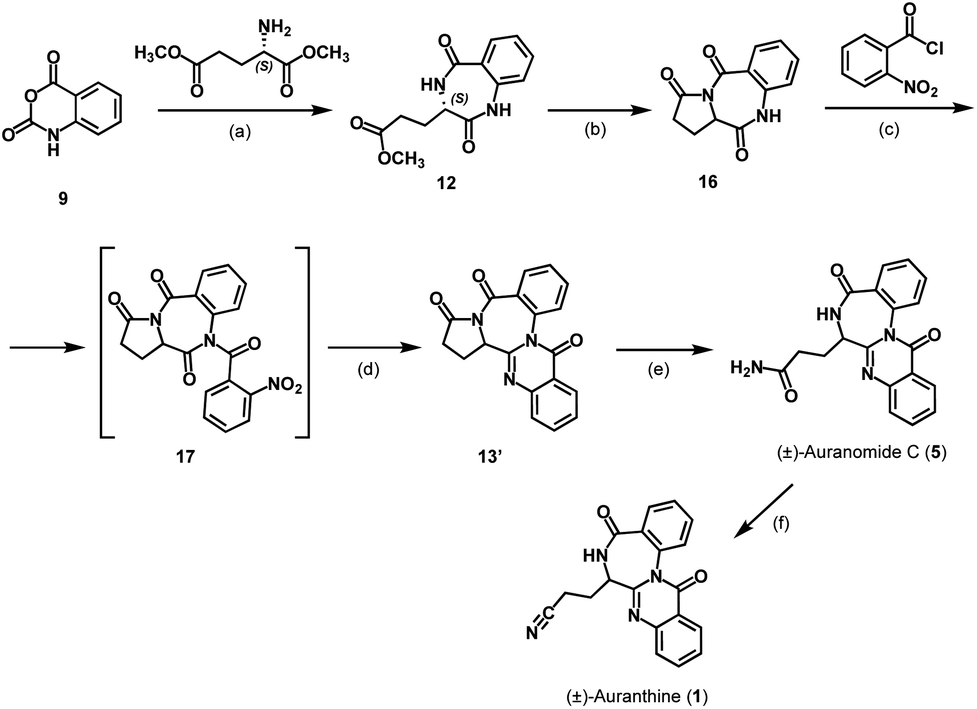 | ||
| Scheme 3 Synthesis of racemic auranthine (1): (a) pyridine, 120 °C, 16 h, 17%; (b) DMA, 180 °C, 20 h, 61%; (c) DCM, DMAP, Et3N, 0 °C, 0.5 h; (d) DCM, Zn, AcOH, −20 °C to r.t., 3 h, 25% over two steps; (e) THF-NH3 (0.5 M), 50 °C, 18 h, 76%; (f) DMF, pyridine, TsCl, r.t., 24 h, 34%. | ||
This defined the subsequent intermediates and the final product as racemates and the synthesis as non-stereospecific (Scheme 3). While this was somewhat disappointing, it wasn't a critical setback because the primary aim of this synthesis was to verify the structure of the refined auranthine (1), rather than to establish a new synthetic pathway. The physicochemical properties of synthetic (±)-auranthine, such as 1H-NMR, 13C-NMR, and IR spectra, are expected to be indistinguishable from those of natural (−)-auranthine. The alignment of these spectral characteristics will ultimately allow to confirm the structure of natural (−)-auranthine. We, therefore, proceeded with the racemate of 16. In the next synthetic steps, tricyclic intermediate 16 was acylated with 2-nitrobenzoyl chloride to afford 17, the nitro group of which was then reduced to facilitate further intramolecular cyclization reaction yielding quinazolinobenzodiazepine 13′. The lactam ring opening of 13′ with ammonia yielded (±)-auranomide C (5) as reported previously.14 Finally, (±)-auranomide C (5) was treated with p-toluenesulfonyl chloride in a DMF/pyridine mixture to dehydrate its primary amide group and produce desired (±)-auranthine (1) in 34% yield with 95% purity. To our knowledge, this reaction has not been documented in the literature for auranomide C (5).
