
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective fluorescence turn-on detection of combination cisplatin–etoposide chemotherapy based on N-CDs/GSH-CuNCs nanoprobe†
Khalid Alhazzania,
Ahmed Z. Alanazia,
Aya M. Mostafabc,
James Barkerb,
Mohamed M. El-Wekilc and
Al-Montaser Bellah H. Ali *c
*c
aDepartment of Pharmacology and Toxicology, College of Pharmacy, King Saud University, Riyadh, Saudi Arabia
bSchool of Life Sciences, Pharmacy, and Chemistry, Kingston University, Kingston-upon-Thames, London KT1 2EE, UK
cDepartment of Pharmaceutical Analytical Chemistry, Faculty of Pharmacy, Assiut University, Assiut, Egypt. E-mail: Almontaser_bellah@aun.edu.eg
First published on 11th January 2024
Abstract
Cisplatin (CIS) and etoposide (ETP) combination therapy is highly effective for treating various cancers. However, the potential for pharmacokinetic interactions between these drugs necessitates selective sensing methods to quantitate both CIS and ETP levels in patient's plasma. This work develops a dual fluorescence probe strategy using glutathione-capped copper nanoclusters (GSH-CuNCs) and nitrogen-doped carbon dots (N-CDs) for the simultaneous analysis of CIS and ETP. The fluorescence signal of GSH-CuNCs at 615 nm increased linearly with CIS concentration while the N-CD emission at 480 nm remained unaffected. Conversely, the N-CD fluorescence was selectively enhanced by ETP with no interference with the CuNC fluorescence. Extensive materials characterization including UV-vis, fluorescence spectroscopy, XRD, and TEM confirmed the synthesis of the nanoprobes. The sensor showed high sensitivity with limits of detection of 6.95 ng mL−1 for CIS and 7.63 ng mL−1 for ETP along with excellent selectivity against potential interferences in rabbit plasma. Method feasibility was demonstrated with application to real rabbit plasma samples. The method was further applied to estimate the pharmacokinetic parameters of CIS before and after ETP coadministration. The dual nanoprobe sensing strategy enables rapid and selective quantitation of CIS and ETP levels to facilitate therapeutic drug monitoring and optimization of combination chemotherapy regimens.
1. Introduction
Cisplatin (CIS) is a widely used chemotherapeutic agent for treating various solid malignancies including testicular, ovarian, bladder, and lung cancers.1,2 CIS contains a platinum(II) center that forms nuclear DNA crosslinks, blocking replication and transcription to induce cytotoxicity.3 Meanwhile, etoposide (ETP) is a topoisomerase II inhibitor employed for treating testicular cancer, lung cancer, lymphoma, and leukemia.4,5 Due to their different mechanisms of cytotoxicity, CIS and ETP are often combined as part of chemotherapy regimens for refractory germ cell tumors and small cell lung cancer.6–9 The two agents work synergistically to induce cell cycle perturbations and DNA damage leading to apoptotic cell death.7,9 CIS exhibits dose-dependent elimination, meaning its clearance slows with increasing doses. This necessitates careful dose adjustments to avoid dose-limiting nephrotoxicity, which occurs in nearly one-third of patients exceeding 100 mg m−2. Although its distribution half-life is short (25–49 min), its elimination half-life is greater than 3 days which contributes to prolonged side effects like nausea, vomiting, and ototoxicity. A standard dose of 75–100 mg m−2 is administered intravenously for 3 weeks, with dose modifications guided by renal function and individual tolerability.10 ETP's pharmacokinetics demonstrate elements of non-linearity, with greater than proportional increases in drug exposure at higher doses. This biphasic elimination features an initial rapid clearance followed by a slower terminal phase with a half-life of 4–11 hours. The typical dosing range is 50–100 mg per m2 per day for 3–5 days, repeated every 3–4 weeks, but varies depending on the specific treatment regimen. Myelosuppression, particularly neutropenia, is the primary dose-limiting toxicity, with the incidence of severe cases depending on the specific dose and schedule used, not simply exceeding a certain threshold.11However, both drugs share certain pharmacokinetic properties that raise concerns about potential interactions. Specifically, CIS and ETP are extensively protein-bound in plasma and undergo partly overlapping metabolic pathways including CYP3A4 metabolism.12–16 These similarities create risk for displacement interactions at protein binding sites and metabolic enzyme inhibition between the two agents when given concurrently.17,18 Such interactions can lead to excessive free drug levels and systemic buildup of CIS when co-administered with ETP. This may lead to toxicity including myelosuppression, nephrotoxicity, peripheral neuropathy, and ototoxicity.19,20 Careful monitoring of CIS pharmacokinetics and adverse effects is warranted to ensure optimal efficacy while avoiding potentiation of toxicity. Dose adjustments may be required to mitigate harm from interactions between these first-line chemotherapeutics. The studied drugs were previously analyzed utilizing various analytical techniques, including HPLC (High-Performance Liquid Chromatography),21–25 spectrophotometric,26,27 spectrofluorometric,28–34 and electrochemical methods.35–39
Fluorescent metal nanoclusters are emerging as a new class of nanoscale fluorophores.40 Metal nanoclusters with particle sizes below 10 nm exhibit bright photoluminescence emission due to quantum confinement effects.41 Their ultra-small sizes, large Stokes shifts, good photostability, and biocompatibility make them ideal for a wide range of sensing and imaging applications.40 Different metal nanoclusters have been developed including gold, silver, copper, and platinum nanoclusters.42,43 These can be stabilized using various ligands and capping agents such as DNA, glutathione (GSH), proteins, polymers, and dendrimers.40 Reported applications include chemical sensing, bioimaging, drug delivery, photodynamic therapy, and optoelectronics. For example, DNA-stabilized silver nanoclusters have been applied for metal ion detection44 and GSH-protected gold nanoclusters for live cell imaging.45 The choice of ligands for stabilizing metal nanoclusters can significantly impact their properties.40 GSH-coated copper nanoclusters in particular offer advantages such as ease of preparation, low toxicity, good water solubility, and long-term stability.46–48 They exhibit strong photoluminescence and high quantum yields while maintaining small hydrodynamic sizes under 5 nm.48 The carboxyl, thiol, and amino groups of GSH allow excellent surface passivation and water dispersibility.49 Additionally, the copper centers provide multifunctionality for catalysis and sensing.50
Glutathione-protected copper nanoclusters (GSH-CuNCs) were applied as fluorescent sensors for the determination of CIS. The strong coordination between copper and the sulfur groups of glutathione provides stabilization and aqueous dispersibility. Additionally, the carboxyl and amino groups of GSH allow effective surface passivation to yield highly fluorescent copper nanoclusters.51 CIS can interact with GSH-stabilized copper nanocluster system leading to aggregation-induced emission enhancement (AIEE).52 However, ETP preclude such interactions with copper nanoclusters. Therefore, fluorescent carbon dots were used as probes for ETP sensing. Carbon dots (CDs) offer many advantages like low toxicity, chemical stability, and tunable photoluminescence.53 They have diverse applications in biosensing, bioimaging, photocatalysis, and drug delivery.54 Hydrothermal approaches provide a simple route for synthesizing carbon dots from various precursors.55 To prepare nitrogen-doped carbon dots (N-CDs) in this study, a hydrothermal method was employed using a mixture of citric acid and triphenylamine.
Fluorescent nanomaterials like metal nanoclusters and carbon dots have previously been applied for multichannel sensing of various analytes. For example, combined carbon dots and gold nanoclusters were utilized for selective quantitation of multiple metal ions.56 Dual emissive carbon dots and copper nanoclusters were developed for arginine and acetaminophen sensing.57 In this work, we pioneer the first use of dual emissive GSH-CuNCs and N-CDs for fluorometric quantitation of the important chemotherapy agents; CIS and ETP. CIS and ETP are first-line treatment for various tumors, but co-administration raises concerns of pharmacokinetic interactions impacting efficacy and safety. However, analytical methods to investigate such interactions have been limited. This work addresses the need for a simple technique to study CIS and ETP pharmacokinetics. GSH-CuNCs and N-CDs were rationally designed as probes selective to CIS and ETP, respectively. By enabling rapid, mix-and-detect analysis of both drugs, this dual nanoprobe can unlock new pharmacokinetic insights to guide combination chemotherapy.
