
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZinc and sulfur functionalized biochar as a peroxydisulfate activator via deferred ultraviolet irradiation for tetracycline removal†
Yixue Qinab,
Sheng Wang*b,
Bingbing Zhangbc,
Weijie Chen bc,
Mingze Anb,
Zhao Yangb,
Hairong Gaoab and
Shuhao Qin*ab
bc,
Mingze Anb,
Zhao Yangb,
Hairong Gaoab and
Shuhao Qin*ab
aCollege of Materials and Metallurgy, Guizhou University, Guiyang 550025, China. E-mail: pec.shqin@gzu.edu.cn
bNational Engineering Research Center for Compounding and Modification of Polymer Materials, Guiyang, 550014, China. E-mail: 1191117410@qq.com
cResources and Environmental Engineering Department, Guizhou University, Guiyang 550025, China
First published on 13th February 2024
Abstract
To enhance the degradation of tetracycline class (TC) residuals of high-concentration from pharmaceutical wastewater, a novel zinc (Zn) and sulfur (S) functionalized biochar (SC-Zn), as a peroxydisulfate (PDS) activator, was prepared by two-step pyrolysis using ZnSO4 accumulated water-hyacinth. Results showed that the removal rate of 50, 150, and 250 mg per L TC reached 100%, 99.22% and 94.83% respectively, by the SC-Zn/PDS system at a dosage of 0.3 g per L SC-Zn and 1.2 mM PDS, via the deferred ultraviolet (UV) irradiation design. Such excellent performance for TC removal was due to the synergetic activation of PDS by the biochar activator and UV-irradiation with biochar as a responsive photocatalyst. The functionalization of the co-doped Zn and S endowed the biochar SC-Zn with a significantly enhanced catalytic performance, since Zn was inferred to be the dominant catalytic site for SO4˙− generation, while S played a key role in the synergism with Zn by acting as the primary adsorption site for the reaction substrates. The employed SC-Zn/PDS/UV system had excellent anti-interference under different environmental backgrounds, and compared with the removal rate of TC by adsorption of SC-Zn, the increasing rate in the SC-Zn/PDS/UV system (18.75%) was higher than the sum of the increases in the SC-Zn/PDS (9.87%) and SC-Zn/UV systems (3.34%), furtherly verifying the systematic superiority of this synergy effect. This study aimed to prepare a high-performance functionalized biochar activator and elucidate the rational design of deferred UV-irradiation of PDS activation to efficiently remove high-concentration antibiotic pollutants.
1. Introduction
The tetracycline class (TC) was discovered and reported as natural products from actinomycetes soil bacteria in the 1940s, and soon after, commercialized for clinical applications.1 It is a group of broad-spectrum, well-tolerated, and easy-to-administer antibiotic compounds, with the highest use in human and veterinary medicine, aquaculture and agricultural purposes.2,3 Since it is poorly metabolized and absorbed by humans and animals, TC is discharged into wastewater in the form of the parent compounds. With undesirable accumulation in the environment, it has been frequently detected in many types of water and soil leading to potential adverse effects such as chronic toxicity and dissemination into antibiotic-resistant genes, and further posing a threat to the ecosystem and human health in the long term.4 Meanwhile, it is noteworthy that the global consumption of antibiotics including TC witnessed a significant upsurge from the 2000s to date, with the improving standards of living and affordability of care in low- and middle-income countries.5 The rising quantity of pharmaceutical wastewater containing TC residuals has posed a larger threat to the ecosystem and human health.6 Thus, it is really crucial to develop an effective and low-cost technology for TC removal in wastewater.The main challenge for TC removal from water comes from its variable state, low biodegradability and complex molecular structure. Over the past few years, the persulfate-based advanced oxidation processes (AOPs) were considered as highly competitive technologies in the field of water treatment, because the diverse activation methods of persulfates (PSs) allow its application in water over a wide pH-range. And other advantages such as convenient transportation and storage of related reagents, low-cost catalysts originating from abundant biomass have also received widespread attention.7 Persulfates are divided into peroxydisulfate (PDS) and peroxymonosulfate (PMS), and PDS-based AOPs established for TC removal in water have been widely studied, because of the greater cost-efficiency, eco-sustainability, and storage stability of PDS compared to PMS.8 However, researchers mainly focused on TC residuals below 50 mg L−1, but paid less attention on pharmaceutical wastewater of TC up to 200 mg L−1.6 To remove TC of high-concentration, the better performance of PDS-based AOPs is urgently needed. It was proposed a new way to solve mentioned problems by designing a synergistic activation method of PDS, for instance, the biochar/PDS/UV system.9 The synergistic system has exhibited more excellent performance in removing refractory organic compounds by being activated through biochar and UV irradiation. The generation of sulfate radical (SO4˙−) and hydroxyl radical (˙OH) with stronger oxidation capacity was strongly corresponding to the performance of biochars.10 Therefore, under condition of biochars of excellent performance, a well-designed biochar/PDS/UV synergistic system can be regarded as an effective approach to remove high-concentration TC residual from wastewater.
So far, various biochars has been prepared by pyrolysis of renewable resources and waste materials such as straw, wood chips and animal manure.11 On purpose to obtain the optimized the performance of biochars, functionalization by doping heteroatoms (such as N, S, P) or/and loading transition metals (such as Zn, Fe, La) have been employed indispensably.12–14 Thereinto, Guo et al.15 synthesized sulfur-doped activated carbon with thiophene as the precursor, and its catalytic activity for PDS was higher than that of sulfur-free activated carbon. Tian et al.16 used sulfur as a co-dopant to further improve the performance of nitrogen-doped graphene in PS activation and considered sulfur to be a promising co-dopant. Recent studies have shown that Zn2+ has fast charge transfer kinetics, and Wang et al.17 used this advantage to synthesize zinc–nitrogen-doped zinc–metal–organic framework-derived porous carbon (ZMDPC) materials, which have excellent electrochemical properties such as high specific capacitance and cycling stability. Xi et al.18 further demonstrated that the combined interaction of radical pathways and nonradical pathways is a feasible method with practical application value. Their results show that Fe@N co-doped biochar prepared by doping Fe and N has porous, large specific surface area, abundant nitrogen/oxygen-containing functional groups, and graphite structural defects, the formation of which is conducive to charge transfer and provides active sites, showing excellent catalytic degradation performance. This is due to the fact that the doped heterogeneous Fe and N together initiate free radicals and nonradical pathways. Afterwards, Zhong et al.19 used wood chips as raw materials, urea as the nitrogen source, and sulfur powder as the sulfur source, and uniformly mixed urea, sulfur powder, and BC for grinding and pyrolysis to prepare a nitrogen sulfur co doped biochar (NS-BC). Wang et al.20 prepared S, N co-doped magnetic biochar (IBC) from Dianthus chinensis stem by simple immersion pyrolysis. The research reports show that the method of achieving heterometal/non-metal co-doping is still limited to the commonly used methods. The most common way of biochar functionalization is the impregnation methods aided by different equips, such as vacuum or ultrasonic or hydrothermal treatment devices.21,22 However, these methods may induce metal aggregation, interface inhomogeneity and metal leaching that cause secondary pollution.23–25 To gain biochars with good dispersity, homogeneity and leaching resistance, using prominent accumulator plants as biochar precursors has become a wise strategy, in which case target metal or/and nonmetal elements can be controllably and directly introduced into carbon atomic structure of biochars.26–28
Water-hyacinth, an alien invasive floating plant of the aggressive growth rate among the highest of any known plants, has been considered as a major weed species in more than 50 countries. It has caused environmental damages for decades, including lowering dissolved oxygen concentration in aqua, damaging fish populations, decrease biodiversity and clogging river.29 Thus, strict management and treatment for this species has been conducted, and resource utilization of such species are strongly encouraged as well. Based on its intrinsic characteristics, water-hyacinth can be regarded as an ideal biochar precursor.
In this study, a novel Zn and S (metal and non-metallic) co-doped functionalized biochar (SC-Zn) was prepared. For functionalization of biochar, water-hyacinth was accumulated by ZnSO4 in hydroponics process and then went through a two-step pyrolysis. The common TC was selected as a model antibiotic pollutant to study the synergistic activation performance of functionalized SC-Zn in the biochar/PDS/UV system. The synergism of doped Zn and S on SC-Zn was explored. A deferred UV irradiation design was employed in the system to enhance the removal of TC, and the rationality of such design was elucidated. Besides, main factors that effected on the removal of TC in this well-designed system were systematically discussed.
2. Materials and methods
2.1 Materials
Water hyacinth was collected from a natural lake in Shantou, Guangdong. Zinc sulfate (ZnSO4, ≥99.5%), hydrochloric acid (HCl, 36–38%), potassium hydroxide (KOH, ≥85%) and sodium thiosulfate (Na2O3S2, ≥99%) were purchased from China Shanghai Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Tetracycline hydrochloride (TC, 99.9%), sodium persulfate (Na2S2O8) (AR, 99%), sodium bicarbonate (NaHCO3, ≥99.5%), humic acids (HA, ≥90%), 5,5-dimethyl-1-pyrrolidine N-oxide (DMPO, ≥97%) and 2,2,6,6-tetramethylpiperidine (TEMP, ≥97%) were purchased from Macklin Biochemical Co., Ltd (Shanghai, China). Absolute ethanol (EtOH, 99.7%) (AR), tert-butyl alcohol (TBA, ≥99%) and L-histidine (L-H, ≥99%) were purchased from Aladdin (Shanghai, China). Sodium sulfate (Na2SO4, ≥99%) (AR) and sodium chloride (NaCl, ≥99.5%) (AR) were purchased from Chongqing Chuandong Chemical (Group) Co., Ltd. Sodium hydroxide (NaOH, ≥96%) (AR) was purchased from Chengdu Kelong Chemical Reagent Factory. Disodium hydrogen phosphate (Na2HPO4·12H2O, ≥99%) was purchased from Tianjin Damao Chemical Reagent Factory. All chemicals were analytical grade and can be used without further purification. Unless otherwise noted, deionized water was used for all solutions.2.2 Biochar preparation
Water-hyacinths in the growing phase were collected from lakes, washed with deionized water, wiped off the surface water after washing, and transferred to the ZnSO4 solution at concentrations of 200 mg L−1 for 24 days. The growth of water-hyacinth plants was recorded every 3 days (see in Fig. S1a†).The pristine water hyacinth samples (17.64 ± 0.2 g per sample) has been cultured for 24 days by 200 mg per L ZnSO4 solution in flowerpots. From each flowerpot, 20.7 ± 0.3 g of water hyacinth sample was collected. The collected samples were then dried at 80 °C for 24 hours and 8.34 ± 0.2 g of each dried precursor sample was obtained. The collected ZnSO4 accumulated water-hyacinth as precursor went through a two-step pyrolysis process (before two-step pyrolysis, the tube furnace was purified with nitrogen to remove oxygen): firstly, and the dried precursor was heated to 600 °C at a heating rate of 5 °C min−1 in the tube furnace and the yield rate of the obtained parent charcoal was about 40.58%; secondly, a mixture of the obtained parent charcoal, KOH powders (four times weight of the parent charcoal) and 30 ml deionized water was stirred and heated in a water bath at 100 °C for spontaneous evaporation, dried at 80 °C for 12 h in the oven, heated to 800 °C at a heating rate of 5 °C min−1 in the tube furnace and naturally cooled down to the room temperature. During the above pyrolysis and cooling process, protective gas (nitrogen) with purity of 99.99% was introduced into the tube furnace at a flow rate of 60 ml min−1. Finally, the functionalized biochar, with 58.58% yield rate, was gained after the product of two-step pyrolysis process was washed with 0.1 M HCl and deionized water until the washing solution was nearly neutral, dried at 120 °C for 24 h, and recorded as SC-Zn.