Now, having synthetic (±)-auranthine in hands, we recorded its spectral characteristics to compare them with a sample of natural (−)-auranthine isolated from Penicillium aurantiogriseum as we reported previously.6 Recorded 1H-NMR spectra (DMSO-d6) of synthetic (upper spectrum) and natural auranthine (lower spectrum) appeared to be nearly identical (Fig. 2). Both 1H-NMR spectra feature characteristic proton signals of structure 1, which include one amide proton at 8.89 ppm, eight aromatic protons between 7.58 and 8.20 ppm, one methine proton at the asymmetric C-atom (4.25 ppm), and three signals of two methylene groups of propanenitrile moiety at 2.70 ppm, 2.44 ppm, and 2.29 ppm (Fig. 2).
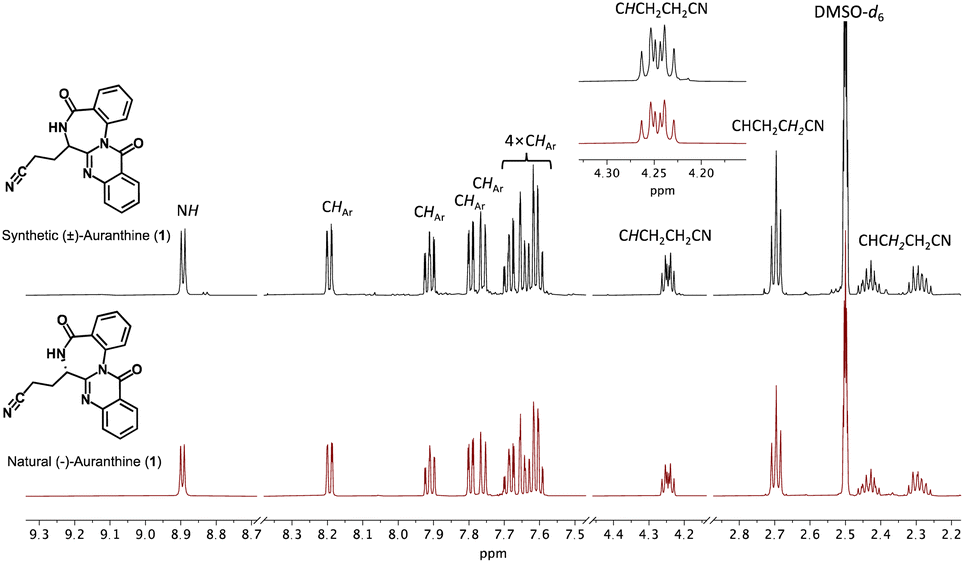 | ||
| Fig. 2 Comparison of 1H-NMR spectra in DMSO-d6 of synthetic (upper spectrum, black) and natural auranthine (lower spectrum, red). The fragment of 1H-NMR spectra with assigned proton signals is given. | ||
Similarly, the 13C-NMR spectra (DMSO-d6) of synthetic and natural auranthine appeared to be identical featuring characteristic signals, among which the C-atom of the –CN moiety appeared in both spectra at 120.1 ppm (Fig. 3). Also, the IR spectra of synthetic and natural auranthine matched exhibiting, for instance, the absorption band of the –CN moiety at 2249.00 cm−1 in both spectra (ESI†). Considering the identical spectral characteristics of synthetic and natural auranthine, we concluded that the synthesis of racemic auranthine was successful, and, more importantly, that the structure of natural auranthine (1) was correctly refined by us previously.6
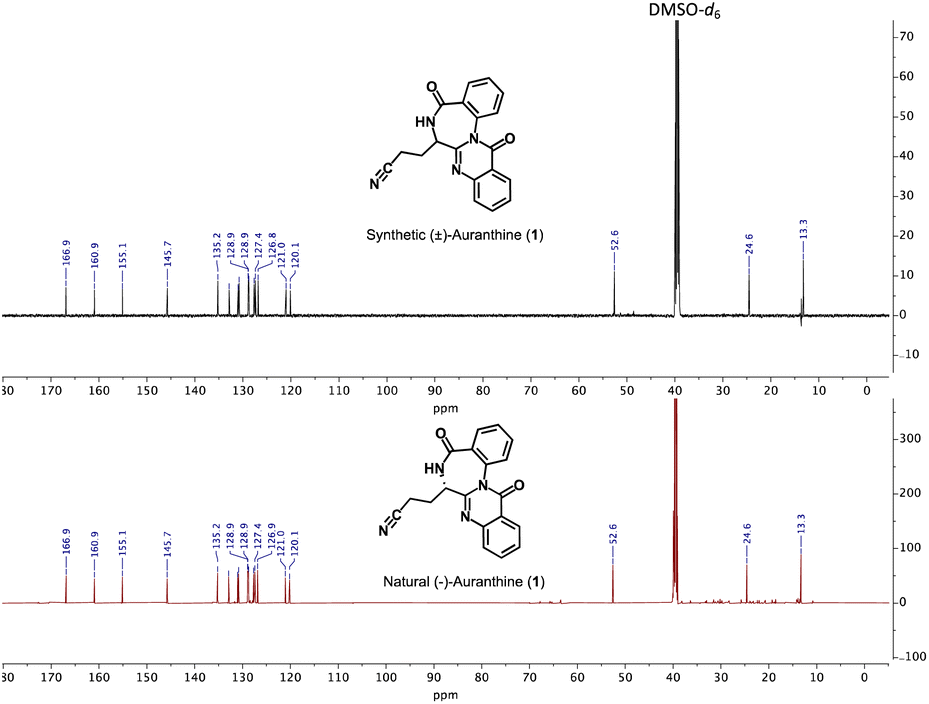 | ||
| Fig. 3 Comparison of 13C-NMR spectra in DMSO-d6 of synthetic (upper spectrum, black) and natural auranthine (lower spectrum, red). | ||
Conclusions
In summary, we successfully accomplished the total synthesis of (±)-auranthine 1, thus enabling us to unambiguously confirm the refined structure of natural (−)-auranthine isolated from Penicillium aurantiogriseum. It is now established that natural (−)-auranthine is indeed a fused quinazolino benzodiazepinedione 1 with an acyclic aliphatic nitrile moiety, dispelling the previously proposed structure (compound 2) from 1986. This finding allows for further investigations into the biosynthetic pathway of auranthine. Additionally, the reported synthesis will facilitate the preparation of synthetic analogs of auranthine for the purpose of studying their biological activity.Experimental
General experimental procedures
