
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards an efficient methodology for the synthesis of functionalized dihydropyrans by silyl-Prins cyclization: access to truncated natural products†
Laura F. Peña,
Paula González-Andrés and
Asunción Barbero *
*
Department of Organic Chemistry, Faculty of Science, 47011 Valladolid, Spain. E-mail: asuncion.barbero@uva.es
First published on 3rd January 2024
Abstract
We herein present a selective methodology for the synthesis of disubstituted dihydropyrans by silyl-Prins cyclization of Z-vinylsilyl alcohols mediated by trimethylsilyl trifluoromethanesulfonate (TMSOTf). The reaction features broad substrate scope, short reaction times and ease of process scale-up. Moreover, to showcase the applicability of the proposed method, we also report a facile and linear synthesis of analogues of rhopaloic acid and natural doremox fragrance.
Introduction
Marine metabolites are well recognized to be a prolific reservoir for drug development. Within them, the relevance of heterocyclic marine structures is of particular note due to the plethora of pharmacological properties associated with them (such as anticancer, anti-inflammatory, antibacterial or antiviral). Special interest has been focused on marine compounds with 2H or 4H-pyranyl moieties in their structure, due to their relatively large abundance and promising biological properties.1 For instance, rhopaloic acid C,2 isolated from the marine sponge Rhopaloeides sp., exhibits potent inhibitory action on the gastrulation of the starfish (Asterina pectinifera) embryo and induces apoptosis and autophagy in human bladder cancer (Fig. 1), whereas aspergillide, which is a 14-membered macrolide isolated from the marine-derived fungus Aspergillus ostianus strain 01F313, shows cytotoxic activity against mouse lymphocytic leukemia cells.3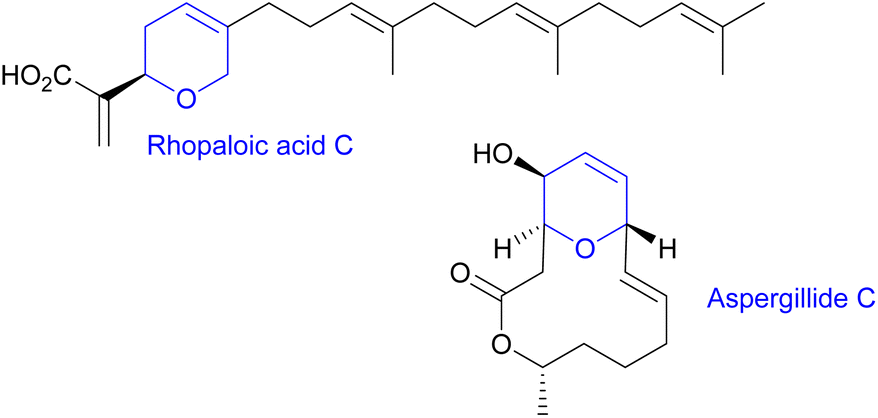 | ||
| Fig. 1 Marine drugs bearing a dihydropyran unit. | ||
As is known, the search for bioactive compounds has been a source of inspiration for the scientific community for decades. Two main strategies have attracted the interest of synthetic chemist: the first one is natural product synthesis which aims at the preparation of an identical structure to the naturally occurring product and usually implies a large number of steps, and low overall yield, to reach the desired target. A more recently developed approach is the diversity-oriented synthesis, which focuses on the preparation of natural product analogues in which structural variation is aimed at as a means to achieve higher diversity in the final target. Moreover, natural product-like compounds have shown great potential for pharmaceutical drug discovery programs.
Over the years, a great number of synthetic methodologies have been described for the synthesis of dihydropyrans. Among others, Prins cyclization has emerged as a powerful tool for the preparation of saturated six membered oxacycles.4,5 A convenient modification of this methodology, which employs electron-rich alkenyl silanes as nucleophiles, has allowed the synthesis of oxacycles bearing a double bond in their structure.6 An important advantage of these Prins-type cyclizations is that they proceed with high reaction rates and selectivity.