As a control, blank samples prepared products under the same conditions, and as-prepared product was denoted as BC. For verification the leaching resistance of ZnSO4 accumulated water-hyacinth derived biochar, the impregnated biochar was prepared, too. First, the dried and smashed water-hyacinth was sinking in 200 mg per L ZnSO4 solution 12 h for direct impregnation. The obtained precursor then went through the above-mentioned two-step pyrolysis process. The as-prepared product was denoted as BC-Zn.
2.3 Experimental procedure of TC removal
Different systems were designed for TC removal experiments. Simulated wastewater of containing TC was prepared under different concentrations. All experiments are performed in 50 ml brown centrifuge tubes for 30 min in the first place to approach the adsorption equilibrium of TC on biochar. For the darkness procedure, 50 ml brown centrifuge tubes were engaged, while for the UV-irradiation procedure, 100 ml double-layer jacketed beakers were engaged.The SC-Zn/PDS/UV system via deferred UV-irradiation was designed as: (1) the first stage was to add SC-Zn of certain dosages in 40 ml TC wastewater of different concentration in darkness during recorded −30 to 0 min; (2) and then certain amount of PDS was added in for activation procedure at the recorded 0 min; (3) at last, UV-irradiation was carried out by a xenon lamp (300 W) from the recorded 60 to 180 min (the end). Experimental parameters including initial concentration of TC, the starting time of UV lighting, dosages of SC-Zn and PDS, initial pH value, influence of the variety and amount of natural organic matter (HA) and co-existing ions (CO32−, HPO42−, Cl− and HCO3−), have been explored to determine the optimal parameter in final. For comparison, The BC/PDS/UV system with the optimal parameters was conducted in same procedures.
For further comparison, other systems were employed. The SC-Zn/PDS system was conducted in total darkness from the recorded −30 to 180 min, while the SC-Zn/UV system was conducted in darkness during the recorded −30 to 0 min and in UV lighting from 0 to 180 min. Adsorption procedure was skipped in systems without SC-Zn, including PDS/UV, PDS and UV-irradiation systems. With PDS added in the simulated wastewater at 0 min, the PDS/UV system was conducted in UV-irradiation from 0–180 min, while the PDS system was conducted in total darkness; and the UV system was under lighting from 0–180 min without any PDS. The simplex adsorption system was conducted in total darkness from −30 to 180 min by SC-Zn.
In all designed systems, the reactive subjects were stirred at 200 rpm at room temperature. The initial pH value of simulated wastewater was adjusted by 0.1 M HCl and 0.1 M NaOH. Removal experiments were performed in triplicate and repeated three times with similar results, average value and standard deviation were calculated.
2.4 Characterization
The specific surface area and pore structure of all biochar catalysts were analyzed by fully automatic specific surface area and porosity analyzer (Quantachrome, NOVA-1000e, USA), and the specific surface area and pore size distribution were calculated by density functional theory (DFT) method. Materials morphologies were observed by scanning electron microscopy (SEM: Quanta FEG 250, American FEI) and transmission electron microscopy (TEM: JEM-F200, URP), and the elemental distribution of functionalized biochar were determined by element mapping. Raman spectroscopy was used to analyze the defect structure and graphitization degree of biochar. Fourier transform infrared spectroscopy (FT-IR: Nicolet NEXUS670, USA) was used to determine the surface functional groups of all biochars in the range of 400–4000 cm−1. X-ray diffractometer (XRD: X'Pert PRO) was used to verify the crystal structure and chemical composition of biochar. The scanning rate was 2° min−1 and the scanning range was 10–80°. X-ray photoelectron spectroscopy (XPS: Thermo Scientific K-Alpha) was used to analyze the elemental composition and chemical state of biochars. The Zn2+ concentration in the reaction solution of the system was determined by inductively coupled plasma-mass spectrometry (ICP-MS).To monitor the change of removal rate of TC, 1 ml samples were removed from the reaction solution at different sampling time points (0, 10, 20, 30, 45, 60, 65, 70, 80, 90, 120 and 180 min) by syringes, quickly quenched in 0.2 M sodium thiosulfate solution, filtered by 0.45 μm polyether sulfone resin (PES) filters, and analyzed by a 752 N UV-vis spectrophotometer. All experiments were averaged in three parallel sets of experiments.
To further understand the mechanism of PDS activation in designed systems, electron paramagnetic resonance spectroscopy (EPR) was used to identify sulfate radicals, hydroxyl radicals and singlet oxygen in designed systems, with 5,5-dimethyl-1-pyrrolidine N-oxide (DMPO) and 2,2,6,6-tetramethylpiperidine (TEMP) as free radical traps. Liquid chromatography-mass spectrometry (LC-MS) (experimental conditions: mobile phase A: 0.1% formic acid aqueous solution; mobile phase B: acetonitrile solution, flow rate: 0.3 ml min−1) and total organic carbon (TOC) were used to determine TC degradation intermediates.
3. Results and discussion
3.1 Characterization
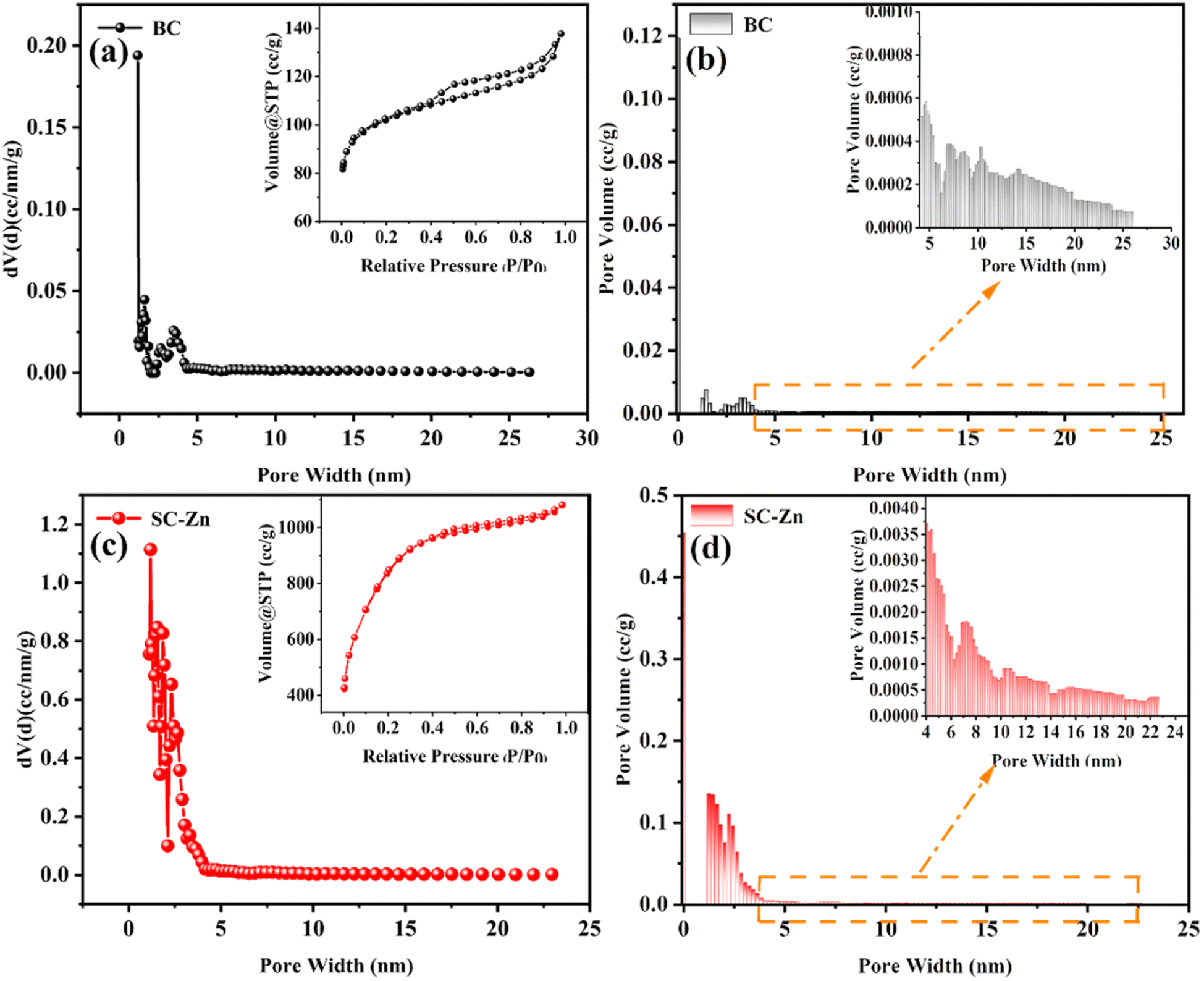 | ||
| Fig. 1 N2 adsorption–desorption isotherm, pore size distribution curve and pore size distribution histogram (a) and (b) BC, (c) and (d) SC-Zn. | ||
| Sample | Surface area (m2 g−1) | Average pore diameter (nm) | Total pore volume (cm3 g−1) |
|---|---|---|---|
| BC | 340.345 | 1.178 | 0.187 |
| SC-Zn | 2007.812 | 1.178 | 1.494 |
 | ||
| Fig. 2 SEM image of (a) BC, (b) SC-Zn, TEM images of (c) BC, (d) SC-Zn and (e) C, O, N, S and Zn element distribution of SC-Zn. | ||
At the same time, the SC-Zn prepared in this study was compared with the Zn-BC prepared by chemical impregnation method, and it was found that SC-Zn had good metal stability and would not cause serious secondary pollution (Fig. S1c†). The method for determining the zinc content was described in Text S1.†
| Sample | D1 peak (%) | S3 peak (%) | A4 peak (%) | G2 peak (%) | ID/IG (area ratio) |
|---|---|---|---|---|---|
| BC | 40.47 | 18.48 | 15.05 | 26.00 | 1.56 |
| SC-Zn | 32.36 | 14.74 | 34.71 | 18.19 | 1.78 |
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1089 cm−1 (CC), 1413 and 1455 cm−1 (C–O bond from CO3-group)36 on the surface of biochars. In presence of Zn2+, the C–H bending for aromatic out of plane deformation (873 cm−1) become apparent in SC-Zn. On the spectrum of SC-Zn, the bending for vibrations of Zn2+–O2− (464 cm−1) could be observed indicating that ZnO is the main form of Zn2+ on its surface.37 The surface hydrophilicity of catalysts BC and SC-Zn were studied by water contact angle test (Fig. S4†). Because of these new surface functional groups emerging on biochars, the hydrophilicity of SC-Zn was slightly weaker than that of pristine BC.