2. Experimental
2.1. Reagents and chemicals
CIS (99.0%) and ETP (99.0%) were retrieved from Hikma Pharmaceuticals (Cairo, Egypt). Unistin® vial containing 100 mg CIS per vial and Etopul® vial containing 100 mg ETP per vial were obtained from local drug store. Citric acid and triphenylamine were acquired from Sigma-Aldrich (Steinheim, Germany). Glutathione (GSH), sodium hydroxide (NaOH) and copper sulphate pentahydrate (CuSO4·5H2O) were obtained from Fluka (Buchs, Switzerland). For the preparation of all aqueous solutions in this study, double distilled water was used.2.2. Instrumentation
A Shimadzu RF-5301PC fluorescence spectrometer (Tokyo, Japan) was utilized for the fluorescence emission spectrum measurements. The instrument was equipped with a 1 cm quartz cell and employed a 5 nm slit width. UV-visible measurements were performed on Shimadzu UV-1601 (Tokyo, Japan). Transmission electron microscopy (TEM) images were captured from (JEOL, JEM-1400, 200 kV, Japan). FT-IR spectral studies were performed using a Nicolet 6700 FT-IR series spectrophotometer with KBr pellets as the source material, covering a spectral range from 4000 cm−1 to 400 cm−1. The X-ray diffraction pattern was performed on Philips PW 1700 X-ray diffractometer (Eindhoven, Netherlands). X-ray photoelectron spectroscopy (XPS) data were acquired using an ESCALAB 250 XI instrument (Thermo Scientific) attached with an Al Kα X-ray radiation as excitation source. Particle size measurements were conducted using a Zetasizer Nano analyzer.2.3. Preparation of N-CDs/GSH-CuNCs
To ensure the cleanliness of all glassware employed in the synthesis of GSH-CuNCs, they were immersed in a freshly prepared aqua regia solution for a duration of 10 hours, followed by thorough rinsing with double distilled water. The proposed procedure was conducted following previously reported method with minor modifications.58 In a 50 mL round-bottomed flask, 0.1 M solution of GSH (1 mL) was added to a 0.1 M solution of CuSO4 (0.1 mL) in a 50 mL round-bottomed flask while continuously stirred. This transparent solution underwent a transformation, resulting in a white suspension. Subsequently, a 1.0 M NaOH solution was cautiously added drop by drop until the previously turbid liquid became transparent and acquired a light-yellow color. The resulting nanoclusters were subjected to centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 minutes. Subsequently, the supernatant was collected and lyophilized. The obtained powder of GSH-CuNCs (yield = 32% ± 3%, n = 3 syntheses) stored at 4 °C for further experimentation.
000 rpm for 10 minutes. Subsequently, the supernatant was collected and lyophilized. The obtained powder of GSH-CuNCs (yield = 32% ± 3%, n = 3 syntheses) stored at 4 °C for further experimentation.
N-CDs were synthesized using a one-step hydrothermal method. To initiate the process, a mixture containing 3.0 g of citric acid and 3.0 g of triphenylamine was added to 25 mL of a mixture comprising an equal ratio of double distilled water. The mixture was thoroughly mixed until complete dissolution was achieved. Subsequently, the obtained solution was moved into a Teflon-lined stainless-steel autoclave with a capacity of 50 mL and subjected to heating at 200 °C for duration of 8 hours. After the reaction, the solution was centrifuged at 5000 rpm for 10 minutes, and the resulting supernatant was subsequently filtered through a 0.22 μm syringe filter to remove any impurities. To purify the N-CDs and remove unreacted reagents or molecular fluorescent impurities, the filtered solution underwent dialysis for 48 hours using 1000 Da dialysis bag. The N-CDs particles was acquired by lyophilization. The prepared N-CDs (yield = 28% ± 5%, n = 5 syntheses) was stored at 4 °C until use.
2.4. Fluorescence quantum yields (QYs) of N-CDs/GSH-CuNCs
For detailed procedures, please refer to the ESI file.†2.5. Simultaneous fluorescence detection of ETP and CIS using N-CDs/GSH-CuNCs
To prepare N-CDs/GSH-CuNCs dual sensor, 1 mL of N-CDs (1 mg mL−1) and 4 mL of GSH-CuNCs (0.5 mg mL−1) were added to 5.0 mL double distilled water. The resulting mixture was stirred thoroughly at room temperature. The fluorescence spectrum of the N-CDs/GSH-CuNCs was measured at excitation wavelength of 380 nm and emission wavelengths of 480 nm and 615 nm. In the experimental procedure, a 1.0 mL solution of N-CDs/GSH-CuNCs was combined with 100 μL of ETP and 100 μL of CIS, each with varying concentrations. The prepared solution was then diluted to a total volume of 10 mL in a volumetric flask using Britton–Robinson buffer (pH 4.0, 10 mmol L−1). After incubating the solution for 120 seconds to facilitate interaction, the fluorescence spectra were measured and recorded.2.6. Sample preparation
The precipitation method for plasma sample preparation is a cost-effective approach that has been demonstrated to yield comparable results when compared to the conventional plasma ultrafiltration technique, as reported in the literature.59 For the measurement of unbound CIS concentrations, proteins in 1 mL of plasma samples were precipitated by adding 1 mL of 20% cold trichloroacetic acid. Following a 10 minute incubation on ice, the samples were centrifuged at 10000 rpm for 10 minutes at a temperature of 4 °C. The resulting supernatant was then analyzed for unbound concentrations of both CIS and ETP.
2.7. Animals
Assiut University Ethics Committee has granted the approval for the experimental procedures involving the care and use of animals, and all the experiments were performed in accordance with the principles outlined in the Declaration of Helsinki. Eight male New Zealand white rabbits, with an average weight of approximately 2.5 ± 0.2 kg, were procured from an authorized animal supplier located in Assiut, Egypt. These rabbits were housed in a controlled environment free from pathogens and provided with unlimited access to food. The room temperature was maintained at 20 ± 2 °C, while the relative humidity was maintained at 40 ± 5%. Prior to the initiation of the experiment, the rabbits underwent a fasting period of approximately 12 hours, during which they were allowed access to water.2.8. Determination of plasma pharmacokinetics