Typically, allylsilyl alcohols have shown great potential for the synthesis of tetrahydropyrans bearing an exocyclic double bond (methylene tetrahydropyrans). On the other hand, the still underdeveloped silyl-Prins cyclization which employs vinylsilyl alcohols permits the preparation of 6-membered oxacycles bearing an endocyclic double bond (dihydropyrans).7 The pioneering work of Dobbs in this area has opened the access to cis-2,6-disusbstituted dihydropyrans in good yields, by reaction of Z-(trimethyl)silyl homoallylic alcohols with aliphatic aldehydes under the mediation of InCl3. However, only a limited number of aromatic aldehydes (with p-NO2 or p-CF3 electron withdrawing groups) provide the desired oxacycle (Scheme 1a).8,9
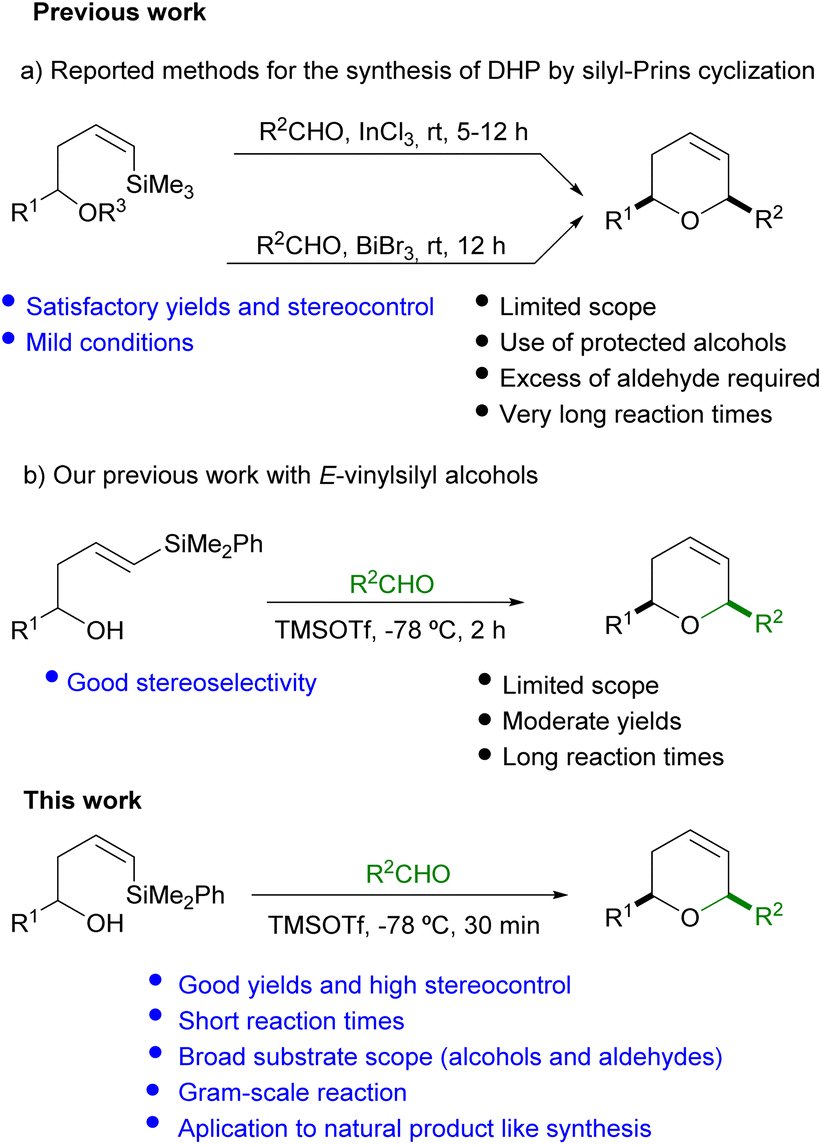 | ||
| Scheme 1 Towards the synthesis of dihydropyrans by silyl-Prins cyclization. | ||
In another approach, Hinkle10 has reported the reaction of TES protected Z-(trimethyl)silyl alcohols with aldehydes, in the presence of BiBr3. The corresponding 2,6-cis-dihydropyrans are produced in good yield and selectivity when alkylic aldehydes or p-CF3C6H5CHO are used. However, lower yields and selectivities are observed for other aromatic aldehydes. Moreover, the need of a significant excess of aldehyde (2 equiv.) and the additional alcohol protection step also imply a relevant drawback (Scheme 1a).
Despite the mild conditions employed in the precedent publications, a common drawback is the rather long reaction time (up to 12 hours) required for completion. Moreover, the discrete number of examples reported, together with the scarce variety of substituents (R1 and R2) on the alcohol and aldehyde compatible with this cyclization, prompted us to explore and expand the scope of this methodology (Scheme 1).