C), 1089 cm−1 (CC), 1413 and 1455 cm−1 (C–O bond from CO3-group)36 on the surface of biochars. In presence of Zn2+, the C–H bending for aromatic out of plane deformation (873 cm−1) become apparent in SC-Zn. On the spectrum of SC-Zn, the bending for vibrations of Zn2+–O2− (464 cm−1) could be observed indicating that ZnO is the main form of Zn2+ on its surface.37 The surface hydrophilicity of catalysts BC and SC-Zn were studied by water contact angle test (Fig. S4†). Because of these new surface functional groups emerging on biochars, the hydrophilicity of SC-Zn was slightly weaker than that of pristine BC.XRD (Fig. S3b†) showed broad diffraction peaks on BC and SC-Zn, which were attributed to the formation of amorphous carbon (002) and graphitic carbon (100), respectively.19 SiO2 (PDF #46-1441) and CaCO3 (PDF #72-1652) appeared in all two catalysts because the plants themselves contain Si and Ca, and gradually form some stable mineral crystal compositions (such as SiO2 and CaCO3) after pyrolysis.38 The ZnO diffraction peaks appear at 2θ at 36.25°, 62.86°, and 67.96°, respectively, corresponding to (101), (103), and (112) (PDF #36-1451). Because of the conversion of enriched ZnO to ZnS when the pyrolysis temperature exceeds 450 °C, during the high-temperature activation process, ZnO will be reduced by C to form elemental Zn.39 SC-Zn also has peaks of ZnS at 26.92° and 63.56°, corresponding to lattices (100) and (202) (PDF #36-1450), respectively. Peaks of Zn appear at 43.22° and 77.05°, corresponding to lattices of (101) and (004) (PDF #87-0713), respectively.
O), 289.0 eV (OC–O) and 290.5 eV (π–π*),40 while the spectra of O 1s were divided into three peaks CO (531.5 eV), C–O (532.8 eV) and OC–O (533.5 eV), respectively.41 The high-resolution N 1s spectra were decomposed into four sections that corresponded to pyridinic-N (398.1 eV), pyrrolic-N (399.6 eV), graphitic-N (400.4 eV) and oxidized-N (403.9 eV).42 The graphitic-N on SC-Zn, which was conducive to the activation of PDS and participates in the non-radical pathway to generate the active site 1O2.43 On SC-Zn, the fine spectra of S 2p show four peaks corresponding to thiophene sulfur (–C–S–C–) and sulfur oxide groups (–C–SOx–C–), respectively.44
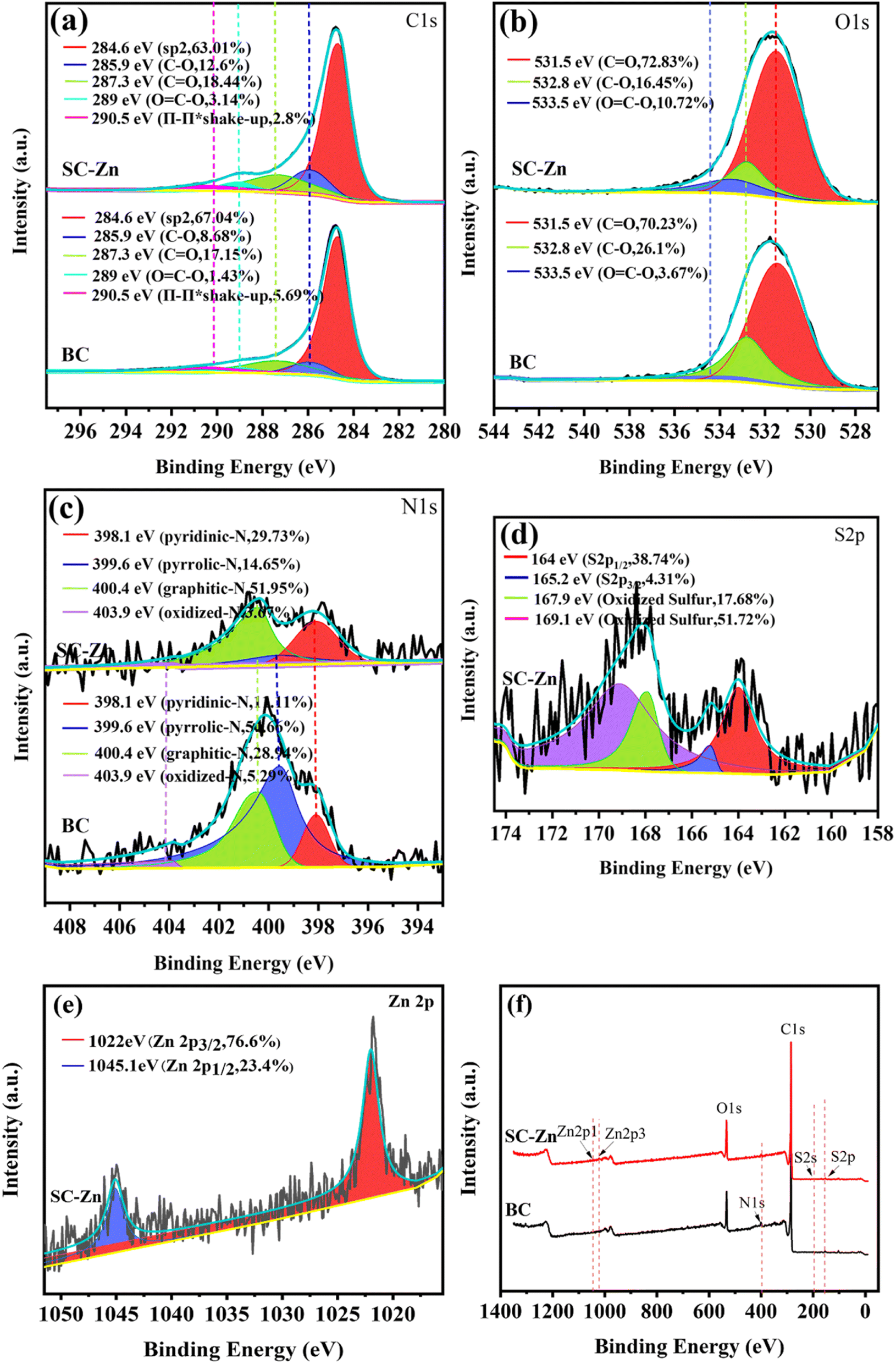 | ||
| Fig. 3 Fine XPS spectra of BC and SC-Zn before reaction (a) C 1s, (b) O 1s, (c) N 1s, (d) S 2p and (e) Zn 2p, (f) XPS full spectrum of BC and SC-Zn. | ||
On the Zn 2p fine spectrum of SC-Zn, peaks appear at 1022 eV and 1045 eV correspond to Zn 2p3/2 and Zn 2p1/2, respectively. They were attributed to lattice zinc in zinc oxide, and their spacing (23.1 eV) was within the standard value of ZnO. Such binding energy and peak spacing indicate that zinc exists mainly in the +2 oxidation status,45 which have also been validated in FTIR and XRD. As one of the main forms of existence, ZnO participates in the degradation process, and its rapid electron transfer rate (200–300 cm2 V−1 s−1)46 favors PDS activating into SO4˙−.
3.2 Design of synergy system for TC removal
Various systems have been designed for TC removal, including the unitary systems (simplex adsorption of SC-Zn, single PDS system and single UV lighting system), the dual systems (SC-Zn/PDS, SC-Zn/UV and PDS/UV), and the ternary systems (SC-Zn/PDS/UV system with or without deferred UV lighting). Details of those systems were given in “2.3 Experimental procedure of TC removal”.Fig. 4a shows the removal trends of high-concentration TC in various designed systems. As for systems without SC-Zn, the removal rates of TC were 13.53%, 8.93% and 19.41% in single PDS, single UV lighting system and PDS/UV system, respectively, indicating the difficult to remove TC of high-concentration without functionalized biochar as an activating media. In contrary, as for systems with SC-Zn, 64.07% of TC has been removed from aqua by the simplex adsorption in the first 30 min (recorded as −30 to 0 min), due to the abundant sulfur oxide groups on the large surface of SC-Zn acting as efficient adsorptive sites, which was consistent with the previous characterization. In the designed simplex adsorption of SC-Zn (from −30 to 180 min), the removal rate of TC can reach 76.08%. When dual systems (SC-Zn/PDS and SC-Zn/UV) were built based on SC-Zn, the removal rate of TC reach 85.95% and 79.42%, increasing 9.87% and 3.34% compared to that of the SC-Zn simplex adsorption system. When the ternary system, SC-Zn/PDS/UV system with deferred UV lighting, was employed, 94.83% TC of such high concentration was removed. The functionating of Zn and S endowed SC-Zn with good performance for both PDS activation and UV photocatalysis. It was notably that, compared with the removal rate of TC by simplex adsorption of SC-Zn, the increasing rate in SC-Zn/PDS/UV (deferred) system (18.75%) was higher than the sum of the increases in SC-Zn/PDS (9.87%) and SC-Zn/UV system (3.34%), verifying the systematic superiority of synergy effect of such designed ternary system based on SC-Zn.
