
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the selectivity of the CO2 hydrogenation reaction using boron-doped cobalt-based catalysts†
Jiaqi Wangab,
Kaihong Liub,
Jingjing Zhaob,
Xiuping Lib,
Bolin Yinb,
Bo Jiangb and
Hexing Li *ab
*ab
aSchool of Chemistry and Molecular Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China
bChinese Education Ministry Key Lab, Joint International Research Lab of Resource Chemistry, Shanghai Frontiers Science Center of Biomimetic Catalysis, College of Chemistry and Materials Science, Shanghai Normal University, Shanghai 200234, China. E-mail: hexing-li@shnu.edu.cn
First published on 21st February 2024
Abstract
Direct CO2 hydrogenation to value-added chemicals is a promising path toward realizing the “carbon-neutral” goal. However, controlling the selectivity of CO2 hydrogenation toward desired products (e.g., CO and CH4) using non-precious metal-based catalysts is important but challenging. It is imperative to explore catalysts with high activity and stability. Herein, boron-doped cobalt nanoparticles supported on H-ZSM-5 were devised for CO2 hydrogenation to produce CO in a gas–solid flow system. Our results demonstrate that boron doping not only increases the CO2 adsorption capability of the catalyst but also optimizes the electronic state of Co for CO desorption during hydrogenation process. As a result, the boron-doped cobalt catalysts displayed an enhanced CO selectivity of 94.5% and a CO2 conversion rate of 45.6%, which is much higher than that of Co-ZSM-5 without boron doping. This study shows that the strategic design of metal borides is important for controlling the selectivity of desired products in the CO2 hydrogenation reaction.
1. Introduction
Heterogeneous CO2 hydrogenation to value-added chemicals using green H2 is a promising approach for alleviating the energy crisis and environmental pollution.1–4 This is because carbon dioxide functions as a chemical raw material in this process, producing renewable chemicals without using fossil fuels.5–7 The hydrogenation of CO2 usually produces C1 compounds, such as CO, CH4, and CH3OH.8–11 Among C1 products, CO is a resource for the synthesis of alcohols, liquid hydrocarbons, and organic acids using existing syngas conversion infrastructure.12,13 In CO2 hydrogenation to CO (eqn (1)), namely, reverse water gas conversion reaction (RWGS), achieving high CO selectivity is a challenge due to the simultaneous formation of CH3OH and CH4 as by-products.| CO2 + H2 ↔ CO + H2O | (1) |
Great efforts have been made to design catalysts that maximize selectivity and understand the underlying mechanisms of CO formation.12,13 When supported metal catalysts are used for CO2 hydrogenation, the noble metal usually determines the efficiency of the catalyst. However, the price of noble metals often restricts catalysts from being used in the experimental stage. Therefore, cost-effective non-precious metal catalysts are promising alternatives.14–18 Among them, cobalt-based catalysts have been developed for the hydrogenation of CO2. However, Co nanoparticles favor methane formation, whereas single-atom Co catalysts favor CO formation. For example, the Co catalyst with atomic dispersion on a ZrO2 support achieved a CO selectivity of more than 95% during hydrogenation.19 In contrast, the Co nanoparticle/ZrO2 catalyst showed 100% methane selectivity. The preparation procedure of single-atom catalysts is very complex, and the poor stability of single-atom catalysts during the reaction cannot be neglected. Therefore, it is important to explore a simple method for developing cobalt-based catalysts with high CO selectivity.
Incorporating transition metal-based materials with boron offers opportunities for electrocatalytic hydrogen evolution reactions, electrocatalytic oxygen evolution reactions, CO2 hydrogenation reduction, etc.20–26 For example, a boron-doped Ru(Co, Ni) metal catalyst exhibited a 578 μmol g−1 h−1 production rate and 99% selectivity to HCOOH during liquid-phase CO2 hydrogenation owing to the coordination-unsaturated, electron-enriched, and synergistic active sites, which promoted the adsorption, activation, dissociation, and surface reaction of CO2 and H2.27 In the RWGS reaction, CO2 adsorption capacity is one of the most critical factors that can improve the reaction efficiency. However, in liquid-phase reactions, the strong adsorption of CO2 on the catalyst surface does not occur perfectly due to the low solubility of CO2 in water. Boron can play a significant role in solving this issue.28 Boron-doped metal catalysts have unparalleled potential in improving heterogeneous CO2 hydrogenation reactions.