THF was dried with sodium/benzophenone and was freshly distilled before use. Thin layer chromatography (TLC): silica gel 60 F254 plates (Merck). Flash chromatography (FC): silica gel 60, 40–63 μm (Macherey-Nagel). Automatic flash column chromatography: Isolera One (Biotage); brackets include eluent, cartridge-type. Melting point (m.p.): melting point apparatus SMP 3 (Stuart Scientific), uncorrected. 1H NMR (400 MHz), 1H NMR (600 MHz), and 13C NMR (151 MHz): Agilent DD2 400 and 600 MHz spectrometers; chemical shifts (δ) are reported in ppm against the reference substance TMS and calculated using the solvent residual peak of the undeuterated solvent. IR: IR Prestige-21 (Shimadzu). HRMS: MicrOTOF-QII (Bruker). The microwave irradiation experiments: single-mode cavity system CEM Discover LabMate (CEM Corporation, NC). HPLC method to determine the purity of compounds: equipment 1: pump: L-7100, degasser: L-7614, autosampler: L-7200, UV detector: L-7400, interface: D-7000, data transfer: D-line, data acquisition: HSMS software (all from LaChrom, Merck Hitachi); equipment 2: pump: LPG-3400SD, degasser: DG-1210, autosampler: ACC-3000T, UV detector: VWD-3400RS, interface: Dionex UltiMate 3000, data acquisition: Chromeleon 7 (Thermo Fisher Scientific); column: LiChrospher 60 RP-select B (5 μm), LiChroCART 250–4 mm cartridge; flow rate: 1.0 mL min−1; injection volume: 5.0 μL; detection at λ = 210 nm; solvents: (A) demineralized water with 0.05% (v/v) trifluoroacetic acid, (B) acetonitrile with 0.05% (v/v) trifluoroacetic acid; gradient elution (% A): 0–4 min: 90%; 4–29 min: gradient from 90 to 0%; 29–31 min: 0%; 31–31.5 min: gradient from 0 to 90%; 31.5–40 min: 90%.2-(2-Aminobenzamido)benzoic acid (10)
Synthesis was performed as previously reported.13 Isatoic anhydride (2.51 g, 15.4 mmol) and anthranilic acid (2.30 g, 16.8 mmol) were dissolved in H2O (50 mL) and stirred under reflux for 2 h. After cooling to r.t., the precipitate was filtered off, washed with H2O und dried under reduced pressure. Product 10 was obtained as an off white solid (2.98 g, 11.6 mmol, 76%). M.p.: 203 °C. TLC: Rf = 0.66 (EtOAc). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 11.92 (s, 1H), 8.65 (dd, J = 8.5/1.2 Hz, 1H), 8.04 (dd, J = 7.9/1.7 Hz, 1H), 7.63 (ddd, J = 8.8/7.3/1.7 Hz, 1H), 7.60 (dd, J = 8.1/1.5 Hz, 1H), 7.24 (ddd, J = 8.4/7.0/1.5 Hz, 1H), 7.17 (ddd, J = 8.2/7.4/1.2 Hz, 1H), 6.80 (dd, J = 8.4/1.2 Hz, 1H), 6.62 (ddd, J = 8.1/7.0/1.2 Hz, 1H). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 170.0, 167.4, 150.6, 141.4, 134.2, 132.8, 131.2, 127.3, 122.5, 119.9, 117.1, 116.3, 115.2, 114.0. IR (neat): ṽ [cm−1] = 1600, 1649, 2980, 3356, 3468. HRMS (APCI): m/z = 257.0906, calcd 257.0921 for C14H13N2O3 [M + H]+. HPLC: tR = 16.55 min; purity: 95.3%.Methyl 2-(2-aminobenzoylamino)benzoate (14)