A group of eight male New Zealand white rabbits (n = 8) received a single intravenous dose of 5 mg per kg CIS. Following a washout period of 14 days to ensure clearance of any remaining CIS from the blood of the test rabbits, the same rabbits were administered CIS (5.0 mg kg−1, intravenous infusion) in combination with ETP at a dose of 5.0 mg kg−1 via intravenous infusion. Blood was drawn from rabbits through the marginal ear vein. Blood samples were drawn before the start of infusion (blank plasma) and at 0.25, 0.5, 1, 1.5, 3, 5, 7, 10, 14, 18, and 24 h after the first infusion. Heparinized tubes were used to receive the collected blood samples, and plasma was obtained by centrifuging at 5000 rpm for 10 minutes. The plasma samples were frozen at −20 °C until analysis. Plasma concentrations of CIS were individually plotted versus time, and a pharmacokinetic analysis was conducted utilizing a two-compartment model with Win-Nonlin Professional software, provided by Scientific Consulting, Inc., based in Lexington, KY, USA. Several key parameters were examined in this pharmacokinetic study to characterize the drug concentration–time profile. These included the total drug exposure quantified by area under the curve (AUC) from 0 to 24 hours and extrapolated to infinity, peak plasma level (Cmax), the first-order elimination rate constant (kel), distribution half-life representing the initial rapid distribution phase (t1/2α), terminal elimination half-life (t1/2β), mean residence time (MRT), volume of distribution in the central (Vc), and peripheral compartments (Vp) denoting drug tissue partitioning, and total body clearance (CL). The pharmacokinetic parameters were reported as mean plus or minus the standard deviation to convey the central tendency and variability in the measured values. Pharmacokinetic calculations for the samples were conducted by Graph Pad Prism (version 3.02, Graph Pad Software, San Diego, CA, USA).3. Results and discussions
3.1. Characterizations and morphology of N-CDs/GSH-CuNCs
The morphology of N-CDs and GSH-CuNCs was characterized by TEM. As depicted in Fig. 1A, the prepared N-CDs were uniformly distributed and had good dispersion. The inset of Fig. 1A presented the particle size distribution of N-CDs with an average diameter of 4.4 ± 0.50 nm. As shown in Fig. 1B, GSH-CuNCs were spherical and had monodispersed dispersion. The particle-size distribution of GSH-CuNCs is shown in the inset of Fig. 1B (The average particle diameter is 2.51 ± 0.30 nm).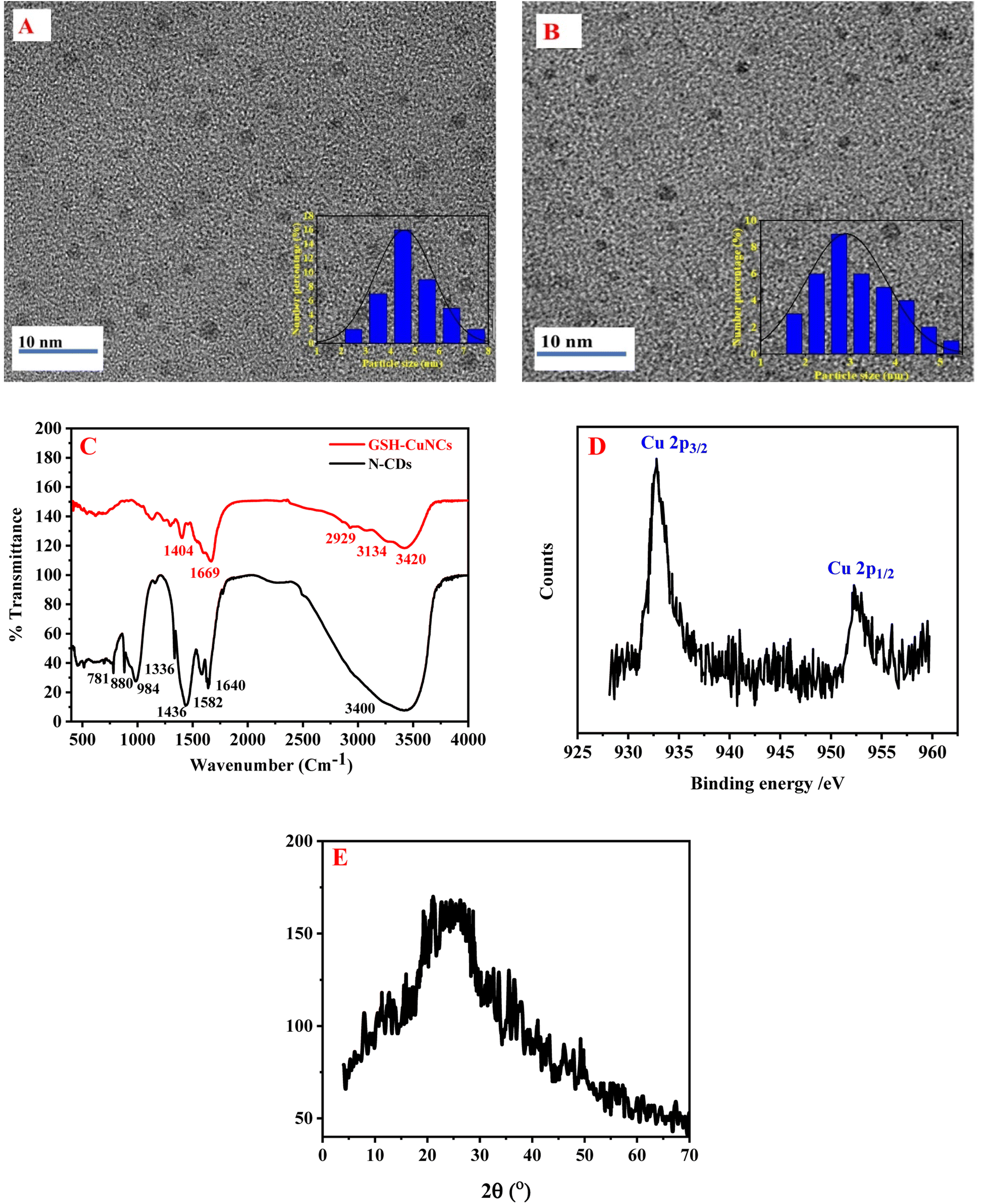 | ||
| Fig. 1 (A) TEM image of N-CDs, (B) TEM image of CuNCs, (C) FT-IR of N-CDs and GSH-CuNCs, (D) high resolution XPS spectra of the Cu 2p in CuNCs (E) XRD pattern of N-CDs. | ||
The FT-IR spectra of N-CDs and GSH-CuNCs are depicted in Fig. 1C. The FT-IR spectrum of N-CDs displayed a peak at 3400 cm−1, which can be assigned to O–H and N–H stretching vibrations. The sharp peak at 1640 cm−1 corresponds to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of carbonyl groups, likely from carboxylic acids and amides. The peak at 1582 cm−1 can be associated with CC stretching vibrations of aromatic rings. The bending vibrations of methyl and methylene groups resulted in sharp peak at 1436 cm−1. The peak at 1336 cm−1 can be allocated to C–N stretching vibrations, implying that nitrogen functionalities like amines or amides are present. The peak at 984 cm−1 is indicative of C–O stretching in carboxylic acids. Finally the peaks at 880 cm−1 and 781 cm−1 correspond to out-of-plane C–H bending of aromatics, further confirming the presence of aromatic groups.60 The FTIR spectrum suggests the N-CDs contain hydroxyl, carbonyl, carboxyl, amine/amide, aromatic, and aliphatic functional groups.
O stretching of carbonyl groups, likely from carboxylic acids and amides. The peak at 1582 cm−1 can be associated with CC stretching vibrations of aromatic rings. The bending vibrations of methyl and methylene groups resulted in sharp peak at 1436 cm−1. The peak at 1336 cm−1 can be allocated to C–N stretching vibrations, implying that nitrogen functionalities like amines or amides are present. The peak at 984 cm−1 is indicative of C–O stretching in carboxylic acids. Finally the peaks at 880 cm−1 and 781 cm−1 correspond to out-of-plane C–H bending of aromatics, further confirming the presence of aromatic groups.60 The FTIR spectrum suggests the N-CDs contain hydroxyl, carbonyl, carboxyl, amine/amide, aromatic, and aliphatic functional groups.
The FT-IR spectrum of GSH-CuNCs displayed a peak at 3420 cm−1 attributable to O–H stretching vibrations. Additionally, a peak at 1669 cm−1 corresponded to CO stretching. These results indicate that carboxylic acid groups are present on the synthesized GSH-CuNCs surfaces. Peaks were also observed at 2929 cm−1 and 1404 cm−1, related to C–H stretching and bending vibrations, respectively. A shoulder peak at 3134 cm−1 was associated with N–H stretching, suggesting amine functionalities. The FT-IR spectrum of the GSH-CuNCs confirmed the presence of carboxyl, hydroxyl, and amine surface groups derived from the capping GSH ligands. Notably, the spectrum lacked a peak at 2522 cm−1 related to S–H stretching vibrations of free GSH.46 The absence of free thiol groups implies covalent Cu–S bonds are formed between the copper core and GSH capping agents.
The copper oxidation state in CuNCs was studied using X-ray photoelectron spectroscopy (XPS) (Fig. 1D). The XPS measurements revealed two intense peaks at 932.8 and 952.5 eV, which can be credited to the Cu 2p3/2 and Cu 2p1/2 signals of metallic copper (Cu0), respectively. Critically, the absence of an additional satellite peak around 942 eV indicates that Cu2+ species are not present. The lack of Cu2+ suggests that the Cu2+ precursor used in the nanoclusters synthesis was fully reduced to Cu0 during the preparation method. Therefore, the XPS results provide evidence that the one-pot reaction successfully generated CuNCs containing only zerovalent copper.