In our study, B-doped Co catalysts supported on ZSM-5 (CoB/ZSM-5) with 1–25 wt% of Co were synthesized using a simple impregnation method and applied to enhance the CO2 hydrogenation reaction. Compared with the Co/ZSM-5 catalysts, which produce CH4 mainly, CO2 was highly selectively converted to CO by B-doped Co supported on ZSM-5. At the same time, the CO2 conversion rate of CoB/ZSM-5 was greatly improved. The structure of the catalyst was clarified using various characterization methods. The catalytic activity was measured under different reaction conditions and correlated with the catalytic performance. Under the optimal reaction conditions, the CO2 conversion rate of the 20% CoB/ZSM-5 catalyst reached 48.56% with a high CO selectivity of 94.17%. This work sheds light on the use of boron to modulate the electronic state of metals to design highly product-selective and stable catalysts for CO2 hydrogenation.
2. Experimental section
2.1 Materials and reagents
H-ZSM-5 zeolites with different molar ratios of SiO2/Al2O3 of 25, 50, 130, and 300 were obtained from Nankai University Catalyst. Unless specifically mentioned, H-ZSM-5 with a SiO2/Al2O3 ratio of 300 was used to support CoB. Cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O), sodium hydroxide, and sodium borohydride were obtained from Aladdin Reagents. Unless specified otherwise, all reagents in this study were analytical-grade products and used without further purification.2.2 Catalyst preparation
To synthesize CoB/ZSM-5, a certain amount of Co(NO3)2·6H2O solution (0.85 M) was added to a 10 mL suspension solution containing 100 mg H-ZSM-5, followed by impregnation for 12 h. The solution was dried at 80 °C, and the obtained powders were calcined at 200 °C for 2 h in air. Then, the calcined samples were re-dispersed in 10 mL H2O, followed by the dropwise addition of a NaBH4/NaOH (2 M/0.2 M) aqueous solution at 0 °C under vigorous stirring. The samples were then washed thoroughly with a mixture of water and ethanol. Finally, the catalysts were dried at 60 °C for 12 h. The as-prepared samples were designated as x% CoB/ZSM-5, where x refers to the theoretical mass ratio of Co deposited on H-ZSM-5. The loading amount of Co was adjusted by changing the added amount of Co(NO3)2 solution. CoB/SiO2 and CoB/Al2O3 were synthesized using a similar method where H-ZSM-5 was replaced with SiO2 or Al2O3, respectively.For comparison, a Co–H-ZSM-5 catalyst with 20 wt% Co was synthesized using the following method. H-ZSM-5 was dispersed in 30 mL H2O and Co(NO3)2·6H2O was added. The mixture was stirred at room temperature for 1 h. Water was evaporated under reduced pressure, and the powder was dried at 150 °C for 12 h, followed by calcination at 500 °C for 3 h in the air. This catalyst was named Co/ZSM-5.
Pure CoB was synthesized by chemical reduction. Briefly, an aqueous mixture of NaBH4/NaOH (2 M/0.2 M) was added dropwise to the Co(NO3)2·6H2O solution (0.85 M) at 0 °C under vigorous stirring. The solution was stirred for 0.5 h until no bubbles were released. An excess amount of NaBH4 was used to ensure the complete reduction of the Co2+ ions. The obtained black precipitate was thoroughly washed with distilled water to remove soluble boron species and Na+ ions. Finally, the sample was dried at 60 °C for 12 h.
2.3 Catalyst characterization
The N2 adsorption–desorption isotherms were obtained at 77 K using a Micromeritics ASAP 2010 instrument, and the specific surface area (SBET) and pore volume (VP) were calculated by applying the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) models. Wide-angle X-ray diffraction (XRD) patterns were recorded on Rigaku Ultimate IV operated at 40 kV and 40 mA using CuKα X-ray (λ = 1.54 Å). X-ray photoelectron spectroscopy (XPS) was performed using a Thermo Scientific K-Alpha instrument. Charge correction was performed by adjusting the carbon peak to 284.8 eV. TEM and STEM images were obtained using a JEOL JEM 2100F atomic resolution electron microscope at an acceleration voltage of 200 kV. Temperature-programmed desorption of the catalysts was evaluated in the presence of mixed CO2–He (CO2 = 10%) on a Micromeritics AutoChem II Chemisorption Analyzer equipped with a TCD detector. Before measurement, the samples were pretreated with He at 300 °C for 30 min, followed by CO2 adsorption at room temperature for 30 min. After purging with He for 30 min, TPD was performed at a ramp rate of 10 °C min−1.2.4 Activity measurement
The catalytic activity of the materials toward CO2 hydrogenation was evaluated in a stainless-steel fixed bed flow reactor system. The products were analysed using an online GC (Agilent, GC7890B) equipped with TCD and FID detectors. Typically, the catalyst and 2 g quartz sand were evenly mixed and placed in a self-made quartz tube (∅ = 5 mm) held by quartz wool. A thermocouple was inserted into the reactor to monitor the bed temperature. In a typical test, the catalyst was arranged in the raw gas environment at room temperature, and a mass flowmeter was used to control the flow rate of the raw gas. After 30 min of purging with the raw gas, the reactor temperature was raised to the target reaction temperature. The total flow rate of the reactant gas was 50 mL min−1 to maintain the space velocity at 3000 mL h−1 g−1. The reaction was carried out by reducing the amount of catalyst and increasing the flow rate under different space velocities. All feedstock gases contained 25% argon as an internal standard. The liquid products were analyzed offline on a gas chromatograph equipped with FID and liquid chromatography. Unless otherwise stated, the reaction conditions were as follows: 20% CoB/ZSM-5, 500 °C reaction temperature, 3000 mL g−1 h−1, the stoichiometric ratio of CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 = 1:4, and 3 h reaction period.