Results and discussion
Following our interest in the synthesis of polisubstituted heterocycles using the silyl-Prins cyclization,11,12 we now present a general, selective and short time protocol for the synthesis of a variety of cis-2,6-dihydropyrans, by siyl-Prins cyclization (mediated by trimethylsilyl trifluoromethanesulfonate) (TMSOTf) of Z-vinylsilyl alcohols which bear the phenyldimethylsilyl group.To find the optimal conditions for this cyclization, we choose the reaction of racemic Z-vinylsilyl alcohol 1a with phenylacetaldehyde. The results are shown in Table 1.
|
||||
|---|---|---|---|---|
| Entry | Lewis acid (equiv.) | Reaction conditionsa | drb | Yieldc (%) |
a Conditions: 1a (0.36 mmol), phenylacetaldehyde (0.43 mmol), under N2.b Diastereomeric ratio (dr) of 2,6-cis-tetrahydropyran![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2,6-trans-tetrahydropyran was determined by 1H NMR analysis.c Isolated yields.d n.r. stands for no reaction. :2,6-trans-tetrahydropyran was determined by 1H NMR analysis.c Isolated yields.d n.r. stands for no reaction. |
||||
| 1 | TMSCl (1.2)/BiCl3 (0.05) | 0 °C, 40 min | >95:5 |
51 |
| 2 | TMSCl (1.2) | 0 °C, 60 min | — | n.r.d |
| 3 | BiCl3 (0.5) | 0 °C, 110 min | 90:10 |
39 |
| 4 | TMSBr (1.2) | −78 °C, 60 min | — | n.r.d |
| 5 | TMSI (1.0) | −78 °C, 100 min | 70:30 |
53 |
| 6 | BF3·OEt2 (1.0) | 0 °C, 60 min | >95:5 |
56 |
| 7 | BF3·OEt2 (1.0) | −78 °C, 60 min | — | n.r.d |
| 8 | BF3·OEt2 (0.5) | −78 °C, 60 min | — | n.r.d |
| 9 | TMSOTf (1.0) | −78 °C, 20 min | >95:5 |
74 |
| 10 | TMSOTf (0.5) | −78 °C, 25 min | >95:5 |
66 |
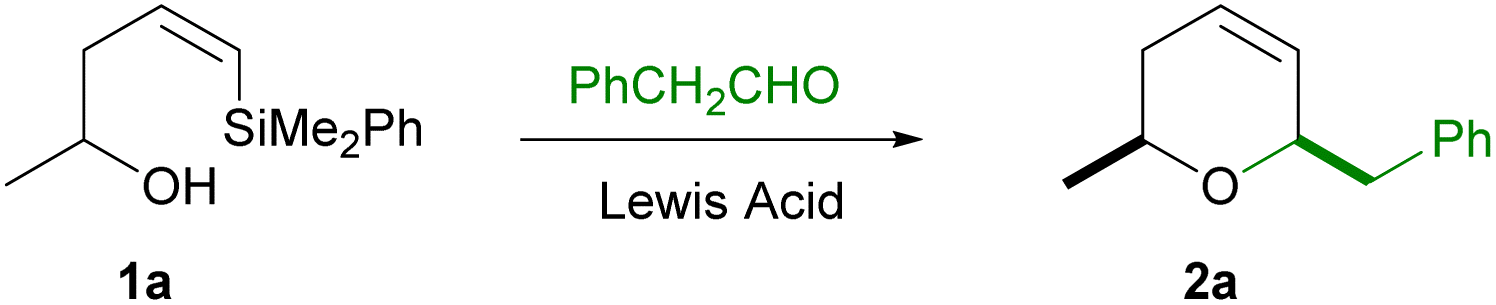
As shown in Table 1, neither trimethylsilyl halides (TMSCl, TMSBr or TMSI) nor BiCl3 were efficient Lewis acids for this cyclization, providing either low yields, low stereoselectivities or no reaction (entries 1–5). Pleasingly, both BF3·OEt2 at 0 °C (entry 6) and TMSOTf at −78 °C (entries 9 and 10) showed to be efficient initiators for this process. From both acids, we choose TMSOTf for additional studies, due to the shorter reaction times and the slightly better yields (entry 9 vs. 6). Interestingly, the reaction was also effective and highly diastereoselective when 0.5 equiv. of the activator was used (entry 10). However, slightly lower yields of the dihydropyran were obtained, which prompted us to use stoichiometric amounts of the acid in further reactions.
Encouraged by these results, we decided to explore the generality of this reaction using different types of aldehydes and various R1 substituents on the alcohol. The results are shown in Table 2.