XPS analysis was conducted to further explore the chemical compositions of the N-CDs. The XPS results, as shown in Fig. S1A,† revealed the presence of three main element peaks: C 1s, N 1s, and O 1s. The C 1s spectrum (Fig. S1B†) exhibited three distinct bands at 283.93 eV (representing C–C/CC bonds), 284.67 eV (indicating C–N/C–O bonds), and 285.92 eV (corresponding to CN bonds). The N 1s peak (Fig. S1C†) was resolved into three peaks at 399, 399.5, and 400.2 eV, which were assigned to pyridinic N, pyrrolic N, and amino N, respectively. The O 1s spectrum (Fig. S1D†) displayed peaks corresponding to CO at 531.2 eV, C–O at 531.93 eV, and O–H groups at 533.12 eV.61–64
The X-ray diffraction (XRD) pattern shown in Fig. 1E displays a diffraction peak at 25.5°, which is associated with the (0 0 2) diffraction angle of carbon. This peak indicates that the N-CDs possess an amorphous structure.60
3.2. Photoluminescence properties of N-CDs/GSH-CuNCs
N-CDs and GSH-CuNCs were characterized using UV-visible absorption spectroscopy and fluorescence emission spectroscopy. Fig. 2A shows broad absorption peaks around 241 nm and 300 nm for N-CDs. Additionally, noticeable fluorescence excitation and emission signals are observed at 377 nm and 480 nm, respectively. Under excitation at 365 nm using a UV lamp, N-CDs exhibit blue-emitting fluorescence, as depicted in the insets of Fig. 2A. Fig. 2B shows that the emission wavelengths of N-CDs do not depend on the excitation wavelengths. GSH-CuNCs exhibited an absorption maximum at 300 nm in the UV-visible spectrum (Fig. 2C). Excitation of the CuNCs at 380 nm resulted in strong fluorescence emission peaked at 615 nm, as revealed by the excitation and emission profiles (Fig. 2C). Under 365 nm UV illumination, the synthesized GSH-CuNCs displayed bright red fluorescence, visually confirming their emissive behavior (Fig. 2C insets). Fig. 2D presents the fluorescence emission of GSH-CuNCs under several excitation wavelengths. It is shown that GSH-CuNCs probe exhibits slight shift in emissions under various excitation wavelengths, which may be because of the slight difference in sizes of CuNCs. Excitation of GSH-CuNCs at 380 nm resulted in dual emission peaks at 480 nm and 615 nm. The quantum yield of GSH-CuNCs was measured to be 34%, while N-CDs exhibited a quantum yield of 29%.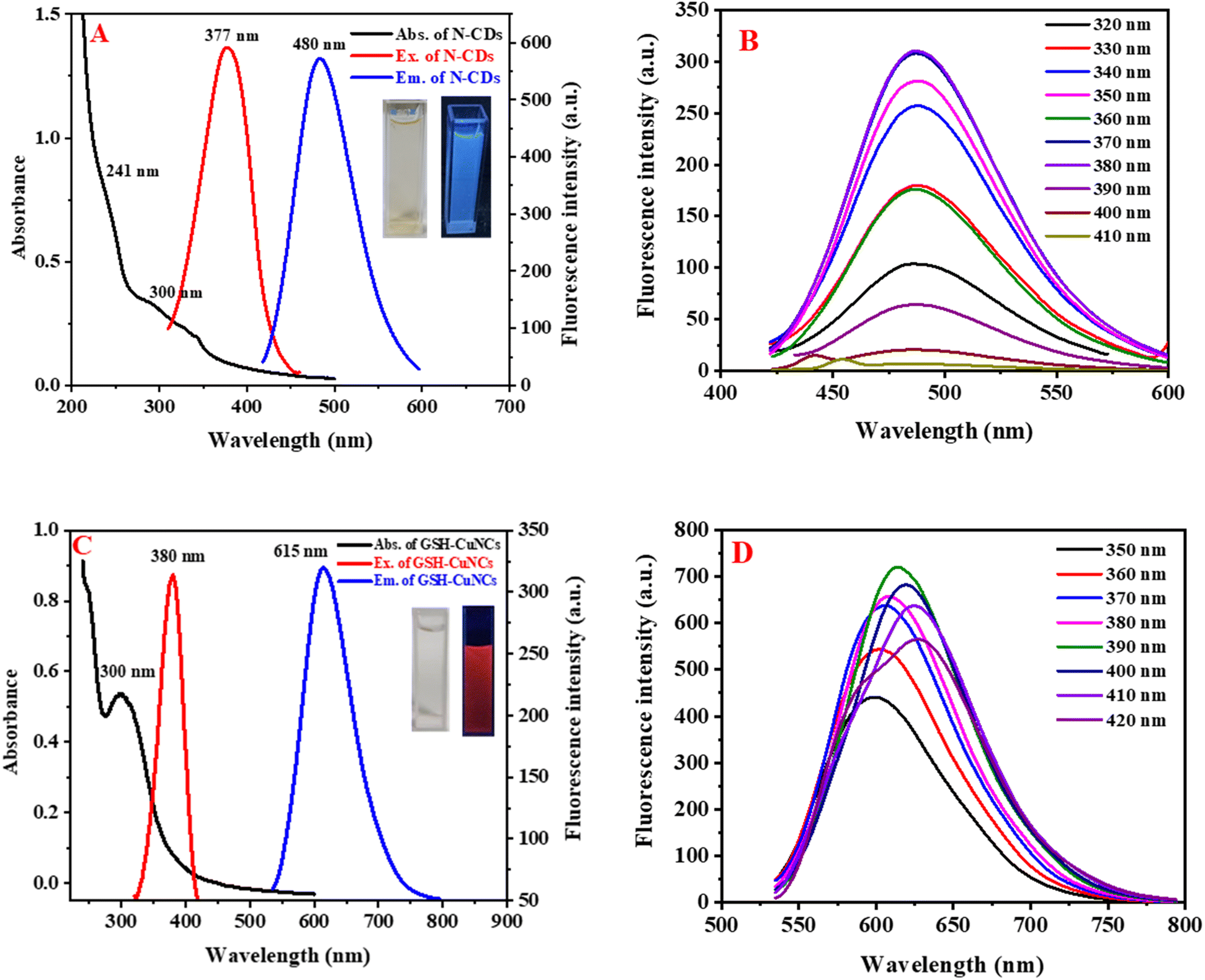 | ||
| Fig. 2 (A) UV-visible absorption (black), fluorescence excitation (red), and emission (blue) spectra of the N-CDs. Inset displays photographs under ambient light and 365 nm UV illumination. (B) Emission spectra of the N-CDs at excitation wavelengths from 320 to 410 nm. (C) UV-vis absorption (black), excitation (red) and emission (blue) spectra for the glutathione-capped copper nanoclusters. Inset shows images under visible and UV light. (D) Fluorescence emission spectra of the Cu nanoclusters obtained with excitation wavelengths ranging from 350 to 410 nm. | ||
3.3. Fluorescence stability of N-CDs/GSH-CuNCs
Various parameters were tested to evaluate the spectroscopic properties of N-CDs and GSH-CuNCs, including pH values (ranging from 2 to 11), NaCl concentrations (0.01 to 2.0 mol L−1), storage times (up to 60 days), UV irradiation (up to 3 hours), and different temperatures (20 to 60 °C). Fig. S2† illustrates the results obtained. The fluorescence intensity of N-CDs reached its maximum at pH 6 (Fig. S2A†), while the intensity of GSH-CuNCs decreased as the pH shifted from acidic to alkaline (Fig. S2B†). This pH-induced variation in fluorescence can be associated with the protonation and deprotonation of surface groups. Furthermore, the fluorescence intensity showed no significant reduction as the NaCl concentration increased from 0.01 to 2.0 mol L−1 (Fig. S2C and D†), and even after 3 hours of UV irradiation (Fig. S2E and F†). After being stored at 4 °C for 60 days (Fig. S2G and H†) and exposed to high temperature (Fig. S2I and J†), GSH-CuNCs exhibited a slight decrease in fluorescence compared to N-CDs. These findings suggest that GSH-CuNCs and N-CDs demonstrated excellent storage stability.3.4. Optimization of CDs/GSH-CuNCs for CIS and ETP determination