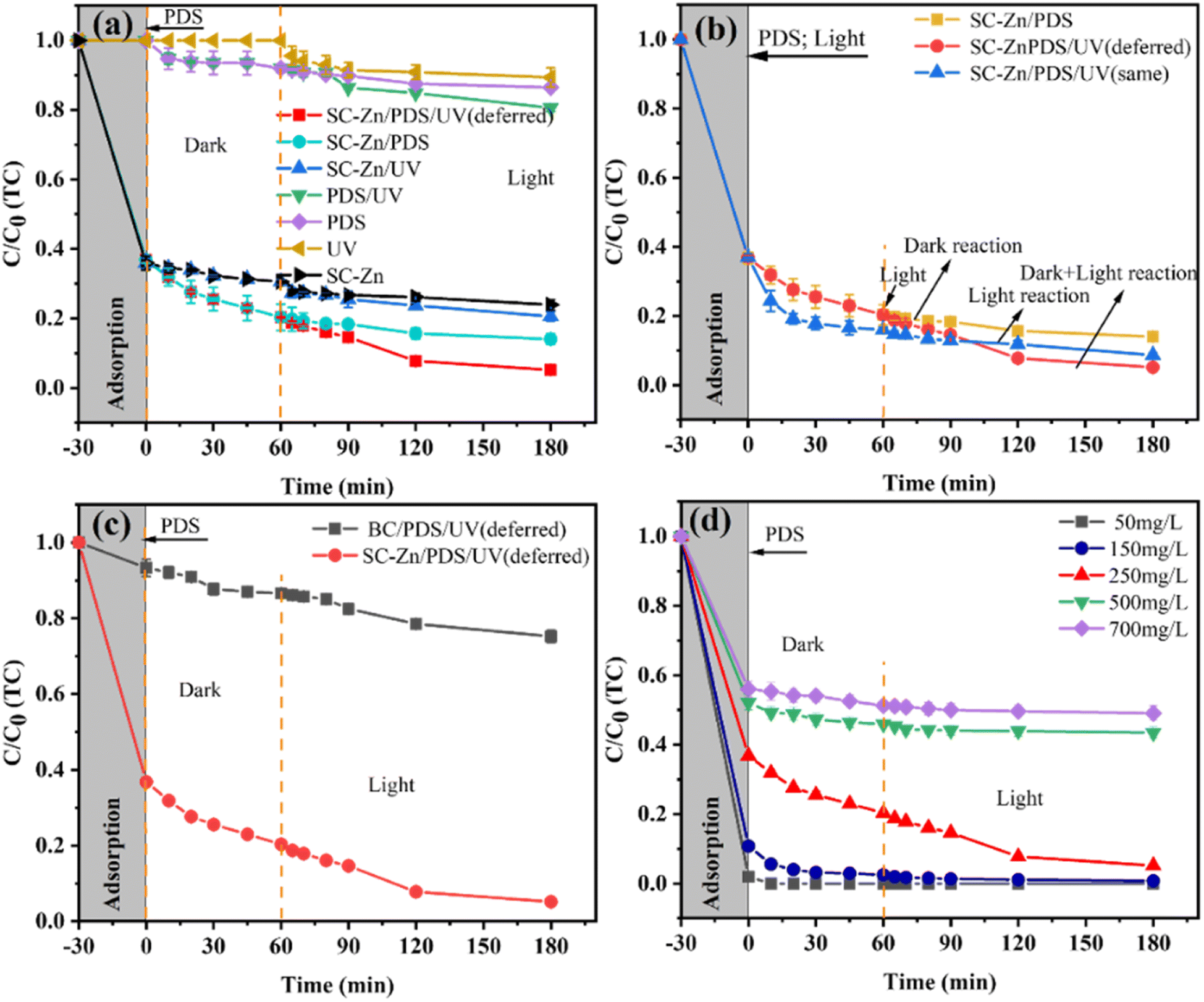 | ||
| Fig. 4 Removal experiments of 250 mg per L TC (a) in various designed systems, (b) in designed SC-Zn/PDS/UV system via different the starting time of UV lighting, (c) in designed system based on BC and SC-Zn; (d) removal experiments of TC with different initial concentrations in designed SC-Zn/PDS/UV (deferred) system (all experimental conditions: activator: 0.3 g L−1, PDS: 1.2 mM, T = 25 °C, pH = 7.12). | ||
The starting time of UV lighting for the SC-Zn/PDS/UV system was discussed afterwards. The curves of TC removal rate in SC-Zn/PDS/UV system via UV-lighting at recorded 0 min (same), at the same time of PDS adding, and 60 min (deferred) was shown in Fig. 4b, and the SC-Zn/PDS system without UV lighting was also shown. The pattern of curves of TC removal rate in SC-Zn/PDS system and SC-Zn/PDS/UV (same) system were similar with an amplification of the latter about 5.36% at 180 min. When the deferred UV-lighting was employed, the curve for SC-Zn/PDS/UV (deferred) decreased rapidly from 60 min, with an amplification about 8.88% at 180 min compared with the SC-Zn/PDS system, meanwhile showing a cross point with the curve for SC-Zn/PDS/UV (same) system. The SC-Zn/PDS/UV (deferred) system showed better performance than the SC-Zn/PDS/UV (same) system. It was implied that the time interval between pre-adsorption of PDS on SC-Zn and UV-irradiation could be beneficial to the synergism of PDS activation by biochar and UV, since the pre-adsorbed PDS would act as a receptor for SC-Zn to transfer electrons to restrain the recombination of photogenerated holes and electron pairs.43 In the optimal SC-Zn/PDS/UV (deferred) system, after the near adsorption equilibrium of TC on SC-Zn during the recorded −30 to 0 min, the presence of PDS would cause a competitive adsorption equilibrium process between PDS and TC on SC-Zn accompanying with oxidant free radicals generation by PDS activation in 60 min, while the deferred UV-irradiation broke the balance and led to a secondary chemical equilibrium in a positive direction. Thus, in the designed SC-Zn/PDS/UV (deferred) system, the deferred UV-irradiation has enhanced synergic activation of PDS by biochar and UV for exhibiting an excellent performance.
To elucidate the role of functionalized biochar in the designed ternary system, as-prepared BC and SC-Zn were applied in the similar ternary systems under the same conditions, and the experimental results of both BC/PDS/UV (deferred) and SC-Zn/PDS/UV (deferred) systems were shown in Fig. 4c. It can be seen that the BC/PDS/UV (deferred) system can only remove 24.77% TC of 250 mg L−1 at 180 min, while the system based on SC-Zn shown a high removal rate of 94.83%, demonstrating Zn, S co-doped functionalized biochar plays the key role. The extra-high surface area and total pore volume of functionalized SC-Zn provided enough interface area for synergism of adsorption, PDS activation, photocatalysis and electron transfer. With abundant oxygen-containing functional groups (CO, C–O, –COOH and –OH), SC-Zn provided more active sites for PDS.47 The functionalization of the co-doped Zn and S endowed the biochar SC-Zn with a significantly enhanced catalytic performance, since Zn was inferred to be the dominant catalytic site for SO4˙− generation, while S played a key role to the synergism with Zn by acting as the primary adsorption site for the reaction substrates. Thus, the synergism of designed SC-Zn/PDS/UV (deferred) system was an integrated effects including synergic activation of PDS by biochar and UV-irradiation, and the synergy effect of co-doped Zn and S on biochar surface as adsorption and catalytic sites.
The SC-Zn/PDS/UV (deferred) system was used to remove TC of different initial concentrations, and the results (see in Fig. 4d) showed that TC of 50 mg L−1 was completely removed just in 10 min after adding PDS. At the recorded 180 min, the removal rates of TC of 150, 250, 500 and 700 mg L−1 were 99.22%, 94.83%, 56.57% and 50.92%, respectively, which proved that the designed SC-Zn/PDS/UV (deferred) system has excellent performance for removing TC of high concentration.
3.3 Effects of various factors on TC degradation
The PDS concentration of 0.8–1.6 mM were selected to investigate the degradation efficiency of catalysts as shown in Fig. S5b,† It can be seen that when the dosage of SC-Zn increased from 0.8 mM to 1.2 mM, the removal rates of TC were 80.82% (0.8 mM), 82.71% (1 mM) and 94.83% (1.2 mM), respectively, indicating that with more amount of PDS, more free radicals could be produced for effective TC removal. However, when excessive PDS was added to designed systems, an inhibitory effect was showed. With 1.4 mM and 1.6 mM PDS in the designed system, the removal rates of TC have decreased to 84.76% and 90.10%, respectively. Such inhibitory effect may be due to the quenching of SO4˙− and ˙OH radicals by excessive PDS during the following reaction shown in eqn (1) and (2):49
| S2O82− + SO4˙− → SO42− + S2O8˙− | (1) |
| S2O82− + ˙OH → OH− + S2O8˙− | (2) |
Based on the above experimental results and consideration, 1.2 mM was determined as the optimal amount of PDS for the final ternary system.
| SO4˙− + OH− → SO42− + ˙OH | (3) |
| SO4˙− + SO4˙− → S2O82− | (4) |
| SO4˙− + S2O82− → SO42− + S2O8˙− | (5) |
In general, the system showed that it could adapt to a wide range of pH values, considering that the natural water body was closer to neutral, the initial pH value of TC solution was finally adjusted to 7.12.
| SO4˙− + Cl− → SO42− + Cl˙ | (6) |
| Cl− + ˙OH → ClOH˙− | (7) |
| ClOH˙− + Cl− → Cl2˙− + OH− | (8) |
| Cl− + Cl˙ → Cl2˙− | (9) |
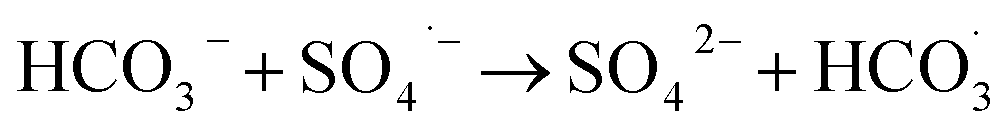 | (10) |
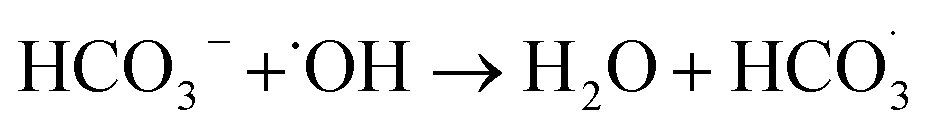 | (11) |
The effect of different concentrations of HA on TC removal was shown in Fig. S5f.† It can be seen that with the increase of HA concentration, the removal rate of TC gradually decreases, when the HA concentration was 0 mg L−1, 2 mg L−1, 5 mg L−1 and 8 mg L−1, the removal rate of TC was 94.83%, 88.73%, 86.76% and 83.23%, respectively, the inhibitory effect may be due to the presence of competing active substances between the added HA and the TC. In addition, some researchers57 have shown that HA has strong π–π stacking, which was easy to adsorb on the surface of SC-Zn and cover the active sites on the surface, thereby affecting the interaction between TC and PDS. After further research, it was believed that both inorganic anions and HA have different degrees of inhibition on the removal of TC in the system.