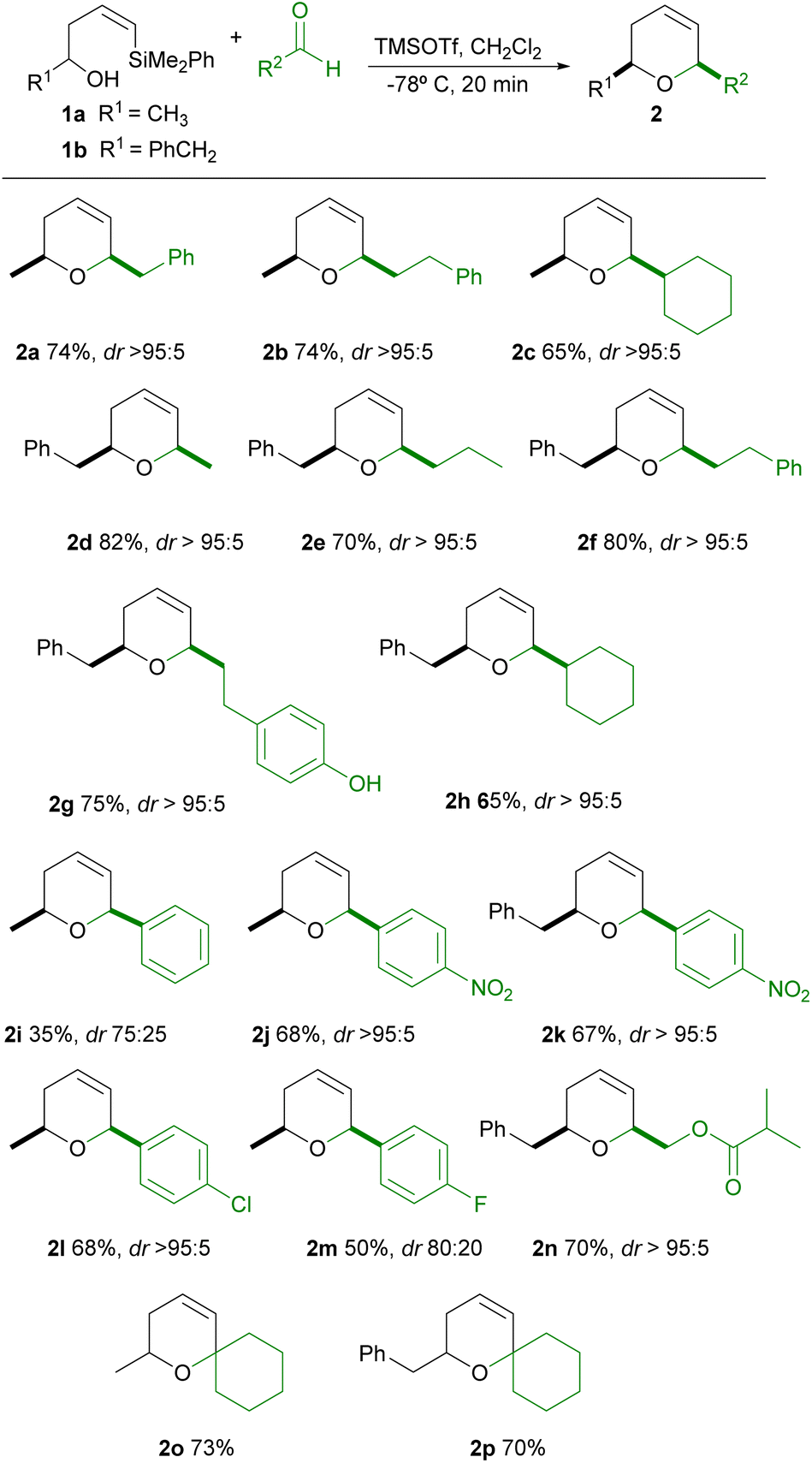
As can be seen in the Table 2, the reaction of Z-phenyldimethylsilyl alkenols 1a–b (being R1 an alkylic substituent) with alkylic aldehydes provide good yields of the corresponding dihydropyrans (2a–h). Moreover, excellent diastereoselectivitity towards the cis-2,6-disubstituted dihydropyran is observed, since a single stereoisomer is always obtained. Interestingly, the appropriate election of R1 and R2 substituents on the starting alcohol and aldehyde permits the selective formation of regioisomers, such as 2a and 2d, both in excellent stereoselectivity and yield. We then attempted the synthesis of 2b in larger scale (starting from 1.08 g of 1a) and to our delight the desired dihydropyran was obtained in reproducible yield and selectivity (690 mg, 74%). Aiming to expand the possibilities of this methodology, we then decided to use aromatic aldehydes in the cyclization. Satisfactorily, we could observe the formation of the corresponding dihydropyran (2j–l), in good yield and excellent stereoselectivity, when deficient aromatic aldehydes are used. However, yields and stereoselectivities decrease for other aromatic aldehydes (2i, 2m).13 In addition, the reaction seems to be compatible with functional groups, such as esters (2n), maintaining good yield and excellent diasteroselectivity, which opens a convenient tool for further synthetic elaborations. Worthy of note is the reaction of 1a–b with ketones, which gives in good yield spiro-dihydropyran structures 2o–p, despite the poor ability of ketones to condense with alcohols during the rate-limiting step of Prins cyclization.14 However, other cyclic ketones (such as cyclopentanone and cycloheptanone) were unreactive under these cyclization conditions. It has to be noted, as we have recently reported,15 that this cyclization is readily influenced by the geometry of the vinylsilane, since the corresponding silyl-Prins cyclization with E-vinylsilyl alcohols is limited to alkylic aldehydes, requires longer reaction times (1–2 hours), and proceeds in lower yields.