To obtain the strongest fluorescence signal, pH of the solution (Fig. S3A†) and incubation time (Fig. S3B†) were studied to achieve the best reaction conditions. The fluorescence quenching of GSH-CuNCs induced by the addition of CIS was found to be virtually constant across a wide pH range from 3 to 11. This pH-independent quenching suggests that the interaction between CIS and the nanoclusters is not driven by protonation or deprotonation of the GSH thiol groups. While acidic pH can potentially compete with ligand binding at the platinum center, GSH has a high affinity and can still bind and displace chloride of CIS.65 This coordination likely causes structural changes or aggregation of the nanoclusters, disrupting the metal–thiolate bonds critical for fluorescence emission. The interaction between N-CDs and ETP led to an enhancement in the fluorescence intensity of N-CDs. Interestingly, this fluorescence enhancement was noted to be at its greatest value at pH 4. It is important to mention that the fluorescence of N-CDs alone was relatively low at this pH. Despite this, pH 4 was chosen as the preferred pH for the subsequent investigations due to the significant fluorescence enhancement observed after the addition of ETP. This result suggests that the interaction between N-CDs and ETP is pH-dependent, with pH 4 providing optimal conditions for the fluorescence enhancement effect. The underlying mechanism behind this pH-dependent fluorescence enhancement could be associated with the changes in the surface properties due to hydrogen bond formation and charge distribution of the N-CDs at variable pH levels. Therefore, pH 4 is used for all subsequent measurements. The effect of incubation time on the interaction between N-CDs and ETP was investigated. The results indicated that the optimum incubation time was found to be 1.0 minute. The optimal incubation time for the interaction between CIS and nanoclusters was determined to be 120 s. Based on these findings, a simultaneous determination method for both drugs was developed, utilizing an incubation time of 120 s.3.5. Evaluation of analytical performance
The fluorescence reduction of GSH-CuNCs towards CIS suggested that it was possible to utilize GSH-CuNCs probe for CIS sensing. As displayed in Fig. S4A,† the fluorescence signal of GSH-CuNCs at 615 nm was increased with increasing the concentration of CIS while the signal at 480 nm of N-CDs is not affected. Moreover, the fluorescence intensity increased linearly with increasing the concentration of CIS from 20 to 160 ng mL−1 (Fig. S4A inset†). The linear regression between (F/F0) and CIS concentration was described as F/F0 = 0.017 [CIS] + 1.10 (R2 = 0.9964). Based on this equation, the LOD was calculated to be 5.2 ng mL−1. Where F and F0 represent the fluorescence intensities of the sensor with and without CIS, respectively. The fluorescence enhancement of N-CDs towards ETP suggested that it was possible to apply N-CDs for ETP sensing. As displayed in Fig. S4B,† the fluorescence intensity of N-CDs at 480 nm was increased with raising the concentration of ETP while the signal at 615 nm of GSH-CuNCs is not affected. The fluorescence intensity increased linearly with increasing the concentration of ETP from 25 to 200 ng mL−1 (Fig. S4B inset†). The linear correlation between (F/F0) and ETP concentration was described as F/F0 = 0.0175 [ETP] + 1.00 (R2 = 0.9964). Based on this equation, the LOD was calculated to be 7.9 ng mL−1. As a result, CIS and ETP can be simultaneously quantified using N-CDs/GSH-CuNCs as a fluorescent probe (Fig. 3A). Reliable linearity was reflected from the plotted calibration curves across the concentration range of (20–160 ng−1 for CIS and 25–200 ng mL−1 for ETP) as illustrated in Fig. 3B. The calculated regression equations were as follows:| F/F0 = 0.018 [CIS] + 1.10 (R2 = 0.9983) |
| F/F0 = 0.016 [ETP] + 1.11 (R2 = 0.9976) |
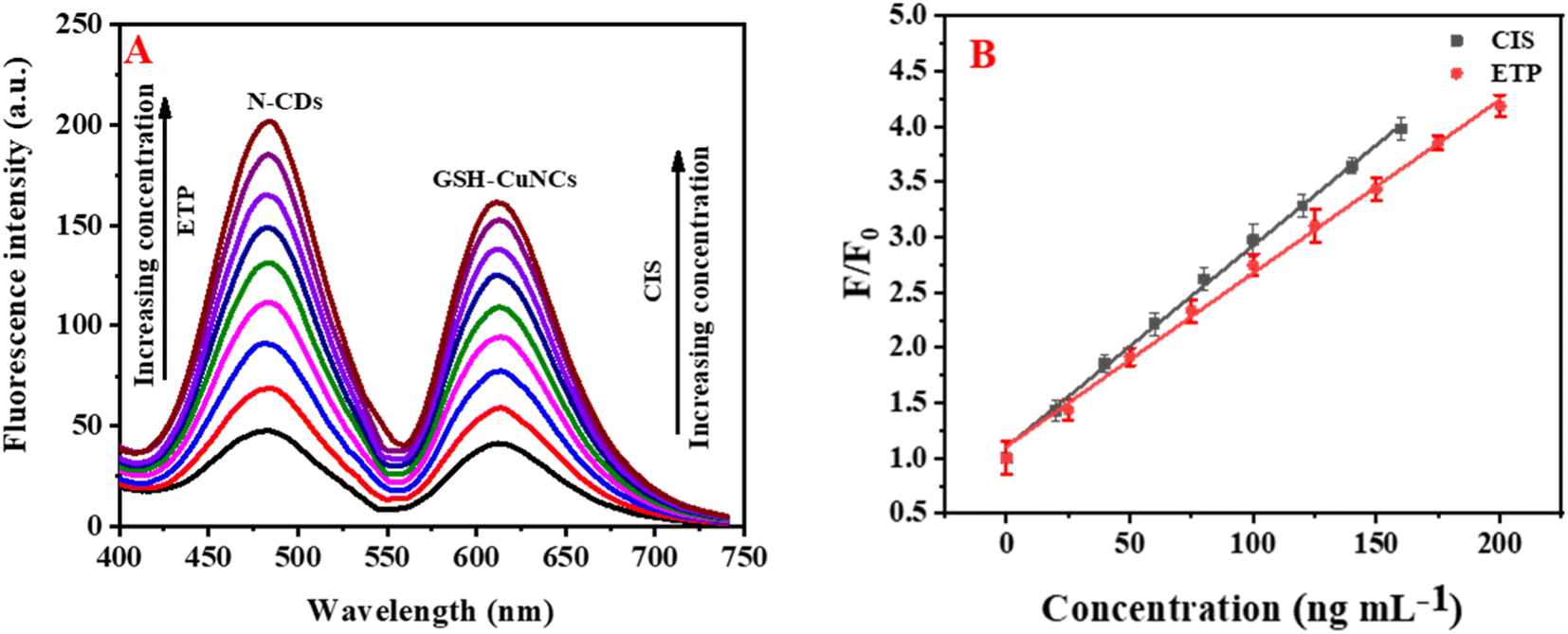 | ||
| Fig. 3 (A) The influence of variation of concentrations of CIS (20–160 ng mL−1) and ETP (25–200 ng mL−1) on the emission intensity of N-CDs/GSH-CuNCs. (B) Relationship of (F/F0) against concentration of CIS/ETP. | ||
The limits of detection (LOD) for CIS and ETP were determined to be 5.08 and 7.57 ng mL−1, respectively. Additionally, the limits of quantification (LOQ) were depicted as 15.56 and 22.93 ng mL−1 for CIS and ETP, respectively.