In general, the optimal conditions of the system were screened out by investigating the effects of SC-Zn dosage, PDS dosage, initial pH value, inorganic anions and natural organic matter on TC removal. Under the optimal conditions of SC-Zn dosage of 0.3 g L−1, PDS dosage of 1.2 mM, initial pH of 7.12 and TC concentration of 250 mg L−1, the degradation rate of TC 180 min was 94.83%, and the SC-Zn/PDS/UV (deferred) system showed excellent anti-interference under optimal conditions.
3.4 TC degradation mechanism using the SC-Zn/PDS/UV (deferred) system
ln(qe − qt) = ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) qe − k1t qe − k1t
| (12) |
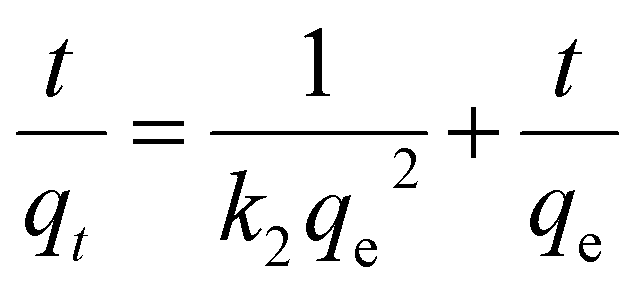 | (13) |
| qt = kintt1/2 + b | (14) |
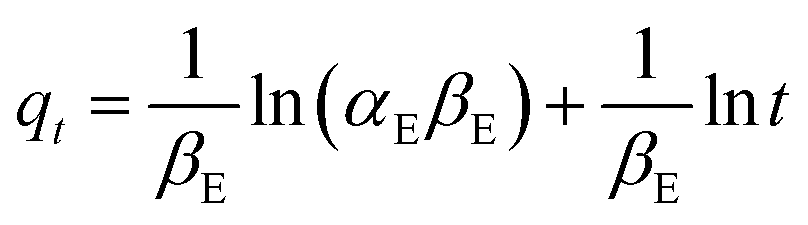 | (15) |
| C0 (mg L−1) | qe (mg g−1) | PFO | PSO | Elovich | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| qe,cal | K1 | R2 | qe,cal | K2 | R2 | αE | βE | R2 | ||
| 50 | 134.19 | 1.34 | 0.0038 | 0.2361 | 200 | 1 | 1 | 0 | 0 | 0 |
| 150 | 483.14 | 35.82 | 0.0306 | 0.9634 | 476.19 | 0.0025 | 0.9999 | 2.90 × 1024 | 0.1224 | 0.9594 |
| 250 | 844.25 | 284.23 | 0.0158 | 0.8577 | 833.33 | 0.0002 | 0.9924 | 39196.24 |
0.0141 | 0.8906 |
| 500 | 1170.24 | 116.20 | 0.0319 | 0.9517 | 1111.11 | 0.0010 | 0.9999 | 4752 × 109 | 0.0258 | 0.8970 |
| 700 | 1302.18 | 117.87 | 0.0241 | 0.9505 | 1250 | 0.0007 | 0.9998 | 1.42 × 1014 | 0.0260 | 0.9305 |
| C0 (mg L−1) | Step 1 | Step 2 | ||||
|---|---|---|---|---|---|---|
| Kint1 (min−1) | Intercept 1 | R2 | Kint2 (min−1) | Intercept 2 | R2 | |
| 50 | 0.5731 | 131.56 | 0.8417 | 0 | 134.19 | 0 |
| 150 | 8.0176 | 431.31 | 0.9763 | 1.4054 | 466.1 | 0.7583 |
| 250 | 22.511 | 565.35 | 0.9929 | 23.564 | 537.55 | 0.9513 |
| 500 | 17.387 | 1021.2 | 0.9581 | 2.5133 | 1138 | 0.9441 |
| 700 | 8.2242 | 1183.2 | 0.9630 | 5.2100 | 1236.6 | 0.7449 |
After the four models fit different concentrations of TC, the kinetic correlation coefficients were R2 > 0.0038 (PFO), R2 > 0.9924 (PSO), R2 > 0.8906 (Elovich) and R12 > 0.8417 (IPD), R22 > 0 (IPD), respectively, and the PSO model showed better correlation coefficient values. Combined with experimental and fitting results, it was found that PSO can better characterize the degradation process of TC. In this model, the rate-limiting step is surface adsorption, which involves chemisorption that originates from PS activation and photocatalysis on the biochar surface, where the removal of TC from water is due to a physicochemical interaction between biochar solids and aqueous solutions.
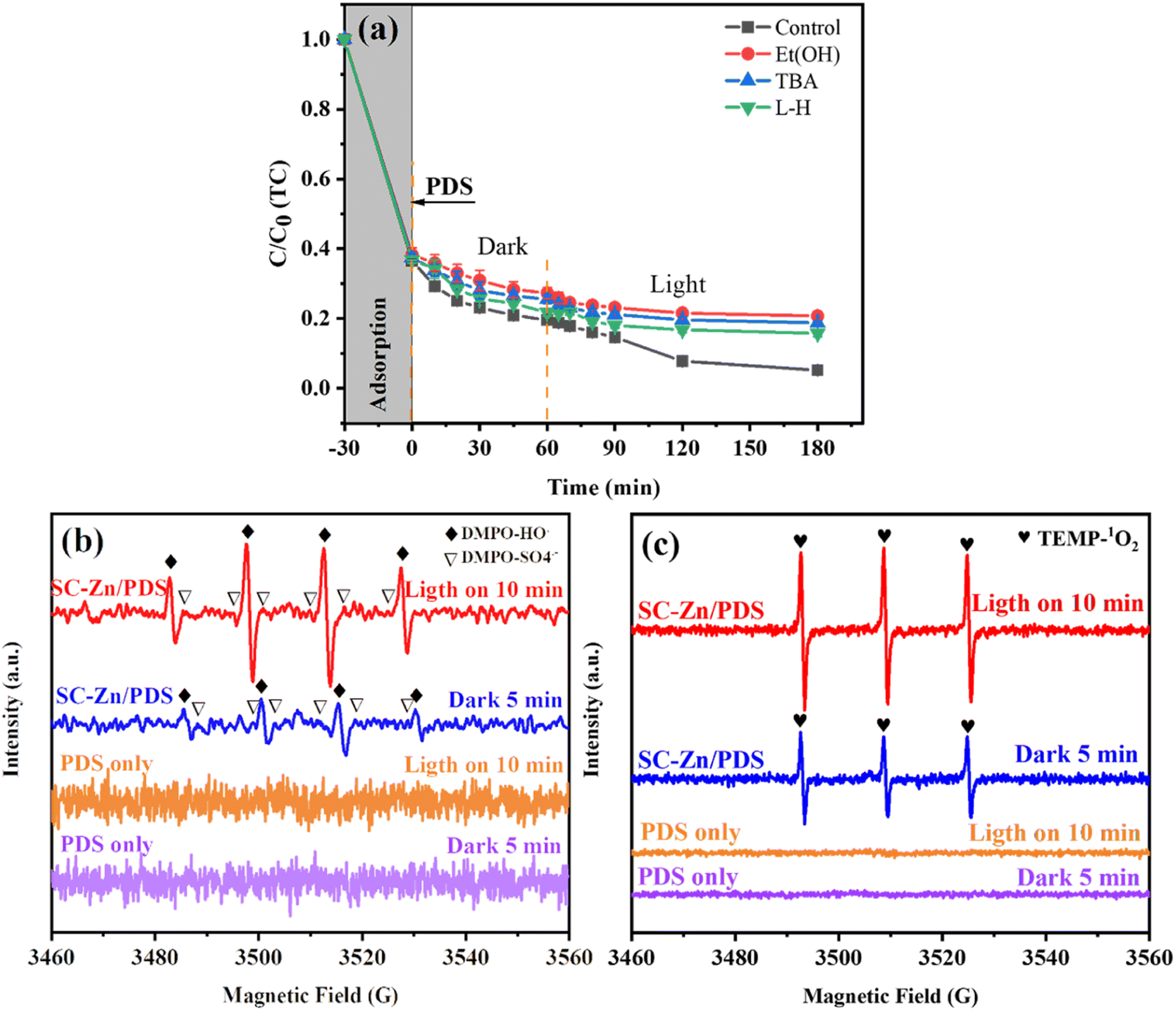 | ||
| Fig. 5 (a) Quenching experiment (experimental conditions: catalyst: 0.3 g L−1, PDS: 1.2 mM, TC: 250 mg L−1, T = 25 °C, [Et (OH)]0 = 100 mM, [TBA]0 = 100 mM, [L-H]0 = 100 mM). EPR spectra in SC-Zn/PDS/UV (deferred) system (b) SO4˙− and ˙OH (c) 1O2. | ||
Further verification with EPR spectra, the results are shown in Fig. 5b, there were almost no characteristic peaks produced in the pure PDS system, and after adding SC-Zn, the typical EPR signals of DMPO-SO4˙− and DMPO-˙OH can be clearly observed, indicating that there were ˙OH and SO4˙− in the SC-Zn/PDS/UV (deferred) system, and with the passage of reaction time, the characteristic peaks of SO4˙− were not obvious at 10 min, which may be ˙OH and SO4˙− transformation occurs during the reaction, the peak intensity of ˙OH stronger than SO4˙−, but it was not determined that it plays a dominant role in the degradation of TC.61 In Fig. 5c, pure PDS did not present with any EPR typical peaks, only when the catalyst was added, the typical three-line peak (intensity ratio 1:1:1) of TEMP-1O2 can be clearly observed, indicating the presence of 1O2 in the SC-Zn/PDS/UV (deferred) system, and the EPR signal intensity of these two figures was getting stronger, indicating that SC-Zn can continuously activate PDS to decompose active substances.62 In general, SO4˙− and ˙OH and 1O2 play a leading role in the degradation of TC.