The high stereoselectivity observed for the formation of 2,6-cis-dihydropyrans can be explained by the initial formation of an E-oxocarbenium ion, which will readily undergo 6-endo cyclization through a chair like transition state in which the substituents attached to C2 and C6 will adopt the most stable equatorial conformation. The corresponding β-silyl carbocation thus obtained will then undergo elimination of the silyl group to provide the final dihydropyran (Scheme 2).
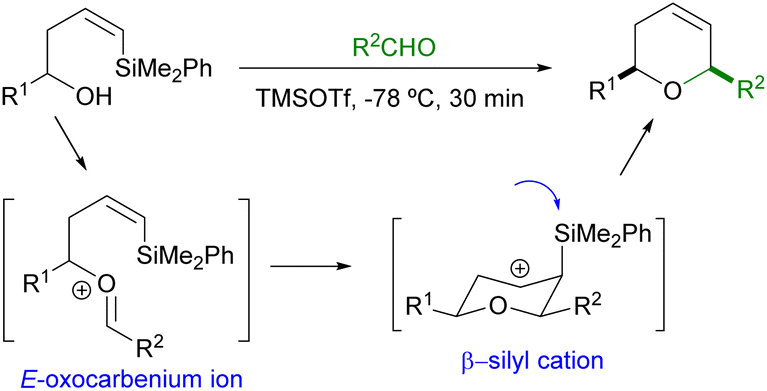 | ||
| Scheme 2 Mechanistic proposal. | ||
Having stablished a wide scope of tolerable aldehydes in this process, we next explored the starting alcohol scope. For this purpose, we choose an alcohol bearing an R1 aromatic ring substituent, in order to study the influence of such substituent in the outcome of the reaction. It's known that the oxonia-Cope rearrangement is a secondary process which is usually observed in Prins cyclizations when the starting alkenyl alcohol bears a substituent (such as phenyl) which is able to stabilize the corresponding oxocarbenium ion.16 We envisaged the possibility of suppressing this competing oxonia-Cope rearrangement process under our conditions, since the fast reaction rates of this process could help to minimize the chance of occurrence of this side reaction. The results are shown in Table 3.
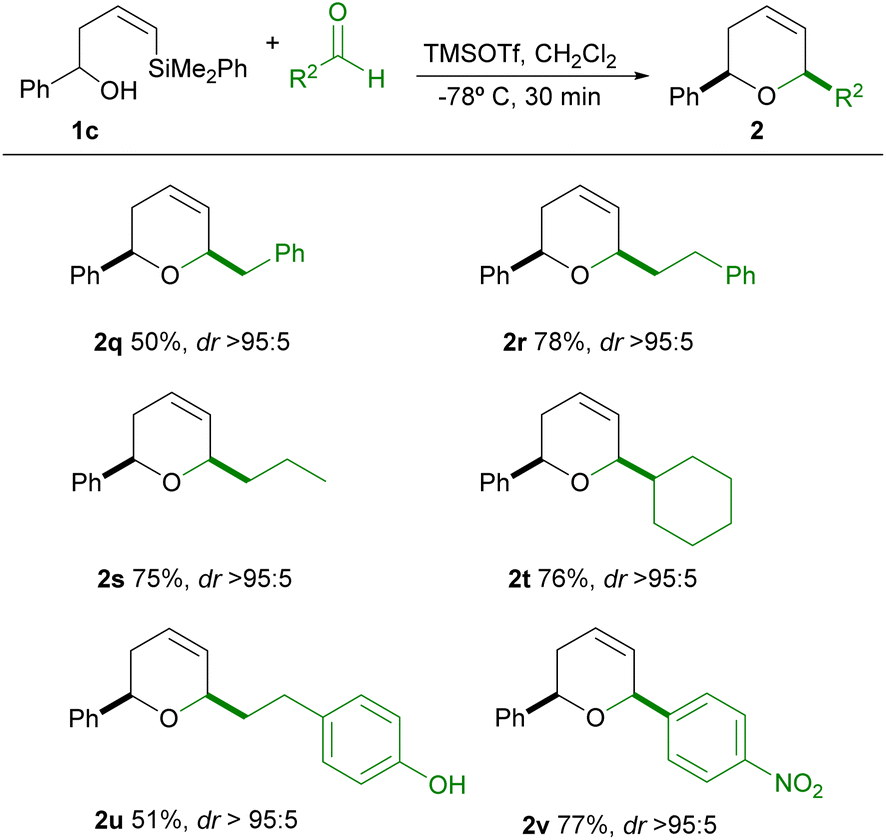
As can be seen in Table 3, the reaction of 1c with either alkylic aldehydes or electron-deficient aromatic aldehydes in general proceeds in good yield and excellent stereoselectivity (2q–2v). In no case the symmetrical product of oxonia-Cope rearrangement could be observed in the reaction mixture.