Synthesis was performed as previously reported.13 Compound 10 (2.82 g, 11.00 mmol) was dissolved in MeOH (50 mL), conc. sulfuric acid (3.20 mL) was added, and the reaction mixture was stirred at reflux for 96 h. The solvent was removed, the residue was dissolved in H2O (20 mL), the pH was adjusted to 8 using 3 M NaOH upon cooling. The resulting mixture was extracted with DCM, the combined organic layers were dried over Na2SO4, and the solvent was removed in vacuo. After purification with flash column chromatography (cyclohexane/EtOAc = 100/0 → 50/50), product 14 was obtained as an off-white solid (2.04 g, 7.55 mmol, 69%). M.p.: 114 °C. TLC: Rf = 0.33 (cyclohexane/EtOAc = 9/1). 1H-NMR (400 MHz, DMSO-d6): δ (ppm) = 11.39 (s, 1H), 8.52 (dd, J = 8.5/1.2 Hz, 1H), 8.00 (dd, J = 8.0/1.6 Hz, 1H), 7.68–7.64 (m, 1H) 7.64–7.60 (m, 1H), 7.25 (ddd, J = 8.1/6.9/1.3 Hz, 1H), 7.23–7.18 (m, 1H), 6.80 (dd, J = 8.3/1.2 Hz, 1H), 6.65 (ddd, J = 8.1/7.0/1.2 Hz, 1H), 6.56 (s, 2H), 3.88 (s, 3H). 13C-NMR (101 MHz, DMSO-d6): δ (ppm) = 168.1, 167.4, 150.5, 140.6, 134.2, 132.8, 130.7, 127.4, 122.9, 120.7, 117.1, 116.8, 115.2, 113.9, 52.6. IR (neat): ṽ [cm−1] = 1655, 1682, 3337, 3474. HRMS (APCI): m/z = 271.1077, calcd 271.1064 for C15H15N2O3 [M + H]+. HPLC: tR = 19.58 min; purity: 98.2%.Methyl (S)-2-(2-(5-amino-2-(((benzyloxy)carbonyl)amino)-5-oxopentanamido)benzamido)benzoate (15)
To a solution of 14 (3.51 g, 13.0 mmol) in DCM (50 mL), DMAP (3.17 g, 26 mmol), EDCI (4.98 g, 26 mmol), and N-carbobenzoxy-L-glutamine (5.46 g, 19.5 mmol) was added. The reaction mixture was stirred at r.t. under N2-atmosphere for 18 h. The resulting precipitate was filtrated off and washed with 1 M HCl and H2O and dried under reduced pressure. The filtrate was extracted (3×) with DCM and 1 M HCl, the combined organic layers were dried over Na2SO4, and the solvent evaporated in vacuo. The residue was purified twice by flash chromatography (cyclohexane/EtOAc = 50/50 → 0/100 and DCM/MeOH = 70/30) and combined with the solids. The product 15 was obtained as a white solid (4.28 g, 8.0 mmol, 62%). M.p.: 207 °C. TLC: Rf = 0.56 (EtOAc). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 11.41 (s, 1H), 11.25 (s, 1H), 8.41 (dd, J = 8.4/1.2 Hz, 1H), 8.35 (dd, J = 8.4/1.2 Hz, 1H), 8.00 (dd, J = 7.9/1.6 Hz, 1H), 7.96 (d, J = 7.0 Hz, 1H), 7.90 (dd, J = 8.0/1.6 Hz, 1H), 7.61 (ddd, J = 8.7/7.3/1.5 Hz, 1H), 7.54 (ddd, J = 8.6/7.4/1.7 Hz, 1H), 7.38–7.28 (m, 6H), 7.28–7.24 (m, 2H), 6.76 (s, 1H), 5.01 (d, J = 12.5 Hz, 1H), 4.93 (d, J = 12.5 Hz, 1H), 4.02 (ddd, J = 9.7/7.0/4.7 Hz, 1H), 3.86 (s, 3H), 2.23–2.14 (m, 2H), 2.12–2.02 (m, 1H), 1.86–1.71 (m, 1H). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 173.5, 171.0, 167.7, 166.7, 156.2, 139.3, 138.4, 136.7, 133.9, 132.7, 130.7, 128.3 (2C), 127.82, 127.77, 127.7 (2C), 124.0, 123.6, 122.3, 121.9, 121.2, 118.6, 65.8, 56.0, 52.6, 31.4, 26.7. IR (neat): ṽ [cm−1] = 1657, 1695, 3289, 3412. HRMS (APCI): m/z = 533.2042, calcd 533.2031 for C28H29N4O7 [M + H]+. HPLC: tR = 19.88 min; purity: 95.3%.Methyl (S)-2-(2-(2,5-diamino-5-oxopentanamido)benzamido)benzoate (11)