The sensitivity of the proposed dual nanoprobe strategy is improved compared to previous fluorescence-based techniques for quantitation of CIS and ETP, as evidenced by the LODs achieved in this work. The LOD of 6.95 ng mL−1 for CIS detection is considerably lower than LODs ranging from 24.0 to 216.0 ng mL−1 reported by other fluorescence methods (Table S1†). For ETP, the LOD of 7.63 ng mL−1 demonstrates much higher sensitivity versus the prior fluorescence approach relying on native ETP emission, which had an LOD of 23 ng mL−1.28
3.6. Selectivity of N-CDs/GSH-CuNCs system
To assess the selectivity of N-CDs/GSH-CuNCs nanoprobe, the influence of various coexisting species was examined, including K+, Na+, Fe2+, Zn2+, Mg2+, Mn2+, Ca2+, Cu2+, Al3+, Co2+, Ni2+, dopamine, ascorbic acid, glucose, cysteine, adenine, guanine, uric acid, and bovine serum albumin (BSA). In addition, the selectivity study was conducted to assess the impact of commonly co-administered pharmaceutical compounds, including penicillin, tetracycline, aspirin, ibuprofen, and paracetamol, on the performance of the N-CDs/GSH-CuNCs nanoprobe. The inclusion of these pharmaceutical compounds is crucial as they are frequently co-administered with anticancer drugs in therapeutic treatments. The final concentration for the studied drugs and possible interferences was 50.0 and 500.0 ng mL−1, respectively. Notably, the presence of these interfering species did not exhibit a remarkable effect on the fluorescence signal of the N-CDs/GSH-CuNCs nano system. However, when CIS was introduced, a pronounced variation in the fluorescence response was observed, indicating the selectivity of the N-CDs/GSH-CuNCs nanoprobe for CIS sensing (Fig. 4A). Moreover, the selectivity of the N-CDs/GSH-CuNCs nanoprobe towards ETP was investigated (Fig. 4B). The results demonstrated that the nanoprobe displayed excellent selectivity for ETP even in the presence of the aforementioned coexisting species. This finding confirms the ability of the N-CDs/GSH-CuNCs nanoprobe to selectively detect CIS and ETP while maintaining minimal interference from other substances.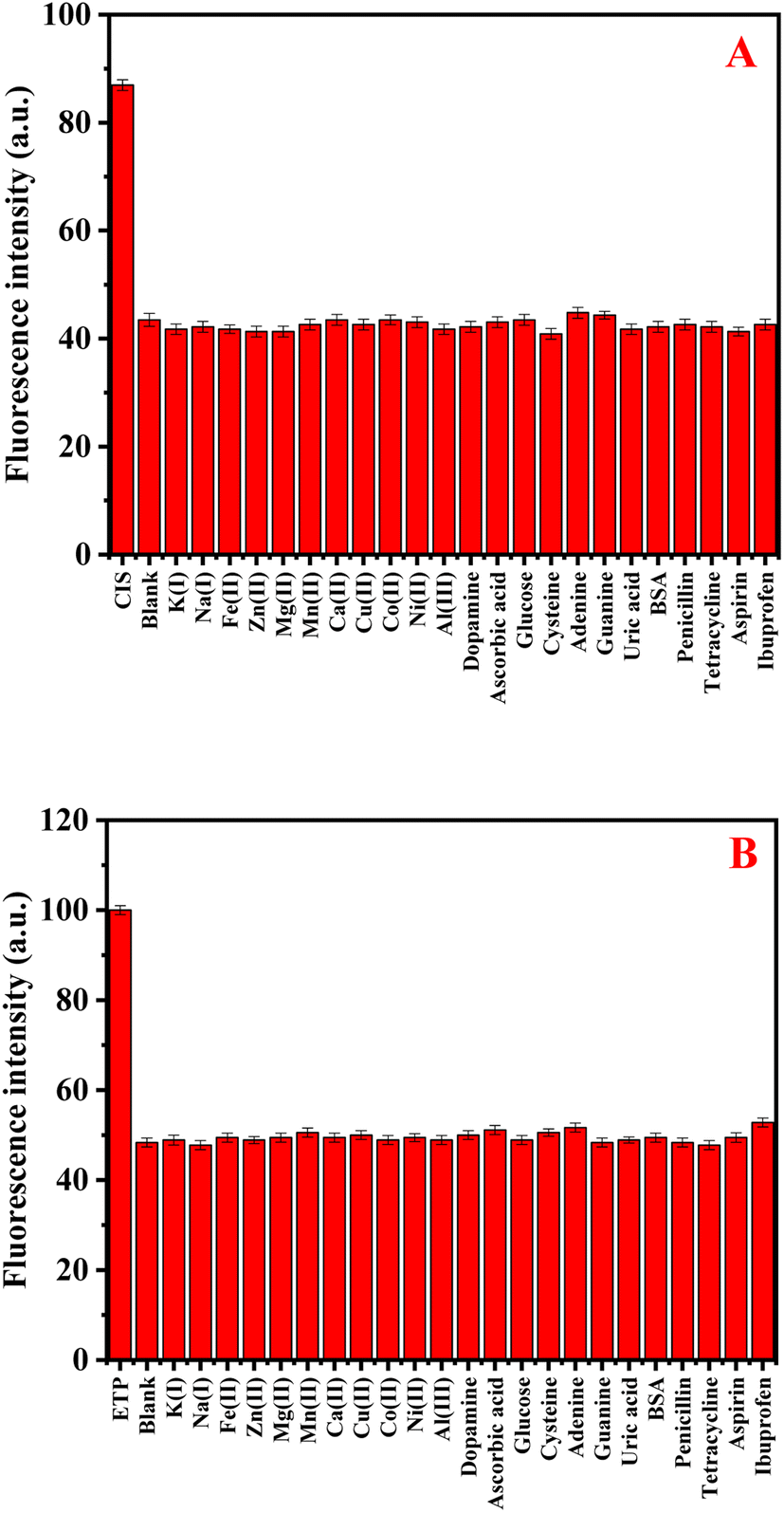 | ||
| Fig. 4 (A) The selectivity GSH-CuNCs towards CIS in the presence of different interferents species. (B) The selectivity of N-CDs towards ETP in the existence of different interferents. | ||
3.7. Sensing mechanism
CIS has been shown to selectively react with the GSH capping ligands on the surface of CuNCs. Cisplatin [PtCl2(NH3)2] contains two labile chloride ligands that can be readily substituted by sulfur donors like the cysteine residue of GSH. The thiol group of cysteine forms a coordinate bond with the platinum center of CIS, displacing a chloride ligand.65 The CIS–GSH causes aggregation of CuNCs, which has been directly visualized by TEM images. The TEM images reveal clear formation of aggregated nanocluster species upon CIS addition (Fig. S5A†). This controlled aggregation process turns on the strong fluorescence of the CuNCs, leading to a marked enhancement in the emission intensity. The fluorescence activation is attributed to several factors related to aggregation. Firstly, aggregation minimizes non-radiative relaxation pathways thus reducing exciton quenching effects. Secondly, exciton migration between closely spaced nanoclusters in aggregates enables excitation energy to funnel into emissive trap states. Finally, the CIS-induced structural changes on the nanocluster surface may improve passivation and reduce electronic defects that can favor fluorescence.66The fluorescence enhancement of N-CDs triggered by ETP addition can be attributed to specific intermolecular interactions between the nanoparticles and drug.67,68 N-CDs possess abundant oxygen-containing functional groups like carboxylates and hydroxyls along with amine moieties on their surface. These groups provide sites for hydrogen bonding with the multiple hydroxyl substituents present in ETP's structure. The binding of ETP to N-CDs also induces a noticeable decrease in the hydrodynamic diameter of the nanoparticles, as shown in TEM images (Fig. S5B and C†). The disaggregation effect provides further evidence for specific intermolecular interactions driving the fluorescence enhancement. ETP binding appears to disrupt weak attractive forces between adjacent N-CDs in aggregated dots. This reduces interparticle contacts, thereby decreasing the ensemble particle size. Relatedly, disaggregation helps spatially isolate N-CDs and prevents fluorescent quenching mechanisms that arise from direct dot–dot interactions. The changes in both optical properties and physical dimensions indicate ETP intercalates within existing N-CD aggregates, interacting with multiple dot surfaces to lower interparticle cohesion. Breaking up of N-CD assemblies into smaller, discrete dots underlies the fluorescence turn-on by eliminating aggregation-induced quenching pathways.
3.8. Method applications
| F/F0 = 0.019 [CIS] + 1.02 (R2 = 0.9939) |
| F/F0 = 0.017 [ETP] + 1.05 (R2 = 0.9973) |
The estimated LOD for CIS and ETP were calculated to be 6.95 and 7.63 ng mL−1, respectively, while LOQ were determined to be 21.01 and 23.12 ng mL−1, correspondingly.