We then decided to apply this efficient methodology to the synthesis of analogues of natural rhopaloic acid C.2 For this purpose, we prepared vinylsilyl alcohol 1d (from Citronellal), which was obtained as an inseparable mixture of diastereoisomers. Under the optimized conditions for this silyl-Prins cyclization, the reaction of 1d with two different functionalyzed aldehydes, such as 2-(isobutyryloxy)acetaldehyde and methyl 3,3-dimethoxypropionate, were performed. As shown the corresponding dihydropyrans 2w and 2x, bearing an ester functionality appended to C2, were obtained in moderate yields and excellent diastereoselectivity towards the 2,6-cis-dihydropyran. Both substrates bear a convenient functionalised group at C2, which could allow the preparation of more complex structures. For instance, further saponification of 2x, under standard conditions, provided in high yield truncated rhopaloic acid 3 in high yield (Scheme 3).
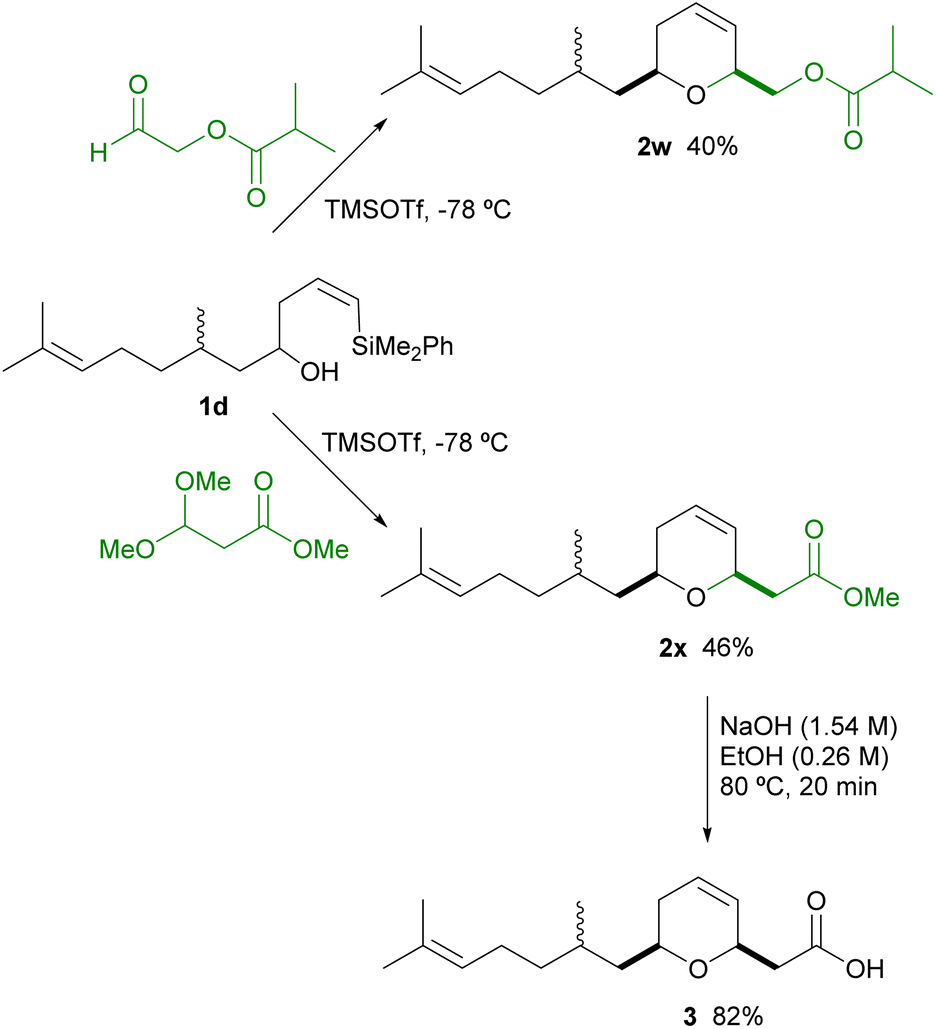 | ||
| Scheme 3 Access to truncated natural rhopaloic acid. | ||
Finally, we also prepared a tetrahydropyranyl analog 4 of natural doremox,17 a rose oxide replacement fragrance, by standard hydrogenation of the corresponding dihydropyran 2b, as shown in Scheme 4. Interestingly, 4 is also a regiosomeric analog of 2,2,5-trimethyl-5-phenyltetrahydropyran, which has been described as a synthetic odorant with a fragrance profile of fresh methyl pamplemousse odor and woody and camphoraceous notes.18
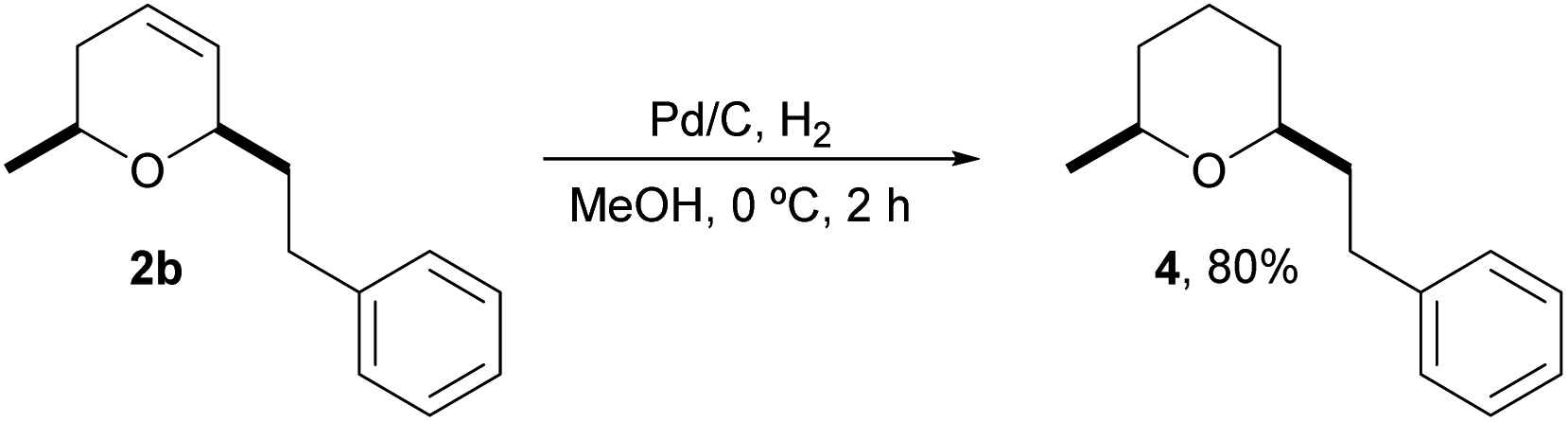 | ||
| Scheme 4 Access to truncated natural doremox. | ||
Conclusions