Compound 15 (1.00 g, 1.88 mmol) and Pd(OH)2/C (0.15 g, 15 wt%) were dissolved in THF/MeOH (4/1, 50 mL) under N2-atmosphere. H2-atmosphere was applied, and the reaction mixture stirred for 3 h at r.t. The solution was then filtered through Celite washing with MeOH. The filtrate was evaporated under reduced pressure to obtain product 11 as a white solid (0.71 g, 1.79 mmol, 95%). M.p.: 98–99 °C. TLC: Rf = 0.36 (DCM/MeOH = 9.5/0.5). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 11.35 (s, 1H), 8.52 (dd, J = 8.4/1.2 Hz, 1H), 8.39 (dd, J = 8.3/1.2 Hz, 1H), 7.99 (dd, J = 7.9/1.6 Hz, 1H), 7.85 (dd, J = 7.8/1.6 Hz, 1H), 7.70 (ddd, J = 8.2/7.3/1.7 Hz, 1H), 7.58 (ddd, J = 8.6/7.3/1.5 Hz, 1H), 7.31–7.23 (m, 3H), 6.72 (s, 1H), 3.86 (s, 3H), 3.33–3.28 (m, 1H), 2.23–2.10 (m, 2H), 2.00–1.93 (m, 1H), 1.67 (dtd, J = 13.6/8.4/6.3 Hz, 1H). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 174.4, 174.1, 167.7, 166.4, 139.5, 138.3, 134.1, 132.3, 130.6, 127.7, 123.9, 123.1, 122.9, 121.7, 120.9, 118.4, 55.4, 52.6, 31.6, 30.4. IR (neat): ṽ [cm−1] = 1649, 1682, 3333, 3385. HRMS (APCI): m/z = 399.1656, calcd 399.1663 for C20H23N4O5 [M + H]+. HPLC: tR = 14.75 min; purity: 96.3%.Methyl (S)-3-(2,5-dioxo-2,3,4,5-tetrahydro-1H-1,4-benzodiazepin-3-yl) propanate (12)
Synthesis was performed as previously reported.14 L-Glutamic acid dimethyl ester hydrochloride (6.02 g, 28.4 mmol) and isatoic anhydride (9) (4.64 g, 28.4 mmol) were dissolved in pyridine (60 mL) and refluxed at 120 °C for 16 h. Pyridine was evaporated under reduced pressure and the residue was purified by column chromatography (cyclohexane/EtOAc = 35/65 → 30/70). Product 12 was obtained as a white solid (1.28 g, 4.88 mmol, 17%). M.p.: 180 °C. TLC: Rf = 0.69 (MeOH/DCM = 0.5/9.5). 1H-NMR (400 MHz, DMSO-d6): δ (ppm) = 10.40 (s, 1H), 8.46 (d, J = 5.6 Hz, 1H), 7.74 (dd, J = 7.8/1.6 Hz, 1H), 7.51 (ddd, J = 8.1/7.3/1.7 Hz, 1H), 7.26–7.18 (m, 1H), 7.10 (dd, J = 8.1/1.1 Hz, 1H), 3.70 (dt, J = 8.0/6.1 Hz, 1H), 3.56 (s, 3H), 2.46–2.35 (m, 2H), 2.18–1.96 (m, 1H), 1.91–1.77 (m, 1H). 13C-NMR (101 MHz, DMSO-d6): δ (ppm) = 172.9, 171.3, 167.8, 136.6, 130.4, 126.2, 124.0, 120.9, 51.3, 50.9, 29.6, 23.1. IR (neat): ṽ [cm−1] = 1447, 1485, 1659, 1678, 1728, 3059. HRMS (APCI): m/z = 263.1022, calcd 263.1026 for C13H15N2O4 [M + H]+. HPLC: tR = 11.13 min; purity: 96.2%.(±)-1,11a-Dihydro-3H-benzo[e]pyrrolo[1,2-a][1,4]diazepine-3,5,11(2H,10H)-trione (16)
Synthesis was performed as previously reported.14 Compound 12 (0.47 g, 1.79 mmol) was dissolved in DMA (2.40 mL) and stirred under reflux at 180 °C for 20 h. The solvent was removed in vacuo, the residue was dissolved in EtOAc and H2O, and layers were separated. The organic layer was dried with Na2SO4, and the solvent was removed under vacuum. After purification with flash column chromatography (DCM/MeOH = 98/2 → 95/5), product 16 was obtained as a beige solid (0.25 g, 1.10 mmol, 61%). M.p.: 133–134 °C. TLC: Rf = 0.65 (DCM/MeOH = 9.8/0.2). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 10.71 (s, 1H), 7.81 (dd, J = 7.9/1.6 Hz, 1H), 7.61 (ddd, J = 8.1/7.3/1.6 Hz, 1H), 7.31–7.27 (m, 1H), 7.17 (dd, J = 8.2/1.1 Hz, 1H), 4.66 (dd, J = 8.4/1.0 Hz, 1H), 2.60–2.51 (m, 2H), 2.49–2.43 (m, 1H), 2.14–2.04 (m, 1H). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 173.3, 169.6, 164.1, 136.6, 133.7, 131.3, 125.8, 124.4, 121.6, 56.0, 31.0, 17.8. IR (neat): ṽ [cm−1] = 1477, 1667, 1690, 1759, 2978. HRMS (APCI): m/z = 231.0751, calcd 231.0764 for C12H11N2O3 [M + H]+.(±)-6,7-Dihydro-8H,10H-benzo[6,7]pyrrolo[2′,1′:3,4][1,4]diazepino[2,1-b]quinazoline-8,10,16(5bH)-trione (13′)
Synthesis was performed as previously reported.14 Compound 16 (0.559 g, 2.43 mmol) was dissolved in DCM (5.6 mL) and cooled down to 0 °C. Triethylamine (0.68 mL), DMAP (0.119 g, 0.97 mmol) and 2-nitrobenzoyl chloride (0.39 mL, 2.91 mmol) were added, and the mixture was stirred for 0.5 h at room temperature. The reaction mixture was cooled down to −20 °C and Zn dust (1.57 g, 24.0 mmol, 9.9 eq) and acetic acid (5.60 mL) was added. The reaction mixture was stirred for 1 h before slowly warming up to −5 °C and stirring for another 2 h at this temperature before letting it warm up to room temperature. The mixture was filtered through Celite and the residue was washed with DCM. The filtrate was washed with saturated NaHCO3, the combined organic layers dried with Na2SO4 and the solvent evaporated. Product 13′ was obtained after flash chromatography (DCM/MeOH = 99/1 → 95/5) as a white solid (0.198 g, 0.60 mmol, 25%). M.p.: 259–260 °C. TLC: Rf = 0.58 (DCM/MeOH = 9.8/0.2). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 8.19 (dd, J = 7.9/1.5 Hz, 1H), 7.90 (ddd, J = 8.4/7.1/1.6 Hz, 1H), 7.85 (dd, J = 7.8/1.6 Hz, 1H), 7.78–7.72 (m, 2H), 7.66–7.60 (m, 3H), 5.10–5.07 (m, 1H), 2.96–2.89 (m, 1H), 2.79 (ddd, J = 17.5/11.7/9.4 Hz, 1H), 2.56–2.51 (m, 1H), 2.32–2.22 (m, 1H). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 173.2, 163.6, 161.1, 152.7, 145.7, 135.0, 132.8, 131.9, 131.3, 130.3, 129.2, 128.8, 127.7, 127.6, 126.8, 121.5, 58.5, 31.7, 19.0. IR (neat): ṽ [cm−1] = 1593, 1759, 1690, 2978. HRMS (APCI): m/z = 332.1014, calcd 332.1030 for C19H14N3O3 [M + H]+.(±)-Auranomide C (5)
Synthesis was performed as previously reported with minor variations.14 To compound 13′ (0.118 g, 0.36 mmol) THF-NH3 (0.5 M, 3.50 mL) was added. The reaction mixture was heated to 50 °C for 18 h. The solvent was evaporated, and auranomide C (5) was obtained after purification with flash column chromatography (DCM/MeOH = 97/3 → 95/5) as a white solid (0.094 g, 0.27 mmol, 76%). M.p.: 220–221 °C. TLC: Rf = 0.44 (DCM/MeOH = 9.5/0.5). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 8.81 (d, J = 6.2 Hz, 1H), 8.19 (dd, J = 7.9/1.5 Hz, 1H), 7.91 (ddd, J = 8.4/7.2/1.5 Hz, 1H), 7.80–7.74 (m, 2H), 7.69–7.63 (m, 2H), 7.63–7.57 (m, 2H), 7.24 (s, 1H), 6.74 (s, 1H), 4.19–4.13 (m, 1H), 2.38–2.22 (m, 3H), 2.20–2.10 (m, 1H). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 173.7, 166.9, 161.0, 155.8, 145.9, 135.1, 133.0, 131.2, 130.7, 128.85, 128.81, 128.6, 127.6, 127.4, 126.8, 121.0, 53.2, 30.8, 24.1. IR (neat): ṽ [cm−1] = 1647, 1678, 2920, 3071, 3318, 3418. HRMS (APCI): m/z = 349.1321, calcd 349.1222 for C19H17N4O3 [M + H]+. HPLC: tR = 12.54 min; purity: 94.3%. Spectra are in agreement with previously published data.14(±)-Auranthine (1)
(±)-Auranomide C (5) (0.094 g, 0.27 mmol) was dissolved in DMF (1.20 mL), then pyridine (0.11 mL) and 4-toluenesulfonyl chloride (0.113 g, 0.59 mmol) was added, and the mixture stirred at room temperature. After 24 h, the solvent was evaporated, the residue was dissolved in EtOAc and washed with aqueous 10% citric acid. The organic layer was dried with Na2SO4 and the solvent evaporated. After purification with flash column chromatography (DCM/MeOH = 97/3 → 90/10 and then cyclohexane/EtOAc = 50/50 → 0/100), (±)-auranthine (1) was obtained as a white solid (0.03 g, 0.09 mmol, 34%). M.p.: 157–159 °C. 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 8.89 (d, J = 6.3 Hz, 1H), 8.19 (ddd, J = 7.9/1.6/0.5 Hz, 1H), 7.91 (ddd, J = 8.2/7.2/1.6 Hz, 1H), 7.79 (dd, J = 7.6/1.6 Hz, 1H), 7.76 (dd, J = 8.3/1.1 Hz, 1H), 7.69 (ddd, J = 8.2/7.1/1.6 Hz, 1H), 7.65 (dd, J = 8.1/1.4 Hz, 1H), 7.63–7.58 (m, 2H), 4.25 (dt, J = 8.7/5.9 Hz, 1H), 2.70 (t, J = 7.6, 2H), 2.47–2.38 (m, 1H), 2.33–2.25 (m, 1H). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 166.9, 160.9, 155.1, 145.7, 135.2, 132.9, 131.0, 130.8, 128.94, 128.91, 128.7, 127.7, 127.4, 126.8, 121.0, 120.1, 52.6, 24.6, 13.3. IR (neat): ṽ [cm−1] = 3174, 3067, 2924, 2249, 1686, 1655, 1613, 1597, 1454, 1393, 1250, 775, 694. HRMS (APCI): m/z = 331.1190, calcd 331.1117 for C19H15N4O2 [M + H]+. HPLC: tR = 15.00 min; purity: 94.6%. Spectra are in agreement with those of natural auranthine.6Author contributions
The manuscript was written through contributions of all authors. All authors have given approval.Conflicts of interest
There are no conflicts to declare.Abbreviations
| APCI | Atmospheric pressure chemical ionization |
| CD | (Circular dichroism) spectroscopy |
| DCM | Dichloromethane |
| DFT | Density functional theory |
| DMA | Dimethylacetamide |
| DMAP | 4-Dimethylaminopyridine |
| DMF | Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| EDCI | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide, |
| HRMS | High-resolution mass spectrometry |
| THF | Tetrahydrofuran |
| TLC | Thin layer chromatography |
Acknowledgements
We acknowledge support from the open access publication fund of the University of Münster.References
- S. E. Yeulet, P. G. Mantle, J. N. Bilton, H. S. Rzepa and R. N. Sheppard, J. Chem. Soc., Perkin Trans. 1, 1986, 1891–1894, 10.1039/p19860001891.