:H2 = 1:4, and 3 h reaction period.
3. Results and discussion
3.1 Synthesis and characterization of the catalysts
The morphology and structure of the as-prepared samples were first characterized by transmission electron microscopy (TEM). As shown in Fig. 1, 20% CoB/ZSM-5 mainly comprised nanoparticles in the size range of 15–30 nm (Fig. 1a and inside 1a). No crystal fringe was observed at the edge of the nanoparticles, which is a typical feature of an amorphous structure (Fig. 1b). The EDS spectrum showed uniformly dispersed Si, Al, and O elements in the ZSM-5 support, and the Co element was mainly found in the nanoparticles supported on ZSM (Fig. 1c–i). | ||
| Fig. 1 (a and b) TEM image with particle size distribution (inset), and (c–i) elemental mappings of 20% CoB/ZSM-5. | ||
The phase structure of the prepared samples was determined by wide-angle X-ray diffraction (XRD). As shown in Fig. 2a, the diffraction peaks of the CoB/ZSM-5 catalysts impregnated with different concentrations of Co(NO3)2 were consistent with those of H-ZSM-5, indicating that the incorporation of CoB did not change the crystal structure of the carrier ZSM-5. The absence of Co diffraction peaks in the pattern may be due to the amorphous structure of CoB and the high crystallinity of ZSM-5. The CoB catalysts supported on ZSM-5 carriers with different Si/Al ratios exhibited similar crystal structures (Fig. S1†). The contrast samples CoB/SiO2 and CoB/Al2O3 were also measured, displaying an amorphous CoB structure. The electronic states of Co and B in 20% CoB/ZSM-5 were then studied by X-ray photoelectron spectroscopy (XPS). In the XPS spectra of 20% CoB/ZSM-5 (Fig. 2b), the Co peaks could be fitted to the Lorentzian–Gaussian model, and the two fitted peaks corresponding to Co3+ 2p3/2 and Co3+ 2p1/2 were located at 780.27 eV and 795.64 eV, respectively. The Co(II) peaks corresponding to Co2+ 2p3/2 and Co2+ 