In conclusion, a general, effective and selective methodology for the synthesis of a variety of disubstituted dihydropyrans is described. The process is compatible with different substituents and functional groups both in the alcohol and the aldehyde. Moreover, excellent stereoselectivity towards the cis-2,6-disubstituted dihydropyrans are obtained in most cases. The applicability of this methodology has been shown by the synthesis of analogs of natural rhopaloic acid and natural doremox fragrance.Author contributions
L. F. P. for project administration, investigation and methodology development, P. G.-A. for investigation and A. B. for conceptualization, supervision and writing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Ministry of Science and Innovation, Spain for financial support by grants TED2021-131705B-C21 and PID2021-125909OB-I00. L. F.-P. and P. G.-A. acknowledge predoctoral Grants funded by either the European Social Fund and the “Junta de Castilla y León” or the University of Valladolid.Notes and references
- D. Kumar, P. Sharma, H. Singh, K. Nepali, G. K. Gupta, S. K. Jain and F. Ntie-Kang, The value of pyrans as anticancer scaffolds in medicinal chemistry, RSC Adv., 2017, 7, 36977–36999 RSC.
- M. Yanai, S. Ohta, E. Ohta and S. Ikegami, Novel norsesterpenes, which inhibit gastrulation of the Starfish embryo, from the marine sponge Rhopaloeides sp, Tetrahedron, 1998, 54, 15607–15612 CrossRef CAS.
- H. Kobayashi, M. Kanematsu, M. Yoshida and K. Shishido, Efficient access to a dihydropyran-containing macrolide via a transannular oxy-Michael reaction: total synthesis of (+)-aspergillide C, Chem. Commun., 2011, 47, 7440–7442 RSC.
- A. Budakoti, P. K. Mondal, P. Verma and J. Khamrai, Prins cyclization-mediated stereoselective synthesis of tetrahydropyrans and dihydropyrans: an inspection of twenty years, Beilstein J. Org. Chem., 2021, 17, 932–963 CrossRef CAS.