| Catalyst (g L−1) | Raw material | Antibiotic | Concentrations (mg L−1) | Time (min) | Removal rate (%) | Mechanism | References |
|---|---|---|---|---|---|---|---|
| ZnO/BC (0.1) | Water-hyacinth | Tetracycline (TC) | 50 | 50 | 44.98 | SO4˙− and ˙OH | 67 |
| CNS-800 (0.1) | Pine needles | Tetracycline (TC) | 20 | 20 | 85.6 | O2˙−, 1O2 and electron transfer | 68 |
| Cu/BC700 (0.5) | Corn stalks | Tetracycline (TC) | 120 | 330 | 55.4–72.6 | SO4˙− and ˙OH | 69 |
| MMBC (0.2) | Bamboo residue | Tetracycline (TC) | 20 | 180 | 93 | SO4˙− and ˙OH | 70 |
| N-BC800 (0.2) | Straw | Tetracycline (TC) | 20 | 120 | 100 | Electron transfer | 34 |
| PBC800 (0.5) | The raw bagasse | Tetracycline (TC) | 100 | 90 | 100 | SO4˙−, ˙OH O2˙− and 1O2 | 71 |
| CBs (0.1) | Cyanobacterial | Antibiotic norfloxacin (NOR) | 5 | 300 min | 100 | Electron transfer | 72 |
| MBC (1) | — | Sulfadiazine (SDZ) | 40 | 60 | 91.79 | SO4˙−, ˙OH and 1O2 | 73 |
| BC (0.09) | Spent malt rootlets | Sulfamethoxazole (SMX) | 0.25 | 90 | 94 | Ultrasound and radicals | 74 |
| SC-Zn (0.3) | Water-hyacinth | Tetracycline (TC) | 250 | 180 | 94.83 | SO4˙−, ˙OH and 1O2 | This study |
 | ||
| Fig. 6 Proposed reaction mechanisms for TC degradation in the SC-Zn/PDS/UV (deferred) system. | ||
It has also been suggested that the functional groups of biochar can play an indispensable role in the PDS activation process as mediation and induction.63 Previous studies have shown that C3 atoms adjacent to graphite N can accept electrons from PS to generate 1O2 through proton-facilitated disproportionation due to the lack of electrons, and thus are considered to be effective sites for activating PS.64 It can be seen from the fine analysis of N 1s of XPS spectrum that the proportion of graphite-N decreased from 51.95% to 14.43%, indicating that the configuration of graphite was reconstituted during the catalytic degradation reaction, which affected the electron density of adjacent carbon, which was similar to the previous results, indicating that graphite-N was involved in the nonradical pathway to generated 1O2 in this study. In addition, previous studies had also shown that carbon-based materials have abundant functional groups, defect structures, and porous structures, which were important active sites for PDS activation.65 Combined with the characterization analysis, it was found that the doping of Zn and S in this study made SC-Zn have ultra-high specific surface area and total pore volume, abundant oxygen-containing functional groups, and induce the formation of more amorphous carbon, defect sites and irregular layered stacked loose structures, which are conducive to electron transfer and provide more active sites. From the O 1s of XPS spectra (Fig. S7b†), it can be seen that the proportion of CO of fresh SC-Zn was 72.83%, and the proportion of CO of used SC-Zn had decreased to 17.8% significantly, indicating that it was consumed in the process of catalytic degradation, it was considered to be effective in the activation of PDS. In addition, unlike most other systems, sulfur oxide played a role in the activation of PDS in this study. From the S 2p of XPS spectra (Fig. S7d†), the proportion of oxidized sulfur before and after the reaction decreased from 69.4% to 38.76%. The results of this study were similar to those of Liu66 and show that −C−SOx−C− was involved in the adsorption process to form sulfides and acts as the adsorption site of the reaction substrate, assists the free radical pathway (Zn2+ and pyridinic-N) to achieve the effective activation of PDS, and indirectly participates in the degradation of TC. The O2˙− produced in the system eventually reacts with 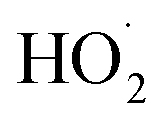 to form 1O2 (eqn (19)). It can also be seen from Raman that the amorphous carbon structure and single-layer graphene structure in the catalyst can accelerate the transfer of electrons, which was conducive to the activation of PDS.
to form 1O2 (eqn (19)). It can also be seen from Raman that the amorphous carbon structure and single-layer graphene structure in the catalyst can accelerate the transfer of electrons, which was conducive to the activation of PDS.
| S2O82− + 2H2O → HO2− + 2SO42− + 3H+ | (16) |
| S2O82− + HO2− → SO4˙− + SO42− + H+ + O2˙− | (17) |
| SO4˙− + H2O → SO42− + ˙OH + H+ | (18) |
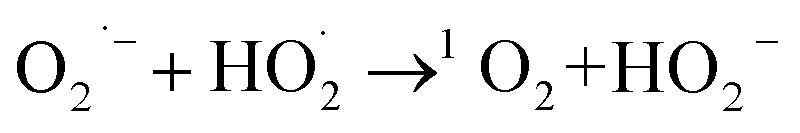 | (19) |
According to the above analysis, the functionalization of co-doping Zn and S makes the biochar SC-Zn have significantly enhanced catalytic performance, because (1) Zn is the main catalytic site for the formation of SO4˙−, and S, as the main adsorption site of the reaction substrate, plays a key role in the synergy with Zn, (2) the SC-Zn/PDS/UV (delay) system designed by SC-Zn was used to realize the co-activation of PDS through biochar activator and ultraviolet irradiation with biochar as a response photocatalyst. (3) SO4˙− and ˙OH were generated through the radical pathway because of the role of electron transfer of Zn2+ on SC-Zn, the photocatalytic process of ZnO and the role of pyridinic-N in promoting free electron transfer on SC-Zn pyridinic-N. 1O2 was generated by nonradical pathway related to graphite-N, CO, oxidized sulfur and defective structure present in SC-Zn.
During the degradation of TC, double bond, phenolic group and amine group are easily attacked by SO4˙− and ˙OH.75 In this study, for pathway I, compared to TC parent molecular, the formation of a product with m/z = 476 was due to the reactivity of hydroxyl radicals, multiple hydroxylation reactions occurred during preliminary oxidation.76 For pathway II, this pathway occurs due to TC was demethylated and hydroxylated at sensitive reaction sites, which leads to structures of the kind of product with m/z = 449.77,78 For pathway III, this pathway was dominated by ring-opening reactions, the formation of m/z = 361, 244 and 158 were the gradual opening of the loop.79 As mentioned earlier, TC had three functional groups that were vulnerable to free radical attack, among them, double bond was the most easily attacked.80 For pathway IV, since the double bond on TC was extremely unstable and easy to be attacked by active substances SO4˙− and ˙OH, hydroxylation and demethylation of TC after attack were carried out to form intermediate product of m/z = 432.81,82 For pathway V, the resulting m/z = 431 and 416 intermediates were formed by N-methylation of the TC matrix during oxidation because of the weak bond energy of N–C, the intermediate m/z = 358 was formed by shearing the amino and acylamino group because of m/z = 416.83 For pathway VI, as the reaction progresses, TC was oxidized by reactive radicals (SO4˙−, ˙OH or 1O2) causing the C–N structure to fall off, releasing a by-product of m/z = 416,84,85 the by-product of m/z = 416 by ring-opening hydroxylation,86 losing the N-methyl group or hydroxyl group,87 removals of hydroxyl group and amino group, to produce a smaller molecular structure of m/z = 300, 402, 344, 338 and 304,88,89 these intermediates may in turn undergo hydroxylation, dealkylation, dehydration, and ring-opening degradation pathways to form smaller molecular structures of m/z = 256, 274 and 194.85 For pathway VII, the TC matrix was attacked by reactive free radicals, and dihydroxylation, demethylation and ring-opening reactions occur to release smaller compounds with m/z of 427, 410, 388, 320, 415, 362 and 318,90 another possibility was to start with a dihydroxylation and deamination reaction, followed by a ring-opening reaction and a cleavage of C–C bonds to form a molecular structure with m/z of 362, 318, 302 and 274.91 The last possible pathway, dihydroxylation, demethylation and ring-opening reactions occur continuously after the TC matrix was attacked by active free radicals, which was the cause of the formation of intermediate m/z = 282.92 As the degradation process progresses, the key groups of TC were oxidized to low-toxicity intermediates, gradually forming products with m/z of 310, 268, 229, 226, 174 and 162, which further open and break the chain to form smaller molecules with m/z = 119, 118, 116, 113 and 97. Finally, TC was mineralized to H2O and CO2 under continuous attack by SO4˙−, ˙OH and 1O2.93 This can be demonstrated in TOC.
In order to explore the cause of inactivation, BET characterization was performed for catalysts after three cycles, and FTIR, XRD and XPS were characterized for catalysts after the first reaction. It can be seen from the N2 adsorption isotherm and pore size distribution (Fig. S10a–d†) that the TC molecules adsorbed on the surface of the catalyst and the intermediates released by the reaction cover the active site and effective pores (pores less than 15 Å in diameter were ineffective for TC molecules).94 Then FTIR (Fig. S10e†), XRD (Fig. S10f†) and XPS (Fig. S7†) results showed that after the reaction, the main active material Zn+ on the surface of the catalyst was severely lost, which may be related to the surface residual intermediate explored earlier. And the loss of Zn+ leads to a reduction in the reduction surface of SC-Zn, which limits the transfer of electrons from SC-Zn surface to PDS, and the loss of sulfur oxide (C–SOx–C) affects the adsorption capacity of the catalyst and the activation site of PDS decreases, therefore, the catalyst showed poor adsorption capacity and degradation effect on TC in the experiment of continuous circulation.
Therefore, in order to restore the pore structure and surface chemistry of the catalyst after three cycles, different treatment methods were explored, in the past many researchers used heat treatment, which was a good option but in their studies still showed that the catalyst can only partially recover, the activity was still poor, and the method of high heat treatment consumes high energy.15,95 This study used acid bubbles (with a concentration of 0.1 mol per L HCl solution soaked the catalyst after the reaction, and then washed repeatedly with ethanol and water, and the reaction conditions were adjusted to be consistent with the initial conditions), the study showed that the removal rate of TC within 180 min of the treated catalyst could only be restored to 39.97%, indicating that the method could only restore part of the activity of the catalyst, and the reason why it could not be completely restored may be because the pore structure and lost active material could not be fully restored, but its method was simpler and had lower energy consumption. In addition, the carbon material recycling treatment method needs to be further explored.