2p1/2 were located at 781.37 eV and 797.16 eV, respectively. Then, the Co(II)/Co molar ratio in 20% CoB/ZSM-5 was calculated to be 79.74% from the fitted peak area ratio. In the B 1s spectrum (Fig. 2c), the peak of the B element was found at 192.1 eV, indicating the formation of B–O.23,25,29–31
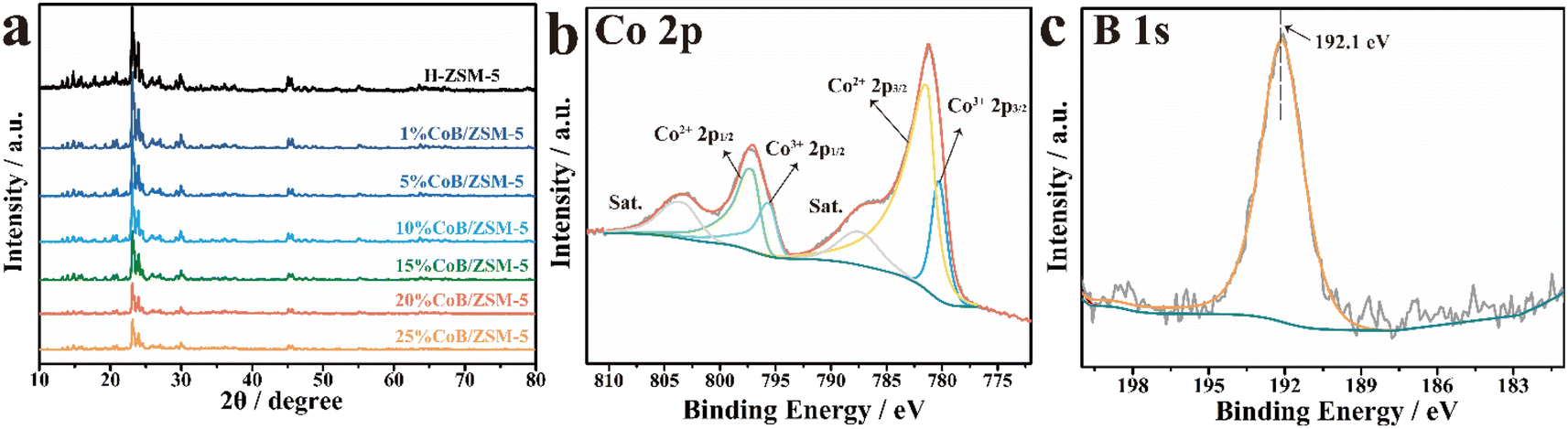 | ||
| Fig. 2 (a) XRD patterns of different catalysts; XPS spectra of the (b) Co 2p and (c) B 1s levels of 20% CoB/ZSM-5. | ||
The specific surface areas of the as-prepared samples were also measured from the N2 adsorption and desorption isotherms (Fig. 3a and Table S1†), revealing a high specific surface area for H-ZSM-5 and a low specific surface area for CoB. The large specific surface area of the H-ZSM-5 carrier is beneficial to improving CO2 adsorption in gas–solid CO2 hydrogenation reactions. After CoB loading, the specific surface area of the CoB/ZSM-5 sample decreased because CoB partially blocked the micropores of ZSM-5.
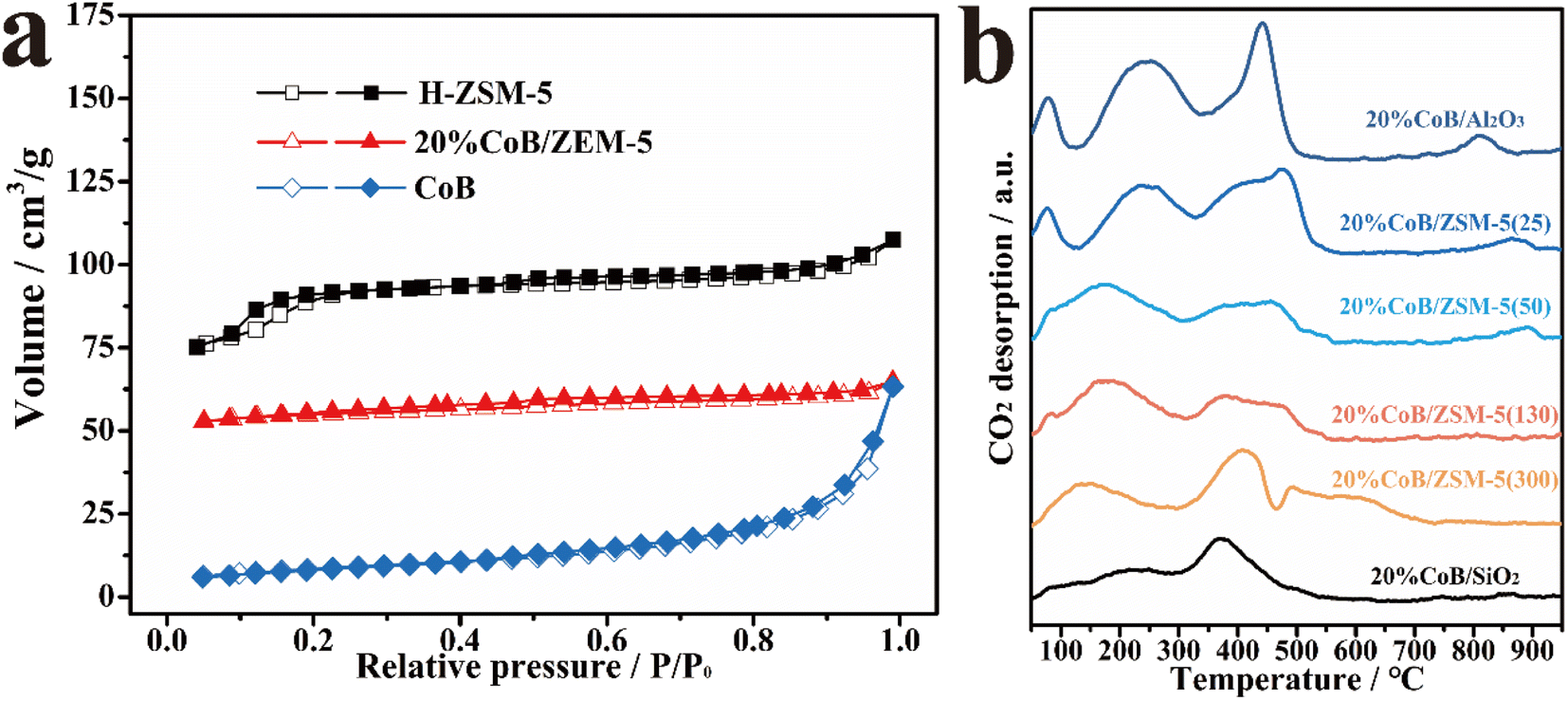 | ||
| Fig. 3 (a) N2 adsorption–desorption isotherms of H-ZSM-5, 20% CoB/ZSM-5, and CoB; (b) CO2-TPD curves of the different as-prepared catalysts. The number in brackets in the catalyst name denotes the SiO2/Al2O3 ratio in the H-ZSM-5 zeolites. | ||
Fig. 3b shows the temperature-programmed CO2 desorption spectra (CO2-TPD) of the supported catalysts. When CoB was introduced into ZSM-5, several peaks at both low and high-temperature regions were observed. The desorption peak in the low-temperature region (100 to 300 °C) is assigned to physically adsorbed CO2. The desorption intensity increased continuously with increasing amount of CoB, and a strong CO2 adsorption peak (>500 °C) appeared. The results show that CoB has a strong CO2 adsorption capacity, which benefits CO2 hydrogenation.
3.2 Catalytic performance evaluation