| Parameters | CIS | CIS in presence of ETP |
|---|---|---|
| a Indicates statistically significant differences between parameter values of CIS and CIS/ETP (p < 0.05). | ||
| Cmax (ng mL−1) | 700 ± 81.07 | 720 ± 45.12 |
| t1/2α (h) | 0.35 ± 0.074 | 0.41 ± 0.058 |
| t1/2β (h) | 5.98 ± 0.87 | 6.73 ± 0.18a |
| Vc (L kg−1) | 0.094 ± 0.031 | 0.087 ± 0.024 |
| Vp (L kg−1) | 21.5 ± 0.83 | 20.2 ± 0.55 |
| kel (h−1) | 0.12 ± 0.024 | 0.103 ± 0.054 |
| CL (L kg−1 h−1) | 0.046 ± 0.051 | 0.041 ± 0.041 |
| MRT (h) | 6.42 ± 0.15 | 7.18 ± 0.72a |
| AUC0–24 (ng h mL−1) | 8700 ± 7955.56 | 9450 ± 647.02 |
| AUC0–∞ (ng h mL−1) | 9300 ± 9850.81 | 10200 ± 735.45 |
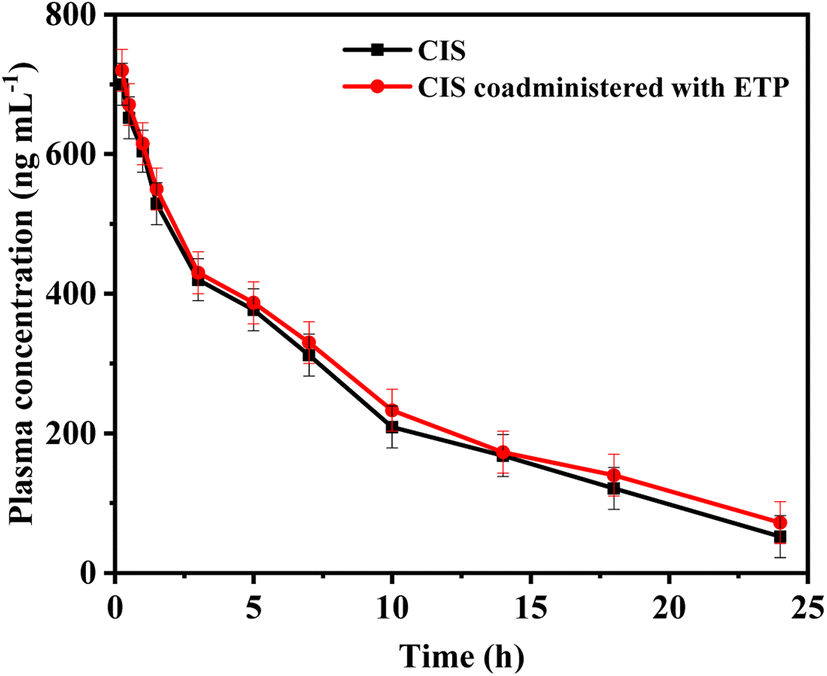 | ||
| Fig. 5 Comparison between mean CIS concentrations over time with and without ETP administration. | ||
3.9. Real sample analysis and potential limitations
While this dual nanoprobe strategy demonstrated excellent sensitivity and selectivity for quantitation of CIS and ETP at pharmacologically relevant concentrations in rabbit plasma, analysis at the lower clinically relevant levels in human patient samples may be challenging without further optimization. The current detection limits of CIS and ETP may require preconcentration or analyte enrichment steps for application to human plasma samples. Testing CIS and ETP spiked into pooled human plasma at these lower clinically relevant concentrations represents an important future direction, along with optimization of the assay sensitivity if needed via enhanced fluorescence signaling or reduced matrix interference. Variations in matrix composition and physiological conditions between rabbit and human plasma may influence the sensor's performance. Additionally, the pharmacokinetic parameters estimated in rabbit plasma may not precisely mirror those in humans, necessitating further validation studies. The potential interferences in human plasma, distinct from those encountered in rabbit plasma, should be systematically investigated to ensure the method's robustness and reliability. Addressing these challenges will be crucial in establishing the clinical utility of the dual fluorescence probe strategy for simultaneous analysis of CIS and ETP in human samples.4. Conclusions
In conclusion, this study highlights the significance of monitoring combination therapy using cisplatin (CIS) and etoposide (ETP) in cancer treatment. To address this, a fluorescence-based probe utilizing glutathione-stabilized nanoclusters and nitrogen-doped carbon dots (N-CDs/GSH-CuNCs) was developed for their simultaneous sensing. The proposed probe exhibited excellent selectivity and sensitivity. Moreover, the application of the proposed method to spiked rabbit plasma samples further validated its robustness. By estimating the pharmacokinetic parameters of CIS before and after coadministration with ETP, this study provided valuable insights into potential pharmacokinetic interactions and the necessity of dose adjustment. The results revealed a minor pharmacokinetic interaction with ETP slightly prolonging the half-life and clearance time of CIS. This indicates combination of CIS and ETP therapy requires monitoring but no dose adjustments. Compared to conventional methods like HPLC or LC-MS, this rapid and simple fluorescence strategy for the quantification of CIS and ETP offers significant advantages in terms of cost-effectiveness, ease of operation, and high throughput analysis, making it promising for clinical applications requiring frequent drug monitoring. This research contributes to the development of effective therapeutic strategies and analytical methods for optimizing combination therapies in cancer treatment.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors would like to extend their appreciation to the Researchers Supporting Project number (RSPD2023R563) at King Saud University, Riyadh, Saudi Arabia.References
- M. E. Campany, P. A. Reck Dos Santos, B. B. Donato, C. M. Alwardt, V. Ernani, J. D'Cunha and S. E. Beamer, J. Thorac. Dis., 2023, 15, 5064–5073 CrossRef PubMed.
- J. Enríquez Pérez, S. Fritzell, J. Kopecky, E. Visse, A. Darabi and P. Siesjö, Sci. Rep., 2019, 9, 5632 CrossRef PubMed.
- S. Dasari and P. B. Tchounwou, Eur. J. Pharmacol., 2014, 740, 364–378 CrossRef CAS.
- D. M. Gershenson, G. M. Lentz, F. A. Valea and R. A. Lobo, Comprehensive Gynecology, Elsevier, St. Louis (MO), 8th edn, 2022, pp. 618–636 Search PubMed.
- K. Agrawal, in xPharm: The Comprehensive Pharmacology Reference, ed. S. J. Enna and D. B. Bylund, Elsevier, New York, 2007, pp. 1–5 Search PubMed.
- G. Giaccone, A. Ardizzoni, A. Kirkpatrick, M. Clerico, T. Sahmoud and N. van Zandwijk, J. Clin. Oncol., 1996, 14, 814–820 CrossRef CAS PubMed.
- Z. Wang, S. Mai, P. Lv, L. Xu and Y. Wang, Am. J. Transl. Res., 2021, 13, 12825–12833 CAS.
- A. Ardizzoni, G. Antonelli, F. Grossi, L. Tixi, M. Cafferata and R. Rosso, Ann. Oncol., 1999, 10(suppl. 5), S13–S17 CrossRef PubMed.
- S. A. Funt, D. J. McHugh, S. Tsai, A. Knezevic, D. O'Donnell, S. Patil, D. Silber, M. Bromberg, M. Carousso, V. E. Reuter, B. S. Carver, J. Sheinfeld, R. J. Motzer, D. F. Bajorin, G. J. Bosl and D. R. Feldman, Oncologist, 2021, 26, 483–491 CrossRef CAS PubMed.
- A. Gouyette, A. Apchin, M. Foka and J. M. Richard, Eur. J. Cancer Clin. Oncol., 1986, 22, 257–263 CrossRef CAS PubMed.
- G. Toffoli, G. Corona, R. Sorio, I. Robieux, B. Basso, A. M. Colussi and M. Boiocchi, Br. J. Clin. Pharmacol., 2001, 52, 511–519 CrossRef CAS PubMed.
- A. A. Miller, G. L. Rosner, M. J. Ratain, D. R. Hollis, M. R. Green and R. L. Schilsky, Clin. Cancer Res., 1997, 3, 719–725 CAS.
- R. A. Fleming, W. E. Evans, S. G. Arbuck and C. F. Stewart, J. Pharm. Sci., 1992, 81, 259–264 CrossRef CAS.
- B. Liu, H. M. Earl, C. J. Poole, J. Dunn and D. J. Kerr, Cancer Chemother. Pharmacol., 1995, 36, 506–512 CrossRef CAS PubMed.
- A. I. Ivanov, J. Christodoulou, J. A. Parkinson, K. J. Barnham, A. Tucker, J. Woodrow and P. J. Sadler, J. Biol. Chem., 1998, 273, 14721–14730 CrossRef CAS PubMed.
- J. Wang, J. Tao, S. Jia, M. Wang, H. Jiang and Z. Du, Pharmaceuticals, 2021, 14(2), 1–16 Search PubMed.
- H. D. Thomas, D. J. Porter, I. Bartelink, J. R. Nobbs, M. Cole, S. Elliott, D. R. Newell, A. H. Calvert, M. Highley and A. V. Boddy, Br. J. Clin. Pharmacol., 2002, 53, 83–91 CrossRef CAS PubMed.
- A. Gouyette, A. Deniel, J.-L. Pico, J.-P. Droz, D. Baume, M. Ostronoff, N. Le Bail and M. Hayat, Eur. J. Cancer Clin. Oncol., 1987, 23, 1627–1632 CrossRef CAS.
- C. Tang, M. J. Livingston, R. Safirstein and Z. Dong, Nat. Rev. Nephrol., 2023, 19, 53–72 CrossRef CAS PubMed.
- R. P. Miller, R. K. Tadagavadi, G. Ramesh and W. B. Reeves, Toxins, 2010, 2, 2490–2518 CrossRef CAS.
- C. M. Riley, L. A. Sternson and A. J. Repta, J. Pharm. Sci., 1983, 72, 351–355 CrossRef CAS PubMed.
- D. R. Newell, Z. H. Siddik and K. R. Harrap, in Drug Determination in Therapeutic and Forensic Contexts, ed. E. Reid and I. D. Wilson, Springer US, Boston, MA, 1984, pp. 145–153 Search PubMed.