4. Conclusion
In this work, we prepared a novel Zn and S functionalized biochar (SC-Zn) as PDS activator by two-step pyrolysis. The modified SC-Zn catalyst can completely remove 50 mg per L TC within 10 min, and the removal rate of higher concentrations of TC (150 mg L−1 and 250 mg L−1) can reach 99.22% and 94.83% with 180 min, respectively, such excellent performance for TC removal was due to the synergetic activation of PDS by biochar activator and UV-irradiation with biochar as a responsive photocatalyst and the functionalization of the co-doped Zn and S endowed the biochar SC-Zn with a significantly enhanced catalytic performance. In addition, SC-Zn dosage, PDS dosage, pH value, coexisting inorganic anions and natural organic matter had little effect on the removal of TC by SC-Zn/PDS/UV (deferred) system, it showed excellent anti-interference. EPR tests confirmed that SO4˙−, ˙OH and 1O2 were effective active species in the system, and the pathways by which TC was degraded and mineralized into CO2 and H2O were explored by LC-MS, meanwhile quenching experiments and further mechanistic studies showed that SC-Zn activated PDS to produce SO4˙−, ˙OH and 1O2 through both radical and nonradical pathways. Among them, the doped Zn, S element, SC-Zn large specific surface area, total pore volume, loose sheet structure, defect structure (sp2-C), pyridine-N, graphite-N and CO played a role. In summary, SC-Zn had the advantages of significant activation ability, good mineralization capacity and environmental protection, and was a promising and green catalyst for advanced treatment of high-concentration antibiotics.
Data availability
Data will be made available on request.Author contributions
Yixue Qin conceived, designed, performed the experiments, analysed data and calculate and prepared the manuscript. Sheng Wang analysed data and directed the writing of manuscripts. Bingbing Zhang, Weijie Chen, Mingze An, Zhao Yang and Shuhao Qin guided experiments. Hairong Gao help with experiments.Conflicts of interest
The authors state that they have no known competing financial interests or personal relationships that could affect the work covered in this paper.Acknowledgements
This work was financially supported by Guizhou Provincial Basic Research Program (Natural Science) (QKHJC-ZK[2023]YB158); Guizhou Science and Technology Support Program [2022] General 219; Guizhou Science and Technology Joint Service Enterprise 2021[9]; The Innovation Capacity Building Project of Scientific Research Institutions in Guizhou Province (Grant No. [2023] 035); Qiandongnan Prefecture Science and Technology Support Program (Grant No. J [2022]59).References
- X. Chen, Y. Yang, Y. Ke, C. Chen and S. Xie, Sci. Total Environ., 2022, 814, 152852 CrossRef CAS PubMed.
- Y. Amangelsin, Y. Semenova, M. Dadar, M. Aljofan and G. Bjørklund, Antibiotics, 2023, 12, 440 CrossRef CAS PubMed.
- S. Shao, M. Wang, J. Zhong and X. Wu, Geomicrobiol. J., 2023, 40, 399–412 CrossRef CAS.
- A. N. Ngigi, Y. S. Ok and S. Thiele-Bruhn, J. Hazard. Mater., 2019, 364, 663–670 CrossRef CAS PubMed.
- J. Scaria, K. V. Anupama and P. V. Nidheesh, Sci. Total Environ., 2021, 771, 145291 CrossRef CAS PubMed.
- Z. Song, Y. Ma and C. Li, Sci. Total Environ., 2019, 651, 580–590 CrossRef CAS PubMed.
- H. Wang, W. Guo, B. Liu, Q. Si, H. Luo, Q. Zhao and N. Ren, Appl. Catal., B, 2020, 279, 119361 CrossRef CAS.
- D. Ouyang, Y. Chen, J. Yan, L. Qian, L. Han and M. Chen, Chem. Eng. J., 2019, 370, 614–624 CrossRef CAS.
- J. M. Monteagudo, A. Durán, I. San Martín and P. Carrillo, Chem. Eng. J., 2019, 364, 257–268 CrossRef CAS.
- R. Tang, D. Gong, Y. Deng, S. Xiong, J. Zheng, L. Li, Z. Zhou, L. Su and J. Zhao, J. Hazard. Mater., 2022, 423, 126944 CrossRef CAS PubMed.
- X. Tan, Y. Liu, G. Zeng, X. Wang, X. Hu, Y. Gu and Z. Yang, Chemosphere, 2015, 125, 70–85 CrossRef CAS PubMed.
- C. Gan, Y. Liu, X. Tan, S. Wang, G. Zeng, B. Zheng, T. Li, Z. Jiang and W. Liu, RSC Adv., 2015, 5, 3517–35115 Search PubMed.
- J. Lee, U. von Gunten and J. Kim, Environ. Sci. Technol., 2020, 54, 3064–3081 CrossRef CAS PubMed.
- J. Pan, H. Yang, L. Liu, B. Li, X. Tang, X. Wu, L. Zhang and G. Ying, Environ. Sci.: Water Res. Technol., 2022, 8, 2873–2883 RSC.
- Y. Guo, Z. Zeng, Y. Zhu, Z. Huang, Y. Cui and J. Yang, Appl. Catal., B, 2018, 220, 635–644 CrossRef CAS.
- W. Tian, H. Zhang, X. Duan, H. Sun, M. O. Tade, H. M. Ang and S. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 7184–7193 CrossRef CAS PubMed.
- D. Wang, Z. Li, D. Guo and M. Sun, Electrochim. Acta, 2022, 427, 140854 CrossRef CAS.
- M. Xi, K. Cui, M. Cui, Y. Ding, Z. Guo, Y. Chen, C. Li and X. Li, Chem. Eng. J., 2021, 420, 129902 CrossRef CAS.
- S. Zhong, J. Pan, K. Tian, J. Qin, T. Qing and J. Zhang, Process Saf. Environ. Prot., 2023, 169, 437–446 CrossRef CAS.
- J. Wang, P. Zhang, J. Peng, Q. Zhang, J. Yao, X. Wu and Y. Li, Environ. Res., 2023, 216, 114745 CrossRef CAS PubMed.
- L. Lin, W. Qiu, D. Wang, Q. Huang, Z. Song and H. W. Chau, Ecotoxicol. Environ. Saf., 2017, 144, 514–521 CrossRef CAS PubMed.
- T. Sizmur, T. Fresno, G. Akgül, H. Frost and E. Moreno-Jiménez, Bioresour. Technol., 2017, 246, 34–47 CrossRef CAS PubMed.
- S. Mondal, K. Aikat and G. Halder, Appl. Water Sci., 2017, 7, 4479–4495 CrossRef CAS.
- K. Zoroufchi Benis, A. Motalebi Damuchali, J. Soltan and K. N. McPhedran, Sci. Total Environ., 2020, 739, 139750 CrossRef CAS PubMed.
- F. Yu, F. Tian, H. Zou, Z. Ye, C. Peng, J. Huang, Y. Zheng, Y. Zhang, Y. Yang, X. Wei and B. Gao, J. Hazard. Mater., 2021, 415, 125511 CrossRef CAS PubMed.
- J. Zhou, H. Fei, Q. He, P. Li, Y. Pan and X. Liang, Sci. Total Environ., 2023, 882, 163670 CrossRef CAS PubMed.
- R. Cai, X. Wang, X. Ji, B. Peng, C. Tan and X. Huang, J. Environ. Manage., 2017, 187, 212–219 CrossRef CAS PubMed.
- C. M. Hung, C. W. Chen, C. P. Huang and C. D. Dong, Environ. Res., 2022, 211, 113076 CrossRef CAS PubMed.
- Q. He, C. Zhao, L. Tang, Z. Liu, B. Shao, Q. Liang, T. Wu, Y. Pan, J. Wang, Y. Liu, S. Tong and T. Hu, Chemosphere, 2023, 326, 138326 CrossRef CAS PubMed.
- Q. Liu, S. Jiang, X. Su, X. Zhang, W. Cao and Y. Xu, Chemosphere, 2021, 275, 129966 CrossRef CAS PubMed.
- T. Guo, L. Jiang, K. Wang, Y. Li, H. Huang, X. Wu and G. Zhang, Appl. Catal., B, 2021, 286, 119883 CrossRef CAS.
- J. Yu, L. Tang, Y. Pang, Y. Zhou, H. Feng, X. Ren, J. Tang, J. Wang, L. Deng and B. Shao, J. Cleaner Prod., 2022, 374, 133995 CrossRef CAS.
- Z. Wang, Y. Lin, H. Zhou, Z. Liu, R. Miao, X. Xu, L. He and Q. Guan, Chem. Eng. J., 2023, 455, 140795 CrossRef CAS.
- Q. Zhong, Q. Lin, W. He, H. Fu, Z. Huang, Y. Wang and L. Wu, Sep. Purif. Technol., 2021, 276, 119354 CrossRef CAS.
- D. G. Kim, V. L. Pham and S. O. Ko, J. Environ. Chem. Eng., 2022, 10, 107179 CrossRef CAS.
- W. Gao, G. Li, Q. Wang, L. Zhang, K. Wang, S. Pang, G. Zhang, L. Lv, X. Liu, W. Gao, L. Sun, Y. Xia, Z. Ren and P. Wang, Chem. Eng. J., 2023, 464, 142694 CrossRef CAS.
- R. Xu, M. Li and Q. Zhang, Chem. Eng. J., 2022, 429, 132294 CrossRef CAS.
- X. Cao and W. Harris, Bioresour. Technol., 2010, 101, 5222–5228 CrossRef CAS PubMed.
- I. Jones, J. Preciado-Hernandez, M. Zhu, J. Zhang, Z. Zhang and D. Zhang, Waste Manage., 2021, 126, 549–558 CrossRef CAS PubMed.
- Z. Wang, Y. Lin, H. Zhou, Z. Liu, R. Miao, X. Xu, L. He and Q. Guan, Chem. Eng. J., 2023, 455, 140795 CrossRef CAS.
- X. Hou, H. Dong, Y. Li, J. Xiao, Q. Dong, S. Xiang and D. Chu, J. Environ. Chem. Eng., 2023, 11, 109348 CrossRef CAS.