- (a) C. Olier, M. Kaafarani, S. Gastaldi and M. P. Bertrand, Synthesis of tetrahydropyrans and related heterocycles via prins cyclization; extension to aza-prins cyclization, Tetrahedron, 2010, 66, 413–445 CrossRef CAS; (b) X. Han, G. Peh and P. E. Floreancig, Prins-Type Cyclization Reactions in Natural Product Synthesis, Eur. J. Org Chem., 2013, 2013, 1193–1208 CrossRef CAS.
- (a) A. Barbero, A. Diez-Varga, M. Herrero and F. J. Pulido, From Silylated Trishomoallylic Alcohols to Dioxaspiroundecanes or Oxocanes: Catalyst and Substitution Influence, J. Org. Chem., 2016, 81, 2704–2712 CrossRef CAS; (b) A. Diez-Varga, H. Barbero, F. J. Pulido, A. González-Ortega and A. Barbero, Competitive Silyl-Prins Cyclization versus Tandem Sakurai-Prins Cyclization: An Interesting Substitution Effect, Chem.–Eur. J., 2014, 20, 14112–14119 CrossRef CAS PubMed.
- A. P. Dobbs, S. J. J. Guesne, S. Martinovic, S. J. Coles and M. B. Hursthouse, J. Org. Chem., 2003, 68, 7880–7883 CrossRef CAS.
- F. K. Chio, J. Warne, D. Gough, M. Penny, S. Green, S. J. Coles, M. B. Hursthouse, P. Jones, L. Hassall, T. M. McGuire and A. P. Dobbs, On the choice of Lewis acid for Prins reaction; two total syntheses of Civet, Tetrahedron, 2011, 67, 5107–5124 CrossRef CAS.
- A. P. Dobbs and S. Martinovic, The silyl-Prins reaction: a novel method for the synthesis of dihydropyrans, Tetrahedron Lett., 2002, 43, 7055–7057 CrossRef CAS.
- Y. Lian and R. J. Hinkle, BiBr3-initiated tandem addition/silyl-Prins reactions to 2,6-disubstituted dihydropyrans, J. Org. Chem., 2006, 71, 7071–7074 CrossRef CAS PubMed.
- C. Díez-Poza and A. Barbero, Unexpected Domino Silyl-Prins/Aryl Migration Process from Geminal Vinylsilyl Alcohols, Org. Lett., 2021, 23, 8385–8389 CrossRef PubMed.
- C. Díez-Poza, L. Fernández-Peña, P. González-Andrés and A. Barbero, Changing the Reaction Pathway of Silyl-Prins Cyclization by Switching the Lewis Acid: Application to the Synthesis of an Antinociceptive Compound, J. Org. Chem., 2023, 88, 6776–6783 CrossRef PubMed.
- The reaction with electron-rich aldehydes (p-MePhCHO and p-MePhCHO) doesn't proceed.
- D. A. Cruz, V. Sinka, V. S. Martín and J. I. Padrón, Iron-Catalyzed Prins-Peterson Reaction for the Direct Synthesis of Δ4-2,7-Disubstituted Oxepenes, J. Org. Chem., 2018, 83, 12632–12647 CrossRef CAS PubMed.
- L. F. Peña, E. López, A. Sánchez-González and A. Barbero, Diastereoselective Synthesis of cis-2,6-Disubstituted dihydropyrane Derivatives through a Competitive Silyl-Prins Cyclization versus Alternative Reaction Pathways, Molecules, 2023, 28, 3080 CrossRef.
- (a) R. Jasti and S. D. Rychnovsky, Racemization in Prins Cyclization Reactions, J. Am. Chem. Soc., 2006, 128, 13640–13648 CrossRef CAS; (b) R. Jasti, C. D. Anderson and S. D. Rychnovsky, Utilization of an Oxonia-Cope Rearrangement as a Mechanistic Probe for Prins Cyclizations, J. Am. Chem. Soc., 2005, 127, 9939–9945 CrossRef CAS; (c) S. R. Crosby, J. R. Harding, C. D. King, G. D. Parker and C. L. Willis, Oxonia-Cope Rearrangement and Side-Chain Exchange in the Prins Cyclization, Org. Lett., 2002, 4, 577–580 CrossRef CAS PubMed.
- P. Kraft, J. A. Bajgrowicz, C. Denis and G. Frater, Odds and trends: recent developments in the chemistry of odorants, Angew. Chem., Int. Ed., 2000, 39, 2980–3010 CrossRef CAS.
- B. Förster, R. Bertermann, P. Kraft and R. Tacke, Sila-rhubafuran and Derivatives: Synthesis and Olfactory Characterization of Novel Silicon-Containing Odorant, Organometallics, 2014, 33, 338–346 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07520f |
| This journal is © The Royal Society of Chemistry 2024 |