The performances of 20% CoB/ZSM-5 and the reference samples were evaluated in catalyzing the thermal CO2 hydrogenation reaction in a stainless-steel fixed-bed reactor. First, all potential products of thermal CO2 hydrogenation obtained in the presence of 20% CoB/ZSM-5 were investigated, and only CO and CH4 were found to be formed (Fig. S2†). The carbon source in the product was tracked through 20% CoB/ZSM-5 activity tests under different environments (Table S2†). The results suggest that catalysts and other possible carbon sources triggering the products of CO2 hydrogenation were excluded. The impact of supports with different molar ratios of Ai: Si on the performance was also investigated, as shown in Fig. S3,† suggesting that the Si/Al ratio greatly affects the CO selectivity (36.17–48.56%). However, there were no obvious changes in the CO2 conversion rate. According to the above study, we selected H-ZSM-5(300) as the optimal carrier.We further investigated the catalytic performance of the x% CoB/ZSM-5 catalysts with different amounts of CoB. The CO2 conversion rate was only 8.53%, and the CO selectivity was 98.84% at 500 °C when 1% CoB/ZSM-5 was applied as the catalyst (Fig. 4a). The CO2 conversion rate and CO yield gradually increased as the amount of CoB increased, and the 20% CoB/ZSM-5 catalyst exhibited the highest conversion rate and CO STY of 48.56% and 2.99 μmol g−1 s−1, respectively. However, CO selectivity decreased slightly to 94.17%. Moreover, excess CoB loading on ZSM-5 also resulted in poor CO2 conversion and CO STY owing to the aggregation of the CoB nanoparticles at high temperatures. It should be noted that methane was the main product when the Co/ZSM-5 catalyst was not doped with B, implying that B optimizes the Co electronic structure to support CO generation. In addition, the CO2 conversion rate of the Co/ZSM-5 catalyst is also lower than that of CoB/ZSM-5, probably owing to the enhanced CO2 adsorption capability.