- S. E. Yeulet and P. G. Mantle, FEMS Microbiol. Lett., 1987, 41, 207–210 CrossRef CAS.
- N. Arora, P. Dhiman, S. Kumar, G. Singh and V. Monga, Bioorg. Chem., 2020, 97, 103668 CrossRef CAS PubMed.
- A. Witt, A. Gustavsson and J. Bergman, J. Heterocycl. Chem., 2003, 40, 29–35 CrossRef CAS.
- U. A. Kshirsagar, V. G. Puranik and N. P. Argade, J. Org. Chem., 2010, 75, 2702–2705 CrossRef CAS PubMed.
- S. A. Kalinina, D. V. Kalinin, Y. Hovelmann, C. G. Daniliuc, C. Muck-Lichtenfeld, B. Cramer and H. U. Humpf, J. Nat. Prod., 2018, 81, 2177–2186 CrossRef CAS PubMed.
- F. H. Song, B. A. Ren, K. Yu, C. X. Chen, H. Guo, N. Yang, H. Gao, X. T. Liu, M. Liu, Y. J. Tong, H. Q. Dai, H. Bai, J. D. Wang and L. X. Zhang, Mar. Drugs, 2012, 10, 1297–1306 CrossRef CAS PubMed.
- M. A. Goetz, M. Lopez, R. L. Monaghan, R. S. L. Chang, V. J. Lotti and T. B. Chen, J. Antibiot., 1985, 38, 1633–1637 CrossRef CAS PubMed.
- R. S. L. Chang, V. J. Lotti, R. L. Monaghan, J. Birnbaum, E. O. Stapley, M. A. Goetz, G. Albersschonberg, A. A. Patchett, J. M. Liesch, O. D. Hensens and J. P. Springer, Science, 1985, 230, 177–179 CrossRef CAS PubMed.
- B. K. Joshi, J. B. Gloer, D. T. Wicklow and P. F. Dowd, J. Nat. Prod., 1999, 62, 650–652 CrossRef CAS PubMed.
- H. H. Sun, C. J. Barrow, D. M. Sedlock, A. M. Gillum and R. Cooper, J. Antibiot., 1994, 47, 515–522 CrossRef CAS PubMed.
- F. He, B. M. Foxman and B. B. Snider, J. Am. Chem. Soc., 1998, 120, 6417–6418 CrossRef CAS.
- M. C. Tseng, C. Y. Lai, Y. W. Chu and Y. H. Chu, Chem. Commun., 2009, 4, 445 RSC.
- K. Sorra, K. Mukkanti and S. Pusuluri, Synthesis, 2014, 46, 189–194 CrossRef.
- T. Sugimori, T. Okawa, S. Eguchi, A. Kakehi, E. Yashima and Y. Okamoto, Tetrahedron, 1998, 54, 7997–8008 CrossRef CAS.
- P. E. Zhichkin, X. M. Jin, H. L. Zhang, L. H. Peterson, C. Ramirez, T. M. Snyder and H. S. Burton, Org. Biomol. Chem., 2010, 8, 1287–1289 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07483h |
| This journal is © The Royal Society of Chemistry 2024 |