- S. Annamalai and W. S. Shin, Chem. Eng. J., 2022, 440, 135815 CrossRef CAS.
- Z. Jiang, J. Wei, Y. Zhang, X. Niu, J. Li, Y. Li, G. Pan, M. Xu, X. Cui, N. Cui and J. Li, J. Cleaner Prod., 2023, 384, 135641 CrossRef CAS.
- M. Huang, X. Wang, C. Liu, G. Fang, J. Gao, Y. Wang and D. Zhou, J. Environ. Chem. Eng., 2021, 9, 106536 CrossRef CAS.
- J. H. Zheng, Q. Jiang and J. S. Lian, Appl. Surf. Sci., 2011, 257, 5083–5087 CrossRef CAS.
- M. Amir, T. Fazal, J. Iqbal, A. A. Din, A. Ahmed, A. Ali, A. Razzaq, Z. Ali, M. S. U. Rehman and Y. Park, J. Ind. Eng. Chem., 2022, 115, 171–182 CrossRef CAS.
- L. Midya, A. S. Patra, C. Banerjee, A. B. Panda and S. Pal, J. Hazard. Mater., 2019, 369, 398–407 CrossRef CAS PubMed.
- Y. Lee, S. Lo, J. Kuo and C. Huang, J. Hazard. Mater., 2013, 261, 463–469 CrossRef CAS PubMed.
- Q. Zhong, Q. Lin, R. Huang, H. Fu, X. Zhang, H. Luo and R. Xiao, Chem. Eng. J., 2020, 380, 122608 CrossRef CAS.
- J. Yan, Y. Chen, L. Qian, W. Gao, D. Ouyang and M. Chen, J. Hazard. Mater., 2017, 338, 372–380 CrossRef CAS PubMed.
- W. Wang, M. Gao, M. Cao, J. Dan and H. Yang, Sci. Total Environ., 2021, 759, 143542 CrossRef CAS PubMed.
- J. He, Y. Xiao, J. Tang, H. Chen and H. Sun, Sci. Total Environ., 2019, 690, 768–777 CrossRef CAS PubMed.
- Q. Huang, C. Chen, X. Zhao, X. Bu, X. Liao, H. Fan, W. Gao, H. Hu, Y. Zhang and Z. Huang, J. Environ. Chem. Eng., 2021, 9, 105800 CrossRef CAS.
- Z. Zhou, X. Liu, K. Sun, C. Lin, J. Ma, M. He and W. Ouyang, Chem. Eng. J., 2019, 372, 836–851 CrossRef CAS.
- H. Fu, P. Zhao, S. Xu, G. Cheng, Z. Li, Y. Li, K. Li and S. Ma, Chem. Eng. J., 2019, 375, 121980 CrossRef CAS.
- H. Song, Q. Li, Y. Ye, F. Pan, D. Zhang and D. Xia, Sep. Purif. Technol., 2021, 272, 118971 CrossRef CAS.
- J. Liu, S. Jiang, D. Chen, G. Dai, D. Wei and Y. Shu, Chem. Eng. J., 2020, 381, 122637 CrossRef CAS.
- R. Yin, W. Guo, H. Wang, J. Du, X. Zhou, Q. Wu, H. Zheng, J. Chang and N. Ren, Chem. Eng. J., 2018, 334, 2539–2546 CrossRef CAS.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef CAS PubMed.
- M. Ahmadi and F. Ghanbari, J. Environ. Manage., 2018, 228, 32–39 CrossRef CAS PubMed.
- Z. Yan, F. He, J. Zhang, J. Fang, J. Wang and H. Zhou, J. Environ. Chem. Eng., 2022, 10, 108860 CrossRef CAS.
- X. Xie, Y. Liu, Y. Li, J. Tao, C. Liu, J. Feng, L. Feng, Y. Shan, S. Yang and K. Xu, J. Taiwan Inst. Chem. Eng., 2023, 146, 104891 CrossRef CAS.
- S. Liu, J. Wang, Z. Zhang, M. Dou, K. Huo, G. Ding, Y. Zhou and C. Qiao, Environ. Sci. Pollut. Res., 2023, 30, 45259–45273 CrossRef CAS PubMed.
- O. S. Furman, A. L. Teel and R. J. Watts, Environ. Sci. Technol., 2010, 44, 6423–6428 CrossRef CAS PubMed.
- J. Song, Q. Zhang, Y. Zhang, H. Guo and L. Wang, Environ. Sci. Pollut. Res. Int., 2023, 30, 49632–49643 CrossRef CAS PubMed.
- M. Liu, X. Liu, Z. Wu, Y. Zhang, Q. Meng and L. Yan, J. Environ. Manage., 2022, 322, 115900 CrossRef CAS.
- R. Xu, M. Li and Q. Zhang, Chem. Eng. J., 2022, 429, 132294 CrossRef CAS.
- S. Zhong, J. Pan, K. Tian, J. Qin, T. Qing and J. Zhang, Process Saf. Environ. Prot., 2023, 169, 437–446 CrossRef CAS.
- J. Chen, X. Yu, C. Li, X. Tang and Y. Sun, Chem. Eng. J., 2020, 382, 122916 CrossRef CAS.
- D. Huang, Q. Zhang, C. Zhang, R. Wang, R. Deng, H. Luo, T. Li, J. Li, S. Chen and C. Liu, Chem. Eng. J., 2020, 391, 123532 CrossRef CAS.
- R. Zhang, X. Zheng, D. Zhang, X. Niu, J. Ma, Z. Lin, M. Fu and S. Zhou, Sci. Total Environ., 2021, 768, 144281 CrossRef CAS PubMed.
- C. Wang, P. E. Holm, M. L. Andersen, L. G. Thygesen, U. G. Nielsen and H. Hansen, Bioresour. Technol., 2023, 388, 129785 CrossRef CAS PubMed.
- F. Dong, L. Yan, S. Huang, J. Liang, W. Zhang, X. Yao, X. Chen, W. Qian, P. Guo, L. Kong, W. Chu and Z. Diao, Process Saf. Environ. Prot., 2022, 157, 411–419 CrossRef CAS.
- L. Kemmou, Z. Frontistis, J. Vakros, I. D. Manariotis and D. Mantzavinos, Catal. Today, 2018, 313, 128–133 CrossRef CAS.
- J. Wang, D. Zhi, H. Zhou, X. He and D. Zhang, Water Res., 2018, 137, 324–334 CrossRef CAS PubMed.
- N. Barhoumi, H. Olvera-Vargas, N. Oturan, D. Huguenot, A. Gadri, S. Ammar, E. Brillas and M. A. Oturan, Appl. Catal., B, 2017, 209, 637–647 CrossRef CAS.
- J. Cao, L. Lai, B. Lai, G. Yao, X. Chen and L. Song, Chem. Eng. J., 2019, 364, 45–56 CrossRef CAS.
- Y. Liu, X. He, Y. Fu and D. D. Dionysiou, J. Hazard. Mater., 2016, 305, 229–239 CrossRef CAS PubMed.
- B. Huo, F. Meng, J. Yang, Y. Wang, J. Qi, W. Ma, Z. Wang, J. Wang and Z. Wang, Chem. Eng. J., 2022, 436, 135173 CrossRef CAS.
- Y. Zhang, J. Shi, Z. Xu, Y. Chen and D. Song, Chemosphere, 2018, 202, 661–668 CrossRef CAS PubMed.
- H. Xu, T. Zhang, D. Wang, D. Cai, S. Chen, H. Wang, S. Shu and Y. Zhu, Sep. Purif. Technol., 2022, 300, 121833 CrossRef CAS.
- H. Wang, B. Liao, T. Lu, Y. Ai and G. Liu, J. Hazard. Mater., 2020, 385, 121552 CrossRef CAS PubMed.
- W. Liu, C. Wei, R. Peng, R. Chu, H. Sun, X. Zhang and F. Xie, New J. Chem., 2022, 46, 10854–10862 RSC.
- S. Zhong, C. Li, M. Shen, C. Lv and S. Zhang, J. Mater. Res. Technol., 2019, 8, 1849–1858 CrossRef CAS.
- N. Amaly, A. Y. EL-Moghazy, N. Nitin, G. Sun and P. K. Pandey, Chem. Eng. J., 2022, 430, 133077 CrossRef CAS.
- X. Zhan, Y. Zeng, H. Zhang, X. Wang, D. Jin, H. Jin, S. Luo, L. Yang and B. Hong, J. Environ. Chem. Eng., 2023, 11, 109201 CrossRef CAS.
- D. Jiang, T. Wang, Q. Xu, D. Li, S. Meng and M. Chen, Appl. Catal., B, 2017, 201, 617–628 CrossRef CAS.
- J. Qin, Y. Ruan, L. Yi, H. Sun, Q. Qi, L. Zhao, Y. Xiong, J. Wang and D. Fang, Chem. Eng. Process.: Process Intensif., 2022, 172, 108801 CrossRef CAS.
- W. Gao, G. Li, Q. Wang, L. Zhang, K. Wang, S. Pang, G. Zhang, L. Lv, X. Liu, W. Gao, L. Sun, Y. Xia, Z. Ren and P. Wang, Chem. Eng. J., 2023, 464, 142694 CrossRef CAS.
- F. Deng, L. Zhao, X. Luo, S. Luo and D. D. Dionysiou, Chem. Eng. J., 2018, 333, 423–433 CrossRef CAS.
- D. Wang, F. Yin, B. Cheng, Y. Xia, J. Yu and W. Ho, Rare Met., 2021, 40, 2369–2380 CrossRef CAS.
- X. Zhang, B. Xu, S. Wang, X. Li, C. Wang, B. Liu, F. Han, Y. Xu, P. Yu and Y. Sun, Chem. Eng. J., 2022, 431, 133477 CrossRef CAS.
- X. Xie, Y. Liu, Y. Li, J. Tao, C. Liu, J. Feng, L. Feng, Y. Shan, S. Yang and K. Xu, J. Taiwan Inst. Chem. Eng., 2023, 146, 104891 CrossRef CAS.
- X. Zhu, C. Li, J. Li, B. Xie, J. Lü and Y. Li, Bioresour. Technol., 2018, 263, 475–482 CrossRef CAS PubMed.
- C. Shi, K. Hu, L. Nie, H. Wang, L. Ma, Q. Du and G. Wang, Inorg. Chem. Commun., 2022, 146, 110160 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07923f |
| This journal is © The Royal Society of Chemistry 2024 |