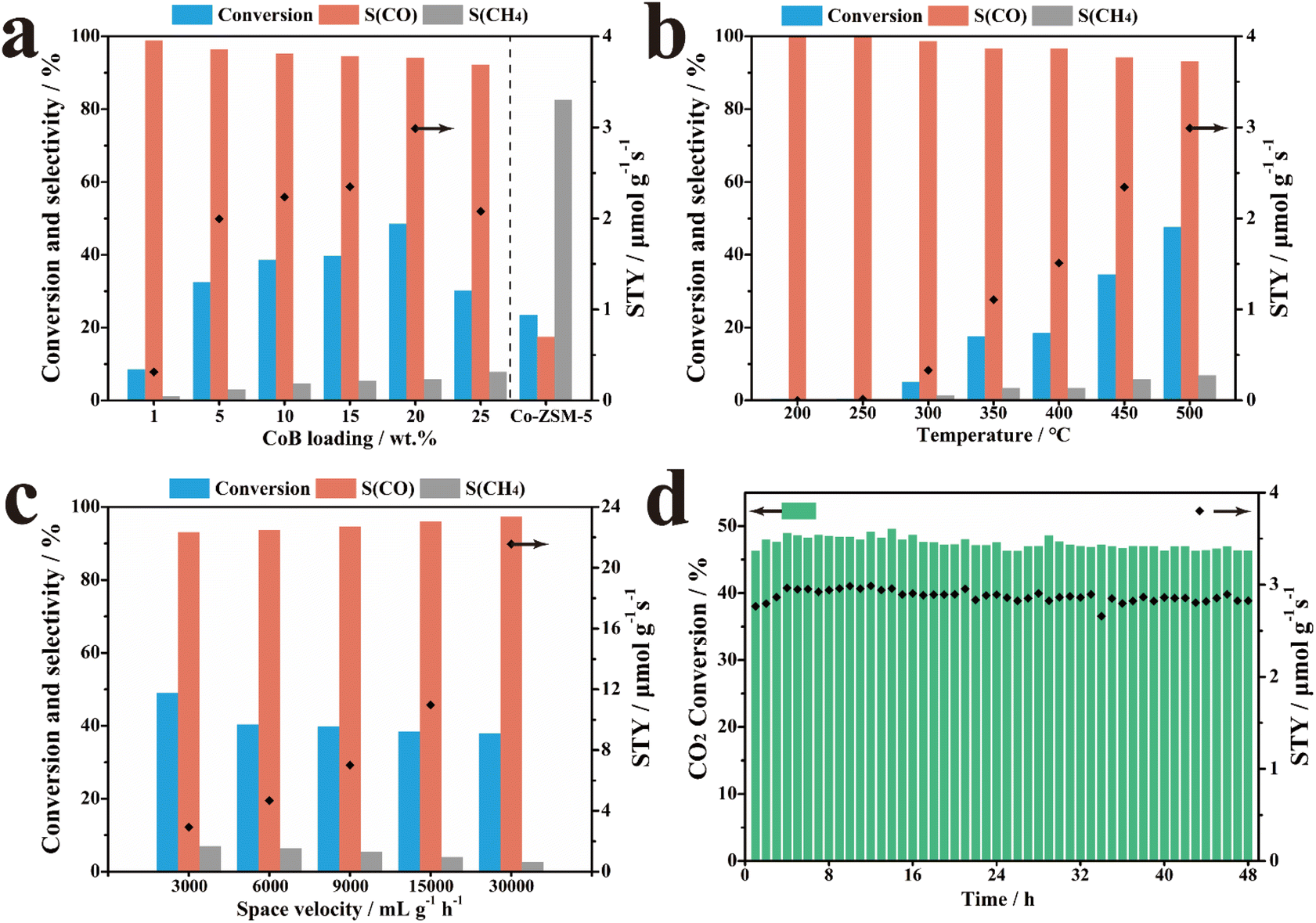 | ||
| Fig. 4 Performance of the catalysts in CO2 hydrogenation. (a) Conversion rate, selectivity, and space-time yield (STY) with x% CoB/ZSM-5 catalysts and reference sample at different reaction temperatures; (b) performance of 20% CoB/ZSM-5 at different temperatures, (c) activity of 20% CoB/ZSM-5 at different space velocities, and (d) stability of 20% CoB/ZSM-5. | ||
The performance of the 20% CoB/ZSM-5 catalyst was also investigated as a function of the reaction temperature from 200 to 500 °C. As shown in Fig. 4b, the reaction temperature not only affected the product selectivity of the reaction but also the conversion rate. At lower temperatures, only 0.36–5.03% of the conversion rate was achieved below 300 °C. With a continuous rise in temperature, the conversion rate of CO2 and CO STY significantly increased in the range of 300–500 °C. At lower temperatures, the dispersed CoB particles selectively formed CO on the catalyst, whereas, at higher temperatures, further hydrogenation resulted in methane.
Fig. 4c shows the effect of space velocity on the catalytic performance of 20% CoB/ZSM-5, indicating strong tolerance toward high space velocity, which is attributed to the high intrinsic activity of CoB.