- K. H. Kaushik, V. K. Sripuram, S. Bedada, N. Y. Reddy, G. I. Priyadarshini and K. R. Devarakonda, Clin. Res. Regul. Aff., 2010, 27, 1–6 CrossRef.
- F. H. Shirazi, G. Bahrami, D. J. Stewart, E. Tomiak, F. Delorme, D. Noel and R. Goel, J. Pharm. Biomed. Anal., 2001, 25, 353–356 CrossRef CAS.
- A. H. Algan, M. Gumustas, A. Karatas and S. A. Ozkan, J. Pharm. Biomed. Anal., 2016, 124, 382–389 CrossRef CAS.
- I. Bennani, M. A. Chentoufi, A. Cheikh, M. E. Karbane and M. Bouatia, J. Oncol. Pharm. Pract., 2021, 27, 99–107 CrossRef CAS.
- H. Tafazoli, M. Safaei and M. R. Shishehbore, Anal. Sci., 2020, 36, 1217–1221 CrossRef CAS PubMed.
- M. Snehalatha and R. N. Saha, Pharmazie, 2006, 61, 664–666 CAS.
- J. X. Ong, H. V. Le, V. E. Y. Lee and W. H. Ang, Angew Chem. Int. Ed. Engl., 2021, 60, 9264–9269 CrossRef CAS PubMed.
- T. Jantarat, S. Chuaychob, C. Thammakhet-Buranachai, P. Thavarungkul, P. Kanatharana, W. Srisintorn and C. Buranachai, Sens. Actuators, B, 2021, 326, 128764 CrossRef CAS.
- F. Fan, L. Zhang, F. Mu and G. Shi, ACS Sens., 2021, 6, 1400–1406 CrossRef CAS PubMed.
- L. Mitchell, C. Shen, H. C. Timmins, S. B. Park and E. J. New, ACS Sens., 2021, 6, 1261–1269 CrossRef CAS PubMed.
- H. Yang, H. Cui, L. Wang, L. Yan, Y. Qian, X. E. Zheng, W. Wei and J. Zhao, Sens. Actuators, B, 2014, 202, 714–720 CrossRef CAS.
- J. X. Ong, C. S. Q. Lim, H. V. Le and W. H. Ang, Angew. Chem., Int. Ed. Engl., 2019, 58, 164–167 CrossRef CAS PubMed.
- B. Atay, G. Önal and A. Levent, Monatsh. Chem., 2023, 154, 765–773 CrossRef CAS.
- B. Bozal-Palabiyik, B. Dogan-Topal, B. Uslu, A. Can and S. A. Ozkan, J. Solid State Electrochem., 2013, 17, 2815–2822 CrossRef CAS.
- A. E. Radi, N. Abd-Elghany and T. Wahdan, Chem. Pharm. Bull., 2007, 55, 1379–1382 CrossRef CAS.
- S. Khumngern, J. Choosang, P. Kanatharana, P. Thavarungkul and A. Numnuam, Talanta, 2024, 267, 125147 CrossRef CAS.
- P. Shearan, J. M. Fernández Alvarez and M. R. Smyth, J. Pharm. Biomed. Anal., 1990, 8, 555–561 CrossRef CAS.
- J. Li, J.-J. Zhu and K. Xu, TrAC, Trends Anal. Chem., 2014, 58, 90–98 CrossRef CAS.
- L. Zhang and E. Wang, Nano Today, 2014, 9, 132–157 CrossRef CAS.
- J. Xu and L. Shang, in Fluorescent Materials for Cell Imaging, ed. F.-G. Wu, Springer Singapore, Singapore, 2020, pp. 97–128 Search PubMed.
- M. F. Matus and H. Häkkinen, Nat. Rev. Mater., 2023, 8, 372–389 CrossRef CAS.
- C. Song, J. Xu, Y. Chen, L. Zhang, Y. Lu and Z. Qing, Molecules, 2019, 24(22), 1–16 Search PubMed.
- Y. Bai, Y. Zhou, H. Liu, L. Fang, J. Liang and S. Xiao, ACS Appl. Nano Mater., 2018, 1, 969–976 CrossRef CAS.
- Z. Cai, L. Wu, K. Qi, C. Deng and C. Zhang, Spectrochim. Acta, Part A, 2021, 247, 119145 CrossRef CAS PubMed.
- R. Rajamanikandan, B. Aazaad, S. Lakshmipathi and M. Ilanchelian, Microchem. J., 2020, 158, 105253 CrossRef CAS.
- Y. Luo, H. Miao and X. Yang, Talanta, 2015, 144, 488–495 CrossRef CAS PubMed.
- H.-B. Wang, B.-B. Tao, N.-N. Wu, H.-D. Zhang and Y.-M. Liu, Spectrochim. Acta, Part A, 2022, 271, 120948 CrossRef CAS PubMed.
- S. Rasaily, D. Sharma, S. Pradhan, N. Diyali, S. Chettri, B. Gurung, S. Tamang and A. Pariyar, Inorg. Chem., 2022, 61, 13685–13699 CrossRef CAS PubMed.
- M. Ahmed, World Appl. Sci. J., 2014, 29, 1357–1362 Search PubMed.
- N. Goswami, Q. Yao, Z. Luo, J. Li, T. Chen and J. Xie, J. Phys. Chem. Lett., 2016, 7, 962–975 CrossRef CAS PubMed.
- F. Belal, M. Mabrouk, S. Hammad, H. Ahmed and A. Barseem, J. Fluoresc., 2023, 33, 1–20 CrossRef.
- G. Magdy, H. Elmansi, F. Belal and A. K. El-Deen, Curr. Pharm. Des., 2023, 29, 415–444 CrossRef CAS PubMed.
- N. Nammahachak, K. K. Aup-Ngoen, P. Asanithi, M. Horpratum, S. Chuangchote, S. Ratanaphan and W. Surareungchai, RSC Adv., 2022, 12, 31729–31733 RSC.
- S. Wang, G. Deng, J. Yang, H. Chen, W. Long, Y. She and H. Fu, Sens. Actuators, B, 2022, 369, 132194 CrossRef CAS.
- X. Bu, Y. Fu, X. Jiang, H. Jin and R. Gui, Microchim. Acta, 2020, 187, 154 CrossRef CAS PubMed.
- L. Li, M. Fu, D. Yang, Y. Tu and J. Yan, Spectrochim. Acta, Part A, 2022, 267, 120563 CrossRef CAS PubMed.
- S. Urien and F. Lokiec, Br. J. Clin. Pharmacol., 2004, 57, 756–763 CrossRef CAS PubMed.
- K. Alhazzani, A. Z. Alanazi, A. M. Mostafa, J. Barker, M. M. El-Wekil and H. A. A. M. Bellah, RSC Adv., 2023, 13, 28940–28950 RSC.
- A. Z. Alanazi, K. Alhazzani, A. M. Mostafa, J. Barker, M. M. El-Wekil and A.-M. B. H. Ali, J. Pharm. Biomed. Anal., 2024, 238, 115862 CrossRef CAS PubMed.
- S. A. Alkahtani, A. M. Mahmoud, Y. S. Alqahtani, A.-M. B. H. Ali and M. M. El-Wekil, Spectrochim. Acta, Part A, 2023, 303, 123252 CrossRef CAS PubMed.
- A.-M. B. H. Ali, F. A. M. Abdel-aal, A. H. Rageh and A.-M. I. Mohamed, J. Sep. Sci., 2022, 45, 4187–4197 CrossRef CAS PubMed.
- A. M. I. Mohamed, N. A. Mohamed and A. M. B. H. Ali, Thai J. Pharm. Sci., 2020, 44, 82–90 CAS.
- Y. Kasherman, S. Sturup and D. Gibson, J. Med. Chem., 2009, 52, 4319–4328 CrossRef CAS PubMed.
- X. Guo, A. Zafar, H. Nan, Y. Yu, W. Zhao, Z. Liang, X. Zhang and Z. Ni, Appl. Phys. Express, 2016, 9, 055502 CrossRef.
- J. Chen, H. Du, Y. Xu, B. Ma, Z. Zheng, P. Li and Y. Jiang, J. Mater. Sci.: Mater. Electron., 2021, 32, 13581–13587 CrossRef CAS.
- J. Hua, Y. Jiao, M. Wang and Y. Yang, Microchim. Acta, 2018, 185, 137 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07844b |
| This journal is © The Royal Society of Chemistry 2024 |