Finally, the stability of the 20% CoB/ZSM-5 catalyst was evaluated under the optimum condition of 3000 mL h−1 g−1 at 500 °C. As shown in Fig. 4d, both the CO2 conversion rate and STY were stable over a time of 48 h. Therefore, the high performance of the supported CoB catalysts under various conditions is attractive. The activity of 20% CoB/ZSM-5 is better than those of the non-noble-metal catalysts reported in the literature and is comparable to those of noble-metal-based catalysts (Table S3†).
4. Conclusions
In conclusion, we have presented a simple approach for exploring highly efficient CO2 hydrogenation catalysts using the impregnation method. Compared with 20% Co-ZSM-5, 20% CoB/ZSM-5 exhibited enhanced CO2 conversion and high CO selectivity. Under the optimal condition of 3000 mL h−1 g−1 at 500 °C, the CO2 conversion rate of the 20% CoB/ZSM-5 catalyst reached 48.56%, and the CO selectivity was 94.17%. The key to this improved performance is B incorporation, which results in the formation of an amorphous phase and modulates the electronic state of Co, providing abundant active sites for CO2 adsorption and avoiding strong coordinate bonding to CO, respectively. This work highlights that boron doping is important to maximize CO2 conversion and selectivity to desired CO2 hydrogenation products.Author contributions
Jiaqi Wang: data curation, formal analysis, investigation, writing – original draft. Kaihong Liu: formal analysis and software. Jingjing Zhao: formal analysis and validation. Xiuping Li: formal analysis. Bolin Yin: data curation. Bo Jiang: data curation, formal analysis, writing, review and editing. Hexing Li: conceptualization, funding acquisition, supervision, writing, review, and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2020YFA0211004), the National Natural Science Foundation of China (22236005, 22106106), and the Foundation from the Shanghai Local Government (22dz1205400, 21ZR1446600, 22010503400, 23520711100), and “111” Innovation and Talent Recruitment Base (D18020). This work was also sponsored by the Shanghai Engineering Research Center of Green Energy Chemical Engineering and Energy Science and Technology under the Shanghai Class IV Peak Disciplinary Development Program.Notes and references
- A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Chem. Rev., 2017, 117, 9804–9838 CrossRef PubMed.
- T. N. Do, C. You and J. Kim, Energy Environ. Sci., 2022, 15, 169–184 RSC.
- Y. Li, L. Zeng, G. Pang, X. Wei, M. Wang, K. Cheng, J. Kang, J. M. Serra, Q. Zhang and Y. Wang, Appl. Catal., B, 2023, 324, 122299 CrossRef CAS.
- G. Varvoutis, A. Lampropoulos, E. Mandela, M. Konsolakis and G. E. Marnellos, Energies, 2022, 15, 4790 CrossRef CAS.
- H. Kang, J. Ma, S. Perathoner, W. Chu, G. Centi and Y. Liu, Chem. Soc. Rev., 2023, 52, 3627–3662 RSC.
- H. Li, W. Fang, L.-X. Wang, Y. Liu, L. Liu, T. Sun, C. Liao, Y. Zhu, L. Wang and F.-S. Xiao, The Innovation, 2023, 4, 100445 CrossRef CAS PubMed.
- S. Zou, L. Wang, H. Wang, X. Zhang, H. Sun, X. Liao, J. Huang and A. R. Masri, Energy Environ. Sci., 2023, 1754–5692 Search PubMed.
- J. Bao, G. H. Yang, Y. Yoneyama and N. Tsubaki, ACS Catal., 2019, 9, 3026–3053 CrossRef CAS.
- J. Li, Y. He, L. Tan, P. Zhang, X. Peng, A. Oruganti, G. Yang, H. Abe, Y. Wang and N. Tsubaki, Nat. Catal., 2018, 1, 787–793 CrossRef CAS.
- W. Z. Li, J. X. Liu, J. Gu, W. Zhou, S. Y. Yao, R. Si, Y. Guo, H. Y. Su, C. H. Yan, W. X. Li, Y. W. Zhang and D. Ma, J. Am. Chem. Soc., 2017, 139, 2267–2276 CrossRef CAS PubMed.
- Y. Li, W. Gao, M. Peng, J. Zhang, J. Sun, Y. Xu, S. Hong, X. Liu, X. Liu, M. Wei, B. Zhang and D. Ma, Nat. Commun., 2020, 11, 61 CrossRef CAS PubMed.
- J. A. Buss, G. A. Bailey, J. Oppenheim, D. G. VanderVelde, W. A. Goddard and T. Agapie, J. Am. Chem. Soc., 2019, 141, 15664–15674 CrossRef CAS PubMed.
- R. Li, Y. Li, Z. Li, S. Ouyang, H. Yuan and T. Zhang, Adv. Mater., 2023, 2307217 CrossRef CAS PubMed.
- A. M. Bahmanpour, M. Signorile and O. Kröcher, Appl. Catal., B, 2021, 295, 120319 CrossRef CAS.
- J. Cui, Y. Q. Liu, Y. A. Mei, J. M. Li, H. Zhang, S. H. Zhu, Y. L. Niu and T. S. Deng, Appl. Catal., B, 2023, 339, 123099 CrossRef.
- A. Fedorov, H. Lund, V. A. Kondratenko, E. V. Kondratenko and D. Linke, Appl. Catal., B, 2023, 328, 122505 CrossRef CAS.
- F. M. Simons, T. J. de Heer, R. C. J. van de Poll, V. Muravev, N. Kosinov and E. J. M. Hensen, J. Am. Chem. Soc., 2023, 145, 20289–20301 CrossRef PubMed.
- L. Wang, M. S. Bootharaju, J. H. Kim, Y. Wang, K. Wang, M. Zhao, R. Zhang, J. Xu, T. Hyeon, X. Wang, S. Y. Song and H. J. Zhang, J. Am. Chem. Soc., 2023, 145, 2264–2270 CrossRef PubMed.
- H. M. Dostagir, R. Rattanawan, M. Gao, J. Ota, J. Y. Hasegawa, K. Asakura, A. Fukuoka and A. Shrotri, ACS Catal., 2021, 11, 9450–9461 CrossRef.
- K. Bai, Y. Wu, X. C. Zhou, Y. F. Ye, K. Q. Nie, J. O. Wang, M. Xie, Z. X. Zhang, Z. J. Liu, T. Cheng and C. B. Gao, Nat. Commun., 2022, 13, 6094 CrossRef PubMed.
- B. Qi, Y. Zhang, L. Yang, Y. H. Zhao, Y. H. Zhu, H. L. Jiang and C. Z. Li, Nat. Commun., 2022, 13, 4602 CrossRef PubMed.
- J. Wu, M. L. Hou, Z. L. Chen, W. J. Hao, X. L. Pan, H. Y. Yang, W. L. Cen, Y. Liu, H. Huang, P. W. Menezes and Z. H. Kang, Adv. Mater., 2022, 34, 2202995 CrossRef CAS PubMed.
- M. Q. Yu, C. Weidenthaler, Y. Wang, E. Budiyanto, E. O. Sahin, M. M. Chen, S. DeBeer, O. Rüdiger and H. Tüysüz, Angew. Chem., Int. Ed., 2022, 61, e202211543 CrossRef CAS PubMed.
- H. Zhang, Y. T. Jia, J. Y. Zhan, G. M. Liu, G. H. Liu, F. Li and F. S. Yu, Angew. Chem., Int. Ed., 2023, 62, e202303483 CrossRef PubMed.
- S. Zhang, Z. M. Xia, Y. Zou, M. K. Zhang and Y. Q. Qu, Nat. Commun., 2021, 12, 3382 CrossRef CAS PubMed.
- S. Zhou, F. L. Che, M. Liu, C. Q. Zou, Z. Q. Liang, P. De Luna, H. F. Yuan, J. Li, Z. Q. Wang, H. P. Xie, H. M. Li, P. N. Chen, E. Bladt, R. Quintero-Bermudez, T. K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef PubMed.
- J. Zhao, J. Q. Wang, Y. Bai, H. R. Du, J. J. Yang, B. L. Yin, B. Jiang and H. X. Li, J. CO2 Util., 2023, 74, 2212–9820 Search PubMed.
- G. Liu, X. G. Meng, H. B. Zhang, G. X. Zhao, H. Pang, T. Wang, P. Li, T. Kako and J. H. Ye, Angew. Chem., Int. Ed., 2017, 56, 5570–5574 CrossRef CAS PubMed.
- W. Guo, J. H. Bi, Q. G. Zhu, J. Ma, G. Y. Yang, H. H. Wu, X. F. Sun and B. X. Han, ACS Sustainable Chem. Eng., 2020, 8, 12561–12567 CrossRef CAS.
- H. He, Z.-H. Li, Z.-Y. Wang, K. Wang, Y.-C. Sun, S.-W. Wang, W.-T. Wang, Y. Yang and Z.-T. Liu, Green Chem., 2021, 23, 5775–5785 RSC.
- H. Xiong, Z. J. Du, H. Li, J. Y. Xu, J. J. Li, X. J. Zhao and L. F. Liu, ACS Sustainable Chem. Eng., 2019, 7, 1493–1501 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07488a |
| This journal is © The Royal Society of Chemistry 2024 |